

Abstract
Hypertrophic lysosomes are critical for tumor progression and drug resistance; however, effective and specific lysosome-targeting compounds for cancer therapy are lacking. Here we conducted a lysosomotropic pharmacophore-based in silico screen in a natural product library (2,212 compounds), and identified polyphyllin D (PD) as a novel lysosome-targeted compound. PD treatment was found to cause lysosomal damage, as evidenced by the blockade of autophagic flux, loss of lysophagy, and the release of lysosomal contents, thus exhibiting anticancer effects on hepatocellular carcinoma (HCC) cell both in vitro and in vivo. Closer mechanistic examination revealed that PD suppressed the activity of acid sphingomyelinase (SMPD1), a lysosomal phosphodieserase that catalyzes the hydrolysis of sphingomyelin to produce ceramide and phosphocholine, by directly occupying its surface groove, with Trp148 in SMPD1 acting as a major binding residue; this suppression of SMPD1 activity irreversibly triggers lysosomal injury and initiates lysosome-dependent cell death. Furthermore, PD-enhanced lysosomal membrane permeabilization to release sorafenib, augmenting the anticancer effect of sorafenib both in vivo and in vitro. Overall, our study suggests that PD can potentially be further developed as a novel autophagy inhibitor, and a combination of PD with classical chemotherapeutic anticancer drugs could represent a novel therapeutic strategy for HCC intervention.
Keywords: polyphyllin D, hepatocellular carcinoma, lysosome hypertrophy, lysosomal cell death, sorafenib resistance, lysophagy
Graphical abstract
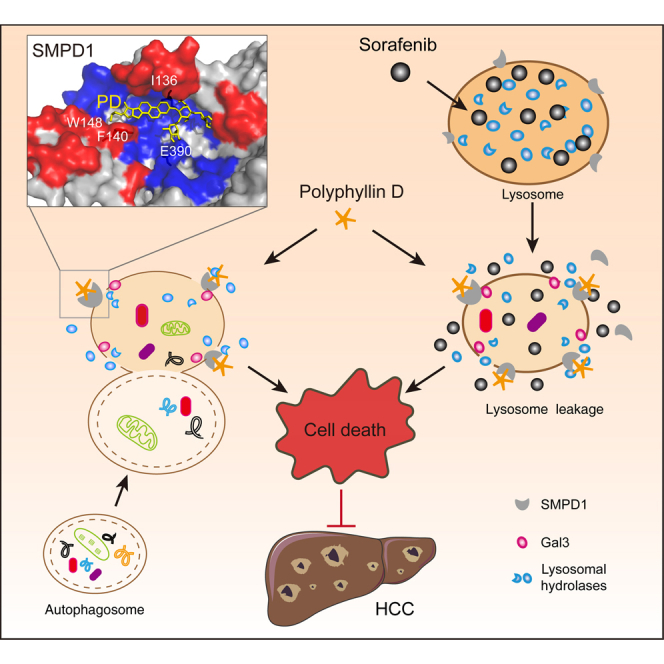
Wang and colleagues identify polyphyllin D as a novel lysosome-targeted compound that causes lysosome damage and thus suppresses cancer growth. Polyphyllin D directly interacts with SMPD1 and inhibits its activity to trigger lysosome injury, which releases sorafenib trapped by lysosome to amplify its anticancer effect on hepatocellular carcinoma.
Introduction
Lysosomes are membrane-enclosed organelles containing various hydrolytic enzymes that degrade intracellular and extracellular materials via endocytosis, phagocytosis, or autophagy.1,2 Lysosomes are critical in maintaining cellular homeostasis and cell survival. Cancer cells frequently display lysosome hypertrophy with high hydrolytic activity to meet their high energetic demands and to trap toxic compounds, which contributes to cancer cell survival and drug resistance.3 Interestingly, hypertrophy renders lysosomes fragile, increasing the tendency for greater lysosomal membrane permeabilization (LMP).4 High LMP enhances the release of lysosomal contents such as cathepsins and sphingomyelinases into the cytosol, triggering a cascade of events that lead to a programmed form of cell death known as lysosomal cell death (LCD).5 This implies that targeting the hypertrophic lysosome has a great therapeutic potential against cancer.
Hepatocellular carcinoma (HCC) is one of the most prevalent life-threatening cancers worldwide. Due to intrinsic or acquired resistance to chemotherapeutic agents, HCC has a poor prognosis. For many first-line anticancer drugs, such as tyrosine kinase inhibitors (sorafenib and sunitinib), their antitumor potency is proved to be attenuated by several mechanisms, such as increased efflux of drugs by ATP-binding cassette transporters6 and sequestered by the acidic environment of lysosomes.7 Interestingly, certain lysosomotropic compounds trapped within the lysosomes can increase LMP.8 For instance, the cationic amphiphilic drug siramesine, has been reported to detach acid sphingomyelinase (ASM)/SMPD1 from the lysosomal membrane to promote lysosomal destabilization.9,10 Although these lysosomotropic drugs can disrupt lysosomal function and have shown anticancer activity in mice,11 they do not improve patient overall survival when used in combination with chemotherapy.12 In this study, we aimed to search for novel lysosomotropic compounds with anticancer effects to improve the efficacy of first-line drugs in HCC.
Amphiphilic drugs have been reported to preferentially accumulate within lysosomes and have the potential to induce lysosomal destabilization.13 Here, we established a lysosomotropic pharmacophore model based on known amphiphilic drugs to screen for lysosome-targeted compounds from a natural product library, and identified polyphyllin D (PD) as a novel anticancer agent that specifically disrupts lysosomal function in cancer cells. PD, a steroid saponin isolated from the Paris polyphylla rhizome, was not previously demonstrated to affect lysosomes. In this study, PD was found to suppress lysosomal function and increase the LMP in HCC cells by interacting with the sphingomyelinase SMPD1 at residue Tryptophan 148 (Trp148, W148). By performing a series of functional assays, we demonstrated that PD triggers LCD, and more importantly, potentiates the anticancer efficacy of sorafenib by directly targeting hypertrophic lysosomes to disrupt lysosomotropism. Our study collectively provides experimental evidence illustrating the significant anticancer effect of PD combined with sorafenib to overcome lysosomal hypertrophy-associated drug resistance in HCC.
Results
HCC cells display lysosome hypertrophy
Lysosomes are functionally indispensable for malignant transformation; however, whether lysosomes undergo cancer-specific alteration in HCC remained unclear. We compared the morphology of lysosomes between paired HCC and paracancerous tissues using transmission electron microscope (TEM), and found that the lysosomes were significantly larger in diameter in cancer tissues vs. paracancerous tissues (Figure 1A). In addition, larger lysosomes were observed in HCC cells (Hep3B and HepG2) than in the normal liver LO2 cells (Figure 1B). To further confirm this phenomenon in clinical samples, we determined the expression levels of TMEM192, a transmembrane protein that stably resides at the lysosomal membrane,14 in 80 pairs of HCC tumor tissues and matched adjacent noncancerous tissues by immunohistochemistry (IHC). As shown in Figure 1C, 57.5% (46 of 80) of tumor tissues showed high TMEM192 expression (moderate and strong staining), whereas only 43.75% (35 of 80) of normal tissues had high TMEM192 expression. In contrast, 42.5% of tumor tissues displayed low TMEM192 expression (weak and negative staining), whereas 56.25% of normal tissues showed low TMEM192 expression. Furthermore, we found a significant correlation between tumor TMEM192 expression and T stage (p = 0.0122), as well as pathological stage (Ⅰ and Ⅱ vs. Ⅲ and Ⅳ; p = 0.0018) in 80 HCC patients (Table S1). Interestingly, punctate TMEM192 staining dots with elevated number and enlarged size were observed in the cytoplasm of HCC cells in tumor tissues, as compared with normal cells in paracancerous tissues (Figure 1C). The findings demonstrated that lysosome hypertrophy is a significant feature of HCC, representing a cancer-specific target for drug discovery.
Figure 1.

HCC cells display lysosomal hypertrophy
(A) Representative TEM images of lysosomal morphology and quantification of lysosomal diameter from three HCC and paired paracancerous tissues. (B) The morphology of lysosomes in HepG2, Hep3B, and LO2 cells stained with anti-LAMP1 antibody (fixed cells) or LysoTracker (living cells) were imaged by confocal microscope, and the lysosome diameter was measured and statistically analyzed. Five random fields with at least 50 lysosomes were measured for lysosome diameter in each cell type. All data are represented as mean ± SD; significance was determined by unpaired Student’s t test for (A) and (B); ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001. (C) Representative images and expression pattern of TMEM192 in cancer tissues and paired paracancerous tissues from 80 HCC patients. Scale bar, 20 μm.
PD specifically targets lysosomes
To identify novel compounds that specifically target lysosomes, we established an in silico library screening strategy by collecting 14 known amphiphilic drugs (Table S2) as a training set to establish a lysosomotropic pharmacophore, to screen a natural product library containing 2,212 compounds (Figures 2A and 2B). After filtering those compounds with low solubility and high hepatotoxicity, the top 20 hits were employed for further cell viability determination. As shown in Figure 2C, PD with significant anticancer effect (inhibitory rate: 11.16% ± 3.72%) on HCC was chosen for further investigation. To verify the specificity of PD in targeting lysosomes, we subsequently labeled PD with the Cy3 fluorophore, and observed that PD-Cy3 was colocalized with the lysosomal marker TMEM192-EGFP, LAMP1-EGFP, and LysoTracker-Green, but not with Mito-Tracker (mitochondria) in cells (Figures 2D and 2E). Furthermore, excess PD competed with PD-Cy3 for lysosome binding (Figures S1A and S1B), suggesting that PD is a novel amphiphilic agent that specifically targets lysosomes to exhibit anticancer effect on HCC.
Figure 2.

Identification of PD as a lysosome-targeting compound
(A) Schematic diagram depicting the strategy for the identification of amphiphilic drugs by virtual screening with a natural product library (2,212 compounds). (B) Pharmacophore mapping of fit compounds in the test set with 14 known amphiphilic drugs. Pharmacophore features: Green, HB acceptor; cyan, hydrophobic; orange, Ring aromatic; red, POS ionizable. (C) Cell viability measurement of HepG2 cells treated with the top 20 compounds at 10 μM for 24 h by WST-1 assay. Polyphyllin D (PD) with the highest inhibitory ratio on cell viability was marked in red. (D) Colocalization of PD with lysosomes. PD-Cy3 (1 μM) loaded HepG2 cells were stained with LysoTracker (green) or Mito-Tracker (green) or transfected with TMEM192-EGFP and LAMP1-EGFP, and subjected to confocal imaging. (E) The intensity profile of PD-Cy3 and TMEM192-EGFP along the white line was plotted in (D), with the colocalization sites marked with black arrows.
PD exhibits a significant anticancer effect on HCC in vitro and in vivo
We then examined the anticancer effect of PD on HCC in detail. It was found that PD decreased the viability of both HepG2 and Hep3B cells in a dose- and time-dependent manners, but only had a little cytotoxic effect on the normal liver cell line LO2 (Figure 3A) after 48 h treatment. PD and PD-Cy3 were found to have similar inhibitory ratios in both HepG2 and Hep3B cells (Figure S1C), indicating that the addition of Cy3 group did not affect the bioactivity of PD in both HCC cells. Moreover, utilizing propidium iodide (PI) influx as a marker of cell death demonstrated that PD caused a pronounced increase in the number of PI-positive cells in HCC cells (Figure 3B). Additionally, we established a subcutaneous xenograft tumor model to assess the anticancer activity of PD in vivo. Mice orally administrated (i.g. 4 mg/kg) or intraperitoneally injected (i.p. 2 mg/kg) with PD presented with considerable tumor growth inhibition, as evidenced by the decrease of tumor volume and Ki-67 staining (Figures 3C–3E). There was no significant change in body weight (Figure S2A), blood biochemical alanine aminotransferase (ALT) and aspartate aminotransferase (AST) (Figure S2B), or morphology of vital organs such as lung, heart, liver, and kidney (Figure S2C). Furthermore, we developed an in vivo MiniPDX assay based on capsular implantation to assess the effects of PD on tumors derived from clinical HCC specimens (Figure 3F), and found that PD exhibited a significant anticancer effect on the MiniPDX tumors (Figure 3G). Taken together, these data indicate that PD strongly suppresses HCC tumor growth with negligible toxicity.
Figure 3.

PD inhibits HCC cell growth both in vitro and in vivo
(A) Cell viability measurement of HepG2, Hep3B, and LO2 cells treated with PD at increasing concentrations (up to 6 μM) for 12, 24, and 48 h by WST-1 assays. (B) The cell death of HepG2 and Hep3B cells stimulated with PD (0–6 μM, 24 h) was detected by PI staining, and the PI-positive cells were quantified. (C) Representative images of excised Hep3B tumors from mice with intragastric administration (i.g.) at 4 mg/kg or intraperitoneal injection (i.p.) at 2 mg/kg (n = 6). (D) Tumor growth curves; data are represented as mean ± SEM. (E) IHC staining for Ki-67. (F) An overview of the establishment of HCC MiniPDX model. (G) Scatterplot showing the relative proliferation rate of the HCC MiniPDX model upon treatment with PD (i.g. 4 mg/kg). n = 6; data are represented as mean ± SD; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
PD impairs cellular lysosomal pathway
To systematically analyze the protein alteration in HCC cells induced by PD, data-independent acquisition (DIA)-based quantitative proteomics was carried out (Figure 4A). We observed high reproducibility and similar distribution of the quantified proteins in the three biological replicates by the power law global error model and violin plot analysis (Figures S3A and S3B). The fold change (FC) distribution upon comparison of the PD-treated and control groups is displayed in a volcano plot (Figure 4B) containing 247 (73.95%) upregulated and 87 (26.04%) downregulated proteins (with the criteria FC > 1.5 and p < 0.05; Table S3), and the expression levels of differentially expressed proteins (DEPs) were demonstrated by cluster analysis (Figure S3C). Kyoto Encyclopedia of Genes and Genomes pathway and gene ontology cellular component analyses showed that these upregulated proteins are mainly related to lysosomes, endocytosis, apoptosis, and autophagy (Figures 4C and S3D), suggesting that PD induces lysosomal dysfunction and thereby disrupts the protein degradation system. To confirm this finding, we determined alterations in the levels of intracellular ubiquitinated proteins and observed that PD stimulation not only increased global ubiquitin-tagged protein levels in HCC cells (Figure 4D), but also delayed the lysosomal degradation of cargo labeled with ubiquitin (Figure 4E).
Figure 4.

PD induces lysosomal injury and delays global cargo degradation
(A) Workflow of DIA-based proteomics for the identification of PD-regulated proteins. (B) Volcano plot of DEPs in the PD group vs. the control group. Red dots, upregulated proteins; Blue dots, downregulated proteins. (C) Kyoto Encyclopedia of Genes and Genomes pathway analysis of the upregulated proteins in the PD group. (D, E) HCC cells were treated with PD (0–6 μM) for 24 h, and the ubiquitin levels in whole-cell lysates (D) and lysosomes (E) were determined by western blotting. The HCC cells expressing GFP tagged TMEM192 for the lysosome extraction. TMEM192-GFP was used as loading control for lysosome fraction. (F) Heatmap showing the KFERQ motif-bearing proteins regulated by PD. (G) HCC cells expressing the KFERQ-PA-mCherry1 reporter were subjected to photoconversion at 405 nm for 10 min, and then treated with PD (1.5 μM), Baf-A1 (200 nM; an inhibitor of vacuolar-type H+-ATPase), or Torin1 (1 μM; an inhibitor of mTOR), respectively. Fluorescence (emitted at 535 nm) was measured with a multifunctional microplate reader at the indicated time points. (H) HepG2 cells expressing mCherry-Gal3 and LAMP1-EGFP were treated with or without PD (1.5 μM) for 12 h, and subjected to confocal imaging. At least 30 random cells were measured for Gal3-positive dots. All data are represented as mean ± SD; ns, no significance; ∗∗∗p < 0.001.
Proteins bearing KFERQ-like motif can be selected as substrates of chaperone-mediated autophagy (CMA) for degradation in lysosomes.15 Therefore, we extracted proteins bearing the classic KFERQ-like motif from our list of proteins that are regulated by PD, and found that they were significantly enriched in PD-stimulated groups (Figure 4F). To confirm this finding, we employed the KFERQ-PA-mCherry1 reporter to assess the effect of PD on the substrate-degradation activity of lysosomes. As shown in Figure 4G, both PD and Baf-A1 (positive control) blocked the degradation of the PA-mCherry1 fluorescence signal over time, as compared with Torin1 and control, suggesting that PD induces global cargo accumulation by impairing lysosome function. Lysophagy is an intracellular membrane repair process that occurs in response to lysosomal injury.16 Here, staining with a lysosome permeabilization sensor, galectin-3 (Gal3),16 we observed that Gal3 (mCherry) translocated to lysosomes (LAMP1) in HCC cells upon treatment with PD (Figure 4H). The increase in the number of Gal3-mCherry puncta and the accumulation of ubiquitinated cargo induced by PD demonstrated that lysosomal injury is triggered by PD.
PD impairs autophagic flux in HCC
Intracellular macromolecules reach the lysosomes by various routes, and autophagy is a key pathway that delivers cargo for lysosomal degradation.17 We observed that PD induced punctate EGFP-LC3 in the cytoplasm of HCC cells (Figure S4A), and that LC3 expression in xenograft tumors were considerably increased with PD treatment compared with that in the control group (Figure S4B). By using TEM, we detected numerous enlarged membrane-bound autophagosomes in the cytoplasm of PD-treated HCC cells (Figure S4C). Moreover, treatment with PD in both HCC cell lines resulted in a dose- and time-dependent accumulation of ATG5, LC3-II, and p62 (Figures S4D and S4E). We then employed the mCherry-EGFP-LC3 reporter to further investigate the effect of PD on cellular autophagic flux, and found that the EGFP signal persistently colocalized with the mCherry signal and was not quenched in autophagolysosomes after 24 h of PD treatment (Figure 5A). Moreover, the EGFP-LC3 (a marker of autophagolysosomes) and LAMP1-mCherry (a marker of lysosomes) signals did not separate after PD stimulation (Figure 5B), suggesting that autophagic flux is blocked at the late stage after autophagosome-lysosome fusion. We found that PD increased the relative fluorescein isothiocyanate (FITC) intensity in HCC cells stained with FITC-dextran, suggesting that PD altered lysosomal pH in HCC cells (Figure 5C). Moreover, analysis of the chimeric protein in both HCC cell lines expressing EGFP-LC3 by western blotting showed that PD treatment resulted in the accumulation of free EGFP fragments, as well as EGFP-LC3 (Figure 5D). These results suggest that PD injures lysosomes accompanied with autophagy flux disruption.
Figure 5.

PD blocks autophagic flux in HCC cells
(A) HepG2 cells transfected with mCherry-EGFP-LC3 were treated with PD (3 μM, 24 h), and photographed using confocal microscopy. The intensity profiles of EGFP and mCherry along the white line are plotted in the lower panels, with the colocalization sites marked with black arrows. (B) HepG2 cells co-transfected with the EGFP-LC3 and LAMP1-mCherry plasmids were incubated with PD (3 μM, 24 h), and then detected by confocal microscopy. The intensity profiles of EGFP-LC3 and LAMP1-mCherry along the white line were plotted in the lower panels, with the colocalization sites marked by black arrows. (C) Both HCC cell lines were loaded with FITC-dextran for 24 h and then treated with increasing concentrations of PD (0–6 μM); FITC fluorescence was detected by flow cytometry. Data are represented as the mean ± SD, n = 3; ∗∗p < 0.01; ∗∗∗p < 0.001. (D) Western blot analysis of EGFP-LC3 and free EGFP levels in EGFP-LC3 expressing HCC cells treated with increasing concentrations of PD for 24 h. (E) Cells pretreated with PD (3 μM, 12 h) were incubated with EGF (200 ng/mL, 2 h), then the medium was washed out and samples were collected at the indicated time points for IB analysis. (F) After HCC cells were treated with PD (3 μM, 12 h), then western blot analysis was performed to measure the expression of p62 and LC3-II at the indicated time points after PD washed out. (G) HepG2 cells expressing TMEM192-EGFP was incubated with DMSO (Ctrl), PD (3 μM), or Baf-A1 (200 nM, an inhibitor of vacuolar-type H+-ATPase) for 24 h, and then imaged upon photobleaching at the indicated time points. The decay of intensity was calculated in each frame, from the first frame after photobleaching to the end of the acquisition.
Epidermal growth factor receptor (EGFR) is reported to undergo rapid internalization through endocytosis and is targeted for lysosomal degradation or recycling after stimulation by EGF,18 thus, detection of EGFR degradation is another strategy to evaluate lysosome activity. Here, we found that PD inhibited EGF-triggered EGFR degradation in HCC cells (Figure 5E), indicating that lysosomal proteolytic activity is impaired in the PD-treated HCC cells. Next, we examined whether the lysosomal impairment induced by PD was reversible. Apparently, the accumulation of p62 and LC3-II was not relieved 6 h after PD was removed from the medium (Figure 5F). We also employed fluorescence recovery after photo bleaching (FRAP) experiments to determine the influence of PD on the lysosomal membrane fluidity by transfecting HepG2 cell with TMEM192-EGFP, an abundant protein stably expressed on the lysosomal membrane. Upon PD stimulation, EGFP fluorescence in lysosomes recovered slower than that in the control groups (Figure 5G). We then employed lysosomotropic reagent L-leucyl-L-leucine methyl ester (LLOMe), which induces specific and acute lysosomal damage.19 We monitored the clearance of Gal3-labeled damaged lysosomes over time after LLOMe washout, and found that compared with control cells, PD-treated cells showed impaired lysosome clearance after LLOMe treatment. (Figure S5A). Together, these data confirmed that PD impairs the integrity of lysosomes in HCC cells by disrupting lysosomal pH, lysosomal membrane fluidity, and repairment.
PD induces lysosome-dependent cell death
LMP has been shown to trigger a cascade of regulated cell demise mediated by the release of lysosomal enzymes (SMPD1, cathepsins, etc.) into the cytosol to induce LCD, sometimes accompanied with apoptosis.2 Here, we found that PD not only induced the release of SMPD1 and CTSB (cathepsin B) from lysosome to cytoplasm (Figure 6A), but also increased the activity of cathepsin B (Figure 6B) and augmented cleaved caspase-3 and cleaved PARP protein levels (Figure S5B). To further confirm the PD-induced LCD is linked to apoptosis, we performed caspase-3/7 activity and Annexin V/PI staining assays. As shown in Figures S5C and S5D, the activity of caspase-3/7 and the apoptotic percentage of HCC cells were gradually increased in response to PD treatment (0–6 μM, 24 h). Meanwhile, the employment of zVAD-FMK (a pan-caspase inhibitor) not only reduced the HCC cell apoptosis and growth inhibition induced by PD (Figures S5E and S5F), but also suppressed the expression of cleaved caspase-3 (Figure S5G), suggesting that PD induces HCC apoptosis-like LCD. In addition, the specific cathepsin B inhibitor CA-074 Me reversed the PD-induced cell growth inhibition and apoptosis-like LCD in HCC cells (Figures 6C–6E). In addition, we enhanced the cellular autophagic flux by inducing serum starvation and mammalian target of rapamycin (mTOR) signaling inhibition, and found that nutrient deprivation or Torin1 (a mTOR inhibitor) treatment augmented the lethal effect of PD (Figures 6F–6H), as evidenced by WST-1 assay (Figure 6F), colony formation experiment (Figure 6G), and caspase 3/7 activity measurement (Figure 6H). These data suggest that autophagic flux induced by PD aggravates the LCD in HCC on the basis of lysosomal damage (Figure 6I).
Figure 6.

PD triggers LMP to release lysosomal contents augmented by autophagic flux burden
(A) Western blot analysis of the SMPD1 and cathepsin B levels in lysosome and cytoplasm fractions after PD treatment. (B–E) HepG2 and Hep3B cells were treated with PD (3 μM, 24 h) in the presence or absence of CA-074 methyl ester (50 μM), a cathepsin B inhibitor. Then the activity of cathepsin B was analyzed by flow cytometry (B), the cell viability was tested by WST-1 assay (C), death cells were analyzed by PI staining assay (D), and the expression of pro-caspase 3, cleaved caspase-3 was determined by western blotting (E). (F–H) The anticancer effects of PD were augmented by starvation and Torin1 treatment. HCC cells were treated with PD (3 μM), serum deprived or Torin1 (1 μM, a specific mTOR inhibitor) as indicated. The cell viability was then tested by WST-1 assay (F), the ability to form colonies was determined by the colony formation assay (G), and caspase 3/7 activity was measured by flow cytometry (H). (I) A proposed model summarizing how PD triggers lysosome leakage, contributing to cell death, which can be amplified by the intracellular autophagic flux burden. All data are represented as mean ± SD, n = 3; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
PD induces sorafenib release to potentiate the anticancer effect on HCC cells
Sorafenib is a first-line drug approved by the Food and Drug Administration that is used for the treatment of HCC,20 but can be sequestered in the lumen of lysosomes due to its lysosomotropic property, resulting in lysosome-mediated drug resistance.7 Here, we tested whether sorafenib can be released from the lysosomes damaged by PD and found that PD treatment decreased the concentration of sorafenib within lysosomes, suggesting that it induces the release of trapped sorafenib from lysosomes (Figure 7A). This phenomenon inspired us to examine the efficacy of combined treatment of PD and sorafenib. PD potentiated the anticancer effect of sorafenib on HCC cells in vitro (Figures 7B–7D). Moreover, we used the HCC MiniPDX model for experiment and found that combined treatment of PD with sorafenib exhibited a more significantly suppressive effect on primary HCC cells than the treatments with single-agent PD or sorafenib (Figure 7E). In addition, our xenograft model also supported that the administration of PD amplified the anticancer effect of sorafenib on xenograft tumors (Figures 7F–7I). During this combined therapy, we found no significant change in body weight (Figure S6A), blood biochemical ALT and AST (Figure S6B) or morphology of vital organs such as lung, heart, liver, and kidney (Figure S6C). IHC staining of Ki-67 demonstrated that the combination of sorafenib and PD decreased the cell proliferation. Accumulation of Gal3 puncta was observed in both PD and PD + sorafenib treatment groups, suggesting that PD induces lysosomal injury in tumor xenograft. Sorafenib has been reported to exert its anticancer effect by inhibiting vascular endothelial growth factor receptor (VEGFR)-mediated angiogenesis and targeting the MEK/ERK signaling pathway.21 Here, by CD31 staining, we observed that PD treatment alone did not alter angiogenesis or ERK signaling in xenograft tumor tissues, but did augment the suppressive effects of sorafenib on anti-angiogenesis and ERK phosphorylation to potentiate its anticancer effects (Figure 7J). These data illustrate that PD, as a specific lysosome-targeted compound, can selectively act on cancer cells with hypertrophic lysosomes to potentiate the anticancer effects of sorafenib.
Figure 7.

PD amplifies the anticancer effects of sorafenib on HCC in vitro and in vivo
(A) The concentration of sorafenib in lysosomes with or without PD treatment (3 μM) was analyzed by LC-MS/MS, showing that PD induced sorafenib release from lysosomes. (B–D) HCC cells were treated with PD (1.5 μM) in the presence or absence of sorafenib (10 μM) as indicated. Then, caspase 3/7 activity was measured by flow cytometry (B), cell viability was tested by WST-1 assay (C), and the cell growth was determined by colony formation assay (D). (E) The therapeutic response of PD and sorafenib combined treatment on the HCC MiniPDX model. The relative proliferation rate of the HCC MiniPDX model upon treatment with PD (i.g. 4 mg/kg) and sorafenib (i.g. 10 mg/kg) was measured (n = 6). (F–J) Nude mice bearing HepG2-derived xenografts received PD (4 mg/kg), sorafenib (10 mg/kg), or vehicle control by oral gavage every 3 days, respectively (n = 6) (F). The tumor photos (G), tumor volume curves (H), tumor weights (I), and IHC staining for Ki-67, CD31, p-ERK, and Gal3/DAPI in the indicated tumor xenografts (J) are shown. All data are presented as the mean ± SD; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ns, no significance.
PD directly interacts with SMPD1 and inhibits its activity
Many amphiphilic drugs, such as desipramine, have been reported to reduce SMPD1 activity by displacing it from the lysosomal membrane.22 Here, we found that SMPD1 activity was remarkably inhibited by PD (Figure 8A), which leads us to speculate that there is an interaction between SMPD1 and PD. To address this hypothesis, we established two SMPD1-knockout (KO) HCC cell lines and found that both SMPD1-KO HepG2 and Hep3B cell lines showed a decrease in cell growth and resistance to PD stimulation, as compared with the corresponding control cells (Figures S7A, S7C, and S7D). Moreover, overexpression of SMPD1 restored the inhibitory effect of PD (Figure S7E), suggesting that SMPD1 is critical for the anticancer effect of PD. We then performed a fluorescence titration assay (FTA) and cellular thermal shift assay (CETSA) to analyze the interaction between SMPD1 and PD. The FTA showed that the addition of PD to recombinant SMPD1 caused a significant fluorescence quenching with an equilibrium dissociation constant (Kd) = 0.37 × 10−6 M (Figures 8B and 8C). The CETSA showed that treatment with PD led to thermal shifts of SMPD1 in the denaturation temperature range of 58–67°C (Figure 8D), importantly, cells expressing SMPD1-GFP displayed significant colocalization with PD-Cy3 (Figure 8E), indicating that PD directly binds to SMPD1 and suppresses its activity. Since SMPD1 is acid sphingomyelinase that converts sphingomyelin (the substrate of SMPD1) to ceramide, we compared the cellular level of sphingomyelin and found that HCC cells (HepG2 and Hep3B) displayed a dramatically higher level of sphingomyelin than noncancerous cells (LO2), as shown in Figure S7F. Cancer cells with a high level of sphingomyelin are associated with lysosome hypertrophy, with increasing tendency of LMP4; this could explain why HepG2 and Hep3B with larger lysosomes in size are sensitive to PD treatment.
Figure 8.

PD inhibits HCC cell growth by targeting the surface groove of SMPD1 at W148
(A) Enzymatic activity of SMPD1 in HCC cells treated with increasing concentrations of PD (0–3 μM) or desipramine (10 μM, positive control) was determined by SMPD1 activity assay. (B) The interaction between PD and SMPD1 was determined by fluorescence titration experiments. (C) The fluorescence quenching of SMPD1 at 340 nm vs. the PD concentration was fitted with Hill plot. (D) CETSA curves comparing the change in the thermal stability of recombinant SMPD1 upon treatment with PD and DMSO. (E) Confocal microscope of cells expressing SMPD1-GFP after treatment with PD-Cy3 (1 μM), the colocalization of SMPD1 and PD-Cy3 was imaged. Scale bar, 10 μm. (F) The favorable counts for each amino acid of SMPD1 bound with PD. (G) Proposed binding pose depiction of the potential interaction mode between PD and SMPD1. Red, hydrophobic region; blue, hydrophilic region. (H) SMPD1WT, SMPD1I136/P140A, SMPD1W148A, or SMPD1E390A was introduced into SMPD1-KO cells, and the enzymatic activity of SMPD1 was detected by SMPD1 activity assay. (I) SPR sensorgram for the association of PD with immobilized SMPD1WT and SMPD1W148A. (J–L) Tumors formed by HepG2-SMPD1-KO cells expressing WT or W148 mutant SMPD1 proteins were treated with or without PD (i.g 4 mg/kg), then the tumor photos (J), tumor growth curves (K), and tumor weight (L) were compared (n = 6). All data are represented as the mean ± SD; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001. (M) Schematic diagram summarizing how PD overcomes sorafenib resistance in HCC.
We then performed molecular docking analysis to investigate the interaction between PD and SMPD1 (Protein DataBank ID: 5i85). A total of 200 possible docking conformations were used to analyze the non-bond interactions between PD and SMPD1, and we found that PD was surrounded by the hydrophobic residues Ile136, Phe140, and Trp148, and the hydrophilic residue Glu390 in SMPD1 (Figures S8A–S8C and 8F). Based on the 3D binding mode, PD binds these amino acid residues in a pocket of SMPD1 at its surface (Figures 8G and S8D), by forming hydrophobic and hydrogen bonds (Figure S8E). To identify the residues most critical for PD binding, we individually mutated several of these interacting residues in SMPD1, and reintroduced the mutants SMPD1WT, SMPD1I136/F140A, SMPD1W148A, or SMPD1E390A in SMPD1-KO HCC cells (Figure S7B). It was found that only re-expression of SMPD1W148A could not restore cell proliferation of HCC, which was similar to the effect of SMPD1-KO (Figures S7G and S7H). Importantly, HCC cells with SMPD1-KO as well as SMPD1W148A displayed lysosomal damage, as evidenced by accumulated Gal3 puncta (Figure S7I). Furthermore, the W148A mutant had greater inhibitory effect than the other mutants on the enzymatic activity of SMPD1 (Figure 8H). Surface plasmon resonance (SPR) analysis revealed dose-dependent PD binding with SMPD1WT; however, the SMPD1W148A mutant protein showed comparatively low PD-interaction signal (Figure 8I). These data illustrated that W148 is a critical amino acid residue for both SMPD1 enzymatic activity and the binding of SMPD1with PD.
To further evaluate the role of W148 in the anti-HCC activity of PD, we performed WST-1 and colony formation assays and confirmed that HCC cells bearing SMPD1W148A were resistant to PD-induced growth inhibition (Figures S9A and S9B). Moreover, our animal experiments revealed that the growth of xenograft tumors with SMPD1W148A was slow, but they were refractory to PD treatment, as compared to tumors in the SMPD1WT group that were significantly suppressed by PD treatment (Figures 8J–8L). Taken together, our results illustrated that PD suppresses the activity of SMPD1 by directly interacting with the residue W148 in SMPD1, which induces lysosomal leakage, then causes sorafenib release and augments the anticancer efficacy of sorafenib (Figure 8M).
Discussion
Hypertrophic lysosomes endow cancer cells with high abilities in metabolism, survival, and drug resistance. However, lysosome hypertrophy is also a vulnerability in which the fragility of lysosomes provides an opportunity for drug design. In this study, we identified PD as a specific lysosome-targeted compound with in vitro and in vivo antitumor effects that disrupt lysosomal function via increasing LMP. We then demonstrated that PD directly binds and inhibits SMPD1, breaking hypertrophic lysosomes to release trapped sorafenib and thus augments the anticancer efficacy of sorafenib, implicating a novel therapeutic strategy to overcome sorafenib resistance in HCC through a combination therapy of sorafenib with PD (Figure 8M).
As the “garbage-disposal system” of mammalian cells, lysosomes were recently reported to be dramatically altered during cancer progression, increasing in size, number, and hydrolase abundance.23 This may attribute to the increased level of cellular sphingomyelin in malignancies, which modifies the composition of lysosomal membranes and decreases their fusion with other intracellular vesicles, resulting the enlargement of lysosome volume.24 The hypertrophic feature of lysosomes in HCC were characterized and confirmed in this work, and it was found to be associated with advanced stage of HCC patients (Figure 1 and Table S1). Hypertrophic lysosomes provide a ground for drug resistance via a mechanism known as lysosomal sequestration.25 The low pH in lysosomes enables a spectrum of chemotherapeutics formulated as weak bases accumulate within the lysosomal lumen. These chemotherapeutics include many anticancer drugs, such as gefitinib26 and sorafenib,7 that have been reported to have high affinity for the lysosomal compartment due to their chemical properties. The accumulation of these drugs in lysosomes strongly handicaps the outcome of their anticancer effects in clinics, suggesting that targeting the lysosomal compartment to achieve lysosomal membrane rupture can be a feasible strategy to overcome drug resistance. Here, we demonstrated that the lysosomotropic compound PD can specifically “puncture” the hypertrophic lysosomes of HCC cells to eliminate lysosomal sequestration, resulting in the release of sorafenib from lysosomes and thus potentiating its anticancer efficacy. Sorafenib inhibits angiogenesis by targeting B-Raf and receptor tyrosine kinases such as VEGFR.27,28 Our in vivo study revealed that the antiangiogenic effects of sorafenib, presenting as a reduction in microvessel density, were amplified upon PD and sorafenib combination treatment, confirming the role of PD in potentiating sorafenib against drug sequestration by hypertrophic lysosomes.
PD has been reported to trigger cell-cycle arrest, DNA damage, and apoptosis in multiple cancers.29,30 The current work unexpectedly revealed that PD executes its anticancer function by inducing lysosomal membrane permeabilization and thus LCD, a form of lytic cell death. It was known that the autophagy-lysosome pathway includes the formation of autophagosomes (early stage) and the fusion of autophagolysosomes (late stage).31 Our mCherry-EGFP-LC3 tandem reporter indicated that the autophagic flux in PD-treated HCC cells is impaired, while the stable colocalization of LC3 with LAMP1 after PD treatment suggested that PD did not disturb autophagosome-lysosome fusion (Figures 5A and 5B). Intact lysosomes are acidic, and the appropriate pH is maintained by lysosomal membrane integrity and lysosomal hydrolase activity.32 Upon PD treatment, decreased lysosomal acidification and the accumulation of the LC3, p62, and ATG5 proteins were observed (Figures 5C and S4D), suggesting that PD causes lysosomal dysfunction and the loss of membrane integrity. Correspondingly, in the route of endocytosis toward lysosome, EGF-triggered lysosomal degradation of EGFR was inhibited by treatment with PD (Figure 5E). Based on Gal3 staining, a well-recognized indicator for lysosomal injury,16 accumulation of Gal3-positive lysosomes induced by PD treatment was observed in vitro and in vivo, supporting the effect of PD on LMP. We speculated that PD is accumulated on the lysosomes to slow lysosomal membrane fluidity (Figure 5G), which subsequently impaired the integrity of cellular lysosomes. Note that PD-mediated LCD displayed apoptotic features such as the cleavage of PARP and caspase 3.
SMPD1 is a lysosomal enzyme that catalyzes the conversion of sphingomyelin to phosphorylcholine and ceramide.33 The inactivation of SMPD1 reduces the generation of ceramides and accumulation of sphingomyelin, which improves the tendency of LMP.9,34 We revealed that PD directly interacts with SMPD1 to induce lysosomal injury, which was observed as LMP and the release of the lysosomal contents, including SMPD1 itself and cathepsin B. Amphiphilic PD attaches to the pocket of the SMPD1 surface containing hydrophobic and hydrophilic regions. Among the amino acids in this SMPD1 pocket, Trp148 is the main residue for PD binding and SMPD1 activity, as evidenced by the finding that the mutation of Trp148 to Ala decreased SMPD1 activity and tumor growth (Figures 8H–8L). This result is similar to our previous finding where curcumol binds to NQO2, blocking the catalytic activity of the enzyme.35 Obviously, PD functions via direct interaction with SMPD1 in lysosomes, resulting in increased lysosomal fragility, LMP, and cell death. Importantly, the cellular sphingomyelin level in cancer cells (HepG2 and Hep3B) is dramatically higher than that in noncancerous cells (LO2), SMPD1 inhibition (PD treatment) results in sphingomyelin accumulation and subsequently LMP. This could explain why HepG2 and Hep3B cells with larger lysosomes are sensitive to PD treatment.
Clinically, the treatment of HCC remains unoptimistic with unfavorable prognosis in HCC.36 Acquired resistance to sorafenib is frequently observed, and the long-term effectiveness of the extensive application of sorafenib is still controversial. Among the mechanisms that account for sorafenib resistance, lysosomal sequestration by PD is notable and fundamental. Based on the occurrence of lysosome hypertrophy in HCC, we identified PD as a specific lysosomotropic compound that disrupts lysosomal function by inducing LMP to release destructive hydrolases and sequestered sorafenib, and thus augmenting the anticancer effect of sorafenib. This discovery highlights that combination of sorafenib and PD could be a novel strategy for the treatment of sorafenib-resistant HCC.
Materials and methods
Cell lines and cell culture
Human HCC cell lines (HepG2 and Hep3B) were obtained from the American Type Culture Collection (ATCC, Rockville, MD, USA). Human normal hepatocyte cell line (LO2) was obtained from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). Cell lines were kept in complete DMEM (Life Technologies, Gaithersburg, MD, USA) containing 10% fetal bovine serum (Gibco-Invitrogen Corporation, CA, USA) with 1% penicillin/streptomycin (GBCBIO Technologies, Guangzhou, China) at 37°C in 5% CO2.
Chemicals and reagents
Polyphyllin D (Polyphyllin I, CAS 50773-41-6, PubChem CID: 44425145, ≥99.0% purity) was obtained from Must Bio-Technology Co. (Chengdu, China). Desipramine and CA-074 methyl ester were purchased from TargetMol (Shanghai, China). Polyphyllin D-Cy3 was synthesized in Xinqiao Biotechnology Co., Ltd. (Hangzhou, China). zVAD-FMK was purchased from Beyotime (Jiangsu, China). Torin1 and Bafilomyclin A1 (Baf-A1) were purchased from Selleck Chemicals (Houston, TX, USA). The antibodies including PARP, LC3, and ATG5 were purchased from Cell Signaling Technology (Danvers, MA, USA). Acid sphingomeyelinase (ASMase)/SMPD1 was from Bioss (Beijing, China). Ubiquitin, p62, LAMP1, SMPD1, EGFP, EGFR, TMEM192, β-actin, and the secondary antibody against rabbit/mouse were from Proteintech (Wuhan, China). Caspase-3, cleaved caspase-3, cathepsin B (CTSB), and Tubulin were obtained from ABclonal (Wuhan, China).
Tissue microarray and IHC
IHC was performed as we previously described.37 A human HCC tissue microarray containing 80 pairs of HCC tissues and matched adjacent para-cancer tissues (Outdo Biotech, Shanghai, China), as well as tumor xenograft sections, were deparaffinized in xylene, rehydrated in a graded series of ethanol solutions, and processed for IHC. The slides were blocked with normal serum and then incubated with TMEM192, CD31, Galectin-3, LC3, p-ERK, or Ki-67 antibodies (Dako, Mississauga, ON, Canada) overnight at 4°C, followed by matching biotinylated secondary antibodies and peroxidase-conjugated avidin-biotin complex (Dako). Immunostaining was visualized using 3,3′-diaminobenzidine (Dako), which served a chromogen, and the sections were counterstained with hematoxylin. A scale of 1 (negative) to 2 (weak) representing low expression and a scale of 3 (moderate) to 4 (strong) representing high expression were used to grade the intensity of TMEM192 staining. All experiments were approved by Committee on the Shanghai Outdo Biotech Company (Project number: YB M-05-01, YB M-05-02).
Pharmacophore-based virtual screening
Virtual screening was performed by using Discovery Studio 4.5 software (DS4.5, Accelrys Inc., San Diego, CA, USA) on a natural product library including 2,122 compounds. First, we collected 14 known amphiphilic drugs as a training set to establish amphiphilic pharmacophore to screen compounds. The ADMET tool was used to select the compounds with optimal absorption, distribution, metabolism, excretion, and toxicity properties at the early stage of virtual screening.38 The ADMET-filtered natural products were subjected to high-throughput virtual screening using the screen library module, and the top-ranked 20 molecules were selected for cell viability assay.
Cell viability assay and colony formation
Cells were seeded in 96-well plates and cultured for 12 h, then treated with indicated concentrations of PD for the indicated duration (12, 24, and 48 h). Then, the cell viability was assessed by a WST-1 kit (Beyotime) at 450 nm using a microplate spectrophotometer (BioTek Instruments, Winooski, VT, USA). For colony formation assay, HCC cells were seeded in 12-well or 6-well plates at a density of 300 or 600 cells per well for 48 h, and treated with increasing concentrations of PD (0, 0.75, 1.5, 3, 6 μM) for 2 weeks. After fixing with methanol, cells were stained with 0.1% crystal violet for 0.5 h. Colonies were quantified by ImageJ software.
Cell death assay and cathepsin B activity measurement
After HCC cells were stimulated as indicated, Annexin V and PI (KeyGen, Jiangsu, China) were added, and cells were incubated in the dark for 15 min according to the manufacturer’s instructions. The PI-positive percentage (indicating death cells) was measured by C6 flow cytometer (BD Biosciences, San Diego, CA, USA). The activity of caspase 3/7 was determined by the Magic RedCaspase-3/7 Assay Kit (Immuno Chemistry Technologies, Bloomington, MN, USA) according to the manufacturer’s instructions. The Magic RedCathepsin B Assay Kit (ImmunoChemistry Technologies) was used to detect the Cathepsin B activity according to the method described previously.34
Western blot assay
Western blot was performed as we previously described.39 Both HepG2 and Hep3B cells were prepared with RIPA lysis buffer (Cell Signaling Technology), then BCA kit (Thermo Fisher Scientific, Waltham, MA, USA) was applied to determine the concentration of protein. All samples were separated by SDS-PAGE, and subsequently electro-transferred to PVDF membrane (BioRad, Hercules, CA, USA). After blocking with 5% nonfat milk for 1 h, the membrane was incubated with primary antibodies at 4°C overnight, and then with corresponding secondary antibodies for another 2 h at room temperature. The membrane was visualized in Tannon 5200-Multi (Tanon Science & Technology Co. Ltd, Shanghai, China) using ECL reagent (BioRad, Hercules, CA, USA).
Plasmids, transfection, and infection
TMEM192-EGFP, Galectin 3 (Gal3)-mCherry, SMPD1-EGFP, LAMP1-GFP, and LAMP1-mCherry were amplified by PCR and cloned into the N or C terminal of fluorescent protein (EGFP, PA-mCherry1, and mCherry), and then subcloned to EcorRⅠ of pLVX-puro lentiviral vector using Clone Express Ⅱ One Step Cloning Kit (Vazyme, Nanjing, China). We performed CRISPR-CAS9 technology to knockout of human SMDP1 using lentiCRISPR v2 vector (blasticidin addgene #52961). The single guide RNA against SMPD1 was 5′-ACATCCCCGCACATGATGTC-3′. The plasmids EGFP-tagged SMPD1-I136A/F140A, W148A, and E390A were conducted using Mut Express Ⅱ Fast Mutagenesis Kit (Vazyme) according to the manufacturer’s instructions. The stable overexpression or knockdown cells were generated by transduction with lentivirus. The viral particles were produced in 293T cells. Infected cells were selected with puromycin or blasticidin for 7–14 days and confirmed at the protein level. Lipofectamine 3000 reagent was used as the transfection reagent (Thermo Fisher Scientific) according to the manufacturer’s method.
Lysosome staining and isolation
Lysosomes were detected by LysoTracker red/green staining (Beyotime) according to the manufacturer’s instructions. In brief, after pretreatment with PD for 24 h, HCC cells were stained with LysoTracker red for 0.5 h, washed twice with PBS, and subjected to C6 flow cytometer analysis. For confocal microscopy analysis, 300 cells were seeded on the slides in 24-well plates overnight. After rinsing twice with PBS and fixing with 4% paraformaldehyde for 0.5 h at 25°C, the cells were permeabilized with 0.2% Triton X-100 for 10 min, and fluorescence images were captured by laser scanning confocal microscopy (LSM710 or LSM900; Carl Zeiss, Jena, Germany). The lysosome extraction kit (BestBio, Shanghai, China) was used to isolate lysosomes according to the manufacturer’s instructions. For TMEM192-EGFP-based lysosome isolation, briefly, HCC cells stably expressing TMEM192-EGFP were administered the indicated treatment, harvested, washed, and homogenized in extraction buffer (5 mM MOPS, pH 7.65, 0.25 M sucrose, 1 mM EDTA, 0.1% ethanol, and protease inhibitors). The lysates were incubated with a specific EGFP antibody at 4°C for 4 h, followed by the addition of protein A agarose beads and incubation for 2 h. The bound lysosomes were washed three times with extraction buffer and subjected to western blotting.14
Tumorgenicity in nude mice
The Ethics Committee for Animal Experiments of Jinan University approved all the studies (Project number: 20181112-010). Four- to 6-week-old female BALB/c nude mice were fed based on the institutional guidelines for animal care. In brief, Hep3B or HepG2 cells (1 × 106 cells/mice) were suspended in a 1:1 mixture of PBS/Matrigel (Corning Inc., NY, USA) and then subcutaneously injected into the flanks of the mice. Tumor size and body weight of the mice were monitored every 3 days, and the tumor volume was calculated as V(mm3) = (length × width2) × 0.5. In the treatment experiments, mice were randomly divided into control and treatment groups after tumor reached ∼5-mm diameter. The treatment groups included intragastric of 4 mg/kg (i.g. 4 mg/kg) and intraperitoneal injection of 2 mg/kg (i.p. 2 mg/kg) every other day. For PD/sorafenib combinational treatment experiment, treatment groups received oral gavage of PD (4 mg/kg) or/and sorafenib (10 mg/kg) every 2 days, whereas the control group received vehicle. For the experiment of comparing the effect of SMPD1W148A and SMPD1WT in PD treatment, the indicated cell lines were subcutaneously injected into the axilla of each mouse. After solid tumor reached ∼5-mm diameter, treatment groups received oral gavage of PD (4 mg/kg) every 2 days, whereas the control group received vehicle. At the end of the experiment, the tumors, lungs, hearts, livers, and kidneys were excised and either immediately fixed in formalin or frozen in liquid nitrogen. These tissues were used for immunohistochemical analysis with Ki-67, CD31, Gal3, and p-ERK, as well as H&E staining. Meanwhile, mice serum was measured for the levels of ALT and AST.
MiniPDX assay
MiniPDX models were established using freshly removed tumor tissue from an HCC patient, according to the method described previously.40 Drug sensitivity was examined using the OncoVee-Mini PDX assay (LIDE Biotech, Shanghai, China). Briefly, HCC sample was washed and digested with collagenase at 37°C for 1–2 h to prepare HCC cell suspensions. Cells with removal of blood cells and fibroblasts were washed with HBSS and transferred into the capsules (hollow fiber culture system, LIDE Biotech), which were embedded into the subcutaneous tissue of BALB/c nude mice (GemPharmatech, Jiangsu, China; animal project number: LDIACUC007 and LWIACUC002P.). Each mouse received three capsules. Mice were administered orally with PD (4 mg/kg) and/or sorafenib (10 mg/kg) for 7 days, respectively, and the cell viability was evaluated based on relative fluorescence units (RFUs) measured using the CellTiter-Glo Luminescent Cell Viability Assay (Promega, Madison, WI, USA). Relative proliferation rate was calculated according to the following formula: relative proliferation rate = (RFUdrug day7 − RFUdrug day0) ∕ (RFUvehicle day7 − RFUvehicle day0) × 100%. The experiment was approved by institutional review board of The First Affiliated Hospital of Jinan University (Approval number: KY-2023-099).
Transmission electron microscope
To determine the morphology of intracellular lysosomes, the cells were fixed with TEM fixative (Wuhan Servicebio Technology, Wuhan, China) at 4°C for 2 h, then pre-embedded in 1% agarose, and fixed with 1% osmium tetroxide. After dehydration at room temperature using ethanol, the cells were embedded in Poly/Bed resin, followed by polymerization at 65°C. The ultrathin section was stained with 2% uranium acetate-saturated alcohol solution and 2.6% lead citrate. TEM (HT7700, HITACHI, Fukuoka, Japan) was used to take images for morphological analysis.
DIA-based proteomics and bioinformatics analyses
To determine the global protein alteration induced by PD, DIA-based proteomics was performed. Cells with or without PD treatment were lysed with RIPA buffer, and then subjected to trypsin digestion and desalination, according to the method described by Sun et al.41 Next, Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific) was used to detect the samples, and raw data were analyzed by using Proteome Discoverer (Thermo Fisher Scientific) and Spectronaut (Omicsolution Co., Ltd., Shanghai, China) software. With a false discovery rate of 1%, proteins were identified through Uniprot database. The PD-regulated proteins were clustered according to their molecular function, and presented by bubble chart. Proteins bearing KFERQ-like motif were generated by KFERQ finder,42 extracted and presented using Heatmap.
Sphingomyelin measurement
We detected the cellular sphingomyelin level by using Amplite Fluorimetric sphingomyelin assay kit (AAT Bioquest, Sunnyvale, CA) according to the manufacturer’s instructions. Briefly, for each well (96-well plate), 50 μL of HCC cell lysates were incubated with 50 μL SMase working solution at 37°C for 2 h, and then mixed with 50 μL of sphingomyelin assay mixture for 1–2 h at room temperature (protected from light). The fluorescence change was monitored with a fluorescence microplate reader (Synergy H1, BioTek) at Ex/Em = 540/590 nm.
FRAP experiments
For FRAP experiments, HepG2 cells stably expressing TMEM192-EGFP seeded on 20-mm glass bottom cell culture dishes (NEST, Jiangsu, China) were treated with PD (3 μM), Baf-A1 (200 nM), or DMSO for 24 h, respectively. Then the cells were imaged using a confocal microscopy with a 488-nm laser through a ×63 oil immersion. A region of interest comprising the whole cytosol was selected and movie acquisition was started. Cells were photobleached with three consecutive 488-nm pulses, each spaced out by three acquisition frames, and then imaged for 6 min. All images were then analyzed using ImageJ software for the calculation of EGFP intensity. The decay of intensity was calculated in each frame, starting from the first frame after photobleaching to the end of the acquisition.
Lysosomal pH measurement
FITC-Dextran (TargetMol) was used for lysosomal pH measurement. In brief, both HCC cells were seeded onto 12-well plates and incubated with FITC-dextran (0.1 mg/mL) for 24 h, then the cells were treated with indicated concentrations of PD for 24 h. The fluorescence (excited at 488 nm and emitted at 530 nm) was measured with a multifunctional microplate reader (Synergy H1, BioTek).
CMA activity measurement
KFERQ-PA-mCherry1 reporter was used to monitor CMA activity43 in cultured cells stimulated with PD. In brief, both HepG2 and Hep3B cells expressing the KFERQ-PA-mCherry1 reporter were seeded on 12-well plates and subjected to photoconversion at 405 nm for 10 min. More than 90% of the cells were viable after the photoconversion, and these cells were then treated with PD (1.5 μM), Baf-A1 (200 nM), and Torin1 (1 μM) for 72 h. The fluorescence (emitted at 535 nm) was measured with a multifunctional microplate reader (Synergy H1, BioTek) at indicated time points.
Determination of sorafenib content in lysosomes
Liquid chromatography-tandem mass spectrometry (LC-MS/MS) was performed to determine the sorafenib content in lysosomes. Lysosomes were isolated from 2 × 107 HCC cells treated with sorafenib or PD/sorafenib using the lysosome extraction kit (BestBio, Shanghai, China). Then the samples were lysed with three freeze-thaw cycles using liquid nitrogen and a 25°C water bath. Soluble fractions were separated and collected, acetonitrile solution (TBTM 40 ng/mL) was added, and the mixture was then centrifuged at 13000 × g for 5 min. Then, 100 μL of supernatant was diluted with 100 μL of pure water. After uniform vortexing, LC-MS/MS detection was performed. The sorafenib level in the supernatants was analyzed by AB SCIEX 4500 mass spectrometry system with an Agilent ZORBAX Eclipse XDB-C18 (3.5 μm, 50 mm × 2.1 mm), and the concentration was calculated according to a standard curve.
Molecular docking
Molecular docking was performed by using AutoDock v4.2 and AutoDock Tools MGL v1.5.6. For SMPD1 structure (PDB: 5i85) preparation, all water molecules were removed, and hydrogen atoms were added. The grid box size was 98 × 100 × 100 with a spacing of 0.375 Å, calculated by AutoGrid program. During the docking process, Lamarckian genetic algorithm was performed, SMPD1 was rigid, PD was flexible, trials of 200 runs were set, and other parameters were maintained at their default setting. The conformer with the lowest binding free energy was visualized and rendered by using Discovery Studio 4.5 software.
Protein purification
SMPD1 was constructed into the GST fusion vector pGEX-4T-1 (GE Healthcare, Piscataway, NJ, USA), and expressed by Escherichia coli BL21. When the optical density at 600 nm (OD600) reached at 0.6, 0.5 mM of isopropyl-β-D-thiogalactoside (IPTG) was added to induce protein expression. After an additional 6 h of induction, BL21 was harvested and the GST-fused proteins were purified by glutathione Sepharose 4B resin (Beyotime) according to the manufacturer’s instructions. The GST-fused proteins were eluted, and then digested by PreScission Protease (Beyotime) to remove GST-tag.
SMPD1 enzymatic activity assay
Total cellular SMPD1 activity was measured by the cleavage of HMU-PC essentially as described previously.9 Briefly, both HCC cells were harvested in water on ice and sonicated. The samples were then spun down and adjusted to equal concentration. HMU-PC was dissolved in the substrate buffer (0.1 M sodium acetate buffer pH 4.5, containing 0.2% [w/v] sodium taurocholate and 0.02% sodium azide) and mixed with the samples, which were incubated at 37°C for 2 h. The reaction was quenched with 0.2 M glycine-NaOH buffer (pH 10.7) containing 0.2% (w/v) sodium dodecyl sulfate and 0.2% (w/v) Triton X-100. The fluorescence of the formed HMU (ex. = 360 nm, em. = 465 nm) was measured by employing multifunctional microplate reader (Synergy H1, BioTek).
Fluorescence spectroscopy
Fluorescence spectroscopy was performed as we previously described.35 Briefly, recombination SMPD1 protein was diluted to 1 M in PBS and the PD stock solution was dissolved to 0.6 mM in PBS. Aliquots of PD were gradually added to SMPD1 protein solution and the fluorescence spectra were collected at 37°C by using fluorescence spectrometry (Hitachi F7000).
Surface plasmon resonance
To examine the binding of PD to SMPD1, SPR was performed on an OpenSPR system (Nicoya Lifesciences Inc., Kitchener, Canada). In brief, the recombinant WT SMPD1 or W148A protein was diluted to 25 μg/mL in immobilization buffer (PBS, pH 7.4), then immobilized on sensor chips (Nicoya). Subsequently, PD in running buffer (1% DMSO PBS, pH 7.4) was injected to flow cell of channel at a flow rate of 20 μL/min for an association of 240 s, followed by 300-s dissociation. Five cycles were performed according to PD concentrations in ascending order.
Statistical analysis
Student’s t tests, one-way ANOVA analysis, and two-way ANOVA analysis were performed using GraphPad Prism software v.5.01. All in vitro experiments were repeated at least three times, and the values were presented as the mean ± SEM or mean ± SD. The differences with ∗p < 0.05, ∗∗p < 0.01, or ∗∗∗p < 0.001 were considered statistically significant.
Data availability
Data are available on reasonable request. All data relevant to the study are included in the article or uploaded as supplemental information.
Acknowledgments
This work was supported by the National Key Research and Development Program of China (no. 2022YFA1304600 and 2020YFE0202200), the National Natural Science Foundation of China (no. 82103208, 82002948 and 82227802), and the Guangdong Basic and Applied Basic Research Foundation (no. 2022A1515220212 and 2023A1515030115). We thank Mr. Xing-Feng Yin (Jinan University) and Mr. Zheng-Hua Sun (Jinan University) for their technical assistance on mass spectrometry.
Author contributions
Y.W., J.Z., and Q.Y.H. designed the study and wrote the manuscript. G.G.B., J.Z., Y.Y.C., N.N.Y., SJ.H., Y.H.Z., Q.Z., and S.Y.W. performed the experiments. Y.W., G.G.B., J.Z., Y.Y.C., L.O., and N.L. analyzed the data. M.R.C. and L.X.L. provided the advisement. Y.W., J.Z., S.X.Z., and Q.Y.H. supervised the research and revised the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2023.05.015.
Contributor Information
Yang Wang, Email: wangyang0507@jnu.edu.cn.
Shuixing Zhang, Email: zsx7515@jnu.edu.cn.
Jing Zhang, Email: zj6410@jnu.edu.cn.
Qing-Yu He, Email: tqyhe@email.jnu.edu.cn.
Supplemental information
References
- 1.Schaaf M.B., Houbaert D., Meçe O., To S.K., Ganne M., Maes H., Agostinis P. Lysosomal pathways and autophagy distinctively control endothelial cell behavior to affect tumor vasculature. Front. Oncol. 2019;9:171. doi: 10.3389/fonc.2019.00171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Serrano-Puebla A., Boya P. Lysosomal membrane permeabilization in cell death: new evidence and implications for health and disease. Ann. N. Y. Acad. Sci. 2016;1371:30–44. doi: 10.1111/nyas.12966. [DOI] [PubMed] [Google Scholar]
- 3.Piao S., Amaravadi R.K. Targeting the lysosome in cancer. Ann. N. Y. Acad. Sci. 2016;1371:45–54. doi: 10.1111/nyas.12953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gulbins E., Kolesnick R.N. It takes a CAD to kill a tumor cell with a LMP. Cancer Cell. 2013;24:279–281. doi: 10.1016/j.ccr.2013.08.025. [DOI] [PubMed] [Google Scholar]
- 5.Mrschtik M., Ryan K.M. Lysosomal proteins in cell death and autophagy. FEBS J. 2015;282:1858–1870. doi: 10.1111/febs.13253. [DOI] [PubMed] [Google Scholar]
- 6.Narayanan S., Cai C.Y., Assaraf Y.G., Guo H.Q., Cui Q., Wei L., Huang J.J., Ashby C.R., Jr., Chen Z.S. Targeting the ubiquitin-proteasome pathway to overcome anti-cancer drug resistance. Drug Resist. Updat. 2020;48:100663. doi: 10.1016/j.drup.2019.100663. [DOI] [PubMed] [Google Scholar]
- 7.Gavini J., Dommann N., Jakob M.O., Keogh A., Bouchez L.C., Karkampouna S., Julio M.K.d., Medova M., Zimmer Y., Schläfli A.M., et al. Verteporfin-induced lysosomal compartment dysregulation potentiates the effect of sorafenib in hepatocellular carcinoma. Cell Death Dis. 2019;10:749. doi: 10.1038/s41419-019-1989-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gotink K.J., Broxterman H.J., Labots M., de Haas R.R., Dekker H., Honeywell R.J., Rudek M.A., Beerepoot L.V., Musters R.J., Jansen G., et al. Lysosomal sequestration of sunitinib: a novel mechanism of drug resistance. Clin. Cancer Res. 2011;17:7337–7346. doi: 10.1158/1078-0432.CCR-11-1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Petersen N.H.T., Olsen O.D., Groth-Pedersen L., Ellegaard A.M., Bilgin M., Redmer S., Ostenfeld M.S., Ulanet D., Dovmark T.H., Lønborg A., et al. Transformation-associated changes in sphingolipid metabolism sensitize cells to lysosomal cell death induced by inhibitors of acid sphingomyelinase. Cancer Cell. 2013;24:379–393. doi: 10.1016/j.ccr.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 10.Ostenfeld M.S., Høyer-Hansen M., Bastholm L., Fehrenbacher N., Olsen O.D., Groth-Pedersen L., Puustinen P., Kirkegaard-Sørensen T., Nylandsted J., Farkas T., et al. Anti-cancer agent siramesine is a lysosomotropic detergent that induces cytoprotective autophagosome accumulation. Autophagy. 2008;4:487–499. doi: 10.4161/auto.5774. [DOI] [PubMed] [Google Scholar]
- 11.Yang S., Wang X., Contino G., Liesa M., Sahin E., Ying H., Bause A., Li Y., Stommel J.M., Dell'antonio G., et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011;25:717–729. doi: 10.1101/gad.2016111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karasic T.B., O'Hara M.H., Loaiza-Bonilla A., Reiss K.A., Teitelbaum U.R., Borazanci E., De Jesus-Acosta A., Redlinger C., Burrell J.A., Laheru D.A., et al. Effect of gemcitabine and nab-paclitaxel with or without hydroxychloroquine on patients with advanced pancreatic cancer: a phase 2 randomized clinical trial. JAMA Oncol. 2019;5:993–998. doi: 10.1001/jamaoncol.2019.0684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van der Horst G., van de Merbel A.F., Ruigrok E., van der Mark M.H., Ploeg E., Appelman L., Tvingsholm S., Jäätelä M., van Uhm J., Kruithof-de Julio M., et al. Cationic amphiphilic drugs as potential anticancer therapy for bladder cancer. Mol. Oncol. 2020;14:3121–3134. doi: 10.1002/1878-0261.12793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abu-Remaileh M., Wyant G.A., Kim C., Laqtom N.N., Abbasi M., Chan S.H., Freinkman E., Sabatini D.M. Lysosomal metabolomics reveals V-ATPase- and mTOR-dependent regulation of amino acid efflux from lysosomes. Science. 2017;358:807–813. doi: 10.1126/science.aan6298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kirchner P., Bourdenx M., Madrigal-Matute J., Tiano S., Diaz A., Bartholdy B.A., Will B., Cuervo A.M. Proteome-wide analysis of chaperone-mediated autophagy targeting motifs. PLoS Biol. 2019;17:e3000301. doi: 10.1371/journal.pbio.3000301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Radulovic M., Schink K.O., Wenzel E.M., Nähse V., Bongiovanni A., Lafont F., Stenmark H. ESCRT-mediated lysosome repair precedes lysophagy and promotes cell survival. EMBO J. 2018;37:e99753. doi: 10.15252/embj.201899753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Y., Zhang J., Huang Z.H., Huang X.H., Zheng W.B., Yin X.F., Li Y.L., Li B., He Q.Y. Isodeoxyelephantopin induces protective autophagy in lung cancer cells via Nrf2-p62-keap1 feedback loop. Cell Death Dis. 2017;8:e2876. doi: 10.1038/cddis.2017.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tan X., Lambert P.F., Rapraeger A.C., Anderson R.A. Stress-induced EGFR trafficking: mechanisms, functions, and therapeutic implications. Trends Cell Biol. 2016;26:352–366. doi: 10.1016/j.tcb.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Y., Zhang J., Li B., He Q.Y. Advances of proteomics in novel PTM discovery: applications in cancer therapy. Small Methods. 2019;3:1900041. [Google Scholar]
- 20.Wu F.Q., Fang T., Yu L.X., Lv G.S., Lv H.W., Liang D., Li T., Wang C.Z., Tan Y.X., Ding J., et al. ADRB2 signaling promotes HCC progression and sorafenib resistance by inhibiting autophagic degradation of HIF1alpha. J. Hepatol. 2016;65:314–324. doi: 10.1016/j.jhep.2016.04.019. [DOI] [PubMed] [Google Scholar]
- 21.Llovet J.M., Montal R., Sia D., Finn R.S. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2018;15:599–616. doi: 10.1038/s41571-018-0073-4. [DOI] [PubMed] [Google Scholar]
- 22.Saftig P., Sandhoff K. Cancer: killing from the inside. Nature. 2013;502:312–313. doi: 10.1038/nature12692. [DOI] [PubMed] [Google Scholar]
- 23.Fehrenbacher N., Jäättelä M. Lysosomes as targets for cancer therapy. Cancer Res. 2005;65:2993–2995. doi: 10.1158/0008-5472.CAN-05-0476. [DOI] [PubMed] [Google Scholar]
- 24.Utermöhlen O., Herz J., Schramm M., Krönke M. Fusogenicity of membranes: the impact of acid sphingomyelinase on innate immune responses. Immunobiology. 2008;213:307–314. doi: 10.1016/j.imbio.2007.10.016. [DOI] [PubMed] [Google Scholar]
- 25.Zhitomirsky B., Assaraf Y.G. Lysosomes as mediators of drug resistance in cancer. Drug Resist. Updat. 2016;24:23–33. doi: 10.1016/j.drup.2015.11.004. [DOI] [PubMed] [Google Scholar]
- 26.Kazmi F., Hensley T., Pope C., Funk R.S., Loewen G.J., Buckley D.B., Parkinson A. Lysosomal sequestration (trapping) of lipophilic amine (cationic amphiphilic) drugs in immortalized human hepatocytes (Fa2N-4 cells) Drug Metab. Dispos. 2013;41:897–905. doi: 10.1124/dmd.112.050054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wilhelm S.M., Carter C., Tang L., Wilkie D., McNabola A., Rong H., Chen C., Zhang X., Vincent P., McHugh M., et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64:7099–7109. doi: 10.1158/0008-5472.CAN-04-1443. [DOI] [PubMed] [Google Scholar]
- 28.Dai W., Xu L., Yu X., Zhang G., Guo H., Liu H., Song G., Weng S., Dong L., Zhu J., et al. OGDHL silencing promotes hepatocellular carcinoma by reprogramming glutamine metabolism. J. Hepatol. 2020;72:909–923. doi: 10.1016/j.jhep.2019.12.015. [DOI] [PubMed] [Google Scholar]
- 29.Watanabe S., Suzuki T., Hara F., Yasui T., Uga N., Naoe A. Polyphyllin D, a steroidal saponin in Paris polyphylla, induces apoptosis and necroptosis cell death of neuroblastoma cells. Pediatr. Surg. Int. 2017;33:713–719. doi: 10.1007/s00383-017-4069-4. [DOI] [PubMed] [Google Scholar]
- 30.Yang Q., Chen W., Xu Y., Lv X., Zhang M., Jiang H. Polyphyllin I modulates MALAT1/STAT3 signaling to induce apoptosis in gefitinib-resistant non-small cell lung cancer. Toxicol. Appl. Pharmacol. 2018;356:1–7. doi: 10.1016/j.taap.2018.07.031. [DOI] [PubMed] [Google Scholar]
- 31.Condello M., Pellegrini E., Caraglia M., Meschini S. Targeting autophagy to overcome human diseases. Int. J. Mol. Sci. 2019;20:725. doi: 10.3390/ijms20030725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Settembre C., Ballabio A. Lysosomal adaptation: how the lysosome responds to external cues. Cold Spring Harb. Perspect. Biol. 2014;6:a016907. doi: 10.1101/cshperspect.a016907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meyer N., Henkel L., Linder B., Zielke S., Tascher G., Trautmann S., Geisslinger G., Münch C., Fulda S., Tegeder I., et al. Autophagy activation, lipotoxicity and lysosomal membrane permeabilization synergize to promote pimozide- and loperamide-induced glioma cell death. Autophagy. 2021;17:3424–3443. doi: 10.1080/15548627.2021.1874208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Taniguchi M., Ogiso H., Takeuchi T., Kitatani K., Umehara H., Okazaki T. Lysosomal ceramide generated by acid sphingomyelinase triggers cytosolic cathepsin B-mediated degradation of X-linked inhibitor of apoptosis protein in natural killer/T lymphoma cell apoptosis. Cell Death Dis. 2015;6:e1717. doi: 10.1038/cddis.2015.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang J., Zhou Y., Li N., Liu W.T., Liang J.Z., Sun Y., Zhang W.X., Fang R.D., Huang S.L., Sun Z.H., et al. Curcumol overcomes TRAIL resistance of non-small cell lung cancer by targeting NRH:quinone oxidoreductase 2 (NQO2) Adv. Sci. 2020;7:2002306. doi: 10.1002/advs.202002306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eggert T., Greten T.F. Current standard and future perspectives in non-surgical therapy for hepatocellular carcinoma. Digestion. 2017;96:1–4. doi: 10.1159/000464282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Y., Zhang J., Li Y.J., Yu N.N., Liu W.T., Liang J.Z., Xu W.W., Sun Z.H., Li B., He Q.Y. MEST promotes lung cancer invasion and metastasis by interacting with VCP to activate NF-kappaB signaling. J. Exp. Clin. Cancer Res. 2021;40:301. doi: 10.1186/s13046-021-02107-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang J., Sun Y., Zhong L.Y., Yu N.N., Ouyang L., Fang R.D., Wang Y., He Q.Y. Structure-based discovery of neoandrographolide as a novel inhibitor of Rab5 to suppress cancer growth. Comput. Struct. Biotechnol. J. 2020;18:3936–3946. doi: 10.1016/j.csbj.2020.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Y., Zhang J., Zhong L.Y., Huang S.J., Yu N.N., Ouyang L., Niu Y.L., Chen J.X., Lu C.H., He Q.Y. Hsa-miR-335 enhances cell migration and invasion in lung adenocarcinoma through targeting Copine-1. MedComm. 2021;2:810–820. doi: 10.1002/mco2.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhan M., Yang R.M., Wang H., He M., Chen W., Xu S.W., Yang L.H., Liu Q., Long M.M., Wang J. Guided chemotherapy based on patient-derived mini-xenograft models improves survival of gallbladder carcinoma patients. Cancer Commun. 2018;38:48. doi: 10.1186/s40880-018-0318-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun Y., Yang Y.M., Hu Y.Y., Ouyang L., Sun Z.H., Yin X.F., Li N., He Q.Y., Wang Y. Inhibition of nuclear deacetylase Sirtuin-1 induces mitochondrial acetylation and calcium overload leading to cell death. Redox Biol. 2022;53:102334. doi: 10.1016/j.redox.2022.102334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kirchner P., Bourdenx M., Madrigal-Matute J., Tiano S., Diaz A., Bartholdy B.A., Will B., Cuervo A.M. Correction: Proteome-wide analysis of chaperone-mediated autophagy targeting motifs. PLoS Biol. 2022;20:e3001550. doi: 10.1371/journal.pbio.3001550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koga H., Martinez-Vicente M., Macian F., Verkhusha V.V., Cuervo A.M. A photoconvertible fluorescent reporter to track chaperone-mediated autophagy. Nature communications. 2011;2:386. doi: 10.1038/ncomms1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available on reasonable request. All data relevant to the study are included in the article or uploaded as supplemental information.