

Abstract
Protein citrullination (or deimination), an irreversible post-translational modification, has been implicated in several physiological and pathological processes, including gene expression regulation, apoptosis, rheumatoid arthritis, and Alzheimer’s disease. Several research studies have been carried out on citrullination under many conditions. However, until now, challenges in sample preparation and data analysis have made it difficult to confidently identify a citrullinated protein and assign the citrullinated site. To overcome these limitations, we generated a mouse hyper-citrullinated spectral library and set up coordinates to confidently identify and validate citrullinated sites. Using this workflow, we detect a four-fold increase in citrullinated proteome coverage across six mouse organs compared with the current state-of-the art techniques. Our data reveal that the subcellular distribution of citrullinated proteins is tissue-type-dependent and that citrullinated targets are involved in fundamental physiological processes, including the metabolic process. These data represent the first report of a hyper-citrullinated library for the mouse and serve as a central resource for exploring the role of citrullination in this organism.
Keywords: posttranslational modification, protein database, citrullination, mass spectrometry
Graphical Abstract
INTRODUCTION
Citrullinated proteins are generated upon the deimination of arginine into the nonstandard amino acid citrulline. The reaction is catalyzed by Ca2+-dependent protein arginine deiminase (PAD), a family consisting of five enzyme isoforms.1 This irreversible post-translational modification (PTM) leads to a small increase in mass of 0.9840 Da and the loss of a positive charge (pI ∼11.41 for arginine versus pI ∼5.91 for citrulline).2 The loss of positive charge can have a substantial effect on the overall charge distribution, isoelectric point, and hydrophobicity.1 Therefore, citrullination can cause the destabilization of hydrogen-bond formation and can affect the interaction with other amino acid residues of the same protein, thus altering the protein structure.3 Citrullination can also impact charged interactions with other proteins that result in the alteration of cell signaling,4,5 immune response,6–8 and gene regulation.9,10 Moreover, this protein modification can generate neoepitopes, thus causing the formation of neo-(auto)antigens, and after presentation to autoreactive B and T cells, an immune response initiation event is triggered.11,12 Despite the use of biochemical enrichment and antibody detection methods,13,14 relatively few unambiguously assigned endogenous citrullinated residues have been identified.15 Whereas anticitrulline antibodies can detect protein citrullination, the definition of the exact site of modification, especially in global screens, is best suited for mass spectrometry (MS) approaches.
The fundamental challenges for mapping endogenous citrullinated sites are (1) robust detection of citrullination and (2) quantification of the PTM. Each current approach, as discussed below, has limitations, making it technically challenging to quantify citrullination and thus reducing the number of investigations in which citrullination is studied. Even though citrullinated proteins can be characterized directly by tandem mass spectrometry (MS/MS) based on the 0.9840 Da mass difference between citrulline and arginine, the same mass increment of 0.9840 Da for modified arginine (citrullination) frequently occurs for deamidation of the amino acid asparagine (Asn/N) and glutamine (Gln/Q) residues. Moreover, the potential for substoichiometric levels of this modification16 could lead to more interference during precursor ion isolation and result in incorrect assignments of modified amino acids (i.e., coisolation of other species), which lead to mixed or contaminated MS/MS spectra. An extra complication in these analyses is the 13C isotope event; especially when in the database search, the parent mass tolerance is more than 5 parts per million (ppm). Another alternative is to combine reversed-phase liquid chromatography (LC) to separate the peptides with CID MS/MS. CID of citrullinated peptides produces fragment ion series corresponding to b (−43 Da) and y (−43 Da) ions.17–19 However, all citrullinated peptides should produce the −43 Da ion series, and if more than one arginine residue exists in the peptide, then a full ion series (all b and y ions of −43 Da) is required to localize the residue position of citrulline within a peptide. This approach was taken recently by Lee et al., where 375 citrullination sites on 209 human proteins were identified and validated by a data-mining approach.19
Another MS approach is a chemical tagging strategy, which allows for the selective pinpointing of citrullinated peptides in a complex mixture after LC–MS analysis.20,21 The tagging reaction is based on a covalent reaction between 2,3-butanedione and citrulline residues, which results in a 50 Da shift in singly charged mass. Similarly, chemical tagging with 2,3-butanedione and antipyrine (238 Da shift)22 or 4-hydroxyphenylglyoxal (132 Da shift)23 has been developed; however, sensitivity and lack of specificity are major limitations for these technologies. As an alternative approach, Bicker et al. developed a chemical probe, rhodamine-phenylglyoxal, which is based on the reaction between glyoxals and either citrulline or arginine residue under acidic or basic conditions.14 The methodology requires the reduction of sample complexity by immunoprecipitation and gel electrophoresis to increase label specificity. Furthermore, the Rh-PG labeling is performed in 20% trichloroacetic acid at 37 °C, which may cause protein precipitation and loss of material during the sample preparation steps. The limited enrichment strategies24–26 all mentioned above are not specific and sensitive enough to be used in complex mixtures. Raijmakers et al. used a different approach and characterized citrullinated proteins present in the synovial fluid of rheumatoid arthritis (RA) patients based on trypsin’s inability to cut after citrullinated arginine residue, as trypsin should no longer cleave after a neutral amino acid. In this case, the “missed cleavage” will be the read out for citrullination.27,28 However, the protease performance is never 100% efficient; therefore, a low level of nonspecific cleavage and missed cleavage are typically observed. In particular, C-terminal citrulline residues are more commonly observed, as has been shown by Zhao et al.29,30 Additionally, peptides with a parent charge higher than two (often a reduction of the charge) should be seen for the citrullinated peptide, as discussed by Raijmakers.27 The existence of potential false-positives demands the careful manual verification of MS/MS spectra of each candidate peptide. As a result, the analysis is very time-consuming and labor-intensive and is not practical for large-scale proteomic studies.
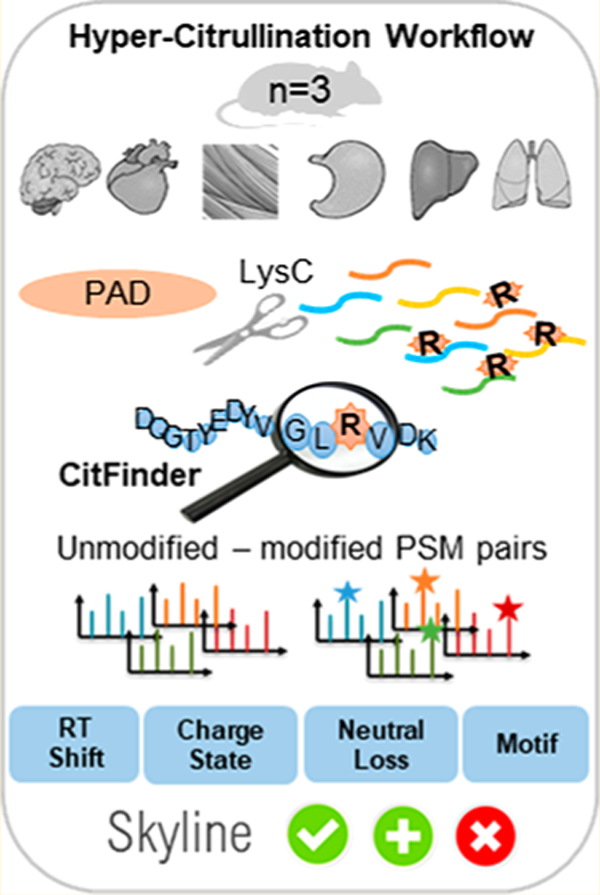
Here we present a novel approach for the analysis of citrullination by the generation of a generic large-scale mouse hyper-citrullinated spectral library that can be used to enable the robust detection of citrullinated peptides using DIA–MS. The step-by-step library generation workflow is based on spectral matching of modified peptides to their noncitrullinated forms, along with the delta retention time shift (ΔRT), as a signature for citrullination. The validation steps of citrullinated peptides include the detection of the neutral loss of isocyanic acid in peptides in collision-induced dissociation (CID) spectra and Skyline validation. It is further demonstrated that the created library reveals a complementary citrullinome expression profile that supports each tissue’s unique physiology. The library has been available as a resource for SWATH or data-independent acquisition (DIA) analysis at SWATHAtlas.org to aid the analysis of existing biological data and to inspire future biological investigations.
METHODS
Citrullinated Peptide Standards
Standard peptides: synthetic peptides composed of the amino acid sequences SAVRARSSVPGVR, SAVRA[Cit]SSVPGVR, and SAV[Cit]A[Cit]SSVPGVR were obtained from GenScript and solubilized in water/0.1% formic acid (v/v) as a neat solution (aliquoted and stored at −80 °C). Peptides (66 fmol) were loaded on a nanoLC 425 HPLC system (SCIEX) operating in microflow mode. Peptides were first loaded onto a trap column (10 × 0.3 mm, C18CL, 5 μm, 120 Å, SCIEX) for 3 min at 10 μL/min of solvent A (0.1% formic acid [FA] in water), followed by separation on an analytical column (ChromXP C18CL, 150 × 0.3 mm, 3 μm, 120 Å, SCIEX) at a flow rate of 5 μL/min using a linear A/B gradient of 3–35% solvent B (0.1% FA in acetonitrile (ACN)) for 60 min, 35–85% B for 2 min, holding at 85% B for 5 min, then re-equilibrating at 3% B for 7 min. Peptides were analyzed using a TripleTOF 6600 system (SCIEX) operating in data-dependent acquisition (DDA) mode. A MS1 scan covering 400–1250 m/z was acquired for 250 ms, followed by MS/MS of the top 30 precursor ions. The source voltage was set to 5500 V, Gas 1 was set to 15, Gas 2 was set to 20, curtain gas was set to 25, and the source temperature was set to 100 °C.
Sample Preparation for Hyper-Citrullinated Spectral Libraries
Wild-type mice (C57BL/6, 12 weeks old) were sacrificed, and the following organs, brain (n = 3), heart (n = 3), liver (n = 3), lung (n = 3), kidney (n = 2), and skeletal muscle (n = 3), were immediately harvested, washed in cold PBS buffer, and snap-frozen in liquid nitrogen. The tissues from each animal were independently and mechanically homogenized using a 7 mm steel grinding ball (Retsch) and lysed with 0.1% RapiGest/50 mM Tris/150 mM NaCl/protein inhibitor buffer for 60 cycles at 30 000 psi using a NEP 2320 barocycler (Pressure Biosciences, South Easton, MA). After centrifugation for 15 min at 14 000 rpm at 4 °C, the protein concentration in each supernatant was quantified using the Pierce BCA protein assay kit (Thermo Scientific). Each sample (200 μg) was divided into two tubes (twin samples). One twin sample was treated with PAD cocktail (cocktail of the five PAD isoforms: PAD1, PAD2, PAD3, PAD4, and PAD6; 1:20 ratio, SignalChem), whereas the second sample was treated with H2O at the same ratio. All samples were incubated in deimination buffer (100 mM Tris, 10 mM CaCl2, 5 mM DTT) for 3 h at 37 °C. The reaction was stopped by the addition of EDTA to a final concentration of 5 mM. The samples were subsequently reduced with 10 mM DTT (Sigma-Aldrich) for 60 min at 55 °C and alkylated with 15 mM 2-iodoacetamide (AppliChem) for 30 min at room temperature in the dark. LysC protease (1:50 enzyme/sample ratio) was added to each sample and incubated at 37 °C for 4 h. An additional portion of LysC was added (1:50 ratio) and the incubation was continued at 37 °C for 18 h. All samples were cleaned on an Oasis HLB plate (Waters) prior to LC–MS analysis.
Liquid Chromatography–Mass Spectrometry for the Citrullination Data-Dependent Acquisition (DDA–MS) Library
The separation of LysC digests of mouse organ tissue samples was performed on a NanoLC 425 System (SCIEX) operating in trap elute mode at microflow rates on a hybrid triple quadrupole time-of-flight mass spectrometer 6600 (TripleTOF 6600). A sample was loaded onto a 0.3 × 10 mm trap cartridge and washed with mobile phase A for 3 min at 10 μL/min. The trap valve was then switched, and the sample was eluted off the trap through a 0.3 × 150 cm column using a 100 min gradient (3–35% solvent B, mobile phase A: 100% water in 0.1% FA; mobile phase B: 100% ACN in 0.1% FA in water) at 5 μL/min (total run time 120 min). Both the column and the trap were packed with ChromXP C18CL 3 μm, 120 Å media (SCIEX). Three to six μL of sample was injected, and each cycle consisted of a 250 ms TOF MS scan, followed by 30 MS/MS scans at 100 ms each, resulting in a total cycle time of 3.3 s. Rolling collision energy for peptides was used along with a collision energy spread of 5. Precursors between m/z of 400 and 1250 were selected for the majority of DDA–MS runs acquired for the library generation. All samples injected contained a peptide standard for retention time calibration, as previously described by Escher.31
Generation of Ion Library Generation via DDA–MS Analysis
All raw files were converted to mzML format using the SCIEX Data Converter (in Protein Pilot mode) and then reconverted to mzXML format using ProteoWizard v.3.0.6002 for peak list generation.32,33 The MS/MS spectra were queried against the reviewed canonical Swiss-Prot mouse complete proteome database as of February 28, 2018 and appended with iRT protein sequence and shuffled sequence decoys.34 All data were searched using X!Tandem Native v.2013.06.15.1, X!Tandem Kscore v.2013.06.15.1,35 and Comet v.2014.02 rev.2.36 The search parameters included the following criteria: static modifications of Carbamidomethyl (C) and variable modifications of Oxidation (M), Deamidation (NQ), and Citrullination (R). The parent mass tolerance was set to be 50 ppm, and the monoisotopic fragment mass tolerance was 100 ppm (which was further filtered to be <0.05 Da for building the spectral library); LysC peptides with up to two missed cleavages were allowed. The identified peptides were processed and analyzed through Trans-Proteomic Pipeline v.4.833 and were validated using the PeptideProphet37 scoring, and the PeptideProphet results were statistically refined using iProphet.38 All of the peptides were filtered at a false discovery rate (FDR) of 1% with a peptide probability cutoff of ≥0.99. The raw spectral libraries were generated from all valid peptide spectrum matches and then refined into the nonredundant consensus libraries39 using SpectraST v.4.0.40 All citrullinated peptides and their unmodified forms were analyzed in pairs for physiochemical properties such as delta retention time shift (ΔRT), charge state, and neutral loss. (See Supplemental Table 1.) In cases where the unmodified peptide forms were absent, other modified forms (e.g., oxidized, deamidated, or singly/doubly citrullinated peptide forms) were used to represent the modified–unmodified peptide pairs.
Skyline Validation
The final step was quality control of the peak groups performed using Skyline-daily version 42. In brief, chromatograms were extracted for the +2, +3, +4, and +5 charged precursors of each peptide through 50–2000 m/z range. For each peptide precursor, chromatograms were extracted for the precursor, precursor [M+1], and precursor [M+2] ions from the MS data. Endoproteinase Lys-C with a maximum of two missed cleavages and a minimum peptide length of 6 to a maximum peptide length up to 50 residues was used. Additionally, Isotope dot product, Retention Time Drift, Total Area, Average Mass Error, And Full Width at Half Maximum were used to verify the correct chromatographic peak integration into the following three categories: “Good”, “Okay”, and “Bad”. (See Supplemental Figure 2 for examples.) Chromatographic Peak Groups with Confirmed Peptide Identity, Isotope Dot Product ≥ 0.9, Retention Time Drift ≤ ±0.2, Total Area ≥ 100 000, TotalArea/FWHM ≥ 1 000 000, and Average Mass Error PPM ≤ ±5 were defined as “Good”. Chromatographic Peak Groups with Confirmed Peptide Identity and meeting four of the following five quality measures, Isotope Dot Product ≥ 0.7, Retention Time Drift ≤ ±0.4, Total Area ≥ 10 000, TotalArea/FWHM ≥ 100 000, and Average Mass Error PPM ≤ ±10, were defined as “Okay”. All other Peak Groups that did not meet the above thresholds or had poor peak quality measures such as Isotope Dot Product < 0.7 or Retention Time Drift ≥ ±1 were automatically flagged as “Bad” and discarded from any further analyses. (See Supplemental Table 1.)41,42
Bioinformatics Analysis
The sequence logo, R-based package, and ggseqlogo were used to investigate and visualize the amino acid consensus sequence of the citrullinated sites, which were organ-specific. On the basis of the list of validated citrullinated peptides, the amino acid frequencies ranging from −10 to +10 residues around the citrullination site were analyzed with respect to its relative frequency at a given position.43 The GO annotation proteome was derived from http://www.pantherdb.org,44 and the significantly changing proteins were classified by GO annotation based on two categories: molecular function and biological process. Proteins containing citrullinated residues were mapped to KEGG pathways using STRING (https://string-db.org/), and the whole genome was used as background. Subcellular localization analysis was performed using Functional Enrichment analysis tool, FunrichR, version 3.0.
The MS proteomics data have been deposited to SWATHAtlas.org and can be accessed at http://www.peptideatlas.org/PASS/PASS01334. The algorithm can be accessed at https://github.com/Citrullinome/CitFinder.
Single Nucleotide Polymorphism Analysis
The single nucleotide polymorphism (SNP) information (protein accession number and SNP ID) of the detected citrullinated proteins gene was retrieved from the UniProt database, and the amino acid sequence homologies of the residues around the citrullinated sites were compared between human and mice.
RESULTS
Hyper-Citrullinated Library Workflow
Figure 1 shows the overall workflow of the hyper-citrullinated library. The novelty of the workflow is how we apply the characteristics of citrullinated residues (ΔRT shift, neutral loss, and Skyline validation) into the data-processing steps. The ΔRT shift that occurs with citrullination was investigated using commercially available non-, mono-, and double-citrullinated synthetic peptides. The synthetic peptides were pooled and analyzed on a TripleTOF 6600 apparatus, and the samples were eluted with either of the two following gradients: Gradient 1: linear gradient from 3–35% phase B solution; Gradient 2: linear gradient from 20 to 35% phase B solution over increasing gradient lengths (45 to 120 min) (n = 3, technical MS runs). The ΔRT shifts between the elution of the non-, mono-, and double-citrullinated peptides are shown in Figure 2. For example, the ΔRT between the monocitrullinated versus the noncitrullinated peptide for Gradient 1, over a 45 min run (0.711%/min), was 2.655 min (1.88% ACN), whereas the ΔRT between the double-citrullinated and the monocitrullinated peptide was 3.125 min (2.22% ACN) (Figure 2c). Slowing down the gradient improved the peptide resolution. The ΔRT between monocitrullinated and noncitrullinated peptides for Gradient 2 over 45 min (0.333%/min) was 5.308 min (1.75% ACN), whereas the ΔRT between the double-citrullinated and the monocitrullinated peptide was 4.778 min (1.57% ACN). Thus our analysis corroborates the previous finding that a modified peptide elutes later due to the change of the peptide charge and hydrophobicity. On the basis of the information, we decided to use a linear gradient of acetonitrile (3–35% over 120 min, 1.9% ACN monocitrullinated versus the noncitrullinated peptide, and 1.7% for double-citrullinated and the monocitrullinated peptide) to target citrullinated peptides in complex biological samples. To validate our results, we explored the gas-phase fragmentation pathways of citrullinated peptides by electrospray tandem mass spectrometry, where we looked for a characteristic neutral loss of 43 Da from an isocyanic acid (HN=CO) moiety from the citrulline ureido group.17 We first acquired MS/MS and on-the-fly neutral-loss-triggered MS/MS/MS spectra for the commercially available mono- and double-citrullinated peptide and compared its fragmentation behavior with the arginine-containing counterpart (Supplemental Figure 1).
Figure 1.
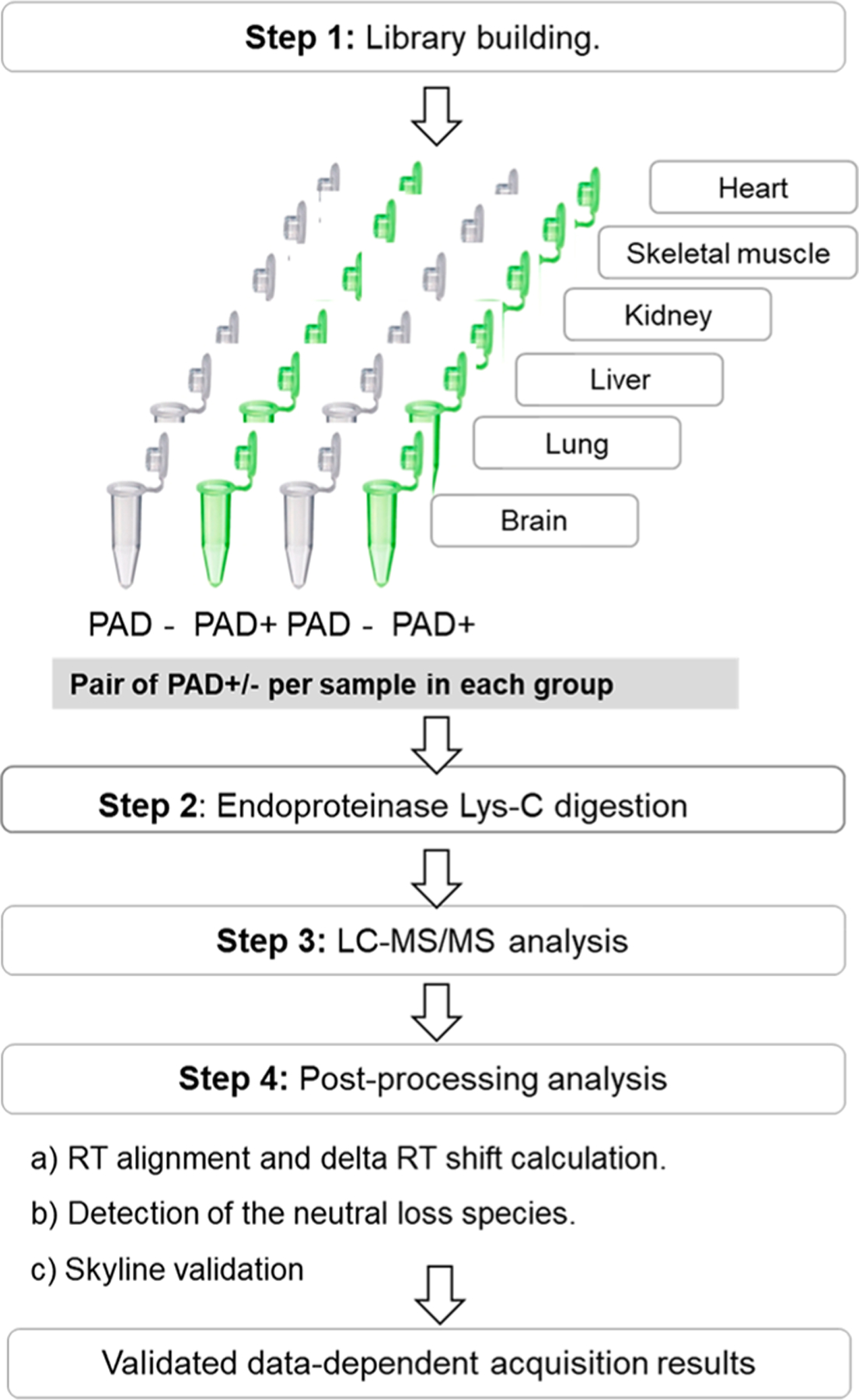
Flowchart outlining the workflow for the hyper-citrullinated library generation pipeline. A total of six tissues were isolated from three male wild-type mice. The tissues were snap-frozen, homogenized, and solubilized, and the total protein concentration was determined using a BCA protein assay kit. Step 1. Library building. In brief, each sample was split into two samples; one was treated with PAD cocktail (five isoforms, ratio 1:20) to cover all potential citrullinated targets. The corresponding second sample was treated with water under the same condition. Step 2. Digestion with LysC (ratio 1:20), followed by LC–MS/MS (TripleTOF 5600). Step 3. Postprocessing analysis with algorithm that examines the MS runs for the evidence of citrullinated site and Skyline validation.
Figure 2.
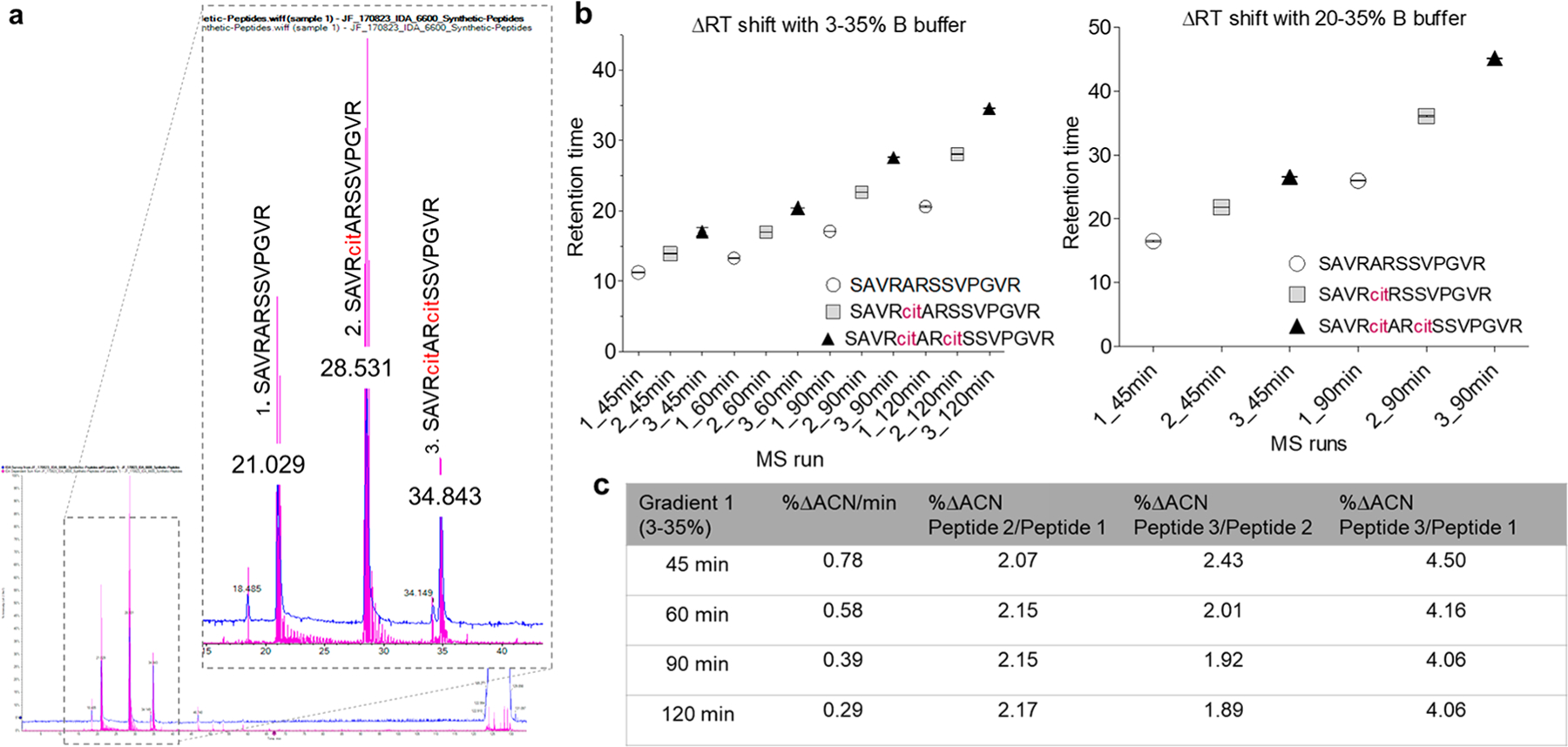
Overview of the ΔRT shift based on the recombinant citrullinated peptides. (a) Base-peak chromatograms of nonmodified, monomodified, and double-modified citrullinated peptide with (b) different times and gradients of ACN. Each peptide was run three times under the same condition (c) Table of ΔRT calculations between each peptide with final gradients of ACN from 3 to 35% at different time increments.
Mass Spectrometry Hyper-Citrullinated Library Generation and ΔRT Shift Application
To ensure the maximal usability of DDA–MS data for the detection of citrullinated peptides, we created a hyper-citrullinated library. We propose three major changes to the common spectral library generation pipeline, as outlined in Figure 1. First, to enhance the viability of the citrullinated peptides in the library, 200 μg of protein per each organ and biological replicate was treated with PAD enzyme (a cocktail of the five PAD isoforms: PAD1, PAD2, PAD3, PAD4, and PAD6) to cover all potentially citrullinated targets. Second, LysC was used for the digestion of proteins to minimize type-I errors and miscleavages caused by modified peptides. Third, an iRT alignment is critical because the identification of citrullinated peptides is based on a comparison between modified and unmodified peptide pairs and the ΔRT shift between these peptides. Modified peptides will only be identified if they are derived from an already identified, unmodified peptide. Importantly, all forms of the peptide should be incorporated in the ΔRT shift analysis, including potentially deamidated N/Q residues and all charge states. We developed an algorithm to apply the ΔRT rule and to accurately select the modified peptides. Even though chromatographic behaviors of peptides can be complicated and different LC conditions can utilize a different modification status, our study demonstrated that the ΔRT shift for citrullinated peptides is constant and additive for each citrullination site on a peptide (Figure 3a). In addition, we also collected information on the MS3 analysis of the neutral loss species eliminated from citrulline peptides (see −43 Da peak in Supplemental Figure 1). We confirmed that out of the total 3026 validated peptides, 2950 citrullination peptides (∼98%) were detected with at least one neutral loss ion of the isocyanic acid (Figure 3b and Supplemental Table 1). Furthermore, we checked if modified peptides with a parent charge higher than two experienced the reduction of the charge, as suggested by Raijmakers et al.27 Our analysis showed that >21% of the identified citrullinated peptides have not lost charge state due to the modification (Figure 3c). Further validation of the citrullinated peptides was performed using Skyline-daily version 4.2. with detailed examination of peak prosperity using our algorithm. On the basis of the chromatographic peak groups in Skyline, we removed an additional 195 bad peptides (6%) that did not meet the chromatographic peak criteria. 36% of the citrullinated peptides were classified as Good, 58% citrullinated were classified as Okay.
Figure 3.
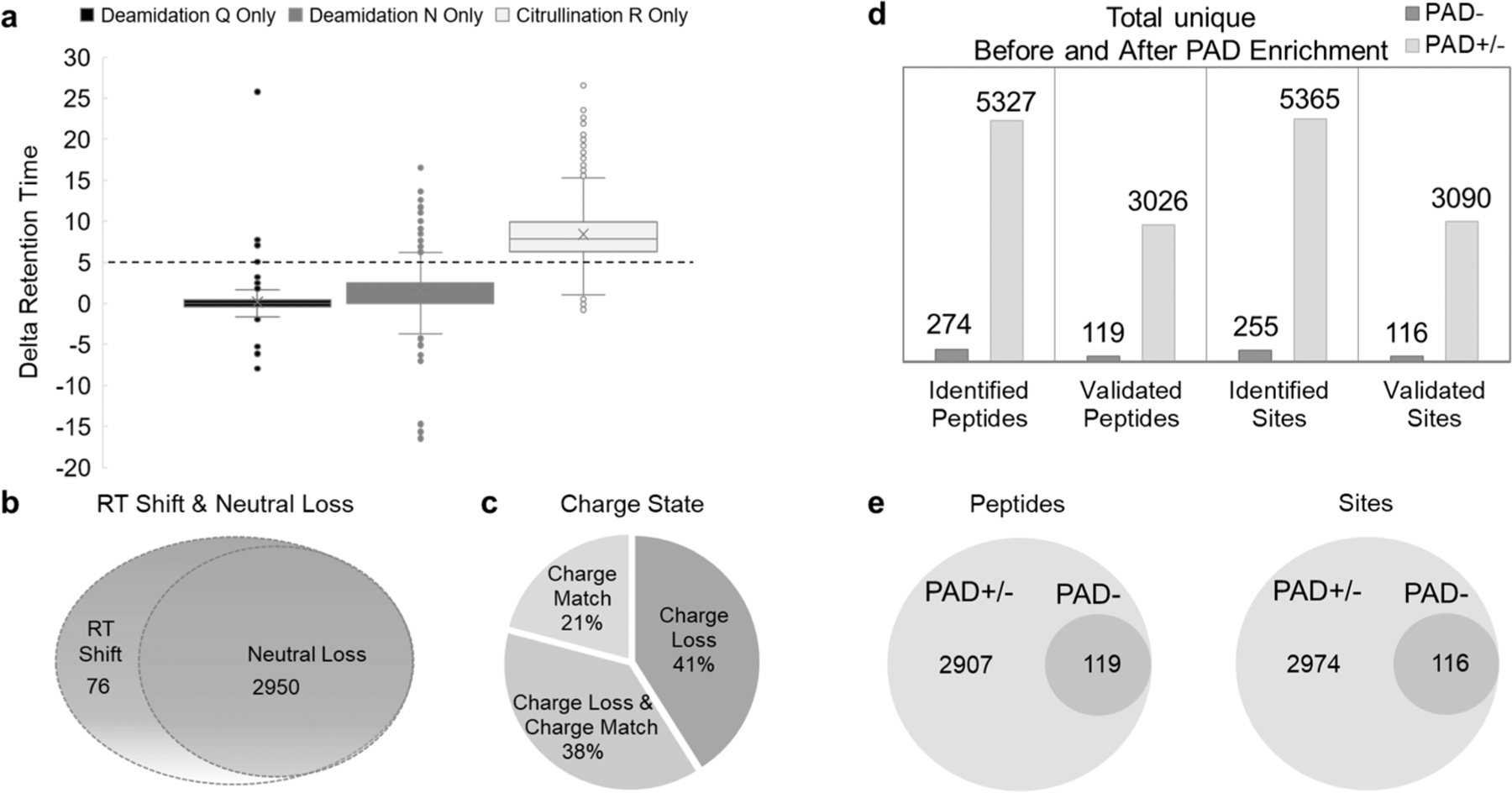
Physicochemical properties of citrullinated peptides. (a) Comparing the differences in ΔRT, modified to unmodified pairs for peptides containing deamidated amino acid asparagine (N), amino acid glutamine (Q), and citrullinated amino acid arginine (R). (b) Neutral loss distribution of 3026 validated peptides with a 5 min ΔRT shift. A peptide was considered to have a neutral loss if at least one product ion on MS/MS/MS spectra showed loss of HNCO (43 Da). (c) Charge distribution in 3026 validated peptides with a 5 min ΔRT shift. Prior to checking the charge state loss from peptides with a charge state higher than 3 observed with citrullination, we compared the charge status of all peptide match pairs. Only peptides that have match pairs were considered. (d) Bar graph summarizes the number of citrullinated peptides and sites identified with the enrichment method before and after validation. (e) Venn diagrams showing the distribution of identified citrullinated peptides and citrullinated residues across the hyper-citrullinated library (PAD±) and PAD− library.
In summary, combining the enrichment step with the PAD cocktail incubation and validation algorithm led to the validation of 3090 citrullination sites on 1037 mice proteins. The 25-fold increase in the citrullinated proteome of our hyper-citrullinated library is a promising enrichment method to study the full extent of cellular protein citrullination. Further analysis and validation steps improve the specificity and sensitivity of citrullination identification and confirmation (Figures 3d and 4 and Supplemental Table 1).
Figure 4.
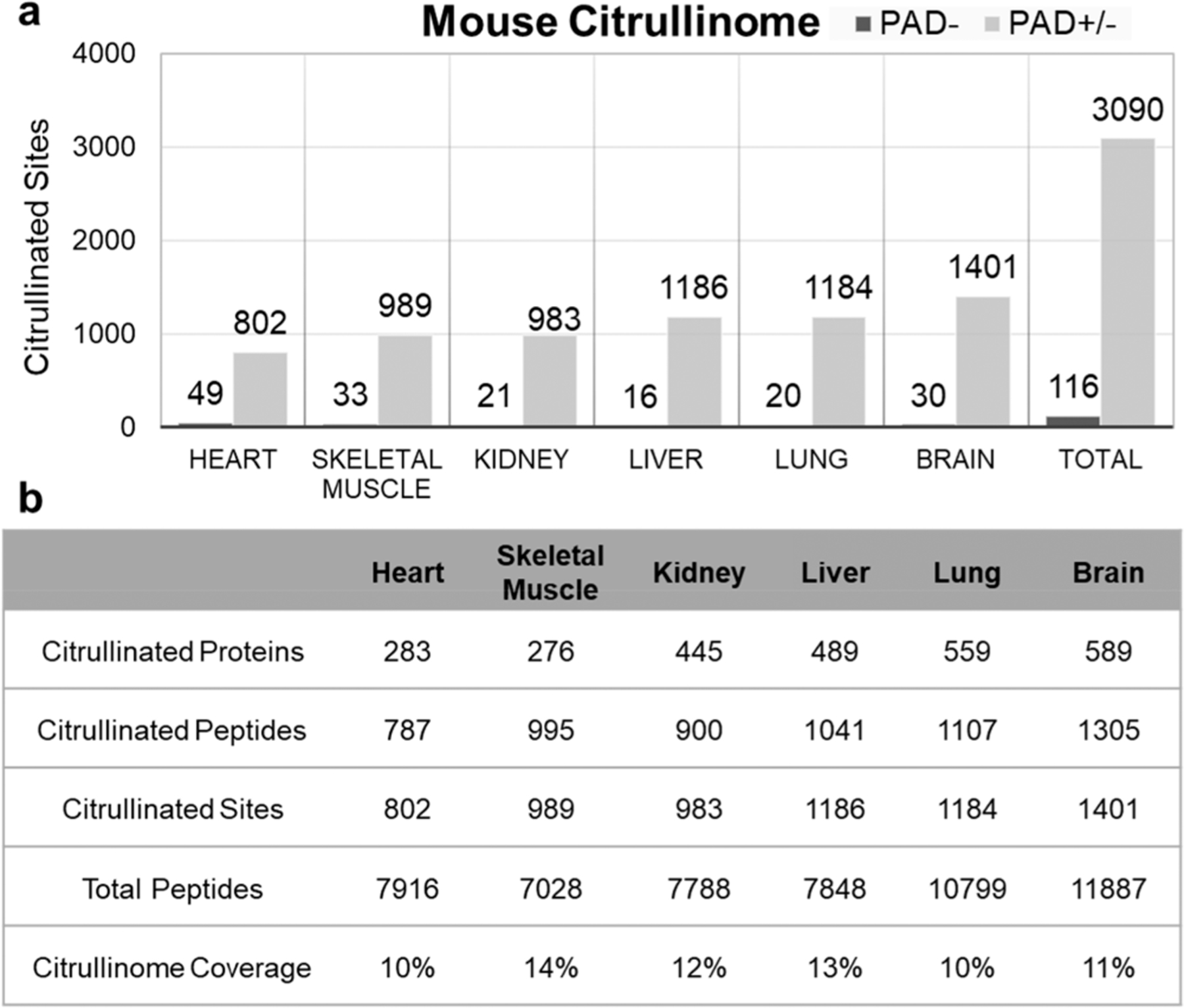
Tissue distribution of citrullinated sites in the hyper-citrullinated mouse library. (a) Distribution of citrullinated sites across the mouse organ. (b) Numbers of citrullinated proteins, peptides, and sites per organ are provided in the table together with the total number of proteins detected per organ.
Distribution of Citrullinated Sites Across Mouse organs
Over 90% of observed sites and citrullinated proteins were previously unreported in the literature (Supplemental Table 1). The lowest numbers of citrullinated sites were identified in the heart, falling short of 802 total citrullinated residues compared with 1401 total citrullinated sites in the most abundant organ, the brain. The kidney, the lung, and the liver each contained ∼500 citrullinated proteins and ∼1100 localized residues (Figure 4). Subcellular localization analysis revealed the stoichiometric distribution of citrullinated peptides when analyzing proteins from different cellular compartments (Figure 5a). The study showed that ∼20% of citrullinated proteins were in the cytoplasm. Furthermore, some proteins were predicted to be distributed in the mitochondrion (3–23%), membrane (16–24%), and nucleus (8–14%) depending on the organ (Figure 5a). To better understand the citrullination, GO functional classification of all citrullinated proteins was investigated based on their molecular functions. The largest group of citrullinated proteins consists of enzymes that are associated with catalytic activity (41.4%) and proteins related to binding (37.6%) in the molecular function classification, and others were assigned to structural molecular activity (9.1%), transporter activity (5.7%), and molecular function regulator (4.6%) (Figure 5b). These data together suggest that citrullination has a unique cellular diversity and specialized signaling networks.
Figure 5.
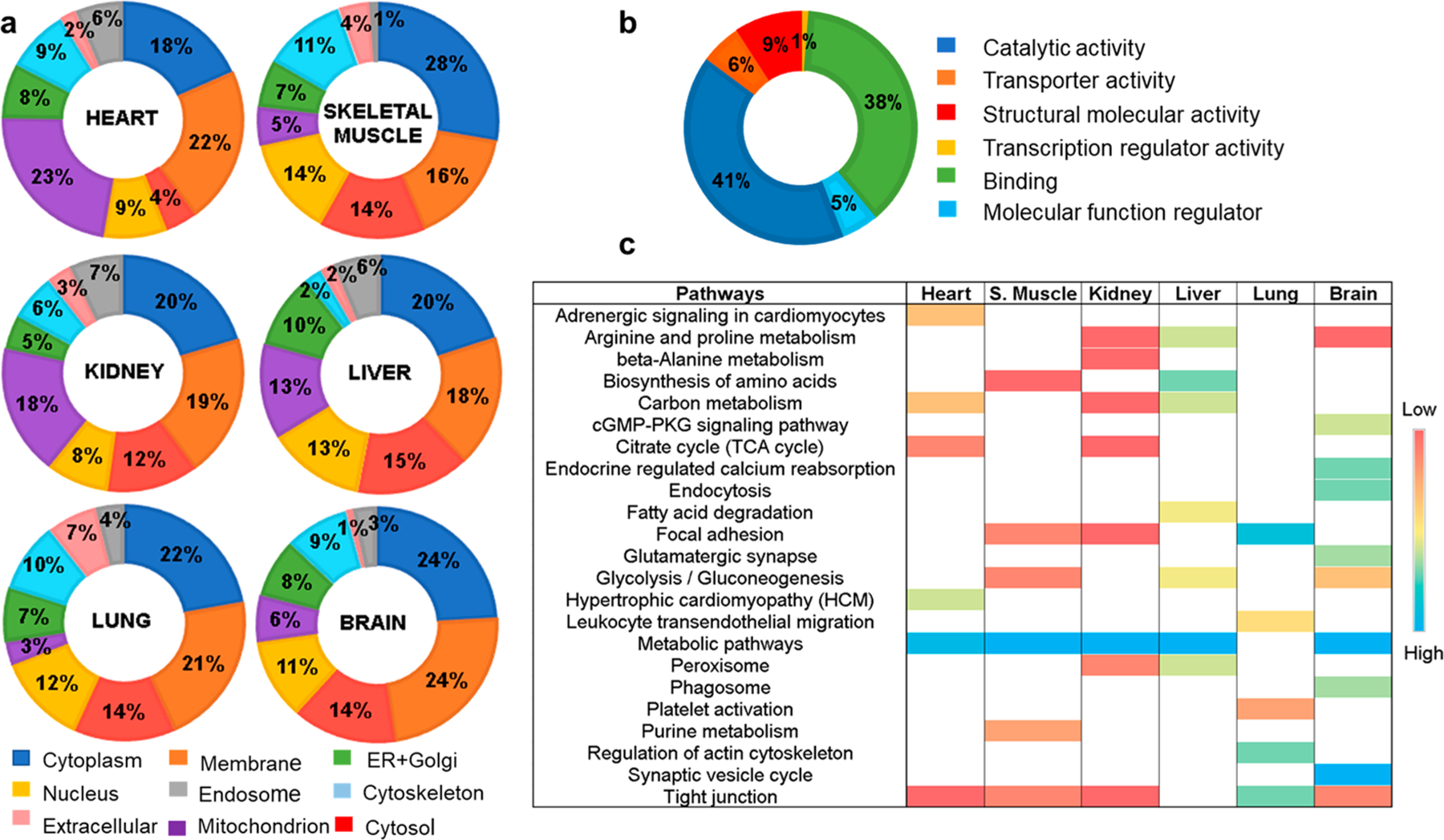
Functional classification of citrullinated proteins in the hyper-citrullinated mouse library. (a) Classification of the citrullinated proteins based on subcellular localization. (b) Classification of the citrullinated proteins based on molecular function. (c) Classification of the citrullinated proteins based on the KEGG enrichment pathway for each organ.
Clustering of Citrullinated Proteins on Pathway Analysis
To explore the differences underlying the tissue-specific patterns of citrullinated proteins, we performed gene ontology and KEGG pathway enrichment analyses for each tissue. The hierarchical clustering presented in Figure 5c shows pathways where proteins carrying citrullinated residue(s) were identified. For example, the citrullinated proteins that are more abundant or exclusive in the brain are primarily involved in neuronal signal transmission (e.g., glutamatergic synapses or synaptic vesicle cycle), whereas the specific citrullinated proteins of the kidney and liver are enriched for proteins involved in the metabolism of fatty acids and amino acids (Figure 5c). Interestingly, as evident from the hierarchical clustering, there is one major process, the metabolic pathway, where citrullinated proteins are predominate in five out of the six tissues characterized. To investigate the biological under-pinnings for this process, we focused on a protein–protein interaction network of citrullinated proteins. We found that oxidoreductases and glycolytic enzymes are the major class of enzymes citrullinated across the analyzed tissues (data not shown). Other pathways are more specific for each organ.
Conserved Citrullination Amino Acid Sequence Motifs
To disentangle the influence of local amino acid content around the citrullination, tissue-based consensus sequence motifs were analyzed for all of the verified citrullinated sites identified in our tissue database. Recent studies have shown that different PAD isoforms have different restrictions in the selection of arginine substrates. For example, a study by Stensland et al. indicated that approximately one-fifth of the PAD4 substrates contained an RG/RGG motif in human tissue and plasma.45,46 We did not find this consensus sequence for any of the analyzed tissues; however, we found that the glutamic acid residues are enriched on the N-terminus of the citrullinated site in heart and lung tissue, whereas preference for lysine residues on the N-terminus of the citrullinated site was found in muscle tissue (Supplemental Figure 3). Discovering the tissue-specific preferences of citrullinated sites is important, as citrullination has been found to be critical in many physiological and pathological processes.1,4,47–49 However, the procedure used in our workflow required treatment with all PAD isoforms together. The simplification of the treatment came with the limitation that it does not allow for the investigation of structural trends and site-specific motifs for each PAD enzyme. The identification of the compartment-specific sequence motifs with individual PAD isoforms should be performed to confirm PAD isoform specificity, as described by others.46
DISCUSSION
False-positive results are always a concern in MS analysis of PTMs, especially when limitations in available methodology to robustly enrich, detect, and localize the modification exist. As a result, even though citrullination has been implicated under many physiological and pathological conditions,1 there is a limited number of citrullination sites that have been identified with high confidence.19,48–50 To assist the research community in mapping out the citrullinated proteins in their studies, we developed this workflow and present a generic large-scale mouse hyper-citrullinated spectral library for high-confidence citrullinated peptide identification. The workflow incorporates automated elements to cope with library quality and false-positives. We identified a total of 3026 citrullinated peptides in 1037 citrullinated proteins, which are involved in a wide variety of biological processes. These citrullinated proteins are localized to multiple cellular compartments and belong to diverse functional groups, suggesting that arginine citrullination plays important roles in regulating numerous cellular processes in mouse. Interestingly, the distribution of citrullinated proteins is tissue-specific. The database of citrullinated peptides reported in this study, combined with the tissue-centric KEGG pathway analysis of citrullinated proteins, showed involvement in multiple physiological functions with which the modification has not previously been associated. The largest number of citrullinated proteins was found in the brain, which is consistent with previous studies.51 Interestingly, lung tissue is the second most enriched organ for citrullinated proteins in our database, exhibiting a different citrullinome pattern compared with all other tissues. The tissue is enriched in citrullinated proteins associated with extracellular exosomes and proteins involved in local adhesion, some major signal mediators between cells, and shuttling cargo in health and disease. Our data also showed that the kidney is the third organ most enriched in the detected citrullinated proteins. Previous publications confirmed the presence of PAD2 and PAD4 in this organ as well as the presence of citrullinated proteins.52 These data represent the first comprehensive view of the citrullinome in multiple organs of mouse. Moreover, many of the arginine residues, found to be citrullinated in our database, have been previously identified as a component of several SNPs (Supplemental Table 2). 79 citrullinated sites were mapped to known SNPs in the UniProt database. Most often when this occurred, the SNP was a missense mutation and was replaced with an amino acid with neutral charge, similar to what happens when arginine is citrullinated. This suggests that changing arginine into citrulline could mimic the disease-inducing effects of these missense variants and thus influence biological processes by inducing protein structure disorders,3 disrupting protein–protein or protein–macromolecules interactions,4 or affecting other PTMs.53 For example, R212Q mutation on actin (ACTA2; actin alpha 2, aortic smooth muscle; P62736) may be mimicked by citrullination on the same residue (R212) that was identified in our study. Alteration of this site was found to be involved in aortic aneurysm, familial thoracic 6 ((AAT6) [MIM:611788]: (dbSNP:rs397516685)). Likewise, the missense mutation on Enoyl-CoA hydratase (ECHS1; Enoyl-CoA hydratase, mitochondrial UniProtKB - P30084), R54H (dbSNP:rs375266808), causes mitochondrial encephalopathy in newborns and was also found to be citrullinated in our database. Future work remains to model these variations.
To contrast our results with those from available resources, we compared citrullinated peptides in our database to citrullinated peptides identified by Lee et al., where 375 citrullination sites on 209 human proteins were validated.19 We observed an overlap of only 34 sites between mouse and human data (Supplemental Figure 4). Taking a closer look at the 341 citrullinated sites unique to human, we found that 179 sites were associated with proteins not detected in our mouse data set, 91 sites were not conserved between mouse and human, and 71 sites were conserved, yet undetected, in mouse. Additionally, a large proportion of the missing sites could also be explained by the fact that out of the total 209 human citrullinated proteins associated with the 375 sites, 38 proteins were secreted, a part of the mouse proteome that was not included in our study.
Better knowledge of PAD substrates and the effect of citrullination on their functions would improve our knowledge and understanding of biological and pathological processes that are citrullination-based and in the future will help to develop better diagnostics and treatments. Notably, a recent study showed that citrullination inactivates nicotinamide-N-methyl-transferase (NNMT), an enzyme that uses S-adenosyl methionine (SAM) as a cofactor to catalyze the N-methylation of nictotinamide (NAM) to form N-methyl nicotinamide (MeNAM).48 Likewise, an example from Tilvawala et al. study showed that serpin citrullination abolishes its ability to inhibit its cognate proteases and consequently modulates serpin-regulated pathways including the fibrinolysis, complement pathway, and cell motility.47
In summary, our workflow using the hyper-citrullinated spectral library provides a rich resource of candidates for hypothesis generation and will open new avenues for large-scale investigations of citrullinated proteins in clinical research. 90% of the validated citrullinated residues and proteins have not been previously reported. In addition, the citrullination data set shows that most citrullinated proteins are tissue-specific and that the dominant cellular function/compartments may also differ. Finally, these data provide evidence that citrullinated proteins may be involved in multiple physiological functions with which the modification has not been previously associated.
Supplementary Material
Figure S1. Neutral loss of HNCO from model citrullinated peptides. Figure S2. Schematic demonstration on how to define citrullinated and noncitrullinated peptide sequence in the Skyline. Figure S3. Properties of arginine citrullinated sites. Figure S4. Comparison of two citrullinated data sets in human and mouse (PDF)
Table S1. List of detected citrullinated sites across the mouse organs (TXT)
Table S2. UniProt list of genes that have at least one SNP at the arginine residue that matches the residue identified in our hyper-citrullinated library (TXT)
ACKNOWLEDGMENTS
We thank Sarah J. Parker and Irina Tchernyshyov for discussions and help with the workflow. The study was funded by the National Institutes of Health (National Institute of Arthritis and Musculoskeletal and Skin Diseases: R01 AR050026-12A1; National Heart, Lung, and Blood Institute: R01HL111362).
ABBREVIATIONS
- ACN
acetonitrile
- Asn/N
amino acid asparagine
- Gln/Q
amino acid glutamine
- ACPAs
anticitrullinated protein antibodies
- Arg
arginine amino acid
- CID
collision-induced dissociation
- DDA–MS
data-dependent acquisition mass spectrometry
- DIA–MS
data-independent acquisition mass spectrometry
- Da
Dalton
- FA
formic acid
- LC
liquid chromatography
- Lys-C
endoproteinase
- PTM
post-translational modification
- PAD
protein arginine deiminase
- RA
rheumatoid arthritis
- RT
retention time
- iRT
internal retention time standard
- MS
mass spectrometry
- Cit
citrullinated amino acid
- SNP
single nucleotide polymorphism
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jproteome.9b00118.
The authors declare no competing financial interest.
The mass spectrometry proteomics data have been deposited to SWATHAtlas.org and can be accessed at http://www.peptideatlas.org/PASS/PASS01334. The algorithm can be accessed at https://github.com/Citrullinome/CitFinder. All other data supporting the findings of this study are available from the corresponding author on request.
REFERENCES
- (1).Gyorgy B; Toth E; Tarcsa E; Falus A; Buzas EI Citrullination: a posttranslational modification in health and disease. Int. J. Biochem. Cell Biol 2006, 38, 1662–1677. [DOI] [PubMed] [Google Scholar]
- (2).Orgován G; Noszál B The complete microspeciation of arginine and citrulline. J. Pharm. Biomed. Anal 2011, 54, 965–971. [DOI] [PubMed] [Google Scholar]
- (3).Ordonez A; et al. Effect of citrullination on the function and conformation of antithrombin. FEBS J 2009, 276, 6763–6772. [DOI] [PubMed] [Google Scholar]
- (4).Stadler SC; et al. Dysregulation of PAD4-mediated citrullination of nuclear GSK3beta activates TGF-beta signaling and induces epithelial-to-mesenchymal transition in breast cancer cells. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 11851–11856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Sun B; et al. Citrullination of NF-kB p65 enhances its nuclear localization and TLR-induced expression of IL-1β and TNFα. Science immunology 2017, 2, eaal3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Shelef MA; et al. Peptidylarginine Deiminase 4 Contributes to Tumor Necrosis Factor α–Induced Inflammatory Arthritis. Arthritis Rheumatol 2014, 66, 1482–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Sohn DH; et al. Local joint inflammation and histone citrullination provides a murine model for the transition from preclinical autoimmunity to inflammatory arthritis. Arthritis Rheumatol 2015, 67, 2877–2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Romero V; et al. Immune-mediated pore-forming pathways induce cellular hypercitrullination and generate citrullinated auto-antigens in rheumatoid arthritis. Sci. Transl. Med 2013, 5, 209ra150–209ra150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Li P; et al. Coordination of PAD4 and HDAC2 in the regulation of p53-target gene expression. Oncogene 2010, 29, 3153–3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Li P; et al. Regulation of p53 target gene expression by peptidylarginine deiminase 4. Molecular and cellular biology 2008, 28, 4745–4758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Travers TS; et al. Extensive Citrullination Promotes Immunogenicity of HSP90 through Protein Unfolding and Exposure of Cryptic Epitopes. J. Immunol 2016, 197, 1926–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Valesini G; Gerardi MC; Iannuccelli C; Pacucci VA; Pendolino M; Shoenfeld Y; et al. Citrullination and autoimmunity. Autoimmun. Rev 2015, 14, 490–497. [DOI] [PubMed] [Google Scholar]
- (13).Hensen SMM; Pruijn GJM Methods for the Detection of Peptidylarginine Deiminase (PAD) Activity and Protein Citrullination. Mol. Cell. Proteomics 2014, 13, 388–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Bicker KL; Subramanian V; Chumanevich AA; Hofseth LJ; Thompson PR Seeing citrulline: development of a phenylglyoxal-based probe to visualize protein citrullination. J. Am. Chem. Soc 2012, 134, 17015–17018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Hagiwara T; Hidaka Y; Yamada M Deimination of histone H2A and H4 at arginine 3 in HL-60 granulocytes. Biochemistry 2005, 44, 5827–5834. [DOI] [PubMed] [Google Scholar]
- (16).Verheul MK; et al. Pitfalls in the detection of citrullination and carbamylation. Autoimmun. Rev 2018, 17, 136–141. [DOI] [PubMed] [Google Scholar]
- (17).Hao G; et al. Neutral loss of isocyanic acid in peptide CID spectra: a novel diagnostic marker for mass spectrometric identification of protein citrullination. J. Am. Soc. Mass Spectrom 2009, 20, 723–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Jin Z; et al. Identification and characterization of citrulline-modified brain proteins by combining HCD and CID fragmentation. Proteomics 2013, 13, 2682–2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Lee CY; et al. Mining the human tissue proteome for protein citrullination. Mol. Cell. Proteomics 2018, 17, 1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).De Ceuleneer M; et al. Modification of citrulline residues with 2,3-butanedione facilitates their detection by liquid chromatography/mass spectrometry. Rapid Commun. Mass Spectrom 2011, 25, 1536–1542. [DOI] [PubMed] [Google Scholar]
- (21).Stensland M; Holm A; Kiehne A; Fleckenstein B Targeted analysis of protein citrullination using chemical modification and tandem mass spectrometry. Rapid Commun. Mass Spectrom 2009, 23, 2754–2762. [DOI] [PubMed] [Google Scholar]
- (22).Holm A; et al. Specific modification of peptide-bound citrulline residues. Anal. Biochem 2006, 352, 68–76. [DOI] [PubMed] [Google Scholar]
- (23).Choi M; Song J-S; Kim H-J; Cha S; Lee EY Matrix-assisted laser desorption ionization–time of flight mass spectrometry identification of peptide citrullination site using Br signature. Anal. Biochem 2013, 437, 62–67. [DOI] [PubMed] [Google Scholar]
- (24).Clancy KW; Weerapana E; Thompson PR Detection and identification of protein citrullination in complex biological systems. Curr. Opin. Chem. Biol 2016, 30, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Tutturen AE; Holm A; Fleckenstein B Specific biotinylation and sensitive enrichment of citrullinated peptides. Anal. Bioanal. Chem 2013, 405, 9321–9331. [DOI] [PubMed] [Google Scholar]
- (26).Tutturen AEV; Holm A; Jørgensen M; Stadtmüller P; Rise F; Fleckenstein B; et al. A technique for the specific enrichment of citrulline-containing peptides. Anal. Biochem 2010, 403, 43–51. [DOI] [PubMed] [Google Scholar]
- (27).Raijmakers R; et al. Elevated levels of fibrinogen-derived endogenous citrullinated peptides in synovial fluid of rheumatoid arthritis patients. Arthritis research & therapy 2012, 14, R114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Bennike T; Lauridsen KB; Olesen MK; Andersen V; Birkelund S; Stensballe A Optimizing the Identification of Citrullinated Peptides by Mass Spectrometry: Utilizing the Inability of Trypsin to Cleave after Citrullinated Amino Acids. J. Proteomics Bioinf 2013, 6, 288–295. [Google Scholar]
- (29).Vandermarliere E; Mueller M; Martens L Getting intimate with trypsin, the leading protease in proteomics. Mass Spectrom. Rev 2013, 32, 453–465. [DOI] [PubMed] [Google Scholar]
- (30).Zhao X; et al. Circulating immune complexes contain citrullinated fibrinogen in rheumatoid arthritis. Arthritis research & therapy 2008, 10, R94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Escher C; et al. Using iRT, a normalized retention time for more targeted measurement of peptides. Proteomics 2012, 12, 1111–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Chambers MC; et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol 2012, 30, 918–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Keller A; Eng J; Zhang N; Li XJ; Aebersold R A uniform proteomics MS/MS analysis platform utilizing open XML file formats. Mol. Syst. Biol 2005, 1 (2005), E1–E8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Elias JE; Gygi SP Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 2007, 4, 207–214. [DOI] [PubMed] [Google Scholar]
- (35).Craig R; Beavis RC TANDEM: matching proteins with tandem mass spectra. Bioinformatics 2004, 20, 1466–1467. [DOI] [PubMed] [Google Scholar]
- (36).Eng JK; Jahan TA; Hoopmann MR Comet: an open-source MS/MS sequence database search tool. Proteomics 2013, 13, 22–24. [DOI] [PubMed] [Google Scholar]
- (37).Keller A; Nesvizhskii AI; Kolker E; Aebersold R Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem 2002, 74, 5383–5392. [DOI] [PubMed] [Google Scholar]
- (38).Shteynberg D; Deutsch EW; Lam H; Eng JK; Sun Z; Tasman N; Mendoza L; Moritz RL; Aebersold R; Nesvizhskii AI; et al. iProphet: multi-level integrative analysis of shotgun proteomic data improves peptide and protein identification rates and error estimates. Mol. Cell. Proteomics 2011, 10 (12), M111.007690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Collins BC; et al. Quantifying protein interaction dynamics by SWATH mass spectrometry: application to the 14–3–3 system. Nat. Methods 2013, 10, 1246–1253. [DOI] [PubMed] [Google Scholar]
- (40).Lam H; et al. Development and validation of a spectral library searching method for peptide identification from MS/MS. Proteomics 2007, 7, 655–667. [DOI] [PubMed] [Google Scholar]
- (41).Bereman MS; MacLean B; Tomazela DM; Liebler DC; MacCoss MJ The development of selected reaction monitoring methods for targeted proteomics via empirical refinement. Proteomics 2012, 12, 1134–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Urisman A; et al. An Optimized Chromatographic Strategy for Multiplexing In Parallel Reaction Monitoring Mass Spectrometry: Insights from Quantitation of Activated Kinases. Mol. Cell. Proteomics 2017, 16, 265–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Wagih O ggseqlogo: a versatile R package for drawing sequence logos. Bioinformatics 2017, 33, 3645–3647. [DOI] [PubMed] [Google Scholar]
- (44).Mi H; Muruganujan A; Casagrande JT; Thomas PD Large-scale gene function analysis with the PANTHER classification system. Nat. Protoc 2013, 8, 1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Tanikawa C; et al. Citrullination of RGG Motifs in FET Proteins by PAD4 Regulates Protein Aggregation and ALS Susceptibility. Cell Rep 2018, 22, 1473–1483. [DOI] [PubMed] [Google Scholar]
- (46).Stensland ME; Pollmann S; Molberg O; Sollid LM; Fleckenstein B Primary sequence, together with other factors, influence peptide deimination by peptidylarginine deiminase-4. Biol. Chem 2009, 390, 99–107. [DOI] [PubMed] [Google Scholar]
- (47).Tilvawala R; Nguyen SH; Maurais AJ; Nemmara VV; Nagar M; Salinger AJ; Nagpal S; Weerapana E; Thompson PR; et al. The Rheumatoid Arthritis-Associated Citrullinome. Cell Chemical Biology 2018, 25, 691–704. e696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Nemmara VV; et al. Citrullination Inactivates Nicotinamide-N-methyltransferase. ACS Chem. Biol 2018, 13, 2663–2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Lazarus RC; et al. Protein Citrullination: A Proposed Mechanism for Pathology in Traumatic Brain Injury. Frontiers in neurology 2015, 6, 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Sipila KH; et al. Joint inflammation related citrullination of functional arginines in extracellular proteins. Sci. Rep 2017, 7, 8246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Pritzker LB; Nguyen TA; Moscarello MA The developmental expression and activity of peptidylarginine deiminase in the mouse. Neurosci. Lett 1999, 266, 161–164. [DOI] [PubMed] [Google Scholar]
- (52).Feng D; et al. Citrullination preferentially proceeds in glomerular Bowman’s capsule and increases in obstructive nephropathy. Kidney Int 2005, 68, 84–95. [DOI] [PubMed] [Google Scholar]
- (53).Clancy KW; et al. Citrullination/Methylation Crosstalk on Histone H3 Regulates ER-Target Gene Transcription. ACS Chem. Biol 2017, 12, 1691–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Neutral loss of HNCO from model citrullinated peptides. Figure S2. Schematic demonstration on how to define citrullinated and noncitrullinated peptide sequence in the Skyline. Figure S3. Properties of arginine citrullinated sites. Figure S4. Comparison of two citrullinated data sets in human and mouse (PDF)
Table S1. List of detected citrullinated sites across the mouse organs (TXT)
Table S2. UniProt list of genes that have at least one SNP at the arginine residue that matches the residue identified in our hyper-citrullinated library (TXT)