

Abstract
Glioblastoma (GBM), the most prevalent brain tumor, is one of the least treatable malignancies due to its propensity for intracranial spread, high proliferative potential, and innate resistance to radiation and chemotherapy. Current GBM therapy is limited due to unfavorable, non-specific therapeutic effects in healthy cells and the difficulty of small molecules to penetrate the blood brain barrier (BBB) and reach the tumor microenvironment. Adding PARP-1 inhibitors inhibit DNA repair enzymes thereby increasing the cytotoxicity of anticancer agents. Hence, we aimed to discover potential naturally occurring PARP-1 inhibitors that can be utilized in the treatment of glioma by using multiple in silico tools like molecular docking, absorption, distribution, metabolism, and excretion (ADME) profile, pharmacophore modeling, and molecular dynamic (MD) simulations. Among 43 phytocompounds we screened, two of them (Ellagic acid and Naringin) were discovered to be bound to the catalytic site of PARP-1 with an affinity more remarkable than commercially available PARP-1 inhibitors (Talazoparib, Niraparib, and Rucaparib) except Olaparib. The molecular interactions were analyzed, and data shows that bound entity attained a conserved domain via hydrogen bond interactions, polar interactions, and π-π stacking. Pharmacophore modeling studies showed electronic and steric features of ligands responsible for supramolecular interaction with PARP-1. ADME properties were studied, to assess drug-likeness, hydrophilic nature, hydrophobicity, brain permeability, and oral bioavailability of the natural PARP-1 inhibitors. The simulation study demonstrated the development of a stable complex between Naringin, Ellagic acid, and PARP-1 protein. Moreover, cell culture studies and animal investigations are essential to determine pharmacokinetics and pharmacodynamics.
Keywords: PARP inhibitors, Cancer, Phytomedicines, In silico, Pharmacophore, Molecular Dynamic Simulation
1. Introduction
Cancer is one of the most devastating disease, and it is still the leading cause of mortality worldwide. (Siegel et al., 2018). Increased understanding of the molecular processes underlying cancer progression has resulted in a profusion of anticancer drugs (Siddiqui et al., 2022). Even then, cancer chemotherapy faces principal disadvantages: cancer recurrence, drug resistance, and harmful effects on normal healthy cells; all of these factors may limit the use of chemotherapeutic drugs and hence diminish the life expectancy of cancer patients (X. Wang et al., 2019). To address the shortcomings of current therapies, the pursuit of novel, prospective anticancer drugs with higher potency and fewer adverse effects continues.
Phytomedicines, naturally occurring plant molecules, act as crucial elements for breakthrough drugs and as alternatives for cancer care. They act on various molecular signal pathways, including protein kinases, downstream tumor suppressors, transcriptional factors, cyclin, caspases, micro RNAs and other molecular targets (Choudhari et al., 2019). New techniques and innovative chemopreventive medicines are required to increase the efficacy of current cancer therapies (Dias et al., 2012).
Malignant glioma, is the deadliest form of adult brain tumor. The conventional treatment is maximal safe resection preceded by adjuvant radiotherapy and oral temozolomide, adds patients' lives by 16 to 18 months (Wen et al., 2020). The tumor microenvironment has numerous biological and physical barriers that make it challenging to treat GBM successfully and pose the risk of recurrence even after following the recommended treatment plan (Cha et al., 2020). Many drugs cannot be used in glioma patients due to poor physicochemical qualities, lack of targeting capability, and inability to penetrate the BBB and blood–brain tumor barrier. Some of these shortcomings can be overcome by targeted formulation approaches using nanotechnology.
PARP is an intriguing target in GBM because it is a DNA repair protein essential for the nucleotide or base excision repair of cellular DNA damage, including chemotherapy-induced DNA breaks. (Morales et al., 2014). PARP-1, the most typical PARP member, has DNA-binding, self-modifying, and catalytic areas. The catalytic domain transfers ADP-ribose from NAD+ to the substrate protein. PARP-1 decreases NAD+ and utilizes intracellular ATP after DNA damage. ATP depletion causes cell impairment and necrosis. PARP-1 helps DNA cell repair and survival, as shown by many PARP-1 knockout mouse experiments (Lu et al., 2022, Zhou et al., 2019a). PARP-1 catalyzes the PARylation process and plays a vital role in regulating chromatin shape and promoting DNA repair, among other functions. Glioma cells with isocitrate dehydrogenase (IDH) mutations were eliminated by PARP-1 inhibitors, but not cells with normal IDH. In addition, a PARP-1 inhibitor increased the toxicity of chemotherapy on IDH-mutant cells. Adding PARP-1 inhibitors or inhibiting DNA repair enzymes augments the cytotoxicity of genotoxic treatments (Zhang et al., 2020). Olaparib, Rucaparib, Niraparib, and Talazoparib are FDA- and EMA-approved PARP-1 inhibitors (Slade, 2020). Many times, PARP-1 inhibitors have been used in combination with DNA-damaging treatments such as TMZ, topoisomerase inhibitors, and radiation. These agents promote PARP-1 activity for the repair of DNA damage, which makes tumor cells more sensitive to the effects of the DNA-damaging agents (Chen, 2011, Javle and Curtin, 2011). Small molecule PARP-1 inhibitors Veliparib, Talazoparib and Niraparib are currently being tested in combination with temozolomide (TMZ) to treat primary Glioblastoma (Bai et al., 2011). However, the adverse events (C. Wang & Li, 2021) of PARP-1 inhibitors and the development of resistance to medications with an extended treatment of existing treatment, points to the necessity of discovering new naturally occurring PARP-1 inhibitors against glioma.
By taking into account the anticancer effects of phytoconstituents, the present study advocated visually screening the finest phytomedicines that can function as effective PARP-1 inhibitors by applying computational procedures. There is an urgent requirement for novel PARP-1 inhibitors with high potency and favorable pharmacodynamic and pharmacokinetic profiles. Until now, only a small number of publications have attempted to model ligand interactions with the PARP-1 receptor employing simulation or pharmacophore techniques (Revathi et al., 2021, Zhou et al., 2019a). The Naturally Occurring Plant Based Anticancerous compound Activity Target (NPACT) database comprises 1574 entries that detail the structure, physical, elemental, and topological properties of compounds, as well as their in vitro and in vivo biological activity, cancer type, cell lines, inhibitory values, molecular targets, commercial suppliers, and drug likeness. Additionally it represents protein targets that have been proven to be inhibited by phytomedicines in cancer cell lines (Mangal et al., 2013). We tested the PARP-1 inhibitory activity of phytomedicines against 5DS3 protein (crystal structure of constitutively active PARP-1) and specifically performed structure-based in silico screening of 43 phyto-ligands. In addition, we performed MD simulation for the identified lead hits to validate the stability of the complexes over 200 ns of MD. As a result, it is able to undergo additional processing for use in pharmacological research to treat glioblastoma.
2. Materials and methods
2.1. Phytoconstituent data
Phytoconstituents such as alkaloids, flavonoids, polyphenols, terpenoids, saponins, lignans, steroids, polyketides, and other organic chemicals were collected from research articles and (NPACT) database (https://crdd.osdd.net/raghava/npact), and all these drugs are reported for anticancer activities, and structures are illustrated in Table 1.
Table 1.
List of Flavonoids, Alkaloids, Polyphenols, Terpenoids, Saponins, Lignans, Steroids, Polyketides, organic chemicals, and FDA-approved PARP-1 inhibitors.
| Flavonoids |  |
| Alkaloids |  |
| Polyphenols |  |
| Terpenoids |  |
| Saponins | 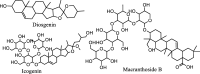 |
| Lignans |  |
| Steroids |  |
| Polyketides | |
| Organic chemicals |  |
| FDA approved PARP inhibitors |
 |
2.2. Platform for in silico studies
To predict in silico AMDE profile, pharmacophore model, and drug-receptor interactions were performed using Schrödinger application the Maestro V-12.3, programmed on a LENOVO Inc.17″ workstation machine running on Intel core i-7 processor with 3.60 GHz of clock speed with octa-core, a processor with 8 GB random access memory and 1024 Gigabyte hard drive with Linux -x86 64 bit as the OS (operating system) was utilized. The MD simulation studies were performed on Dell Prec. 3650 tower with the 11th generation Intel core i-7–11700 16 MB Cache octacore 2.5 GHz to 4.9 GHz clock speed 32 GB (2x16) RAM DDR4, 1 TB PCIe Gen4 M.2 Solid State Drive with 12 GB NVIDIARTX A2000 Graphic Card.
2.3. Molecular docking interactions study and MM-GBSA analysis
Using Chem sketch, a diagram of the two-dimensional structure of each ligand molecule was created. In order to convert the 2D molecular structures into 3D, Ligprep was utilized in this process, that is a part of the Schrodinger. SMILES were produced, structures were imported, and most stable conformation of ligand were considered for study. From the Research Collaboratory for Structural Bioinformatics (RCSB) protein data library, the X-ray structure of crystal PARP-1 with the protein data bank identification number of 5DS3 (R-Value Observed: 0.202, R-Value Free: 0.251, Resolution: 2.60 Å, R-Value Work: 0.200) was obtained and analyzed (Dawicki-McKenna et al., 2015). The preparation of PARP-1 was accomplished with the assistance of Schrodinger's protein preparation wizard. The procedure for the protein preparation concluded with a minimization that had been refined, optimized, and constrained. Schrodinger's Receptor Grid Generation modules were utilized for the process that is typically utilized to generate grids. During grid creation, the active site of the protein and the most favorable interaction between ligand molecules are primarily identified. After that, these ligands were docked with the help of Glide's XP (extra precision) scoring tools. The binding energy (MM-GBSA) of the ligand-receptor complex was estimated using Schrodinger's Prime module.
2.4. ADME and physicochemical properties
Since computational technology has decreased the number of experimental drug trials and increased the success rate, it has become an indispensable instrument for drug candidate identification. The Schrodinger program's QikProp module was utilized to estimate the ligand molecules' PK(Pharmacokinetic) properties as well as ADME. The same software predicted ligand molecule’s physicochemical or drug-likeness properties. Drug likeness properties analyzed are molecular weight, Donor Hb, Dipole, QP log o/w, and Acceptor HB calculation. Lipinski's rule of five was used to make predictions about the potential drug-likeness of the compounds.
2.5. Generation of pharmacophore modeling
The Phase add-on to Schrodinger program was used to create the models in this investigation. A receptor-based pharmacophore was employed in the procedure, and it describes the spatial configuration of molecules required for the bioactivity of the ligands being seen with the targeted receptor (Kaserer et al., 2015).
2.6. Molecular dynamic simulation
The stability of the best-docked interaction for ellagic acid and naringin with 5DS3 was assessed by an all-atom 100 ns MD simulation on Gromacs version 2021.6. Protein was stabilized, and its topology was generated via charmm36 forcefield using the pdb2gmx module of gromacs. The proteins were solvated using 3-point water model by using a dodecahedron box with 1 nm dimensions on all sides. Further, sodium and chloride ions were used to stabilize the system. To get the least energy confirmation, energy reduction was carried out utilizing a steepest descent integrator with a verlet cutoff scheme for a maximum of 50,000 steps. Isobaric (NPT) and Canonical (NVT) were used to equilibrate the system for 100 ps. Constant temperature and volume at 300 K was achieved by integrating V-rescale thermostat. Similarly, a constant pressure of 1 bar was maintained via C-rescale coupling algorithm. Computing coulomb, van der Waals, and long-range electrostatic interactions required the use of the particle mesh Ewald approximation, with a cut-off value of 1 nm; bond length was constrained by LINCS algorithm. Built-in gromacs utilities were used to examine the obtained trajectories. On completion of molecular dynamic run, Root mean square fluctuation (RMSF), Root mean square deviation (RMSD), Radius of gyration (RoG), Solvent assessable surface area (SASA), and number of H- bonds were analyzed. In addition, DSV (Discovery Studio Visualizer) was used to visualize the complex at the start and end of the MD run (Berendsen et al., 1995, Dwivedi et al., 2021, Khanal et al., 2022).
2.7. MM-PBSA analysis
The gmx_MMPBSA module was used to analyze the energy contribution parameters like Vander Waals, total relative binding, electrostatic molecular mechanics, and total energy contribution per residue.
A total of 1,000 frames were used for the MMPBSA run, with a 10-frame interval. A PBSA internal solver in a sander was used to investigate the Poisson Boltzmann computations. The gmx MMPBSA run results were visualized using the MMPBSA ana module. (Valdés-Tresanco et al., 2021).
3. Results
3.1. Molecular docking interactions study and MM-GBSA analysis
Docking interactions of natural and FDA-approved PARP-1 inhibitors were illustrated in Table 2 & Supplementary Sheet 1 as well as Fig. 1 & Supplementary Sheets 2&3. More details have been provided in Supplementary Sheet 4. Polyphenols:- Ellagic acid and flavonoid-Naringin showed better PARP inhibitory activity than the FDA-approved PARP-1 inhibitors (Talazoparib, Niraparib, and Rucaparib) except Olaparib. Ellagic acid showed excellent binding interactions with PARP-1 (5DS3) with a docking score of −11.024 kcal/mol. Amino acids such as GLU988, GLY888, and GLY863 showed hydrogen bonding interaction. Other amino acids (TYR896, PHE 897, ALA898, TYR889, MET 890, PHE891, TYR907) were involved in the PARP-1 inhibition (5DS3), indicating lipophilic interaction with the ligand and π-π bonding with TYR 907. While the amino acid complexes (SER 904, SER 864, HIS 862) of the enzyme PARP-1inhibitors (5DS3) showed polar interaction with ligands. Naringin showed better affinity towards 5DS3 protein, having a bound score of −10.793 kcal/mol. This molecule developed H- bond interaction with (TYR896, GLY894, ARG878, and SER 904) amino acid complexes of PARP-1 inhibitor protein. In addition, lipophilic interaction (TYR 907, TYR 889, ALA 898, TRP861, TYR896, ILE895, LEU877, ILE879, ALA880, PHE897, and ILE872), polar interaction (SER904, HIS862, ASN868), pi- pi stacking (TYR907) were observed. Moreover, FDA-approved PARP-1 inhibitors showed significant activity toward the 5DS3 protein. Apart from the four different FDA-approved PARP-1 inhibitors, Olaparib showed the highest affinity with a binding score of −10.545 kcal/mol and established H-bonding interaction with (SER 904, GLY 863) amino acid residues of PARP-1 protein. It is found that hydrophobic interactions (TYR 989, TYR 907, ALA898, TRP 861, TYR896, TYR889, PHE897, and ILE895), polar interactions (SER 864, HIS 862, SER 904) and Pi-Pi stacking (TYR907) with amino complexes of 5DS3 protein. Among the 43 natural PARP-1 inhibitors screened, the top two leads were selected for further studies (pharmacophore generation and simulation studies).
Table 2.
Molecular docking scores of natural and FDA-approved PARP-1 inhibitors.
| Sl No. | Drugs | Glide Score | Glide EvdW | Glide e Model | Glide ecoul | G rotatable bonds | XP Hbond |
|---|---|---|---|---|---|---|---|
| 1 | Ellagic acid | −11.024 | −37.147 | −81.446 | −17.073 | 4 | −4.443 |
| 2 | Naringin | −10.793 | −40.984 | −80.325 | −18.185 | 14 | −1.859 |
| 3 | Icogenin | −10.232 | −36.886 | −74.941 | −24.513 | 23 | −5.822 |
| 4 | EGCG | −10.072 | −49.769 | −91.458 | −17.546 | 12 | −3.904 |
| 5 | Silymarin | −10.051 | −48.812 | −85.121 | −9.151 | 9 | −2.683 |
| 6 | Macranthoside B | −10.048 | −42.519 | −47.046 | −10.721 | 25 | −2.673 |
| 7 | Curcumin | −8.839 | −45.529 | −80.529 | −9.703 | 10 | −3.32 |
| 8 | Quercetin | −8.819 | −31.717 | −63.237 | −14.093 | 6 | −2.488 |
| 9 | Poncirin | −8.789 | −40.697 | −74.371 | −15.784 | 14 | −2.4 |
| 10 | Baicalein | −8.762 | −34.268 | −61.18 | −13.675 | 4 | −2.835 |
| Molecular docking scores of FDA-approved PARP-1 inhibitors | |||||||
| 1 | Olaparib | −12.840 | −53.005 | −105.69 | −13.940 | 6 | −2.800 |
| 2 | Talazoparib | −10.545 | −40.674 | −64.57 | −8.531 | 2 | −1.761 |
| 3 | Niraparib | −7.911 | −37.699 | −76.205 | −12.253 | 3 | −1.89 |
| 4 | Rucaparib | −6.678 | −41.864 | −60.681 | −1.309 | 3 | −0.554 |
Fig. 1.

(A& B) 2D and 3D interactions of Ellagic acid with 5DS3. (C & D) 2D and 3D interactions of Naringin with 5DS3.
The (MM-GBSA) binding free energy of both natural and FDA-approved PARP inhibitors towards 5DS3 protein were listed in Supplementary sheet 5. Natural PARP-1 inhibitors have ΔGbind values ranging from −108.62 to −42.24 kcal/mol for 5DS3 proteins. With FDA-approved PARP-1 drugs, the ΔGbind ranges from −55.5 to −106.99 kcal/mol for the relevant protein 5DS3. It has been found that the primary donors to the interactions of natural PARP-1 inhibitors with 5DS3 are (ΔGLipo (non-polar solvation), −14.22 to −78.35 kcal/mol and ΔGvdW (van der Waals); −25.42 to −63.43 kcal/mol). Because of the significant negative values produced by all-natural PARP-1 inhibitors in the MM-GBSA experiment, the energies that affect ligand binding in the binding pocket of 5DS3 are ΔG vdW and ΔG Lipo. Other energies, such as covalent energy (ΔG Cov) and (ΔG bind H bond), should not prefer receptor binding. The ΔG Coul moderately favors both natural and FDA-approved PARP-1 inhibitors.
3.2. Generation of pharmacophore modeling
Ellagic acid and naringin, two naturally occurring compounds that inhibit PARP-1 through distinct chemical interactions, were the focus of pharmacophore modeling analyses to decipher the molecular mechanisms behind their PARP-1inhibiting effects. The pharmacophore elucidates the crucial characteristics accountable for biological functions, proving the H-bonding interaction to be the most important property (Fig. 2 and Supplementary sheet 6). The receptor-based pharmacophore hypothesis of Ellagic acid with 5DS3 (Fig. 2A) consists of two aromatic rings (R14 and R15) and four donor groups (D9, D10, D11, and D12). D11 develops an H-bond with amino acids GLU988,
Fig. 2.

Pharmacophore model complex of A) Ellagic acid and B) Naringin with 5DS3.
D9, and D10 with GLY863, and D12 with GLY888, respectively. In the pharmacophore hypothesis of Naringin-5DS3, one aromatic ring (R25) and four donors (D15, D17, D18, D19, and D21) are present, in which D15 hydrogen bonds with amino acid SER904, D17 had an H bond interaction with TYR896 and GLY894, and finally, three different donors (D18, D19, D21) made H bond interaction with a single amino acid ARG878 (Fig. 2B). Pharmacophore properties of FDA-approved PARP-1 inhibitors are described in (Supplementary sheets 6 and 7).
3.3. Physicochemical properties of both natural and FDA-approved PARP-1 inhibitors
QikProp scrutinized the drug-likeness properties of both natural and FDA-approved PARP-1 inhibitors, which assessed the molecular weight of all the natural PARP-1 inhibitors. Ellagic acid and Naringin levels are within the usual range. The molecular weights of Icogenin and Macranthoside B exceed the allowed range. Supplementary sheet 8 illustrates the physicochemical properties of natural and FDA-approved PARP inhibitors. Naringin, Icogenin, EGCG, Macranthoside B, and Poncirin have hydrogen donors above five, and Naringin, Icogenin, Macranthoside B, and Poncirin have hydrogen acceptors above ten. In addition, analysis of QPlog o/w predicts the lipophilicity of the natural PARP-1 inhibitors. The n-octanol/water partition coefficient measures lipophilicity. The lipophilicity of substances affects membrane permeability. Lipophilicity can reduce it, while hydrophilic substances can't diffuse passively. Squamocin and garcinol are beyond the allowed range (-2.0 to 6.5), but other drugs are within.
3.4. ADME studies
Ellagic acid and Naringin showed poor permeability and human oral absorption. Supplementary sheet 9 demonstrated the ADME properties of natural and FDA-approved PARP-1 inhibitors. QPlogBB assessed the access to the CNS. As per the recommended range (-3 to 1.2), Ellagic acid and other natural PARP inhibitors can penetrate the blood–brain barrier except for Naringin, Icogenin, EGCG, Macranthoside B, Ponicirin, and Narirutin, respectively. All FDA-approved drugs are within the limit. QikProp used a scale from −2 (inactive) to + 2 (active) to foretell the state of the central nervous system. In this study, values indicated that both natural PARP-1 and FDA-approved drugs are normal in range. QikProp analyzed the total number of metabolic reactions of each natural PARP-1 inhibitors and FDA-approved drugs. Ellagic acid’s metabolic reactions rate is within the recommended limit (1–8) except for Narinigin, Icogenin, EGCG, Macranthoside B, Ponicirin, Narirutin, Garcinol, and Cepharanthine. The SASA, FISA, and FOSA values are shown in Supplementary sheet 9. Icogenin, Macranthoside B, and Squamocin were the only phytocompounds with SASA values above 1000 Å; all others fell below the normal limits. Icogenin and Squamocin have FOSA values greater than 750, indicating their hydrophobic portion of SASA (solvent-accessible surface area) is greater. Antofine and Cepharanthine exhibit scores under seven, indicating the presence of a portion of SASA that is less hydrophilic (FISA). Naringin, Icogenin, Macranthoside B, and Narirutin all received scores greater than 330, indicating a stronger aqueous component of solvent accessible surface area.
3.5. Molecular dynamic simulation
3.5.1. Complex of Ellagic acid with 5DS3
The RMSD of complex displayed a maximum fluctuation value upto ∼ 8.8 Å; the RMSD of the complex showed deviations less than 0.5 Å indicating the stability of the complex throughout the run. The RMSD was stable after 20 ns of stabilization time and thereafter displayed deviations less than 0.3 Å. The RMSF of the c-alpha atoms displayed a fluctuation in the range of ∼ 0.5 Å to ∼ 5.5 Å, and residues glycine 784 and glutamic acid 883 showed the highest RMSF of 5.3 Å and 4.8 Å respectively; identical with the residues possessing maximum fluctuation in olaparib-5DS3 complex (Supplementary sheet 10; Movie 3). This indicates the interaction of ellagic acid with PARP-1 to be a stable complex and interaction is at the active site of the protein. The Radius of Gyration (RoG) for the backbone and complex showed fluctuation in the range of ∼ 17.7 Å to ∼ 18.3 Å with minor deviations indicating the compactness of the protein throughout the run. The SASA showed fluctuation in the range of ∼ 123 nm2 to ∼ 139 nm2, which was unstable for the initial ∼ 25 ns of md run and thereafter a gradual increase in SASA was observed; this may be due to the number of bonds being formed and deformed during the MD run. A highest of 7 hydrogen bonds were seen throughout the MD run in which a minimum of 2 bonds were constant; however, formation and deformation of hydrogen bonds was seen throughout the run. The total energy decomposition per residue showed tyrosine 907 and 896 to possess the minimum energy contribution of −1.80 and −1.07 kcal/mol, respectively. The total energy contribution of the ligand was found to be −4.98 kcal/mol for the 100 ns of MD run (Fig. 3, Movie 1).
Fig. 3.

Represents the stability of the complex (Ellagic acid-5DS3) with respect to, (a) Root Mean Square Deviation (RMSD) of the complex, (b) Root Mean Square Fluctuation (RMSF) of the c-alpha atoms, (c)Radius of Gyration (RoG) of the backbone (black) and complex (red), (d) Solvent Assessable Surface Area (SASA) for the protein(black) and complex (red) (e) Number of Hydrogen bonds, (f) Total energy decomposition per residue.
3.5.2. Complex of Naringin with 5DS3
The RMSD of the complex showed a maximum fluctuation of ∼ 0.5 Å within the range ∼ 11.5 Å to 12.0 Å throughout the MD run of 200 ns indicating the complex stability. The RMSD of the complex was seen to stabilize after ∼ 20 ns of MD run and thereafter showed RMSD deviations of less than 0.25 Å. The RMSF showed fluctuation in the range of ∼ 1Å to ∼ 7 Å; the residues glycine 784 and serine 783 showed maximum RMSF of 7.0 Å and 6.9 Å respectively, which is identical with the standard olaparib-5DS3 complex (Supplementary sheet 10; Movie 3). The Radius of Gyration (RoG) for the complex and backbone showed fluctuation in the range of ∼ 17.5 Å to ∼ 18.3 Å; the RoG showed deviations for the initial ∼ 30 ns of md run which thereafter was seen to be stable with deviations less than ∼ 0.5Å, this suggests that the docked complex is stable and that the protein is compact. The SASA showed fluctuation in the range of ∼ 123 nm2 to ∼ 140 nm2, which was unstable for the initial 20 ns, and thereafter gradual increase in the SASA was showed with minor fluctuation after ∼ 60 ns of simulation. A maximum of 7 hydrogen bonds were shown throughout the run; initially, for ∼ 40 ns the bonds were not stable and gained stability after ∼ 60 ns of MD run. A minimum of 2 hydrogen bonds were constantly seen after ∼ 60 ns. The total energy decomposition per residue revealed proline 881 and asparagine 868 to be in the favor of interaction possessing the minimum energy contribution of −1.42 and −0.73 kcal/mol respectively (Fig. 4; Movie 2).
Fig. 4.

Represents the stability of the complex (Naringin-5DS3) with respect to, (a) Root Mean Square Deviation (RMSD) of the backbone (black) and complex (red), (b) Root Mean Square Fluctuation (RMSF) of the c-alpha atoms, (c)Radius of Gyration (RoG) of the backbone (black) and complex (red), (d) Solvent Assessable Surface Area (SASA) for the protein(black) and complex (red) (e) Number of Hydrogen bonds, (f) Total energy decomposition per residue.
3.5.3. MM-PBSA analysis
MMPBSA analysis (Table 3) revealed the Ellagic acid-5DS3 complex to possess the least total energy decomposition of −2039.24 ± 58.76 kcal/mol. The naringin-5DS3 complex possessed the least VDWAALS, EPB, and GSOLV of −1692.26 ± 7.3, −3254.58 ± 35.44, and −2386.63 ± 35.14 kcal/mol, respectively. Similarly, Ellagic acid-5DS3 complex possessed the least electrostatic, non-polar contribution of solute–solvent interactions to the solvation energy and total gas phase molecular mechanics energy with energy decomposition of −15874.26 ± 42.68, 1943.75 ± 4.84, and 274.96 ± 49.63 kcal/mol respectively. The standard Olaparib showed a total relative binding energy of −1927.69 ± 58.61 kcal/mol which is lower than the naringin-5DS3 complex however greater than ellagic acid-5DS3 complex indicating ellagic acid complex to be more stable with greater binding affinity with the protein.
Table 3.
MM-PBSA analysis of Ellagic acid, Naringin & Olaparib with PARP-1.
| Bioactive | VDWAALS | EEL | EPB | ENPOLAR | EDISPER | GGAS | GSOLV | GTotal |
|---|---|---|---|---|---|---|---|---|
| Ellagic acid | −1660.78 ± 6.58 | −15874.26 ± 42.68 | −3165.66 ± 30.74 | 1943.75 ± 4.84 | −1092.30 ± 4.57 | 274.96 ± 49.63 | −2314.21 ± 31.45 | −2039.24 ± 58.76 |
| Naringin | −1692.26 ± 7.3 | −15680.59 ± 46.05 | −3254.58 ± 35.44 | 1963.55 ± 4.49 | −1095.60 ± 3.96 | 475.92 ± 45.15 | −2386.63 ± 35.14 | −1910.71 ± 14.67 |
| Olaparib* | −1683.65 ± 6.88 | −15803.39 ± 42.21 | −3177.41 ± 31.60 | 1965.65 ± 4.67 | −1103.91 ± 6.43 | 387.98 ± 48.71 | −2315.67 ± 32.58 | −1927.69 ± 58.61 |
All the data are presented in mean ± SEM (n = 100) and unit for each parameter is Kcal/mol.
4. Discussion
Molecular docking is an integral aspect of the drug discovery procedure, and it is utilized in this work to assess the binding interactions of the hit molecules with the target 5DS3 protein (Deshpande et al., 2021). In addition to the docking score, the binding free energy, binding modes, and interactions of each ligand with the functional residues of the targeted protein (including H- bonding, lipophilic and polar interactions, and - π-π stacking) were investigated. The model features interact directly with critical amino acids, which inhibit PARP-1 (Li et al., 2016). Because of this, we can look to these characteristics as crucial chemical traits in the search for new PARP-1 inhibitors (Tabrez et al., 2022). The docking outcomes by the MM-GBSA approach were confirmed by the binding free energy calculations of the ligand–protein complexes (Forouzesh & Mishra, 2021). Van der Waals and non-polar solvation energies are the vibrant forces required for natural PARP inhibitors to interact with the protein (Thorsell et al., 2017). Pharmacophore is a set of steric and electronic properties that validate optimal supramolecular interactions during the virtual screening of massive chemical databases. It is a more effective and efficient technique than autodock for locating molecules which can activate or inhibit macromolecular activity(Zhou et al., 2019b). The molecule with similar or relevant characteristics should possess the same or a greater level of activity than the search molecule (Rohilla et al., 2017). Molecular weight is an essential characteristic in the drug discovery process for small compounds. During compound optimization, this feature is often researched since it affects many molecular changes, including uptake, bile excretion rate, BBB invasion, and target interactions (Lagorce et al., 2017). QikProp analysis gives information about the rule of five, predicting the drug-like nature of the molecules. The rules must satisfy the limits of molecular weight MW less than 500, QPlogPo/w less than 5, donor HB ≤ 5, accept HB ≤ 10 (Lipinski et al., 1997). Some drugs (Naringin, Icogenin, EGCG, Macranthoside B, Poncirin) disobeyed the Lipinskis’s rule mainly due to their highly hydrophobic nature. Based on docking studies, best interacted natural PARP-1 inhibitors, Naringin has three rule of five violations, while Ellagic acid has zero violations.
ADME studies were conducted based on the molecular weight (MW), G-rotatable bonds, H-bond acceptors and donors, surface area, Intestinal absorption, BBB permeability, number of Lipinski violations, solvent accessible area, metabolism, and, Plasma protein binding (Gürdere et al., 2021). Bioavailability predictions based on the Caco-2 cell permeability predicted apparent cell permeability in nm/sec., and estimated human oral absorption on a 0 to 100% scale (Deshpande et al., 2021). The BBB is the primary factor impeding the development of novel therapies for brain disorders. Certain small molecule medications can penetrate the BBB via lipid-mediated free diffusion if their molecular weight is less than 400 Da and they form less than eight hydrogen bonds. Most small and large-molecule therapeutics lack these characteristics (Pardridge, 2012). These large molecules can be re-engineered with different nano-delivery systems such as liposomes, polymeric nanoparticles, micelles and protein nanoparticles. Several studies point to hydrogen bonding significantly influencing drug permeability (Raevsky et al., 2000).Therapeutic medicines that are designed to interact with their molecular targets in the CNS must cross the BBB, while medications that function only on the periphery should not cross the BBB to prevent unwanted CNS adverse events (Zhu et al., 2018). Drugs and xenobiotics are metabolized to be excreted. Poor bioavailability can be caused by metabolic liability due to fast clearance, potential toxicity from reactive metabolites, and drug-drug interactions, including enzyme inhibition, induction, and mechanism-based inactivation (Y. Wang et al., 2015). Phase I and II enzymes predominantly catalyze metabolic processes in the liver (A. Smith, 2011). Following the path of the probe sphere's center allows one to calculate the SASA. The rolling ball algorithm is commonly used to compute the surface area of a biomolecule, and the area is typically referred to using the unit of square angstroms. When a biomolecule goes from a polar media to a nonpolar medium, the SASA provides information regarding the free energy transfer that occurs during this process (Dasari et al., 2017). The surface of a water molecule has a typical radius of 1.4 A°. SASA is usually in the range of 300.0–1000.0 Å. FOSA refers to the lipophilic component of the SASA, while FISA indicates the aqueous components of the SASA(Divyashri et al., 2021).
In addition, we also performed MD simulation for the lead complex which revealed the ellagic acid complex with 5DS3 to be the most stable. The RMSD showed fluctuation of less than ∼ 0.4 Å indicating it to be stable complex when compared to the standard which showed deviations of ∼ 10Å. Also, the naringin complex with 5DS3 showed better stability than the standard. For both the complexes, i.e., naringin and ellagic acid with 5DS3, showed 4–5 constant hydrogen bonds in a 100 ns of MD run which was observed to be 2 for the standard. The total energy decomposition per residue for ellagic acid-5DS3 complex showed TYR907 and TYR896 to possess the minimum energy contribution of −1.80 and −1.07 kcal/mol, respectively. The ligand showed a total energy contribution of −4.98 kcal/mol for the 100 ns of MD run. Additionally, the complete energy decomposition per residue for the naringin-5DS3 complex revealed PRO881 and ASP868 to favor interaction possessing the minimum energy contribution of −1.42 and −0.73 kcal/mol respectively. These stability parameters indicate that the interaction of the ligands ellagic acid and naringin with 5DS3 was stable throughout the MD run and provides validation to the molecular docking studies.
5. Conclusion
In this study, forty-three phytomedicines were virtually analyzed for their activity against PARP-1 inhibition via molecular docking with PARP-1 inhibitor (5DS3); majority of them showed good interaction at the binding site. In addition, Ellagic acid and Naringin showed a greater stable interaction trajectory with the PARP than with the olaparib-5DS3 complex. Discovering new medicines relies heavily on the drug molecule's pharmacophore. Molecular structure is crucial for pharmacological and biological actions. Hydrogen bonds (donor and acceptor) and aromatic rings were discovered to be involved in the chemical interactions between the natural PARP-1 inhibitors (Ellagic acid and Naringin) and the protein 5DS3. Any new drug molecule that can outperform currently available cancer treatments and move the field in exciting new directions is always appreciated. The compounds' drug-likeness and bioavailability were highlighted through the use of physicochemical parameters as well as the ADME program. Poor oral bioavailability was observed, and the pharmacokinetic profile revealed that ellagic acid followed the Rule of 5. The natural compounds shown to be most effective in the virtual screening procedure could potentially serve as lead molecules in the fight against glioma cancer.
CRediT authorship contribution statement
Arunraj Tharamelveliyil Rajendran: Methodology, Data curation, Investigation, Writing - original draft. Gupta Dheeraj Rajesh: Methodology, Formal analysis. Pankaj Kumar: Supervision, Validation. Prarambh Shivam Raju Dwivedi: Methodology, Data curation, Formal analysis. Chakrakodi Shashidhara Shastry: Supervision, Resources. Anoop Narayanan Vadakkepushpakath: Conceptualization, Supervision, Methodology.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
We acknowledge NITTE (Deemed to be University), Mangaluru, for providing software and technical support for the smooth conduction of the research work.
Footnotes
Peer review under responsibility of King Saud University.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.sjbs.2023.103698.
Contributor Information
Arunraj Tharamelveliyil Rajendran, Email: arunraj.21phdp102@student.nitte.edu.in.
Gupta Dheeraj Rajesh, Email: dheeraj.21phdp111@student.nitte.edu.in.
Pankaj Kumar, Email: pankajpgr@nitte.edu.in.
Prarambh Shivam Raju Dwivedi, Email: prarambh.21phdp109@student.nitte.edu.in.
Chakrakodi Shashidhara Shastry, Email: principal.ngsmips@nitte.edu.in.
Anoop Narayanan Vadakkepushpakath, Email: anoopnarayanan@nitte.edu.in.
Appendix A. Supplementary material
The following are the Supplementary data to this article:
References
- Bai R.Y., Staedtke V., Riggins G.J. Molecular targeting of glioblastoma: drug discovery and therapies. Trends Mol. Med. 2011;17(6):301. doi: 10.1016/J.MOLMED.2011.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendsen H.J.C., van der Spoel D., van Drunen R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995;91(1–3):43–56. doi: 10.1016/0010-4655(95)00042-E. [DOI] [Google Scholar]
- Cha G.D., Kang T., Baik S., Kim D., Choi S.H., Hyeon T., Kim D.H. Advances in drug delivery technology for the treatment of glioblastoma multiforme. J. Control. Release. 2020;328:350–367. doi: 10.1016/J.JCONREL.2020.09.002. [DOI] [PubMed] [Google Scholar]
- Chen A. PARP inhibitors: its role in treatment of cancer. Chin. J. Cancer. 2011;30(7):463. doi: 10.5732/CJC.011.10111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhari A.S., Mandave P.C., Deshpande M., Ranjekar P., Prakash O. Phytochemicals in cancer treatment: From preclinical studies to clinical practice. Front. Pharmacol. 2019;10 doi: 10.3389/FPHAR.2019.01614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawicki-McKenna J.M., Langelier M.F., DeNizio J.E., Riccio A.A., Cao C.D., Karch K.R., McCauley M., Steffen J.D., Black B.E., Pascal J.M. PARP-1 activation requires local unfolding of an autoinhibitory domain. Mol. Cell. 2015;60(5):755–768. doi: 10.1016/J.MOLCEL.2015.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasari, T., Kondagari, B., Dulapalli, R., Abdelmonsef, A. H., Mukkera, T., Padmarao, L. S., Malkhed, V., & Vuruputuri, U. (2017) Design of novel lead molecules against RhoG protein as cancer target – a computational study, Journal of Biomolecular Structure and Dynamics, 35:14, 3119-3139, DOI: 10.1080/07391102.2016.1244492 [DOI] [PubMed]
- Deshpande, N. S., Gowdru, S., Mahendra, N. N., Aggarwal, B., Felicity, D., Gatphoh, B., & Chandrashekharappa, R. (2021). Insilico design, ADMET screening, MM-GBSA binding free energy of novel 1,3,4 oxadiazoles linked Schiff bases as PARP-1 inhibitors targeting breast cancer. Future Journal of Pharmaceutical Sciences 7:1, 7(1), 1–10. 10.1186/S43094-021-00321-4. [DOI]
- Dias D.A., Urban S., Roessner U. A historical overview of natural products in drug discovery. Metabolites. 2012;2(2):303. doi: 10.3390/METABO2020303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divyashri G., Murthy T.P.K., Sundareshan S., Kamath P., Murahari M., Saraswathy G.R., Sadanandan B. In silico approach towards the identification of potential inhibitors from Curcuma amada Roxb against H. pylori: ADMET screening and molecular docking studies. Bioimpacts. 2021;11(2):119. doi: 10.34172/BI.2021.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwivedi P.S.R., Patil R., Khanal P., Gurav N.S., Murade V.D., Hase D.P., Kalaskar M.G., Ayyanar M., Chikhale R.V., Gurav S.S. Exploring the therapeutic mechanisms of Cassia glauca in diabetes mellitus through network pharmacology, molecular docking and molecular dynamics. RSC Adv. 2021;11(62):39362–39375. doi: 10.1039/D1RA07661B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forouzesh N., Mishra N. An effective MM/GBSA protocol for absolute binding free energy calculations: A case study on SARS-CoV-2 spike protein and the human ACE2 receptor. Molecules. 2021;26(8) doi: 10.3390/MOLECULES26082383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gürdere M.B., Budak Y., Kocyigit U.M., Taslimi P., Tüzün B., Ceylan M. ADME properties, bioactivity and molecular docking studies of 4-amino-chalcone derivatives: new analogues for the treatment of Alzheimer, glaucoma and epileptic diseases. In Silico Pharmacol. 2021;9(1):34. doi: 10.1007/S40203-021-00094-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javle, M., & Curtin, N. J. 2011. The role of PARP in DNA repair and its therapeutic exploitation. British Journal of Cancer 105(8), 1114–1122. 10.1038/bjc.2011.382. [DOI] [PMC free article] [PubMed]
- Kaserer T., Beck K.R., Akram M., Odermatt A., Schuster D., Willett P. Pharmacophore models and pharmacophore-based virtual screening: Concepts and applications exemplified on hydroxysteroid dehydrogenases. Molecules. 2015;20(12):22799. doi: 10.3390/MOLECULES201219880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanal P., Patil V.S., Bhandare V.V., Dwivedi P.S.R., Shastry C.S., Patil B.M., Gurav S.S., Harish D.R., Roy S. Computational investigation of benzalacetophenone derivatives against SARS-CoV-2 as potential multi-target bioactive compounds. Comput. Biol. Med. 2022;146 doi: 10.1016/J.COMPBIOMED.2022.105668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagorce, D., Douguet, D., Miteva, M. A., & Villoutreix, B. O. (2017). Computational analysis of calculated physicochemical and ADMET properties of protein-protein interaction inhibitors. Scientific Reports. 7(1), 1–15. 10.1038/srep46277. [DOI] [PMC free article] [PubMed]
- Li J., Zhou N., Cai P., Bao J. In silico screening identifies a novel potential PARP1 inhibitor targeting synthetic lethality in cancer treatment. Int. J. Mol. Sci. 2016;17(2) doi: 10.3390/IJMS17020258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski C.A., Lombardo F., Dominy B.W., Feeney P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997;23(1–3):3–25. doi: 10.1016/S0169-409X(96)00423-1. [DOI] [PubMed] [Google Scholar]
- Lu G., Nie W., Xin M., Meng Y., Gu J., Miao H., Cheng X., Chan A.S.C., Zou Y. Design, synthesis, biological evaluation and molecular docking study of novel urea-based benzamide derivatives as potent poly(ADP-ribose) polymerase-1 (PARP-1) inhibitors. Eur. J. Med. Chem. 2022;243 doi: 10.1016/J.EJMECH.2022.114790. [DOI] [PubMed] [Google Scholar]
- Morales J.C., Li L., Fattah F.J., Dong Y., Bey E.A., Patel M., Gao J., Boothman D.A. Review of poly (ADP-ribose) polymerase (PARP) mechanisms of action and rationale for targeting in cancer and other diseases. Crit. Rev. Eukaryot. Gene Expr. 2014;24(1):15–28. doi: 10.1615/CRITREVEUKARYOTGENEEXPR.2013006875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardridge W.M. Drug transport across the blood–brain barrier. J. Cereb. Blood Flow Metab. 2012;32(11):1959. doi: 10.1038/JCBFM.2012.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raevsky O.A., Fetisov V.I., Trepalina E.P., McFarland J.W., Schaper K.J. Quantitative estimation of drug absorption in humans for passively transported compounds on the basis of their physico-chemical parameters. Quant. Struct.-Act. Relat. 2000;19(4):366–374. doi: 10.1002/1521-3838(200010)19:4<366::AID-QSAR366>3.0.CO;2-E. [DOI] [Google Scholar]
- Revathi P., Kanth S.S., Gururaj S., Chander O.S., Rajender P.S. Understanding structural characteristics of PARP-1 inhibitors through combined 3D-QSAR and molecular docking studies and discovery of new inhibitors by multistage virtual screening. Struct. Chem. 2021;32(5):2035–2050. doi: 10.1007/S11224-021-01765-3/METRICS. [DOI] [Google Scholar]
- Siddiqui A.J., Jahan S., Singh R., Saxena J., Ashraf S.A., Khan A., Choudhary R.K., Balakrishnan S., Badraoui R., Bardakci F., Adnan M. Plants in anticancer drug discovery: From molecular mechanism to chemoprevention. Biomed Res. Int. 2022;2022 doi: 10.1155/2022/5425485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel R.L., Miller K.D., Jemal A. Cancer statistics, Cancer J. Clin. 2018;68(1):7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- Slade D. PARP and PARG inhibitors in cancer treatment. Genes Dev. 2020;34(5–6):360. doi: 10.1101/GAD.334516.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohilla, A., Khare, G., & Tyagi, A. K. 2017. Virtual Screening, pharmacophore development and structure based similarity search to identify inhibitors against IdeR, a transcription factor of Mycobacterium tuberculosis. Scientific Reports 7 (1), 1–14. 10.1038/s41598-017-04748-9. [DOI] [PMC free article] [PubMed]
- Mangal, M., Sagar, P., Singh, H., Raghava, G. P. S., & Agarwal, S. M. 2013. NPACT: Naturally Occurring Plant-based Anti-cancer Compound-Activity-Target database. Nucleic Acids Research, 41, D1124. 10.1093/NAR/GKS1047 [DOI] [PMC free article] [PubMed]
- A. Smith, D. 2011. Discovery and ADMET: Where are we now. Current Topics in Medicinal Chemistry, 11(4), 467–481. 10.2174/156802611794480909 [DOI] [PubMed]
- Tabrez S., Hoque M., Suhail M., Khan M.I., Zughaibi T.A., Khan A.U. Identification of anticancer bioactive compounds derived from Ficus sp. by targeting Poly[ADP-ribose]polymerase 1 (PARP-1) J. King Saud Univ. - Sci. 2022;34(5) doi: 10.1016/J.JKSUS.2022.102079. [DOI] [Google Scholar]
- Thorsell A.G., Ekblad T., Karlberg T., Löw M., Pinto A.F., Trésaugues L., Moche M., Cohen M.S., Schüler H. Structural basis for potency and promiscuity in poly(ADP-ribose) polymerase (PARP) and tankyrase inhibitors. J. Med. Chem. 2017;60(4):1262. doi: 10.1021/ACS.JMEDCHEM.6B00990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdés-Tresanco M.S., Valdés-Tresanco M.E., Valiente P.A., Moreno E. Gmx_MMPBSA: A new tool to perform end-state free energy calculations with GROMACS. J. Chem. Theory Comput. 2021;17(10):6281–6291. doi: 10.1021/ACS.JCTC.1C00645/SUPPL_FILE/CT1C00645_SI_001.PDF. [DOI] [PubMed] [Google Scholar]
- Wang C., Li J. Haematologic toxicities with PARP inhibitors in cancer patients: an up-to-date meta-analysis of 29 randomized controlled trials. J. Clin. Pharm. Ther. 2021;46(3):571–584. doi: 10.1111/JCPT.13349. [DOI] [PubMed] [Google Scholar]
- Wang Y., Xing J., Xu Y., Zhou N., Peng J., Xiong Z., Liu X., Luo X., Luo C., Chen K., Zheng M., Jiang H. In silico ADME/T modelling for rational drug design. Q. Rev. Biophys. 2015;48(4):488–515. doi: 10.1017/S0033583515000190. [DOI] [PubMed] [Google Scholar]
- Wang X, Zhang H, Chen X. Drug resistance and combating drug resistance in cancer. Cancer Drug Resist. 2019;2(2):141-160. doi: 10.20517/cdr.2019.10. Epub 2019 Jun 19. PMID: 34322663; PMCID: PMC8315569. [DOI] [PMC free article] [PubMed]
- Wen P.Y., Weller M., Lee E.Q., Alexander B.M., Barnholtz-Sloan J.S., Barthel F.P., Batchelor T.T., Bindra R.S., Chang S.M., Antonio Chiocca E., Cloughesy T.F., DeGroot J.F., Galanis E., Gilbert M.R., Hegi M.E., Horbinski C., Huang R.Y., Lassman A.B., Le Rhun E., van den Bent M.J. Glioblastoma in adults: a Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) consensus review on current management and future directions. Neuro Oncol. 2020;22(8):1073–1113. doi: 10.1093/NEUONC/NOAA106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Shih D.J.H., Lin S.Y. Role of DNA repair defects in predicting immunotherapy response. Biomarker Res. 2020;8(1) doi: 10.1186/S40364-020-00202-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, Y., Tang, S., Chen, T., & Niu, M. M. (2019b). Structure-Based Pharmacophore Modeling, Virtual Screening, Molecular Docking and Biological Evaluation for Identification of Potential Poly (ADP-Ribose) Polymerase-1 (PARP-1) Inhibitors. Molecules, 24(23). https://doi.org/10.3390/MOLECULES24234258 [DOI] [PMC free article] [PubMed]
- Zhou Y., Tang S., Chen T., Niu M.M. Structure-based pharmacophore modeling, virtual screening, molecular docking and biological evaluation for identification of potential poly (ADP-Ribose) polymerase-1 (PARP-1) inhibitors. Molecules. 2019;24(23) doi: 10.3390/MOLECULES24234258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L., Zhao J., Zhang Y., Zhou W., Yin L., Wang Y., Fan Y., Chen Y., Liu H. ADME properties evaluation in drug discovery: in silico prediction of blood-brain partitioning. Mol. Divers. 2018;22(4):979–990. doi: 10.1007/S11030-018-9866-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.