

Abstract
PARylation plays critical roles in regulating multiple cellular processes such as DNA damage response and repair, transcription, RNA processing, and stress response. More than 300 human proteins have been found to be modified by PARylation on acidic residues, i.e., Asp (D) and Glu (E). We used the deep-learning tool AlphaFold to predict protein-protein interactions (PPIs) and their interfaces for these proteins based on coevolution signals from joint multiple sequence alignments. AlphaFold predicted 260 confident PPIs involving PARylated proteins, and about one quarter of these PPIs have D/E-PARylation sites in their predicted PPI interfaces. AlphaFold predictions offer novel insights into the mechanisms of PARylation regulations by providing structural details of the PPI interfaces. D/E-PARylation sites have a preference to occur in coil regions and disordered regions, and PPI interfaces containing D/E-PARylation sites tend to occur between short linear sequence motifs in disordered regions and globular domains. The hub protein PCNA is predicted to interact with more than 20 proteins via the common PIP box motif and the structurally variable flanking regions. D/E-PARylation sites were found in the interfaces of key components of the RNA transcription and export complex, the SF3a spliceosome complex, and H/ACA and C/D small nucleolar ribonucleoprotein complexes, suggesting that systematic PARylation have a profound effect in regulating multiple RNA-related processes such as RNA nuclear export, splicing, and modification. Finally, PARylation of SUMO2 could modulate its interaction with CHAF1A, thereby representing a potential mechanism for the cross-talk between PARylation and SUMOylation in regulation of chromatin remodeling.
Keywords: D/E-PARylation, protein-protein interaction, interaction interface, coevolution
1. Introduction
Poly-ADP-ribosylation (PARylation) is a protein posttranslational modification (PTM) process that results in covalent attachment of ADP-ribose polymers to proteins. PARylation plays critical roles in regulation of multiple cellular processes including DNA replication and repair, transcription, stress response and RNA biogenesis [1, 2]. The mono- or poly-ADP-ribose modifications (linear or branched repeats of ADP-ribose up to 200 units) are catalyzed by a family of enzymes called poly-ADP-ribose polymerases (PARPs). Among the 17 members of this family in human, PARP1 is the founding member and is an abundant nuclear protein with a plethora of substrates [3]. PARP1, with several DNA-binding domains, is recruited to the DNA strand breaks and is activated to modify of a variety of target proteins through PARylation, a crucial step to initiate DNA repair [4]. Due to the important roles of PARPs in DNA repair, PARP inhibitors have been developed to treat certain cancers where their DNA repair capacities have been compromised by genetic alterations (e.g., genetic variants in BRCA1 and BRCA2) [5].
PARylation can modify the sidechains of a variety of amino acid acceptors, including charged residues (Asp, Glu, Lys and Arg), Ser, Cys, His, and Tyr [6]. PARylation is reversed by several PAR-degrading enzymes, such as PARG (poly-ADP-ribose glycohydrolase) [7]. PARylation can lead to drastic change of electrostatic and topological properties of an acceptor protein, thus potentially affecting their molecular function through disruption of protein-protein interactions (PPIs) and protein-DNA/RNA interactions. On the other hand, poly-ADP-ribose resembles DNA and RNA and could serve as a scaffold for recruiting other proteins to form functional molecular complexes. Several PAR-binding domains have been identified, including PBZ (PAR-binding zinc fingers), WWE, BRCT, macrodomain, and some OB-fold domains [8, 9].
PPIs are the foundation of functional protein complexes and play essential roles in all cellular processes [10]. Large-scale high-throughput experiments using techniques such as affinity purification and chemical cross-linking have been used to detect PPIs in human and other organisms at the whole proteome level [11–13]. However, high-throughput PPI datasets often have high false positive and false negative rates and show considerable inconsistencies between studies [14, 15]. Recently developed deep-learning based methods such as AlphaFold and RoseTTAFold have allowed accurate modeling of protein 3D structures and their complexes by exploring coevolutionary information in multiple sequence alignments (MSAs) [16, 17]. We showed that application of these methods to yeast and human mitochondrial proteins can detect interacting proteins on a proteome-wide scale, and the protein complex models provided mechanistic insights to a wide range of core eukaryotic functions [18, 19].
A number of studies have been performed to identify PARylation sites at the whole-proteome level, resulting in a large collection of PARylated proteins [20–23]. In this study, we focus on a specific type of PARylation that occurs on acidic residues Asp and Glu (D/E-PARylation). We report the use of the deep-learning and coevolution-based method, AlphaFold [24], for large scale predictions of PPIs for D/E-PARylated human proteins. Mapping D/E-PARylation sites onto the 3D structures of these PPIs allowed us to identify PARylation sites residing on the PPI interfaces. PARylation to these sites are expected to disrupt these protein complexes and thus regulate a variety of cellular processes related to DNA repair and RNA processing.
2. Materials and methods
2.1. Identification of D/E-PARylated proteins and their potential interactions partners
The list of D/E-PARylated proteins was obtained from the combined datasets of our previous proteomics studies [25, 26]. We mapped these proteins to 360 UniProt entries with 1,215 D/E-PARylation sites[27]. Furthermore, we obtained human PPIs identified in experimental studies from BIOGRID [13], IntAct [28], DIP [29], and MINT [30] databases. These databases link each PPI to a set of PubMed IDs that represent the experimental studies supporting this interaction. We collected the PubMed IDs associated with each PPI from all databases. We mapped the PPIs documented by these databases to UniProt entries, thus we can identify putative PPIs involving D/E-PARylated proteins: a total of 72,431 putative PPIs were identified, mostly from large-scale studies.
Because large-scale PPI studies tend to contain a high fraction of false positives, they do not offer as strong support as dedicated studies focusing on a couple of PPIs. To weigh low-throughput studies more, we transformed the support from a literature to a PPI to a support score defined as the reciprocal of the number of PPIs associated with this literature. We summed the support scores of each PPI from all associated literatures and obtained 5,859 more confident PPIs involving PARylated proteins that satisfy either of the following criteria: (1) summed support score >= 0.1 if it is supported by a single paper; (2) summed support score >= 0.01 if it is supported by multiple papers. Out of these candidate PPIs, 5,361 were further evaluated and modeled by AlphaFold; 498 (8.5%) were not modeled because we were not able to obtain joint MSAs (see 2.2) or the protein complexes could not fit into GPU memory (see 2.3).
2.2. Generation of protein sequence alignments
The eukaryotic proteomes were downloaded from the NCBI genome database. It consists of 49,102,568 proteins from 2,568 representative or reference proteomes (one proteome per species). We attempted to find orthologs from these proteomes for each reviewed human entry in the UniProt database [27]. For each human protein (referred to as the target protein below), the corresponding orthologous group at the eukaryotes level in OrthoDB [31] was identified. Human proteins that were not classified in OrthoDB or belonged to the Zinc finger C2H2-type group (OrthoDB group: 1318335at2759), the Reverse transcriptase, RNase H-like domain group (OrthoDB group: 583605at2759) and the Pentatricopeptide repeat group (Orthodb group: 1344243at2759) were ignored because these groups contain a large number (over 58000) of paralogous sequences resulting from gene duplications. The set of OrthoDB orthologous proteins for a target protein were clustered by CD-HIT [32] at 40% identity level. For each CD-HIT cluster, we selected one representative sequence that showed the lowest BLAST [33] e-value to the target protein. The representative sequences were then used as queries to search against the eukaryotic proteomes to identify homologous proteins to the target protein (e-value cutoff: 0.001).
For each target protein, we identified a single best hit from each organism, if available, that satisfies two criteria: (1) its sequence identity to the target protein is above 35%; (2) it shows the highest sequence similarity to the target protein among the combined BLAST hits found by multiple representative sequences. These best hits together with the target protein were aligned by MAFFT (with the --auto option) [34]. We labeled each protein by the organism it belongs in the resulting multiple sequence alignments (MSAs) and removed positions that are gaps in the human proteins. To construct the joint MSA for any two human proteins, we concatenated the aligned sequences of the same organism from two MSAs. In cases where a sequence is missing from an organism, we used gaps to replace that sequence and added these cases at the end of the joint MSA.
2.3. Modeling protein complexes with AlphaFold
One limitation of AF modeling is that large protein complexes cannot fit in the memory of our GPUs (≤ 48GB). To solve this problem, we parsed proteins into domains based on DeepMind’s models of monomeric human proteins (https://alphafold.ebi.ac.uk/). In addition to the predicted 3D structure, DeepMind also provided the predicted aligned error (PAE) for each residue pair in a protein. PAE reflects AF’s confidence in the distance between two residues, and it is suggested to be useful in defining domains of a protein. Residues within the same domain are tightly packed together to form a globular and rigid 3D structure, and thus PAEs between them are expected to be low. In contrast, since the relative orientation and distance between different domains can be variable, a pair of residues from different domains frequently shows high PAE.
We wrote an in-house script to iterate the following procedure to split proteins into segments until all segments are shorter than 500 amino acids or cannot be further split. We split a protein or a segment into two segments if (1) it had more than 500 residues and (2) the density of residue pairs showing low PAE (≤12Å) within each segment (Dintra) was much higher than the density of residue pairs showing low PAE (≤12Å) between two segments (Dinter). We found the split site that maximize the Dintra / Dinter ratio, and we required this ratio to be at least 10 for proteins or segments larger than 1000 residues and at least 20 for proteins or segments with 500–1000 residues.
As described previously, we deployed AF to model protein complexes by feeding it with joint MSAs of two proteins and introducing a gap of 200 residues in between. Protein pairs with combined length ≤ 1500 were directly modeled. Larger proteins were split into domains as described above and all pairs of domains were modeled if none of them has combined length > 1500. AF produces probabilities for the Cß – Cß distances of residue pairs to fall into a series of distance bins. Residue-residue contact probability was calculated as the sum of the probabilities for the distance bins under 12Å. For a pair of proteins, the matrix () of contact probability is of the shape by , where and are the length of the first and second proteins, respectively. We extracted the submatrix (the first rows and the last columns) that contains the inter-protein contact probabilities. The highest residue-residue contact probability in this matrix is used as the contact probability for a pair of proteins.
2.4. Analysis of D/E-PARylated proteins and their interactions with other proteins
For D/E-PARylated proteins, we obtained secondary structure predictions by PSIPRED [35] and disordered prediction by IUPred2A [36]. We retrieved predicted local distance difference test scores (pLDDT, a confidence metric) from AlphaFold models [37] of these proteins and calculated residue-level relative solvent accessibilities using DSSP [38]. Sequence conservation was calculated by using AL2CO [39]. Enrichment analysis of functional categories and domain types in these proteins was conducted by the Database for Annotation, Visualization and Integrated Discovery (DAVID) web server [40]. We loaded 261 PPIs involving D/E-PARylated proteins with AlphaFold contact probability above 0.9 to the Cytoscape software [41], where proteins were represented as nodes and nodes were connected by edges if they interact (AlphaFold contact probability ≥ 0.9). We used the community cluster method, Glay [42], to cluster the network formed by these PPIs assuming undirected edges. We analyzed the function for proteins in each cluster with >= 3 proteins using the DAVID web server [40]. We annotated each cluster by the predominant functional group. We mapped the D/E-PARylation sites to the predicted complex structures and recorded those interfaces (defined as residues with predicted contacts probabilities above 0.5) that contains such sites. Interesting biological examples from this PPI network were studied manually with the help of Pymol and literature.
3. Results and discussion
3.1. The collection of D/E-PARylated protein and their functional and structural features
We collected 360 proteins modified with D/E-PARylation from two previous proteome-wide studies (supplementary Table S1) [25, 26]. The majority (235 out of 360) of these PARylated proteins contain one or two D/E-PARylation sites (Figure 1A). The most heavily modified protein is PARP1, with 46 D/E-PARylation sites. Five other proteins have 20 or more D/E-PARylation sites: DDX21 (DExD-Box Helicase 21), HNRNPU (heterogeneous nuclear ribonucleoprotein U), YLPM1 (YLP Motif Containing 1), MKI67 (marker of proliferation Ki-67), and RFC1 (Replication Factor C Subunit 1). Functional enrichment analysis by the DAVID server [43] suggests that D/E-PARylated proteins are significantly enriched in a number of biological processes, including transcription, transcription regulation, DNA damage and repair, mRNA processing and splicing, and cell division.
Figure 1. Statistics about D/E-PARylated proteins and their interactions with other proteins.
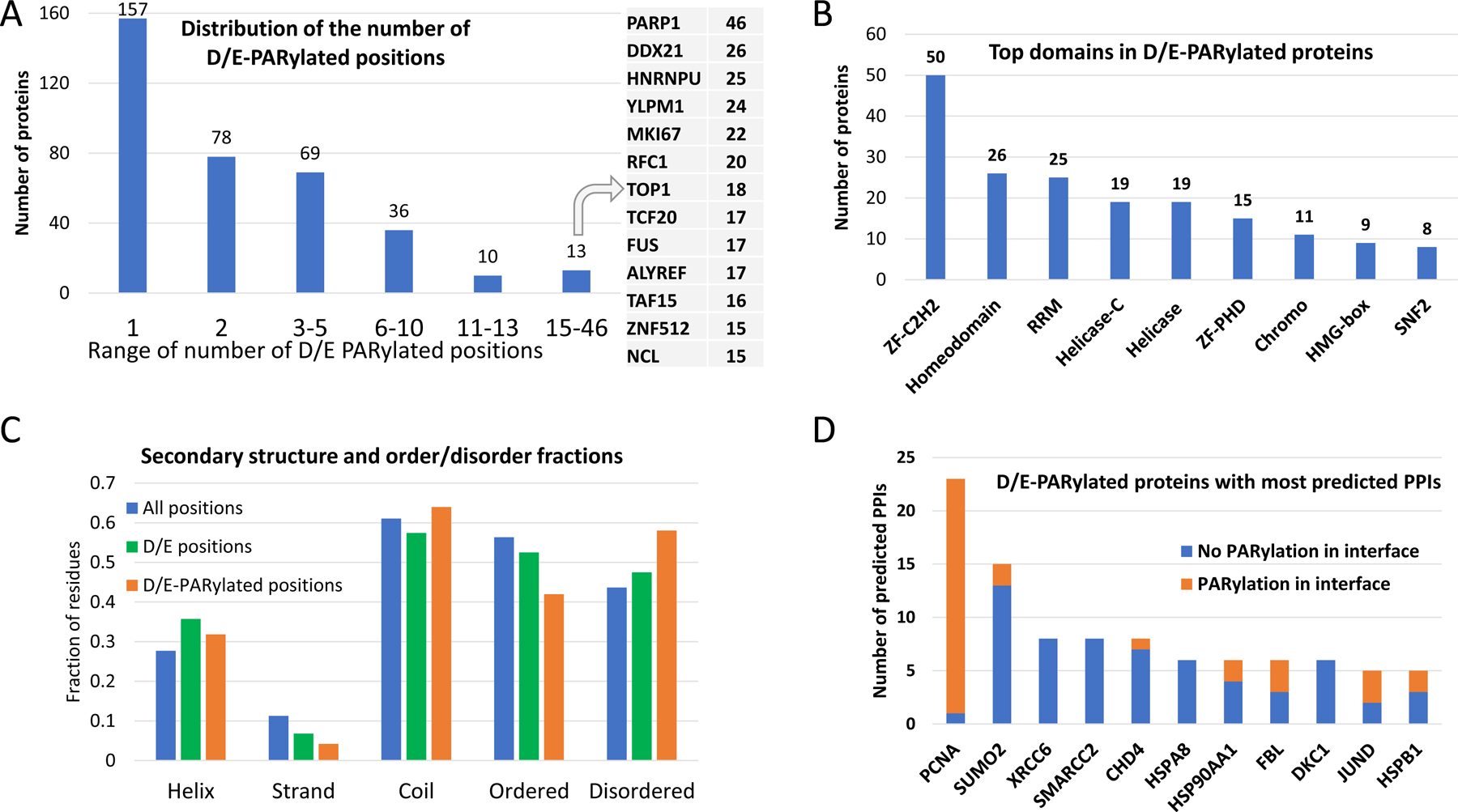
(A) Distribution of the number of D/E-PARylation sites among D/E-PARylated proteins. Proteins with the largest numbers of D/E-PARylation sites are listed to the right. (B) Frequently observed domains among D/E-PARylated proteins. ZF-C2H2 - C2H2 type zinc finger domain, ZF-PHD: PHD-type zinc finger domain, RRM -RNA recognition motif. (C) Secondary structure contents and ordered/disordered states of PARylated proteins and PARylation sites. The blue, green and orange bars show fraction of different secondary structure and order/disorder states of all positions in PARylated proteins, positions with D/E, and D/E-PARylation sites, respectively. Statistical significance of comparing the fractions is indicated by the number of stars: 4 stars - p-value<0.00001, 3 stars - 0.00001<p-value<0.0001, 2 stars - 0.0001<p-value<0.001, and 1 star - 0.01<p-value<0.04. (D) D/E-PARylated proteins with the largest numbers of predicted PPIs. The orange and blue portions of the bars correspond to PPI interfaces with and without D/E-PARylation sites, respectively.
Over half of these D/E-PARylated proteins are also modified with ubiquitin-like protein conjugation, suggesting the importance of interplay between different types of posttranslational modifications such as PARylation, ubiquitination, and SUMOylation [44, 45]. D/E-PARylated proteins are enriched in a number of domain types (e-value < 0.001 after Benjamini correction for multiple statistical tests), including RNA recognition motif domain (RRM), zinc finger domain (C2H2 type and PhD type), helicase domain, chromo domain, and homeodomain (Figure 1B), consistent with the general functions of these proteins in DNA and RNA-related processes.
We analyzed the secondary structure content and ordered/disordered state of the D/E-PARylation sites. Compared to all positions in D/E-PARylated proteins and positions with Asp or Glu, D/E-PARylation sites have slight preference in coils (p-value < 0.04 by Fisher’s exact test) and disordered regions (p-value < 0.0001) and are disfavored in beta-strands (p-value < 0.0001) (Figure 1C). Only 4% of D/E-PARylation sites are found in beta-strand as opposed to 6.8% for sites with Asp/Glu and 11% for all positions. Such statistics are consistent with the observation that D/E-PARylation sites tend to be exposed [25], for example, in flexible loops, for easy access by PARP enzymes. Indeed, D/E-PARylation sites exhibit on average larger relative solvent accessibility (RSA) (mean: 0.739 and median: 0.799) and lower pLDDT scores (a residue-level confidence metric) (mean: 59.6 and median: 52.7) compared to all D/E-containing positions (RSA mean: 0.654 and median: 0.701; pLDDT mean: 62.7 and median: 62.0) in AlphaFold models.
3.2. Prediction of protein-protein interactions of D/E-PARylated proteins
We used the deep learning-based method, AlphaFold, to predict PPIs between D/E-PARylated proteins and their potential interaction partners (see Materials and methods). Among the 5,361 pairs of proteins, AlphaFold supported 260 PPIs (294 domain-level interactions from 260 protein pairs, supplementary Table S2) with contact probability above 0.9, among which 98 are supported by experimental structures. Of the 181 PPIs with AlphaFold scores above 0.95, 90 are supported by experimental structures. We mapped the D/E-PARylation sites to the predicted complex structures with AlphaFold contact probability above 0.9 and recorded the occurrences of these positions in PPI interfaces (AlphaFold contact probability > 0.5). In total, we identified 69 PPI interfaces containing D/E PARylation sites. D/E-PARylation sites in general exhibit lower sequence conservation compared to all D/E-containing positions (supplementary Figure S1), possibly due to the preference of D/E-PARylation sites occurring in disordered regions. However, D/E-PARylation sites in the interfaces of predicted complexes have a higher fraction in the category of high conservation (supplementary Figure S1).
Eleven D/E-PARylated proteins have 5 or more AlphaFold-predicted PPIs (Figure 1D). PCNA (Proliferating cell nuclear antigen) has the largest number of predicted PPIs, and 22 out of the 23 predicted PPIs of PCNA have D/E-PARylation sites occurring in the PPI interface. The network formed by AlphaFold-predicted PPIs was visualized using Cytoscape [41]. We performed clustering analysis [42] of proteins involving these PPIs to partition the network into groups of functionally related proteins linked by PPIs. The resulting clusters (excluding clusters with only two proteins) are shown in Figure 2, where proteins are represented as dots and interacting proteins are connected by edges (solid line - with experimental structure; dashed line - without experimental structures; green line - with PARylation sites mapped to the interface). The most predominant clusters involve DNA damage and repair, DNA replication, transcription, chromatin regulation, and RNA processing. These clusters are consistent with the well-studied roles of PARylation in these functional categories [46, 47]. Because many of these PARylation events occurred under genotoxic stress conditions, it is conceivable that PARylation could modulate the assembly of the involved complexes, serving a mechanism to regulate the relevant biological processes during DNA damage conditions. As an example, it is well recognized that PARylation plays a critical role in regulating many steps in RNA metabolism [48]. Consistent with this notion, we found that several components of the protein complexes involved in RNA splicing, processing, and transportation (e.g., the splice factor 3a complex and the TREX complex) were PARylated [49]. Importantly, these PARylation events could regulate the assembly of these complexes, pointing to the intriguing hypothesis that PARylation could regulate RNA metabolism under genotoxic conditions.
Figure 2. Clusters of D/E-PARylated proteins and their interaction partners.
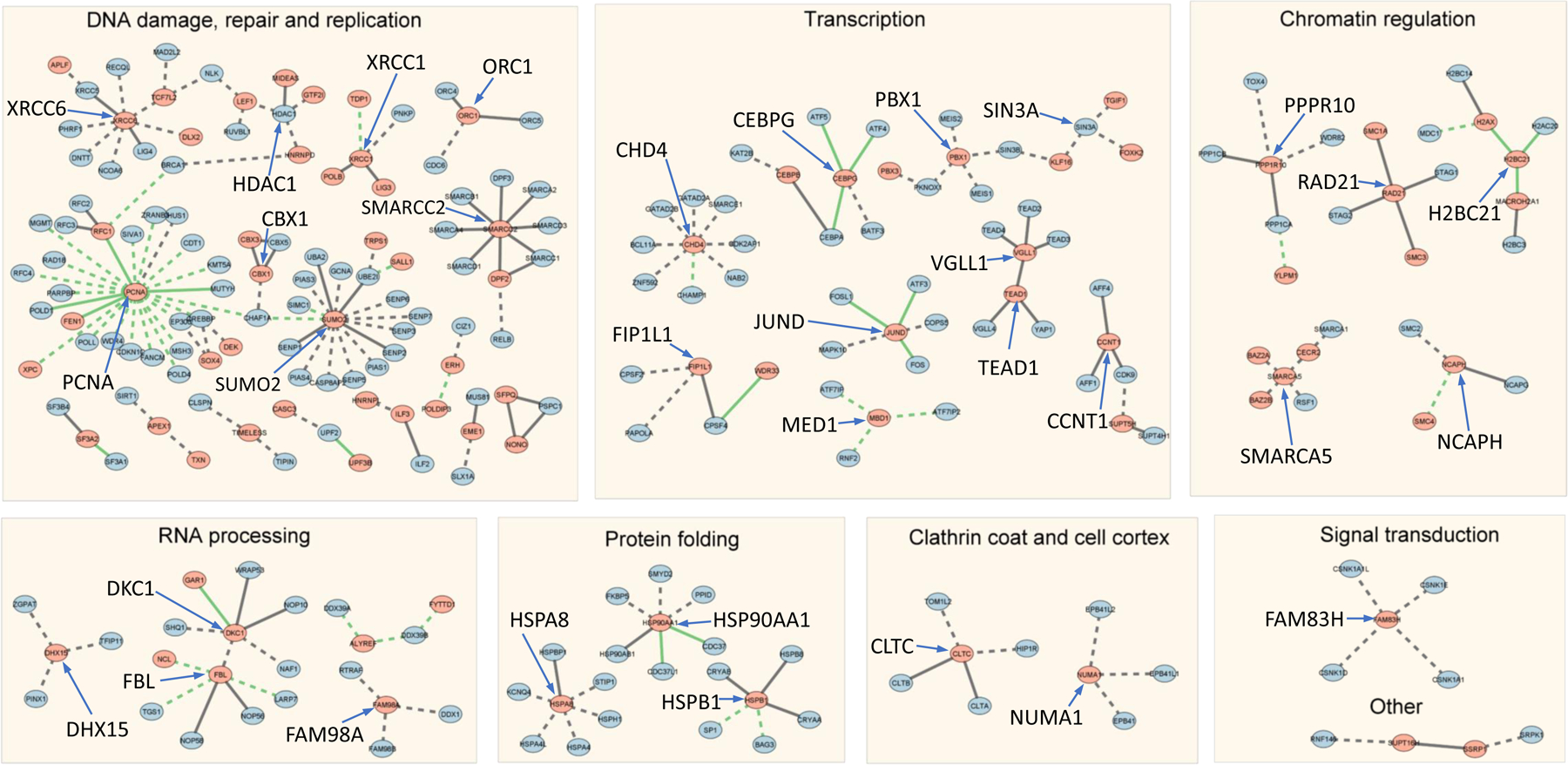
Dots represent proteins and edges connect proteins that are predicted to interact (AlphaFold contact probability > 0.9). Clusters are labeled by the predominant functional category. Solid lines represent interactions with experimental structures. Dashed lines represent interactions without experimental structures. Green lines represent interactions where PARylation sites are mapped to the PPI interfaces.
3.3. PCNA - a hub of PPIs with PARylation sites in the interaction interfaces
PCNA is a cellular hub protein functioning in an array of DNA-related processes such as DNA replication and repair, chromatin remodeling, DNA methylation, and cell cycle regulation [50, 51]. PCNA interacts with a diverse set of proteins, mainly through a short linear sequence motif in its binding partners known as the PIP (PCNA-interaction protein) box. Canonical PIP box adopts a consensus sequence of Qxxhxxaa (x: any amino acid, h: a hydrophobic residue, a: an aromatic residue). A single turn of helix positions the hydrophobic and aromatic positions in this motif into a binding pocket of PCNA. Variations of this motif have been increasingly found in other proteins [52].
Among D/E-PARylated proteins, PCNA has the largest number of PPIs (23 in total) supported by AlphaFold (contact probability > 0.9). Binding partners of PCNA mostly use the PIP box to interact with one end of the central beta-sheet in PCNA (Figure 3), consistent with experimental studies and available structures. However, regions flanking the PIP box could adopt different conformations for different PCNA binding partners. These flanking regions have been shown to significantly contribute to binding affinity of PIP boxes [52]. Some of them adopts extended loop or beta-strand (Figure 3C-3H), such as FANCM and CHAF1A. Others form extended helices around the PIP box, such as XPC and SIVA1 (Figure 3I and 3J). The PIP boxes mostly locate in disordered regions of the PCNA-interacting partners. However, for FEN1 and POLD1, other regions from a globular domain also contribute to the interface (Figure 3A and 3B). AlphaFold predicted the interaction between PCNA and ZRANB3 through the C-terminal region (Figure 3K), consistent with the experimental structure [53].
Figure 3. AlphaFold models of PCNA and its binding partners.

PCNA is shown in green, and its interaction partners are shown in cyan, and the gene names of the interacting partners are labeled by the structure. Predicted contacts are shown in sticks (yellow: contact probability ≥ 0.9; orange: 0.5 ≤ contact probability < 0.9). Sidechains of D/E-PARylation sites are shown in magenta sphere representations. ZRANB3_C and ZRANB3_RING represents C-terminal region and the RING finger domain, respectively.
Interestingly, AlphaFold also predicted an interface formed by ZRANB3 RING finger domain in the opposite site of the PCNA-PIP box interface (Figure 3L) where PCNA binds DNA in the experimental structure [54]. Whether this predicted interface is real remains to be explored experimentally, and it may reveal new insights into PCNA function. PCNA possesses 11 D/E-PARylation sites. Several of them are in the interface between PCNA and PIP boxes, including four from the middle of the protein (D120, D122, E124 and E130) and three from the C-terminal end of the protein (D257, E258 and E259) (colored magenta in Figure 3). Multiple D/E-PARylation sites in the PCNA binding interfaces suggest that PARylation is important for regulation of the function of this PPI hub.
3.4. DDX39B-ALYREF and DDX39B-FYTTD1 interactions
The transcription and export complex (TREX), consisting of the THO complex, DDX39B (ortholog of yeast protein UAP56), and ALYREF (also named THOC4), is responsible for loading the messenger ribonucleoprotein complexes (mRNPs) for nuclear export [55]. Recently the structure of the THO-DDX93B complex was solved by Cryo-EM [56]. However, information of how ALYREF fits in this complex remains missing. It has been reported that ALYREF uses both the N-terminal [57] and C-terminal regions [58] for the interaction with DDX39B. AlphaFold predicted the interaction between DDX39B and ALYREF for both an N-terminal motif (score: 0.923) and a C-terminal motif (score: 0.823), consistent with previous experimental studies. DDX39B is a DEAD/DEAH-box helicase with an N-terminal catalytic domain (light green in Figure 4A) and a C-terminal Helicase_C domain (green in Figure 4). In the AlphaFold model, the N-terminal motif of ALYREF (residues 1–20) forms an alpha hairpin that interacts with two helices (residues 270–284 and residues 296–308) from the C-terminal Helicase_C domain of DDX39B, whereas the C-terminal motif of ALYREF (residues 238–257) forms a helix that interacts with two helices from the N-terminal domain of DDX39B (residues 204–221 and residues 233–245). Interestingly, both N- and C-terminal motifs of ALYREF contain multiple PARylated D/E residues (Figure 4A), suggesting that PARylation could play an important role in the modulation of such an interaction and nuclear mRNP export. Interestingly, it has been previously shown that the depletion of the TREX complex could lead to profound defects in transcription elongation and mRNA export [59]. Specifically, the mRNA levels in cells with ALYREF depletion were decreased to 19% of those in the control cells. It is conceivable that PARylation could function through the TREX complex to regulate mRNA maturation under genotoxic conditions. Furthermore, it was shown that the TREX complex is also a key player for genome integrity. It was shown that depletion of ALYREF also caused genomic instability (as shown by high levels of DNA breaks and replication alteration) [59], a phenomenon that could be coupled with the defective mRNA processing pathway in these cells. The role of ALYREF PARylation in regulating mRNA processing and genomic instability warrants future studies.
Figure 4. AlphaFold models of the (A) DDX39B-ALYREF and (B) DDX39B-FYTTD1 complexes.
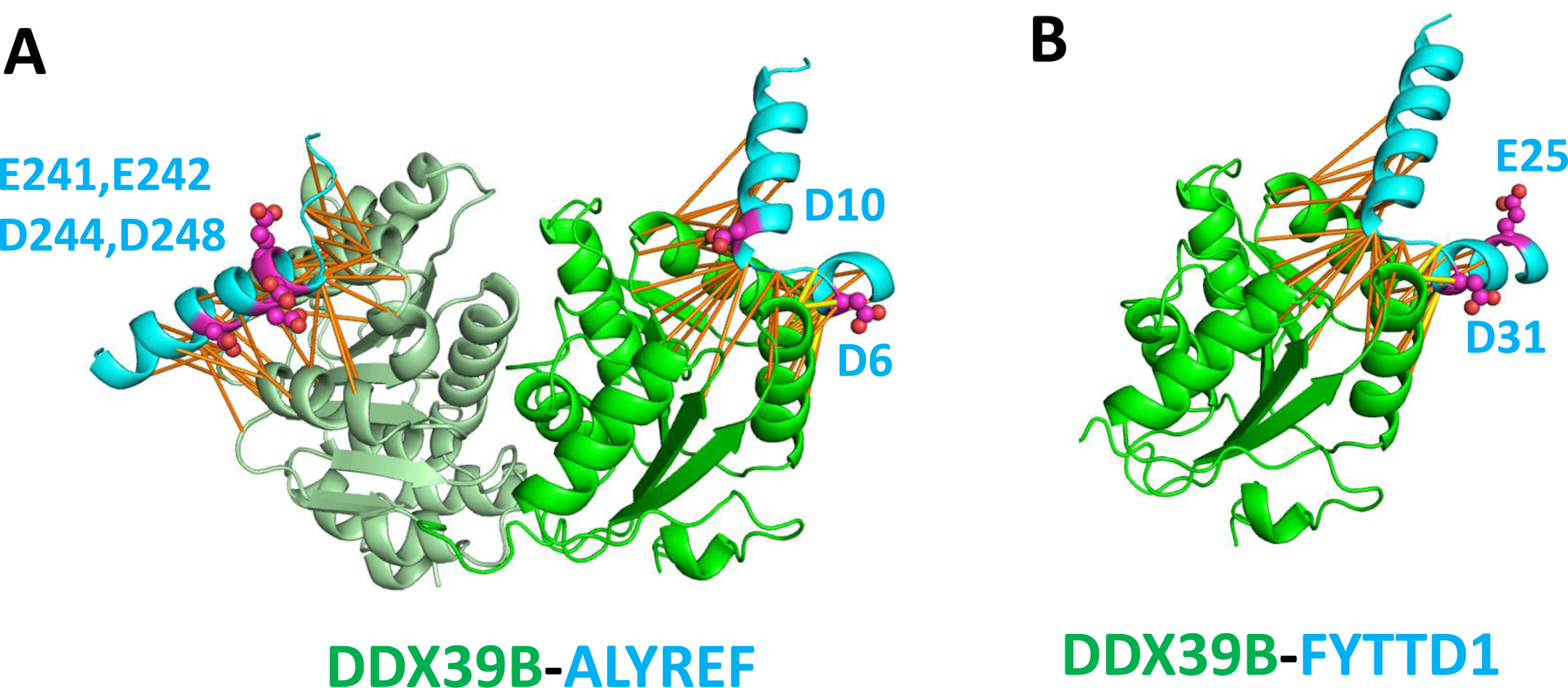
DDX39B has two domains, the helicase domain is shown in light green and the C-terminal Helicase_C domain is shown in green. Predicted contacts and D/E-PARylation residues are shown in the same way as in Figure 3.
The N-terminus of another DDX39B-interacting protein FYTTD1 (also named UIF) contains a sequence motif that is homologous to that of ALYREF [60]. AlphaFold also predicted the interaction between DDX39B and FYTTD1 through this motif (Figure 4B). This motif in FYTTD1 also contains several D/E-PARylation sites, suggesting that PARylation could have a profound effect on the export of mRNPs through modulating multiple interactions in this process.
3.5. ERH-POLDIP3 and ERH-CIZ1 interactions
ERH (Enhancer of Rudimentary Homolog) is a multifunctional conserved small protein (104 aa) involved in an array of cellular and developmental processes such as pyrimidine metabolism, DNA repair, mRNA splicing, microRNA maturation, cell cycle regulation, and erythroid differentiation [61, 62]. One of ERH’s interaction partner is POLDIP3 [63], which is a binding partner and robust activator of DNA polymerase d [64]. POLDIP3 was found to also play a crucial role in activation of DNA damage checkpoint [65]. The structural details of the ERH-POLDIP3 interaction have not been revealed before. The ERH-POLDIP3 interaction was strongly supported by AlphaFold (contact probability > 0.99) (Figure 5A). The C-terminus of POLDIP3 (residues 389–421) interacts with ERH by using a linear motif with an alpha-helix and a C-terminal beta-strand that forms a parallel beta-sheet with another beta-strand from ERH (residue 49–53). Both ERH and POLDIP3 are D/E-PARylated proteins and have D/E-PARylation sites in the interface (Figure 5A). The PARylation site on POLDIP3 (E416) occurs at the beginning of the beta-strand that interacts with ERH, and the PARylation site on ERH (E15) occurs at the opposite end near the alpha-helix of POLDIP3. PARylation at these sites could regulate the formation of the ERH-POLDIP3 complex and its function in DNA damage responses.
Figure 5. AlphaFold models of the ERH-POLDIP3 and ERH-CIZ1 complexes.

Predicted contacts and D/E-PARylation residues are shown in the same way as in Figure 3.
CIZ1, a nuclear zinc finger protein, is another interaction partner of ERH [66]. AlphaFold predicted the ERH-CIZ1 interaction with a high contact probability (> 0.99). The interacting motif (534–566) from CIZ1 is predicted to be in the disordered region and outside the zinc finger domain (residues 595–617). The range of this region is consistent with the region found by experiments (531–644). AlphaFold model provided structural details of this interaction (Figure 5B). CIZ1 interacts with ERH through a beta-hairpin (residues 547–562) that forms a beta-sheet with a beta-strand (residues 49–53) from ERH, the same region as involved in the ERH-POLDIP3 interface. Unlike the ERH-POLDIP3 interface, the PARylation site in ERH is distal to the interface between ERH and CIZ1, and thus this interaction is probably not regulated by PARylation.
3.6. DKC1-GAR1-WRAP53 complex
DKC1 is the catalytic subunit of the H/ACA small nucleolar ribonucleoprotein (H/ACA snoRNP) complex that is responsible for pseudouridylation of ribosomal and spliceosomal RNAs [67, 68]. It is also involved in the processing and trafficking of the RNA component of the telomerase reverse transcriptase holoenzyme [69]. AlphaFold predicted the interactions of DKC1-GAR1, DKC1-WRAP53, and DKC1-NOP10, consistent with a recent experimental structure complex of the telomerase holoenzyme [70]. WRAP53, a WD40-domain containing protein, can stimulate the catalytic activity of the H/ACA snoRNA complex [71]. GAR1, another component of H/ACA snoRNA complex, has two D/E-PARylation sites (E81 and D82) that lie in the interface between DKC1, GAR1, and WRAP53 (Figure 6). It is thus likely that D/E-PARylation at these GAR1 sites could prevent the association of WRAP53 with the H/ACA snoRNA complex and negatively regulate its catalytic activity. PARylation may also affect the activity of DKC1 in this complex in other ways, as DKC1 itself contains multiple D/E-PARylation sites in its C-terminal disordered region.
Figure 6. AlphaFold models of the DKC1-GAR1-WRAP53 complex.

Predicted contacts and D/E-PARylation sites are shown in the same way as in Figure 3.
3.7. D/E-PARylation sites in other PPI interfaces
We also identified D/E-PARylation sites in a number of other protein complexes predicted by AlphaFold. One recurring scheme of interactions is the utilization of a beta-strand from a nonglobular region in one protein to interact with an edge beta-strand from a globular domain in its binding partner. Such interactions are observed between SF3A2 and SF3A1, between FBL (fibrillarin) and NCL (nucleolin), between HSP90AA and CDC37, and between SUMO2 and CHAF1A (Figure 7). Such an observation may be due to the polar nature of D/E residues causing them to be more likely found at the rim of the interaction interface than to be completely buried inside the interface.
Figure 7. AlphaFold models of the protein complexes with D/E-PARylation sites in their interfaces.
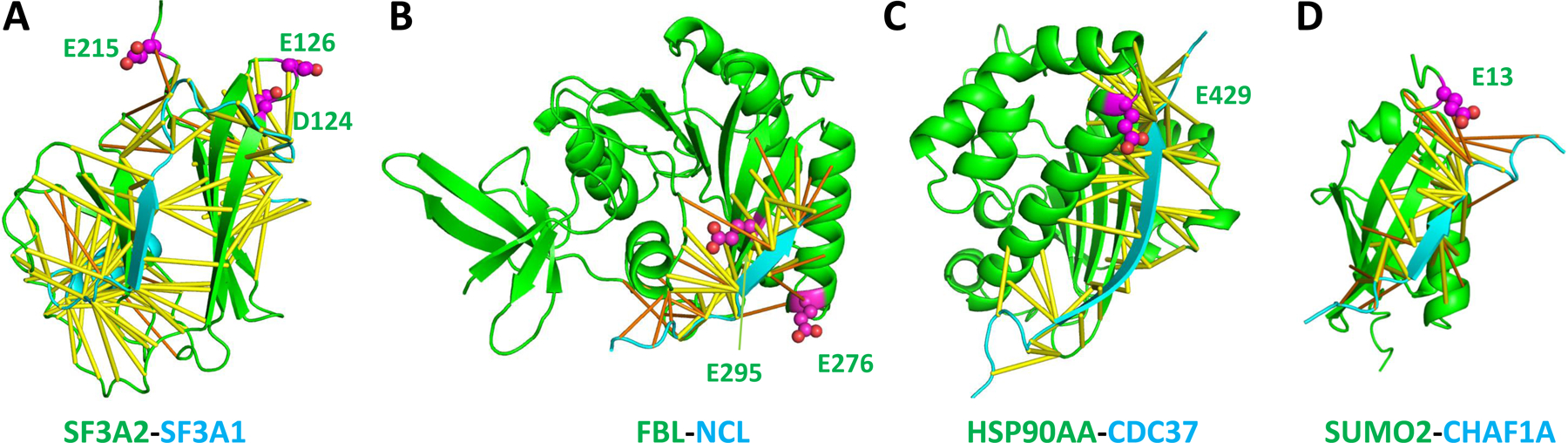
(A) SF3A2 and SF3A1 of the SF3a spliceosome complex. (B) FBL and NCL. (C) HSP90AA and CDC37. (D). SUMO2 and CHAF1A. Predicted contacts and D/E-PARylation sites are shown in the same way as in Figure 3.
SF3A2 and SF3A1 are components of the splice factor 3a complex [72]. FBL is a catalytic component of C/D box snoRNPs that catalyzes 2′-O-methylation of rRNAs [73]. HSP90AA is a molecular chaperone that interacts with its co-chaperone CDC37 in regulation of stress response [74]. D/E-PARylation sites are observed near these interaction interfaces involving edge beta strands, suggesting that PARylation could have diverse effects on RNA splicing, RNA modification, and stress response by modulating these interactions in a variety of ways. The PAR moiety could sterically disrupt PPIs and prevent the formation of protein complexes. The negative charges on the PAR moiety near the interaction interface may stabilize or destabilize the interactions depending on the electrostatic properties of the interface. While non-specific interactions with PAR-binding proteins may be responsible for abolishing existing PPIs, the PAR moiety may help establish new protein interaction scaffolds by interacting with various PAR-binding proteins. Future development of structural modeling tools incorporating the PAR moiety could further shed light on the functional mechanisms of PARylation events.
SUMO2, a SUMO (small ubiquitin-like modifier) family protein, contains a D/E-PARylation site (E13) in its N-terminus. AlphaFold predicted the interaction between SUMO2 and CHAF1A (chromatin assembly factor 1 subunit A, also named the P150 subunit), which is a histone chaperone and a primary component of the CAF-1 complex that mediates chromatin assembly in DNA replication and DNA repair [75]. The AlphaFold model shows that CHAF1A utilizes a beta-strand in a disordered region (residues 110–125) to interact with an edge beta-strand of SUMO2 (Figure 7D). The AlphaFold predicted interaction region in CHAF1A is consistent with the experimentally identified region of interaction [76]. The PARylation site of SUMO2 is in the SUMO2-CHAF1A interface (Figure 7D), suggesting that this interaction could be modulated by PARylation and represent a mechanism of cross-talk between PARylation and SUMOylation in chromatin remodeling.
4. Concluding remarks
We applied AlphaFold to predict and model PPIs for 360 human proteins with D/E-PARylation based on coevolution signals from joint multiple sequence alignments. D/E-PARylation sites in these proteins tend to occur in coil and disordered regions. These regions frequently contain short linear sequence motifs that interact with globular domains of other proteins. In particular, more than 20 interactions were found for the hub protein PCNA that involves the binding of the PIP box motif and its flanking regions. Detailed analysis of interaction interfaces revealed one recurring pattern of interactions that involves binding of short motifs to edge beta-strands in globular domains. D/E-PARylation sites are mapped to several interfaces in important protein complexes involved in RNA export, splicing, and enzymatic modifications. PARylation plays essential roles in DNA damage response and DNA repair by recruiting factors to DNA damage site. Synergistically, to assist the DNA repair process, PARylation could negatively affect other processes in genotoxic stress response in the nucleus such as transcription and RNA processing by disrupting critical interaction interfaces.
Supplementary Material
Supplementary Table S1. D/E-PARylated proteins and their PARylation sites.
Supplementary Table S2. Top-scoring protein-protein interactions by AlphaFold (contact probability score ≥ 0.9).
Supplementary Figure S1. Sequence conservation distribution of D/E positions.
Statement of significance.
Protein-protein interactions are the foundations of functional complexes and networks in most cellular processes. An essential role of protein posttranslational modifications is to modulate protein-protein interactions. PARylation, the covalent attachment of ADP-ribose units to protein sidechains, has been identified as a critically important posttranslational modification in biological processes related to DNA repair, transcription, and stress response. Recent advances in deep learning methods have led to a breakthrough in predicting protein structures and their complexes with high precision. We used one such method, AlphaFold, to systematically predict protein-protein interactions for 360 human proteins modified with PARylation on Asp and Glu residues. The resulting network revealed a number of protein complexes whose 3D structures have not been determined experimentally. Detailed structural analysis of predicted complexes coupled by mapping of D/E-PARylation sites to 3D structures helps us to identify a number of PARylation events expected to disturb the protein-protein interactions, revealing the functional consequences of PARylation in a number of cellular processes such as RNA nuclear export, splicing, and modification.
Acknowledgements
Qian Cong is a Southwestern Medical Foundation endowed scholar. J.Z. is supported by a training grant RP210041 from Cancer Prevention and Research Institute of Texas. This research is supported in part by grant I-2095-20220331 to Q.C. from Welch Foundation. This work was supported in part by grants R35GM134883, R01NS122533, and R21CA261018 to Y.Y. National Institutes of Health. The authors thank Dr. Payal Kapur and Hua Zhong for providing the UT Southwestern Kidney Cancer Program WES datasets and Dr. Nick V. Grishin for helpful discussions.
Footnotes
Conflict of interest: none declared.
Data availability
Structural models of predicted complexes involving D/E-PARylated proteins can be downloaded at: https://conglab.swmed.edu/DE_PARylation/.
References
- 1.Gupte R, Liu Z, Kraus WL, PARPs and ADP-ribosylation: recent advances linking molecular functions to biological outcomes. Genes Dev 31, 101–126 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eleazer R, Fondufe‐Mittendorf YN, The multifaceted role of PARP1 in RNA biogenesis. Wiley Interdisciplinary Reviews: RNA 12, e1617 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Langelier M-F, Eisemann T, Riccio AA, Pascal JM, PARP family enzymes: regulation and catalysis of the poly (ADP-ribose) posttranslational modification. Current opinion in structural biology 53, 187–198 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krishnakumar R, Kraus WL, The PARP side of the nucleus: molecular actions, physiological outcomes, and clinical targets. Molecular cell 39, 8–24 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mateo J et al. , A decade of clinical development of PARP inhibitors in perspective. Ann Oncol 30, 1437–1447 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Larsen SC, Hendriks IA, Lyon D, Jensen LJ, Nielsen ML, Systems-wide analysis of serine ADP-ribosylation reveals widespread occurrence and site-specific overlap with phosphorylation. Cell reports 24, 2493–2505. e2494 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Min W, Wang Z-Q, Poly (ADP-ribose) glycohydrolase (PARG) and its therapeutic potential. Frontiers in Bioscience-Landmark 14, 1619–1626 (2009). [DOI] [PubMed] [Google Scholar]
- 8.Gibson BA, Kraus WL, New insights into the molecular and cellular functions of poly (ADP-ribose) and PARPs. Nature reviews Molecular cell biology 13, 411–424 (2012). [DOI] [PubMed] [Google Scholar]
- 9.Liu C, Vyas A, Kassab MA, Singh AK, Yu X, The role of poly ADP-ribosylation in the first wave of DNA damage response. Nucleic acids research 45, 8129–8141 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stelzl U, Wanker EE, The value of high quality protein-protein interaction networks for systems biology. Curr Opin Chem Biol 10, 551–558 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Rolland T et al. , A proteome-scale map of the human interactome network. Cell 159, 1212–1226 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rual JF et al. , Towards a proteome-scale map of the human protein-protein interaction network. Nature 437, 1173–1178 (2005). [DOI] [PubMed] [Google Scholar]
- 13.Oughtred R et al. , The BioGRID interaction database: 2019 update. Nucleic Acids Res 47, D529–D541 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuchaiev O, Rasajski M, Higham DJ, Przulj N, Geometric de-noising of protein-protein interaction networks. PLoS Comput Biol 5, e1000454 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mackay JP, Sunde M, Lowry JA, Crossley M, Matthews JM, Protein interactions: is seeing believing? Trends Biochem Sci 32, 530–531 (2007). [DOI] [PubMed] [Google Scholar]
- 16.Evans R et al. , Protein complex prediction with AlphaFold-Multimer. bioRxiv, 2021.2010.2004.463034 (2022).
- 17.Bryant P, Pozzati G, Elofsson A, Improved prediction of protein-protein interactions using AlphaFold2. Nat Commun 13, 1265 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Humphreys IR et al. , Computed structures of core eukaryotic protein complexes. Science 374, eabm4805 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pei J, Zhang J, Cong Q, Human mitochondrial protein complexes revealed by large-scale coevolution analysis and deep learning-based structure modeling. bioRxiv, (2021). [DOI] [PubMed]
- 20.Larsen SC, Hendriks IA, Lyon D, Jensen LJ, Nielsen ML, Systems-wide Analysis of Serine ADP-Ribosylation Reveals Widespread Occurrence and Site-Specific Overlap with Phosphorylation. Cell Rep 24, 2493–2505 e2494 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Ayyappan V et al. , ADPriboDB 2.0: an updated database of ADP-ribosylated proteins. Nucleic acids research 49, D261–D265 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Daniels CM, Ong S-E, Leung AK, The promise of proteomics for the study of ADP-ribosylation. Molecular cell 58, 911–924 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhen Y, Yu Y, Proteomic analysis of the downstream signaling network of PARP1. Biochemistry 57, 429–440 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jumper J et al. , Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Y, Wang J, Ding M, Yu Y, Site-specific characterization of the Asp-and Glu-ADP-ribosylated proteome. Nature methods 10, 981–984 (2013). [DOI] [PubMed] [Google Scholar]
- 26.Zhen Y, Zhang Y, Yu Y, A cell-line-specific atlas of PARP-mediated protein Asp/Glu-ADP-ribosylation in breast cancer. Cell reports 21, 2326–2337 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.UniProt C, UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res 49, D480–D489 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hermjakob H et al. , IntAct: an open source molecular interaction database. Nucleic Acids Res 32, D452–455 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xenarios I et al. , DIP: the database of interacting proteins. Nucleic Acids Res 28, 289–291 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zanzoni A et al. , MINT: a Molecular INTeraction database. FEBS Lett 513, 135–140 (2002). [DOI] [PubMed] [Google Scholar]
- 31.Kriventseva EV et al. , OrthoDB v10: sampling the diversity of animal, plant, fungal, protist, bacterial and viral genomes for evolutionary and functional annotations of orthologs. Nucleic Acids Res 47, D807–D811 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fu L, Niu B, Zhu Z, Wu S, Li W, CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Altschul SF et al. , Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25, 3389–3402 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Katoh K, Standley DM, MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30, 772–780 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McGuffin LJ, Bryson K, Jones DT, The PSIPRED protein structure prediction server. Bioinformatics 16, 404–405 (2000). [DOI] [PubMed] [Google Scholar]
- 36.Meszaros B, Erdos G, Dosztanyi Z, IUPred2A: context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res 46, W329–W337 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tunyasuvunakool K et al. , Highly accurate protein structure prediction for the human proteome. Nature 596, 590–596 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kabsch W, Sander C, Dictionary of protein secondary structure: pattern recognition of hydrogen‐bonded and geometrical features. Biopolymers: Original Research on Biomolecules 22, 2577–2637 (1983). [DOI] [PubMed] [Google Scholar]
- 39.Pei J, Grishin NV, AL2CO: calculation of positional conservation in a protein sequence alignment. Bioinformatics 17, 700–712 (2001). [DOI] [PubMed] [Google Scholar]
- 40.Huang da W, Sherman BT, Lempicki RA, Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4, 44–57 (2009). [DOI] [PubMed] [Google Scholar]
- 41.Shannon P et al. , Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13, 2498–2504 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Su G, Kuchinsky A, Morris JH, States DJ, Meng F, GLay: community structure analysis of biological networks. Bioinformatics 26, 3135–3137 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sherman BT et al. , DAVID: a web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res, (2022). [DOI] [PMC free article] [PubMed]
- 44.Wang L et al. , The cross-talk between PARylation and SUMOylation in C/EBPβ at K134 site participates in pathological cardiac hypertrophy. International journal of biological sciences 18, 783 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vivelo CA, Ayyappan V, Leung AK, Poly (ADP-ribose)-dependent ubiquitination and its clinical implications. Biochemical pharmacology 167, 3–12 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wei H, Yu X, Functions of PARylation in DNA damage repair pathways. Genomics, proteomics & bioinformatics 14, 131–139 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Páhi ZG, Borsos BN, Pantazi V, Ujfaludi Z, Pankotai T, PARylation during transcription: insights into the fine-tuning mechanism and regulation. Cancers 12, 183 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ke Y, Zhang J, Lv X, Zeng X, Ba X, Novel insights into PARPs in gene expression: regulation of RNA metabolism. Cellular and Molecular Life Sciences 76, 3283–3299 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhen Y, Zhang Y, Yu Y, A Cell-Line-Specific Atlas of PARP-Mediated Protein Asp/Glu-ADP-Ribosylation in Breast Cancer. Cell Rep 21, 2326–2337 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Maga G, Hubscher U, Proliferating cell nuclear antigen (PCNA): a dancer with many partners. Journal of cell science 116, 3051–3060 (2003). [DOI] [PubMed] [Google Scholar]
- 51.González-Magaña A, Blanco FJ, Human PCNA structure, function, and interactions. Biomolecules 10, 570 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Prestel A et al. , The PCNA interaction motifs revisited: thinking outside the PIP-box. Cellular and Molecular Life Sciences 76, 4923–4943 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hara K et al. , Structure of proliferating cell nuclear antigen (PCNA) bound to an APIM peptide reveals the universality of PCNA interaction. Acta Crystallographica Section F: Structural Biology Communications 74, 214–221 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.De March M et al. , Structural basis of human PCNA sliding on DNA. Nature communications 8, 1–7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gromadzka AM, Steckelberg A-L, Singh KK, Hofmann K, Gehring NH, A short conserved motif in ALYREF directs cap-and EJC-dependent assembly of export complexes on spliced mRNAs. Nucleic acids research 44, 2348–2361 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pühringer T et al. , Structure of the human core transcription-export complex reveals a hub for multivalent interactions. Elife 9, e61503 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Golovanov AP, Hautbergue GM, Tintaru AM, Lian L-Y, Wilson SA, The solution structure of REF2-I reveals interdomain interactions and regions involved in binding mRNA export factors and RNA. Rna 12, 1933–1948 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Luo M-J et al. , Pre-mRNA splicing and mRNA export linked by direct interactions between UAP56 and Aly. Nature 413, 644–647 (2001). [DOI] [PubMed] [Google Scholar]
- 59.Dominguez-Sanchez MS, Barroso S, Gomez-Gonzalez B, Luna R, Aguilera A, Genome instability and transcription elongation impairment in human cells depleted of THO/TREX. PLoS Genet 7, e1002386 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hautbergue GM et al. , UIF, a New mRNA export adaptor that works together with REF/ALY, requires FACT for recruitment to mRNA. Current Biology 19, 1918–1924 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Graille M, Rougemaille M, ERH proteins: connecting RNA processing to tumorigenesis? Current Genetics 66, 689–692 (2020). [DOI] [PubMed] [Google Scholar]
- 62.Pang K et al. , ERH Gene and Its Role in Cancer Cells. Front Oncol 12, 900496 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Smyk A, Szuminska M, Uniewicz KA, Graves LM, Kozlowski P, Human enhancer of rudimentary is a molecular partner of PDIP46/SKAR, a protein interacting with DNA polymerase δ and S6K1 and regulating cell growth. The FEBS journal 273, 4728–4741 (2006). [DOI] [PubMed] [Google Scholar]
- 64.Liu L, Rodriguez-Belmonte EM, Mazloum N, Xie B, Lee MY, Identification of a novel protein, PDIP38, that interacts with the p50 subunit of DNA polymerase δ and proliferating cell nuclear antigen. Journal of Biological Chemistry 278, 10041–10047 (2003). [DOI] [PubMed] [Google Scholar]
- 65.Zhang S, Lee EY, Lee MY, Zhang D, POLDIP3 facilitates the activation and maintenance of DNA damage checkpoint in response to replication stress (2022). [DOI] [PMC free article] [PubMed]
- 66.Łukasik A, Uniewicz KA, Kulis M, Kozlowski P, Ciz1, a p21Cip1/Waf1‐interacting zinc finger protein and DNA replication factor, is a novel molecular partner for human enhancer of rudimentary homolog. The FEBS journal 275, 332–340 (2008). [DOI] [PubMed] [Google Scholar]
- 67.Schwartz S et al. , Transcriptome-wide mapping reveals widespread dynamic-regulated pseudouridylation of ncRNA and mRNA. Cell 159, 148–162 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Meier UT, The many facets of H/ACA ribonucleoproteins. Chromosoma 114, 1–14 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Venteicher AS et al. , A human telomerase holoenzyme protein required for Cajal body localization and telomere synthesis. Science 323, 644–648 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ghanim GE et al. , Structure of human telomerase holoenzyme with bound telomeric DNA. Nature 593, 449–453 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen L et al. , An activity switch in human telomerase based on RNA conformation and shaped by TCAB1. Cell 174, 218–230. e213 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhan X, Yan C, Zhang X, Lei J, Shi Y, Structures of the human pre-catalytic spliceosome and its precursor spliceosome. Cell research 28, 1129–1140 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Padilla PI et al. , Association of guanine nucleotide-exchange protein BIG1 in HepG2 cell nuclei with nucleolin, U3 snoRNA, and fibrillarin. Proceedings of the National Academy of Sciences 105, 3357–3361 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Verba KA et al. , Atomic structure of Hsp90-Cdc37-Cdk4 reveals that Hsp90 traps and stabilizes an unfolded kinase. Science 352, 1542–1547 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hoek M, Stillman B, Chromatin assembly factor 1 is essential and couples chromatin assembly to DNA replication in vivo. Proceedings of the National Academy of Sciences 100, 12183–12188 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Uwada J et al. , The p150 subunit of CAF-1 causes association of SUMO2/3 with the DNA replication foci. Biochemical and biophysical research communications 391, 407–413 (2010). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S1. D/E-PARylated proteins and their PARylation sites.
Supplementary Table S2. Top-scoring protein-protein interactions by AlphaFold (contact probability score ≥ 0.9).
Supplementary Figure S1. Sequence conservation distribution of D/E positions.
Data Availability Statement
Structural models of predicted complexes involving D/E-PARylated proteins can be downloaded at: https://conglab.swmed.edu/DE_PARylation/.