

Abstract
Acute traumatic coagulopathy is a complex phenomenon following injury and a main contributor to hemorrhage. It remains a leading cause of preventable death in trauma patients. This phenomenon is initiated by systemic injury to the vascular endothelium that is exacerbated by hypoperfusion, acidosis, and hypothermia and leads to systemic activation of the coagulation cascades and resultant coagulopathy. Many previous studies have focused on endotheliopathy with targeted markers such as syndecan-1, soluble thrombomodulin, and plasma adrenaline as potential culprits for initiation and propagation of this state. However, in more recent studies, hyperadhesive von Willebrand factor (VWF), which is released following endothelial injury, and its cleaving metalloprotease ADAMTS13 have emerged as significant targets of the downstream effect of endothelial breakdown and coagulation dysregulation. Elucidation of the mechanism by which the dysregulated VWF-ADAMTS13 axis leads to endothelial dysfunction and coagulopathy after trauma can help identify new targets for therapy and sites for intervention. Much of what is known mechanistically regarding VWF stems from work done in traumatic brain injury. Following localized brain injury, brain-derived extracellular vesicles are released into circulation where they induce a hypercoagulable state that rapidly turns into consumptive coagulopathy. VWF released from injured endothelial cells binds to these extracellular vesicles to enhance their activity in promoting coagulopathy and increasing endothelial permeability. However, there are numerous gaps in our knowledge of VWF following injury, providing a platform for further investigation.
Keywords: A2 protein, ADAMTS13, coagulopathy, endothelial dysfunction, hemorrhagic shock, lactadherin, traumatic brain injury
VON WILLEBRAND FACTOR (VWF): FROM HEALTH TO DISEASE
VWF is the largest plasma multimeric glycoprotein encoded by the VWF gene on the short arm of chromosome 12 (1). VWF is primarily synthesized in megakaryocytes and endothelial cells first as pre-pro-VWF protein (2813 amino acids) that is comprised of a signal peptide (22 amino acids), a propeptide (741 amino acids), and mature polypeptide (2050 amino acids) (2, 3). In the endoplasmic reticulum, the pro-VWF monomers first form dimers through the C-terminal disulfide bonds in the CK domain. These VWF dimers then form multimers through the N-terminal disulfide bonds after the large signal peptide is enzymatically removed (4–6). During the transition through these intracellular granules, VWF undergoes extensive N-linked and O-linked glycosylation, including ABO antigens (3). Each mature VWF monomer contains multiple domains with defined structure and function (7). The D’/D3 domain binds to factor VIII to prevent its enzymatic degradation in blood, the A1 domain contains the binding site for the GP Ib-IX-V complex on platelets, the A3 domain binds to the subendothelial collagen, and the C1 domain contains a RGD sequence that interacts with integrins such as αIIbβ3 on platelets and αvβ5 on endothelial cells. The A2 domain contains the peptide bond that is cleaved by the metalloprotease ADAMTS13 (a disintegrin and metalloprotease with thrombospondin type 1 repeats, number 13).
After synthesis, VWF multimers are either released constitutively into the circulation or stored in the Wiebel-Palade bodies in endothelial cells and in alpha granules of platelets, where multimerization continues to form ultra-large VWF (ULVWF) multimers. When endothelial cells are activated, they release stored ULVWF multimers that are rapidly cleaved into small multimers by ADAMTS13, which is primarily synthesized by the liver, before being released into circulation. However, stretching and unfolding of these multimers occurs with hydrodynamic forces found in blood circulation to expose the A2 domain for cleavage by ADAMTS13 and to expose the A1 domain for binding and activating platelets (8, 9). It is not known which of these two processes is predominant in trauma patients who may suffer hemorrhagic shock that drastically changes blood hydrodynamics.
While VWF plays a crucial role in facilitating normal hemostasis and has been widely used as the reliable marker for endothelial cell activation and injury, recent evidence suggests it may propagate endotheliopathy and coagulopathy after trauma. The ULVWF multimers released by activated endothelial cells or platelets in response to traumatic, ischemic, and inflammatory stimulations (2, 3, 10) differ from circulating plasma VWF in molecular mass and in adhesive activity and will be the focus of the current review.
VWF IN INFLAMMATION
VWF has been associated with inflammation and endothelial damage such as in glomerulonephritis (11, 12), arteritis, diabetes, and sepsis, which led to its early characterization as an acute-phase reactant (13). Recently, VWF antigen levels were found to be elevated in COVID-19 patients and significantly correlated with mortality (14). Its role in inflammation is thought to be due to endothelial activation by inflammatory mediators such as IL-1, IL-8, and TNF-α to release ULVWF multimers, while IL-6 has been shown to inhibit the cleavage of ULVWF-platelet strings on endothelial cells (1, 15). VWF bound to activated endothelial cells is also able to attract leukocytes and platelets to propagate vascular inflammation and endothelial injury (16–19). Similarly, VWF antigen was higher and ADAMTS13 significantly lower in patients with severe sepsis than in patients with organ failure unrelated to sepsis or in healthy subjects. Additionally, ADAMTS13 was negatively correlated with VWF antigen, thrombomodulin, IL-6, APACHE-II score, shock, and acute renal injury (20). Interestingly, while patients with higher ADAMTS13 activity had improved survival, VWF did not show the same association. This finding raises the possibility that VWF multimers found in these patients may be resistant to cleavage, a phenomenon described in multiple settings. For example, under oxidative stress conditions, oxidized VWF was found to resist cleavage by ADAMTS13 (21). Similarly, extracellular hemoglobin was found to bind multimeric VWF and prevent cleavage (22). On the other hand, several potential mechanisms have been suggested for the low levels of ADAMTS13 in sepsis. First, the large amount of newly released ULVWF from the activated endothelium may lead to a consumptive deficiency of ADAMTS13. This was demonstrated in both healthy individuals and individuals with von Willebrand Disease, where the administration of DDAVP stimulated the release of VWF from endothelial cells, and resulted in the transient appearance of ULVWF in plasma and decreased in ADAMTS13 activity (23), indicating that an overwhelming release of ULVWF may exhaust ADAMTS13 activity (24), at least transiently. Second, ADAMTS13 activity can be decreased by inflammatory cytokines such as IL-6 (20). Similarly, ADAMTS13 synthesis can be inhibited by the inflammatory cytokines IFN-γ, IL-4, and TNF-α (25). Finally, proteases released from neutrophils have been suggested to cleave circulating ADAMTS13, as recombinant ADAMTS13 is degraded by purified granulocyte elastase (26). Similar to the inflammation seen in sepsis, evaluation of the VWF-ADAMTS13 axis in disseminated intravascular coagulation also demonstrates lower ADAMTS13 plasma activity in non-survivors of hemorrhagic complications, with a cut-off level of 42% predicting risk of these complications (27).
VWF AFTER INJURY
VWF in trauma
As an acute phase reactant, VWF is released from injured endothelial cells and activated platelets during acute injury, significantly increasing plasma levels of VWF, as extensively reported in clinical observational studies. Russell et al. (28) demonstrated in severely injured pediatric patients that VWF antigen and activity were significantly elevated on admission as compared with uninjured controls. Similar findings were seen in adult trauma patients in a prospective observational study by Tang et al. (29). Plasma levels of VWF were above normal in 85% of patients following initial resuscitation and remained elevated for the first 3 days (29). Interestingly, VWF levels remained elevated at day seven in non-survivors, suggesting ongoing endothelial dysfunction.
In patients with severe trauma complicated by acute lung injury (ALI), Siemiatkowski et al. (30) found plasma VWF to be significantly elevated for the first 7 days compared with less severely injured and controls. Furthermore, VWF antigen levels were increased at all times tested, up to 10 days, in ICU patients who developed adult respiratory adult syndrome (ARDS) and in non-survivors compared with patients without ARDS and survivors, respectively (31). VWF levels correlated with injury severity scores (ISS) and APACHE II scores. These findings are consistent with previous reports of VWF elevation in ALI/ARDS not associated with trauma (32), with some studies finding early elevation and a greater than 450% increase to be prognostic of death (32–34). This significant increase in plasma VWF during acute trauma serves as a reliable marker for endothelial injury and is necessary for trauma hemostasis. However, increasing evidence suggests that the role of VWF goes beyond hemostasis.
Injured patients secrete not only more VWF but also demonstrate an upward shift in VWF multimeric composition (35). The cause of this enhanced VWF adhesive activity remains poorly understood but could result from an imbalance between substantially released VWF and moderately reduced ADAMTS-13. VWF multimers freshly released from injured or activated endothelial cells are enriched in large or ultra large VWF multimers that are known to be prothrombotic as seen in patients with thrombotic thrombocytopenia purpura (36). However, there may be no parallel increase in ADAMTS-13 because the metalloprotease is stored in intracellular storage granules, as VWF is. The ADAMTS13 transcription is not changed after cells being exposed to proinflammatory stimuli such as endotoxin, TNF-α, IL-6, and IL-1β, which could trigger the acute phase reaction (37). In addition, hemorrhagic shock reduces blood flow in the microvasculature and thus reduces shear stress (38), slowing the rate of VWF cleavage. Consistent with this notion, plasma ADAMTS13 antigen and activity are reduced in both injured adults and children (28, 35). In a cohort of coagulopathic patients from the PAMPer trial, Dyer et al. (35) demonstrated a decrease in both ADAMTS13 level and activity on admission and at 24 h. When analyzing by ISS, patients with ISS > 15 had lower ADAMTS13 activity. Interestingly, after multivariable linear regression, ADAMTS activity was independently associated with coagulopathy as defined on admission international normalized units (INR). This was also true in pediatric patients in the study by Russel et al. (28) who demonstrated lower ADAMTS13 in patients with an elevated INR. Despite reports on clinical association between dysregulation of VWF/ADAMTS-13 and poor outcomes of patients with severe trauma, how VWF and ADAMTS-13 contribute to trauma-induced endotheliopathy and coagulopathy remains poorly defined mechanistically.
The CRYOSTAT-1 feasibility study in adults with major traumatic hemorrhage examined the early administration of cryoprecipitate as part of a major hemorrhage protocol and demonstrated sustained fibrinogen levels in the early high-dose cryoprecipitate arm with a suggestion of decreased mortality (39). While the VWF-ADAMTS13 axis was not specifically investigated in this study, it is worthy to note that cryoprecipitate has the highest concentration of VWF and ADAMTS13 of any blood product (40), suggesting cryoprecipitate may be a potential therapeutic to replete ADAMTS13. CRYOSTAT-2 is examining the early use of cryoprecipitate and may shed additional light on this subject (41).
VWF in traumatic brain injury (TBI)
Human data specific to the role of VWF after TBI is limited. In a small human study by Kumar et al. (42) in 32 non-coagulopathic patients with moderate to severe TBI, VWF antigen levels and its activity, measured by its ability to bind collagen III, were significantly increased in TBI patients compared with those in healthy controls. Conversely, ADAMTS13 activity was significantly decreased, resulting in a marked reduced ratio of ADAMTS13 activity to VWF antigen and activity following injury. In a study of 40 non-traumatic subarachnoid hemorrhage patients (SAH), Kumar et al. (43) demonstrated similar findings with elevated plasma VWF antigen and activity and lower ADAMTS13 in SAH patients compared with healthy subjects.
VWF has been shown to play an important role in TBI-induced coagulation abnormalities. Insufficiently cleaved and hyperadhesive VWF multimers released into circulation contribute to trauma-shock-induced coagulopathy that is consumptive in nature, developing from an acute hypercoagulable state. While TBI patients do not lose a significant amount of blood and are therefore rarely in shock, retrospective and observation studies have shown that coagulopathy is as prevalent in isolated TBI as it is in extracranial injuries (44, 45). After TBI, coagulation abnormalities occur early, usually within minutes (46). Elevated D-dimer and fibrinogen degradation products increase within minutes of injury followed by prolonged prothrombin and partial thromboplastin times, a time course consistent with early transition from a hyper- to a hypocoagulable state (47, 48). The initial hypercoagulable states are initiated with the rapid release of brain-derived substances, including brain-derived extracellular vesicles (BDEVs) from the injured brain through the disrupted blood brain barrier. These BDEVs are highly procoagulant as they express tissue factor and the anionic phospholipid phosphatidylserine on their surface and promote thrombin generation (49). Thrombin then cleaves fibrinogen to generate fibrin and activates both platelets and endothelial cells to generate both platelet and endothelial cell EVs. Hyperadhesive VWF contributes to this consumptive coagulopathy by activating platelets and endothelial cells to express procoagulant activity similar to BDEVs. These circulating EVs become circulating platforms to initiate and amplify coagulation that normally occurs on the surface of activated platelets localized to the site of endothelial injury but is now diffuse and exaggerated, culminating in a consumptive coagulopathy.
BDEVs can also activate endothelial cells to promote coagulation and to increase vascular permeability. This BDEV activity is mediated by hyperadhesive VWF multimers that are released from injured or activated endothelial cells and bind to BDEVs and EVs from other cell types. The hyperadhesive VWF can promote endotheliopathy and coagulopathy through different pathways during acute TBI that are highlighted in Table 1. First, it can act as the adhesive ligand to mediate the interaction between EVs and endothelial cells (50, 51). Second, newly released ULVWF multimers can be anchored to endothelial cells through its interaction with different membrane receptors (52–54) and form highly adhesive fibrillary structures that tether platelets, leukocytes, and EVs to the endothelium (16, 55), causing or propagating secondary and inflammatory injury.
Table 1.
Potential mechanisms by which VWF contributes to TBI-induced coagulation abnormalities
| ULVWF functions as an adhesive ligand to mediate the interaction between pathologic BDEVs and endothelial cells. BDEVs with exposed phosphatidylserine bind coagulation factors, leading to a consumptive coagulopathy. |
| ULVWF anchors to endothelial cells and tethers platelets, leukocytes and EVs to the endothelium, worsening inflammatory injury and serving as a nidus for microvascular thromboses. |
| ULVWF released from injured endothelial cells overwhelms the cleavage capabilities of ADAMTS13, resulting in accumulation of uncleaved hyperadhesive VWF in circulation which then activate platelets and occludes the microvasculature. |
VWF indicates von Willebrand factor; ULVWF, ultra-large VWF; BDEVs, brain-derived extracellular vesicles.
There are mechanistic similarities between TBI-induced and hemorrhagic shock-induced microvascular dysfunction and coagulopathy and these are outlined in Table 2 (56). However, whether hemorrhagic shock-induced coagulopathy is also consumptive in nature remains debatable and unanswered. Both lead to exposed endothelial cells following shedding of syndecan-1 and the glycocalyx and subsequent endothelial dysfunction (57–60). Third, the substantial release of VWF from the injured endothelial could overwhelm ADAMTS13, which is not increased in synthesis during acute phase reactions (e.g., trauma), drastically decreasing the ADAMTS13-to-VWF ratio to slow the kinetics of VWF cleavage, resulting in the accumulation of uncleaved hyperadhesive VWF in circulation to activate platelets and to occlude microvasculature, further worsening tissue ischemia induced by shock. This acquired ADAMTS13 deficiency has been detected in severe TBI patients (42), reproduced in mice (50), and is associated with coagulopathy, endothelial damage, and mortality.
Table 2.
Mechanistic comparison between TBI-induced and hemorrhagic shock-induced microvascular dysfunction
| Pathology | TBI | HS |
|---|---|---|
| Excessive hyperadhesive VWF | X | X |
| Low ADAMTS13 | X | X |
| Hyper-to-hypocoagulation (56) | X | X |
| Pathologic EVs | X | ? |
| Syndecan-1 shedding to expose ECs | X | X |
TBI indicates traumatic brain injury; VWF, von Willebrand factor.
However, whether similar pathologic EVs are secreted after hemorrhage remains unknown. Figure 1 reviews a proposed pathway by which hyperadhesive VWF could lead to trauma-induced coagulopathy. First, following shedding of syndecan-1, VWF multimers attract platelets and leukocytes to the surface of exposed endothelial cells, resulting in endothelial inflammation. This process converts the anticoagulant endothelial surface during homeostasis to highly procoagulant by activating endothelial cells to express anionic phospholipids such as phosphatidylserine. Second, hyperadhesive VWF binds and activates platelets to express procoagulant activity and also produce procoagulant extracellular vesicles. Third, the procoagulant microvesicles may serve as platforms on which coagulation is initiated and propagated, leading to systemic consumption of coagulation factors.
Fig. 1.
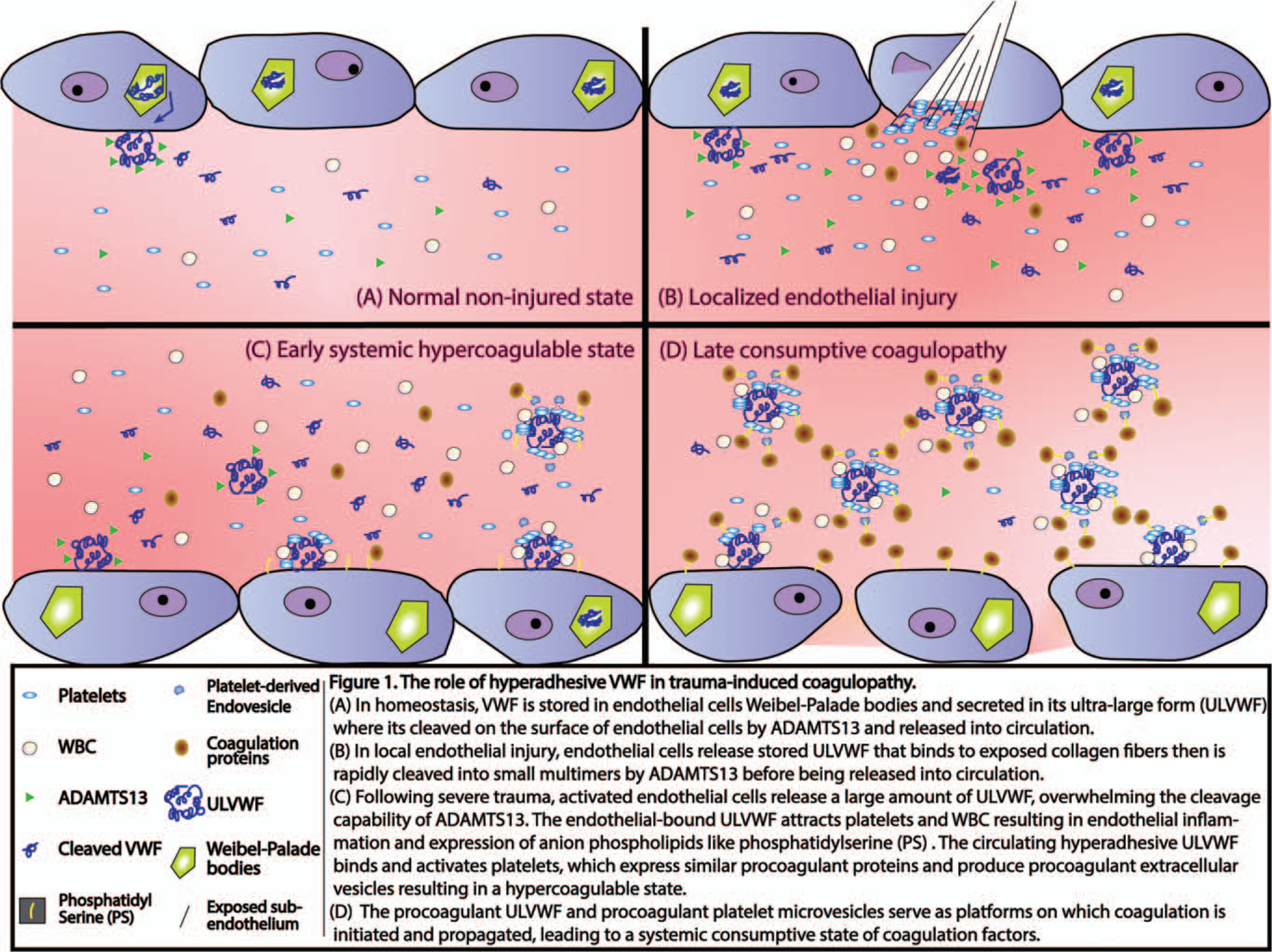
The role of hyperadhesive VWF in trauma-induced coagulopathy.
Potential therapeutics
Several molecules have been tested in mouse models to define the role of VWF in TBI-induced coagulopathy, but also so to identify potential new therapeutics for the condition.
Recombinant ADAMTS13—
We have shown that recombinant ADAMTS13 given to mice pre or post TBI reduced vascular leakage and coagulopathy and improved neurologic recovery and survival by enhancing VWF cleavage without impairing basal hemostasis (50). These protective effects are due to the enhanced ability of ADAMTS13 to cleave hyperadhesive VWF.
VWF A2 domain—
We also have shown that administration of a recombinant VWF A2-domain protein to block the exposed A1 can reduce vascular permeability and cerebral edema, increase cerebral blood flow, prevent TBI-induced coagulopathy, and improve outcomes in a rodent model of TBI (50). This finding suggests that VWF multimers undergo conformational changes that expose the platelet-binding A1-domain, which is hidden in the globular structure of plasma by forming a complex with the adjacent A2 domain. This finding also raises an important question as to whether caplacizumab is effective in preventing or treating TBI-induced coagulopathy. Caplacizumab is an A1-blocking nanobody developed into a humanized single-variable domain immunoglobulin that blocks VWF binding to its receptor GP-Ibα on platelets and has been approved for the treatment of TTP (61, 62).
Lactadherin—
The finding that hyperadhesive VWF can bind and mediate EV-induced endothelial injury and coagulopathy further suggests that increasing EV clearance could also prevent the TBI-induced hypercoagulable state and resultant consumptive coagulopathy. This notion is supported by our mouse study that lactadherin, also known as milk fat globule-epidermal growth factor 8, prevents coagulopathy, reduces cerebral edema, improves neurologic function, and decreases mortality in mice subjected to lateral fluid percussion injury to the brain (42, 63). Lactadherin is an apoptotic scavenge factor that promotes the phagocytosis of apoptotic cells by coupling them to macrophages (64). It achieves this activity by binding both apoptotic cells through its phosphatidylserine-binding domain (procoagulant EVs also express) and macrophages through its integrin-binding RGD sequence. The question remains as whether VWF helps or interferes the EV scavenging process. The study also raises an important question why exogenous lactadherin is needed. The exact reason is not known but similar to baseline levels of ADAMTS13 not being capable of cleaving the massive release of hyperadhesive VWF after TBI, the intrinsic level of lactadherin may be too low to be sufficient for removing the large burden of EVs without supplementation in a timely manner. In this regard, it is also interesting to ask whether plasma levels of lactadherin and other proteins that bind and block anionic phospholipids can serve as a predictive marker for trauma-induced endotheliopathy and coagulopathy.
SUMMARY
Traumatic coagulopathy and uncontrolled hemorrhage remain a significant challenge. Once initiated, it is very difficult to reverse. With the close relationship between coagulopathy and endotheliopathy, studies have identified potential biomarkers, such as VWF and ADAMTS13, for these two processes, thus enhancing our understanding of the instigation and propagation mechanism of these pathologies. VWF and its cleaving enzyme ADAMTS13 may play important roles not only in trauma hemostasis, but also in secondary inflammatory and coagulation dysregulation. The large release of VWF after endothelial injury from trauma, the transient deficiency in its cleaving enzyme ADAMTS13, and the accompanying consumptive coagulopathy seem to be a central role in the pathology that follows injury, at least after traumatic brain injury. Less is known mechanistically regarding the interplay of VWF, ADAMTS13, and coagulation factors following hemorrhagic shock. Further studies into potential therapeutics that can restore balance to the VWF/ADAMTS13 axis, preserving its hemostatic function, and preventing its pathologic hemostatic effects, warrant further investigation.
Acknowledgments
This work was supported by National Institute of Health RO1GM129533 (RAK), and R01 HL119391 and R01HL152200 (J-FD).
Footnotes
The authors report no conflicts of interest.
REFERENCES
- 1.Sadler JE: Biochemistry and genetics of Von Willebrand Factor. Annu Rev Biochem 67(1):395–424, 1998. [DOI] [PubMed] [Google Scholar]
- 2.Chen J, Chung DW: Inflammation, von Willebrand factor, and ADAMTS13. Blood 132(2):141–147, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fowler WE, Fretto LJ, Hamilton KK, Erickson HP, McKee PA: Substructure of human von Willebrand factor. J Clin Invest 76(4):1491–1500, 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wagner DD, Mayadas T, Marder VJ: Initial glycosylation and acidic pH in the Golgi apparatus are required for multimerization of von Willebrand factor. J Cell Biol 102(4):1320–1324, 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vischer UM, Wagner DD: von Willebrand factor proteolytic processing and multimerization precede the formation of Weibel-Palade bodies. Blood 83(12):3536–3544, 1994. [PubMed] [Google Scholar]
- 6.Mayadas TN, Wagner DD: In vitro multimerization of von Willebrand factor is triggered by low pH. Importance of the propolypeptide and free sulfhydryls. J Biol Chem 264:13497–13503, 1989. [PubMed] [Google Scholar]
- 7.Moake JL, Chow TW: Increased von Willebrand factor (vWf) binding to platelets associated with impaired vWf breakdown in thrombotic thrombocytopenic purpura. J Clin Apher 13(3):126–132, 1998. [DOI] [PubMed] [Google Scholar]
- 8.Kroll MH, Hellums JD, McIntire LV, Schafer AI, Moake JL: Platelets and shear stress. Blood 88(5):1525–1541, 1996. [PubMed] [Google Scholar]
- 9.López JA, Dong J: Shear stress and the role of high molecular weight von Willebrand factor multimers in thrombus formation. Blood Coagul Fibrinolysis 16(suppl 1):S11–S16, 2005. [DOI] [PubMed] [Google Scholar]
- 10.Counts RB, Paskell SL, Elgee SK: Disulfide bonds and the quaternary structure of factor VIII/von Willebrand factor. J Clin Invest 62:702–709, 1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ekberg M, Nilsson IM: Factor VIII and glomerulonephritis. Lancet 1(7916): 1111–1113, 1975. [DOI] [PubMed] [Google Scholar]
- 12.Boneu B, Abbal M, Plante J, Bierme R: Letter: factor-VIII complex and endothelial damage. Lancet 1(7922):1430, 1975. [DOI] [PubMed] [Google Scholar]
- 13.Pottinger BE, Read RC, Paleolog EM, Higgins PG, Pearson JD: von Willebrand factor is an acute phase reactant in man. Thromb Res 53(4):387–394, 1989. [DOI] [PubMed] [Google Scholar]
- 14.Goshua G, Pine AB, Meizlish ML, Chang C-H, Zhang H, Bahel P, Baluha A, Bar N, Bona RD, Burns AJ, et al. : Endotheliopathy in COVID-19-associated coagulopathy: evidence from a single-centre, cross-sectional study. Lancet Haematol 7(8):e575–e582, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bernardo A, Ball C, Nolasco L, Moake JF, Dong JF: Effects of inflammatory cytokines on the release and cleavage of the endothelial cell-derived ultralarge von Willebrand factor multimers under flow. Blood 104(1):100–106, 2004. [DOI] [PubMed] [Google Scholar]
- 16.Dong J, Moake JL, Nolasco L, Bernardo A, Arceneaux W, Shrimpton CN, Schade AJ, McIntire LV, Fujikawa K, López JA: ADAMTS-13 rapidly cleaves newly secreted ultralarge von Willebrand factor multimers on the endothelial surface under flowing conditions. Blood 100(12):4033–4039, 2002. [DOI] [PubMed] [Google Scholar]
- 17.Savchenko AS, Borissoff JI, Martinod K, De Meyer SF, Gallant M, Erpenbeck L, Brill A, Wang Y, Wagner DD: VWF-mediated leukocyte recruitment with chromatin decondensation by PAD4 increases myocardial ischemia/reperfusion injury in mice. Blood 123(1):141–148, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chauhan AK, Walsh MT, Zhu G, Ginsburg D, Wagner DD, Motto DG: The combined roles of ADAMTS13 and VWF in murine models of TTP, endotoxemia, and thrombosis. Blood 111(7):3452–3457, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sorvillo N, Mizurini DM, Coxon C, Martinod K, Tilvawala R, Cherpokova D, Salinger AJ, Seward RJ, Staudinger C, Weerapana E, et al. : Plasma peptidylarginine deiminase IV promotes VWF-platelet string formation and accelerates thrombosis after vessel injury. Circ Res 125(5):507–519, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martin K, Borgeri D, Lerolle N: Decreased ADAMTS13 is associated with a poor prognosis in sepsis-induced organ failure. Crit Care Med 35:2375–2382, 2007. [DOI] [PubMed] [Google Scholar]
- 21.Chen J, Fu X, Wang Y, Ling M, McMullen B, Kulman J, Chung DW, Lopez JA: Oxidative modification of von Willebrand factor by neutrophil oxidants inhibits its cleavage by ADAMTS13. Blood 115:706–712, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou Z, Han H, Cruz M, López J, Dong J-F, Guchhait P: Haemoglobin blocks von Willebrand factor proteolysis by ADAMTS-13: a mechanism associated with sickle cell disease. Thromb Haemost 101(06):1070–1077, 2009. [PubMed] [Google Scholar]
- 23.Reiter RA, Knobl P, Varadi K, Turecek PL: Changes in von Willebrand factor-cleaving protease (ADAMTS13) activity after infusion of desmopressin. Blood 101:946–948, 2003. [DOI] [PubMed] [Google Scholar]
- 24.Bianchi V, Robles R, Alberio L, Furlan M, Lammle B: Von Willebrand factor-cleaving protease (ADAMTS13) in thrombocytopenic disorders: a severely deficient activity is specific for thrombotic thrombocytopenic purpura. Blood 100:710–713, 2002. [DOI] [PubMed] [Google Scholar]
- 25.Cao WJ, Niiya M, Zheng XW, Shang DZ, Zheng XL: Inflammatory cytokines inhibit ADAMTS13 synthesis in hepatic stellate cells and endothelial cells. J Thromb Haemost 6(7):1233–1235, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ono T, Mimuro J, Madoiwa S, Soejima K, Kashiwakura Y, Ishiwata A, Takano K, Ohmori T, Sakata Y: Severe secondary deficiency of von Willebrand factor-cleaving protease (ADAMTS13) in patients with sepsis-induced disseminated intravascular coagulation: its correlation with development of renal failure. Blood 107(2):528–534, 2006. [DOI] [PubMed] [Google Scholar]
- 27.Chinen Y, Kuroda J, Ohshiro M, Shimura Y, Mizutani S, Nagoshi H, Sasaki N, Nakayama R, Kiyota M, Yamamoto-Sugitani M, et al. : Low ADAMTS-13 activity during hemorrhagic events with disseminated intravascular coagulation. Int J Hematol 97(4):511–519, 2013. [DOI] [PubMed] [Google Scholar]
- 28.Russell RT, McDaniel JK, Cao W, Shroyer M, Wagener BM, Zheng XL, Pittet JF: Low plasma ADAMTS13 activity is associated with coagulopathy, endothelial cell damage and mortality after severe paediatric trauma. Thromb Haemost 118(4):676–687, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tang N, Yin S, Sun Z, Pan Y: Time course of soluble P-selectin and von Willebrand factor levels in trauma patients: a prospective observational study. Scand J Trauma Resusc Emerg Med 21:70, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Siemiatkowski A, Kłoczko J, Wereszczyńska-Siemiatkowska U, Czaban SL: Effect of severe multiple trauma complicated by acute lung injury on endothelial cell activity. Przeglad Lekarski 58(7–8):767–771, 2001. [PubMed] [Google Scholar]
- 31.Siemiatkowski A, Kloczko J, Galar M, Czaban S: von Willebrand factor antigen as a prognostic marker in posttraumatic acute lung injury. Pathophysiol Haemost Thromb 30(4):189–195, 2000. [DOI] [PubMed] [Google Scholar]
- 32.Flori HR, Ware LB, Milet M, Matthay MA: Early elevation of plasma von Willebrand factor antigen in pediatric acute lung injury is associated with an increased risk of death and prolonged mechanical ventilation. Pediatr Crit Care Med 8(2):96–101, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ware LB, Conner ER, Matthay MA: von Willebrand factor antigen is an independent marker of poor outcome in patients with early acute lung injury. Crit Care Med 29(12):2325–2331, 2001. [DOI] [PubMed] [Google Scholar]
- 34.Ware LB, Eisner MD, Thompson BT, Parsons PE, Matthay MA: Significance of von Willebrand factor in septic and nonseptic patients with acute lung injury. Am J Respir Crit Care Med 170(7):766–772, 2004. [DOI] [PubMed] [Google Scholar]
- 35.Dyer MR, Plautz WE, Ragni MV, Alexander W, Haldeman S, Sperry JL, Guyette FX, Zuckerbraun BS, Rollins-Raval MA, Raval JS, et al. : Traumatic injury results in prolonged circulation of ultralarge von Willebrand factor and a reduction in ADAMTS13 activity. Transfusion 60(6):1308–1318, 2020. [DOI] [PubMed] [Google Scholar]
- 36.Zander CB, Cao W, Zheng XL: ADAMTS13 and von Willebrand factor interactions. Curr Opin Hematol 22(5):452–459, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Claus RA, Bockmeyer CL, Kentouche K, Sieber MW, Oberle V, Kaufmann R, Deigner HP, Lösche W: Transcriptional regulation of ADAMTS13. Thromb Haemost 94(07):41–45, 2005. [DOI] [PubMed] [Google Scholar]
- 38.Chien S: Rheology in the microcirculation in normal and low flow states. Adv Shock Res 8:71–80, 1982. [PubMed] [Google Scholar]
- 39.Curry N, Rourke C, Davenport R, Beer S, Pankhurst L, Deary A, Thomas H, Llewelyn C, Green L, Doughty H, et al. : Early cryoprecipitate for major haemorrhage in trauma: a randomised controlled feasibility trial. Br J Anaesth 115(1):76–83, 2015. [DOI] [PubMed] [Google Scholar]
- 40.Scott EA, Puca KE, Pietz BC, DuChateau BK, Friedman KD: Comparison and stability of ADAMTS13 activity in therapeutic plasma products. Transfusion 47(1):120–125, 2007. [DOI] [PubMed] [Google Scholar]
- 41.Plautz WE, Matthay ZA, Rollins-Raval MA, Raval JS, Kornblith LZ, Neal MD: Von Willebrand factor as a thrombotic and inflammatory mediator in critical illness. Transfusion 60 suppl 3:S158–S166, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kumar MA, Cao W, Pham HP, Raju D, Nawalinski K, Maloney-Wilensky E, Schuster J, Zheng XL: Relative deficiency of plasma a disintegrin and metalloprotease with thrombospondin type 1 repeats 13 activity and elevation of human neutrophil peptides in patients with traumatic brain injury. J Neuro-trauma 36(2):222–229, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kumar M, Cao W, McDaniel JK, Pham HP, Raju D, Nawalinski K, Frangos S, Kung D, Zager E, Kasner SE, et al. : Plasma ADAMTS13 activity and von Willebrand factor antigen and activity in patients with subarachnoid haemorrhage. Thromb Haemost 117(4):691–699, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Harhangi BS, Kompanje EJO, Leebeek FWG, Maas AIR: Coagulation disorders after traumatic brain injury. Acta Neurochir (Wien) 150(2):165–175, 2008. [DOI] [PubMed] [Google Scholar]
- 45.Wafaisade A, Lefering R, Tjardes T, Wutzler S, Simanski C, Paffrath T, Fischer P, Bouillon B, Maegele M: Acute coagulopathy in isolated blunt traumatic brain injury. Neurocrit Care 12(2):211–219, 2009. [DOI] [PubMed] [Google Scholar]
- 46.Maegele M: Coagulopathy after traumatic brain injury: incidence, pathogenesis, and treatment options. Transfusion 53:28S–37S, 2013. [DOI] [PubMed] [Google Scholar]
- 47.Stein SC, Smith DH: Coagulopathy in traumatic brain injury. Neurocrit Care 1(4):479–488, 2004. [DOI] [PubMed] [Google Scholar]
- 48.Lustenberger T, Talving P, Kobayashi L, Inaba K, Lam L, Plurad D, Demetriades D: Time course of coagulopathy in isolated severe traumatic brain injury. Injury 41(9):924–928, 2010. [DOI] [PubMed] [Google Scholar]
- 49.Tian Y, Salsbery B, Wang M, Yuan H, Yang J, Zhao Z, Wu X, Zhang Y, Konkle BA, Thiagarajan P, et al. : Brain-derived microparticles induce systemic coagulation in a murine model of traumatic brain injury. Blood 125(13):2151–2159, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu Y, Liu W, Zhou Y, Hilton T, Zhao Z, Liu W, Wang M, Yeon J, Houck K, Thiagarajan P, et al. : von Willebrand factor enhances microvesicle-induced vascular leakage and coagulopathy in mice with traumatic brain injury. Blood 132(10):1075–1084, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu X, Wang C, Wu Y, Houck KL, Hilton T, Zhou A, Wu X, Han C, Yang M, Yang W, et al. : Conformation-dependent blockage of activated VWF improved outcomes of traumatic brain injury in mice. Blood; 2020. Online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Padilla A, Moake JL, Bernardo A, Ball C, Wang Y, Arya M, Nolasco L, Turner N, Berndt MC, Anvari B, et al. : P-selectin anchors newly released ultralarge von Willebrand factor multimers to the endothelial cell surface. Blood 103(6):2150–2156, 2004. [DOI] [PubMed] [Google Scholar]
- 53.Huang J, Roth R, Heuser JE, Sadler JE: Integrin alpha(v)beta(3) on human endothelial cells binds von Willebrand factor strings under fluid shear stress. Blood 113(7):1589–1597, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fasipe TA, Hong SH, Da Q, Valladolid C, Lahey MT, Richards LM, Dunn AK, Cruz MA, Marrelli SP: Extracellular vimentin/VWF (von Willebrand factor) interaction contributes to VWF string formation and stroke pathology. Stroke 49(10):2536–2540, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li Y, Choi H, Zhou Z, Nolasco L, Pownall HJ, Voorberg J, Moake JL, Dong J-F: Covalent regulation of ULVWF string formation and elongation on endothelial cells under flow conditions. J Thromb Haemost 6(7):1135–1143, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Engelman DT, Gabram SG, Allen L, Ens GE, Jacobs LM: Hypercoagulability following multiple trauma. World J Surg 20:5–10, 1996. [DOI] [PubMed] [Google Scholar]
- 57.Di Battista AP, Rizoli SB, Lejnieks B, Min A, Shiu MY, Peng HT, Baker AJ, Hutchison MG, Churchill N, Inaba K, et al. : Sympathoadrenal activation is associated with acute traumatic coagulopathy and endotheliopathy in isolated brain injury. Shock 46(3 suppl 1):96–103, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rodriguez EG, Cardenas JC, Cox CS, Kitagawa RS, Stensballe J, Holcomb JB, Johansson PI, Wade CE: Traumatic brain injury is associated with increased syndecan-1 shedding in severely injured patients. Scand J Trauma Resusc Emerg Med 26(1):102, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Johansson PI, Stensballe J, Rasmussen LS, Ostrowski SR: A high admission syndecan-1 level, a marker of endothelial glycocalyx degradation, is associated with inflammation, protein C depletion, fibrinolysis, and increased mortality in trauma patients. Ann Surg 254(2):194–200, 2011. [DOI] [PubMed] [Google Scholar]
- 60.Haywood-Watson RJ, Holcomb JB, Gonzalez EA, Peng Z, Pati S, Park PW, Wang W, Zaske AM, Menge T, Kozar RA: Modulation of syndecan-1 shedding after hemorrhagic shock and resuscitation. PLoS One 6(8):e23530, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Scully M, Cataland SR, Peyvandi F, Coppo P, Knöbl P, Kremer Hovinga JA, Metjian A, de la Rubia J, Pavenski K, Callewaert F, et al. : Caplacizumab treatment for acquired thrombotic thrombocytopenic purpura. N Engl J Med 380(4):335–346, 2019. [DOI] [PubMed] [Google Scholar]
- 62.Peyvandi F, Scully M, Kremer Hovinga JA, Cataland S, Knöbl P, Wu H, Artoni A, Westwood J-P, Mansouri Taleghani M, Jilma B, et al. : Caplacizumab for acquired thrombotic thrombocytopenic purpura. N Engl J Med 374(6):511–522, 2016. [DOI] [PubMed] [Google Scholar]
- 63.Zhou Y, Cai W, Zhao Z, Hilton T, Wang M, Yeon J, Liu W, Zhang F, Shi F-D, Wu X, et al. : Lactadherin promotes microvesicle clearance to prevent coagulopathy and improves survival of severe TBI mice. Blood 131(5):563–572, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hanayama R, Tanaka M, Miwa K, Shinohara A, Iwamatsu A, Nagata S: Identification of a factor that links apoptotic cells to phagocytes. Nature 417(6885):182–187, 2002. [DOI] [PubMed] [Google Scholar]