

Abstract
Recent surges in mass spectrometry-based proteomics studies demand a concurrent rise in speedy and optimized data processing tools and pipelines. Although several stand-alone bioinformatics tools exist that provide protein–protein interaction (PPI) data, we developed Protein Interaction Network Extractor (PINE) as a fully automated, user-friendly, graphical user interface application for visualization and exploration of global proteome and post-translational modification (PTM) based networks. PINE also supports overlaying differential expression, statistical significance thresholds, and PTM sites on functionally enriched visualization networks to gain insights into proteome-wide regulatory mechanisms and PTM-mediated networks. To illustrate the relevance of the tool, we explore the total proteome and its PTM-associated relationships in two different nonalcoholic steatohepatitis (NASH) mouse models to demonstrate different context-specific case studies. The strength of this tool relies in its ability to (1) perform accurate protein identifier mapping to resolve ambiguity, (2) retrieve interaction data from multiple publicly available PPI databases, and (3) assimilate these complex networks into functionally enriched pathways, ontology categories, and terms. Ultimately, PINE can be used as an extremely powerful tool for novel hypothesis generation to understand underlying disease mechanisms.
Keywords: proteomics, data visualization, enriched networks, automated bioinformatics tool, post-translational modifications
Graphical Abstract
INTRODUCTION
The aim of mass spectrometry (MS) based proteomics studies is to identify and quantify a wide range of proteins and PTMs in biological samples of interest to uncover disease mechanisms and novel protein biomarkers.1,2 For the last two decades, liquid chromatography–tandem mass spectrometry (LC–MS/MS) of complex mixtures of proteins in data-dependent acquisition (DDA) mode3 has been the standard in proteomics for sampling breadth and discovery. Recent improvements in mass spectrometer design and bioinformatics algorithms have resulted in the development of another peptide sampling method: data-independent acquisition (DIA)4 which aims to combine identification breadth of DDA with the sensitivity and reproducibility of targeted MS assays such as multiple reaction monitoring (MRM) or parallel reaction monitoring (PRM).5,6 These proteomic technological developments have led to a software evolution going from stand-alone software tools for MS data processing to large-scale complex computational pipelines transforming raw data into biological discoveries.7 With these advances, we can now routinely analyze large-scale quantitative differences between samples but more importantly we can also detect and quantify an array of protein PTMs that were previously challenging.8-10
The unambiguous identification and quantitation of PTM sites by classical bottom up methods still relies on proteotypic peptide identification, modified amino acid residue localization, peptide ionization and fragmentation, MS instrument accuracy, as well as its abundance in the biological sample of interest.11,12 Despite these challenges, PTMs can be biologically insightful since they serve to diversify the function of proteins by acting as key regulators of protein function, structure, and PPIs in response to perturbations.13 PTMs can occur on multiple sites of the same protein or the same site of a protein modified by different types of PTMs that could potentially serve as mechanistic switches.14-16 An emerging challenge is therefore to decipher how PTMs, individually or in combination, modulate protein interactions to ultimately inform our understanding of key molecular processes and regulatory functions.17
Several existing bioinformatics tools are available for visualizing and analyzing PPI networks. Databases targeted at collecting and storing PPIs, such as STRING,18 GeneMA-NIA,19 and Reactome20 are publicly available and easily accessible. Similarly, KEGG (Kyoto Encyclopedia of Genes and Genomes),21 Reactome, WikiPathways,22 and Gene Ontology (GO)23,24 act as knowledge bases to provide functional and biological implications of proteins. Ingenuity Pathway Analysis (IPA)25 builds on its comprehensive, manually curated knowledge base to provide biological insights from expression analysis experiments. PTMapper,26 Phospho-Path,27 and PTMOracle28 allow users to covisualize and coanalyze PTM data in the realm of PPI networks and helps improve our understanding of PTM-associated relationships. Analogously, piNET,29 another PTM-centric visualization tool, maps peptides to proteins, performs upstream analysis of PTM sites, and conducts pathway analysis. Despite this, however, there is a sparsity of tools that (1) perform protein-centric PPIs and functional enrichment analysis while maintaining the protein-gene relationship from public databases which predominantly contains gene-centric information, (2) address ambiguity arising from combining data retrieved through various independent databases, and (3) simultaneously explore the regulatory role of PTMs and their involvement in PPIs and biological function.
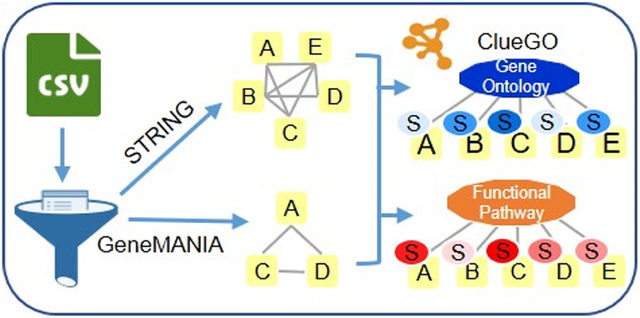
With the goal of addressing these challenges, PINE has been developed as a fully automated, versatile, and easy to use Cytoscape-based tool30 to facilitate mapping, annotation, and visualization of protein and PTM-level quantitative data generated by either targeted, discovery (DDA and DIA), or other proteomics approaches. It allows users to develop systematic searches for proteins of interest and explore complex network visualizations depicting PPIs and functional relationships. In order to improve proteome-wide coverage of PPIs, PINE supports extraction and integration of PPIs from different publicly available databases such as STRING and GeneMANIA with the ability to apply score thresholds based on the nature and quality of the supporting evidence, all while maintaining the protein–gene relationship and resolving ambiguity in the data. PINE utilizes ClueGO31 for functional enrichment analysis over other tools such as BiNGO32 and PIPE33 because of its ability to (1) integrate GO terms and pathway analysis from multiple databases, (2) dynamically create annotation networks based on user query list, and (3) cluster genes and compare functional differences to provide accurate term statistics. PINE also supports overlaying differential expression, statistical significance thresholds, and PTM sites (if applicable) on functionally enriched visualization networks to gain insights into proteome-wide regulatory mechanisms as well as how PTMs might regulate PPIs.
To illustrate how PINE can be used to explore global proteomic signatures, crosstalk between PTMs, and their involvement in PPIs, we present case studies using two mouse models of nonalcoholic steatohepatitis,34 each chosen due to their extreme cellular methylation potential. Nonalcoholic fatty liver disease (NAFLD), the leading cause of chronic liver disease in Western countries, starts as a consequence of defects in diverse metabolic pathways that involve hepatic accumulation of triglycerides. In some cases, this progresses to nonalcoholic steatohepatitis (NASH), a condition characterized by the appearance of inflammation, cellular injury with or without fibrosis, together with steatosis.35 Because there are different causes that can lead to steatosis, we hypothesized that different NASH subtypes may exist, reflecting the variety of mechanisms causing liver fat accumulation, and that identifying each subtype would help us characterize early proteomic changes in NAFLD development as well as key PTMs driving the progression toward NAFLD and subsequent NASH. We assessed this hypothesis by analyzing two mouse models of nonalcoholic steatohepatitis (NASH), (l) the glycine N-methyltransferase knockout (Gnmt −/−) mouse model, which has increased hepatic SAMe and is biologically hypermethylated, and (2) the methionine adenosyltransferase 1A knockout (Mat1a −/−) mouse model, which has hepatic SAMe deficiency and therefore is hypomethylated, to understand the different mechanisms causing liver fat accumulation in NASH.34,36-39 Both animal models spontaneously develop NASH and resemble many aspects of the major subtypes of human disease.34,40 Interestingly, disease in both animal models can be treated by normalization of SAMe levels ,highlighting the importance of maintaining normal hepatic SAMe level and methylation capacity in NASH.34,39,42 This is relevant because it may provide a better understanding of early molecular drivers of NASH and key PTMs that may drive disease progression which would be beneficial in developing early stage biomarkers and therapies for NASH.
METHODS
Sample Preparation for Proteomic Analysis.
Livers were obtained from 10-month old Mat1a −/− male mice in a C57Bl/6 background with hepatic lipid accumulation and their aged-matched wild-type (WT) male sibling littermates,34 and three-month old Gnmt −/− mice in a C57Bl/6 background with hepatic lipid accumulation with age matched WT littermates.36,37 Frozen mouse livers were ground while frozen in a liquid N2-cooled cryohomoginizer (Retsch). Samples were denatured in 8 M urea and 100 mM Tris–HCl, pH 8.0 and ultrasonicated (QSonica) at 4 °C for 10 min in 10 s repeating on/off intervals of 10 s and centrifuged at 16000x for 10 min at 4 °C. The soluble fraction was reduced with DTT (15 mM) for 1 h at 37 °C, alkylated with iodoacetamide (30 mM) for 30 min at room temperature in the dark, diluted to a final concentration of 2 M urea with 100 mM Tris–HCl, pH 8.0, and digested for 16 h on a shaker at 37 °C with a 1:40 ratio of Trypsin/Lys-C mix (Promega). Each sample was desalted using HLB plates (Oasis HLB 30 μm, 5 mg sorbent, Waters).42
Titanium Dioxide Phospho Enrichment.
Titanium dioxide (TiO2) phospho enrichment was performed as previously described.43 Briefly, 400 μg of sample was digested as described above, individually desalted on Oasis HLB cartridges (Water, 10 mg), and eluted in 300 μL of 80% ACN, 5% TFA, 1 mM glycolic acid. Each sample was then incubated in 50 μL of titanium dioxide (TiO2) slurry (30 mg/mL, Glygen Corp, Columbia, MD) at room temperature on a shaker overnight. Then the TiO2 beads were washed twice with 200 μL of 80% ACN, 5% TFA, once with 200 μL of 80% ACN, 0.1% TFA, and eluted in 180 μL of 30% ACN/1% NH4OH and neutralized with 200 μL of 10% FA. Samples were then desalted on Oasis HLB μ-elution plates (Waters) and eluted in 80% ACN, 0.1% FA, dried in a speedvac, and then resuspended in 0.1% FA for LC–MS/MS analysis.
MS Acquisition.
MS data were acquired on Orbitrap Fusion Lumos (Thermo) mass spectrometer connected to an Ultimate 3000 nano LC system (Thermo Scientific) with a 60 min or 120 min gradient operating in DDA-MS or DIA-MS mode.42 Briefly, for DDA analysis, precursor ions were fragmented using HCD and analyzed in the Orbitrap at 15000 K resolution. Monoisotopic precursor selection and dynamic exclusion was enabled and only MS1 signals exceeding 50000 counts triggered the MS2 scans. For DIA analysis, MS1 scans were acquired at 60,000 Hz from mass range 400–1000m/z. DIA MS2 scans were acquired in the Orbitrap at 15,000 Hz with fragmentation in the HCD cell. DIA was performed using 4 Da (150 scan events) windows over the precursor mass range of 400–1000 m/z, and the MS2 mass range was set from 100 to 1500 m/z.
Generation of Spectral Libraries and DIA Analysis.
A lysine PTM enriched peptide assay library42 was used for the DIA analysis and quantitation of individual specimens. Briefly, a hypermethylated DIA assay library was created by DDA acquisitions of 5 SCX fractions from Gnmt −/− mouse livers. This library was supplemented with a DDA acquisition of a methyl-Lys immunoprecipitation from a hepatoma cell line derived from Gnmt −/− mice.41 The library was further supplemented with DDA acquisitions of a “one-pot” acetyl-Lys and succinyl-Lys immunoaffinity enrichment of mitochondrial extract and whole liver lysate as previously described.44
Briefly, raw intensity data for peptide fragments was extracted from DIA files using the open source openSWATH workflow45 against the lysine PTM enriched peptide assay library. Target and decoy peptides were then extracted, scored, and analyzed using the mProphet algorithm46 to determine scoring cut-offs consistent with 1% FDR. Peak group extraction data from each DIA file was combined using the “feature alignment” script, which performs data alignment and modeling analysis across an experimental data set.47 Normalized transition-level data was then processed using the mapDIA software to perform pairwise comparisons between groups at the peptide level.48 All data sets have been made publicly available at Panorama (PXD ID: PXD012621): https://panoramaweb.org/methylation_methods_1.url.
Phospho Enrichment Database Searches and Quantification.
DDA files were converted to mzXML and searched through the Trans Proteomic Pipeline (TPP) using three algorithms, (1) Comet,49 (2) X!tandem! Native scoring,50 and (3) X!tandem! K-scoring51 against a reviewed, mouse canonical protein sequence database, downloaded from the Uniprot52 database on January 24th, 2019, containing 17002 target proteins and 17002 randomized decoy proteins. Target-decoy modeling of peptide spectral matches was performed with PeptideProphet,53 and peptides with a probability score of >95% from the entire experimental data set were imported into Skyline software54 for quantification of precursor extracted ion intensities (XICs). Precursor XICs from each experimental file were extracted against the Skyline library, and peptide XICs with isotope dot product scores >0.8 were filtered for final statistical analysis of proteomic differences.55 Raw peptide intensities were used to calculate pairwise comparisons between experimental groups using the linear mixed effects model built into the open sources MSSTATs (v3.2.2) software suite.56 Peptide abundance differences with an adjusted p-value <0.05 were considered significantly different. The Skyline documents containing precursor XICs from each experimental file are available at Panorama (PXD ID: PXD017324): https://panoramaweb.org/qt1CeZ.url
Software Design and Availability.
PINE is a tool designed to enable visualization of PPI networks and aids in forging biological interpretation through the creation of annotation networks. It allows users to integrate other types of protein data including fold changes, p-values, category, or PTM sites. To facilitate this, all the protein data must be formatted into a comma-separated (CSV) file. Protein data that is formatted into the required input format is parsed and visualized as PPIs networks on the visualization platform, Cytoscape, with nodes representing individual proteins and other protein data mapped onto the nodes. Concurrently, a list of annotation terms is generated from which users can select their terms of interest to create subnetworks depicting annotations linked to their related proteins. The features of PINE are described in more detail in the Results section. PINE is programmed in Python3, and the graphical user interface (GUI) for PINE was created with the Electron framework v6.0.7. Pyinstaller v3.4 was used to wrap PINE’s Python code into an executable for use by the GUI. Prerequisites for using PINE include Cytoscape v3.7 or higher to be installed by users along with Cytoscape apps GeneMANIA and ClueGO. Programmatic access for the PINE workflow is enabled through Cytoscape’s RESTful API methods57 and the py2cytoscape58 utility. The source code for PINE that invoke these respective APIs and the GUI along with additional usage documentation are included in the public GitHub repository, https://github.com/csmc-vaneykjlab/pine, and an executable release is available for Windows 10.
RESULTS
Overview and Features of PINE.
PINE is a visualization tool that provides users with PPI and annotation networks for the integration of different types of protein data. To do so, the PINE framework consists of six primary steps: (1) preprocessing of input, (2) Uniprot mapping for protein to gene transformation, (3) retrieval of STRING PPIs, (4) simultaneous retrieval of GeneMANIA PPIs, (5) merging from both public databases to construct single PPI network, and (6) performing enrichment analysis using Cytoscape plug-in, ClueGO to drive subnetwork creation (Figure 1A).
Figure 1.

Overview of PINE Framework. (A) PPIs derived from public databases (e.g., STRING, GeneMANIA) and functional enrichment-based network annotations from ClueGO are processed by the Cytoscape framework to create PPIs networks. By using PINE, protein or PTM associated differential expression (e.g., fold change) or statistical significance data (e.g., p-value) can also be overlaid onto the Cytoscape networks. (B) Example PPIs network depicting both statistically significant and nonsignificant PTM sites. The rectangular yellow nodes denote proteins (represented by their primary genes), connected to circular nodes representing PTM sites. The color refers to either down-regulated (blue) or up-regulated (orange) sites with statistically significant sites outlined blue. The legend describes the various node types and colors used in visualization of PPI networks
The preprocessing step parses the input containing proteins and their auxiliary data to accurately map protein data onto nodes in the final network. Considerations are made to ensure input data is valid (i.e., fold changes and p-values must be numeric), readily available (i.e., modified peptides must contain PTMs), and nonredundant. This step also allows the user to exclude ambiguous PTM sites with oxidized methionines, missed cleavages, and ragged ends.59,60 Next, the Uniprot mapping step performs an API call to Uniprot to map proteins to their corresponding primary genes. All subsequent steps use the primary gene to represent the input protein in order to remain consistent with various publicly available PPI databases. This step also conducts checks to ensure information is valid (i.e., protein identifiers are truly Uniprot IDs), readily available (i.e., proteins are successfully mapped to a primary gene), and nonredundant. At this level, data ambiguity is resolved by picking the first protein-to-gene match in cases such as (1) many proteoforms mapping to one primary gene and (2) one protein mapping to many primary genes. Additionally, the user also has an option to exclude all ambiguous identifiers from the analysis.
The next step retrieves PPIs from STRING and GeneMANIA and conducts checks to ensure information is valid (i.e., query genes are successfully mapped as a primary interactor), readily available (i.e., query genes are successfully mapped in STRING and/or GeneMANIA) and nonredundant interactions. The merging step consolidates PPI information from STRING and GeneMANIA to create a single, unified PPI network (as shown in Figure 1B). The ClueGO enrichment analysis step identifies enriched gene ontology terms and pathway annotation for all valid (i.e., one-to-one query gene mapping within ClueGO) and readily available (i.e., query genes successfully mapped in ClueGO) genes. Finally, PINE enables selection and visualization of top representative enriched terms into subnetworks using Cytoscape’s RESTful API methods and the py2cytoscape utility. Cytoscape framework supports interactive network visualization through a Zoomable User Interface (ZUI) with customizable features such as zooming, panning, layout, and styling options to manage visual presentation.
Hereby, PINE provides a unique automation solution for robust and thorough data processing to avoid ambiguity, while simultaneously integrating information from various public databases for the creation of functionally enriched visualization networks.
PINE Interface for Visualization.
PINE provides a simple, easy-to-use, graphical user-interface having four main modules: (1) Initial Setup, (2) Parameter Settings, (3) Logging and Data Handling, and (4) Pathway Selection of enriched terms.
The first module is the Setup tab describing a one-time installation guideline required for running the tool. Subsequent uses of PINE automatically register the paths to the necessary packages. Next, the Settings tab (Figure 2A) enables users to customize their analysis with several mandatory and optional parameters. Mandatory parameters include input file containing protein identifiers and related data columns in CSV format, type of run based on the proteomics experiment (for example, PTM-based run type can be selected in the presence of PTM sites), path to output directory and the species of interest. Additionally, in the case of PTM run types, users are required to provide the species database file in FASTA format, digestion enzyme used and modification sites of interest. Optional parameters include: fold change or p-value cutoffs, choice to add blue-colored outline to statistically significant protein or PTM nodes, choice to include or exclude all ambiguous data, choice of PPI databases to be used along with confidence score of interactions, option to include external (nonquery) interactors, functional enrichment analysis based on pathways (comprising of Reactome, KEGG, ClinVar,61 CORUM62 and WikiPathways) or individual GO terms (biological process, cellular component or molecular function), ClueGO grouping level (i.e., representative or specific annotation terms), ClueGO enrichment p-value cutoff, and option to upload a background reference file for the enrichment analysis.
Figure 2.

PINE graphical user interface. (A) Screenshot illustrating the settings tab for easy query upload, providing users the choice to modify parameters for PPIs and annotation analysis. (B) Screenshot illustrating the Pathway Selection tab that lists annotation terms, additionally providing the option to filter, sort, and subsequently select annotation terms of interest in order to generate ontology network.
Once the analysis is submitted using user-defined settings, the interface is redirected to the Log tab which shows the progress of the run as well as records data handling at every step. Once the run is complete, the interface is redirected to the Pathway Selection tab (Figure 2B) which lists enriched annotation terms for the query data set along with term significance, corrected term significance, percent of genes in the cluster associated with the term and total number of term related genes. Top representative terms can be selected based on one or many of the following criterion: (1) terms with adjusted p-value ≤0.01, (2) terms with reasonable coverage based on % genes or number of genes, (3) terms most likely connected to the biological system of interest, and (4) terms comprising of candidate proteins of interest. Filtering and sorting options are also supported on the UI for easy and rapid selection of terms of interest for reanalysis and subnetwork creation which is ultimately rendered using Cytoscape’s network visualization features.
Case Study 1: Identifying and Comparing Enriched Pathways and GO Terms.
Visualization of complex networks to uncover functionally enriched subnetworks can be challenging especially when large clusters of proteins and their PPIs need to be considered. To address this, we performed global quantitative proteomics using DIA across two mouse models to better understand the key commonalities and differences in the underlying functional pathways involved in causing liver fat accumulation in NASH (Figure 3). Using PINE, we first extracted and integrated PPI data from STRING and GeneMANIA for all statistically significant differentially expressed proteins (Bayesian False Discovery Rate ≤0.05) with ≥1.5 log2 fold change by comparing Gnmt −/− mice and Mat1a −/− mice relative to WT littermates. With ClueGO enrichment and Cytoscape operations automated within PINE, we created a subnetwork of top representative enriched terms to reduce visual complexity. Finally, we colored protein nodes based on the observed fold changes (with orange corresponding to upregulation and blue corresponding to downregulation) and enriched functional nodes were colored based on the percentage of upregulated or downregulated protein nodes to indicate activation or inhibition trend for that functional node (Figure 4).
Figure 3.

Hepatic methionine metabolism in different NASH models. Two mouse models of nonalcoholic steatohepatitis (NASH): Gnmt −/− mice (left) have increased hepatic SAMe level and methylation capacity, being biologically hypermethylated, while Mat1a −/− mice (right) are SAMe deficient with reduced methylation capacity and therefore hypomethylated.
Figure 4.

Enriched functional networks generated by PINE across different NASH models. (A) The Gnmt −/− mice model indicates five activated pathways shown as orange central nodes and the one inhibited pathway shown as blue central nodes along with fold-changes from the different protein nodes. (B) The Mat1a −/− mice model indicates two activated pathways shown as orange central nodes and the four inhibited pathway shown as blue central nodes along with fold-changes from the different protein node.
The resulting subnetwork of enriched pathways contained six key functional processes for both NASH mouse models, respectively. The Gnmt −/− mice model showed activation of pathways such as fatty acid metabolism,63 TCA cycle,63,64 glutathione/one carbon metabolism,34,65 peroxisomal protein import,34,66 glycolysis/gluconeogenesis,67,68 and inhibition of pathway such as metabolism of xenobiotics by cytochrome P45034,70 (Figure 4A). On the other hand, The Mat1a −/− mice model showed the reverse trends compared to Gnmt −/− mice model with inhibition of pathways such as fatty acid metabolism,63 TCA cycle,63,64 peroxisomal protein import,34,66 pyruvate metabolism71 and activation of pathways such as glutathione metabolism34,65 and metabolism of xenobiotics by cytochrome P45034,69,70 (Figure 4B). These findings strongly correlate with known functional pathways altered in both NASH mouse models, although their proteomic expression profiles were regulated differently, elucidating key proteins could potentially be used as biomarkers to differentiate subtypes of NASH.34,42,72
With PINE, we also visualized a subnetwork of enriched GO cellular components to identify similarities and differences in the subcellular localization between the two NASH mouse models (Figure 5). In PINE, we can represent differential expression data from multiple comparison groups (for example, Gnmt −/− mice and Mat1a −/− mice relative to their WT littermates) as bar charts wherein the height of the bar represents degree of fold change. Since mitochondria are well-known to play a critical role in the development and pathogenesis of NAFLD,73,74 we focused our cellular subnetwork around the mitochondrial matrix to visualize proteomic signatures between the two mouse models. For example, carbamoyl-phosphate synthase (CPS1), a previously reported candidate biomarker for NASH,75 shown to be enriched in mitochondrial matrix76 was observed to be altered in both NASH mouse models but was significantly downregulated in Mat1a −/− mice relative to WT littermates (log2 fold change of –0.9) and nonsignificantly changed in Gnmt −/− mice relative to WT littermates (log2 fold change of –0.2), indicating that the two mice models might represent different pathophysiologies of NASH disease progression.
Figure 5.

Enriched cellular subnetwork for mitochondrial matrix generated by PINE across different NASH models. Altered cellular components are denoted by central gray nodes. Differential expression data from multiple comparison groups are represented as bar charts and the bar height indicates degree of fold change. (Example: Zoomed view into node CPS1 indicates degree of fold change for Mat1a −/− mice vs WT littermates shown using orange bar and Gnmt −/− mice vs WT littermates shown using blue bar.)
Case Study 2: Exploring Lysine and Arginine PTMs in the Context of Enriched Pathways and GO Terms.
In order to explore PTM crosstalk between methylation, succinylation and acetylation, we performed PTM analysis using DIA across the two NASH mouse models to visualize the relationships between quantified peptide/PTM/protein moieties in the context of NASH pathophysiology. Using PINE, we mapped all statistically significant differentially expressed PTM sites (Bayesian False Discovery Rate ≤0.05) with ≥1.5 log2 fold change between Gnmt −/− mice and Mat1a −/− mice relative to their WT littermates along with their functionally enriched pathways to provide a site specific PTM network view to shed light onto Lysine and Arginine controlled cascades that may be involved (Figure 6).
Figure 6.

Multi-PTM network illustrating crosstalk between methylation, acetylation and succinylation. Functionally enriched pathways are shown by central gray nodes along with gene connections shown by yellow nodes. The PTM site level quantitative differences from each mouse model are shown as a bar chart (Gnmt −/− mice indicated by blue bar and Mat1a −/− mice by orange bar). Sites are indicated with unimod accession in curly brackets (Example, K{34}179 and K{36}179 on gene BHMT would indicate lysine monomethylation and lysine dimethylation events on site 179). Ambiguous sites are indicated using double asterisk (**) symbol next to the site. (Example, K{1}110** on gene GLUD1.)
The resulting subnetwork of enriched pathways contained five key functional processes for both NASH mouse models including amino acid metabolism,34,77 lipid metabolism,34,77-79 PPAR signaling,79 peroxisomal protein import,80,81 and glycolysis/gluconeogenesis,82 some of which were also enriched in the global proteome analysis (Figure 4A,B). PINE also provides the option to exclude ambiguous sites with oxidized methionines, miscleaved versions, and ragged ends.59,60 In cases where PINE found ambiguity in PTM site identity, we indicated it using double asterisk (**) symbol next to the site. For example, BHMT has two dimethylated peptidoforms: one with oxidized methionine and one without. We also observed different PTMs localized to the same protein indicating possibility for modulating biochemical functions through positive and negative regulatory interactions.77 For example, Betaine—homocysteine S-methyltransferase 1 (BHMT) is a well-known target for PTMs such as acetylation and trimethylation, which may be responsible for BHMT localization.83 This enzyme plays an important role in regulation of hepatic lipid metabolism to protect the liver from triacylglycerol accumulation.34,64,84 A closer look at this protein revealed previously unreported lysine monomethylation and dimethylation events at site 179 that are regulated differently across the two NASH models, but their biological significance is yet to be determined. Understanding the crosstalk between these PTMs and regulation of their enzymatic mediators may represent a promising avenue for therapeutics in fatty liver disease.85
Case Study 3: Exploring Protein Phosphorylation in the Context of Enriched Pathways and GO Terms.
Phosphoproteomics, or the large-scale determination of protein phosphorylation status, can be used to characterize and compare a variety of disease states but has been applied to NASH in only a handful of studies. For this use case, we performed global phosphoproteomics analysis using DDA from Gnmt −/− mice relative to WT littermates to look for differentially expressed phosphorylation sites to elucidate the role of kinase activity and regulation in NASH. Using PINE, we identified multiple enriched pathways including TGF-beta signaling,86,87 insulin signaling,88,89 glucagon signaling pathway,88,89 ChREBP mediated metabolic gene expression,88,89 bile acid and bile salt metabolism,89 signaling by Rho GTPases,90 EGFR1 signaling pathway,91 AMPK signaling,92,93 mTORC1-mediated signaling42,94,95 and translation factors.42,96 Since the complex interplay of AMPK and mTORC1 is rapidly gaining clinical relevance in metabolic disease,97-99 we created a subnetwork focused around the crosstalk between mTORC1 and AMPK (Figure 7). AMPK activation has been known to enhance mitochondrial biogenesis and mitophagy and inhibit mTORC1, thus preventing excess-nutrient-induced hepatic lipid accumulation.100
Figure 7.

Phospho-proteome network illustrating significant differentially expressed PTM sites using PINE. Inhibited pathways such as mTORC1-mediated signaling and translation factors are represented as central blue nodes whereas activated pathway such as AMPK signaling pathway are represented as central orange nodes along with gene connections represented as yellow nodes. The PTM site level quantitative differences between Gnmt −/− mice relative to WT littermates are represented as blue for downregulated nodes or orange for upregulated nodes connected to their respective gene nodes. We included both significant and nonsignificant differentially expressed sites indicated by blue node outline and no node outline, respectively.
Our phosphoproteome-based subnetwork confirmed these findings with several upregulated phosphosites from proteins involved in AMPK signaling as well as several downregulated phosphosites from proteins involved in mTORC1-mediated signaling and translation factors.42,94-96 In order to take a complete look at all observed phosphosites associated with these pathways, we included both significant and nonsignificant differentially expressed sites indicated by blue node outline and no node outline, respectively. This network also revealed two significantly differentially expressed phosphosites (Threonine 214 and Threonine 507) of Eukaryotic translation initiation factor 4 gamma proteins (EIF4G1 and EIF4G2) that have been previously reported to be phosphorylated.101,102 We noted that both phosphosites were down-regulated (adjusted p-value ≤0.05 and log2 fold change of −1.0) and have been shown to be involved in mTORC1 regulated translation control, mitosis and protein synthesis. Thus, AMPK could potentially act as a metabolic checkpoint by activating catabolic processes and inhibiting anabolic processes, in part, by negatively regulating mTORC1 signaling and enhancing autophagy flux.100,103,104 Although more clinical research is required to elucidate the role of AMPK and mTORC1 in NASH, novel therapies for metabolic disease could emerge from pharmacological manipulation of this interplay97-99,103
DISCUSSION
We have developed a fully automated, easy-to-use bioinformatics tool called PINE to aid researchers in extracting and visualizing differentially expressed proteins or PTMs and their interaction or functional networks from different types of mass-spectrometry data sets including DDA-, DIA-, and PTM-based experiments. PINE performs accurate protein identifier mapping to resolve ambiguity, retrieve PPIs from multiple publicly available interaction databases, and assimilate these complex networks into meaningful functionally enriched pathways visualized as snapshots of the proteome. Using two mouse models of NASH, we demonstrated that PINE can be used to gain biological insights that can be derived from linking observed protein modification changes to significantly altered protein pathways.
We acknowledge that there are certain areas for improvement within PINE, for example, we can increase support beyond the three commonly used organisms in proteomics profiling, i.e., Homo sapiens, Mus musculus, and Rattus norvegicus, as well, the number of public databases accessed for both interactions and annotations can be expanded along with improved filtering features based on confidence score or interaction sources. An interesting approach would be to compute edge weights based on how many database sources the interactions are present in or to apply the method carried out by Hierarchical HotNet105 which simultaneously combines network interactions and vertex scores to construct a hierarchy of altered subnetworks. In the future extension of PINE, we also plan to improve network coverage by allowing synonym (or secondary) interactors and include upstream regulators to depict enzyme–substrate relationships. Identification of Single Amino Acid Polymorphisms (SAPs) could also be an important addition to this tool, playing a role in exploring how PTM sites containing SAPs affect underlying disease mechanisms. Additionally, due to the challenges involved in the unambiguous identification and localization of PTM, we recommend assessing the validity of these sites using tools such as Ascore,106 PTMProphet,107 LuciPHOr2,108 PIQUed,109 Peptide Collapse plugin for Perseus,110 Inference of Peptidoforms (IPF),111 and Thesaurus112 prior to visualizing these data sets in PINE.
In summary, PINE provides a versatile, user-friendly, automated solution for network and pathway visualization specialized to handle bottom-up proteomics data. Using different case studies, we have demonstrated that PINE can seamlessly combine data from various public databases, while providing a variety of customization options, enabling profound exploration and visualization of global proteome and PTM relationships. Ultimately, PINE can serve as an extremely powerful tool for novel hypothesis generation toward understanding disease mechanisms and potential therapeutics.
ACKNOWLEDGMENTS
We thank Erika Glazer for her generous support through the Erika J. Glazer Endowed Chair in Women’s Heart Health and funds from the Barbra Streisand Women’s Heart Center and the Smidt Heart Institute at Cedars-Sinai Medical Center as well as grants from National Institutes of Health, 5R01HL132075-03, 2R01HL111362-05A1, 1R01HL144509-01, and 5R01DK107288-04. This research was also supported by CSMC proteomics and metabolomics core.
Footnotes
The authors declare no competing financial interest.
Contributor Information
Niveda Sundararaman, Advanced Clinical Biosystems Research Institute, The Smidt Heart Institute, Cedars Sinai Medical Center, Los Angeles, California 90048, United States.
James Go, Advanced Clinical Biosystems Research Institute, The Smidt Heart Institute, Cedars Sinai Medical Center, Los Angeles, California 90048, United States.
Aaron E. Robinson, Advanced Clinical Biosystems Research Institute, The Smidt Heart Institute, Cedars Sinai Medical Center, Los Angeles, California 90048, United States
José M. Mato, CIC bioGUNE, Centro de Investigación Biomédica en Red de Enfermedades Hepáticas y Digestivas (Ciberehd), 48160 Derio, Bizkaia, Spain
Shelly C. Lu, Division of Digestive and Liver Diseases, Cedars-Sinai Medical Center, Los Angeles, California 90048, United States
Jennifer E. Van Eyk, Advanced Clinical Biosystems Research Institute, The Smidt Heart Institute, Cedars Sinai Medical Center, Los Angeles, California 90048, United States
Vidya Venkatraman, Advanced Clinical Biosystems Research Institute, The Smidt Heart Institute, Cedars Sinai Medical Center, Los Angeles, California 90048, United States; Phone: 310-423-2998.
REFERENCES
- (1).Han X; Aslanian A; Yates JR Mass Spectrometry for Proteomics. Curr. Opin. Chem. Biol 2008, 12, 483–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Mcdonald H; Yates JR Shotgun Proteomics and Biomarker Discovery. Dis. Markers 2002, 18, 99–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Zhang Y; Fonslow BR; Shan B; Baek MC; Yates JR Protein analysis by shotgun/bottom-up proteomics. Chem. Rev 2013, 113, 2343–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Doerr A DIA mass spectrometry. Nat. Methods 2015, 12, 35–35. [Google Scholar]
- (5).Liebler DC; Zimmerman LJ Targeted quantitation of proteins by mass spectrometry. Biochemistry 2013, 52, 3797–3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Hu A; Noble WS; Wolf-Yadlin A Technical advances in proteomics: new developments in data-independent acquisition. F1000Research 2016, 5, 419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Lavallée-Adam M; Park SKR; Martínez-Bartolomé S; He L; Yates JR From Raw Data to Biological Discoveries: A Computational Analysis Pipeline for Mass Spectrometry-Based Proteomics. J. Am. Soc. Mass Spectrom 2015, 26, 1820–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Henriksen P; Wagner SA; Weinert BT; Sharma S; Bačinskaja G; Rehman M; Juffer AH; Walther TC; Lisby M; Choudhary C Proteome-wide Analysis of Lysine Acetylation Suggests its Broad Regulatory Scope in Saccharomyces cerevisiae. Mol. Cell. Proteomics 2012, 11, 1510–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Sharma K; D’Souza RCJ; Tyanova S; Schaab C; Wiśniewski JR; Cox J; Mann M Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr-based signaling. Cell Rep. 2014, 8, 1583–1594. [DOI] [PubMed] [Google Scholar]
- (10).Parker BL; Shepherd NE; Trefely S; Hoffman NJ; White MY; Engholm-Keller K; Hambly BD; Larsen MR; James DE; Cordwell SJ Structural Basis for Phosphorylation and Lysine Acetylation Crosstalk in a Kinase Motif Associated with Myocardial Ischemia and Cardioprotection. J. Biol. Chem 2014, 289, 25890–25906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Kim MS; Zhong J; Pandey A Common errors in mass spectrometry-based analysis of post-translational modifications. Proteomics 2016, 16, 700–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Olsen JV; Mann M Status of large-scale analysis of post-translational modifications by mass spectrometry. Mol. Cell. Proteomics 2013, 12, 3444–3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Karve TM; Cheema AK Small changes huge impact: the role of protein posttranslational modifications in cellular homeostasis and disease. J. Amino Acids 2011, 2011, 207691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Nussinov R; Tsai CJ; Xin F; Radivojac P Allosteric post-translational modification codes. Trends Biochem. Sci 2012, 37, 447–455. [DOI] [PubMed] [Google Scholar]
- (15).Seet BT; Dikic I; Zhou MM; Pawson T Reading protein modifications with interaction domains. Nat. Rev. Mol. Cell Biol 2006, 7, 473–483. [DOI] [PubMed] [Google Scholar]
- (16).Bannister AJ; Kouzarides T Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Lothrop AP; Torres MP; Fuchs SM Deciphering post-translational modification codes. FEBS Lett. 2013, 587, 1247–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Szklarczyk D; Gable AL; Lyon D; Junge A; Wyder S; Huerta-Cepas J; Simonovic M; Doncheva NT; Morris JH; Bork P; Jensen LJ STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Montojo J; Zuberi K; Rodriguez H; Kazi F; Wright G; Donaldson SL; Morris Q; Bader GD GeneMANIA Cytoscape plugin: fast gene function predictions on the desktop. Bioinformatics 2010, 26, 2927–2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Fabregat A; Sidiropoulos K; Garapati P; Gillespie M; Hausmann K; Haw R; Jassal B; Jupe S; Korninger F; McKay S; Matthews L; May B; Milacic M; Rothfels K; Shamovsky V; Webber M; Weiser J; Williams M; Wu G; Stein L; Hermjakob H; D’Eustachio P The Reactome pathway Knowledgebase. Nucleic Acids Res. 2016, 44, D481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Kanehisa M; Goto S KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Kutmon M; Riutta A; Nunes N; Hanspers K; Willighagen EL; Bohler A; Mélius J; Waagmeester A; Sinha SR; Miller R; Coort SL; Cirillo E; Smeets B; Evelo CT; Pico AR WikiPathways: capturing the full diversity of pathway knowledge. Nucleic Acids Res. 2016, 44, D488–D494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Ashburner M; Ball CA; Blake JA; Botstein D; Butler H; Cherry JM; Davis AP; Dolinski K; Dwight SS; Eppig JT; Harris MA; Hill DP; Issel-Tarver L; Kasarskis A; Lewis S; Matese JC; Richardson JE; Ringwald M; Rubin GM; Sherlock G Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet 2000, 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).The Gene Ontology Consortium. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019, 47, D330–D338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Krämer A; Green J; Pollard J Jr; Tugendreich S Causal analysis approaches in ingenuity pathway analysis. Bioinformatics 2014, 30, 523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Narushima Y; Kozuka-Hata H; Tsumoto K; Inoue JI; Oyama M Quantitative phosphoproteomics-based molecular network description for high-resolution kinase-substrate interactome analysis. Bioinformatics 2016, 32, 2083–2088. [DOI] [PubMed] [Google Scholar]
- (27).Raaijmakers LM; Giansanti P; Possik PA; Mueller J; Peeper DS; Heck AJR; Altelaar AFM PhosphoPath: Visualization of Phosphosite-centric Dynamics in Temporal Molecular Networks. J. Proteome Res 2015, 14, 4332–4341. [DOI] [PubMed] [Google Scholar]
- (28).Tay AP; Pang CNI; Winter DL; Wilkins MR PTMOracle: A Cytoscape App for Covisualizing and Coanalyzing Post-Translational Modifications in Protein Interaction Networks. J. Proteome Res 2017, 16, 1988–2003. [DOI] [PubMed] [Google Scholar]
- (29).Shamsaei B, Chojnacki S, Pilarczyk M, Najafabadi M, Chen C, Ross K, Matlock A, Muhlich J, Chutipongtanate S, Vidovic D, Sharma V, Vasiliauskas J, Jaffe J, MacCoss M, Wu C, Pillai A, Ma’ayan A, Schurer S, Medvedovic M, Meller J: piNET: a versatile web platform for downstream analysis and visualization of proteomics data. bioRxiv 2019. DOI: 10.1101/607432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Shannon P; Markiel A; Ozier O; Baliga NS; Wang JT; Ramage D; Amin N; Schwikowski B; Ideker T Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Bindea G; Mlecnik B; Hackl H; Charoentong P; Tosolini M; Kirilovsky A; Fridman WH; Pagès F; Trajanoski Z; Galon J ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Maere S; Heymans K; Kuiper M BiNGO: a Cytoscape plugin to assess overrepresentation of Gene Ontology categories in Biological Networks. Bioinformatics 2005, 21, 3448–3449. [DOI] [PubMed] [Google Scholar]
- (33).Ramos H; Shannon P; Aebersold R The protein information and property explorer: an easy-to-use, rich-client web application for the management and functional analysis of proteomic data. Bioinformatics 2008, 24, 2110–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Alonso C; Fernández-Ramos D; Varela-Rey M; Martínez-Arranz I; Navasa N; Van Liempd SM; Lavín Trueba JL; Mayo R; Ilisso CP; de Juan VG; Iruarrizaga-Lejarreta M; delaCruz-Villar L; Mincholé I; Robinson A; Crespo J; Martín-Duce A; Romero-Gómez M; Sann H; Platon J; Van Eyk J; Aspichueta P; Noureddin M; Falcón-Pérez JM; Anguita J; Aransay AM; Martínez-Chantar ML; Lu SC; Mato JM Metabolomic Identification of Subtypes of Nonalcoholic Steatohepatitis. Gastroenterology 2017, 152, 1449–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Cohen JC; Horton JD; Hobbs HH Human fatty liver disease: old questions and new insights. Science 2011, 332, 1519–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Varela-Rey M; Martínez-López N; Fernández-Ramos D; Embade N; Calvisi DF; Woodhoo A; Rodríguez J; Fraga MF; Julve J; Rodríguez-Millán E; Frades I; Torres L; Luka Z; Wagner C; Esteller M; Lu SC; Martínez-Chantar ML; Mato JM Fatty liver and fibrosis in glycine N-methyltransferase knockout mice is prevented by nicotinamide. Hepatology 2010, 52, 105–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Luka Z; Capdevila A; Mato JM; Wagner CA Glycine N-methyltransferase knockout mouse model for humans with deficiency of this enzyme. Transgenic Res. 2006, 15, 393–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Lu SC; Mato JM S-adenosylmethionine in liver health, injury, and cancer. Physiol. Rev 2012, 92, 1515–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Lu SC; Alvarez L; Huang ZZ; Chen L; An W; Corrales FJ; Avila MA; Kanel G; Mato JM Methionine adenosyltransferase 1A knockout mice are predisposed to liver injury and exhibit increased expression of genes involved in proliferation. Proc. Natl. Acad. Sci. U. S. A 2001, 98, 5560–5565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Barr J; Vázquez-Chantada M; Alonso C; Pérez-Cormenzana M; Mayo R; Galán A; Caballería J; Martín-Duce A; Tran A; Wagner C; Luka Z; Lu SC; Castro A; Le Marchand-Brustel Y; Martínez-Chantar ML; Veyrie N; Clément K; Tordjman J; Gual P; Mato JM Liquid Chromatography-Mass Spectrometry (LC/MS)-based parallel metabolic profiling of human and mouse model serum reveals putative biomarkers associated with the progression of non-alcoholic fatty liver disease. J. Proteome Res 2010, 9, 4501–4512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Martínez-López N; García-Rodríguez JL; Varela-Rey M; Gutiérrez V; Fernández-Ramos D; Beraza N; Aransay AM; Schlangen K; Lozano JJ; Aspichueta P; Luka Z; Wagner C; Evert M; Calvisi DF; Lu SC; Mato JM; Martínez-Chantar ML Hepatoma Cells from Mice Deficient in Glycine N-Methyltransferase Have Increased RAS Signaling and Activation of Liver Kinase B1. Gastroenterology 2012, 143, 787–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Robinson AE; Binek A; Venkatraman V; Searle BC; Holewinski RJ; Rosenberger G; Parker SJ; Basisty N; Xie X; Lund PJ; Saxena G; Mato JM; Garcia BA; Schilling B; Lu SC; Van Eyk JE Lysine & Arginine Protein Post-translational Modifications by Enhanced DIA Libraries: Quantification in Murine Liver Disease. bioRxiv 2020, DOI: 10.1101/2020.01.17.910943v2. [DOI] [PubMed] [Google Scholar]
- (43).Stachowski MJ; Holewinski RJ; Grote E; Venkatraman V; Van Eyk JE; Kirk JA Phospho-Proteomic Analysis of Cardiac Dyssynchrony and Resynchronization Therapy. Proteomics 2018, 18, e1800079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Basisty N, Meyer JG, Wei L, Gibson BW, Schilling B: Simultaneous Quantification of the Acetylome and Succinylome by ‘One-Pot’ Affinity Enrichment. Proteomics 18 (2018).1800123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Röst HL; Rosenberger G; Navarro P; Gillet L; Miladinović SM; Schubert OT; Wolski W; Collins BC; Malmström J; Malmström L; Aebersold R OpenSWATH enables automated, targeted analysis of data-independent acquisition MS data. Nat. Biotechnol 2014, 32, 219–223. [DOI] [PubMed] [Google Scholar]
- (46).Reiter L; Rinner O; Picotti P; Hüttenhain R; Beck M; Brusniak MY; Hengartner MO; Aebersold R mProphet: automated data processing and statistical validation for large-scale SRM experiments. Nat. Methods 2011, 8, 430–435. [DOI] [PubMed] [Google Scholar]
- (47).Röst HL; Liu Y; D’Agostino G; Zanella M; Navarro P; Rosenberger G; Collins BC; Gillet L; Testa G; Malmstróm L; Aebersold R TRIC: an automated alignment strategy for reproducible protein quantification in targeted proteomics. Nat. Methods 2016, 13, 777–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Teo G; Kim S; Tsou CC; Collins B; Gingras AC; Nesvizhskii AI; Choi H mapDIA:Preprocessing and Statistical Analysis of Quantitative Proteomics Data from Data Independent Acquisition Mass Spectrometry. J. Proteomics 2015, 129, 108–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Eng JK; Jahan TA; Hoopmann MR Comet: An open-source MS/MS sequence database search tool. Proteomics 2013, 13, 22–24. [DOI] [PubMed] [Google Scholar]
- (50).Craig R; Beavis RC TANDEM: matching proteins with tandem mass spectra. Bioinformatics 2004, 20, 1466–1467. [DOI] [PubMed] [Google Scholar]
- (51).MacLean B; Eng JK; Beavis RC; McIntosh M General framework for developing and evaluating database scoring algorithms using the TANDEM search engine. Bioinformatics 2006, 22, 2830–2832. [DOI] [PubMed] [Google Scholar]
- (52).The UniProt Consortium. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Keller A; Nesvizhskii AI; Kolker E; Aebersold R Empirical Statistical Model To Estimate the Accuracy of Peptide Identifications Made by MS/MS and Database Search. Anal. Chem 2002, 74, 5383–5392. [DOI] [PubMed] [Google Scholar]
- (54).MacLean B; Tomazela DM; Shulman N; Chambers M; Finney GL; Frewen B; Kern R; Tabb DL; Liebler DC; MacCoss MJ Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26, 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Schilling B; Rardin MJ; MacLean BX; Zawadzka AM; Frewen BE; Cusack MP; Sorensen DJ; Bereman MS; Jing E; Wu CC; Verdin E; Kahn CR; MacCoss MJ; Gibson BW Platform-independent and Label-free Quantitation of Proteomic Data Using MS1 Extracted Ion Chromatograms in Skyline. Mol. Cell. Proteomics 2012, 11, 202–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Choi M; Chang CY; Clough T; Broudy D; Killeen T; MacLean B; Vitek O MSstats: an R package for statistical analysis of quantitative mass spectrometry-based proteomic experiments. Bioinformatics 2014, 30, 2524–2526. [DOI] [PubMed] [Google Scholar]
- (57).Otasek D; Morris JH; Bouças J; Pico AR; Demchak B Cytoscape Automation: empowering workflow-based network analysis. Genome Biol. 2019, DOI: 10.1186/s13059-019-1758-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Ono K; Muetze T; Kolishovski G; Shannon P; Demchak B CyREST: Turbocharging cytoscape access for external tools via a RESTful API. F1000Research 2015, 4, 478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Zhai B; Villén J; Beausoleil SA; Mintseris J; Gygi SP Phosphoproteome Analysis of Drosophila melanogaster Embryos. J. Proteome Res 2008, 7, 1675–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Fleitz A; Nieves E; Madrid-Aliste C; Fentress SJ; Sibley LD; Weiss LM; Angeletti RH; Che FY Enhanced Detection of Multiply Phosphorylated Peptides and Identification of Their Sites of Modification. Anal. Chem 2013, 85, 8566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Landrum MJ; Lee JM; Riley GR; Jang W; Rubinstein WS; Church DM; Maglott DR ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014, 42, D980–D985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Ruepp A; Waegele B; Lechner M; Brauner B; Dunger-Kaltenbach I; Fobo G; Frishman G; Montrone C; Mewes HW CORUM: the comprehensive resource of mammalian protein complexes–2009. Nucleic Acids Res. 2010, 38, D497–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Fernández-Tussy P; Fernández-Ramos D; Lopitz-Otsoa F; Simón J; Barbier-Torres L; Gomez-Santos B; Nuñez-Garcia M; Azkargorta M; Gutiírrez-de Juan V; Serrano-Macia M; Rodríguez-Agudo R; Iruzubieta P; Anguita J; Castro RE; Champagne D; Rincón M; Elortza F; Arslanow A; Krawczyk M; Lammert F; Kirchmeyer M; Behrmann I; Crespo J; Lu SC; Mato JM; Varela-Rey M; Aspichueta P; Delgado TC; Martínez-Chantar ML miR-873–5p targets mitochondrial GNMT-Complex II interface contributing to non-alcoholic fatty liver disease. Mol. Metab 2019, 29, 40–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Mato JM; Martínez-Chantar ML; Lu SC S-adenosylmethionine metabolism and liver disease. Ann. Hepatol 2013, 12, 183–189. [PMC free article] [PubMed] [Google Scholar]
- (65).Mato JM; Alonso C; Noureddin M; Lu SC Biomarkers and subtypes of deranged lipid metabolism in non-alcoholic fatty liver disease. World J. Gastroenterol 2019, 25, 3009–3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Moreno-Fernandez ME; Giles DA; Stankiewicz TE; Sheridan R; Karns R; Cappelletti M; Lampe K; Mukherjee R; Sina C; Sallese A; Bridges JP; Hogan SP; Aronow BJ; Hoebe K; Divanovic S Peroxisomal β-oxidation regulates whole body metabolism, inflammatory vigor, and pathogenesis of nonalcoholic fatty liver disease. JCI insight 2018, 3, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Chao HW; Chao SW; Lin H; Ku HC; Cheng CF Homeostasis of Glucose and Lipid in Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci 2019, 20, 298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Hughey CC; Trefts E; Bracy DP; James FD; Donahue EP; Wasserman DH Glycine N-methyltransferase deletion in mice diverts carbon flux from gluconeogenesis to pathways that utilize excess methionine cycle intermediates. J. Biol. Chem 2018, 293, 11944–11954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Murray B; Peng H; Barbier-Torres L; Robinson AE; Li TWH; Fan W; Tomasi ML; Gottlieb RA; Eyk JV; Lu Z; Martínez-Chantar ML; Liangpunsakul S; Skill NJ; Mato JM; Lu SC Methionine Adenosyltransferase α1 Is Targeted to the Mitochondrial Matrix and Interacts with Cytochrome P450 2E1 to Lower Its Expression. Hepatology 2019, 70, 2018–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Fisher C; Lickteig A; Augustine L; Ranger-Moore J; Jackson J; Ferguson S; Cherrington N Hepatic Cytochrome P450 Enzyme Alterations in Humans with Progressive Stages of Non-alcoholic Fatty Liver Disease. Drug Metab. Dispos 2009, 37, 2087–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).van der Veen JN; Lingrell S; da Silva RP; Jacobs RL; Vance DE The concentration of phosphatidylethanolamine in mitochondria can modulate ATP production and glucose metabolism in mice. Diabetes 2014, 63, 2620–2630. [DOI] [PubMed] [Google Scholar]
- (72).Sunny NE; Parks EJ; Browning JD; Burgess SC Excessive Hepatic Mitochondrial TCA Cycle and Gluconeogenesis in Humans with Nonalcoholic Fatty Liver Disease. Cell Metab. 2011, 14, 804–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Nassir F; Ibdah JA Role of Mitochondria in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci 2014, 15, 8713–8742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Wei Y; Rector RS; Thyfault JP; Ibdah JA Nonalcoholic fatty liver disease and mitochondrial dysfunction. World J. Gastroenterol 2008, 14, 193–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Rodriguez-Suarez E; Mato JM; Elortza F Proteomics analysis of human nonalcoholic fatty liver. Methods Mol. Biol 2012, 909, 241–258. [DOI] [PubMed] [Google Scholar]
- (76).Nakagawa T; Lomb DJ; Haigis MC; Guarente L SIRT5 Deacetylates Carbamoyl Phosphate Synthetase 1 and Regulates the Urea Cycle. Cell 2009, 137, 560–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Meyer JG; Softic S; Basisty N; Rardin MJ; Verdin E; Gibson BW; Ilkayeva O; Newgard CB; Kahn CR; Schilling B Temporal dynamics of liver mitochondrial protein acetylation and succinylation and metabolites due to high fat diet and/or excess glucose or fructose. PLoS One 2018, 13, e0208973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Liu L; Zhao X; Zhao L; Li J; Yang H; Zhu Z; Liu J; Huang G Arginine Methylation of SREBP1a via PRMT5 Promotes De Novo Lipogenesis and Tumor Growth. Cancer Res. 2016, 76, 1260–1272. [DOI] [PubMed] [Google Scholar]
- (79).Huang L; Liu J; Zhang XO; Sibley K; Najjar SM; Lee MM; Wu Q Inhibition of protein arginine methyltransferase 5 enhances hepatic mitochondrial biogenesis. J. Biol. Chem 2018, 293, 10884–10894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Sandalio LM; Gotor C; Romero LC; Romero-Puertas MC Multilevel Regulation of Peroxisomal Proteome by Post-Translational Modifications. Int. J. Mol. Sci 2019, 20, 4881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Chen XF; Tian MX; Sun RQ; Zhang ML; Zhou LS; Jin L; Chen LL; Zhou WJ; Duan KL; Chen YJ; Gao C; Cheng ZL; Wang F; Zhang JY; Sun YP; Yu HX; Zhao YZ; Yang Y; Liu WR; Shi YH; Xiong Y; Guan KL; Ye D SIRT5 inhibits peroxisomal ACOX1 to prevent oxidative damage and is downregulated in liver cancer. EMBO Rep. 2018, 19, e45124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Zhang X; Yang S; Chen J; Su Z Unraveling the Regulation of Hepatic Gluconeogenesis. Front. Endocrinol. (Lausanne, Switz.) 2019, DOI: 10.3389/fendo.2018.00802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Pérez-Miguelsanz J; Vallecillo N; Garrido F; Reytor E; Pérez-Sala D; Pajares MA Betaine homocysteine S-methyltransferase emerges as a new player of the nuclear methionine cycle. Biochim. Biophys. Acta, Mol. Cell Res 2017, 1864, 1165–1182. [DOI] [PubMed] [Google Scholar]
- (84).Ab Aziz N Role of Betaine Homocysteine Methyltransferase in Regulating Lipid Metabolism in McArdle RH7777 Cells. ERA 2013, DOI: 10.7939/R32Z12W6W. [DOI] [Google Scholar]
- (85).Puchalska P; Crawford PA Multi-dimensional roles of ketone bodies in fuel metabolism, signaling, and therapeutics. Cell Metab. 2017, 25, 262–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Yang L; Roh YS; Song J; Zhang B; Liu C; Loomba R; Seki E TGF-β Signaling in Hepatocytes Participates in Steatohepatitis Through Regulation of Cell Death and Lipid Metabolism. Hepatology 2014, 59, 483–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Federico A; Trappoliere M; Loguercio C Treatment of patients with non-alcoholic fatty liver disease: current views and perspectives. Dig. Liver Dis 2006, 38, 789–801. [DOI] [PubMed] [Google Scholar]
- (88).Gastaldelli A; Cusi K From NASH to diabetes and from diabetes to NASH: Mechanisms and treatment options. JHEP Reports 2019, 1, 312–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Friedman SL; Neuschwander-Tetri BA; Rinella M; Sanyal AJ Mechanisms of NAFLD development and therapeutic strategies. Nat. Med 2018, 24, 908–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Schierwagen R; Maybüchen L; Hittatiya K; Klein S; Uschner FE; Braga TT; Franklin BS; Nickenig G; Strassburg CP; Plat J; Sauerbruch T; Latz E; Lütjohann D; Zimmer S; Trebicka J Statins improve NASH via inhibition of RhoA and Ras. Am. J. Physiol. Gastrointest. Liver Physiol 2016, 311, G724–G733. [DOI] [PubMed] [Google Scholar]
- (91).Czaja MJ A new mechanism of lipotoxicity: Calcium channel blockers as a treatment for nonalcoholic steatohepatitis? Hepatology 2015, 62, 312–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Martínez-López N; Varela-Rey M; Fernández-Ramos D; Woodhoo A; Vázquez-Chantada M; Embade N; Espinosa-Hevia L; Bustamante FJ; Parada LA; Rodriguez MS; Lu SC; Mato JM; Martínez-Chantar ML Activation of LKB1-Akt pathway independent of PI3 Kinase plays a critical role in the proliferation of hepatocellular carcinoma from NASH. Hepatology 2010, 52, 1621–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Martínez-Chantar ML; Vázquez-Chantada M; Garnacho M; Latasa MU; Varela-Rey M; Dotor J; Santamaria M; Martínez-Cruz LA; Parada LA; Lu SC; Mato JM S–Adenosylmethionine Regulates Cytoplasmic HuR Via AMP–Activated Kinase. Gastroenterology 2006, 131, 223–232. [DOI] [PubMed] [Google Scholar]
- (94).Yen CH; Lin YT; Chen HL; Chen SY; Chen YMA The multi-functional roles of GNMT in toxicology and cancer. Toxicol. Appl. Pharmacol 2013, 266, 67–75. [DOI] [PubMed] [Google Scholar]
- (95).Yen CH; Lu YC; Li CH; Lee CM; Chen CY; Cheng MY; Huang SF; Chen KF; Cheng AL; Liao LY; Lee YHW; Chen YMA Functional characterization of glycine N-methyltransferase and its interactive protein DEPDC6/DEPTOR in hepatocellular carcinoma. Mol. Med 2012, 18, 286–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Wattacheril J; Rose KL; Hill S; Lanciault C; Murray CR; Washington K; Williams B; English W; Spann M; Clements R; Abumrad N; Flynn CR NAFLD Phosphoproteomics: A Functional Piece of the Precision Puzzle. Hepatol. Res 2017, 47, 1469–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Hindupur SK; González A; Hall MN The Opposing Actions of Target of Rapamycin and AMP-Activated Protein Kinase in Cell Growth Control. Cold Spring Harbor Perspect. Biol 2015, 7, a019141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Liang Z; Li T; Jiang S; Xu J; Di W; Yang Z; Hu W; Yang Y AMPK: a novel target for treating hepatic fibrosis. Oncotarget 2017, 8, 62780–62792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Smith BK; Marcinko K; Desjardins EM; Lally JS; Ford RJ; Steinberg GR Treatment of nonalcoholic fatty liver disease: role of AMPK. American Journal of Physiology-Endocrinology and Metabolism 2016, 311, E730–E740. [DOI] [PubMed] [Google Scholar]
- (100).Musso G; Cassader M; Gambino R Non-alcoholic steatohepatitis: emerging molecular targets and therapeutic strategies. Nat. Rev. Drug Discovery 2016, 15, 249–274. [DOI] [PubMed] [Google Scholar]
- (101).Dobrikov MI; Shveygert M; Brown MC; Gromeier M Mitotic Phosphorylation of Eukaryotic Initiation Factor 4G1 (eIF4G1) at Ser1232 by Cdk1:Cyclin B Inhibits eIF4A Helicase Complex Binding with RNA. Mol. Cell. Biol 2014, 34, 439–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Bryant JD; Brown MC; Dobrikov MI; Dobrikova EY; Gemberling SL; Zhang Q; Gromeier M Regulation of Hypoxia-Inducible Factor 1α during Hypoxia by DAP5-Induced Translation of PHD2. Mol. Cell. Biol 2018, DOI: 10.1128/MCB.00647-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Wang H; Liu Y; Wang D; Xu Y; Dong R; Yang Y; Lv Q; Chen X; Zhang Z The Upstream Pathway of mTOR-Mediated Autophagy in Liver Diseases. Cells 2019, 8, 1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Zeng J; Zhu B; Su M Autophagy is involved in acetylshikonin ameliorating non-alcoholic steatohepatitis through AMPK/mTOR pathway. Biochem. Biophys. Res. Commun 2018, 503, 1645–1650. [DOI] [PubMed] [Google Scholar]
- (105).Reyna MA; Leiserson MD; Raphael BJ Hierarchical HotNet: identifying hierarchies of altered subnetworks. Bioinformatics 2018, 34, i972–i980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Beausoleil SA; Villén J; Gerber SA; Rush J; Gygi SP A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol 2006, 24, 1285–1292. [DOI] [PubMed] [Google Scholar]
- (107).Shteynberg DD; Deutsch EW; Campbell DS; Hoopmann MR; Kusebauch U; Lee D; Mendoza L; Midha MK; Sun Z; Whetton AD; Moritz RL PTMProphet: Fast and Accurate Mass Modification Localization for the Trans-Proteomic Pipeline. J. Proteome Res 2019, 18, 4262–4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Fermin D; Avtonomov D; Choi H; Nesvizhskii AI LuciPHOr2: site localization of generic post-translational modifications from tandem mass spectrometry data. Bioinformatics 2015, 31, 1141–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Meyer JG; Mukkamalla S; Steen H; Nesvizhskii AI; Gibson BW; Schilling B PIQED: automated identification and quantification of protein modifications from DIA-MS data. Nat. Methods 2017, 14, 646–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Bekker-Jensen DB; Bernhardt OM; Hogrebe A; Martinez-Val A; Verbeke L; Gandhi T; Kelstrup CD; Reiter L; Olsen JV Rapid and site-specific deep phosphoproteome profiling by data-independent acquisition without the need for spectral libraries. Nat. Commun 2020, 11, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Rosenberger G; Liu Y; Röst HL; Ludwig C; Buil A; Bensimon A; Soste M; Spector TD; Dermitzakis ET; Collins BC; Malmström L; Aebersold R Inference and quantification of peptidoforms in large sample cohorts by SWATH-MS. Nat. Biotechnol 2017, 35, 781–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (112).Searle BC; Lawrence RT; MacCoss MJ; Villén J Thesaurus: quantifying phosphopeptide positional isomers. Nat. Methods 2019, 16, 703–706. [DOI] [PMC free article] [PubMed] [Google Scholar]