

Abstract
Purpose:
Anaplastic Lymphoma Kinase (ALK) aberrations have been identified in pediatric type infant gliomas, but their occurrence across age groups, functional effects, and treatment response have not been broadly established.
Experimental Design:
We performed a comprehensive analysis of ALK expression and genomic aberrations in both newly generated and retrospective data from 371 glioblastomas (156 adult, 205 infant/pediatric and 10 congenital) with in vitro and in vivo validation of aberrations.
Results:
ALK aberrations at the protein or genomic level were detected in 12% of gliomas (45/371) in a wide age range (0–80 years). Recurrent as well as novel ALK fusions (LRRFIP1-ALK, DCTN1-ALK, PRKD3-ALK) were present in 50% (5/10) of congenital/infant, 1.4% (3/205) of pediatric, and 1.9% (3/156) of adult GBMs. ALK fusions were present as the only candidate driver in congenital/infant GBMs and were sometimes focally amplified. In contrast, adult ALK fusions co-occurred with other oncogenic drivers. No activating ALK mutations were identified in any age group. Novel and recurrent ALK rearrangements promoted STAT3 and ERK1/2 pathways and transformation in vitro and in vivo. ALK-fused GBM cellular and mouse models were responsive to ALK inhibitors, including in patient cells derived from a congenital GBM. Relevant to treatment of infant gliomas, we showed that ALK protein appears minimally expressed in the forebrain at perinatal stages and no gross effects on perinatal brain development was seen in pregnant mice treated with the ALK inhibitor ceritinib.
Conclusions:
These findings support use of brain-penetrant ALK inhibitors in clinical trials across infant, pediatric, and adult GBMs.
INTRODUCTION
Glioblastomas and high grade gliomas are aggressive brain tumors with few curative treatment options at any age. Recently, landmark genomic profiling has shown GBMs to encompass molecularly-defined subtypes, with pediatric GBMs harboring distinct driver events compared to their adult counterparts. Indeed, within pediatric GBMs, distinct genetic drivers are observed at different ages. For example, infant glioblastoma (iGBM; occurring in children under two) have recently been shown to harbor ALK, ROS, or NTRK alterations, while older children are more commonly diagnosed with histone-mutant GBMs. In contrast, congenital GBM (cGBM) are rare and less well-studied (1–3). Specifically, it has not been well described whether cGBM identified antenatally or at birth (termed ‘congenital’) may differ from those detected in infants (4).
ALK is an important oncogenic driver in many cancers (5). ALK was first identified as part of the NPM-ALK gene fusion transcript in anaplastic large-cell lymphoma (ALCL) (6) and initial studies suggested one of its primary locations of normal expression was the nervous system (7). Subsequently ALK was found to be fused to echinoderm microtubule-associated protein-like 4 (EML4) in non-small-cell lung cancer (NSCLC) (8–10). To date, over 30 different fusion partners have been described in various cancer types (5). In each described fusion, the C-terminal portion of the protein contains the ALK kinase domain, with its spatial and temporal expression dictated by its N-terminal fusion partner. Gain-of-function mutations in the ALK kinase domain (p.F1174L and R1275Q), have also been described in different pediatric cancers including neuroblastoma (11–13).
ALK fusions have been recently reported in subsets of pediatric brain tumors including high grade gliomas (14–18). Infant glioblastomas in particular have been shown to harbor ALK, ROS1, or MET fusions as their defining alterations and are proposed to be termed ‘infant-type hemispheric gliomas’ to highlight their different biology in the new 2021 WHO CNS classification of pediatric brain tumors(19). Experimental studies suggest glioma ALK fusions are sensitive to targeted therapy in vitro and in mouse models (17). Furthermore, while case reports of ALK inhibitor activity in patients hold promise (18), ALK inhibitor use has been limited due to concerns that ALK inhibitors may have serious side effects on early brain development.
In adult GBM, the role of ALK remains poorly understood. ALK protein overexpression has been reported in GBM and was suggested as a potentially targetable event (20,21) but recurrent ALK rearrangements or activating mutations have not been well described. Preclinical studies have demonstrated that ribozyme targeting (20) and targeted inhibitors (22) of ALK are effective in slowing growth of ALK-expressing GBM cell lines, providing evidence of a clinically relevant pathway to target in this disease. Whether biological differences in ALK activation in adult and pediatric GBM affect treatment response and therapeutic strategies is also not known. These questions are relevant as some GBM patients may receive ALK inhibitors as part of ongoing clinical trials or off-label use when reported in sequencing data (22).
The first-in-class ALK inhibitor crizotinib is approved worldwide for the treatment of ALK-positive NSCLC and ALCL (23–25). Resistance to crizotinib is often mediated by gatekeeper point mutations in the ATP-binding pocket, but is also facilitated by CNS relapse, as crizotinib has poor blood-brain barrier penetrance (26). Second- and third-generation ALK inhibitors developed to combat these resistance mechanisms and to improve blood brain barrier penetration (ceritinib; alectinib; lorlatinib) have shown impressive intracranial responses in NSCLC brain metastases (27,27,28) and neuroblastoma (29,30). However, their safety during brain development in early childhood and efficacy in GBMs, particularly in the infant pediatric population, is not well established.
To address this we analyzed newly generated and pre-existing GBM genomic data using multiple orthogonal approaches to generate a comprehensive analysis of ALK alterations in high grade glioma across all age groups. We identified new patterns of ALK aberrations in pediatric gliomas, including high level focal amplification of rearranged alleles, novel breakpoints, and novel fusion partners. Our data also extend the range of ALK fusions to include adult and congenital GBM and suggest ALK as a promising therapeutic target in this devastating disease.
METHODS
Ethics statement.
The studies here were performed in accordance with the U.S. Common Rule and Declaration of Helsinki where applicable. Ethics approval was granted by relevant human IRB and/or animal research committees (IACUC) of Dana-Farber Cancer Institute (Boston, US), Boston Children’s Hospital (Boston, US), Brigham and Women's Hospital (Boston, US), University of Texas Southwestern Medical Center (Dallas, US), El Paso Children's Hospital (El Paso, US), Centre Hospitalier Sainte Anne (Paris, France). IRB approval from all institutions was obtained, and all patients provided written informed consent prior to collection of samples or were analyzed as de-identified samples if waiver of consent was granted by the IRB.
ALK FISH on FFPE tissue.
FISH was performed on 5 micron formalin-fixed paraffin-embedded (FFPE) tissue sections, which were baked at 60°C for at least two hours then de-paraffinized and digested using methods described previously (Firestein et al., Nature. 2008 Sep 25;455(7212):547–51.). The ALK Break Apart FISH Probe (Abbott Molecular/Vysis, Inc.) was hybridized to tissue sections following manufacturer's directions. Tissues and probes were co-denatured at 80°C, hybridized at least 16 hours at 37°C in a darkened humid chamber, washed in 2X SSC at 71°C for two min, rinsed in room temperature 2X SSC, and counterstained with DAPI (4',6-diamidino-2-phenylindole, Abbott Molecular/Vysis, Inc.). Slides were imaged using an Olympus AX51 fluorescence microscope. Individual images were captured using an Applied Imaging system running CytoVision Genus version 7.5. ALK status was assessed in 50 tumor nuclei per sample.
Immunohistochemistry.
Hematoxylin-eosin (H&E) slides were prepared from 5 microns sections of formalin-fixed, paraffin-embedded tissue (FFPE). Immunohistochemistry for ALK1 (Leica, clone 5A4, predilute) was performed on 5 microns paraffin sections following standard protocol, and positive and negative controls were reviewed. Microscopic examination was performed with an Olympus BX41 microscope and photos were taken with an Olympus DP25 camera. Three additional congenital GBMs from autopsy were excluded from our study for failed hybridization signals (FISH), non-specific ALK staining or absence of GFAP or SOX2-positive cells, most likely due to poor quality specimen.
Samples were classified by a board-certified neuropathologists (K.L.L, S.A and P.V) using W.H.O. 2007 criteria.
Array CGH.
Array-based comparative genomic hybridization (aCGH) was performed on 84 cases, using formalin‐fixed paraffin‐embedded tissue. The modality has been previously described by Ramkissoon et al. In brief, after DNA extraction, patient and reference DNA (Promega, Madison, WI, USA) was fragmented using previously described fragmentation simulation methods and hybridized to Agilent (Santa Clara, CA, USA) SurePrint G3 Human 1 Million feature arrays. Clinical analysis was performed using Agilent Workbench software, and log ratios were normalized using the centralization algorithm, with a threshold score of 6.0 and bin size of 10. The results were interpreted by two cytogeneticists (A.D. and A.L.).
Whole genome sequencing protocol.
HGGs and normal controls from Dana-Farber Cancer Institute, Boston Children’s Cancer Center and UT Southwestern Medical Center were sequenced at UT Southwestern Medical Center - Dallas and The Broad Institute of MIT and Harvard. All analyses were performed as described in Supplemental Text 1.
ALK fusions analysis by Targeted RNA next generation sequencing (NGS) using Anchored Multiplex PCR (AMP).
RNA was isolated from 5 microns FFPE tissue sections (2 slides per tumor sample) using the ALLPrep DNA/RNA FFPE Kit (QIAGEN #80234). Fusion detection was then carried out using the FusionPlex Solid Tumor Kit for Illumina (Archer #AB0027). Briefly, cDNA was synthetized from 100ng RNA using random primers, and a quantitative PCR-based QC assay was performed. Subsequently, the proprietary anchored multiplex PCR procedure was performed. Finally, samples were run on the Illumina MiSeq next-generation sequencing platform at the Broad Institute, with a 151 bp paired-end run including an eight-base index barcode read. Sequencing data were analyzed using the Archer Analysis online tool.
Fusion genes were also detected using the MGH Solid Fusion Assay V2. Exonic and intronic target regions validated for clinical reporting were as follows for the MGH assay: ALK (19–22, intron 19), BRAF (7–12, 15), BRD4 (10, 11), EGFR (2–7 exon skipping/vIII variant, 7–9, 16, 20, 24, 25), EWSR1 (4–14), FGFR2 (2, 8–10, 17), MAML2 (2, 3), MET (exon 14 skipping), NRG1 (1–3, 6), NUTM1 (3), RET (8–13), and ROS1 (31–37). Additional targeted exonic regions as research were as follows: AKT3 (1–3), AR (1–8), ARHGAP26 (2, 10–12), AXL (19,20), BRD3 (9–12), CSF1 (5–9), CSF1R (7,11–13,22), ERG (2–11), ESR1 (3–6), ETV1 (3–13), ETV4 (2, 4–10), ETV5 (2, 3, 7–9), ETV6 (1–7), FGFR1 (2, 8–10, 17), FGFR3 (8–10, 17, intron 17), FGR (2), INSR (12–22), JAZF1 (2–4), MAST1 (7–9, 18–21), MAST2 (2, 3, 5, 6), MET (13, 15), MSMB (2–4), MUSK (7–9, 11–14), MYB (7–9, 11–16), NOTCH1 (2, 4, 26–31, internal exon 3–27 deletion), NOTCH2 (5–7, 26–28), NTRK1 (8,10–13), NTRK2 (11–17), NTRK3 (13–16), NUMBL (3), PDGFRA (7, exon 8 deletion, 10–14), PDGFRB (8–14), PIK3CA (2), PKN1 (10–13), PPARG (1–3), PRKCA (4–6), PRKCB (3), RAF1 (4–7, 9–12), RELA (3, 4), RSPO2 (1, 2), RSPO3 (2), TERT (2), TFE3 (2–8), TFEB (1,2), THADA (24–31,36), and TMPRSS2 (1–6). Analysis pipeline version: 2.5.10.
Processing of previously published single cell RNA-sequencing data.
Single cell human RNA sequencing data was obtained from the Nowakowski et al., (31) spatiotemporal gene expression trajectories. ALK positive cells were defined as any cell in the dataset with an expression value greater than 0. t-SNE plots were obtained from the UCSC cell browser tool (http://cells.ucsc.edu/?ds=cortex-dev) which incorporates the scRNA data. Similarly, single cell mouse RNA sequencing data was obtained from Jessa, S. et al. (32). t-SNE plots of Alk expression and cluster average expression were obtained from the data portal http://cc-shiny-01.functionalgenomics.ca/braindex/clusters/.
RT-PCR.
RNA extracted from patient tumor sample were combined at 200ng/ml with components of the Superscript IV One-Step RT-PCR kit (Thermo Fisher) and custom primers (IDT). RT-PCR DNA products were then separated in 3% agarose at 150V, stained with SYBR Gold Nucleic Acid Gel Stain (Invitrogen) for 20 min., and imaged with the ChemiDoc MP system (Bio-Rad).
Cell line.
HEK293T and NIH3T3 were obtained from ATCC. They were routinely tested for mycoplasma contamination and short tandem repeat profiling.
Generation of neural stem cells.
Embryonic neural stem cells (mNSC) were isolated from C57BL6 wild-type E14.5 dpc mouse embryos as previously described (Nery et al. 2002. Nat. Neurosci) and maintained in culture media with NeuroCult™ Proliferation Kit Mouse and Rat (Stemcell) supplemented with EGF 20ng/uL (Miltenyi Biotec), FGF 10ng/uL (Miltenyi Biotec) and Heparin 0.002% (Stemcell).
Lentiviral vectors.
Wild-type ALK and eGFP constructs were acquired from Addgene (#23917 and #129020 respectively). pECE-M2- PP1Cbeta was a gift from Anne Brunet (Addgene plasmid #31677). They were cloned into the pENTR1a Gateway Entry clone and subsequently flipped into the pLX307 Gateway Destination lentiviral expression clone using the Gateway™ LR Clonase™ II Enzyme (Invitrogen #11791020). PPP1CB-ALK fusion construct was synthesized as a Gateway entry clone and subsequently flipped into pLX307. HEK293T cells were used to generate lentivirus as per standard protocol. Cells were infected with the above viruses for 48 hours followed by puromycin selection (0.5 μg/mL) for at least 3 weeks prior to experimentation.
Western Blot.
Stable cell lines expressing the empty vector pLex307 or the ALK fusions were dissociated and plated at a density of 500 000 cells/well in 6-well plates coated with laminin (50 ug/mL). Cells were allowed to attach to the coating for 24 hours and cells were treated with Crizotinib (PF-02341066) or Lorlatinib (PF-6463922) at 200nM and DMSO (as control) for 3 hours. Cells were then collected, washed with cold PBS and resuspended in RIPA buffer supplemented with phosphatase (PhosSTOP, Roche #04906845001) and protease inhibitors (Complete Mini, Roche #11836153001). Frozen tumor samples were processed in an identical manner.
Protein lysates were subjected to western blot analysis by standard methods and probed with primary antibodies against total ALK (Cell Signaling #3633), phospho-ALK Tyr1604 (Cell Signaling #3341), total ERK (Cell Signaling #9102), phospho-ERK Thr202/Tyr204 (Cell Signaling #4370), total AKT (Cell Signaling #4691), phospho-AKT Ser473 (Cell Signaling #4060), phospho-STAT (Cell signaling #9145), PPP1CB (Invitrogen #PA5–28225), DYKDDDDK Tag (9A3) Mouse mAb (Cell signaling #8146), GAPDH (14C10) Rabbit mAb (Cell Signaling #2118S) and vinculin (Sigma #V9131).
Cell proliferation.
Cell proliferation was quantified by measuring intracellular ATP concentrations via luminescence with CellTiter-Glo (Promega, #G7573). Cells were plated as single cells with a density of 2x103 cells per well (n = 6) in an ultra-low attachment, flat-bottom, 96 well plate (Corning, #3474). Luminescence measurements were performed following the manufacturer’s supplied protocol. A t0 baseline measurement was taken immediately following cell plating, and proceeding measurements were repeated once every 24 hours, until a 72-hour time point was reached.
Soft agar colony assay and quantification.
Anchorage-independent growth of mouse neural stem cells was assayed with the following modifications: mNSC expressing PPP1CB-ALK lentivirus and GFP lentivirus vector as control were plated in 0.35% agar with complete media in 24-well plate in triplicates. Cell colonies were allowed to form for two weeks and fixed with paraformaldehyde 4%. Images were obtained using the Celigo Imaging Cytometer, and colony formation was quantified using a CellProfiler™ image analysis pipeline (McQuin et al., PLoS Biol. 2018). Briefly, images were cropped to include one well in the field of view. The images were gray scale inverted to remove background, and colonies with a diameter larger than ~130um were identified and counted.
Incucyte® Label-Free Cell Proliferation and Drug AUC Metric.
Cellular response to targeted inhibitors was quantified as % confluence using the Incucyte® Live-Cell Analysis System (Sartorius). Briefly, cells were extracted from a fresh patient sample from surgery using the gentleMacs Dissociator and Brain Tumor Dissociation Kit (Miltenyi) according to manufacturer’s instructions. Extracted cells were left in culture to recover for no longer than two weeks for acute testing. Tissue culture-treated, flat bottom 96-well plates (Corning, #353072) were coated with laminin (R&D Systems, 10ug/mL) to coerce adhered growth. Patient-derived cell neurospheres were dissociated using Accutase® (Sigma) and resuspended as a single cell suspension in culture media containing NeuroCult™ Proliferation Kit Human (Stemcell) supplemented with EGF 20ng/uL (Miltenyi Biotec), FGF 10ng/uL (Miltenyi Biotec) and Heparin 0.002% (Stemcell). Cells were plated at 1x103 cells/well and were left to recover and adhere overnight. Ten-fold dilutions of inhibitors were added to wells (n = 3), and plates were imaged every 8 hours in the Incucyte®. Values of confluency (% of well) were obtained using Incucyte® Analysis Software at segmentation of 0.4.
Raw growth curves for each drug condition were analyzed using GraphPad Prism. Area under the curves (AUCs) of growth curves were calculated for each condition, and to account for normal growth rate of each cell line, drug effect of a given concentration was defined as the AUC under a drug condition relative to the AUC of the DMSO condition. The points were plotted in a dose-response manner.
Tumor Formation
Intracranial injections.
Mouse neural stem cells infected with an empty vector or PPP1CB-ALK fusion were injected into the right striatum of 6–8 weeks-old male SCID mice (Jackson Laboratory, Bar Harbor, ME). Mice were anesthetized with isoflurane delivered at 0.2 liters/min, in combination with oxygen, and placed on a stereotactic frame. A suspension of 100,000 cells in 1ul of cell culture medium were delivered via intracranial injection. The coordinates of the injections were obtained as follows: The bregma served as the 0 coordinates (x=0mm, y=0mm, and z=0mm), and a burr hole was made at x=2mm on the right hemisphere of the skull, directly behind the right suture. The needle was slowly dropped to z=2.5mm into the cortical surface, and then lifted for an injection depth of z=2mm. Following the injection, mice were monitored at least 3 times a week for symptoms including ataxia, weight loss, and loss of balance. Moribund animals were euthanized and brains were removed and fixed with 4% paraformaldehyde. After the fixed tissue was paraffin embedded by standard methods, H&E staining was generated on the sections for tumor evaluation.
Mouse flank tumor studies.
NIH3T3 stable cell line were injected subcutaneously into the flanks of 6–8 weeks old male SCID mice (2 million cells per site). Tumor grow and body weight were measured three times a week. The most accurate volume calculations for caliper measurements were obtained using the formula V= ½ (Width2 x Length). When tumor volume reached 100–300 mm3, mice were randomized and treated for 15 days with ceritinib (30mg/kg/d, orally) or vehicle (methylcellulose 0.5% w/v, Tween 80 0.5%, ddH2O filtered water). All animal studies were performed according to Dana-Farber Cancer Institute Institutional Animal Care and Use Committee (IACUC)-approved protocols.
In utero mouse drug studies.
Timed-breeding pairs of C57BL/6 mice were established and monitored closely for successful mating and pregnancy. On days E15.5 – 17.5, pregnant females were given ceritinib (50mg/kg/d, orally) or vehicle (methylcellulose 0.5% w/v, Tween 80 0.5%, ddH2O filtered water). Animals were sacrificed on E18.5. Mouse pups and any unborn embryos were extracted and fixed in 4% paraformaldehyde. After the fixed tissue was paraffin embedded by standard methods, H&E staining was generated on the sections for evaluation of tissue structure and formation. Briefly, to assess the pharmacokinetics of ceritinib, samples were submitted to the Scripps Research Center for Mass Spectrometry and Metabolomics for mass spectrometry on mouse plasma, tissue, and embryo samples. A ceritinib dose was administered to animals two hours before sacrifice. After sacrifice, tissue was removed, and blood was drawn from the mouse and embryos, collected into EDTA-treated anticoagulant tubes, and centrifuged at 1,000g for 10 min under refrigeration. The resulting plasma supernatant was transferred and stored at −80°C until analysis.
Mass spectrometry analysis.
Sample preparation.
Tissue samples were homogenized in a bead homogenizer with 400µL of acetonitrile containing 500nM ceritinib-D7 (IS for ceritinib) or rucaparib (IS for lorlatinib). Plasma samples had 400µL ice-cold acetonitrile added, containing 500nM ceritinib-D7 or rucaparib and were vortexed one minute, then centrifuged. Solvent from all samples was harvested and placed into a speed-vac and evaporated to dryness at 10C. Samples were re-constituted in 100µL acetonitrile and analyzed.
Analytical conditions.
The samples were run on a Waters TQ-S micro triple quadrupole mass spectrometer. The mobile phase was composed of A= H2O/0.1% formic acid and B= ACN/0.1% formic acid. Column used was an Agilent SB-C18 21.x50mm with a 1.7um particle size. Gradient started at 60% A (0–0.5 min) increasing to 100% B in 2min and holding for 0.5min. Gradient was back at 60/40 at 2.7 min and held for 1.3 min to re-equilibrate for a 4 min runtime. Flowrate was 0.4mL/min. The sample injection volume was 5 µL. Operating in positive mode, the source conditions were as follows; capillary V=3.5kV, cone V = 50, desolvation gas flow = 1000L/hr and desolvation temp = 600C.
Data processing.
Data was processed using Waters TargetLynx software. For absolute quantification, calibration curves were generated by comparing the ratio of response of the analyte and its internal standard to its concentration. Concentration of compounds were corrected for the ratio of MS response (peak area) between analyte and IS to account for matrix affects and any losses during prep. Calibration standards ranged from 50fmol/ul – 25pmol/ul.
Statistical analysis.
P-values were calculated using one-way ANOVA to compare means of multiple groups and non-parametric Mann-Whitney test to compare two ranks. Log-rank (Mantel Cox) analysis was performed for animal studies (survival and tumor volume). Error bars show standard error of the mean.
Data availability.
The sequencing data generated in this study have been deposited under dbGAP accession number phs002380.v1.p1. Previously published single cell RNA-Seq data that were re-analyzed here are available under accession code GSE133531 (for the mouse brain dataset) (32) and S-EPMC5991609 (for the human brain dataset) (31,33). Additionally, we used the following publicly available datasets: The Genotype-Tissue Expression (GTEx) dataset (https://gtexportal.org/home) and The Allen Brain Atlas Dataset (https://portal.brain-map.org) All other data supporting the findings of this study are available from the corresponding authors on reasonable request.
RESULTS
Study overview
In order to evaluate the landscape of ALK aberrations in glioblastomas (GBM) across age groups, we evaluated a selected cohort of 371 pediatric and adult patients with a tissue-based pathology diagnosis of a high-grade glioma. These cases were collected from our institutions or publicly accessible pediatric GBM with sequencing data. The cohort included 10 congenital, 20 infant (ages 0–2 years), 185 pediatric (ages 2–21 years) and 156 adult (above 21 years) tumors (Figure 1A, Suppl Table 1). We evaluated tumors for genomic, RNA, and protein aberrations of ALK using newly-generated, clinical, and public data from a range of assays including whole-genome sequencing (n=179 subjects), ALK Fluorescent In-Situ Hybridization (FISH) for rearrangements (n=100), RNA sequencing (n=14), Targeted Exome Sequencing (TES; 500 cancer gene Oncopanel) (n=82), whole genome copy-number arrays (n=84), and ALK immunohistochemistry (IHC) (n=181) (31,32). More than one assay was available on 198 subjects (Figure 1B, Suppl Table 2) and all subjects had clinical data abstracted.
Figure 1. Overview of study and ALK aberrations in GBM.
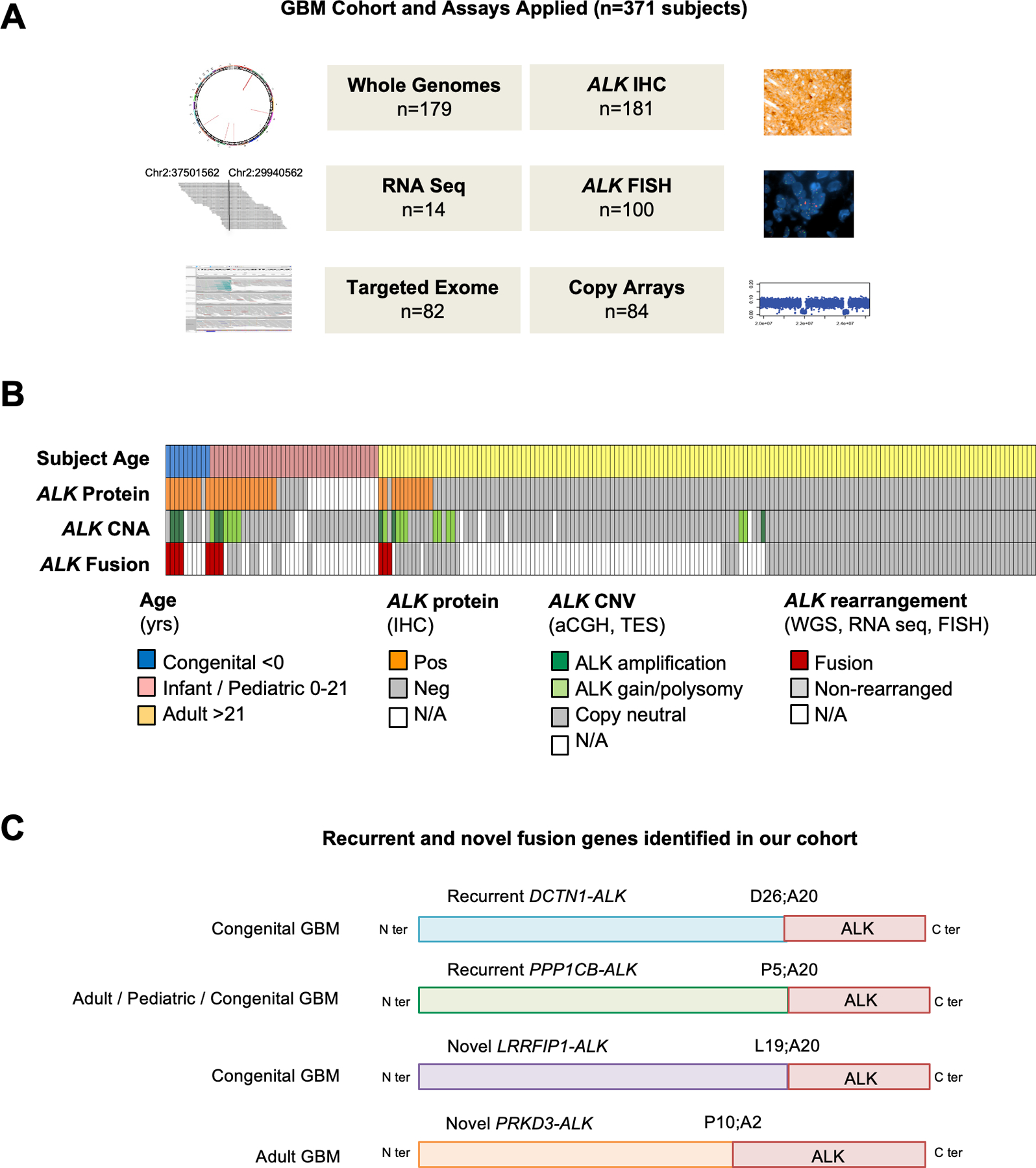
A. ALK aberration screening was performed on 371 GBMs and HGGs using orthogonal assays including Whole Genome Sequencing (WGS), RNA sequencing (RNAS), Targeted Exome Sequencing (TES), immunohistochemistry (IHC), break apart FISH probes flanking 3’ and 5’ ALK regions, and copy number arrays (array CGH and SNP array).
B. Oncoprint summary of ALK alterations in 198 GBMs with sequencing data (WGS or TES). 11/198 GBMs were found to have ALK rearrangements in this cohort. Tumors for which data are unavailable are designated N/A.
C. Schematic structure of ALK fusion proteins identified in our GBM cohort.
Across these 371 GBMs, 45 (12%) exhibited aberrations of ALK, 37 had increased protein expression and 11 had rearrangements/fusions (Figure 1B–C, Suppl Table 3). Most tumors with ALK rearrangement also had protein expression (10/11) or amplification (8/11) concurrently.
Novel and recurrent ALK fusions in congenital glioblastoma
ALK aberration characteristics differed significantly across age groups with aberrations seen most frequently in congenital (documented perinatal or intrauterine tumors) and infant (<2 years) GBMs. Overall, we detected ALK protein by immunohistochemistry in 75% of childhood cases (24/32), including 90% of congenital (9/10) and 68% of pediatric cases (15/22) (Figure 2A–B). Expression showed variable levels of intensity and was generally uniform, but rare cases had focal/heterogeneous staining (Figure 2B).
Figure 2. ALK aberrations are common in congenital and pediatric GBMs.
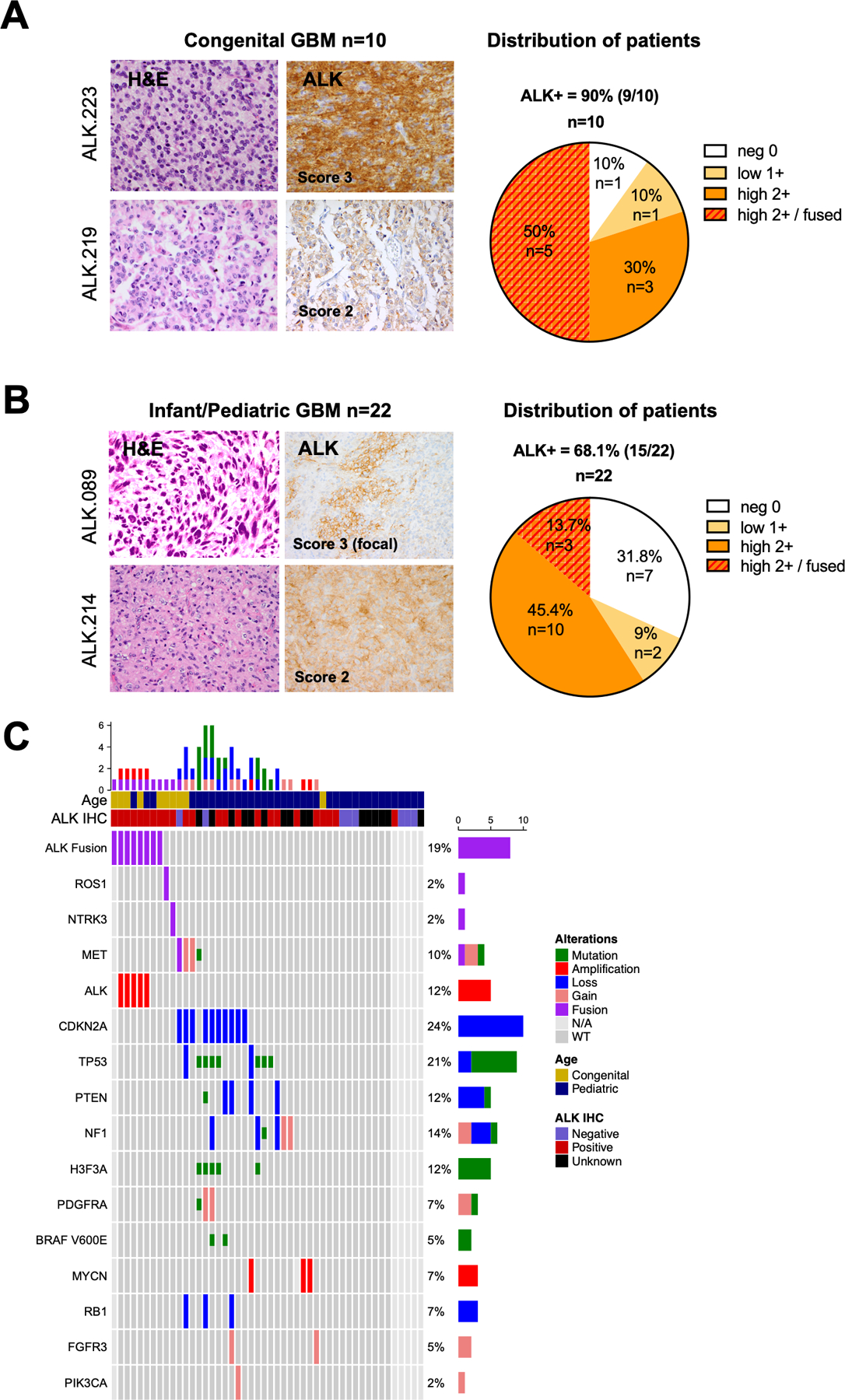
A. H&E staining showing glioma features. ALK immunohistochemistry (IHC) showing typical 3+ (left top) and 2+ (left bottom) staining appearance; pie chart demonstrating the distribution of ALK IHC scores in the congenital GBM cohort (n = 10). See methods for detail scoring schema.
B. H&E staining and ALK IHC of two illustrative pediatric GBMs scored as focally IHC 3+ (left top) and 2+ (left bottom); pie chart demonstrating the distribution of ALK IHC scores in the infant/pediatric GBM cohort (n = 22).
C. Oncoprint of sequenced GBMs illustrating ALK fusion/amplification is the sole candidate driver alteration in pediatric GBMs. The second lane showed the ALK-positive samples by IHC. Genomic aberrations including fusions (red), focal amplifications (>log 2.0, >10Mb), deletions or mutations (green). The tumors ALK.219, ALK.227 and ALK.228 harbored NTRK3, MET and ROS1 fusions respectively but were negative for ALK fusion.
Genomic analysis identified ALK rearrangements as being most prevalent in congenital GBMs with a lower incidence in older children. Overall, evidence of ALK rearrangements was detected in 3.7% of pediatric GBMs (8/215 cases with genomic data), including 50% of congenital cases (5/10) (Figure 2C). Congenital ALK GBM had clinical presentations similar to other congenital tumors, including prenatal/perinatal head U/S abnormalities with minimal mass effect and lesions mimicking hemorrhage or hypoxic ischemic damage (Figure 3A, Case ALK.223). Symptoms tended to occur months after birth, with tumors appearing on MRI requiring surgical resection (Figure 3B). Pathology showed features of GBM with high mitotic rates but often-subtle necrosis or vascular proliferation. All ALK rearranged congenital tumors showed IHC expression of ALK in membranous and cytoplasmic pattern (5/5, Suppl Figure 2) and the glioma markers OLIG2 and GFAP (Suppl Table 4). ALK FISH (n=4) performed on sequence-positive cases showed rearrangement with amplification (double minute pattern) present in a high percentages of tumor nuclei (75%, 3/4 cases) consistent with a clonal driver event (Figure 3F, Case ALK.076). One congenital case harbored an ALK fusion without ALK amplification (Figure 3C, case ALK.223).
Figure 3. Congenital GBMs have novel and recurrent ALK fusions and amplifications as sole oncogenic drivers.
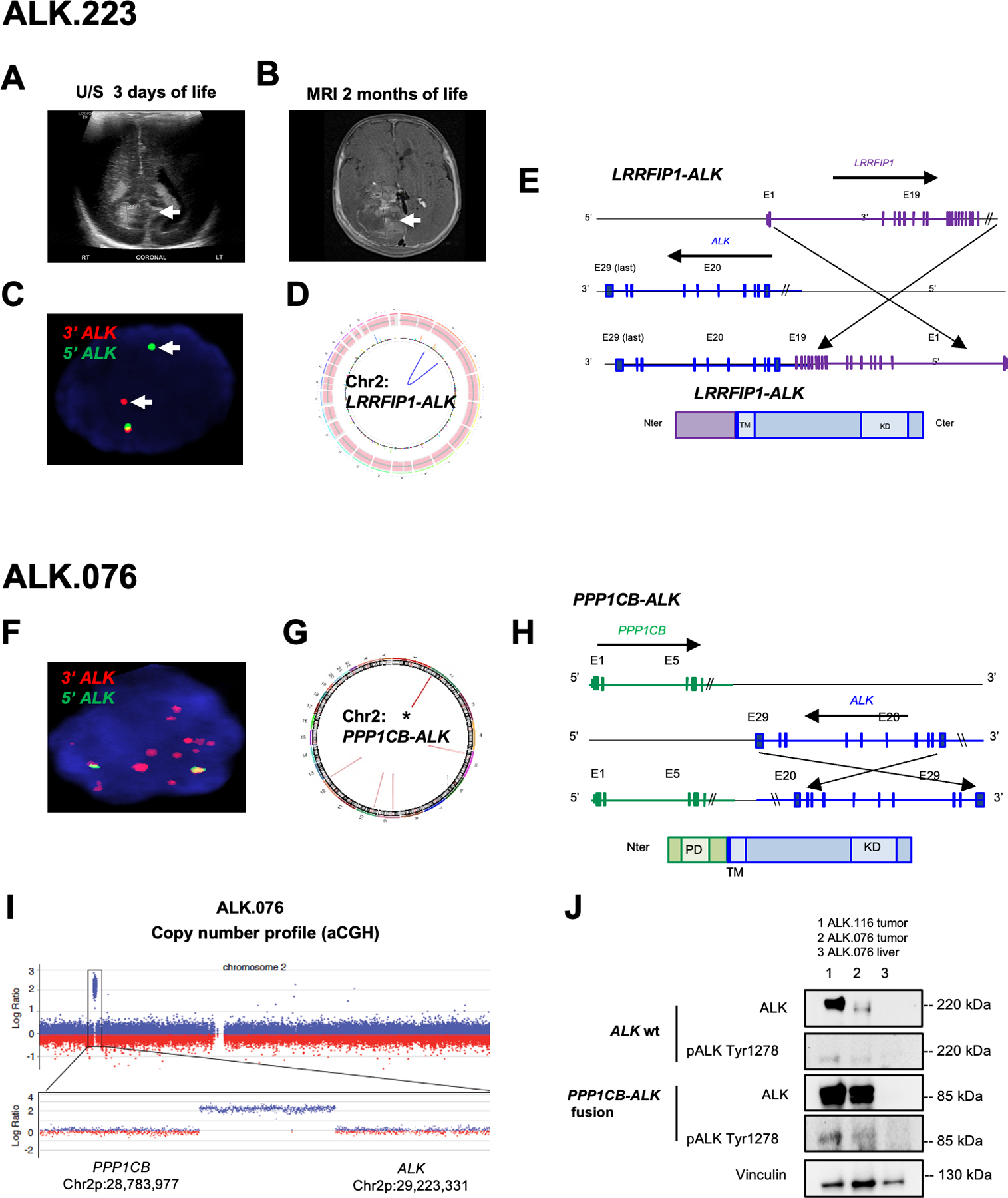
A. Subject #223 head ultrasound at 3 days of life demonstrated periventricular abnormality (arrow) considered as hemorrhage which was monitored.
B. MRI brain at 2 months showing large heterogeneous tumor within the right lateral ventricle (arrow) which was resected and diagnosed as GBM.
C. ALK FISH analysis of ALK.223 tumor nucleus showing probe break apart indicating rearrangement but no evidence of copy change (red, 3’ ALK; green 5’ ALK; overlapping signals in yellow indicate normal locus; nucleus DAPI blue).
D. Circos plot from whole genome sequencing showing intrachromosomal rearrangement of LRRFIP1-ALK and no evidence of other copy aberrations or rearrangements.
E. Schematic representation of LRRFIP1-ALK variant which fused in-frame the first 19 exons of LRRFIP1 (N-ter, Chr 2q) to the last nine exons of ALK (C-ter, Chr 2p).
F. FISH on showing amplification of the 3’ end of the ALK signal (red) in 5 week-old subject ALK.076 with a congenital GBM.
G. Circos plot from WGS identified PPP1CB-ALK fusion as the sole aberration.
H. Schematic representation of the PPP1CB-ALK variant which fuses the N-terminal portion of PPP1CB (including the phosphatase domain PD) to the intracellular region of ALK (containing the tyrosine kinase domain KD).
I. Copy number analysis showing 116kb focal amplification event involving the 3’ end of ALK and the 5’ end of PPP1CB.
J. Western showing PPP1CB-ALK protein expression at the expected molecular weight for the fusion (85 kd) compared to wild type ALK (220 kDa) and the presence of high pALKTyr1278 in fusion positive tumors (column 1 and 2; subjects ALK.116 and ALK.076 respectively). Liver tissue from ALK.076 (comumn 3) did not show expression of ALK wt or ALK fusion.
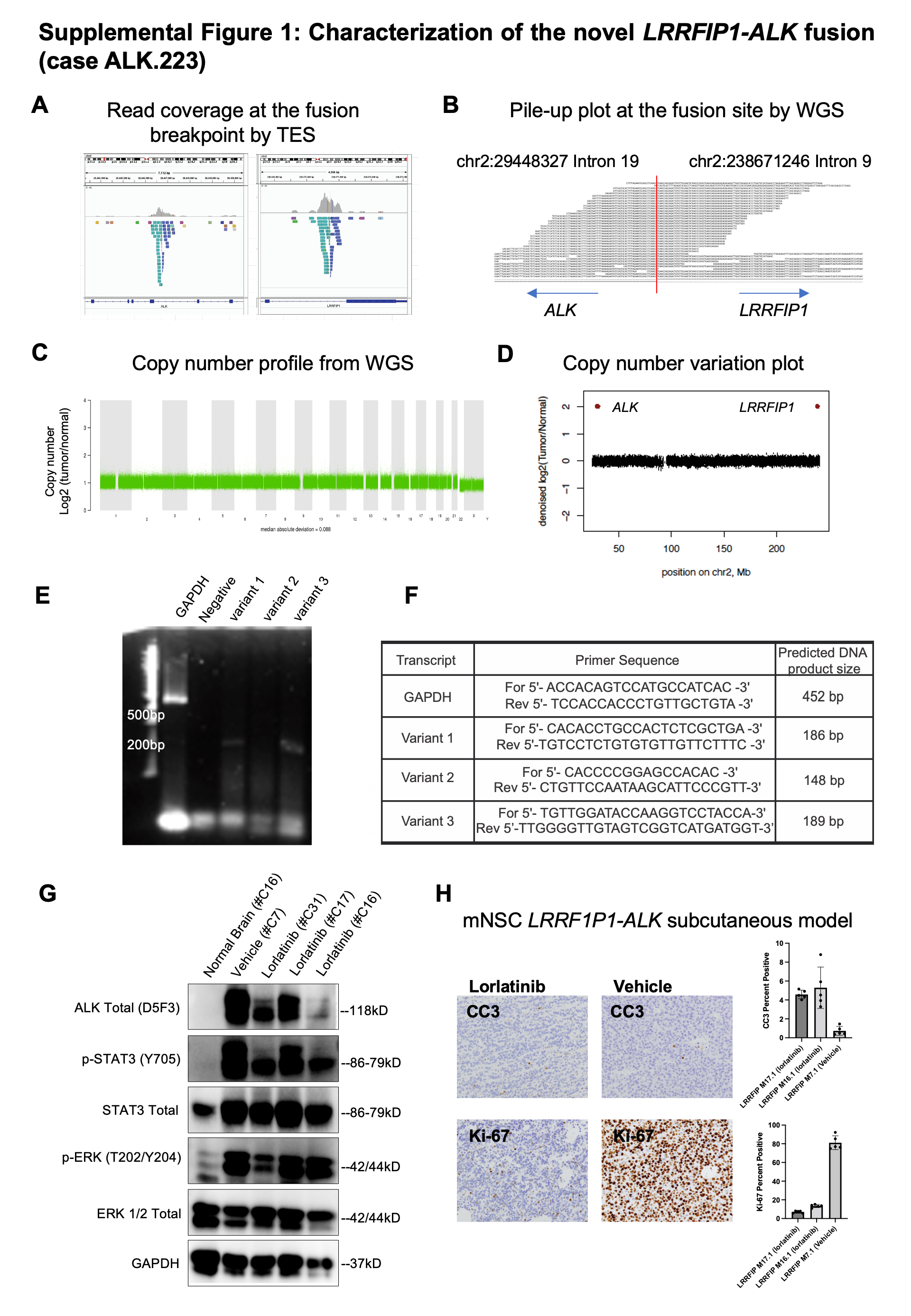
Among the eight congenital, infant, and pediatric cases with evidence of ALK rearrangement, two were novel (ALK-LRRFIP1 and ALK-DCTN1), five had PPP1CB-ALK fusion which was previously reported in pediatric brain tumors (infant GBM (15), low-grade glioma (33) or myxopapillary ependymoma (34)) and one had an unidentified partner. In all eight cases, ALK was the sole candidate oncogenic driver. Whole-genome sequencing and targeted exome sequencing (TES; Oncopanel) identified a novel fusion in the GBM ALK.223, involving the LRRFIP1 and ALK genes (Figure 3D and Suppl Figure 1A). LRRFIP1 (LRR Binding FLII Interacting Protein 1) encodes a transcription factor which regulates platelet function (35). LRRFIP1 is also known to repress TNF, EGFR, and PDGFRA gene transcription (36,37). One LRRFIP1-ALK fusion variant (variant 3) included exons 1–19 of LRRFIP1 (NM_001137550.1; chr2:238668848) fused in frame to C-ter exons 20–29 of ALK (NM_004304.4; chr2:29446394) (Figure 3E). RNA sequencing confirmed the presence of variant 3 with the first 19 exons of LRRFIP1 (N-ter) fused in-frame to exons 20–29 of ALK (Suppl Figure 1B) similar to other ALK fusions across cancers in the literature (10,38). LRRFIP1 (2q37.3) and ALK (2p23.2-p23.1) are located on opposite strands of chromosome 2, suggesting an inversion is responsible for this rearrangement. We did not detect any somatic copy number alterations anywhere in the genome (Suppl Figure 1C–D). An additional unusual variant (variant 1) involved N-ter exons 1–19 of ALK fused to exon 19–24 of LRRFIP1 (Suppl Figure 1E–G).
Three ALK fusions among congenital GBMs were the previously reported PPP1CB-ALK fusion (Figure 3G and Suppl Table 3) (15,16). These co-occurred with high level focal ALK amplifications with extrachromosomal pattern of 15–25 copies in nearly all tumor nuclei (Figure 3F,I). Sequencing showed tumors had the same breakpoints in intron 6 of PPP1CB and intron 19 of ALK (Figure 3H). In two of these, we detected ALK protein expression at the 80 kD mass predicted by the fusion and phosphorylation of both wild-type and fused ALK, indicating constitutive ALK kinase activity (Figure 3J).
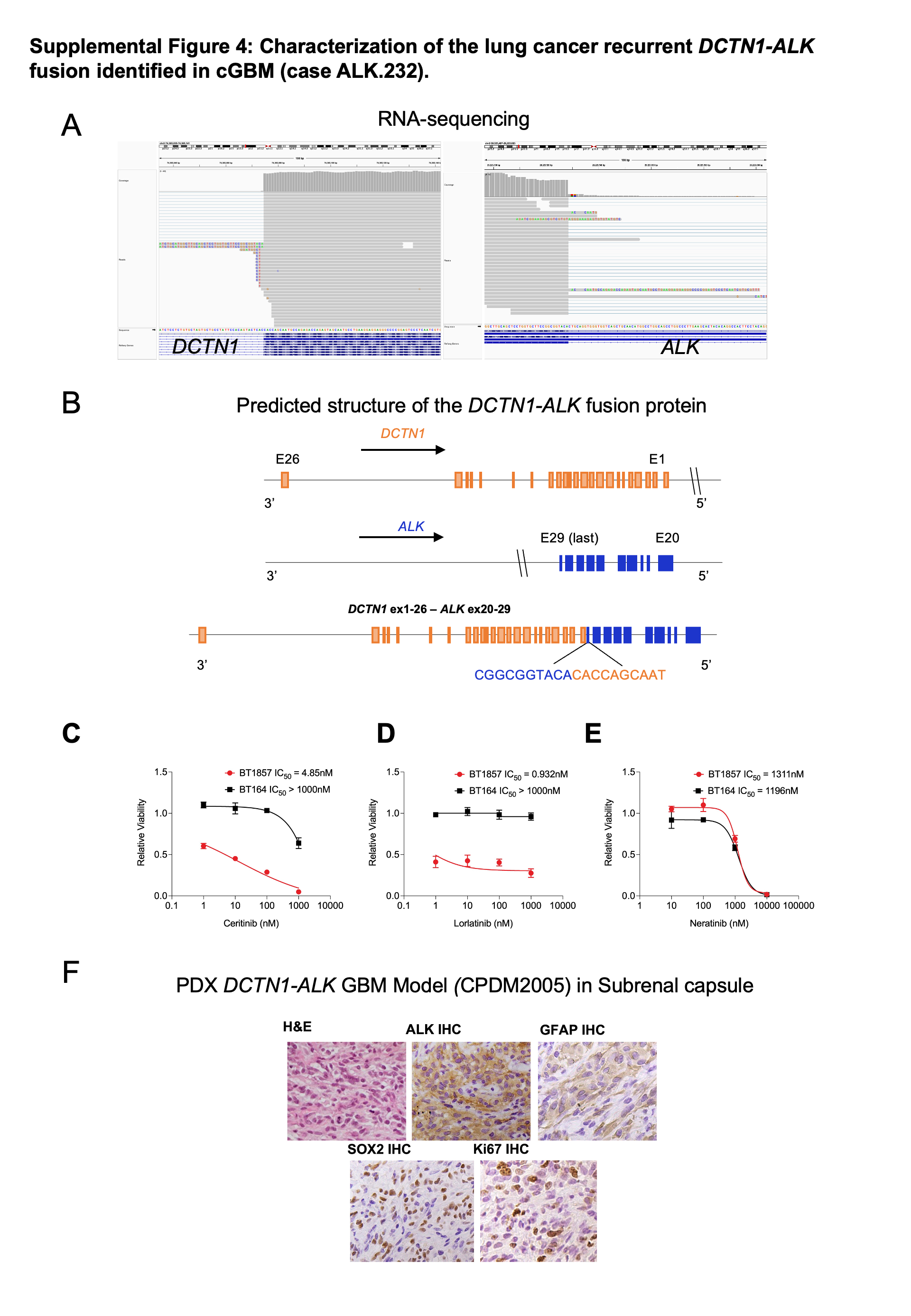
Lastly, we identified one congenital GBM harboring ALK fused to DCTN1, or dynactin subunit 1, a protein involved in transport of vesicles and organelles along microtubules. While not previously reported in gliomas, this event has been seen in rare cases of lung cancer (10) pancreatic cancer (39), inflammatory myofibroblastic tumors (40) and Spitz tumors (41). Sequencing revealed intronic breakpoints in DCTN1 intron 26 (chr2:74591071) and ALK intron 19 (chr2:29446465). Copy number analysis revealed discontiguous gains in chromosome 2; however, no other known tumorigenic variants in this sample were identified (Suppl Table 3).
To assess whether the clinical features can predict the presence of ALK fusion in pediatric GBM, we reviewed MRI data of congenital and pediatric GBM when available. We compared location of the tumor and tumor size for 4 ALK-fused GBM and 12 ALK-negative (no ALK alteration) GBM (Suppl Figure 2 and Suppl Table 5). The most consistent feature is that they are all supratentorial lesions and for the most part they are lobar in location. The average tumor size seems to be smaller in the ALK-negative group (83.2 cm3 ranging from 6.2–184.5 cm3) in comparison to the ALK-fused group (102.9cm3 ranging from 41.6–197cm3) (Suppl Table 5).
Identification of recurrent and novel ALK fusions in adult GBM
The incidence of ALK expression was much lower in the adult cohort, with only 12 of 149 cases (8%) demonstrating ALK IHC positivity (Figure 4A–B). Three of these adult cases demonstrated ALK fusions (Figure 4B), including one with the recurrent PPP1CB-ALK fusion described above. The other two were novel ALK rearrangements based on novel partners and novel fusion breakpoints beyond the prototypical intron 19 location. Case ALK.225 (Figure 4C–D) actually exhibited evidence of two ALK rearrangements, in introns 18 and 20 respectively, both connecting to non-coding loci on chromosome 2 (Figure 4D). This tumor also exhibited high-level ALK amplification (Figure 4C). Another adult tumor (ALK.226) exhibited an in-frame PRKD3-ALK fusion resulting from a 7.5 Mb deletion which led to fusion of exon 1–10 of PRKD3 (protein kinase D) with exons 2–29 of ALK (Figure 4E–I); the fusion transcript was validated at the DNA and RNA level. The chimeric protein was predicted to be 214 kD which preserves both the Pleckstrin homology domain of PRKD3, implicated in protein recruitment to the membrane and trafficking to intracellular compartments, and the kinase domain of ALK (Figure 4G).
Figure 4. Identification of novel ALK fusions in adult GBMs.
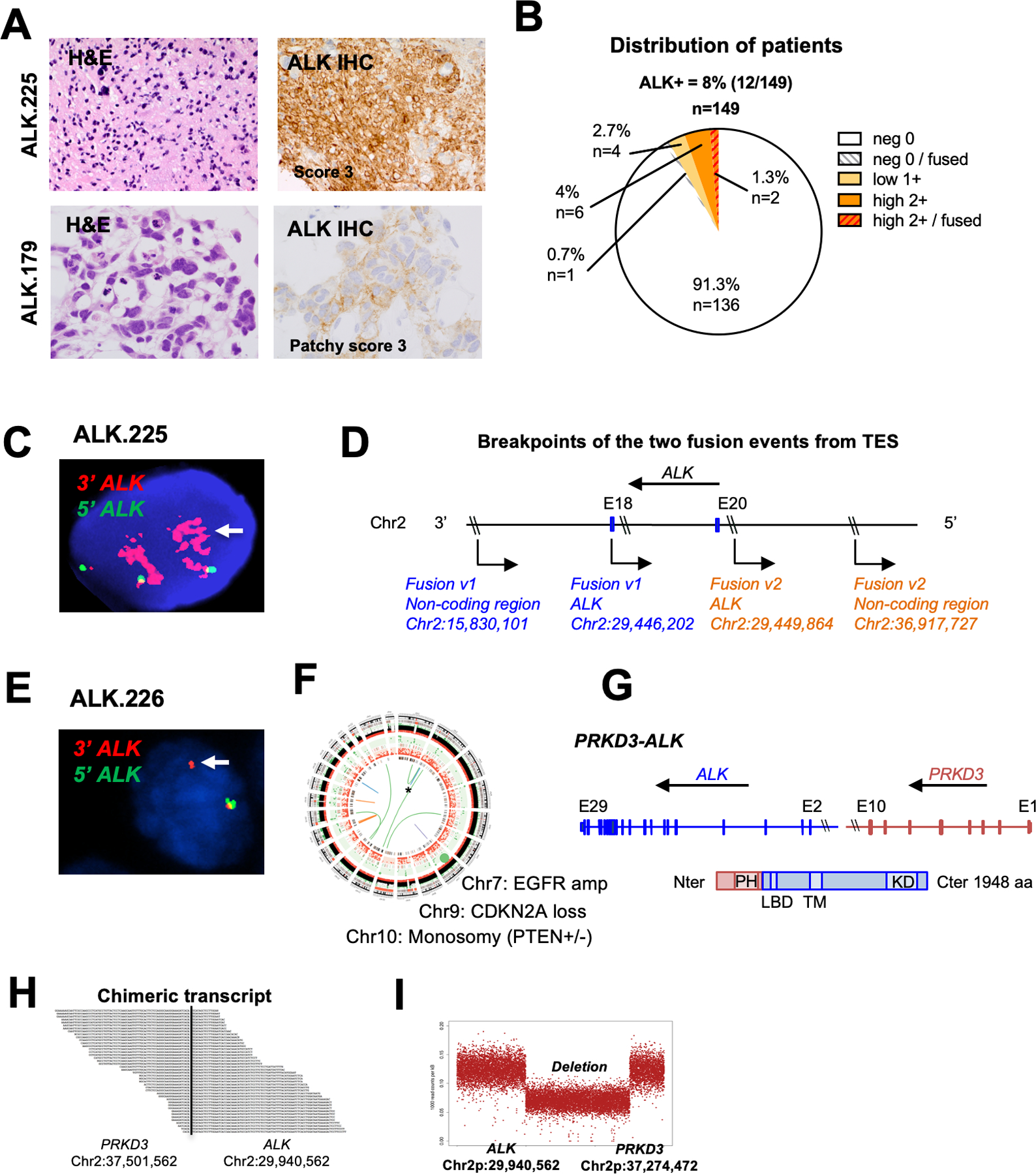
A. H&E staining and ALK IHC of two illustrative adult GBMs scored as IHC 3+ (left top) and 2+ (left bottom); Histology of tumor ALK.225 and ALK.179 showing hallmarks of glioma and strong ALK expression by IHC (score 3+) (ALK.225) and focal moderate staining (ALK.179).
B. Distribution of ALK IHC scores in the adult GBM cohort (n = 149).
C. ALK FISH of tumor ALK.225 showing rearrangement and amplification of the ALK 3’ region (red).
D. Schematic representation of the two ALK-noncoding genomic breakpoints. Fusion variant 1 (blue) occurred between ALK (in intron 20; breakpoint 2:29446202) and a non-coding genomic region on chromosome 2 (breakpoint 2:15830101). Fusion variant 2 (orange) occurred between ALK (exon 18; breakpoint 2:29449864) and a different non-coding region on chromosome 2 (breakpoint 2:36917727).
E. ALK FISH of tumor ALK.226 showing evidence of rearrangement (arrow, single 3’ red signal) and one normal appearing allele (yellow).
F. Circos plot of WGS for ALK.226 confirming novel PRKD3-ALK fusion co-occuring with classic adult GBM aberrations (EGFR amplification, CDKN2A loss, monosomy 10).
G. Schematic representation of the PRKD3-ALK variant, which fuses the N-terminal portion of PRKD3 (including the Pleckstrin homology domain PH implicated in protein membrane recruitment and intracellular trafficking) to the C-terminal portion of ALK starting with exon 2 (including the ligand binding domain LBD, transmembrane TM and kinase KD domains). Amino acid number is indicated on the right.
H. RNA sequencing of the PRKD3-ALK fusion demonstrates the novel breakpoints in intron 2 of ALK (chr2:29940562) and intron 10 of PRKD3 (chr2:37501562).
I. Copy number analysis shows a 7.5 MB interstitial deletion with breakpoints consistent with fusion of PRKD3 and ALK in-frame.
All novel fusions we identified conserved the ALK catalytic domain, consistent with the ALK fusions described in other cancer types. In contrast to pediatric ALK GBM, no adult GBMs exhibited ALK fusion as a sole driver event, and in fact had EGFR amplification as well as numerous other aberrations typical of adult GBM. The analysis of the TumorPortal dataset revealed that the kinase domain of ALK is highly mutated in 7% of neuroblastomas (8/81). No hotspot mutations were reported either in the TumorPortal GBM dataset or in our high-grade glioma cohort. The testing of ALK variants of unknown significance showed no evidence for oncogenic activity as measured in cell growth assays (data not shown).
ALK fusions are oncogenic
We comprehensively evaluated the oncogenic potential of the glioma-specific PPP1CB-ALK fusion and the novel LRRFIP-ALK rearrangement across a range of cell types and lineages including NIH-3T3 mouse fibroblasts and murine embryonic neural stem cells (mNSC) derived from cerebral cortex (CTX), ganglionic eminence (GE) and brainstem (BS) regions at 14.5 dpc (Figure 5A).
Figure 5: Recurrent PPP1CB-ALK and novel LRRFIP1-ALK fusions are oncogenic drivers in vitro and in vivo.
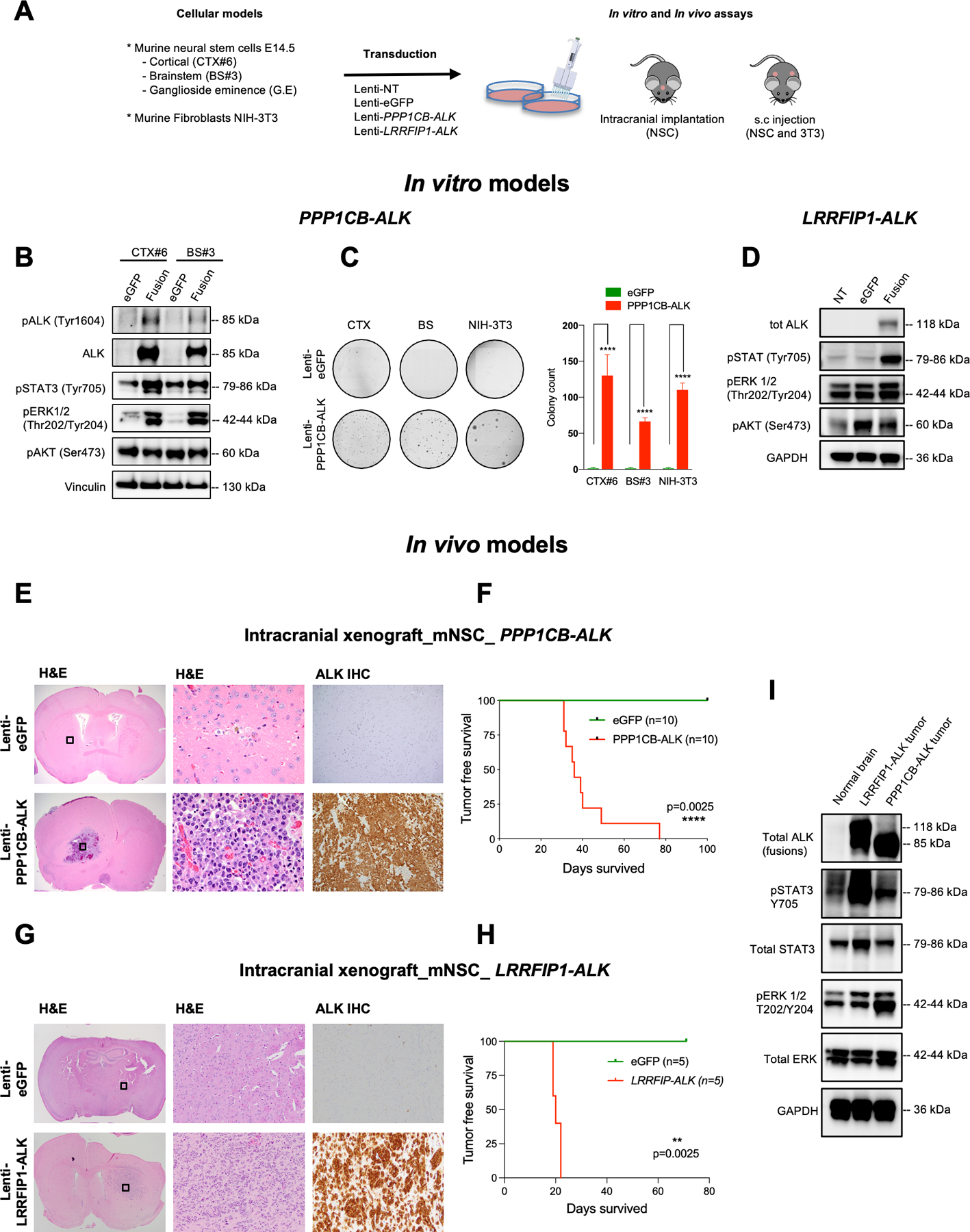
A. Overview of PPP1CB-ALK assessments in vitro and in vivo using murine fibroblasts and mouse NSCs isolated from cortex (CTX), brainstem (BS) and ganglioside eminence (G.E).
B. Representative western blot of PPP1CB-ALK expression and ALK Signaling Pathways in murine neural stem cells (mNSC) isolated from cortex (CTX) and brainstem (BS). Vinculin was used as a loading control.
C. Assessment of the anchorage-independent growth ability of murine NSC and murine fibroblasts expressing PPP1CB-ALK fusion. Quantification of colony formation using CellProfiler. Values represent colony counts ± s.d. The mean of three independent replicates is shown. Error bars show standard error of the mean. Significance between eGFP and PPP1CB-ALK cells is determined by the Mann-Whitney test. ****P < 0.01.
D. Representative western blot of LRRFIP1-ALK expression and ALK Signaling Pathways in murine neural stem cells (mNSC) isolated from cortex (CTX). GAPDH was used as a loading control.
E. H&E and ALK IHC staining of representative mouse brains injected with NSC eGFP (upper row) compared to mNSC PPP1CB-ALK brains (bottom row). H&E boxed areas shown at higher magnification in center.
F. Kaplan-Meier survival curves of SCID mice injected intracranially with mNSC CTX-PPP1CB-ALK (red, n = 10) or mNSC CTX-eGFP (green, n = 10).
G. H&E and ALK IHC staining of representative mouse brains injected with mNSC eGFP (upper row) compared to mNSC LRRFIP1-ALK grafted brains (bottom row). H&E boxed areas shown at higher magnification in center.
H. Kaplan-Meier survival curves of SCID mice injected intracranially with mNSC CTX-PPP1CB-ALK (red, n = 5) or mNSC CTX-eGFP (green, n = 5).
I. Western blot of PPP1CB-ALK and LRRFIP1-ALK tumor lysates from mouse intracranial tumors compared to unrelated normal brain control showing ALK activation effects on STAT and ERK signaling. GAPDH was used as a loading control.
Upon expressing PPP1CB-ALK, neural stem cells from both cortex and brainstem (mNSC CTX#6, BS#3) exhibited constitutive ALK activation as indicated by pALK Tyr1604 and concomitant increases in downstream pSTAT3 Tyr705 and pERK1/2 Thr202/Tyr204 (Figure 5B). In contrast to prior reports of EML4-ALK fusions (42), PPP1CB-ALK did not appear to activate PI3K/AKT signaling as measured by pAKT S473 (Figure 5B). PPP1CB-ALK did not affect proliferation but led to robust colony formation in soft agar while eGFP control cells did not form any colonies (Figure 5C) (43,44). Cortical NSCs ectopically expressing the novel fusion LRRFIP1-ALK also activated pSTAT3 Tyr705 and pERK1/2 Thr202/Tyr204 but have minimal effects on pAKT (Figure 5D).
To test in vivo impact on tumorigenesis, we transduced PPP1CB-ALK into murine fibroblasts (NIH 3T3) and murine neural stem cells (mNSC-CTX#6) and injected the cells into the subcutaneous and intracranial compartments respectively. Both models exhibited robust in vivo tumor formation. PPP1CB-ALK:mNSCs generated intracranial tumors in 100% of mice (10/10). Intracranial tumors had histological features of GBM, with single cell diffuse infiltration of the brain, high proliferation rate, and necrosis Mice were moribund by an average of 47 days post-injection (Figure 5F). Immunohistochemical analysis of intracranial (Figure 5E) and subcutaneous models showed strong cytoplasmic expression of ALK, OLIG2, and GFAP (data not shown). No tumor formation was observed with eGFP controls.
The novel LRRFIP1-ALK was also sufficient to induce formation of subcutaneous and intracranial tumors following injection of mNSCs transduced to express the fusion. All mice injected showed rapid intracranial growth. The mice developed symptoms indicative of intracranial tumor burden more quickly than the PPP1CB-ALK mice within 20 days (Figure 5G. Pathologic examination of the brain revealed that tumors had a close resemblance to patient GBM as well as the PPP1CB-ALK models and displayed high membranous and cytoplasmic ALK expression (Figure 5H). Western blot analysis of the mNSC intracranial mouse tumor models revealed that PPP1CB-ALK and LRRFIP1-ALK both had high levels of pSTAT3 Tyr705 and pERK1/2 Thr202/Tyr204 indicative of activation of these pathways (Figure 5I) consistent with our findings in our in vitro models.
ALK fusions are sensitive to ALK inhibitors
Given the oncogenic properties of the above ALK fusions, we sought to determine their responsiveness to ALK inhibition. In vitro, robust inhibition of ALK phosphorylation and downstream signaling was observed in PPP1CB-ALK mNSC-CTX#6 treated with brain-penetrant ALK inhibitors lorlatinib and ceritinib as compared to vehicle controls (Suppl Figure 3A). PPP1CB-ALK-expressing NIH-3T3 were also sensitive to ceritinib compared to NIH-3T3-GFP controls (IC50=6.4nM and IC50=1442.9nM respectively) (Suppl Figure 3B). Colony formation induced by PPP1CB-ALK was suppressed in a dose-dependent manner (Suppl Figure 3C).
To evaluate in vivo effects of ALK inhibition, we treated PPP1CB-ALK-NIH3T3 subcutaneous tumors for 15 days with ceritinib (30mg/kg/d) or vehicle (Figure 6A). Tumors treated with ceritinib (n=10) all exhibited profound tumor regression, relative to controls (n=7) (Figure 6B – left panel). Ceritinib-treated mice significantly showed the longest survival (Suppl Figure 3H). Immunohistochemical analysis revealed a strong decrease of pSTAT3 Tyr705 and Ki67 index in ceritinib-treated tumors, with no obvious change in markers for pERK (Thr202/Tyr204) expression (Suppl Figure 3D–F) which is consistent with the downstream signaling inhibition effect observed in our PPP1CB-ALK mNSCs in vitro model. Ceritinib treatment did not result in weight loss in mice indicating that the treatment was well tolerated (Suppl Figure 3I).
Figure 6. ALK fusions are sensitive to ALK inhibitors.
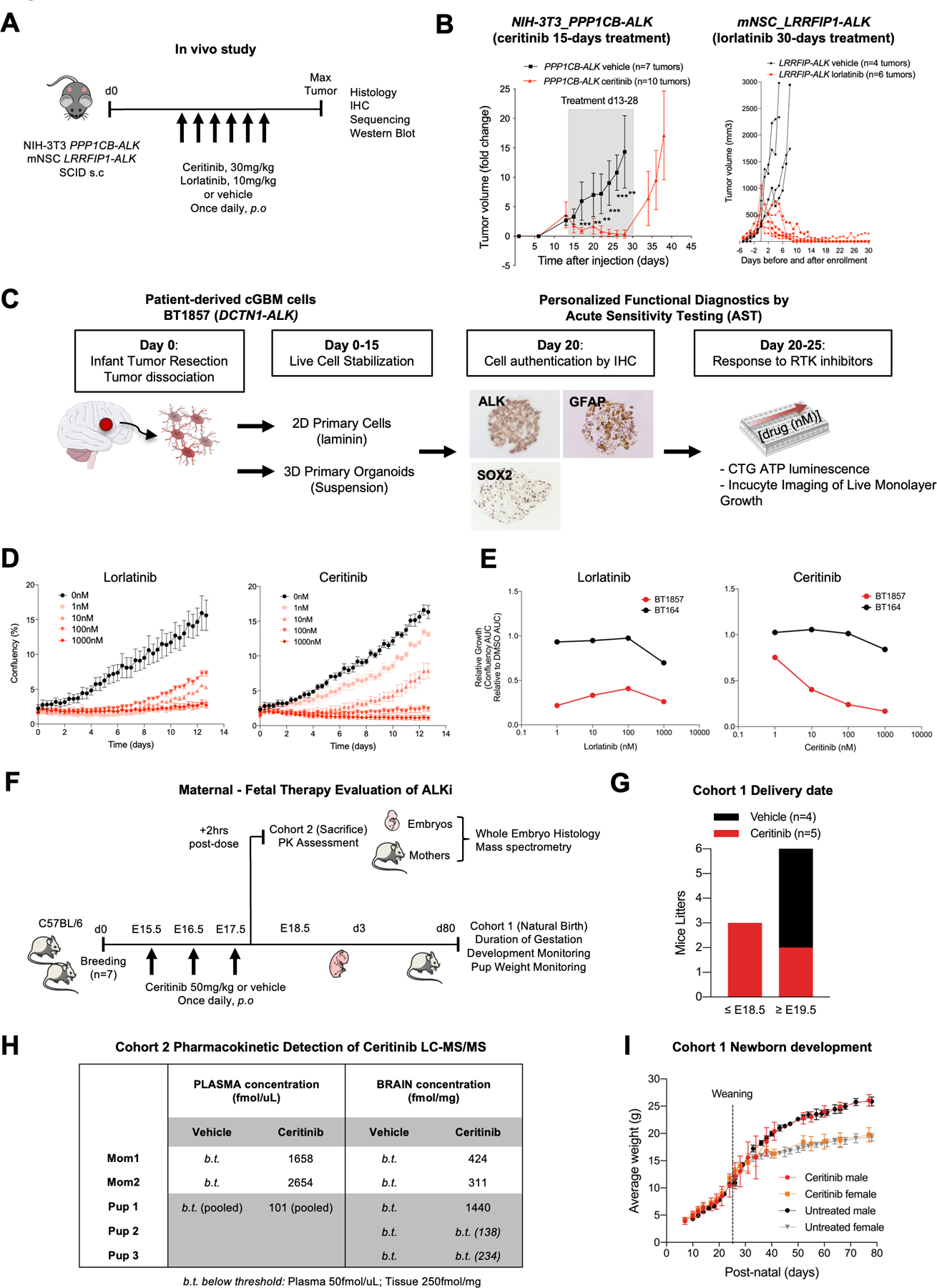
A. Study schematic for subcutaneous transplantation of PPP1CB-ALK-NIH-3T3 and LRRFIP-ALK-mNSC in SCID mice. Ceritinib and Lorlatinib treatments were initiated when tumor volume reached 200 +/− 200mm3 and continued for 15 and 30 consecutive days respectively.
B. Caliper measurements of tumor volume in mice treated with vehicle (black curves) or ALK inhibitors (red curves). Shaded area denotes treatment range. Data are fold change +/− s.e.m of controls.
C. Personalized functional diagnostic approach and acute sensitivity testing of cGBM cells (BT1857), bearing a DCTN1-ALK fusion. Live cells were isolated from the newborn tumor, dissociated, and stabilized for up to 15 days in culture as organoid/spheroid or 2D adherent cultures on laminin prior to authentication by IHC. Cells were tested for sensitivity to ALK and other kinase inhibitors by CellTiterGlo or Incucyte monospheroid/organoid growth assays.
D. Incucyte growth profile of adherent cGBM DCTN1-ALK fused cells (BT1857) showing dose response to ALK inhibitors lorlatinib and ceritinib. Incucyte raw growth curves show confluency (%) over ~13 days of continuous imaging.
E. DCTN1-ALK fused patient cells (BT1857) sensitivity to ALKi compared to an ALK wild-type GBM cell line (BT164). AUC curves of ALKi-treated cells versus DMSO control.
F. Schematic outlining the administration of a brain penetrant ALK inhibitor, Ceritinib, to pregnant C57BL/6 mice to assess pharmacokinetics and neural developmental effects in perinatal period.
G. Delivery dates of live pups for pregnant C57BL/6 mice treated with vehicle or ceritinib from E15.5 to E17.5 embryonic development stages.
H. Quantification of ceritinib concentrations by LC-MS/MS in maternal, neonatal brain, plasma, and liver.
I. Newborn’s weight in control and ceritinib-treated groups.
We also observed in vivo efficacy of Lorlatinib in the setting of LRRFIP-ALK. Lorlatinib is a third generation ALK inhibitor which is reported to be highly brain penetrant and is currently being planned for use in pediatric glioma clinical trials. Mouse NSC-CTX#6 cells were transduced to express the LRRFIP-ALK fusion and injected subcutaneously to establish flank tumors. Mice with tumor sizes of 300–500 mm3 were randomized between vehicle and lorlatinib treatment groups. Mice treated daily with lorlatinib (n=6) all exhibited complete tumor regression on average 10 days after treatment with no change in growth seen in the control vehicle treated mice (n=4) (Figure 6B – right panel). To evaluate the pharmacodynamics of lorlatinib, we acutely treated mice with three doses of lorlatinib (10mg/kg/d) with two doses over 48 hours (n=3) and tumor tissue was harvested 2 hours after the final dose. Downstream signaling inhibition effect was observed with a moderate decrease on Western blots of pSTAT3 (Tyr705) and pERK (Thr202/Tyr204) expression in tumors treated with lorlatinib (Suppl Figure 1G). Furthermore, we saw a marked decreased of proliferation (Ki67) in lorlatinib-treated xenografts and evidence of increased apoptotic cell death (cleaved caspase 3) in line with the strong efficacy results (Suppl Figure 1H). To confirm the potential of lorlatinib to penetrate the blood brain barrier, we also evaluated lorlatinib penetration in the normal brain by mass spectrometry in the same subcutaneous LRRFIP1-ALK tumor bearing mice that received treatment. Lorlatinib was confirmed to show high concentrations of lorlatinib in normal brain while concentrations in vehicle-treated mice were below the lower limit of detection (50nM). Lorlatinib concentrations in normal brains were lower than the concentrations in the corresponding subcutaneous tumors but all concentrations were greater than 1200nM suggesting that Lorlatinib was able to effectively cross the intact blood-brain barrier (Suppl Table 8).
To assess ALK as a key dependency more directly in human GBMs with ALK alterations, we used a novel acute sensitivity testing (AST) assay to examine effects of ALK inhibitor applied to freshly isolated patient tumor cells from the ALK.232 patient with the DCTN1-ALK fusion congenital GBM (Figure 6C). Patient ALK.232 was a two-day old newborn who was found on prenatal testing at 36 week gestational age to have a congenital right hemispheric tumor. Post-delivery, the patient had a tumor biopsy (Surgery #1, S1) resulting in a pathologic diagnosis of GBM. ALK IHC showed strong positivity and Oncopanel sequencing showed a DCTN1-ALK fusion and no other driver aberrations. RNA sequencing confirmed the presence of the variant with the first 26 exons of DCTN1 (N-ter) fused in-frame to exons 20–29 of ALK (Suppl Figure 4A–B). A complete resection of the patient tumor was immediately performed (Surgery #2, S2) and tumor cells were isolated to generate two short-term fresh cell models: a 2D in vitro monolayer (BT1857–2D) and a 3D free floating organoid/spheroid model (BT1857–3D) (Figure 6C). Both models had striking acute sensitivity to clinically relevant doses of targeted ALK inhibitors lorlatinib (IC50=0.932nM) and ceritinib (IC50=4.85nM) which induced sensitivity in a non-small cancer cell line harboring EML4-ALK fusion (45) (Figure 6D–E). These effects appeared to be specific; a concurrently treated ALK-wild type adult GBM patient-derived cell line (BT164) exhibited minimal response (IC50 >1000nM) (Suppl Figure 4C–D), and BT1857–2D and BT1857–3D did not respond to the kinase inhibitor neratinib, an EGFR/HER2/HER4 inhibitor, (IC50 >1000nM) (Suppl Figure 4E). To attempt to develop a long term PDX model for preclinical studies, we then implanted the patient-derived cells of ALK.223 in the sub-renal capsule of a NOD-SCID mouse. Tumor was detected by ultrasound one month after implantation. Immunohistochemistry analysis of the sub renal capsule tumor verified features matching the patient congenital GBM including high expression of ALK, GFAP, SOX2 and Ki67 (Suppl Figure 4F). However, serial passage of the initially formed tumor was unsuccessful.
ALK expression in the developing and perinatal brain
Given that we identified ALK fusion tumors predominantly in the intrauterine or perinatal time frame, a key clinical question is the whether treatment with ALK targeted therapy in infants may be effective and have a favorable safety profile to the current standard of care, surgery and chemotherapy(46). Prior reports had shown evidence of ALK expression in peripheral nervous system and subsets of brainstem neurons at early developmental stages, however, little ALK expression had been described in later gestation and early postnatal periods. We therefore sought to further document the potential impact of targeted ALK inhibitor treatment during early brain development by examining ALK expression and effects of ALK inhibitors during developmental stages.
ALK RNA expression was first examined at the bulk and single cell level in multiple datasets. In analyzing the Genotype-Tissue Expression (GTEx) dataset (https://gtexportal.org/home) we found bulk ALK RNA expression to be low or undetectable (<10TPM) in all mature human tissues, except for testis (n=259, median TPM = 3.081) and pituitary gland (n=183, median TPM = 3.532). No significant ALK expression was detected in multiple brain regions (cortex (n=255, median TPM = 1.199), hypothalamus (n=202, median TPM = 1.650), and cerebellum (n=241, median TPM = 0.2796). Orthogonal data from the Allen Brain Atlas Dataset (https://portal.brain-map.org) also revealed no significant ALK expression across the full course of human brain development (8pcw to 40 years-old) by bulk RNA sequencing or exon expression microarray.
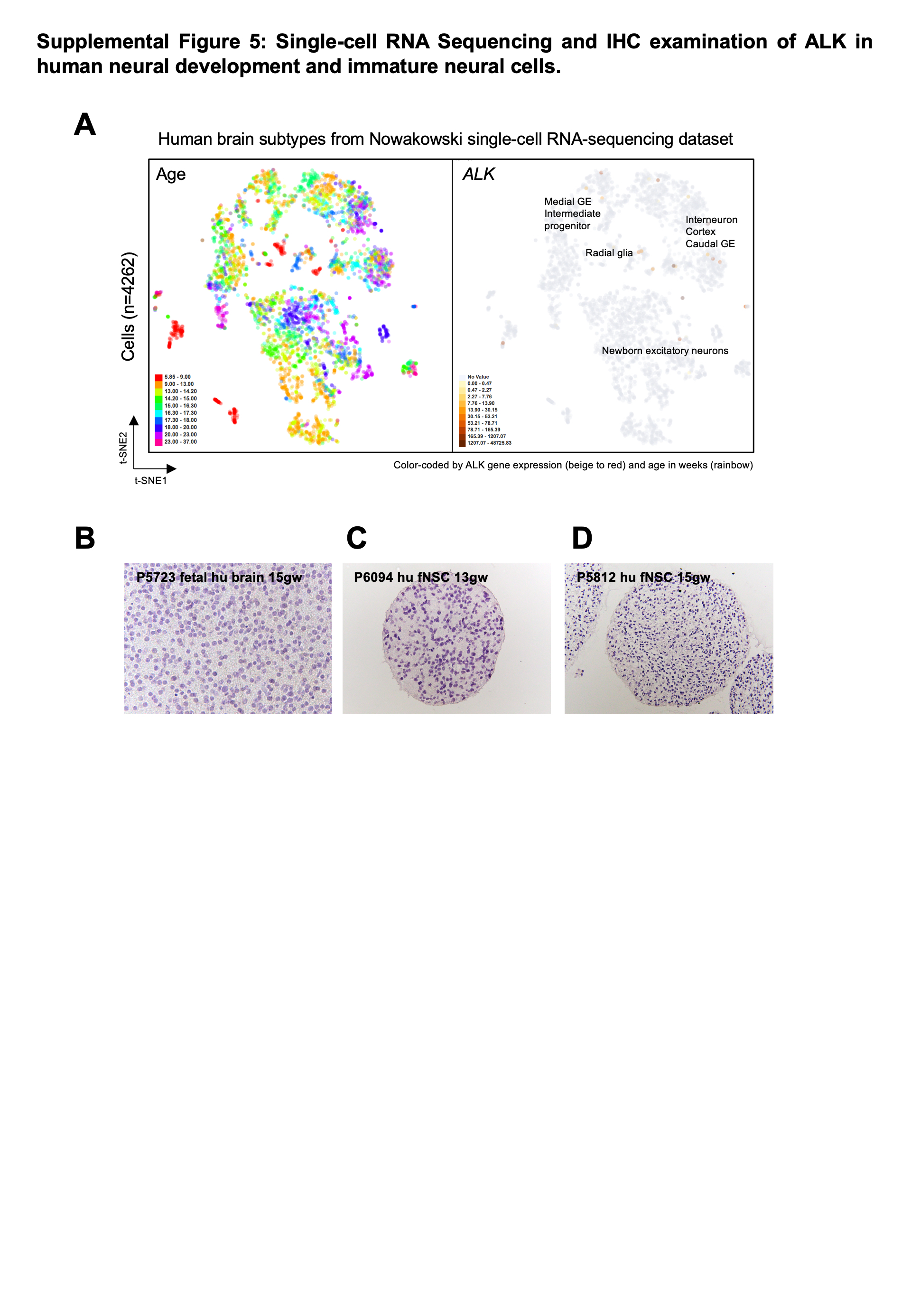
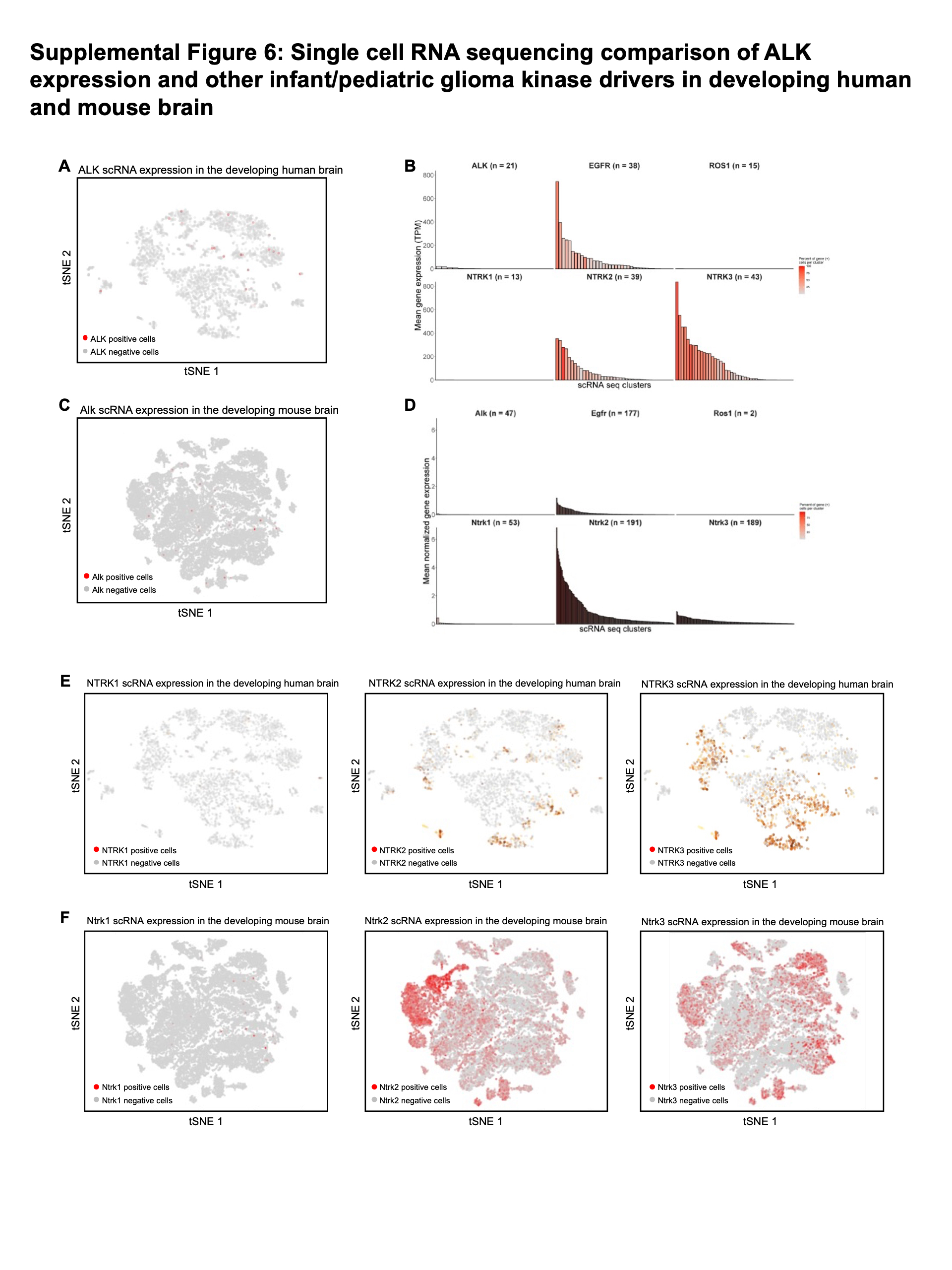
Given that rare subpopulations of ALK-expressing cells may exist in the developing brain at levels below standard bulk sequencing methods, we examined single-cell RNA sequencing data of both the developing human and mouse brain. Human forebrain sequencing data spanning 5–37 gestational weeks was obtained from Nowakowski et al., 2017 (cortex-dev.cells.ucsc.edu). ALK expression was detected in only a small number of cells (31 out of 4261, 0.7%), and showed only a low level of expression (median expression of 30 TPM in ALK positive cells) compared to EGFR, NTRK2, and NTRK3 (Suppl Figures 5A, 6A, 6B). In contrast to EGFR and NTRKs, ALK positive cells did not appear to belong to a specific cluster or cell type, with rare ALK cells distributed across 21/48 (43.8%) clusters. Only 5 of those clusters contained more than 1 ALK positive cell – and no cluster had more than 2 ALK positive cells. Similar results were seen in mouse brain single cell RNA sequencing across forebrain and pons development (E12.5-P6 days, 5 timepoints) (32). Only 72/65,589 (0.1%) cells were identified as Alk positive, and once again normalized Alk expression was extremely low compared to Egfr, Ntrk2, and Ntrk3, and was not localized to a specific cluster or cell type (Suppl Figure 6C, 6D). By comparison, the 2D cluster representation of scRNA sequencing data for NTRK1, NTRK2 and NTRK3 are shown for the developing human (Suppl Figure 6E) and mouse (Suppl Figure 6F) brain from the same corresponding datasets. NTRK1 shows a similar lack of expression in the human and mouse brain as ALK, however NTRK2 and NTRK3 show an increase in both expression values and number of expressing cells across both datasets.
We also extended our analysis to include protein expression, as determined by immunohistochemistry of human embryonic brain at 15 weeks of gestation (Suppl Figure 5B) and human fetal neural stem cells (hu fNSC) at 13 and 15 weeks of gestation (Suppl Figure 5C–D). In each case, we did not detect any evidence of ALK-expressing cell populations. Collectively, these data indicate that ALK appears to be in low abundance or absent in most cell types of the perinatal and late gestation developing forebrain when ALK gliomas present clinically.
Evaluation of normal brain development following maternal-fetal and perinatal exposure to ALK inhibitors
To further explore the potential effects of ALK inhibitors on brain development in the time when cGBM and iGBM develop, we sought to administer ALK inibitors to fetal and perinatal mice (Figure 6F). Given the difficulties with repeated handling and administration of drugs to immediate post-natally born mice, three doses of ceritinib were administered to pregnant mice once daily from E15.5–17.5 dpc to see if drug could be delivered across the placenta and fetal blood-brain barriers. Ceritinib-treated animals delivered one to two days early (n=5, 17.5–18.5 dpc) compared to vehicle (n=4) (Figure 6G) but were born in normal litter sizes, without gross deformities, and developed without maternal neglect. Ceritinib was detected by LC-MS/MS in all samples derived from ceritinib-treated mothers in plasma and brain (Figure 6H and Suppl Table 6) and low but detectable levels of ceritinib were seen in blood and brain tissue of a subset of the pups post-natally indicating some degree of sustained exposure in the immediate post-natal period. We then performed pathologic analysis of the brains and no gross or microscopic defects were detected. Treated mice also continued to develop normally as determined by monitoring of weight from birth to day 80 (Figure 6I). In summary, treatment of perinatal mice with ALK inihibtor ceritinib showed minimal effects on gross brain structure, suggesting more comprehensive and detailed comparison of effects of these agents to other treatments may be warranted.
DISCUSSION
The data we describe here have implications for diagnosis, treatment, and early detection of ALK-rearranged GBM. We describe ALK fusions as recurrent genomic alterations most commonly in congenital GBM (5 of 10 or 50%) and presenting in the infant age range as those tumors now described in the 2021 WHO Classification as ‘infant high-grade gliomas’. However, ALK-rearranged gliomas were also seen in other age groups but specifically were more rare with advancing age including pediatric GBM (3 of 205 or 1.4%) and adult GBM (3 of 156 or 1.9%), the latter of which has not been previously well described. Our study shows that ALK rearrangements seem to be mutually exclusive with other RTK rearrangements in pediatric patients, suggesting it might represent a sole driver event in pediatric high-grade gliomas. However, rearrangement or protein expression in adults seems to co-occur with other common oncogenic drivers in GBM. ALK rearrangements are likely a late event in the course of glioma development as these alterations are not described in pediatric low-grade gliomas (53).
Although ALK fusions are described in several human cancer types, the mechanism by which the fusion occurs is not well-known. In our cohort, we observed that ALK amplification is highly correlated with a fusion event in adult and pediatric GBM with ALK amplification in 54% of ALK-rearranged GBMs (6/11). In contrast analysis of the copy number alterations of 10844 tumors from the TCGA dataset reveals that ALK and the recurrent fusion partners are not focally amplified or located within a focal peak region of amplification in any commonly ALK- rearranged cancer types (e.g. lung cancer). However, high ALK expression has been shown to be tightly associated with ALK amplification in neuroblastoma and rhabdomyosarcoma suggesting the upregulation of ALK expression by increasing DNA dose (11). Non-small cell lung cancer, oesophageal and breast cancers exhibit ALK amplification but this does not correlate with gene rearrangement and increased protein expression by IHC. Our data therefore suggests that amplified fusion protein seen in gliomas may be somewhat unique and could provide novel mechanistic insights on ALK signaling in other cancers.
The fusions of the pediatric cases showed the same fusion location at intron 19 as in the vast majority of other ALK cancers. However, our study also identified potential novel fusion location in the adult PRKD3-ALK. Such fusions seemed to produce similar tumors in patients and in models but some difference in signaling and growth rate of the mNSC models could suggest differences may exist also in patients. NPM1-ALK fusion increases cell proliferation and promotes cell survival through the activation of the ERK1/2 complexes (54) and STAT3 respectively (55). while EML4-ALK fusions are thought to lead to the activation of oncogenic signaling through PI3K/AKT and JAK/STAT (42,56), PPP1CB-ALK and LRRFIP1-ALK seemed to preferentially activate ERK1/2 and STAT3 but we did not see strong activation of PI3K/AKT signaling. This suggests that the utilization of downstream pathways may be unique in the brain with implications for potential combination therapy approaches in congenital/infant high-grade gliomas.
Our study is the first to functionally validate the oncogenic potential of PPP1CB-ALK as well as other novel ALK fusions in glioma, including LRRFIP1-ALK, DCTN1-ALK, and PRKD3-ALK. Furthermore, we show evidence for deep regressions of the tumors in response to multiple inhibitors. This and our acute testing of drugs directly on patient tumor cells provide strong evidence for major potential benefit in the setting of diverse ALK fusions. Our work supports that of Clarke et al. where a patient diagnosed with MAD1L1-ALK fusion infant high-grade glioma (WHO, Grade 4/GBM) at one month of age responded to ceritinib (stable disease for two years) after progression following successive surgery and chemotherapy regimens (18). This combined with prior evidence that early infants presenting with congenital glioma could tolerate surgical resection and chemo- or targeted therapies (63–65) suggests these tumors are highly treatable and shift focus to the minimization of side effects.
Previously normal ALK expression had been reported as being associated with the developing nervous system (58–62) by RNA ISH and protein but had not been well described in the perinatal period and when examined seemed low. However, questions remained as to what the expression might be and the cell types potentially affected if targeted therapy for ALK or other RTKs in infants may be administered. We utilized current effective antibody tools and scRNA studies to more carefully show that ALK seems more highly restricted and is not readily detected at later stages of perinatal brain development when ALK treatment might be considered. As our examination was not exhaustive, it is possible that rare important subtypes of cells may still express ALK, especially given prior reports of brainstem cell types showing expression. Further studies will be needed to determine the exact areas where ALK may affect brain function and development in children. The safety profile of ALK inhibitors has been very favorable for children and adults of older ages with ALK driven cancers to date. However, given the younger ages of infant tumors and planned clinical trials, additional data is clearly needed to optimize safely the timing of treatment. Given the intrauterine recognition of these tumors, the potential for intervention as maternal-fetal therapy also theoretically exists and could provide further impetus for additional studies as to early effects of targeted therapies for ALK, should clinical trials of ALK inhibitors show promise in infant patients.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Translational Relevance.
Glioblastoma (GBM) treatment in children and adults involves surgery, chemotherapy, and radiotherapy, and leads to long-term toxicities including cognitive deficits. The recent discovery of ALK fusions in children is promising as ALK inhibitors are highly effective in other cancers. However, the landscape of ALK expression, mutations, and fusions across adult and pediatric age groups is not well defined. We demonstrate ALK fusions and expression exist across GBM of all ages but are most common in congential/infant GBM. We find that ALK-rearranged GBM animal models and patient-derived cells are highly responsive to ALK inhibitors and normal ALK expression is low in perinatal and adult brain. These findings establish ALK as a relevant target with a potential for minimizing effects on the brain compared to standard therapy. The data support further clinical trials for treatment of ALK-rearranged congenital/infant GBM.
ACKNOWLEDGMENTS
This work was supported by the following funding sources: NCI P50 CA165962 (K.L.L., P.W.), R01CA188228 (K.L.L., R.B.), DFCI Dunkin Donuts Medical Oncology Grant (K.L.L.), National Brain Tumor Society (K.L.L.), Pediatric Brain Tumor Foundation (K.L.L.), and 3000 Miles to the Cure (K.L.L.). We acknowledge the Genotype-Tissues Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. The data used for the analyses described in this manuscript were obtained from the GTEx Portal on 01/05/2020. We thank the BWH Neuropathology Lab (Karen Bryan, Sebastian Valentin) and the BCH Pathology Lab for histology support, and the Scripps Research Center for Metabolomics and Mass Spectrometry (Bill Webb, Elizabeth Billings) for advice on maternal/fetal PK interpretation, and Dr. Kin-Hoe Chow and the staff of the DFCI Center for Patient Derived Models.
Footnotes
Disclosures: K.L.L. founder and equity holder (Travera Inc), consultant (BMS, Integragen), research funding (BMS, Lilly). P.B consultant and research funding (Novartis), R.B. consultant and research funding (Novartis). None of the disclosures are directly related to the study.
REFERENCES
- 1.El-Ayadi M, Ansari M, Kühnöl CD, Bendel A, Sturm D, Pietsch T, et al. Occurrence of high-grade glioma in Noonan syndrome: Report of two cases. Pediatr Blood Cancer 2019;66:e27625. [DOI] [PubMed] [Google Scholar]
- 2.Korostyshevskaya AM, Savelov AA, Papusha LI, Druy AE, Yarnykh VL. Congenital medulloblastoma: Fetal and postnatal longitudinal observation with quantitative MRI. Clin Imaging 2018;52:172–6. [DOI] [PubMed] [Google Scholar]
- 3.Kostadinov S, de la Monte S. A Case of Congenital Brainstem Oligodendroglioma: Pathology Findings and Review of the Literature. Case Rep Neurol Med [Internet] 2017. [cited 2019 Jul 15];2017. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5549497/ [DOI] [PMC free article] [PubMed]
- 4.Anestis D, Tsitsopoulos P, Ble C, Tsitouras V, Tsonidis C. Congenital Glioblastoma Multiforme: An Unusual and Challenging Tumor. Neuropediatrics 2017;48:403–12. [DOI] [PubMed] [Google Scholar]
- 5.Hallberg B, Palmer RH. Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology. Nat Rev Cancer 2013;13:685–700. [DOI] [PubMed] [Google Scholar]
- 6.Bullrich F, Morris SW, Hummel M, Pileri S, Stein H, Croce CM. Nucleophosmin (NPM) Gene Rearrangements in Ki-1-positive Lymphomas. Cancer Res. American Association for Cancer Research; 1994;54:2873–7. [PubMed] [Google Scholar]
- 7.Morris SW, Naeve C, Mathew P, James PL, Kirstein MN, Cui X, et al. ALK, the chromosome 2 gene locus altered by the t(2;5) in non-Hodgkin’s lymphoma, encodes a novel neural receptor tyrosine kinase that is highly related to leukocyte tyrosine kinase (LTK). Oncogene 1997;14:2175–88. [DOI] [PubMed] [Google Scholar]
- 8.Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, et al. Identification of the transforming EML4–ALK fusion gene in non-small-cell lung cancer. Nature 2007;448:561. [DOI] [PubMed] [Google Scholar]
- 9.Stransky N, Cerami E, Schalm S, Kim JL, Lengauer C. The landscape of kinase fusions in cancer. Nat Commun [Internet] 2014. [cited 2018 Jan 31];5. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4175590/ [DOI] [PMC free article] [PubMed]
- 10.Vendrell JA, Taviaux S, Béganton B, Godreuil S, Audran P, Grand D, et al. Detection of known and novel ALK fusion transcripts in lung cancer patients using next-generation sequencing approaches. Sci Rep [Internet] 2017. [cited 2018 Jul 8];7. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5624911/ [DOI] [PMC free article] [PubMed]
- 11.Berry T, Luther W, Bhatnagar N, Jamin Y, Poon E, Sanda T, et al. The ALKF1174L mutation potentiates the oncogenic activity of MYCN in neuroblastoma. Cancer Cell 2012;22:117–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takita J The role of anaplastic lymphoma kinase in pediatric cancers. Cancer Sci 2017;108:1913–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tolbert VP, Coggins GE, Maris JM. Genetic susceptibility to neuroblastoma. Curr Opin Genet Dev 2017;42:81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Olsen TK, Panagopoulos I, Meling TR, Micci F, Gorunova L, Thorsen J, et al. Fusion genes with ALK as recurrent partner in ependymoma-like gliomas: a new brain tumor entity? Neuro-Oncol 2015;17:1365–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aghajan Y, Levy ML, Malicki DM, Crawford JR. Novel PPP1CB-ALK fusion protein in a high-grade glioma of infancy. BMJ Case Rep 2016;2016:bcr2016217189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chmielecki J, Bailey M, He J, Elvin J, Vergilio J-A, Ramkissoon S, et al. Genomic Profiling of a Large Set of Diverse Pediatric Cancers Identifies Known and Novel Mutations across Tumor Spectra. Cancer Res 2017;77:509–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guerreiro Stucklin AS, Ryall S, Fukuoka K, Zapotocky M, Lassaletta A, Li C, et al. Alterations in ALK/ROS1/NTRK/MET drive a group of infantile hemispheric gliomas. Nat Commun [Internet] 2019. [cited 2019 Oct 1];10. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6761184/ [DOI] [PMC free article] [PubMed]
- 18.Clarke M, Mackay A, Ismer B, Pickles JC, Tatevossian RG, Newman S, et al. Infant high grade gliomas comprise multiple subgroups characterized by novel targetable gene fusions and favorable outcomes. Cancer Discov [Internet] American Association for Cancer Research; 2020. [cited 2020 May 17]; Available from: https://cancerdiscovery.aacrjournals.org/content/early/2020/04/04/2159-8290.CD-19-1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro-Oncol 2021;23:1231–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Powers C, Aigner A, Stoica GE, McDonnell K, Wellstein A. Pleiotrophin Signaling through Anaplastic Lymphoma Kinase Is Rate-limiting for Glioblastoma Growth. J Biol Chem 2002;277:14153–8. [DOI] [PubMed] [Google Scholar]
- 21.Karagkounis G, Stranjalis G, Argyrakos T, Pantelaion V, Mastoris K, Rontogianni D, et al. Anaplastic lymphoma kinase expression and gene alterations in glioblastoma: correlations with clinical outcome. J Clin Pathol 2017;70:593–9. [DOI] [PubMed] [Google Scholar]
- 22.Wick W, Dettmer S, Berberich A, Kessler T, Karapanagiotou-Schenkel I, Wick A, et al. N2M2 (NOA-20) phase I/II trial of molecularly matched targeted therapies plus radiotherapy in patients with newly diagnosed non-MGMT hypermethylated glioblastoma. Neuro-Oncol 2019;21:95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lei Y-Y, Yang J-J, Zhang X-C, Zhong W-Z, Zhou Q, Tu H-Y, et al. Anaplastic Lymphoma Kinase Variants and the Percentage of ALK-Positive Tumor Cells and the Efficacy of Crizotinib in Advanced NSCLC. Clin Lung Cancer 2016;17:223–31. [DOI] [PubMed] [Google Scholar]
- 24.Ou S-HI, Bartlett CH, Mino-Kenudson M, Cui J, Iafrate AJ. Crizotinib for the Treatment of ALK -Rearranged Non-Small Cell Lung Cancer: A Success Story to Usher in the Second Decade of Molecular Targeted Therapy in Oncology. The Oncologist 2012;17:1351–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Redaelli S, Ceccon M, Antolini L, Rigolio R, Pirola A, Peronaci M, et al. Synergistic activity of ALK and mTOR inhibitors for the treatment of NPM-ALK positive lymphoma. Oncotarget [Internet] 2016. [cited 2018 Sep 12];7. Available from: http://www.oncotarget.com/fulltext/12128 [DOI] [PMC free article] [PubMed]
- 26.Costa DB, Kobayashi S, Pandya SS, Yeo W-L, Shen Z, Tan W, et al. CSF Concentration of the Anaplastic Lymphoma Kinase Inhibitor Crizotinib. J Clin Oncol 2011;29:e443–5. [DOI] [PubMed] [Google Scholar]
- 27.Gadgeel SM, Shaw AT, Govindan R, Gandhi L, Socinski MA, Camidge DR, et al. Pooled Analysis of CNS Response to Alectinib in Two Studies of Pretreated Patients With ALK-Positive Non–Small-Cell Lung Cancer. J Clin Oncol American Society of Clinical Oncology; 2016;34:4079–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Solomon BJ, Besse B, Bauer TM, Felip E, Soo RA, Camidge DR, et al. Lorlatinib in patients with ALK-positive non-small-cell lung cancer: results from a global phase 2 study. Lancet Oncol 2018;19:1654–67. [DOI] [PubMed] [Google Scholar]
- 29.Heath JA, Campbell MA, Thomas A, Solomon B. Good clinical response to alectinib, a second generation ALK inhibitor, in refractory neuroblastoma. Pediatr Blood Cancer 2018;65:e27055. [DOI] [PubMed] [Google Scholar]
- 30.Guan J, Tucker ER, Wan H, Chand D, Danielson LS, Ruuth K, et al. The ALK inhibitor PF-06463922 is effective as a single agent in neuroblastoma driven by expression of ALK and MYCN. Dis Model Mech 2016;9:941–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Conklin CMJ, Craddock KJ, Have C, Laskin J, Couture C, Ionescu DN. Immunohistochemistry is a Reliable Screening Tool for Identification of ALK Rearrangement in Non–Small-Cell Lung Carcinoma and is Antibody Dependent. J Thorac Oncol 2013;8:45–51. [DOI] [PubMed] [Google Scholar]
- 32.Rogers T-M, Russell PA, Wright G, Wainer Z, Pang J-M, Henricksen LA, et al. Comparison of Methods in the Detection of ALK and ROS1 Rearrangements in Lung Cancer. J Thorac Oncol 2015;10:611–8. [DOI] [PubMed] [Google Scholar]
- 33.Ng A, Levy ML, Malicki DM, Crawford JR. Unusual high-grade and low-grade glioma in an infant with PPP1CB-ALK gene fusion. BMJ Case Rep 2019;12:e228248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lake JA, Donson AM, Prince E, Davies KD, Nellan A, Green AL, et al. Targeted fusion analysis can aid in the classification and treatment of pediatric glioma, ependymoma, and glioneuronal tumors. Pediatr Blood Cancer 2020;67:e28028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Goodall AH, Burns P, Salles I, Macaulay IC, Jones CI, Ardissino D, et al. Transcription profiling in human platelets reveals LRRFIP1 as a novel protein regulating platelet function. Blood 2010;116:4646–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Khachigian LM, Santiago FS, Rafty LA, Chan OL, Delbridge GJ, Bobik A, et al. GC factor 2 represses platelet-derived growth factor A-chain gene transcription and is itself induced by arterial injury. Circ Res 1999;84:1258–67. [DOI] [PubMed] [Google Scholar]
- 37.Suriano AR, Sanford AN, Kim N, Oh M, Kennedy S, Henderson MJ, et al. GCF2/LRRFIP1 represses tumor necrosis factor alpha expression. Mol Cell Biol 2005;25:9073–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cao Z, Gao Q, Fu M, Ni N, Pei Y, Ou W-B. Anaplastic lymphoma kinase fusions: Roles in cancer and therapeutic perspectives. Oncol Lett 2019;17:2020–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shimada Y, Kohno T, Ueno H, Ino Y, Hayashi H, Nakaoku T, et al. An Oncogenic ALK Fusion and an RRAS Mutation in KRAS Mutation‐Negative Pancreatic Ductal Adenocarcinoma. The Oncologist Wiley-Blackwell; 2017;22:158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Subbiah V, McMahon C, Patel S, Zinner R, Silva EG, Elvin JA, et al. STUMP un“stumped”: anti-tumor response to anaplastic lymphoma kinase (ALK) inhibitor based targeted therapy in uterine inflammatory myofibroblastic tumor with myxoid features harboring DCTN1-ALK fusion. J Hematol OncolJ Hematol Oncol 2015;8:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wiesner T, He J, Yelensky R, Esteve-Puig R, Botton T, Yeh I, et al. Kinase fusions are frequent in Spitz tumors and spitzoid melanomas. Nat Commun 2014;5:3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hong S, Chen N, Fang W, Zhan J, Liu Q, Kang S, et al. Upregulation of PD-L1 by EML4-ALK fusion protein mediates the immune escape in ALK positive NSCLC: Implication for optional anti-PD-1/PD-L1 immune therapy for ALK-TKIs sensitive and resistant NSCLC patients. Oncoimmunology [Internet] 2015. [cited 2018 Feb 5];5. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4839382/ [DOI] [PMC free article] [PubMed]
- 43.Hu J, Ho AL, Yuan L, Hu B, Hua S, Hwang SS, et al. Neutralization of terminal differentiation in gliomagenesis. Proc Natl Acad Sci U S A 2013;110:14520–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schonberg DL, Venere M, Rich JN. Changing the fate of cancer, one splice at a time. Proc Natl Acad Sci U S A 2013;110:14510–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zou HY, Friboulet L, Kodack DP, Engstrom LD, Li Q, West M, et al. PF-06463922, an ALK/ROS1 inhibitor, overcomes resistance to 1st and 2nd generation ALK inhibitors in pre-clinical models. Cancer Cell 2015;28:70–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sands SA, Zhou T, O’Neil SH, Patel SK, Allen J, McGuire Cullen P, et al. Long-term follow-up of children treated for high-grade gliomas: children’s oncology group L991 final study report. J Clin Oncol Off J Am Soc Clin Oncol 2012;30:943–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jessa S, Blanchet-Cohen A, Krug B, Vladoiu M, Coutelier M, Faury D, et al. Stalled developmental programs at the root of pediatric brain tumors. Nat Genet 2019;51:1702–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bao X, Wu J, Sanai N, Li J. A liquid chromatography with tandem mass spectrometry method for quantitating total and unbound ceritinib in patient plasma and brain tumor. J Pharm Anal 2018;8:20–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Collier TL, Normandin MD, Stephenson NA, Livni E, Liang SH, Wooten DW, et al. Synthesis and preliminary PET imaging of 11C and 18F isotopologues of the ROS1/ALK inhibitor lorlatinib. Nat Commun [Internet] 2017. [cited 2019 Oct 22];8. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5472746/ [DOI] [PMC free article] [PubMed]
- 50.Johnson TW, Richardson PF, Bailey S, Brooun A, Burke BJ, Collins MR, et al. Discovery of (10 R )-7-Amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro- 2H -8,4-(metheno)pyrazolo[4,3- h ][2,5,11]-benzoxadiazacyclotetradecine-3-carbonitrile (PF-06463922), a Macrocyclic Inhibitor of Anaplastic Lymphoma Kinase (ALK) and c-ros Oncogene 1 (ROS1) with Preclinical Brain Exposure and Broad-Spectrum Potency against ALK-Resistant Mutations. J Med Chem 2014;57:4720–44. [DOI] [PubMed] [Google Scholar]
- 51.Kodama T, Hasegawa M, Takanashi K, Sakurai Y, Kondoh O, Sakamoto H. Antitumor activity of the selective ALK inhibitor alectinib in models of intracranial metastases. Cancer Chemother Pharmacol 2014;74:1023–8. [DOI] [PubMed] [Google Scholar]
- 52.Marsilje TH, Pei W, Chen B, Lu W, Uno T, Jin Y, et al. Synthesis, structure-activity relationships, and in vivo efficacy of the novel potent and selective anaplastic lymphoma kinase (ALK) inhibitor 5-chloro-N2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)-N4-(2-(isopropylsulfonyl)phenyl)pyrimidine-2,4-diamine (LDK378) currently in phase 1 and phase 2 clinical trials. J Med Chem 2013;56:5675–90. [DOI] [PubMed] [Google Scholar]
- 53.Bandopadhayay P, Ramkissoon LA, Jain P, Bergthold G, Wala J, Zeid R, et al. MYB-QKI rearrangements in Angiocentric Glioma drive tumorigenicity through a tripartite mechanism. Nat Genet 2016;48:273–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fujimoto J, Shiota M, Iwahara T, Seki N, Satoh H, Mori S, et al. Characterization of the transforming activity of p80, a hyperphosphorylated protein in a Ki-1 lymphoma cell line with chromosomal translocation t(2;5). Proc Natl Acad Sci U S A 1996;93:4181–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zamo A, Chiarle R, Piva R, Howes J, Fan Y, Chilosi M, et al. Anaplastic lymphoma kinase (ALK) activates Stat3 and protects hematopoietic cells from cell death. Oncogene 2002;21:1038–47. [DOI] [PubMed] [Google Scholar]
- 56.Heuckmann JM, Balke-Want H, Malchers F, Peifer M, Sos ML, Koker M, et al. Differential Protein Stability and ALK Inhibitor Sensitivity of EML4-ALK Fusion Variants. Clin Cancer Res American Association for Cancer Research; 2012;18:4682–90. [DOI] [PubMed] [Google Scholar]
- 57.Shulman DS, DuBois SG. The Evolving Diagnostic and Treatment Landscape of NTRK-Fusion-Driven Pediatric Cancers. Pediatr Drugs 2020;22:189–97. [DOI] [PubMed] [Google Scholar]
- 58.Degoutin J, Brunet-de Carvalho N, Cifuentes-Diaz C, Vigny M. ALK (Anaplastic Lymphoma Kinase) expression in DRG neurons and its involvement in neuron-Schwann cells interaction. Eur J Neurosci 2009;29:275–86. [DOI] [PubMed] [Google Scholar]
- 59.Reiff T, Huber L, Kramer M, Delattre O, Janoueix-Lerosey I, Rohrer H. Midkine and Alk signaling in sympathetic neuron proliferation and neuroblastoma predisposition. Dev Camb Engl 2011;138:4699–708. [DOI] [PubMed] [Google Scholar]
- 60.Iwahara T, Fujimoto J, Wen D, Cupples R, Bucay N, Arakawa T, et al. Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system. Oncogene 1997;14:439–49. [DOI] [PubMed] [Google Scholar]
- 61.Hurley SP, Clary DO, Copie V, Lefcort F. Anaplastic lymphoma kinase is dynamically expressed on subsets of motor neurons and in the peripheral nervous system. J Comp Neurol 2006;495:202–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vernersson E, Khoo NKS, Henriksson ML, Roos G, Palmer RH, Hallberg B. Characterization of the expression of the ALK receptor tyrosine kinase in mice. Gene Expr Patterns 2006;6:448–61. [DOI] [PubMed] [Google Scholar]
- 63.Kuki I, Kawawaki H, Okazaki S, Ehara E, Yoshida Y, Kunihiro N, et al. Efficacy and safety of everolimus in patients younger than 12 months with congenital subependymal giant cell astrocytoma. Brain Dev 2018;40:415–20. [DOI] [PubMed] [Google Scholar]
- 64.Boukas A, Panaretos P, Cowie C, Nicholson C, Jenkins A. Congenital glioblastoma multiforme: complete resection with long-term survival and a novel technique of contralateral cystoventriculostomy. Pediatr Neurosurg 2012;48:327–30. [DOI] [PubMed] [Google Scholar]
- 65.Macy ME, Birks DK, Barton VN, Chan MH, Donson AM, Kleinschmidt-DeMasters BK, et al. Clinical and molecular characteristics of congenital glioblastoma. Neuro-Oncol 2012;14:931–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequencing data generated in this study have been deposited under dbGAP accession number phs002380.v1.p1. Previously published single cell RNA-Seq data that were re-analyzed here are available under accession code GSE133531 (for the mouse brain dataset) (32) and S-EPMC5991609 (for the human brain dataset) (31,33). Additionally, we used the following publicly available datasets: The Genotype-Tissue Expression (GTEx) dataset (https://gtexportal.org/home) and The Allen Brain Atlas Dataset (https://portal.brain-map.org) All other data supporting the findings of this study are available from the corresponding authors on reasonable request.