

Abstract
Dihydropyridines are versatile building blocks for the synthesis of pyridines, tetrahydropyridines, and piperidines. Addition of nucleophiles to activated pyridinium salts allows synthesis of 1,2-, 1,4-, or 1,6-dihydropyridines; however, this process often leads to a mixture of constitutional isomers. Catalyst-controlled regioselective addition of nucleophiles to pyridiniums has the potential to solve this problem. Herein, we report that the regioselective addition of boron-based nucleophiles to pyridinium salts can be accomplished by the choice of a Rh catalyst.
Introduction
Site-selective functionalization of molecules that contain multiple reactive sites remains a significant challenge in organic synthesis.1–7 Along with this challenge comes the opportunity that achieving site-selective functionalization could enable synthesis of a variety of molecules from a small subset of starting materials. While selective functionalization of electrophiles that contain two reactive sites has been significantly developed (e.g., 1,2 vs 1,4 addition to α,β-unsaturated carbonyl compounds), development of methods to functionalize extended π systems has lagged.
In this context, pyridinium salts are multident electrophiles that, in the presence of nucleophiles, can undergo (1,2), (1,4), or (1,6) addition to afford the corresponding dihydropyridines (DHPs).8–31 These scaffolds are useful intermediates for the preparation of nonaromatic nitrogen heterocycles (Figure 1A).32 Directing a nucleophile to a specific site on the ring has proven to be a nontrivial task. This selectivity issue is commonly avoided by using symmetric pyridines, installing blocking groups, or exploiting a pre-existing functionality to direct the addition.11,33,34 Careful selection of the activating group has also been used to influence C2/C6 vs C4 selectivity.33,35 While effective in specific cases, the scope of dihydropyridines that can be accessed via these substrate controlled protocols is intrinsically limited, as they require structural conditions to be met or necessitate modification of the nucleophiles or pyridinium salts to influence the regioselectivity of the addition. These requirements diminish the synthetic utility of the dearomatization approach toward the synthesis of DHPs, tetrahydropyridines, and piperidines.
Figure 1.

A) Reactivity of pyridinium salts with nucleophiles for the synthesis of DHPs. B) Catalyst-controlled regioselec-tive addition of boronic acids. C) Examples of alkaloids and bioactive molecules with 2,6-disubstituted nitrogen heterocycles.
A strategy to overcome these issues is to impart control through the catalyst structure. A specific goal is to enable the addition of the same nucleophiles regioselectively to different positions of heteroarenium salts as a function of the catalyst to yield products with different substitution patterns, which are common in natural products, agrochemicals, and pharmaceuticals. Recently, our group and others have reported Rh-catalyzed addition of aryl boronic acids and aryl boronic acid pinacol esters to pyridinium salts derived from nicotinic acid (Figure 1B).14,17,28 These reactions proceeded in remarkable regioselectivity and delivered the C6 addition products in high yield and ee even when a substituent at the C6 position was present. In addition to the major C6 addition product, C2 and C4 arylation products were also observed in these reactions, albeit in low yield. Inspired by these proof-of-concept studies on catalyst-controlled regioselective functionalization of multident electrophiles, we initiated a study to identify a catalyst that would facilitate selective addition of aryl boron nucleophiles to the C2 position of nicotinic acid-based pyridinium salts. Identification of such a process should enable the synthesis of highly functionalized piperidines that are present in natural products and bioactive molecules such as the ones shown in Figure 1C.
Herein, we describe the identification and analysis of new catalysts for selective C2 addition, which afforded the corresponding 1,2-DHPs in high yield and enantioselectivity. Using statistical modeling tools, including linear regression, logistic regression, and threshold analysis, we identified which ligand parameters are important for achieving high selectivity and yield. This work will more broadly facilitate future efforts to develop site-selective catalysts for the functionalization of multident electrophiles.
Results and Discussions
Our initial studies in this area focused on the regioselective dearomatization reactions of pyridinium 8a (Table 1) with PhBpin as a readily available aryl nucleophile. For initial reaction conditions, we applied those used for the dearomatization of such substrates with Ar-Bpin nucleophiles (Table 1, entry 1).14 We investigated various bis-phosphine ligands with diverse steric and electronic properties.
Table 1.
Ligand screening for identification of a catalyst for C2-selective addition of Aryl-BPin nucleophiles to pyridinium salts 8.[a]

|
Select examples of such ligands are shown in Table 1, and a full list is provided in the Supporting Information. We found that under these conditions, several ligands provided the C6 addition product 10 as the major product, with some ligands even exceeding the selectivity of the previously used BINAP ligand (Table 1,entries 1−4). For example, ligands L2 and L3 gave the C6 addition product 10 in 20:1 and 45:1 selectivity, respectively, while BINAP delivered the same product in an 8:1 selectivity. Perhaps more importantly, we identified several ligands that significantly favored C2 addition (Table 1,entries 5−10). Of these, Me-DuPhos gave the dearomatization products in moderate yield and regioselectivity but high enantioselectivity. P-chiral ligands QUINOX-P* and Benz-P* gave DHPs 9a and 10 in moderate selectivity but in high overall yield and ee. Norphos (L9) yielded 9a and 10 in high regioselectivity, but the overall yield and enantioselectivity were modest. Finally, we identified BOBPHOS (L10), which was originally developed for Rh-catalyzed regioselective hydroformylation reactions, as the optimal ligand with respect to regioselectivity, the yield of the C2 product, and enantioselectivity.36 Using L10 as a ligand and reducing the reaction temperature, conditions were optimized with the goal of maximizing the yield of the C2 product (Table 1,entry 11).
With the optimal reaction conditions in hand, we explored the substrate scope of this reaction with respect to different ArBpins nucleophiles and pyridinium salts. During the scope exploration, we identified that although Quinox-P* had inferior regioselectivity compared to BOBPHOS, in certain cases, it delivered the C2 products in higher yield due to the excellent overall yields of these reactions. Thus, both ligands were used during the scope exploration.
Ar-Bpin nucleophiles containing various electron-neutral and electron-donating substituents at the para position reacted with pyridinium salts containing a C6-Me substituent to give corresponding 1,2-DHPs in high yield, regioselectivity, and enantioselectivity (Table 2, entries 9a−9f). While the regioselectivity and enantioselectivity of the addition of electron-deficient aryl groups was high, the yield of the C2 product was lower than that of electron-neutral and electron rich aryl groups (Table 2,entry 9g). Alkenyl groups can also be added under the standard reaction conditions to give corresponding DHP in moderate yield and high enantioselectivity (Table 2,entry 9i).
Table 2.
Scope of asymmetric C2 selective dearomatization reaction of methyl nicotinate derived pyridinium salts.[a]
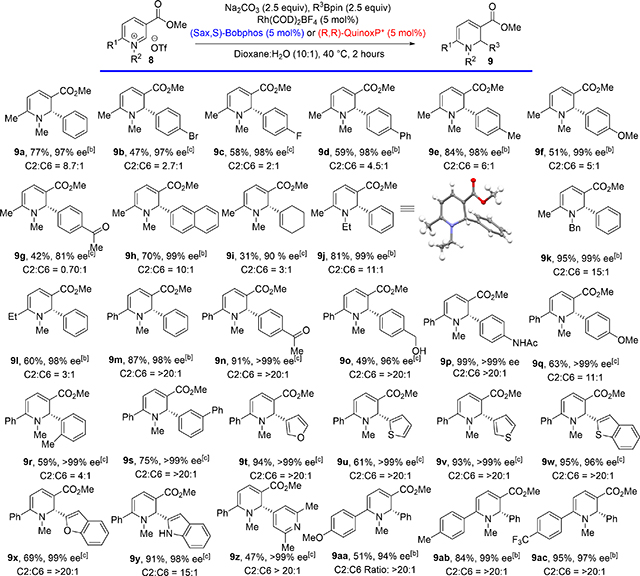
|
Next, we systematically varied substituents on the pyridinium ring to establish the scope o ftolerated substitution patterns. Changing the N-alkyl group from Me to Et or Bn did not significantly impact the reaction outcome, giving the corresponding DHPs in high yield and ee (Table 2,entries 9j and 9k). C6 Et-substituted pyridinium gave the corresponding DHP in decreased yield and regioselectivity compared to those of the C6 Me-substituted pyridinium, while the ee of the product remained high (Table 2,entry 9l).
Next, we explored the dearomatization of C6-arylsubstituted pyridiniums that typically underwent dearomatization to give corresponding 1,2-DHPs as the only detectable products and in high enantioselectivity. Thus, C6 phenyl substituted pyridinium reacted with para-substituted electron neutral, electron-rich, and electron-deficient Aryl-Bpin nucleophiles to deliver 1,2-DHPs 9m−9q (Table 2). Acidic groups such as secondary amide and unprotected primary alcohol are well tolerated under these reaction conditions (Table 2,entries 9o and 9p, respectively). Ortho- and meta-substituted Aryl-Bpin nucleophiles gave the corresponding DHPs 9r and 9s, respectively, in good yield and excellent ee. Five-membered heterocyclic Aryl-Bpin nucleophiles gave the corresponding heterocycle-substituted 1,2-DHPs in good yield and excellent ee (Table 2,entries 9t−9y). While reactions with pyridine-4-Bpin and 2-Me-pyridine-4-Bpin did not deliver any dearomatization product, 2,6-dimethyl-pyridine-substituted DHP was obtained in a 47% yield and >99%ee (see the Selected Unsuccessful Substrates section in the Supporting Information and Table 2, entry 9z, respectively). Electron-rich and electron-deficient C6-aryl-substituted pyridiniums underwent dearomatization in high regioselectivity and yielded the 1,2-DHPs 9aa−9ac, respectively, in >90% ee. A preparative scale reaction (1 mmol) using standard Schlenk line techniques without extensive degassing gave DHP 9w in a 52% yield and a 99% ee. This illustrates that the reaction is robust and can be used without specialized equipment, such as a glovebox (see the Supporting Information for details).
Derivatization of Dearomatization Products.
In order to demonstrate the utility of the reaction products, we evaluated derivatization reactions of these 1,2-DPHs and highlighted the C2-selective dearomatization approach toward the formal synthesis of deoxylasubine II. Product 9m can be partially reduced by treatment with NaCNBH3, delivering tetrahydropyridine 17 as a single diastereomer, while iodine and NaOMe provided a highly functionalized heterocycle 18 in an 84% yield (Scheme 1). The complete reduction could be accomplished to give piperidine 19 as a single diastereomer using a Pd/C-catalyzed hydrogenation.
Scheme 1.

Derivatization of dearomatization products.
The synthesis of deoxylasubine II commenced through a Sonogashira coupling between pyridine 11 and 3-butynol to yield alkyne 12 (Scheme 2). Hydrogenation of alkyne 12 followed by activation of the corresponding primary alcohol triggered an intramolecular cyclization reaction to yield pyridinium 13 after counter-anion exchange. The key dearomatization step was carried out using (R,R)-Ph-BPE as a ligand and gave DHP 14 in a moderate yield and regioselectivity but in high ee. It is worth noting that Bobphos and QuinoxP* gave only a trace amount of dearomatization product 14, presumably due to the increased steric hindrance of the substrate compared to substrates shown in Table 2. Hydrogenation of 1,2-DHP 14 gave piperidine 15. Conversion of piperidine 15 to deoxylasubine II (16) has previously been described.37 It is worth noting that the previous synthesis of piperidine 15 involved 11 synthetic steps, while our dearomatization approach yields this compound in only five steps, demonstrating its potential for streamlining the enantioselective synthesis of substituted piperidines.
Scheme 2.

Dearomative approach toward the formal synthesis of deoxylasubine II. a) (PPh3)2PdCl2 (5 mol%), CuI (10 mol%), Et3N, THF, 30 °C 92%; b) H2 (1 atm), Pd/C (10 mol%), MeOH, 23 °C 72%; c) CBr4, PPh3 then AgOTf, 68% (2 steps); d) Rh(COD)2BF4 (7 mol%), (R,R)-Ph-BPE (8 mol%), K2CO3 10:1 Dioxane:H2O, 80 °C 49%, 1:1 C2:C6, 94% ee; e) H2 (4 atm) Pd/C (10 mol%), MeOH, 23 °C 39%.
Data Science Analysis.
Given their key role in imparting reactivity and selectivity of the N-alkyl nicotinate arylation reaction, we sought to understand what structural features of the bis-phosphine ligands underpin performance (Figure 2). While the ligands also control the enantioselectivity, we were most intrigued with the structure−function relationship that governed the regioselectivity between C2 and C6 arylation products 9a and 10. The identity of the bis-phosphine ligand had a dramatic effect on the reaction outcome, with 9a:10 ratios ranging from 88:12 to 2:98 (Table 1,entries 10 and 3, respectively). This corresponds to a significant ΔΔG‡ range of ~4.1 kcal/mol. Additionally, we were interested in the role that the bis-phosphine ligands played on the reaction yield, as the choice of ligand led to yields ranging from 0 to 94% (Table 1). Since no intuitive trends relating the bis-phosphine structure to the reaction performance were readily identified, we applied data science techniques to correlate the experimental reaction data with computed descriptors of the bis-phosphine ligands. The latter were obtained from one of our group’s recently reported extensive DFT-derived descriptor library of bisphosphine ligands.38,39
Figure 2.

Reactivity and selectivity of Rh-catalyzed arylation correlates with the ligand minimum octant buried volume and bite angle respectively. Plotted circles represent bisphosphine ligands tested in the Rh-catalyzed arylation of 8a to produce 9a or 10.
We began our investigation by using logistic regression to assess the regioselectivity of the Rh-catalyzed arylation of 8a (Figure 2A, left plot). This was accomplished in two steps. First, the C2/C6 product ratio (i.e., 9a:10) was converted to a binary variable by classifying ligands as either C2 or C6 selective (1 or 0, respectively). Next, a logistic regression algorithm was used to correlate the binary selectivity data with a ligand parameter. This protocol resulted in a sigmoidal function (plotted with a solid black line) that related the bisphosphine bite angle to the probability that the ligand will be C2 selective (1.0 and 0.0 reflecting 100 and 0% probability, respectively). This revealed that the C2 arylation product prevails when smaller-bite angle ligands are used, while ligands with wider bite angles favor C6 arylation. The observed importance of bite angle on selectivity is not surprising as similar trends have been well documented, especially for Rh-catalyzed hydroformylation reactions.40–42 Based on these reports, the effect of the bite angle on coordination geometry may underpin the observed correlation (see the Supporting Information for further discussion and a plausible reaction mechanism).
Next, we turned our attention to statistical modeling of reaction yield (Figure 2, right plot). While linear modeling techniques were unsuccessful at correlating yield to the ligand structure, a single-node decision tree classifier proved effective.43 This algorithm functions by classifying each ligand as either reactive or unreactive with a user-defined yield cutoff (10% yield). A parameter is then identified that effectively partitions the ligand classes at an algorithm-defined parameter threshold. This analysis led to the identification of the steric parameter Octant Vburmin– the percent buried volume of the least sterically hindered ligand octant–as being correlated with the reaction yield.44 Bis-phosphines with an Octant Vburmin– value above 13.3% resulted in a significant reduction in reaction yield. This parameter threshold likely reflects a minimum steric profile of the ligand required to facilitate the assembly of the reacting species about the Rh metal center.
The combined results of the reactivity and regioselectivity classification models are graphically depicted in the two-parameter chemical space map, as shown in Figure 2B. In this representation, Octant Vburmin and bite angle are shown on the x-and y-axes, respectively. Each ligand was then plotted, and the corresponding total reaction yield and selectivity were reflected by the circle size color, respectively. The vertical black line at 13.3% is the steric reactivity cliff identified with the single-node decision, while the horizontal line at 92° is the bite angle cutoff found with logistic regression that partitions the C2- and C6-selective ligands. With this representation, it can be readily observed that ligands in the top- and bottom-left quadrants give rise to active catalysts that are C2 and C6 selective, respectively. Additionally, the dramatic decline in yield in the two right-hand quadrants reflects the ligand space that is too sterically encumbered to form functional catalysts.
Following the classification-based modeling of regioselectivity and yield, we questioned whether multivariate linear regression (MLR) models could be constructed to quantitatively predict ligands with improved regioselectivity (Figure 3). Early efforts to build linear statistical models for all ligands were challenging; however, we found that a key curation step facilitated model construction. By removing ligands with Octant Vburmin values above the threshold of 13.3%, a statistically robust three-term MLR model with an R2 of 0.89 and Q2 and fourfold R2 internal validation statistics of 0.84 and 0.83, respectively, was found (Figure 3A). The first two terms were the ligand parameters Octant Vburmax and P−Rback σ*max. The former describes the percent buried volume of the most sterically hindered ligand octant and suggests that more encumbered ligands have a greater preference for C6 arylation. The latter conveys the energy of the σ* orbital between one of the phosphorus atoms and the ligand backbone (see the blue bond in Figure 3B). This term is electronic in nature and is thought to reflect the ability of the ligand to engage in π− backbonding interactions with the Rh metal center. Finally, the third parameter is a substrate classifier wherein N-methyl and N-benzyl nicotinate salts 8a and 8c are assigned values of 1 and 0, respectively.
Figure 3.

A) Multivariate linear regression model correlating the ligand structure to the regioselectivity of the N-alkyl nico-tinate arylation reaction. B) Ligand and substrate parameters used in the MLR model. C) The performance of the most C2 selective ligand, Catasium D, was predicted by the MLR model.
With a statistically robust model in hand, this model was used to predict the performance of untested ligands (Figure 3A,B). Virtual screening suggested that Catasium D L11 was likely to be highly C2 selective, while a handful of ligands were predicted to have a 1.20−1.33 kcal/mol preference for the C6 arylation product (see the Supporting Information for all ligand structures). Several ligands were then procured and tested in the laboratory (Figure 3A, red X’s). Two salient features should be noted from the experimental evaluation of these ligands. First, the model was predictive with a mean average prediction error (MAE) of 0.38 kcal/mol and an R2 of 0.87. Second, L11 was found to provide the highest selectivity for the C2 arylation product of any ligand evaluated in this study, albeit the other reaction outputs were not as compelling.
Conclusions
In conclusion, through conventional ligand screening, we discovered that the regioselectivity of aryl additions to pyridinium salts derived from nicotinic acid derivatives can be controlled by the catalyst used. While Rh/BINAP combination catalyzes the addition of aryl boron nucleophiles to the C6 position of these pyridiniums, smaller-bite angle Bobphos and Quinox P* ligands delivered the C2 addition products. The later catalysts allowed the synthesis ofC2-, C3-, C6-substituted dihydropyridines, which, in turn, can be used for the synthesis of corresponding tetrahydropyridines and piperidines. Using various data science tools, we have identified key parameters that influence the yield and regioselectivity of these reactions. For practical yields, ligands with at least one open quadrant should be used. In addition to the choice of the ligand throughout our optimization studies, we have found that the water content and reaction temperature have a significant impact on the yields of such dearomatization reactions. Logistic regression analysis revealed the bite angle as the key parameter that determines the regioselectivity outcome of these reactions. Currently, our group continues to apply the approach described in this manuscript for the regioselective dearomatization of other heteroarenium salts. The results of these studies will be reported in due course.
Supplementary Material
References
- (1).Norman JP; Neufeldt SR, “The Road Less Traveled: Unconventional Site Selectivity in Palladium-Catalyzed Cross-Couplings of Dihalogenated N-Heteroarenes.” ACS Catal. 2022, 12014–12026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Palani V; Perea MA; Sarpong R, “Site-Selective Cross-Coupling of Polyhalogenated Arenes and Heteroarenes with Identical Halogen Groups.” Chem. Rev. 2022, 122, 10126–10169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Sakakibara Y; Murakami K, “Switchable Divergent Synthesis Using Photocatalysis.” ACS Catal. 2022, 12, 1857–1878. [Google Scholar]
- (4).Najera C; Beletskaya IP; Yus M, “Metal-Catalyzed Regiodivergent Organic Reactions.” Chem. Soc. Rev. 2019, 48, 4515–4618. [DOI] [PubMed] [Google Scholar]
- (5).Hartwig JF, “Catalyst-Controlled Site-Selective Bond Activation.” Acc. Chem. Res. 2017, 50, 549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Giuliano MW; Miller SJ, “Site-Selective Reactions with Peptide-Based Catalysts.” Top. Curr. Chem. 2016, 372, 157–201. [DOI] [PubMed] [Google Scholar]
- (7).Mahatthananchai J; Dumas AM; Bode JW, “Catalytic Selective Synthesis.” Angew. Chem. Int. Ed. Engl. 2012, 51, 10954–10990. [DOI] [PubMed] [Google Scholar]
- (8).Jia J; Hu FD; Xia Y, “Transition-Metal-Catalyzed Nucleophilic Dearomatization of Electron-Deficient Heteroarenes.” Synthesis 2022, 54, 92–110. [Google Scholar]
- (9).Kratena N; Marinic B; Donohoe TJ, “Recent Advances in the Dearomative Functionalisation of Heteroarenes.” Chem. Sci. 2022, 13, 14213–14225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Ding QP; Zhou XL; Fan RH, “Recent Advances in Dearomatization of Heteroaromatic Compounds.” Org. Biomol. Chem. 2014, 12, 4807–4815. [DOI] [PubMed] [Google Scholar]
- (11).Comins DL; Higuchi K; Young DW, “Dihydropyridine Preparation and Application in the Synthesis of Pyridine Derivatives.” Adv. Heterocycl. Chem. 2013, 110, 175–235. [Google Scholar]
- (12).Bull JA; Mousseau JJ; Pelletier G; Charette AB, “Synthesis of Pyridine and Dihydropyridine Derivatives by Regio- and Stereoselective Addition to N-Activated Pyridines.” Chem. Rev. 2012, 112, 2642–2713. [DOI] [PubMed] [Google Scholar]
- (13).Ahamed M; Todd MH, “Catalytic Asymmetric Additions of Carbon-Centered Nucleophiles to Nitrogen-Containing Aromatic Heterocycles.” Eur. J. Org. Chem. 2010, 5935–5942. [Google Scholar]
- (14).Robinson DJ; Ortiz KG; O’Hare NP; Karimov RR, “Dearomatization of Heteroarenium Salts with Arbpin Reagents. Application to the Total Synthesis of a Nuphar Alkaloid.” Org. Lett. 2022, 24, 3445–3449. [DOI] [PubMed] [Google Scholar]
- (15).Nallagonda R; Karimov RR, “Copper-Catalyzed Regio- and Diastereoselective Additions of Boron-Stabilized Carbanions to Heteroarenium Salts: Synthesis of Azaheterocycles Containing Contiguous Stereocenters.” ACS Catal. 2021, 11, 248–254. [Google Scholar]
- (16).Knight BJ; Tolchin ZA; Smith JM, “A Predictive Model for Additions to N-Alkyl Pyridiniums.” Chem. Commun. 2021, 2693–2696. [DOI] [PubMed] [Google Scholar]
- (17).Robinson DJ; Spurlin SP; Gorden JD; Karimov RR, “Enantioselective Synthesis of Dihydropyridines Containing Quaternary Stereocenters through Dearomatization of Pyridinium Salts.” ACS Catal. 2020, 10, 51–55. [Google Scholar]
- (18).Yedoyan J; Wurzer N; Klimczak U; Ertl T; Reiser O, “Regio- and Stereoselective Synthesis of Functionalized Dihydropyridines, Pyridines, and 2h-Pyrans: Heck Coupling of Monocyclopropanated Heterocycles.” Angew. Chem. Int. Ed. Engl. 2019, 58, 3594–3598. [DOI] [PubMed] [Google Scholar]
- (19).Gribble MW; Guo S; Buchwald SL, “Asymmetric Cu-Catalyzed 1,4-Dearomatization of Pyridines and Pyridazines without Preactivation of the Heterocycle or Nucleophile.” J. Am. Chem. Soc. 2018, 140, 5057–5060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Bertuzzi G; Sinisi A; Pecorari D; Caruana L; Mazzanti A; Bernardi L; Fochi M, “Nucleophilic Dearomatization of Pyridines under Enamine Catalysis: Regio-, Diastereo-, and Enantioselective Addition of Aldehydes to Activated N-Alkylpyridinium Salts.” Org. Lett. 2017, 19, 834–837. [DOI] [PubMed] [Google Scholar]
- (21).Bertuzzi G; Sinisi A; Caruana L; Mazzanti A; Fochi M; Bernardi L, “Catalytic Enantioselective Addition of Indoles to Activated N-Benzylpyridinium Salts: Nucleophilic Dearomatization of Pyridines with Unusual C-4 Regioselectivity.” ACS Catal. 2016, 6, 6473–6477. [Google Scholar]
- (22).Lutz JP; Chau ST; Doyle AG, “Nickel-Catalyzed Enantioselective Arylation of Pyridine.” Chem. Sci. 2016, 7, 4105–4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Wang Y; Liu YL; Zhang DD; Wei H; Shi M; Wang FJ, “Enantioselective Rhodium-Catalyzed Dearomative Arylation or Alkenylation of Quinolinium Salts.” Angew. Chem. Int. Ed. Engl. 2016, 55, 3776–3780. [DOI] [PubMed] [Google Scholar]
- (24).Zhang M; Sun WS; Zhu GM; Bao GJ; Zhang BZ; Hong L; Li M; Wang R, “Enantioselective Dearomative Arylation of Isoquinolines.” ACS Catal. 2016, 6, 5290–5294. [Google Scholar]
- (25).Fischer T; Bamberger J; Mancheno OG, “Asymmetric Nucleophilic Dearomatization of Diazarenes by Anion-Binding Catalysis.” Org. Biomol. Chem. 2016, 14, 5794–5802. [DOI] [PubMed] [Google Scholar]
- (26).Mancheno OG; Asmus S; Zurro M; Fischer T, “Highly Enantioselective Nucleophilic Dearomatization of Pyridines by Anion-Binding Catalysis.” Angew. Chem. Int. Ed. Engl. 2015, 54, 8823–8827. [DOI] [PubMed] [Google Scholar]
- (27).Chau ST; Lutz JP; Wu K; Doyle AG, “Nickel-Catalyzed Enantioselective Arylation of Pyridinium Ions: Harnessing an Iminium Ion Activation Mode.” Angew. Chem. Int. Ed. Engl. 2013, 52, 9153–9156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Nadeau C; Aly S; Belyk K, “Rhodium-Catalyzed Enantioselective Addition of Boronic Acids to N-Benzylnicotinate Salts.” J. Am. Chem. Soc. 2011, 133, 2878–2880. [DOI] [PubMed] [Google Scholar]
- (29).Fernandez-Ibanez MA; Macia B; Pizzuti MG; Minnaard AJ; Feringa BL, “Catalytic Enantioselective Addition of Dialkylzinc Reagents to N-Acylpyridinium Salts.” Angew. Chem. Int. Ed. Engl. 2009, 48, 9339–9341. [DOI] [PubMed] [Google Scholar]
- (30).Black DA; Beveridge RE; Arndtsen BA, “Copper-Catalyzed Coupling of Pyridines and Quinolines with Alkynes: A One-Step, Asymmetric Route to Functionalized Heterocycles.” J. Org. Chem. 2008, 73, 1906–1910. [DOI] [PubMed] [Google Scholar]
- (31).Sun ZK; Yu SY; Ding ZD; Ma DW, “Enantioselective Addition of Activated Terminal Alkynes to 1-Acylpyridinium Salts Catalyzed by Cu-Bis(Oxazoline) Complexes.” J. Am. Chem. Soc. 2007, 129, 9300–9301. [DOI] [PubMed] [Google Scholar]
- (32).Sun DB; Confair DN; Ellman JA, “Rhodium-Catalyzed C-H Alkenylation/Electrocyclization Cascade Provides Dihydropyridines That Serve as Versatile Intermediates to Diverse Nitrogen Heterocycles.” Acc. Chem. Res. 2021, 54, 1766–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Grigolo TA; Smith JM, “Regiodivergent Asymmetric Pyridinium Additions: Mechanistic Insight and Synthetic Applications.” Chem. Eur. J. 2022, 28, e202202813. [DOI] [PubMed] [Google Scholar]
- (34).Legault C; Charette AB, “Complexation Promoted Additions to N-Benzoyliminopyridinium Ylides. A Novel and Highly Regioselective Approach to Polysubstituted Piperidines.” J. Am. Chem. Soc. 2003, 125, 6360–6361. [DOI] [PubMed] [Google Scholar]
- (35).Yan XC; Ge L; Reis MC; Harutyunyan SR, “Nucleophilic Dearomatization of N-Heteroaromatics Enabled by Lewis Acids and Copper Catalysis.” J. Am. Chem. Soc. 2020, 142, 20247–20256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Noonan GM; Fuentes JA; Cobley CJ; Clarke ML, “An Asymmetric Hydroformylation Catalyst That Delivers Branched Aldehydes from Alkyl Alkenes.” Angew. Chem. Int. Ed. Engl. 2012, 51, 2477–2480. [DOI] [PubMed] [Google Scholar]
- (37).Zhang Y; Gerasyuto AI; Long QA; Hsung RP, “A General Approach to the Quinolizidine Alkaloids Via an Intramolecular Aza-[3+3] Annulation: Synthesis of (±)-2-Deoxylasubine II.” Synlett 2009, 237–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).It should be noted that the bisphosphine ligand parameters were computed from the corresponding bisphosphine[PdCl2] complexes, however, these parameters have been shown to be applicable to Rh-catalyzed reactions. For further discussion see reference 39.
- (39).Dotson JJ; van Dijk L; Timmerman JC; Grosslight S; Walroth RC; Gosselin F; Puntener K; Mack KA; Sigman MS, “Data-Driven Multi-Objective Optimization Tactics for Catalytic Asymmetric Reactions Using Bisphosphine Ligands.” J. Am. Chem. Soc. 2023, 145, 110–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Birkholz MN; Freixa Z; van Leeuwen PW, “Bite Angle Effects of Diphosphines in C-C and C-X Bond Forming Cross Coupling Reactions.” Chem. Soc. Rev. 2009, 38, 1099–1118. [DOI] [PubMed] [Google Scholar]
- (41).Kamer PC; van Leeuwen PW; Reek JN, “Wide Bite Angle Diphosphines: Xantphos Ligands in Transition Metal Complexes and Catalysis.” Acc. Chem. Res. 2001, 34, 895–904. [DOI] [PubMed] [Google Scholar]
- (42).van Leeuwen PW; Kamer PC; Reek JN; Dierkes P, “Ligand Bite Angle Effects in Metal-Catalyzed C-C Bond Formation.” Chem. Rev. 2000, 100, 2741–2770. [DOI] [PubMed] [Google Scholar]
- (43).Newman-Stonebraker SH; Smith SR; Borowski JE; Peters E; Gensch T; Johnson HC; Sigman MS; Doyle AG, “Univariate Classification of Phosphine Ligation State and Reactivity in Cross-Coupling Catalysis.” Science 2021, 374, 301–308. [DOI] [PubMed] [Google Scholar]
- (44).Falivene L; Cao Z; Petta A; Serra L; Poater A; Oliva R; Scarano V; Cavallo L, “Towards the Online Computer-Aided Design of Catalytic Pockets.” Nat. Chem. 2019, 11, 872–879. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.