

Abstract
Poor intratumoral infiltration is the major challenge for chimeric antigen receptor (CAR)-T cell therapy in solid tumors. Hypofractionated radiotherapy (HFRT) has been reported to induce immune cell infiltration and reshape the tumor immune microenvironment. Here, we showed that HFRT (5 × 5 Gy) mediated an early accumulation of intratumoral myeloid-derived suppressor cells (MDSCs) and decreased infiltration of T cells in the tumor microenvironment (TME) of immunocompetent mice bearing triple-negative breast cancer (TNBC) or colon cancer, which was further confirmed in tumors from patients. RNA sequencing (RNA-seq) and cytokine profiling analysis revealed that HFRT induced the activation and proliferation of tumor-infiltrated MDSCs, which was mediated by the interactions of multiple chemokines and chemokine receptors. Further investigation showed that when combined with HFRT, CXCR2 blockade significantly inhibited MDSCs trafficking to tumors and effectively enhanced the intratumoral infiltration and treatment efficacy of CAR-T cells. Our study demonstrates that MDSCs blockade combined with HFRT is promising for CAR-T cell therapy optimization in solid tumors.
Keywords: hypofractionated radiotherapy, T cell infiltration, myeloid-derived suppressor cells (MDSCs), chimeric antigen receptor T (CAR-T) cells, tumor microenvironment
Graphical abstract
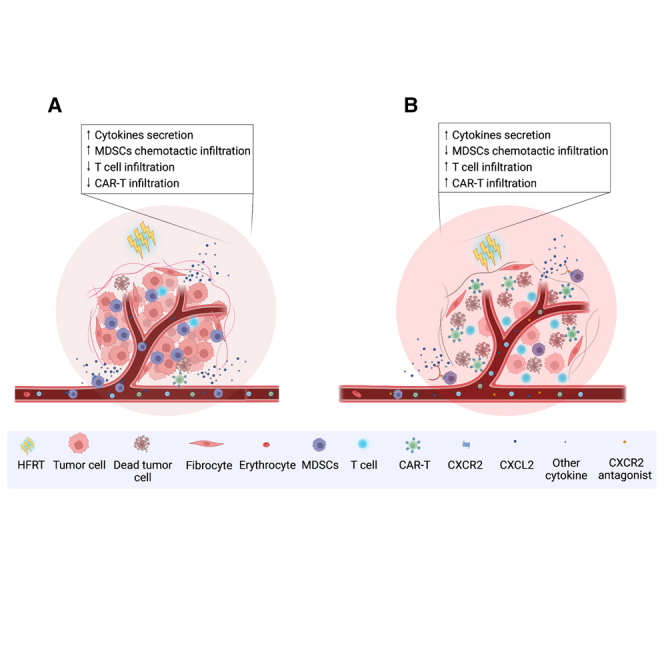
Wang and colleagues demonstrated that HFRT mediated an early accumulation of intratumoral MDSCs via multiple chemokine and chemokine receptors. CXCR2 blockade significantly inhibited MDSC trafficking to tumor and effectively enhanced the intratumoral infiltration and treatment efficacy of CAR-T cells. This provides a new combination strategy for solid tumor.
Introduction
Chimeric antigen receptor (CAR)-T cell therapy has emerged as a promising therapeutic approach for hematological malignancies.1 However, the efficacy of CAR-T cells in solid tumors faces various challenges, including insufficient trafficking into tumors, poor in vivo persistence, and the complex immunosuppressive tumor microenvironment (TME).2 Particularly, the dysregulated chemokine signaling in the TME favors tumor growth and preferentially recruits immunosuppressive cells such as regulatory T (Treg) cells, myeloid-derived suppressor cells (MDSCs), and tumor-associated macrophages (TAMs) over effector T cells.2,3 Therefore, there is a need to promote intratumoral infiltration of CAR-T cells and overcome the suppressive tumor immune microenvironment. To date, encouraging results have been obtained in preclinical studies to address these challenges by amplifying chemokine receptor signaling or targeting stroma and inhibitory immune cells.4,5 Nevertheless, achieving sustained remission is still challenging, and new attempts are needed to improve the efficacy of CAR-T cell therapy in solid tumors.
In recent years, numerous studies have confirmed that focal radiotherapy (RT) can transform a “cold” tumor into a “hot” tumor, which facilitates the entry of immune cells into tumors and reshapes the local immune microenvironment.6,7 In the era of immunotherapy, radiologists are increasingly extending multi-site RT from oligometastatic disease to polymetastatic disease for better tumor control and generation of systemic immune effects.8,9 However, different irradiation doses and fractionations have been reported to exhibit distinct impacts on TME reprogramming by bidirectional and dynamic immune modulations. Clinically, hypofractionated RT (HFRT; 3–20 Gy/day) is frequently used to deliver highly targeted, ablative doses in fewer fractions for multi-site RT of advanced solid tumors.8,9 Compared with traditional RT, HFRT is capable of mobilizing local and systemic immune responses.7,10
Lymphocytes are the most radiosensitive cells in the body, and a single 3 Gy dose could cause tumor-resident T cells to lose 90% viability.11,12,13 Therefore, local RT may provide a new opportunity for adoptive T cells, such as CAR-T cells, to traffic into tumors. RT-mediated intratumoral lymphocyte deletion and potentially increased CAR-T cell infiltration may lead to enhanced antitumor efficiency of CAR-T cells in advanced solid tumors. However, immunosuppressive cells, including MDSCs, TAMs, and Treg cells can also infiltrate into the tumor niche after RT, consequently impairing the infiltration and effector function of CAR-T cells.14,15 Therefore, it is necessary to promote T cell infiltration and simultaneously block immunosuppressive cell accumulation to enhance the therapeutic efficacy of CAR-T therapy. Unfortunately, it is still challenging to figure out RT-induced dynamic changes of immune cells within the TME and the potential mechanism of new immune cell infiltration. Exploring immune cell subsets that re-enter tumors after RT and the underlying mechanism of re-entering tumors will be beneficial for the optimization of CAR-T cell therapeutic strategies.
Here, we report that local HFRT-mediated dynamic changes of intratumoral immune cell infiltration and remodeling of the tumor immune microenvironment. Particularly, HFRT induced an early intratumoral accumulation of MDSCs and decreased T cell infiltration. We found that MDSCs blockade early after HFRT significantly increased CAR-T cell infiltration into tumors and enhanced the therapeutic efficiency of combination therapy with HFRT and CAR-T cells. Our study suggests that MDSCs blockade is necessary when CAR-T cell therapy is used in combination with HFRT to treat solid tumors.
Results
HFRT induces significant alterations of MDSCs and T cells within the TME
To investigate the dynamic changes of tumor-infiltrating immune cells (TIICs) that induced by HFRT, mice bearing colon carcinoma CT26 cells were treated with a schedule of 5 × 5 Gy and sacrificed on days 7, 14, and 21 after the initiation of HFRT for immune cell detection (Figure 1A). To exclude the potential influence of tumor burden, unirradiated mice with a similar tumor size as their irradiated counterparts were used as controls in this study.16,17 In irradiated CT26 tumors, the proportion of CD45+ TIICs increased significantly on day 14 (Figure 1B), whereas no significant changes were observed in tumor-infiltrating CD11c+ dendritic cells (DCs) or CD11b+ F4/80+ macrophages. At the same time, the intratumoral CD49b+ natural killer (NK) cells decreased significantly on day 7 and then gradually returned to the level before HFRT. CD4+ CD25+ Foxp3+ Treg cells significantly increased on day 14, although the proportion was less than 0.5% of all cells (Figures 1B and S1).
Figure 1.

HFRT induces significant alterations of MDSCs and T cells within tumors
(A) Treatment scheme. (B) Bar plots showing fractions of CD45+ tumor-infiltrating immune cells (TIICs), CD11c+ DCs, CD49b+ NK cells, CD11b+ F4/80+ macrophages, or CD4+ CD25+ Foxp3+ Treg cells in CT26 tumors. (C and E) Representative flow plots of CD45+ TIICs, CD3+, CD4+, CD8+ T cells, Gr-1+ CD11b+ MDSCs, Gr-1+ CD11b+ Ly6Ghigh polymorphonuclear (PMN)-MDSCs, or Gr-1+ CD11b+ Ly6Glow M-MDSCs in CT26 tumors (percentage of all live cells) in tumor. (D and F) Bar plots showing fractions of total MDSCs, PMN-MDSCs, M-MDSCs, CD3+, CD4+, or CD8+ T cells (percentage of all live cells) in CT26 tumor. (G) Representative multicolor immunofluorescence images of tumor specimens from rectal cancer patients who received 5 × 5 Gy preoperative HFRT (n = 3; scale bar: 100 μm; magnification 20×; MDSCs: red arrowheads, T cells: white arrowheads). (H–K) Bar plots showing fractions of CD45+ TIICs, CD3+, CD4+, CD8+ T cells, MDSCs, PMN-MDSCs, and M-MDSCs (percentage of all live cells) in 4T1 (H and J) or MC38 (I and K) tumor. The results are presented as mean ± SEM. ∗p < 0.05 and ∗∗p < 0.01; NS, p > 0.05 (unpaired Student’s t test).
On day 7 after HFRT initiation, although the ratio of CD45+ TIICs showed no significant changes compared with the unirradiated counterpart, significantly elevated CD11b+ Gr1+ MDSCs accumulation was detected within the tumor site. In irradiated CT26 tumors, the proportion of tumor-infiltrating MDSCs showed an approximately 2-fold increase, and this trend continued on day 14 (Figures 1C, 1D, and S2A). Further analysis revealed that Gr1+ CD11b+ Ly6Glow M-MDSCs had a remarkable increase on day 7 (Figures 1D and S2A). In parallel, the proportion of intratumoral CD3+, CD4+, and CD8+ T cells decreased significantly on day 7 and then gradually returned to the level before HFRT (Figures 1E, 1F, and S2B). Notably, an increased accumulation of CD11b+ cells and decreased T cell infiltration were further observed in tumor tissues from rectal cancer patients who received 5 × 5 Gy preoperative HFRT compared with the unirradiated counterpart (Figures 1G and S3). In addition, we also investigated the dynamic changes of tumor-infiltrating MDSCs and T cells induced by 3 × 8 Gy, a hypofractionated schedule with a biologically effective dose (BED) similar to that of 5 × 5 Gy, which was reported to induce a strong immune response in mouse tumor models. And a similar tendency was observed in CT26 tumor (Figure S4).
To further confirm our observation, MC38 or 4T1 tumors that received a schedule of 5 × 5 Gy. Consistent with that in CT26 tumors, a significant increase of CD45+ TIICs was observed in irradiated 4T1 and MC38 tumors on day 14 after the initiation of HFRT (Figures 1H, 1I, and S5). In addition, tumor-infiltrating CD3+ T lymphocytes increased significantly on day 14 (Figures 1H, 1I, and S5), and the proportion of MDSCs significantly increased from day 7 compared with the unirradiated counterpart (Figures 1J, 1K, and S6). Overall, these data indicate that after HFRT, the most abundant immune cell infiltration occurred on day 14. Whereas MDSCs were preferentially recruited early on day 7 after the initiation of HFRT, T cells infiltrated later and did not increase significantly until day 14.
Focal HFRT induces systemic alterations in MDSCs and T cells
Given the significant alterations of tumor-infiltrating MDSCs and T cells, we further investigated the dynamic changes of MDSCs and T cells in the peripheral blood, spleen, and bone marrow of CT26, MC38, and 4T1 tumor-bearing mice after HFRT. In peripheral blood, the proportion of MDSCs decreased on day 7 in all three models after the initiation of HFRT and then gradually recovered and even exceeded the level of the unirradiated counterpart (Figure 2A). In mice bearing CT26 or MC38 tumors, the proportion of splenic MDSCs was significantly increased on day 7 and then gradually decreased (Figure 2B). In mice bearing 4T1 tumors, the proportion of splenic MDSCs showed an approximately 3- to 4-fold increase on day 14, and this level was maintained until day 21 (Figure 2B). In bone marrow, the MDSCs proportion in CT26-bearing mice gradually decreased after HFRT (Figure 2C). In contrast, the MDSCs proportion in the 4T1 and MC38 models was significantly increased after HFRT (Figure 2C). Together, these data suggested that focal HFRT led to a systemic mobilization of MDSCs, and the early increased MDSCs accumulation in tumors was accompanied by a decrease in peripheral blood, suggesting that the tumor-infiltrated MDSCs early after HFRT could be recruited from the blood. Then the peripheral blood MDSCs could be supplemented later from the spleen and bone marrow (Figure 2D). Interestingly, the proportions of CD3+, CD4+, and CD8+ T cells in the peripheral blood and bone marrow of irradiated mice were significantly increased early after HFRT (day 7) and gradually returned to the original level (Figures S7A–S7C). Consistent with our findings, an increase in peripheral blood T cells was also reported in non-small-cell lung cancer patients who received HFRT.18
Figure 2.

Focal HFRT induces systemic alterations in MDSCs
Representative flow plots of MDSCs in the blood (A), spleen (B), or bone marrow (C) of mice bearing CT26, 4T1 or MC38 tumors on day 7 after radiation initiation (gated on live cells). Bar plots showing fractions of MDSCs in the blood, spleen, or bone marrow of mice. Results are presented as mean ± SEM. ∗p < 0.05 and ∗∗p < 0.01; NS, p > 0.05 (unpaired Student’s t test). (D) Line chart of the proportion of MDSCs in different tissues of mice.
HFRT promotes intratumoral infiltration of MDSCs and T cells from peripheral blood and induces activation of MDSCs
To further investigate whether tumor-infiltrating MDSCs and T cells were replenished from the peripheral blood, one day before HFRT initiation, MDSCs or T cells (1 × 106) expressing GFP were isolated from EGFP Tg/+ transgenic mice (C57BL/6 background) and intravenously (i.v.) injected into MC38 tumor-bearing mice (Figure 3A). On day 5 after HFRT initiation, significantly increased GFP+ MDSCs and T cells were observed in all tumor-infiltrating MDSCs (Figure 3B) and T cells (Figure 3C), respectively, which proved the successful intratumoral infiltration of exogenously infused immune cells. The spatial and temporal distribution of MDSCs and T cells within tumors was further observed through multicolor immunofluorescence staining, which revealed that MDSCs were preferentially recruited to the tumor site 2 days after the end of HFRT (day 7), while T lymphocytes were significantly increased on day 14. Notably, MDSCs and T cells were frequently observed adjacent to each other (Figure 3D).
Figure 3.

HFRT promotes intratumoral infiltration of MDSCs and T cells from peripheral blood and induces a suppressive phenotype of MDSCs
(A) Experimental scheme. (B and C) Bar plots showing fractions of GFP+ MDSCs and T cells in tumors by flow cytometry on day 5 after radiation initiation. Representative immunofluorescence images show GFP+ MDSCs or T cells in tumors in the indicated groups (n = 3; scale bar: 50 μm). (D) Representative multicolor immunofluorescence images of CT26 tumor (n = 3; scale bar: 100 μm; MDSCs: red arrowheads, T cells: white arrowheads). (E) KEGG enrichment analysis of differentially expressed genes (DEGs) in irradiated CT26 tumors compared with the unirradiated counterpart. (F) GSEA of the Jak-STAT signaling pathway in MDSCs from CT26 tumor. (G) Heatmap of DEGs that encoding genes related to proliferation and activation of MDSCs. (H) GSEA of PD-L1 expression and the PD-1 checkpoint pathway in MDSCs from CT26 tumor. (I) Bar plots showing fractions of PD-L1+ MDSCs in tumors. Results are presented as mean ± SEM, unpaired Student’s t test. (J) Treatment scheme and tumor volume were plotted starting from the day before the initial dose of HFRT (n = 7; results are presented as mean ± SEM; mixed-effects analysis). ∗p < 0.05 and ∗∗p < 0.01; NS, p > 0.05.
To further identify the functional characteristic of HFRT-induced MDSCs, RNA sequencing (RNA-seq) was performed on MDSCs isolated from the tumor, spleen, and bone marrow of CT26-bearing mice day 7 after HFRT initiation. For tumor-infiltrated MDSCs, a total of 3,327 differentially expressed genes (DEGs) were identified (p < 0.05, fold change > 2) compared with the unirradiated counterpart, including 1,310 downregulated and 2,017 upregulated genes. For the bone marrow and splenic MDSCs, 473 and 27 DEGs were identified, respectively (Figure S9A). To further elucidate the functional roles of these DEGs, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis was performed using online analysis from Beijing Genomics Institute (BGI) (https://report.bgi.com/ps/login/login.html). The results indicated that the DEGs in tumor-infiltrated MDSCs, but not bone marrow MDSCs, were mainly enriched in the cytokine-cytokine receptor, chemokine-chemokine receptor interaction pathways, and JAK-STAT signaling pathways, which regulate the function and chemotaxis of MDSCs (Figures 3E and S9B). In particular, the transcriptional modification induced by HFRT in genes involved in JAK/STAT signaling was further confirmed by gene set enrichment analysis (GSEA), which showed positive enrichment of the JAK/STAT gene signature in tumor-infiltrated MDSCs from the irradiated groups (Figures 3F and S9C).
On the basis of cluster analysis, we further analyzed genes related to MDSCs proliferation and activation. In the bone marrow and the spleen, MDSCs proliferation-associated genes (STAT3, IL10, STAT1, Irf1, Myd88, Creb1, Bcl2a1a, Bcl2a1b, and Bcl2a1d) were mostly upregulated in the irradiated group compared with the unirradiated counterpart. However, MDSCs activation-related genes (Nos2, IL6, Tlr8, STAT2, Ncf1, Nrros, Ifng, IL4, S100a9, S100a8, Ncf4, and Ifngr1) were not upregulated considerably (Figure 3G). Interestingly, in the tumor, genes related to the MDSCs proliferation and activation were remarkably upregulated in the HFRT group (Figure 3G). Together, these results suggested that MDSCs were mobilized in the bone marrow and spleen but recruited and activated in the tumor after HFRT. Furthermore, given that the PD-L1/PD-1 pathway plays an important role in the immunosuppressive functions of MDSCs,19,20 we further investigated PD-L1/PD-1 pathway-related gene expression. GSEA revealed enrichment of PD-L1 expression and PD-1 checkpoint pathway-related genes in tumor-infiltrating MDSCs from the HFRT group (Figures 3H and S9D). In addition, upregulated expression of PD-L1 was also observed in tumor-infiltrating MDSCs isolated from irradiated CT26 and MC38 tumors (Figure 3I). Overall, these results suggested that focal HFRT mediated the intratumoral infiltration of MDSCs and T cells from peripheral blood and induced the activation of MDSCs.
On the basis of these findings, we hypothesized that HFRT preconditioning could possibly promote CAR-T cell infiltration into solid tumors. A murine epithelial cell adhesion molecule (EpCAM)-targeting third-generation CAR (EpCAM28.BBz) was constructed previously by our team21 and used in this study. CT26, MC38, and 4T1 cells were screened for EpCAM expression by flow cytometry, and only 4T1 cells showed a high level of EpCAM expression (Figure S9E). Mouse T lymphocytes were transduced with a CAR-encoding retrovirus, and the transfection efficiency was approximately 55% by flow cytometry detection (Figure S9F). The cytotoxicity of EpCAM CAR-T cells was corroborated by significantly elevated IFN-γ release after 24 h co-incubation with 4T1 cells, compared with the untransduced T (UTD) cells (Figure S9G). In vivo, as shown in our previous study, when the tumor volume reached ∼50 mm3, CAR-T cells alone showed a strong antitumor effect.21 Therefore, in this study, HFRT was performed when the 4T1 tumor volume reached ∼90 mm3, and 3 × 106 CAR+ T or UTD cells were administered i.v. on day 7 after HFRT initiation (Figure 3J). Unexpectedly, HFRT combined with EpCAM CAR-T cells did not show significantly enhanced antitumor efficacy, indicating that this combination strategy needs to be further optimized (Figures 3J and S10).
HFRT-mediated MDSC recruitment in a chemokine-chemokine receptor-dependent manner
To further understand the mechanism of HFRT-induced MDSCs accumulation, RNA-seq data were further analyzed on MDSCs isolated from the tumor, spleen, and bone marrow of CT26 tumor-bearing mice on day 7 after HFRT initiation. GSEA showed that genes related to MDSCs chemotaxis were unregulated in the chemokine signaling pathway in tumor-infiltrating MDSCs (Figure 4A). Furthermore, the differential gene expression profile suggested that CCR1, CCR2, CCR5, CXCR1, and CXCR2 were significantly upregulated in MDSCs from the bone marrow and tumor (Figure 4B). Then we investigated the changes of chemokines in CT26 or MC38 tumors that harvested on days 7, 14, and 21 after HFRT initiation. The results indicated that the chemokines CCL1, CCL2, CCL5, CXCL5, and CXCL12 were obviously upregulated on day 7 after HFRT (Figure 4C). To further reverify the changes of chemokines in protein level, total proteins from serum and tumors were harvested in CT26-bearing mice, and 25 chemokine expression levels were detected by commercial cytokine chips. As a result, after HFRT, serum CXCL2 and CCL5 were remarkably elevated (Figure 4D), while in tumors, CXCL2, CXCL5, CCL2, and CCL5 were significantly increased, especially CXCL1, CCL2, and CCL5 (Figure 4E). On the basis of the results of RNA-seq and cytokine chips, three chemokine-chemokine receptor axes, including CCR5-CCL5, CCR2-CCL2, and CXCR2-CXCR2 ligands, were most likely responsible for HFRT-mediated MDSC recruitment. The expression of these chemokine receptors was further detected on tumor-infiltrated MDSCs using flow cytometry, which showed that CCR2, CCR5, and CXCR2 were significantly elevated on day 14 and 21 after HFRT initiation (Figure 4F), indicating that multiple chemokine receptors were involved in HFRT-induced MDSCs migration to tumor site.
Figure 4.

HFRT-mediated MDSC recruitment in a chemokine-chemokine receptor-dependent manner
(A) GSEA of chemokine signaling pathways in MDSCs isolated from the tumor, spleen, and bone marrow (BM) of mice bearing CT26 tumor on day 7 after HFRT. (B) Heatmap of genes encoding chemokine receptors in MDSCs from different tissues. (C) Heatmap of DEGs that encoding chemokines of MDSCs in the tumor bulk. (D and E) Representative cytokine arrays for the proteins of serum and tumors; the boxes indicate the chemokines with significant increases (n = 3). Logarithm of fold change (log2RT/control) ratios for differentially expressed chemokines between the HFRT group and control group in serum and tumor. (F) Fluor cytometric analysis of CCR2+ MDSCs, CCR5+ MDSCs, and CXCR2+ MDSC accumulation in tumors and the quantification are presented. Results are presented as mean ± SEM (n = 5). ∗p < 0.05 and ∗∗p <0.01; NS, p > 0.05 (unpaired Student’s t test). (G) Heatmap of DEGs in tumor bulk encoding chemokines that are chemotactic for T cells. (H) Logarithm of fold change (log2RT/Control) ratios for seven differentially expressed T cell-related chemokines in serum and tumor.
To further identify the mechanism of HFRT-induced early tumor-infiltrated T cell decrease, T cell-associated chemokines were analyzed using tumor bulk RNA-seq data and further confirmed by commercial cytokine chips. The results showed that HFRT induced obvious upregulation of T cell-related chemokines on day 14 after HFRT initiation (Figures 4G and 4H), which furtherly supported our previous findings (Figures 1F, 1H, and 1I).
CXCR2 or CCR2 blockade inhibits the MDSCs mobilization induced by HFRT
Given that HFRT induced the activation of tumor-infiltrated MDSCs, and MDSCs-mediated profound CAR-T cell suppression was reported in patients with lymphoma and leukemia.22 We decided to further optimize the combination strategy by blocking the intratumoral infiltration of MDSCs. As CCL5/CCR5 has been reported to participate in antigen presentation by recruiting conventional type 1 DCs (cDC1),23 we tried to inhibit MDSCs infiltration to tumor site by blocking CXCR2 or CCR2, respectively. We selected MC38 cells to complete the experiments, as CCR2 knockout mice were from C57BL/6 mice. Mice bearing MC38 tumor were administered intraperitoneally (i.p.) 0.5 mg/kg CXCR2 antagonist (SB225002)24,25 on day 5 after the initiation of HFRT, and a significant reduction of MDSCs was detected in the tumor and peripheral blood, which lasted until day 14 (Figures 5A–5C). Importantly, after CXCR2 blockade, a significantly enhanced intratumoral T cell infiltration was detected on day 7 (Figures 5D and 5E). Furthermore, CCR2 knockout mice bearing MC38 tumors were also used to investigate the impact of CCR2 blockade on MDSCs mobilization and T cell infiltration and showed that CCR2 knockout significantly decreased MDSCs accumulation in the tumor, peripheral blood, and spleen on day 7 after HFRT initiation (Figures 5F–5H). In bone marrow, CCR2 knockout significantly decreased the accumulation of MDSCs in the irradiated group (Figure 5H). However, significantly decreased intratumoral T cell infiltration was detected using flow cytometry after HFRT (Figures 5I and 5J), probably because CCR2 is expressed on T cells and contributes to their chemotaxis.26,27 Overall, our results indicated that early intratumoral MDSC accumulation after HFRT was mediated by CCR2 and CXCR2, and CXCR2 blockade inhibited MDSCs trafficking to tumors that induced by HFRT, and enhanced the intratumoral T cell infiltration.
Figure 5.

The effects of CXCR2 and CCR2 blockade on MDSC mobilization and T cell infiltration induced by HFRT
(A) Representative flow plots of MDSCs in MC38 tumors on day 7 after radiation initiation (gated on CD45+ cells). (B and C) Bar plot showing fractions of CD45+ cells and MDSCs in the tumor, blood, spleen, and bone marrow of mice bearing MC38 tumors. (D) Representative flow plots of CD3+(gated on live cells), CD4+, and CD8+ T cells (gated on CD45+ cells) in MC38 tumors. (E) Bar plots showing fractions of CD3+, CD4+, and CD8+ T cells (percentage of all live cells) in MC38 tumors. (F) Representative flow plots of MDSCs in CCR2KO or wild-type (C57BL/6) mice tumors (gated on CD45+ cells). (G and H) Bar plot showing fractions of CD45+ cells and MDSCs in the tumor, blood, spleen, and bone marrow of CCR2KO mice or wild-type mice. (I) Representative flow plots of CD3+ T cells (gated on live cells), CD4+ T cells, and CD8+ T cells (gated on CD45+ cells) in CCR2KO or wild-type (C57 BL/6) mice tumors. (J) Bar plots showing fractions of T cells (percentage of all live cells) in CCR2KO or wild type (C57BL/6) mice. Results are presented as mean ± SEM. ∗p < 0.05 and ∗∗p < 0.01; NS, p > 0.05 (unpaired Student’s t test).
CXCR2 blockade enhances the antitumor efficacy of EpCAM CAR-T cells by impairing MDSCs accumulation in tumor
As decreased intratumoral T cell infiltration was detected in CCR2 knockout mice after HFRT, and CCR2 plays an important role in shaping the activated/memory phenotype of T cells,28,29 we decided to combine prioritized CXCR2 blockade with CAR-T cells for tumor treatment following HFRT. In vivo, BALB/c mice were subcutaneously (s.c.) inoculated with 4T1 cells, and HFRT was initiated when the tumor volume reached ∼150 mm3. On day 5 after the start of HFRT, mice were administered a CXCR2 antagonist (SB225002) or normal saline (NS) by i.p. injection, followed by i.v. administration of 3 × 106 EpCAM CAR+ T or UTD cells 2 days later (Figure 6A). Notably, CXCR2 antagonist or EpCAM CAR-T cells alone only slightly inhibited tumor growth. However, upon further addition of SB225002, the triple combination therapy with a CXCR2 antagonist, HFRT, and EpCAM CAR-T cells showed significantly enhanced antitumor efficacy and prolonged mice survival, compared with the treatment regimen with HFRT and EpCAM CAR-T cells (Figures 6B, 6C, and S11A), and without obvious toxicity (Figures 6D and S11B). Further multicolor immunofluorescence staining analysis showed fewer CD11b+ cells and more T cell infiltration in the TME of mice receiving SB225002 than RT plus CAR-T group (Figure 6E). To investigate whether the combination treatment with HFRT and CXCR2 antagonist enhanced EpCAM CAR-T cell infiltration in tumor, DNA was extracted from tumor tissues, and CAR copies were detected using RT-PCR. The results indicated that although CAR copies in CAR-T cells combined with the CXCR2 antagonist showed a significant increase compared with CAR-T cells alone, a significantly elevated CAR copies were detected in the combination therapy with RT, CAR-T cells, and the CXCR2 antagonist (Figure 6F).
Figure 6.

CXCR2 blockade enhanced efficacy of EpCAM CAR-T cells by impairing MDSC accumulation within the tumor site
(A) Treatment scheme. (B) Left: tumor growth curve of all groups. Right: tumor growth curve of combination groups (n = 7–9 mice pooled). (C) Kaplan-Meier survival curve of 4T1 tumor-bearing mice (n = 11 mice per group). Log rank test. (D) Weight change curve of mice bearing 4T1 tumors (n = 7–9). (E) Representative multicolor immunofluorescence images of tumor from different groups (n = 3; scale bar: 100 μm). CD11b+ cells (red arrowheads) and T cells (white arrowheads). Bar plot showing fractions of CD3+ cells and CD11b+ cells (percentage of total cells) in the tumor. Cell quantification by Halo version 3.6 (unpaired Student’s t test). (F) CAR gene copies in tumor tissue at day 3 after EpCAM CAR-T cell infusion (n = 5). (G) Left: tumor growth curve. Right: weight change curve (n = 7 mice pooled). Results are presented as mean ± SEM. ∗∗p < 0.01; NS, p > 0.05 (mixed-effects analysis).
Additionally, we tried to combine RT with CAR-T cell therapy at different time points during RT therapy. We found that whether CAR-T cells were infused on the day RT ended, or the second day after RT was completed, the combination therapy with HFRT and CAR-T cells did not show enhanced antitumor effect compared with RT alone. Only by adding an MDSC antagonist could the tumor be significantly reduced (Figures 6G and S11C). Overall, after HFRT, CXCR2 blockade significantly enhanced the efficacy of EpCAM CAR-T cells by impairing MDSC accumulation within the tumor site and promoting infiltration of CAR-T cells.
Discussion
The successful eradication of solid tumors with CAR-T cell therapy is contingent upon the critical step of CAR-T cell trafficking into the tumors, which also poses the first challenge. However, the immunosuppressive TME presents another challenge for developing effective CAR-T cell therapy in solid tumors. Combination therapy with RT and CAR-T cells in solid tumors has been considered more effective than CAR-T cell therapy alone.30 Nevertheless, achieving successful treatment of patients with solid tumors requires careful consideration of the optimal dose/fraction of RT and timing of CAR-T cells.31 In our study, we tried to use HFRT as a preconditioning for CAR-T cell therapy owing to the effect of HFRT on tumor ablation and remodeling of immune microenvironment. Besides, the optimal timing of CAR T cell infusion should depend on the dynamics of immune infiltrates after HFRT. Therefore, we investigated the dynamic changes in immune infiltrates in three mouse models following a 5 × 5 Gy HFRT. Our study revealed that MDSCs play a critical role in HFRT-mediated immune suppressive microenvironment and impair T cell infiltration and CAR-T cell efficacy.
MDSCs recruitment into the TME after RT has been considered a host adaptation that decreased the efficacy of RT and the antitumor immune response.32 Studies have shown that HFRT induced intratumoral recruitment of MDSCs, and inhibiting MDSCs can increase the efficacy of RT combined with immunotherapy.33 However, previous studies investigating HFRT-induced MDSCs recruitment in tumors have provided evidence for highly heterogeneous intratumoral MDSCs induced by different doses and fractions. Filatenkov et al.34 found that a single 30 Gy irradiation can recruit antitumor immune cells into tumor and reduce the accumulation of MDSCs in MC38 and CT26 tumor-bearing mice. However, Liang et al.35 found that when MC38 tumor-bearing mice were irradiated with a single 20 Gy dose, intratumoral MDSCs increased significantly on day 3 after irradiation. A study has also found that MDSCs recruitment significantly increased when tumor-bearing mice were irradiated with 3 × 20 Gy HFRT.14 Therefore, further investigation is needed to clarify the dynamics of intratumoral MDSCs following RT.
In this study, we investigated the impact of 5 × 5 Gy HFRT, a recommended dose/fraction of neoadjuvant RT for rectal cancer, on the dynamics of immune cells within tumors. We found that MDSCs were the first immune cells to infiltrate into tumors and significantly increased in number in three different tumor models. In contrast, T cells did not increase or even decreased in tumors as MDSCs began to enter, although an increase in T cells was observed 14 days after HFRT. We also confirmed the increased infiltration of MDSCs in rectal cancer patients who received neoadjuvant 5 × 5 Gy HFRT one week before surgery. More important, we found that MDSCs mobilized systemically and acquired an immunosuppressive phenotype upon entering the tumor, which may be due to metabolites and cytokines in the TME. Yang et al.36 found that RT promoted the activation of MDSCs, which was dependent on enhanced lactate secretion regulated by HIF-1α. In addition, multiple signals such as IL-6, IL-4, and NOS2 are required for myeloid cells to gain immunosuppression function.37 Here, we found that the expression of IL-4 and IL-6 was significantly upregulated in tumors from the irradiated group, which may explain why MDSCs acquired an immunosuppressive phenotype after entering the tumor. Apparently, under this circumstance, the sequential timing of MDSCs and T cell entry into the tumor and the suppressive phenotype of MDSCs are detrimental to intratumoral T cells, including CAR-T cells. In our attempt to combine EpCAM CAR-T cells with HFRT to treat 4T1-bearing mice, we failed to observe significantly enhanced antitumor effects in the absence of lymphodepletion preconditioning. Therefore, it is necessary to block the mobilization of MDSCs or reduce MDSCs-mediated immunosuppression to promote CAR-T cell infiltration.
In this study, we focused on exploring the potential mechanism of MDSCs recruitment induced by HFRT and found that the chemokine signaling pathways CCR5-CCL5, CCR2-CCL2, and CXCR2-CXCL1/2/5 may be related to intratumoral infiltration of MDSCs, which is consistent with existing research results.38,39 Targeting the CCR5-CCL5 axis efficiently controls MDSCs infiltration and prevents tumor growth in preclinical studies.40 However, CCR5 is also expressed on T cells and DCs, and CCR5 blockers may negatively affect the efficacy of adoptive T cell therapy and DC-mediated antigen presentation.23,41 Therefore, the effect of CCR2 or CXCR2 blockade on MDSC recruitment was detected in this study. Consistent with previous research, our study confirmed that CCR2 deficiency significantly reduced intratumoral MDSCs recruitment and systemic mobilization.35,42 However, CCR2 deficiency also induced a reduction in T cell infiltration, which was probably attributed to CCR2 expression on T cells. In this study, we found that a CXCR2 antagonist effectively inhibited the MDSC mobilization and intratumoral accumulation induced by HFRT. More notably, CXCR2 blockade promoted early infiltration and increased accumulation of T cells within tumors. In mice bearing 4T1 breast cancer, focal HFRT combined with a CXCR2 antagonist significantly improved the efficacy of EpCAM CAR-T cells. Therefore, CXCR2 blockade may be an optimal strategy for CAR-T cell combination with HFRT to treat solid tumors.
Chemokines play a crucial role in mediating the recruitment of T cells into tumors. According to the expression of specific chemokines in the TME, CAR-T cells were transduced to express corresponding chemokine receptors, such as CXCR4 and CCR2, to enhance CAR-T cell trafficking. However, chemokines can also promote tumor growth and metastasis by recruiting suppressive immune cells, such as Treg cells. It is essential to understand the expression profile of chemokines in the TME after HFRT to identify which chemokine can promote CAR-T cell intratumoral trafficking, and which chemokine should be blocked to dampen the recruitment of suppressive immune cells. In this study, we revealed that HFRT upregulated the expression of multiple chemokines, such as CXCL1/2, CCL2, CCL5, and CXCL9/11. Among these, CXCL1/2 and CCL2 can recruit MDSCs and Treg cells through CXCR2 and CCR2, while CXCL9/10/11 and CXCR3 may recruit T cells.43,44 Moreover, we observed a significant increase in MDSCs and Treg cells in HFRT tumors. Under these circumstances, the transduction of chemokine receptors may contribute to further increasing the CAR-T cell infiltration by amplifying the chemotactic signaling, which is another optimized strategy we are evaluating.
Although previous investigations have shown that RT has the potential to enhance the efficacy of CAR-T cells in hematological malignancies,45 our data revealed that HFRT alone may not be sufficient to achieve optimal results. Many questions still need to be answered in the future, such as the optimal dose and fractionation of RT. In this study, we also investigated the dynamics of MDSCs and T cells following a 3 × 8 Gy HFRT, as this schedule has been shown to be more capable of inducing an abscopal effect than conventionally fractionated or single-dose RT.46 In addition, lymphodepleting chemotherapy has been shown to increase the efficacy of adoptively transferred T cells.47,48 In our study, we observed an improved efficacy of combination therapy with RT, CAR-T cells, and CXCR2 antagonist without lymphodepletion preconditioning. It remains to be explored whether the addition of lymphodepleting chemotherapy can further improve the antitumor efficacy of CAR-T cells.
In conclusion, our data demonstrate that the early accumulation of MDSCs after HFRT is a common phenomenon in different tumor models, which may inhibit the infiltration and efficacy of CAR-T cell therapy. Thus, MDSC blockade is necessary when CAR-T cell therapy is combined with HFRT to treat solid tumors.
Materials and methods
Cell lines and animals
Mouse 3T3, 4T1 breast cancer, MC38 and CT26 colon adenocarcinoma cell lines, and human HEK293T cells were purchased from the American Type Culture Collection (ATCC). 4T1 and MC38 cells were cultured in RPMI-1640 supplemented with 10% fetal bovine serum, 100 U ml−1 penicillin, and 100 μg mL−1 streptomycin. NIH3T3 cells were cultured in DMEM supplemented with 10% fetal bovine serum, 100 U ml−1 penicillin, and 100 μg mL−1 streptomycin. All cell lines were maintained at 37°C in a humidified 5% CO2 incubator and certified negative for mycoplasma contamination.
Female C57BL/6 and BALB/c mice aged 6–8 weeks were purchased from the Experimental Animal Center (Beijing HFK Bioscience) and maintained in a specific pathogen-free facility. EGFP Tg/+ transgenic C57BL/6 mice were gifts from researchers at Sichuan University. CCR2 knockout mice on the C57BL/6J background (stock #004999; The Jackson Laboratory) were a gift from Prof. Hongxin Deng of Sichuan University. All procedures were approved by the Institutional Animal Care and Use Committee of Sichuan University.
Retrovirus production and CAR-T cell generation
The third-generation mouse EpCAM.28.BBz CAR was generated previously by our group.21 CAR-encoding retrovirus was produced via transfection of HEK293T cells with pMSCV-mEpCAM.28.BBz plasmid, and a pCL-Eco helper plasmid (#12371; Addgene) used HighGene transfection reagent (RM09014; ABclonal). Culture supernatant was collected 48 h after transfection and filtered using a 0.45 mm filter.
T cells were isolated from the lymph nodes of 6- to 8-week-old BALB/c mice, activated, cultured, and transduced as previously described.21 The transduction efficiency of mEpCAM.28.BBz CAR was detected using flow cytometry after staining with recombinant mouse EpCAM protein (CU95; Novoprotein) and anti-human IgG Fc (410722; BioLegend).
Tumor models and treatment
To establish mouse tumor models, 1 × 106 CT26, 4T1, or MC38 cells were injected s.c. into the right proximal hind legs of mice. Tumor volumes were measured every three days by calipers and calculated as length × width2 × 0.5. For the CT26 and MC38 models, HFRT (5 × 5 Gy) was performed on local tumors when the tumor volume reached ∼500 mm3. For the 4T1 model, HFRT was performed on local tumors when the tumor volume reached ∼150 mm3. Before irradiation, mice were anesthetized with pentobarbital (40 mg/kg) and shielded by a lead box with only the xenograft tumor exposed. RT was delivered by an X-ray generator (X-RAD160; Precision X-ray, North Branford, CT) with a dose rate of 1.922 Gy/min; the distance from the X-ray source to the target was 30 cm, and on the first day of RT was designated as day 1.
On day 5 after HFRT, a CXCR2 antagonist (5 mg/kg; SB225002; Tocris) was given by i.p. injection. Two days later, 3 × 106 EpCAM CAR+ or UTD cells were administered by i.v. injection. For ethical reasons, experiments were terminated when the tumor volume reached 2,000 mm3.
Flow cytometry
Tumor samples, blood, spleen, and bone marrow (nonirradiated side) were prepared into suspensions as previously reported.49,50 For surface staining, single-cell suspensions of blood, spleen, bone marrow, or tumor were incubated with FcR blocker and then stained with following antibodies: live/dead dye (32008-T; Invitrogen), FcR blocker (101335; BioLegend), PerCP/CY5.5-conjugated anti-CD45 (103132; BioLegend), fluorescein isothiocyanate (FITC)-conjugated anti-mouse CD3 (100204; BioLegend), PE-conjugated anti-mouse CD4 (100408; BioLegend), APC-conjugated anti-mouse CD8 (100712; BioLegend), mouse MDSC flow cocktail 1 with isotype control (147001; BioLegend), PE/CY7-conjugated anti-mouse PD-L1 (124314; BioLegend), PE/cy7-conjugated anti-mouse CCR2 (150612; BioLegend), PerCP/CY5.5-conjugvated anti-CXCR2 (149606; BioLegend), PE-conjugated anti-mouse CCR5 (107005; BioLegend), PE-conjugated anti-mouse CD49b (103506; BioLegend), APC-conjugated anti-mouse CD107a (121613; BioLegend), APC-conjugated anti-mouse CD11b (101211; BioLegend), FITC-conjugated anti-mouse Gr-1 (108407; BioLegend), PE-conjugated anti-mouse F4/80 (123110; BioLegend), FITC-conjugated anti-mouse MHC II (ant-270-a; BioLegend), PE/CY7-conjugated anti-mouse CD11c (117318; BioLegend), APC-conjugated anti-mouse CD25 (101910; BioLegend). All antibodies were used at the manufacturer’s recommended concentration. Isotype control antibodies were used as controls.
For intracellular cytokine staining, cells were fixed in 2% paraformaldehyde-PBS and permeabilized in permeabilization buffer (Invitrogen) according to the manufacturer’s protocol. Cells were stained with Alexa Fluor 488-conjugated anti-mouse Foxp3 (126405; BioLegend) and PE/CY7-conjugated anti-mouse CD206 (141720; BioLegend). Flow cytometry was performed on an ACEA NovoCyte instrument, and the data were analyzed using NovoExpress.
Cell tracking experiment
The day before RT, MDSCs were isolated from the bone marrow of EGFP Tg/+ transgenic C57BL/6 mice using an MDSCs Isolation Kit (130-094-538; Miltenyi Biotec), and T cells were isolated from the lymph nodes of EGFP Tg/+ transgenic C57BL/6 mice. A total of 1 × 106 isolated MDSCs or T cells were injected into mice bearing MC38 cells by i.v. when the tumor volume reached ∼500 mm3. On day 5 after RT initiation, tumor-infiltrating GFP+ MDSCs, GFP+ T cells, CD45+ cells, CD3+ T cells, and CD11b+ Gr-1+ MDSCs were detected using flow cytometry.
Multicolor immunofluorescence staining
The medical ethics committee of West China Hospital, Sichuan University, approved all patient samples for experimental use in this study. Tumor sections of 4 mm in thickness from mice or patients were fluorescently stained with Opal 7-Color Manual IHC Kit (NEL811001KT) according to the manufacturer’s description (primary antibodies: anti-mouse-CD3 [ab135372; Abcam], anti-mouse-CD8 [ab209775; Abcam], anti-mouse-CD11b [ab184308; Abcam], anti-mouse-Ly6G [ab25377; Abcam] and anti-human-CD3 [ab135372; Abcam], anti-human-CD8 [SI18-01; Huabio], anti-human-CD11b [EM1701-42; Huabio], and anti-human-CD33 [17425-1-AP; Proteintech]). The stained slides were scanned using Vectra 3.0.5 (PerkinElmer). Multispectral images were unmixed using spectral libraries that were built from images of single-stained tissues for each reagent using inform software (inForm 2.1.4; PerkinElmer). The images were analyzed using software (inForm 2.1.4, Halo version 3.6).
RNA-seq analysis
On day 7 after RT initiation, MDSCs were isolated from bone marrow, spleen, and tumors using a mouse MDSC isolation kit (130-094-538; Miltenyi Biotec). The efficiency of MDSCs purification was confirmed using flow cytometry. Then, 1 × 106 purified MDSCs were lysed with TRIzol, and total RNA was extracted. The RNA-seq library was prepared by the BGI in the core facility of microarray.
On days 7, 14, and 21 after RT, tumor bulk was extracted from mice and frozen in liquid nitrogen. The RNA-seq library was prepared by the BGI in the core facility of the microarray. Gene expression and deep sequencing analysis were conducted according to databases derived from the National Center for Biotechnology Information (NCBI) gene annotation system.
Multiplexed protein detection
On days 7 and 14 after HFRT initiation, total proteins were extracted from the serum and tumor tissue of CT26-bearing mice and tested using Ray-Biotech mouse protein array QAM-CHE-1 (Capital Bio, Beijing, China), according to the manufacturer’s description. The signals were visualized using a laser scanner equipped with a Cy3 wavelength and analyzed using Scan Array Express.
Quantification and statistical analysis
Survival was recorded as the number of days from tumor injection until an event. An event was defined as tumor-related death or euthanasia because of protocol-specified tumor burden (>2,000 mm3). Flow cytometric data were gated using NovoExpress 1.4.1. Data analyses were performed using Prism 8 (GraphPad Software), and all data are shown as the mean ± SEM. Significant differences were analyzed using the two-sided unpaired Student’s t test, one-way ANOVA, or the log rank test. A p value <0.05 was considered to indicate statistical significance.
Data availability
All data are available in the main text or the supplementary materials.
Acknowledgments
The authors thank Wenjie Yang for his kind help with statistical analysis, as well as Prof. Qijing Li of Duke University and Sichuan Bright-Tech Company for the professional and technical advice. This work was supported by the National Natural Science Foundation of China (grants 81872489, 82073369, and 8120175).
Author contributions
Y.W. conceptualized the idea of this study. B.Z. and M.H. designed the experiments. B.Z., M.H., Q.M., and K.L. performed most in vitro and in vivo experiments. B.Z. performed data analysis. X.L., X.H., P.S., Y.C., G.G., D.Q., and F.G. helped carry out the in vivo experiments. K.Z. and J.Z. helped with the interpretation of the data. X.W. and N.L. helped obtain and process clinical samples. M.F. and W.L. helped carry out the in vitro experiments. B.Z. and M.H. drafted the manuscript. D.L. and Y.W. revised and edited the manuscript. All authors read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2023.04.008.
Supplemental information
References
- 1.Maude S.L., Frey N., Shaw P.A., Aplenc R., Barrett D.M., Bunin N.J., Chew A., Gonzalez V.E., Zheng Z., Lacey S.F., et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014;371:1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang E., Gu J., Xu H. Prospects for chimeric antigen receptor-modified T cell therapy for solid tumors. Mol. Cancer. 2018;17:7. doi: 10.1186/s12943-018-0759-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kohli K., Pillarisetty V.G., Kim T.S. Key chemokines direct migration of immune cells in solid tumors. Cancer Gene Ther. 2022;29:10–21. doi: 10.1038/s41417-021-00303-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lesch S., Blumenberg V., Stoiber S., Gottschlich A., Ogonek J., Cadilha B.L., Dantes Z., Rataj F., Dorman K., Lutz J., et al. T cells armed with C-X-C chemokine receptor type 6 enhance adoptive cell therapy for pancreatic tumours. Nat. Biomed. Eng. 2021;5:1246–1260. doi: 10.1038/s41551-021-00737-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nalawade S.A., Shafer P., Bajgain P., McKenna M.K., Ali A., Kelly L., Joubert J., Gottschalk S., Watanabe N., Leen A., et al. Selectively targeting myeloid-derived suppressor cells through TRAIL receptor 2 to enhance the efficacy of CAR T cell therapy for treatment of breast cancer. J. Immunother. Cancer. 2021;9 doi: 10.1136/jitc-2021-003237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Formenti S.C., Demaria S. Systemic effects of local radiotherapy. Lancet Oncol. 2009;10:718–726. doi: 10.1016/s1470-2045(09)70082-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee Y., Auh S.L., Wang Y., Burnette B., Wang Y., Meng Y., Beckett M., Sharma R., Chin R., Tu T., et al. Therapeutic effects of ablative radiation on local tumor require CD8+ T cells: changing strategies for cancer treatment. Blood. 2009;114:589–595. doi: 10.1182/blood-2009-02-206870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brooks E.D., Chang J.Y. Time to abandon single-site irradiation for inducing abscopal effects. Nat. Rev. Clin. Oncol. 2019;16:123–135. doi: 10.1038/s41571-018-0119-7. [DOI] [PubMed] [Google Scholar]
- 9.Patel R.R., Verma V., Barsoumian H.B., Ning M.S., Chun S.G., Tang C., Chang J.Y., Lee P.P., Gandhi S., Balter P., et al. Use of multi-site radiation therapy for systemic disease control. Int. J. Radiat. Oncol. Biol. Phys. 2021;109:352–364. doi: 10.1016/j.ijrobp.2020.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dewan M.Z., Galloway A.E., Kawashima N., Dewyngaert J.K., Babb J.S., Formenti S.C., Demaria S. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin. Cancer Res. 2009;15:5379–5388. doi: 10.1158/1078-0432.CCR-09-0265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Van Limbergen E.J., De Ruysscher D.K., Olivo Pimentel V., Marcus D., Berbee M., Hoeben A., Rekers N., Theys J., Yaromina A., Dubois L.J., Lambin P. Combining radiotherapy with immunotherapy: the past, the present and the future. Br. J. Radiol. 2017;90 doi: 10.1259/bjr.20170157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nakamura N., Kusunoki Y., Akiyama M. Radiosensitivity of CD4 or CD8 positive human T-lymphocytes by an in vitro colony formation assay. Radiat. Res. 1990;123:224–227. [PubMed] [Google Scholar]
- 13.Arina A., Beckett M., Fernandez C., Zheng W., Pitroda S., Chmura S.J., Luke J.J., Forde M., Hou Y., Burnette B., et al. Tumor-reprogrammed resident T cells resist radiation to control tumors. Nat. Commun. 2019;10:3959. doi: 10.1038/s41467-019-11906-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crittenden M.R., Cottam B., Savage T., Nguyen C., Newell P., Gough M.J. Expression of NF-κB p50 in tumor stroma limits the control of tumors by radiation therapy. PLoS One. 2012;7 doi: 10.1371/journal.pone.0039295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grapin M., Richard C., Limagne E., Boidot R., Morgand V., Bertaut A., Derangere V., Laurent P.A., Thibaudin M., Fumet J.D., et al. Optimized fractionated radiotherapy with anti-PD-L1 and anti-TIGIT: a promising new combination. J. Immunother. Cancer. 2019;7:160. doi: 10.1186/s40425-019-0634-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee W.C., Wang Y.C., Cheng C.H., Wu T.H., Lee C.F., Wu T.J., Chou H.S., Chan K.M. Myeloid-derived suppressor cells in the patients with liver resection for hepatitis B virus-related hepatocellular carcinoma. Sci. Rep. 2019;9:2269. doi: 10.1038/s41598-019-38785-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Donkor M.K., Lahue E., Hoke T.A., Shafer L.R., Coskun U., Solheim J.C., Gulen D., Bishay J., Talmadge J.E. Mammary tumor heterogeneity in the expansion of myeloid-derived suppressor cells. Int. Immunopharmacol. 2009;9:937–948. doi: 10.1016/j.intimp.2009.03.021. [DOI] [PubMed] [Google Scholar]
- 18.Zhang T., Yu H., Ni C., Zhang T., Liu L., Lv Q., Zhang Z., Wang Z., Wu D., Wu P., et al. Hypofractionated stereotactic radiation therapy activates the peripheral immune response in operable stage I non-small-cell lung cancer. Sci. Rep. 2017;7:4866. doi: 10.1038/s41598-017-04978-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Noman M.Z., Desantis G., Janji B., Hasmim M., Karray S., Dessen P., Bronte V., Chouaib S. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014;211:781–790. doi: 10.1084/jem.20131916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Strauss L., Mahmoud M.A.A., Weaver J.D., Tijaro-Ovalle N.M., Christofides A., Wang Q., Pal R., Yuan M., Asara J., Patsoukis N., Boussiotis V.A. Targeted deletion of PD-1 in myeloid cells induces antitumor immunity. Sci. Immunol. 2020;5 doi: 10.1126/sciimmunol.aay1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qin D., Li D., Zhang B., Chen Y., Liao X., Li X., Alexander P.B., Wang Y., Li Q.J. Potential lung attack and lethality generated by EpCAM-specific CAR-T cells in immunocompetent mouse models. Oncoimmunology. 2020;9 doi: 10.1080/2162402x.2020.1806009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Enblad G., Karlsson H., Gammelgård G., Wenthe J., Lövgren T., Amini R.M., Wikstrom K.I., Essand M., Savoldo B., Hallböök H., et al. A phase I/IIa trial using CD19-targeted third-generation CAR T cells for lymphoma and leukemia. Clin. Cancer Res. 2018;24:6185–6194. doi: 10.1158/1078-0432.Ccr-18-0426. [DOI] [PubMed] [Google Scholar]
- 23.Böttcher J.P., Bonavita E., Chakravarty P., Blees H., Cabeza-Cabrerizo M., Sammicheli S., Rogers N.C., Sahai E., Zelenay S., Reis e Sousa C. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell. 2018;172:1022–1037.e1014. doi: 10.1016/j.cell.2018.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Najjar Y.G., Rayman P., Jia X., Pavicic P.G., Jr., Rini B.I., Tannenbaum C., Ko J., Haywood S., Cohen P., Hamilton T., et al. Myeloid-derived suppressor cell subset accumulation in renal cell carcinoma parenchyma is associated with intratumoral expression of IL1β, IL8, CXCL5, and mip-1α. Clin. Cancer Res. 2017;23:2346–2355. doi: 10.1158/1078-0432.CCR-15-1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen J.Y., Lai Y.S., Chu P.Y., Chan S.H., Wang L.H., Hung W.C. Cancer-derived VEGF-C increases chemokine production in lymphatic endothelial cells to promote CXCR2-dependent cancer invasion and MDSC recruitment. Cancers (Basel) 2019;11 doi: 10.3390/cancers11081120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu P., Zhang F., Chang M.M., Zhong C., Sun C.H., Zhu H.R., Yao J.C., Li Z.Z., Li S.T., Zhang W.C., Sun G.D. Recruitment of γδ T cells to the lesion via the CCL2/CCR2 signaling after spinal cord injury. J. Neuroinflammation. 2021;18:64. doi: 10.1186/s12974-021-02115-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fei L., Ren X., Yu H., Zhan Y. Targeting the CCL2/CCR2 Axis in cancer immunotherapy: one stone, three birds? Front. Immunol. 2021;12 doi: 10.3389/fimmu.2021.771210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carr M.W., Roth S.J., Luther E., Rose S.S., Springer T.A. Monocyte chemoattractant protein 1 acts as a T-lymphocyte chemoattractant. Proc. Natl. Acad. Sci. USA. 1994;91:3652–3656. doi: 10.1073/pnas.91.9.3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moon E.K., Carpenito C., Sun J., Wang L.C.S., Kapoor V., Predina J., Powell D.J., Jr., Riley J.L., June C.H., Albelda S.M. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin. Cancer Res. 2011;17:4719–4730. doi: 10.1158/1078-0432.Ccr-11-0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Minn I., Rowe S.P., Pomper M.G. Enhancing CAR T-cell therapy through cellular imaging and radiotherapy. Lancet Oncol. 2019;20:e443–e451. doi: 10.1016/s1470-2045(19)30461-9. [DOI] [PubMed] [Google Scholar]
- 31.Demaria S., Guha C., Schoenfeld J., Morris Z., Monjazeb A., Sikora A., Crittenden M., Shiao S., Khleif S., Gupta S., et al. Radiation dose and fraction in immunotherapy: one-size regimen does not fit all settings, so how does one choose? J. Immunother. Cancer. 2021;9 doi: 10.1136/jitc-2020-002038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Darragh L.B., Oweida A.J., Karam S.D. Overcoming resistance to combination radiation-immunotherapy: a focus on contributing pathways within the tumor microenvironment. Front. Immunol. 2018;9:3154. doi: 10.3389/fimmu.2018.03154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jiménez-Cortegana C., Galassi C., Klapp V., Gabrilovich D.I., Galluzzi L. Myeloid-derived suppressor cells and radiotherapy. Cancer Immunol. Res. 2022;10:545–557. doi: 10.1158/2326-6066.CIR-21-1105. [DOI] [PubMed] [Google Scholar]
- 34.Filatenkov A., Baker J., Mueller A.M.S., Kenkel J., Ahn G.O., Dutt S., Zhang N., Kohrt H., Jensen K., Dejbakhsh-Jones S., et al. Ablative tumor radiation can change the tumor immune cell microenvironment to induce durable complete remissions. Clin. Cancer Res. 2015;21:3727–3739. doi: 10.1158/1078-0432.Ccr-14-2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liang H., Deng L., Hou Y., Meng X., Huang X., Rao E., Zheng W., Mauceri H., Mack M., Xu M., et al. Host STING-dependent MDSC mobilization drives extrinsic radiation resistance. Nat. Commun. 2017;8:1736. doi: 10.1038/s41467-017-01566-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang X., Lu Y., Hang J., Zhang J., Zhang T., Huo Y., Liu J., Lai S., Luo D., Wang L., et al. Lactate-modulated immunosuppression of myeloid-derived suppressor cells contributes to the radioresistance of pancreatic cancer. Cancer Immunol. Res. 2020;8:1440–1451. doi: 10.1158/2326-6066.Cir-20-0111. [DOI] [PubMed] [Google Scholar]
- 37.Marvel D., Gabrilovich D.I. Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected. J. Clin. Invest. 2015;125:3356–3364. doi: 10.1172/jci80005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Karin N., Razon H. The role of CCR5 in directing the mobilization and biological function of CD11b(+)Gr1(+)Ly6C(low) polymorphonuclear myeloid cells in cancer. Cancer Immunol. Immunother. 2018;67:1949–1953. doi: 10.1007/s00262-018-2245-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Highfill S.L., Cui Y., Giles A.J., Smith J.P., Zhang H., Morse E., Kaplan R.N., Mackall C.L. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci. Transl. Med. 2014;6:237ra67. doi: 10.1126/scitranslmed.3007974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blattner C., Fleming V., Weber R., Himmelhan B., Altevogt P., Gebhardt C., Schulze T.J., Razon H., Hawila E., Wildbaum G., et al. CCR5(+) myeloid-derived suppressor cells are enriched and activated in melanoma lesions. Cancer Res. 2018;78:157–167. doi: 10.1158/0008-5472.Can-17-0348. [DOI] [PubMed] [Google Scholar]
- 41.Spranger S., Bao R., Gajewski T.F. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature. 2015;523:231–235. doi: 10.1038/nature14404. [DOI] [PubMed] [Google Scholar]
- 42.Li K., Shi H., Zhang B., Ou X., Ma Q., Chen Y., Shu P., Li D., Wang Y. Myeloid-derived suppressor cells as immunosuppressive regulators and therapeutic targets in cancer. Signal Transduct. Target. Ther. 2021;6:362. doi: 10.1038/s41392-021-00670-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Foeng J., Comerford I., McColl S.R. Harnessing the chemokine system to home CAR-T cells into solid tumors. Cell Rep. Med. 2022;3 doi: 10.1016/j.xcrm.2022.100543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li C.X., Ling C.C., Shao Y., Xu A., Li X.C., Ng K.T.P., Liu X.B., Ma Y.Y., Qi X., Liu H., et al. CXCL10/CXCR3 signaling mobilized-regulatory T cells promote liver tumor recurrence after transplantation. J. Hepatol. 2016;65:944–952. doi: 10.1016/j.jhep.2016.05.032. [DOI] [PubMed] [Google Scholar]
- 45.Smith E.L., Mailankody S., Staehr M., Wang X., Senechal B., Purdon T.J., Daniyan A.F., Geyer M.B., Goldberg A.D., Mead E., et al. BCMA-targeted CAR T-cell therapy plus radiotherapy for the treatment of refractory myeloma reveals potential synergy. Cancer Immunol. Res. 2019;7:1047–1053. doi: 10.1158/2326-6066.Cir-18-0551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vanpouille-Box C., Alard A., Aryankalayil M.J., Sarfraz Y., Diamond J.M., Schneider R.J., Inghirami G., Coleman C.N., Formenti S.C., Demaria S. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat. Commun. 2017;8:15618. doi: 10.1038/ncomms15618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.North R.J. Cyclophosphamide-facilitated adoptive immunotherapy of an established tumor depends on elimination of tumor-induced suppressor T cells. J. Exp. Med. 1982;155:1063–1074. doi: 10.1084/jem.155.4.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Klebanoff C.A., Khong H.T., Antony P.A., Palmer D.C., Restifo N.P. Sinks, suppressors and antigen presenters: how lymphodepletion enhances T cell-mediated tumor immunotherapy. Trends Immunol. 2005;26:111–117. doi: 10.1016/j.it.2004.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang H., Lin X., Luo Y., Sun S., Tian X., Sun Y., Zhang S., Chen J., Zhang J., Liu X., et al. α-PD-L1 mAb enhances the abscopal effect of hypo-fractionated radiation by attenuating PD-L1 expression and inducing CD8+ T-cell infiltration. Immunotherapy. 2019;11:101–118. doi: 10.2217/imt-2018-0049. [DOI] [PubMed] [Google Scholar]
- 50.Henrich S.E., McMahon K.M., Plebanek M.P., Calvert A.E., Feliciano T.J., Parrish S., Tavora F., Mega A., De Souza A., Carneiro B.A., Thaxton C.S. Prostate cancer extracellular vesicles mediate intercellular communication with bone marrow cells and promote metastasis in a cholesterol-dependent manner. J. Extracell. Vesicles. 2020;10:e12042. doi: 10.1002/jev2.12042. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available in the main text or the supplementary materials.