

Abstract
Objective:
The aim of the study is to investigate the role and mechanisms of tuberous sclerosis complex 1 (TSC1) and mechanistic target of rapamycin complex 1 (mTORC1) in alcohol associated liver disease.
Design:
Liver-specific Tsc1 knockout (L-Tsc1 KO) mice and their matched wild type (WT) mice were subjected to Gao-binge alcohol. Human alcoholic hepatitis (AH) samples were also used for immunohistochemical (IHC) staining, western blot and q-PCR analysis.
Results:
Human AH and Gao-binge alcohol-fed mice had decreased hepatic TSC1 and increased mTORC1 activation. Gao-binge alcohol markedly increased liver/body weight ratio and serum alanine aminotransferase (ALT) levels in L-Tsc1 KO mice compared to Gao-binge alcohol-fed WT mice. Results from IHC staining, western blot and q-PCR analysis revealed that human AH and Gao-binge alcohol-fed L-Tsc1 KO mouse livers significantly increased hepatic progenitor cells, macrophages and neutrophils but decreased HNF4α positive cells. Gao-binge alcohol-fed L-Tsc1 KO mice also developed severe inflammation and liver fibrosis. Deleting Tsc1 in cholangiocytes but not in hepatocytes promoted cholangiocyte proliferation and aggravated alcohol-induced ductular reactions (DR), fibrosis, inflammation and liver injury. Pharmacological inhibition of mTORC1 partially reversed hepatomegaly, DR, fibrosis, inflammatory cell infiltration and liver injury in alcohol-fed L-Tsc1 KO mice.
Conclusion:
Our findings indicate that persistent activation of mTORC1 due to the loss of cholangiocyte TSC1 promotes hepatomegaly, liver cell repopulation, DR, inflammation, fibrosis and liver injury in Gao-binge alcohol fed L-Tsc1 KO mice, which phenocopy the pathogenesis of human AH.
Keywords: Autophagy, cholangiocyte, ductular reactions, progenitor cells, TSC1
Graphical Abstract
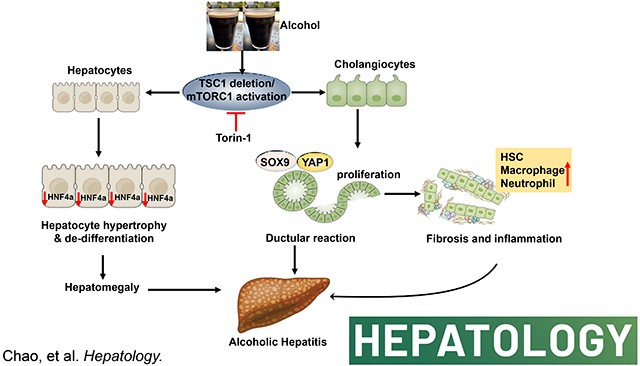
Introduction
Alcohol-associated liver disease (ALD) is a major health problem and a leading cause of chronic liver diseases worldwide, claiming more than 3 million deaths, or 5.9% of all deaths globally per year (1, 2). The latest surveillance report published by the National Institute on Alcohol Abuse and Alcoholism (NIAAA) showed that liver cirrhosis was the 12th leading cause of death in the United States, with a total of 42,443 deaths in 2015, 49.5% of which were estimated to be attributed to ALD (3). ALD comprises a range of disorders and pathologic changes in individuals with acute and chronic alcohol consumption, ranging from simple steatosis to severe forms of liver injury including steatohepatitis, ductular reactions (DR), liver fibrosis/cirrhosis, and hepatocellular carcinoma (HCC) (4–6). Despite the significant progresses on understanding the pathogenesis of ALD, no efficient therapies are available for treating severe ALD other than liver transplantation. The poor therapeutic progress in the field of ALD has, in part, resulted from the lack of suitable experimental models to mimic the advanced pathogenesis of ALD (5). Over the past few decades, investigators have been actively exploring animal models for ALD, studying its pathogenesis and searching for therapeutic drugs for the treatment of ALD (5). However, there are still no animal models that can represent the full spectrum of human ALD yet. Most current animal models involving chronic alcohol feeding only recapitulate some features of the different stages of ALD such as steatosis with low neutrophil infiltration but no obvious fibrosis and DR (7, 8), which are hallmarks of advanced ALD in human AH patients (9). Hence, developing an animal model to recapitulate the most spectrum of ALD will be urgently needed for the development of targeted ALD therapy.
Mechanistic target of rapamycin complex 1 (mTORC1) is a conserved serine-threonine kinase complex that regulates cell growth by coordinating signals from nutrients and growth factors as well as cellular energy levels (10). mTORC1 integrates four major signals: growth factors, energy status, oxygen and amino acids, to regulate many processes that are involved in the promotion of cell growth (11). One of the most important sensors involved in the regulation of mTORC1 activity is the tuberous sclerosis complex (TSC), which is a heterodimer that comprises TSC1 and TSC2 (12). TSC1/2 is a key negative regulator of mTORC1 that functions as a GTPase-activating protein (GAP) for the small Ras-related GTPase Rheb (Ras homolog enriched in brain). The active, GTP-bound form of Rheb directly interacts with and activates mTORC1(10, 13). As a Rheb-specific GAP, TSC1/2 negatively regulates mTORC1 signaling by converting Rheb into its inactive GDP-bound state (14).
Recently, we reported that mTORC1 was significantly activated by alcohol feeding that led to the inhibition of transcription factor EB (TFEB), a master regulator of lysosomal biogenesis, resulting in impaired lysosomal biogenesis and autophagy in mouse livers (15, 16). In addition, administration of Torin1, a specific mTORC1 inhibitor, protected against alcohol-induced liver injury (15). Studies from others also showed that liver-specific Tsc1 knockout (L-Tsc1 KO) mice, which have persistent hepatic mTORC1 activation have increased serum levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST), hepatocyte death, inflammation at the age of 6 months-old, and eventually developed HCC at the age of 9 months-old (17, 18).
Here, we showed that chronic-plus-acute alcohol administration (Gao-binge alcohol) significantly increased hepatomegaly, liver injury, DR, hepatic bile acid retention, fibrosis, inflammation, and liver cell repopulation in L-Tsc1 KO mice, all are hall markers of human AH. Deletion of cholangiocyte but not hepatocyte Tsc1 in mice led to increased DR, inflammation, fibrosis and liver injury after alcohol feeding, highlighting a critical role of cholangiocyte mTORC1 activation in the pathogenesis of ALD. More importantly, inhibition of mTORC1 by its specific inhibitor, Torin1, partially reversed the AH-like pathological changes in mouse livers. In conclusion, we identified a critical role of cholangiocyte mTORC1 in the pathogenesis of severe ALD.
Methods and Materials
Animal Experiments
Mice were specific pathogen free and maintained in a barrier rodent facility under standard experimental conditions. All research was conducted in accordance with both the Declarations of Helsinki and Istanbul. All animal procedures were approved by the Institutional Animal Care and Use Committee of the University of Kansas Medical Center. Tsc1 Flox/Flox (Tsc1flox/flox, C57BL/6J, Jackson Laboratory) mice were crossed with Albumin-Cre mice (Alb-Cre, C57BL/6J, Jackson Laboratory) to generate liver-specific Tsc1 knockout mice (Tsc1flox/flox, Albumin-Cre+, L-Tsc1 KO). Alb Cre+ mice and Alb Cre- (Tsc1flox/flox, Albumin-Cre-, Tsc1 WT) matched littermates were used in this study. Male 8- to 12-week-old mice were subjected to Gao-binge alcohol model (8). Briefly, mice were acclimated to the Lieber-DeCarli liquid control diet (F1259SP; Bio-Serv, Flemington, NJ) for 5 days followed by further feeding with the liquid control or ethanol diet (5% ethanol; F1258SP; Bio-Serv) for 10 days. The diets were isocaloric and ethanol calories were replaced by maltose dextrin in the control liquid diet. The volume of control diet given to mice was matched to the volume of ethanol diet consumed. On the last day of feeding, mice were given ethanol 5 g/kg or maltose dextran 9 g/kg and sacrificed 8 hours later. In some experiments, mTORC1 inhibitor Torin1 (2 mg/kg, i.p.) was given to mice every other day from day 6, and one dose was given right before the gavage on the final day. For acute deletion of Tsc1 in hepatocyte, Tsc1flox/flox mice were given one dose of AAV8-TBG-Cre or AAV-TBG-null (1×1011 GC/mouse, i.v.) and subjected to Gao-binge alcohol feeding 8 weeks later after the injection. For deletion of Tsc1 in cholangiocyte, Tsc1flox/flox mice were given one dose of adenovirus (Ad)-CK19-CreERT2 or Ad-null (2×108 PFU/mouse, i.v.). Mice were then injected with three doses of tamoxifen (75 mg/kg, i.p.) next day for consecutive 3 days and fed with Gao-binge alcohol 8 weeks later. Corn oil was used as vehicle control for tamoxifen. Blood and liver tissues were collected. Liver injury was determined by measuring serum ALT. Hepatomegaly was determined by liver to body weight ratio. Liver cryosections and H & E staining as well as confocal microscopy were performed as described previously (19).
Statistical Analysis
All experimental data were expressed as means ± S.E. and subjected to one-way ANOVA analysis with Bonferroni post hoc test or Student’s t-test where appropriate. A p<0.05 was considered significant.
For additional methods and materials, please refer to supplemental materials.
Results
Decreased hepatic TSC1 protein and increased mTORC1 activity in human alcoholic hepatitis and Gao-binge alcohol-fed mouse livers
Immunohistochemistry (IHC) staining for hepatic TSC1 and phosphorylated S6 (p-S6), a substrate protein that is phosphorylated by mTORC1, showed markedly decreased hepatic TSC1 staining with concomitantly increased p-S6 in AH liver samples compared with liver samples from control healthy donors (Figure 1A). H&E and IHC staining for cytokeratin 19 (CK19), a marker of cholangiocyte, revealed markedly increased CK19 positive ductular structures in human AH samples compared with liver samples from healthy donors (Figure 1B–C). Consistent with the IHC results, western blot analysis showed markedly decreased TSC1 but increased CK19 protein levels in human AH livers compared with control healthy human liver donors (Figure 1D). In line with the findings from human AH, Gao-binge alcohol (EtOH) feeding caused a 30%~40% decrease in TSC1 protein levels but markedly increased levels of p-S6 and 4EBP1 in mouse livers (Figure 1E). These data indicate that decreased hepatic TSC1 protein levels and increased mTORC1 activation are associated with human AH and experimental mouse ALD.
Figure 1. Decreased hepatic TSC1 protein and increased mTORC1 activation in human AH and Gao-binge alcohol fed mouse livers.
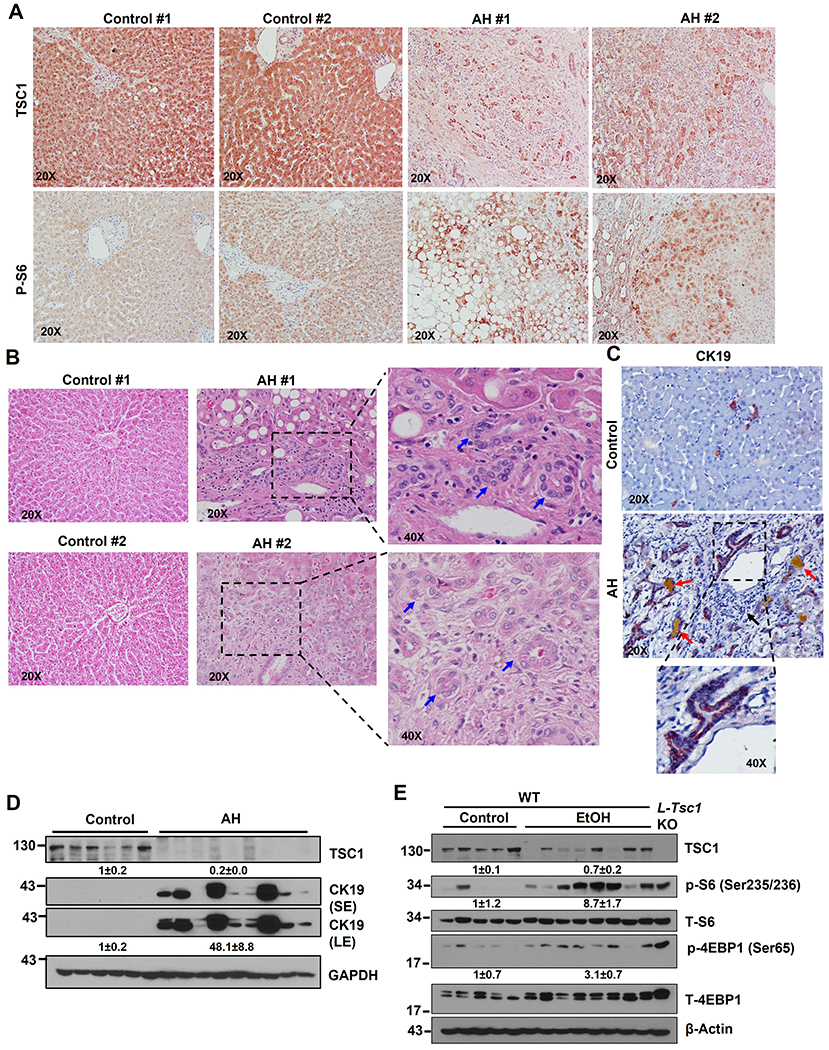
Representative photographs of IHC staining of TSC1 (A), H&E staining (B) & IHC staining of CK19 (C) on liver tissues from AH and healthy donors (control) are shown. Right panel is an enlarged photograph from the boxed area. Arrows denote DR or CK19 positive DR. (D) Total liver lysates from AH or healthy donors (control) were subjected to western blot analysis. (E) Male 8-weeks old WT mice were subjected to Gao-binge alcohol (EtOH) feeding and total liver lysates were subjected to western blot analysis. Liver lysates from a control diet fed L-Tsc1 KO mice were used as a positive control.
Loss of hepatic TSC1 increases mTORC1 activity and promotes hepatomegaly, ductular reaction and liver injury in EtOH-fed mouse livers
To further investigate the impacts of loss of hepatic TSC1 in EtOH-induced liver pathogenesis, L-Tsc1 KO mice and their matched wild type (WT) littermates were subjected to EtOH feeding. Deletion of hepatic TSC1 markedly increased levels of p-S6 and 4EBP1 in mouse livers regardless of EtOH feeding. The levels of p-S6 were comparable between EtOH-fed WT and L-Tsc1 KO mice but levels of p-4EBP1 in EtOH-fed-L-Tsc1 KO mice were higher than WT mice. Interestingly, total levels of S6 and 4EBP1 increased in L-Tsc1 KO mouse livers likely due to increased protein synthesis (Figure 2A). EtOH feeding slightly increased the liver/body weight ratio in WT mice compared with control-diet fed mice. In contrast, loss of hepatic TSC1 significantly increased the liver/body weight ratio in mice irrespective of EtOH feeding (Figure 2B), suggesting hepatic TSC1 deficiency alone is sufficient to cause hepatomegaly and is further aggravated by EtOH. We next determined whether increased liver size resulted from increased cell number or increased cell size in L-Tsc1 KO mice. Immunostaining of Na+/K+ ATPase was performed to indicate the basolateral membrane of hepatocytes. Interestingly, the size of hepatocytes markedly decreased in EtOH-fed WT mouse livers but dramatically increased in both control-diet and EtOH-fed L-Tsc1 KO mouse livers (SFigure 1A–B). The levels of hepatic PCNA and PCNA positive cells increased in L-Tsc1 KO mouse livers compared with WT mice regardless of EtOH feeding (SFigure 1C–E). Interestingly, majority of PCNA positive cells were non-parenchymal cells with only a few hepatocytes in L-Tsc1 KO mice (SFigure 1C, arrow), suggesting a possible cell type repopulation with elevated proliferation of non-parenchymal cells or hepatic progenitor cells in TSC1-deficient mouse livers. Levels of serum ALT activity in control diet-fed L-Tsc1 KO mice were almost identical to that of WT mice (Figure 2C), suggesting deletion of Tsc1 alone in mouse livers do not cause liver injury. However, serum levels of ALT were significantly higher in L-Tsc1 KO mice than in WT mice fed with EtOH (Figure 2C). EtOH feeding increased levels of hepatic triglyceride (TG) and numbers of lipid droplets (LDs) as shown by Oil Red O staining compared with control diet-fed mice but no differences were found between L-Tsc1 KO mice and matched WT mice (Figure 2D–E). Interestingly, H&E and IHC staining revealed increased CK19 positive ductular structures in L-Tsc1 KO mice (Figure 2F–G, arrows). CK19 positive cells also increased in non-portal tract areas in EtOH-fed L-Tsc1 KO mice, which were barely seen in WT mice and control-diet fed L-Tsc1 KO mice (Figure 2G, black arrow). Results from western blot analysis showed markedly increased levels of hepatic CK19 in L-Tsc1 KO mice, which were further increased by EtOH (Figure 2H). These results indicate that loss of hepatic TSC1 promotes hepatomegaly, DR and liver injury in EtOH-fed mice.
Figure 2. Loss of hepatic TSC1 increases mTORC1 activity and promotes hepatomegaly, ductular reaction and liver injury in EtOH-fed mouse livers.
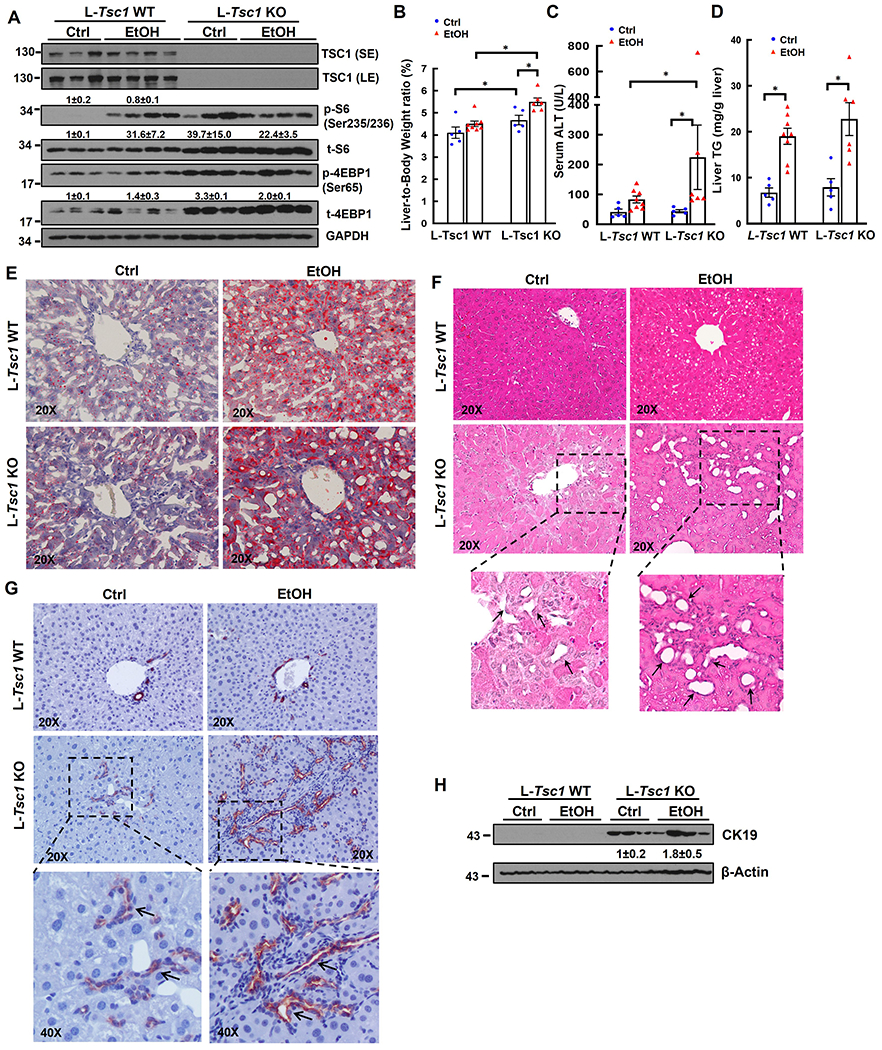
Male Tsc1 WT and L-Tsc1 KO mice were fed with Gao-binge alcohol. (A) Total liver lysates were subjected to western blot analysis. Data are densitometry analysis and presented as means ± SE (n= 3-4). (B) Liver weight versus body weight ratio, (C) Serum alanine aminotransferase (ALT) levels & (D) hepatic triglyceride (TG) levels were analysis. Data are means ± SE (n= 5-10). *p<0.05; One-Way ANOVA analysis with Bonferroni post hoc test. Representative images of Oil Red O (E), H&E staining (F) and IHC staining of CK19 (G). Arrows denote DR or CK19 positive DR. (H) Total liver lysates were subjected to western blot analysis for CK19.
Loss of hepatic TSC1 Promotes Liver Cell Repopulation with Increased Progenitor Cells in human AH and Gao-binge alcohol-fed mice
Gene heatmap of qRT-PCR analysis from normal donor and AH patient liver samples showed a trend of decreased expression of gene signature of hepatocyte, but increased expression of gene signatures of cholangiocyte, hepatic stellate cell (HSC) and KC/infiltrated macrophages (Figure 3A). Liver cell identity and plasticity is regulated by a group of transcription factors and/or co-regulators including HNF4α, yes-associated protein 1 (YAP1) and sex-determining region Y-box (SRY-box) containing gene 9 (SOX9) (20, 21). The protein levels of HNF4α P1 adult isoform decreased but levels of p-YAP1, total YAP1 and SOX9 increased in AH liver samples although there were variations among the AH samples (Figure 3B). Results of qRT-PCR analysis also showed decreased hepatic mRNA levels of adult HNF4A-P1 but increased fetal HNF4A-P2 and decreased ratio of HNF4A-P1 vs HNF4A-P2 (Figure 3C). Moreover, IHC staining showed decreased nuclear HNF4α positive but increased YAP1 and SOX9 positive progenitor/ductular cells in AH samples (Figure 3D). Electron microscopy analysis readily detected accumulation of many non-parenchymal cells including hepatic stellate cells, collagen fibers, cholangiocytes, mast cells, neutrophils and macrophages in AH livers (Figure 3E).
Figure 3. Decreased TSC1 protein levels associated with liver cell population reprogramming featured with increased fetal-like cells and decreased mature hepatocytes in human AH livers.
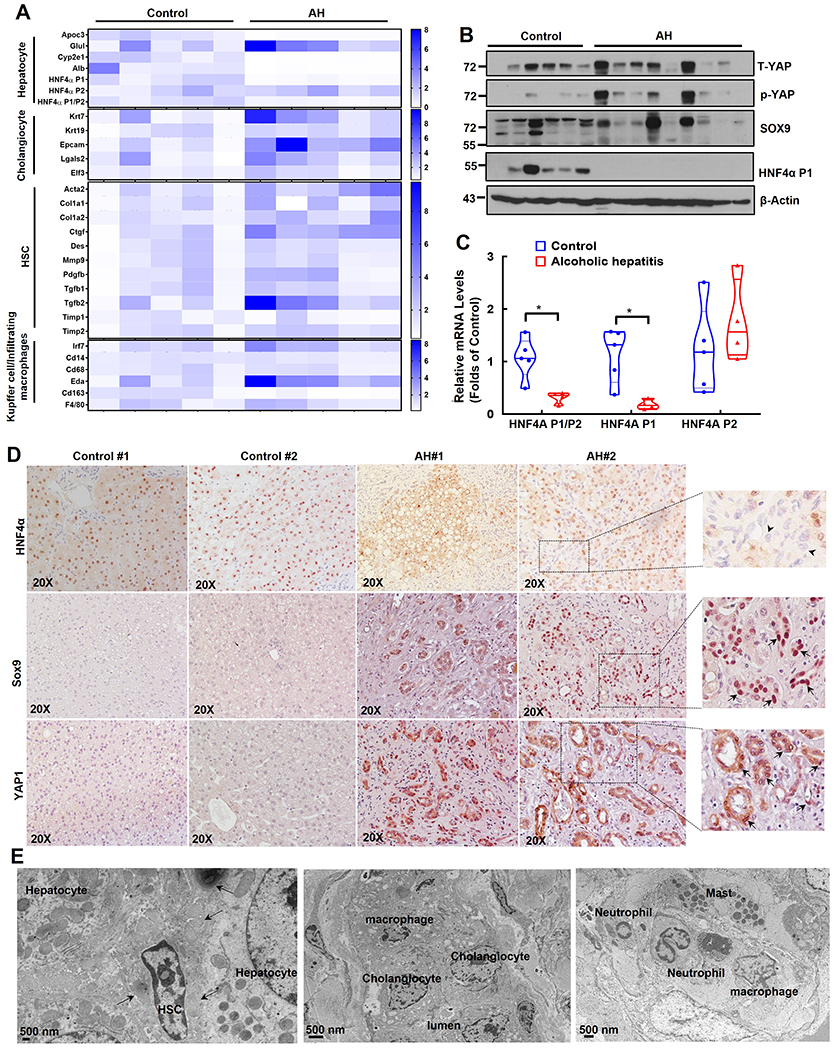
(A) Heat map of gene expression signatures for hepatocyte, cholangiocyte, HSC and Kupffer cell of qRT-PCR analysis. RNA was extracted from healthy donors (Control, n=5) and AH patient livers (n=5). Results were normalized to 18s and expressed as fold change compared with normal donors. (B) Total liver lysates from AH or healthy donors (control) were subjected to western blot analysis. (C) Gene expression of HNF4AP1, HNF4AP2 and HNF4AP1/P2 by qRT-PCR analysis. Data are presented as means ± SE (n=5). *p<0.05; Student t test. (D) Representative images of IHC staining for HNF4α, SOX9 and YAP1 in healthy donor (Control) and AH livers. Arrow heads: HNF4α negative ductular cells; arrows: SOX9 and YAP positive ductular cells. (E) Representative EM images of AH livers. Arrows denote collagen fibers.
Principle component analysis (PCA) and volcano plots of hepatic RNA-seq showed clear separation of gene expression patterns and changes between different genotypes of mice with or without EtOH feeding (SFigure 2A–B). Similar to human AH, heatmap and Gene Set Enrichment Analysis (GSEA) of RNA-seq showed that EtOH-fed L-Tsc1 KO mice also tended to have increased gene expression signatures of cholangiocyte, HSC and KC/infiltrated macrophages but decreased gene signature of hepatocyte (Figure 4A & SFigure 2C–D). Notably, L-Tsc1 KO mice fed with a control diet already showed similar trend of gene expression changes but was milder than EtOH fed mice (Figure 4A & SFigure 2C–D). Western blot analysis showed increased levels of hepatic SOX9, p-YAP1 and YAP1 but decreased HNF4α-P1 in L-Tsc1 KO mice regardless of EtOH feeding (Figure 4B). Increased YAP1 activation was further confirmed by elevated expression levels of several YAP1 target genes including Ctgf, Cyr61 and Areg (Figure 4C). IHC staining showed increased number of SOX9 and YAP1 positive as well as HNF4α negative cells primarily in ductular cells of alcohol-fed L-Tsc1 KO mice (Figure 4D–F). These data indicate that decreased hepatic TSC1 and increased mTORC1 activation leads to marked liver cell repopulation with decreased mature hepatocytes but increased progenitor/fetal hepatocytes and non-parenchyma cells in human AH and alcohol-fed L-Tsc1 mouse livers.
Figure 4. Loss of hepatic Tsc1 causes AH-like cell population reprogramming in EtOH-fed mouse livers.
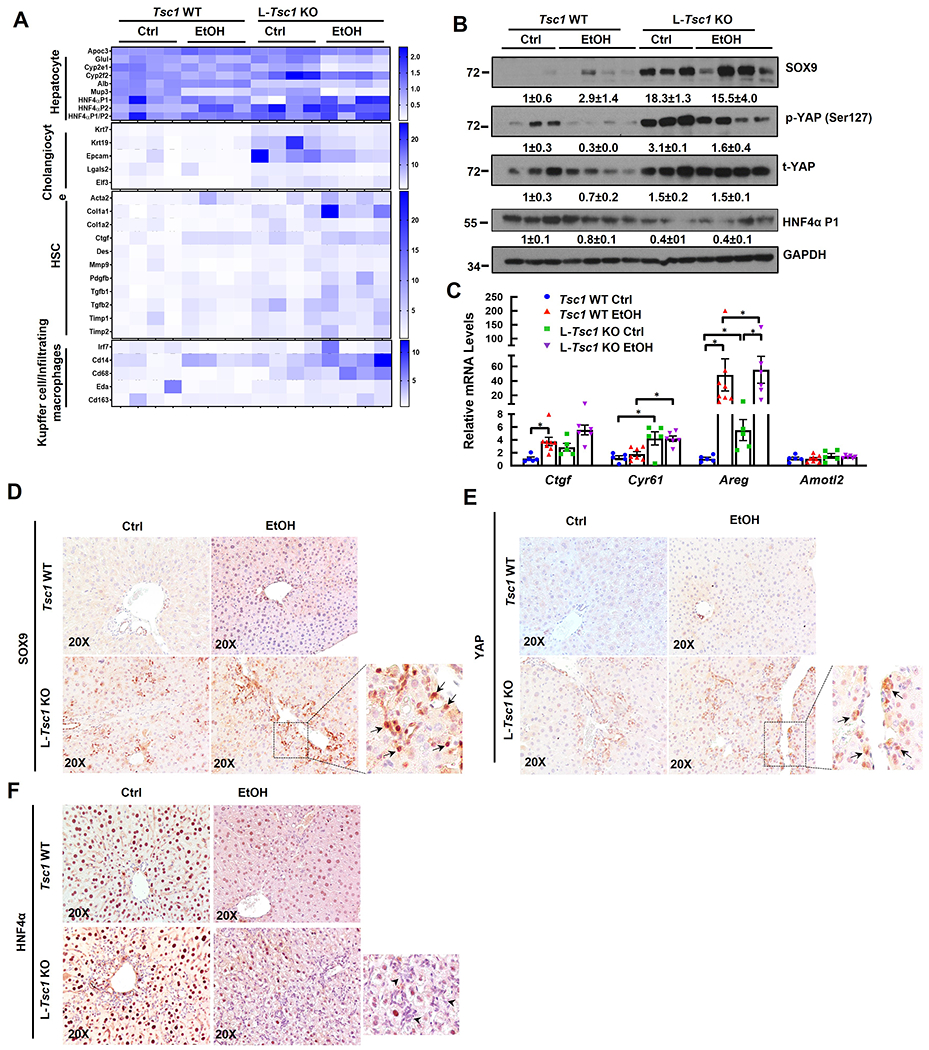
Male Tsc1 WT and L-Tsc1 KO mice were fed with Gao-binge alcohol. (A) Heat map of gene expression signatures for hepatocyte, cholangiocyte, HSC and Kupffer cells from qRT-PCR analysis. RNA was extracted from mouse livers followed by qRT-PCR. Results were normalized to 18s and expressed as fold change compared with Tsc1 WT Ctrl group (n=4). (B) Total liver lysates were subjected to western blot analysis. Data are densitometry analysis and presented as means ± SE (n= 3-4). (C) qRT-PCR analysis of YAP1 target gene expression. Data are presented as means ± SE (n=5-8). *p<0.05; One-Way ANOVA analysis with Bonferroni post hoc test. (D) Representative images of IHC staining for SOX9, YAP1 and HNF4α in mouse livers of indicated genotypes and feeding. Arrow denote SOX9 and YAP1 positive ductular cells, and arrowhead denotes HNF4α negative ductular cells.
Liver-specific Tsc1 KO mice exhibit increased fibrosis, inflammation and cholestasis in response to Gao-binge alcohol feeding
Comparing with normal human donor livers, collagen deposition in the livers of human AH patients dramatically increased, as demonstrated by increased positive Sirius Red staining areas (Figure 5A). Compared with WT mice, positive Sirius Red staining areas also significantly increased in L-Tsc1 KO mice either fed with a control diet or EtOH (Figure 5B–C). Hepatic mRNA levels of fibrogenic genes including Acta2, Col1a, Pdgf and Tgfb1 as well as protein levels of α-SMA significantly increased in EtOH-fed L-Tsc1 KO mice compared with either EtOH-fed WT mice or control-fed L-Tsc1 KO mice (Figure 5D–E). Inflammation is a hall marker of AH, which is characterized by increased infiltration of neutrophils in the liver (22). Compared with control mice, EtOH slightly increased the numbers of KCs and infiltrated neutrophils in WT mouse livers (Figure 5F–H). Loss of hepatic TSC1 further increased the numbers of KCs/infiltrated macrophages and neutrophils as well as the expression levels of hepatic inflammatory genes, indicating increased hepatic inflammation in L-Tsc1 KO mouse livers after EtOH feeding (Figure 5I).
Figure 5. EtOH-fed L-Tsc1 KO mice exhibit increased fibrosis and inflammation.
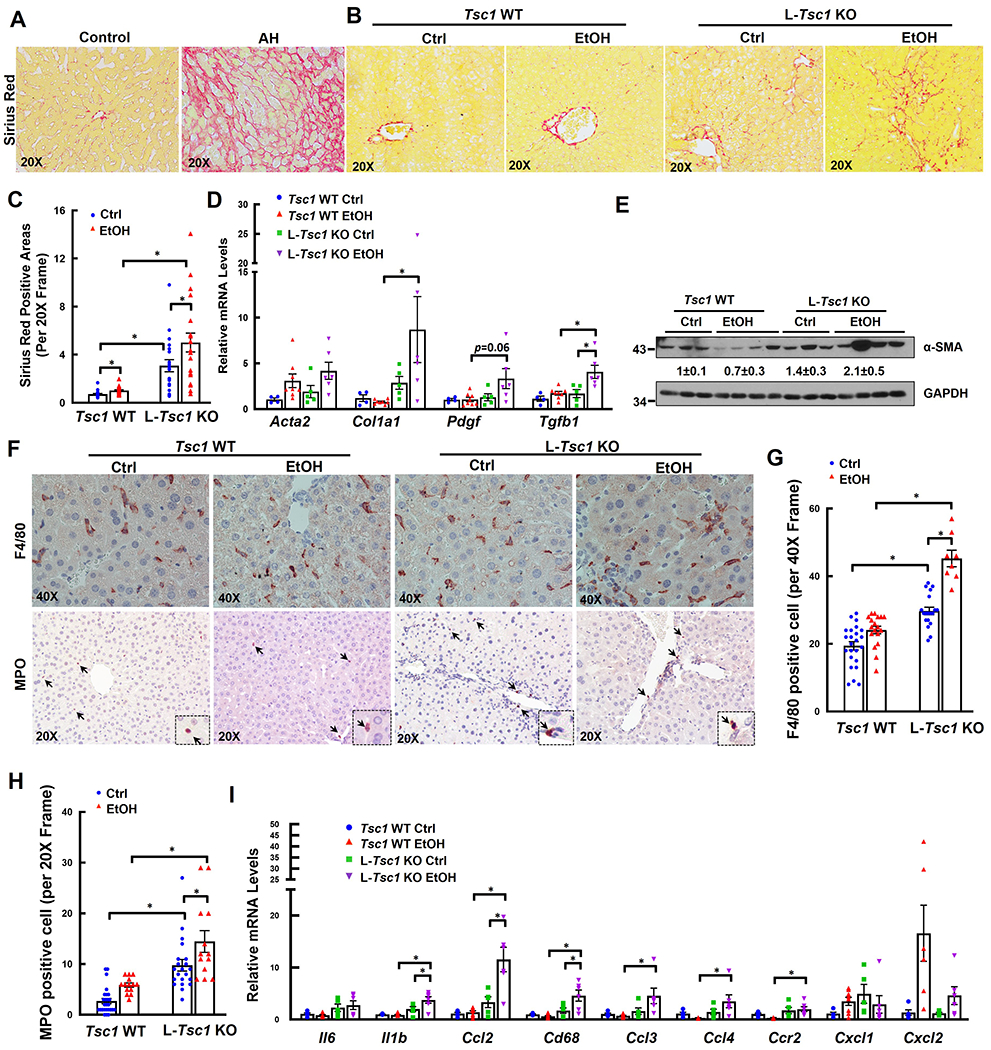
(A) Representative photographs of Sirius Red staining from AH or healthy donor livers (Control) are shown. Male L-Tsc1 WT and L-Tsc1 KO mice were fed with Gao-binge alcohol. (B) Representative photographs of Sirius Red staining are shown. Down panels are enlarged photographs from the boxed area. (C) Quantification of Sirius Red positive areas. 20 different fields (20x) from each mouse were quantified in a blinded fashion using Image J. Data are presented as means ± SE (from n=3-4 mice). (D) qRT-PCR analysis of fibrogenic gene expression. RNA was extracted from mouse livers followed by qRT-PCR. Results were normalized to 18s and expressed as fold change compared with Tsc1 WT Ctrl group. Data are presented as means ± SE (n=4-8). *p<0.05; One-Way ANOVA analysis with Bonferroni post hoc test. (E) Total liver lysates were subjected to western blot and densitometry analysis. Data are presented as means ± SE (n= 3-4). (F) Representative photographs of IHC staining for F4/80 and myeloperoxidase (MPO). Arrows denote F4/80 and MPO positive cells. The numbers of F4/80 (G) and MPO (H) positive cells were quantified. 8 to 27 different fields (40x or 20x) from each mouse were quantified in a blinded fashion. Data are presented as means ± SE (from n=3-4 mice). *p<0.05; One-Way Anova analysis with Bonferroni post hoc test. (I) qRT-PCR analysis of inflammation gene expression. RNA was extracted from mouse livers followed by qRT-PCR. Results were normalized to 18s and expressed as fold change compared with L-Tsc1 WT Ctrl group (n=4-8). *p<0.05; One-Way Anova analysis with Bonferroni post hoc test.
It has been reported that ductular cells display a pro-inflammatory profile and recruit neutrophils in AH (23). A more recent study showed that inflammatory cells interact with cholangiocytes to promote cholestasis in AH (24). We found that F4/80 positive KCs/infiltrated macrophages were mainly adjacent to SOX9+ positive cells, and the number of KCs/infiltrated macrophages associated with SOX9+ cholangiocytes was markedly increased in alcohol fed L-Tsc1 KO mouse livers (SFigure 3A–B).
AH is associated with cholestatic liver injury with impaired hepatocyte bile secretion (25, 26). Consistent with a previous report (27), we found that AH patients had significantly higher hepatic levels of total bile acids (TBAs) than normal donors (SFigure 4A). Levels of hepatic TBAs were moderately increased in WT mice but significantly increased in L-Tsc1 KO mice compared with WT mice after EtOH feeding (SFigure 4B). Further analysis of the BA composition showed that majority of the primary and secondary BAs were elevated in EtOH-fed L-Tsc1 KO mice, except CDCA, αMCA, βMCA, and UDCA (SFigure 4D–E). The heatmaps of RNA-seq analysis revealed that changes on the expression levels of bile acid biosynthesis, receptors and transporter/secretion pathway-related genes were largely heterogenous with both up-and down-regulated genes of these pathways were found in L-Tsc1 KO mice fed with EtOH (SFigure 4F–G). Taken together, these data indicate that loss of hepatic TSC1 exacerbates alcohol-induced liver fibrosis, inflammation and increases immune cell-cholangiocyte interactions to promote cholestasis and liver injury.
Cholangiocyte-specific but not hepatocyte-specific Tsc1 deletion increases liver injury, DR, fibrosis, and inflammatory cell infiltration in Gao-binge alcohol-fed mouse livers
Alb-Cre deletes Tsc1 in both hepatocytes and cholangiocytes in Tsc1flox/flox mouse livers, we next determined the impacts of hepatocyte-specific and cholangiocyte-specific deletion of Tsc1 in ETOH-induced liver injury. Immunoblotting data confirmed that TSC1 was deleted efficiently in mouse livers injected with AAV8-TBG-Cre (SFigure 5A). In contrast, TSC1 protein levels only partially decreased in mouse livers injected with Ad-CK19-CreERt2 plus tamoxifen likely due to the use of whole liver lysates (SFigure 5B). Deleting Tsc1 in hepatocytes led to markedly elevated hepatic levels of p-4EBP1, suggesting hyperactivation of mTORC1 (SFigure 5A). Mice with deletion of Tsc1 in cholangiocytes had higher serum ALT levels than mice with deletion of Tsc1 in hepatocytes fed with EtOH, although the difference was not significant due to variations among the mice (SFigure 5C). No significant differences for the liver to bodyweight ratio and hepatic TG levels in EtOH-fed mice with either hepatocyte-specific or cholangiocyte-specific Tsc1 deletion (SFigure 5D–E). Results from H&E, CK19, Sirius Red, F4/80 and MPO staining revealed that deletion of Tsc1 in cholangiocytes increased DR, fibrosis, numbers of F4/80 and MPO positive cells whereas deletion of Tsc1 in hepatocytes had no significant changes on these parameters in EtOH-fed mouse livers (Figure 6A–H).
Figure 6. Cholangiocyte specific but not hepatocyte specific Tsc1 deletion increases DR, fibrosis and inflammatory cell infiltration in EtOH-fed mouse livers.
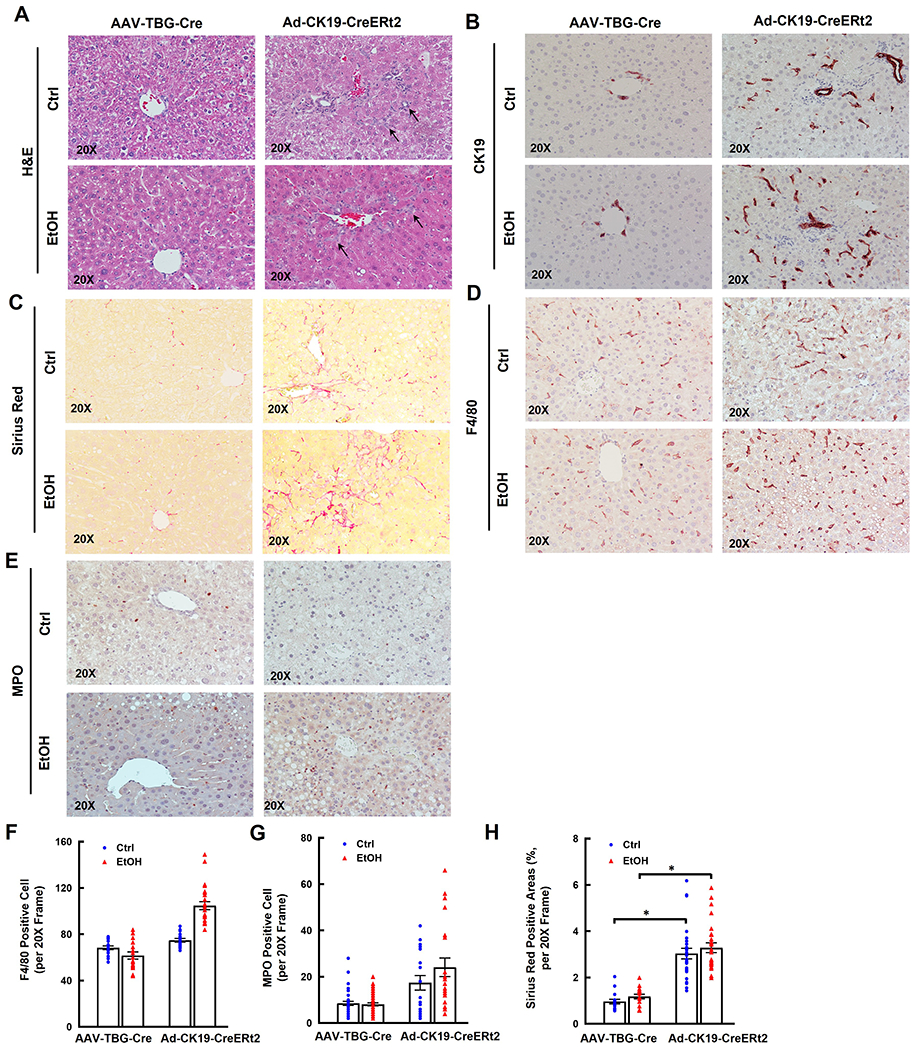
Tsc1flox/flox mice were injected with AAV8-TBG-Cre (1x1011 GC/mouse through tail vein), or Ad-CK19-CreERt2 (2x108 PFU/mouse through tail vein) and followed by tamoxifen (75 mg/kg, i.p.) for 3 days. All mice were subjected to Gao-binge feeding at eight weeks later after virus injection. Representative photographs of H&E staining (A) or IHC staining for CK19 (B), Sirius Red (C), F4/80 (D) and MPO staining (E) are shown. The numbers of F4/80 (F) and MPO (G) positive cells were quantified. More than 20 different fields (20x) from each mouse were quantified in a blinded fashion. Data are presented as means ± SE (from n=3-4 mice). (H) Sirius Red positive areas were quantified. 20 different fields (20x) from each mouse were quantified in a blinded fashion using Image J. Data are presented as means ± SE (from n=3-4 mice). *p<0.05; One-Way ANOVA analysis with Bonferroni post hoc test.
We next determined whether increased cholangiocyte proliferation would contribute to increased ductular cells in EtOH-fed L-Tsc1 KO mice. Compared with WT mice, the number of Ki67 positive cells was significantly increased in L-Tsc1 KO mouse livers, which was further increased by EtOH feeding. Interestingly, majority of the Ki67 positive cells were also EPCAM positive, suggesting increased proliferation rate of biliary epithelial cells (SFigure 6A–B). We next deleted TSC1 using CRISPR-CAS9 approach in an immortalized mouse cholangiocyte cell line. Western blot analysis revealed successful deletion of TSC1 and increased levels of p-S6 and p-4EBP1, indicating increased mTORC1 activation. Interestingly, deletion of TSC1 significantly increased cell proliferation compared with WT parental cells and cells with a control sgRNA (SFigure 6C–D). Collectively, these data suggest that loss of TSC1 in cholangiocytes but not hepatocytes contribute to the pathogenesis of AH featured with DR, fibrosis and inflammation. Increased DR is likely due to increased cholangiocyte proliferation as a result of mTORC1 activation.
Inhibition of mTORC1 partially reverses hepatomegaly, DR, fibrosis, inflammation and liver injury in Gao-binge alcohol-fed L-Tsc1 KO mice
Compared with vehicle, Torin1 treatment significantly decreased serum levels of ALT activity with slightly improved liver to bodyweight ratio and hepatic levels of triglyceride in EtOH-fed L-Tsc1 KO mice (Figure 7A–C). Torin 1 treatment decreased levels of p-4EBP1, CK19, α-SMA but increased levels of HNF4α in EtOH-fed L-Tsc1 KO mouse livers (Figure 7D). Moreover, Torin1 also attenuated DR, fibrosis, macrophage and neutrophil accumulation as indicated by H&E, CK19, Sirius Red staining as well as F4/80 and MPO staining in EtOH-fed L-Tsc1 KO mice (Figure 7E–I). Taken together, pharmacological inhibition of mTORC1 ameliorates AH-like liver pathogenesis in EtOH-fed L-Tsc1 KO mice.
Figure 7. Inhibition of mTORC1 meliorates liver injury, hepatomegaly, DR, fibrosis, and inflammatory cell infiltration in EtOH-fed L-Tsc1 KO mouse livers.
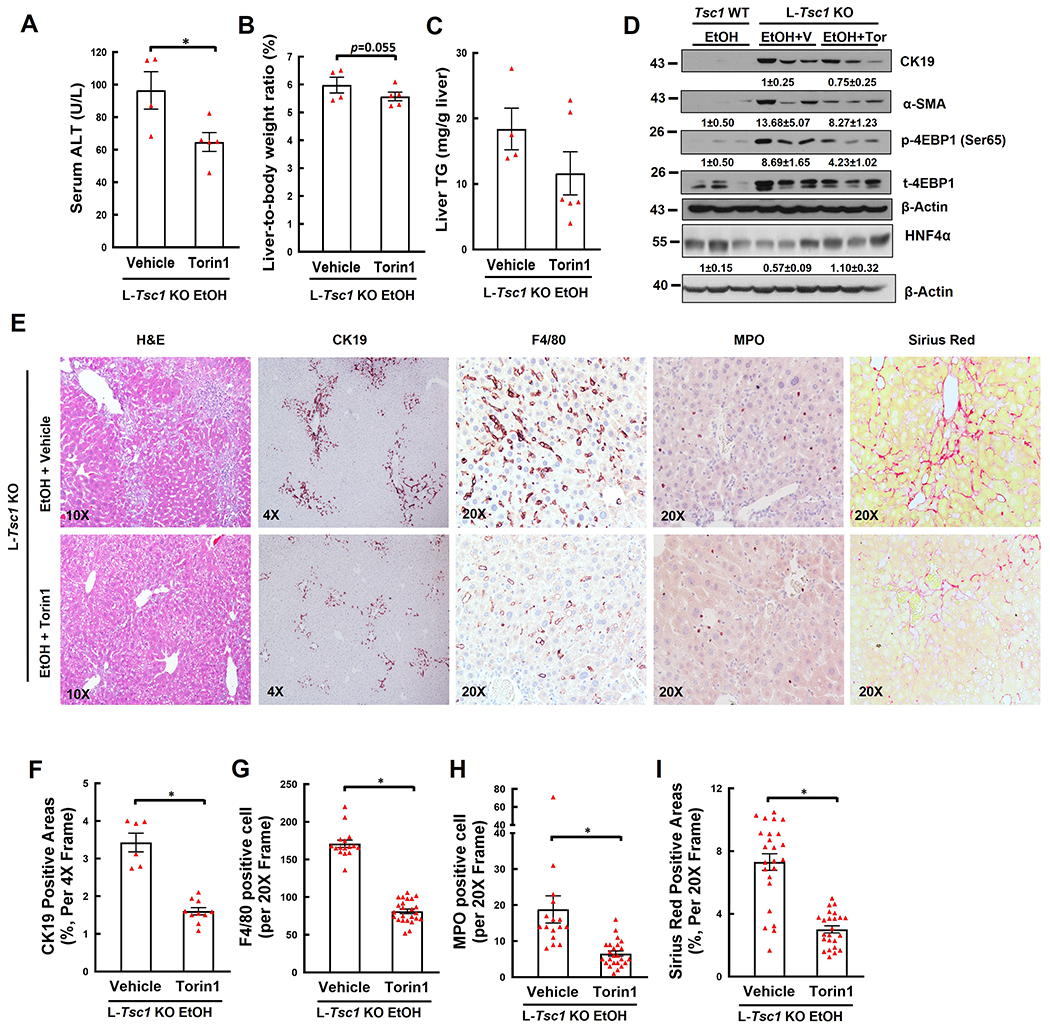
Male L-Tsc1 KO mice were fed with Gao-binge alcohol. Five doses Torin1 or vehicle control were given to mice during the 10 days alcohol feeding (day 1, 3, 5, 7 and 9, i.p. 2 mg/kg), and one dose was given right before the gavage on the final day. (A) Serum ALT activity, (B) liver weight versus body weight ratio, and (C) hepatic TG were analysis. Data are means ± SE (n=4-6). *p<0.05; Student’s t-test. (D) Total liver lysates were subjected to western blot and densitometry analysis. Data are presented as means ± SE (n= 3). (E-I) Representative photographs and quantification data of H&E, CK19, F4/80, MPO and Sirius Red staining are shown. More than 20 different fields (20x) from each mouse were quantified in a blinded fashion. Data are presented as means ± SE (from n=4-6 mice). *p<0.05; Student’s t-test. V: vehicle; Tor: Torin1.
Discussion
AH is a severe liver disease characterized by inflammation and acute liver failure with a short-term mortality around 50% (28). For several decades, corticosteroids remain the only effective treatment for AH but approximately 40% AH patients fail to respond to corticosteroids therapy (29). Therefore, it is urgently needed to develop new therapeutic approaches for this desperate life-threatening disease. However, lack of preclinical AH animal models to mimic the human AH phenotypes has significantly halted the progress on the discovery of novel targets for treating AH. In the present study, we showed that EtOH feeding significantly induced liver injury, steatosis, hepatomegaly, DR, fibrosis, inflammation, and liver cell repopulation, phenocopying the pathological features of human AH. More importantly, pharmacological inhibition of mTORC1 ameliorates human AH-like liver pathologies in EtOH-fed L-Tsc1 KO mice.
It has been suggested that liver failure in AH patients is likely due to excessive liver cell repopulation with increased hepatocyte degeneration and accumulation of fetal-like hepatic progenitor cells and ductular cells or DR (30–32). Hippo-YAP1 signaling pathway as well as SOX9 and HNF4α-mediated transcription programs are critical to maintain hepatocyte fate in the liver (20, 21, 33). AH patient livers have activated YAP1 and SOX9 but decreased HNF4α activation contributing to increased DR and mature hepatocyte degeneration (30–32). Epithelial splicing regulatory protein 2 (ESRP2) converts the fetal splicing variants that encode low kinase activity of neurofibromatosis 2 (NF2) and casein kinase 1 delta (CSNK1D) proteins into adult splicing variants with higher kinase activity to suppress YAP1 (30). Mechanistically, AH-mediated proinflammatory cytokines such as TNFα and IL-1β decreased Esrp2 expression in hepatocytes resulting in YAP1 activation and hepatocyte transdifferentiation to cholangiocyte-like cells (30). In addition, overexpression of the active form of human YAPS127A in hepatocytes also led to increased human hepatocytes transdifferentiation to cholangiocytes in vitro (32). In vivo transduction of YAPS127A using pAAV.CMV.YAPS127A led to increased SOX9 positive biliary cell population upon alcohol feeding (32). In addition to the changes of YAP1 and SOX9, epigenetic changes in DNA methylation and chromatin remodeling in AH livers led to decreased HNF4α expression and hepatocyte degeneration (31). We found increased YAP1 and SOX9 activation and decreased HNF4α expression in EtOH-fed L-Tsc1 KO mice, which are correlated with increased gene expression signatures of cholangicoytes and decreased gene signature of hepatocytes that are also found in human AH. However, we found both YAP1 and SOX9 are highly expressed in biliary epithelial cells or liver progenitor cells but not mature hepatocytes in EtOH-fed L-Tsc1 KO mice and AH human livers. In addition to ESRP2-mediated gene splicing, mTORC1 activation may also directly regulate hepatic levels of YAP1 and SOX9. YAP1 has been identified as a direct substrate and degraded by autophagy (34, 35). As deletion of Tsc1 leads to mTORC1 activation and autophagy inhibition, it is highly likely that increased YAP1 is due to decreased autophagic degradation in EtOH-fed L-Tsc1 KO mice. As reported in skeletogenesis, mTORC1 may directly accelerate the translation of SOX9 in EtOH-fed L-Tsc1 KO mice (36). In addition, SOX9 has been shown to be a downstream target of YAP1 (20, 33), and thus increased SOX9 may also be resulted from YAP1 activation in EtOH-fed L-Tsc1 KO mice. Future studies are needed to further dissect the detail mechanisms by which loss of Tsc1 and mTORC1 activation leads to YAP1 and SOX9 activation in AH.
L-Tsc1 KO mice fed with a high fat diet or NASH diet had similar or even protected against steatosis compared with wildtype mice (37, 38). Mechanistically, loss of hepatic Tsc1 increased VLDL secretion and activated the negative feedback on the mTORC2-AKT-FOXO-SREBP1c lipogenesis axis resulting in decreased hepatic steatosis. This is consistent with our findings that L-Tsc1 KO mice had no effects on alcohol-induced steatosis despite increased cell death and fibrosis. Future studies are needed to determine changes of VLDL secretion and mTORC2-AKT-FOXO-SREBP1c-mediated lipogenesis in alcohol-fed L-Tsc1 KO mice.
AH patients have increased total BA, which is consistent with alcohol-fed L-Tsc1 KO mice. Both the expression of de novo synthesis (CYP7A1) and uptake (SLC10A1, gene name for NTCP) deceased in human AH patients (27), which is consistent with alcohol-fed L-Tsc1 KO mice, suggesting de novo BA synthesis and uptake may not contribute for increased cholestasis in both human AH and alcohol-fed L-Tsc1 KO mice. We found decreased expression of several BA efflux genes (Abcc2, Abcg5 and Abcg8) in alcohol-fed L-Tsc1 KO mice, suggesting decreased BA efflux may contribute to cholestasis in alcohol-fed L-Tsc1 KO mice. However, mRNA levels may not always correlate with protein levels. Future studies are needed to further determine the mechanisms of how alcohol causes cholestasis in L-Tsc1 KO mice.
In our present study, we found increased DR in EtOH-fed L-Tsc1 KO mice. Increased DR could be either due to increased transdifferentiation of hepatocytes to cholangicoytes or increased cholangocyte proliferation in EtOH-fed L-Tsc1 KO mice. However, our results indicate that increased DR is most likely due to increased proliferation of cholangiocytes instead of transdifferentiation of hepatocytes to cholangiocytes. This notion is based on the following observations. First, deletion of Tsc1 in hepatocytes using the AAV-TBG-Cre failed to increased DR in EtOH-fed L-Tsc1 KO mice. Second, deletion of Tsc1 in cholangiocytes using Ad-CK19ERT2 cre increased cholangiocyte proliferation, DR, inflammation and fibrosis, suggesting that activation of mTORC1 in cholangiocytes but not hepatocytes contribute to EtOH-induced DR and liver injury. Third, the number of both Ki67 and EPCAM positive cells was significantly higher in EtOH-fed L-Tsc1 KO mouse livers, indicating a high proliferation rate of cholangiocytes. Finally, CRISPR/CAS9-mediated deletion of Tsc1 in cultured cholangiocytes increased cell proliferation. Future studies are needed to differentiate the specific contributions of YAP1 and SOX9 in hepatocytes vs cholangiocytes in the pathogenesis of AH.
In conclusion, our findings indicate that persistent activation of mTORC1 due to the loss of cholangiocyte TSC1 promotes liver injury, DR, inflammation, and fibrosis after EtOH feeding, which phenocopy the pathogenesis of human AH. Our model faithfully recapitulates human AH pathogenesis, which will have a great value to help to identify novel therapeutic targets such as inhibiting mTORC1 for treating AH in the future.
Supplementary Material
Acknowledgement:
We like to thank Dr. Xin Chen from University of Hawaii to provide us the CK19-CreErt2 plasmid. We like to thank Drs. Heather Francis and Gianfranco Alpini at University of Indiana for providing the mouse immortalized cholangiocyte cell line.
This work was partially supported by NIH grants: R37 AA020518, R01 DK102142 and R01 AG072895 (WXD). R01 DK124612 and R01 DK106540 (WL).
Abbreviations:
- ALD
Alcohol-associated liver disease
- AH
alcoholic hepatitis (AH)
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- CSNK1D
casein kinase 1 delta
- DR
ductular reactions
- ESRP2
Epithelial splicing regulatory protein 2
- GAP
GTPase-activating protein
- GSEA
Gene Set Enrichment Analysis
- HSC
hepatic stellate cell
- HCC
hepatocellular carcinoma
- IHC
Immunohistochemistry
- KC
Kupffer cells
- L-Tsc1 KO
liver-specific Tsc1 knockout
- LDs
lipid droplets
- mTORC1
mechanistic target of rapamycin complex 1
- MPO
myeloperoxidase
- NIAAA
National Institute on Alcohol Abuse and Alcoholism
- p-S6
phosphorylated S6
- qPCR
quantitative real time PCR
- Rheb
ras homolog enriched in brain
- TSC
tuberous sclerosis complex
- TFEB
transcription factor EB
- TG
triglyceride
- WT
wild type
- YAP1
yes-associated protein 1
Footnotes
Conflict of interest: Nothing to disclose.
References
- 1.Liangpunsakul S, Haber P, McCaughan GW. Alcoholic Liver Disease in Asia, Europe, and North America. Gastroenterology 2016;150:1786–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Griswold MG, Fullman N, Hawley C, Arian N, Zimsen SRM, Tymeson HD, Venkateswaran V, et al. Alcohol use and burden for 195 countries and territories, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet 2018;392:1015–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yoon Y- HCC. Liver cirrhosis mortality in the United States: national, state, and regional trends. 2000-2015. Surveillance Report #111; 2018: Available at: https://pubs.niaaa.nih.gov/publications/surveillance111/Cirr115.htm. [Google Scholar]
- 4.Nagy LE, Ding WX, Cresci G, Saikia P, Shah VH. Linking Pathogenic Mechanisms of Alcoholic Liver Disease With Clinical Phenotypes. Gastroenterology 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gao B, Bataller R. Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology 2011;141:1572–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Williams JA, Manley S, Ding WX. New advances in molecular mechanisms and emerging therapeutic targets in alcoholic liver diseases. World J Gastroenterol 2014;20:12908–12933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lamas-Paz A, Hao F, Nelson LJ, Vazquez MT, Canals S, Gomez Del Moral M, Martinez-Naves E, et al. Alcoholic liver disease: Utility of animal models. World J Gastroenterol 2018;24:5063–5075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bertola A, Mathews S, Ki SH, Wang H, Gao B. Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat Protoc 2013;8:627–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lucey MR, Mathurin P, Morgan TR. Alcoholic hepatitis. N Engl J Med 2009;360:2758–2769. [DOI] [PubMed] [Google Scholar]
- 10.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol 2011;12:21–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Laplante M, Sabatini DM. mTOR signaling at a glance. J Cell Sci 2009;122:3589–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shimobayashi M, Hall MN. Making new contacts: the mTOR network in metabolism and signalling crosstalk. Nat Rev Mol Cell Biol 2014;15:155–162. [DOI] [PubMed] [Google Scholar]
- 13.Huang J, Dibble CC, Matsuzaki M, Manning BD. The TSC1-TSC2 complex is required for proper activation of mTOR complex 2. Mol Cell Biol 2008;28:4104–4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003;115:577–590. [DOI] [PubMed] [Google Scholar]
- 15.Chao X, Wang S, Zhao K, Li Y, Williams JA, Li T, Chavan H, et al. Impaired TFEB-Mediated Lysosome Biogenesis and Autophagy Promote Chronic Ethanol-Induced Liver Injury and Steatosis in Mice. Gastroenterology 2018;155:865–879 e812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chao X, Ni HM, Ding WX. Insufficient autophagy: a novel autophagic flux scenario uncovered by impaired liver TFEB-mediated lysosomal biogenesis from chronic alcohol-drinking mice. Autophagy 2018;14:1646–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Menon S, Yecies JL, Zhang HH, Howell JJ, Nicholatos J, Harputlugil E, Bronson RT, et al. Chronic activation of mTOR complex 1 is sufficient to cause hepatocellular carcinoma in mice. Sci Signal 2012;5:ra24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kenerson HL, Yeh MM, Kazami M, Jiang X, Riehle KJ, McIntyre RL, Park JO, et al. Akt and mTORC1 have different roles during liver tumorigenesis in mice. Gastroenterology 2013;144:1055–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ni HM, Bockus A, Boggess N, Jaeschke H, Ding WX. Activation of autophagy protects against acetaminophen-induced hepatotoxicity. Hepatology 2012;55:222–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu YC, Zhuo S, Zhou YX, Ma LC, Sun ZH, Wu XL, Wang XW, et al. Yap-Sox9 signaling determines hepatocyte plasticity and lineage-specific hepatocarcinogenesis. Journal of Hepatology 2022;76:652–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dubois V, Staels B, Lefebvre P, Verzi MP, Eeckhoute J. Control of Cell Identity by the Nuclear Receptor HNF4 in Organ Pathophysiology. Cells 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu MJ, Zhou Z, Parker R, Gao B. Targeting inflammation for the treatment of alcoholic liver disease. Pharmacol Ther 2017;180:77–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aguilar-Bravo B, Rodrigo-Torres D, Arino S, Coll M, Pose E, Blaya D, Graupera I, et al. Ductular Reaction Cells Display an Inflammatory Profile and Recruit Neutrophils in Alcoholic Hepatitis. Hepatology 2019;69:2180–2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takeuchi M, Vidigal PT, Guerra MT, Hundt MA, Robert ME, Olave-Martinez M, Aoki S, et al. Neutrophils interact with cholangiocytes to cause cholestatic changes in alcoholic hepatitis. Gut 2021;70:342–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang Z, Kusumanchi P, Ross RA, Heathers L, Chandler K, Oshodi A, Thoudam T, et al. Serum Metabolomic Profiling Identifies Key Metabolic Signatures Associated With Pathogenesis of Alcoholic Liver Disease in Humans. Hepatol Commun 2019;3:542–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Altamirano J, Miquel R, Katoonizadeh A, Abraldes JG, Duarte-Rojo A, Louvet A, Augustin S, et al. A histologic scoring system for prognosis of patients with alcoholic hepatitis. Gastroenterology 2014;146:1231–1239 e1231-1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brandl K, Hartmann P, Jih LJ, Pizzo DP, Argemi J, Ventura-Cots M, Coulter S, et al. Dysregulation of serum bile acids and FGF19 in alcoholic hepatitis. J Hepatol 2018;69:396–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Louvet A, Mathurin P. Alcoholic liver disease: mechanisms of injury and targeted treatment. Nature Reviews Gastroenterology & Hepatology 2015;12:231–242. [DOI] [PubMed] [Google Scholar]
- 29.Louvet A, Naveau S, Abdelnour M, Ramond MJ, Diaz E, Fartoux L, Dharancy S, et al. The Lille model: A new tool for therapeutic strategy in patients with severe alcoholic hepatitis treated with steroids. Hepatology 2007;45:1348–1354. [DOI] [PubMed] [Google Scholar]
- 30.Hyun J, Sun Z, Ahmadi AR, Bangru S, Chembazhi UV, Du K, Chen T, et al. Epithelial splicing regulatory protein 2-mediated alternative splicing reprograms hepatocytes in severe alcoholic hepatitis. J Clin Invest 2020;130:2129–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Argemi J, Latasa MU, Atkinson SR, Blokhin IO, Massey V, Gue JP, Cabezas J, et al. Defective HNF4alpha-dependent gene expression as a driver of hepatocellular failure in alcoholic hepatitis. Nat Commun 2019;10:3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saleh MB, Louvet A, Ntandja-Wandji LC, Boleslawski E, Gnemmi V, Lassailly G, Truant S, et al. Loss of hepatocyte identity following aberrant YAP activation: A key mechanism in alcoholic hepatitis. Journal of Hepatology 2021;75:912–923. [DOI] [PubMed] [Google Scholar]
- 33.Ding WX, Sancho-Bru P. SOX9 acts downstream of YAP to decide liver cell fate and tumor types. Journal of Hepatology 2022;76:503–505. [DOI] [PubMed] [Google Scholar]
- 34.Liang N, Zhang C, Dill P, Panasyuk G, Pion D, Koka V, Gallazzini M, et al. Regulation of YAP by mTOR and autophagy reveals a therapeutic target of tuberous sclerosis complex. Journal of Experimental Medicine 2014;211:2249–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee YA, Noon LA, Akat KM, Ybanez MD, Lee TF, Berres ML, Fujiwara N, et al. Autophagy is a gatekeeper of hepatic differentiation and carcinogenesis by controlling the degradation of Yap. Nature Communications 2018;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Iezaki T, Horie T, Fukasawa K, Kitabatake M, Nakamura Y, Park G, Onishi Y, et al. Translational Control of Sox9 RNA by mTORC1 Contributes to Skeletogenesis. Stem Cell Reports 2018;11:228–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kenerson HL, Subramanian S, McIntyre R, Kazami M, Yeung RS. Livers with Constitutive mTORC1 Activity Resist Steatosis Independent of Feedback Suppression of Akt. Plos One 2015;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Uehara K, Sostre-Colon J, Gavin M, Santoleri D, Leonard KA, Jacobs RL, Titchenell PM. Activation of Liver mTORC1 Protects Against NASH via Dual Regulation of VLDL-TAG Secretion and De Novo Lipogenesis. Cellular and Molecular Gastroenterology and Hepatology 2022;13:1625–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.