

Abstract
Inhibiting the aggregation of amyloid peptides with endogenous peptides has broad interest due to their intrinsically high biocompatibility and low immunogenicity. Here, we investigated the inhibition mechanism of the prostatic acidic phosphatase fragment SEVI (semen-derived enhancer of viral infection) against Aβ42 fibrillization using atomistic discrete molecular dynamics simulations. Our result revealed that SEVI was intrinsically disordered with dynamic formation of residual helices. With a high positive net charge, the self-aggregation tendency of SEVI was weak. Aβ42 had a strong aggregation propensity by readily self-assembling into β-sheet-rich aggregates. SEVI preferred to interact with Aβ42, rather than SEVI themselves. In the hetero-aggregates, Aβ42 mainly adopted β-sheets buried inside and capped by SEVI in the outer layer. SEVI could bind to various Aβ aggregation species – including monomers, dimers, and proto-fibrils – by capping the exposed β-sheet elongation edges. The aggregation processes Aβ42 from the formation of oligomers to conformational nucleation into fibrils and fibril growth should be inhibited as their β-sheet elongation edges being occupied by the highly charged SEVI. Overall, our computational study uncovered the molecular mechanism of experimentally observed inhibition of SEVI against Aβ42 aggregation, providing novel insights into the development of therapeutic strategies against Alzheimer’s disease.
Graphical Abstract
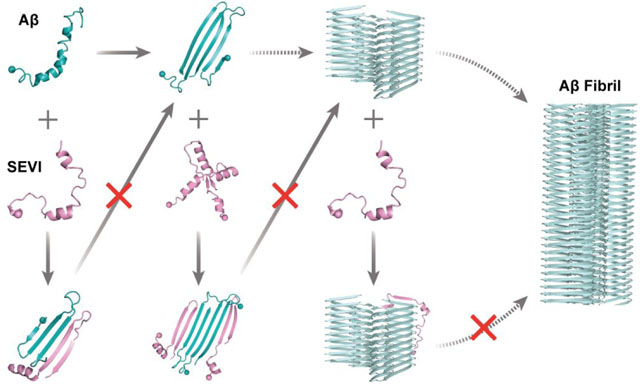
Introduction
The abnormal misfolding and aggregation of proteins to form β-sheet-rich amyloid deposits is a common pathology in numerous neurodegenerative diseases1–3, including amyloid-β (Aβ) and Tau in Alzheimer’s disease (AD)4 and α-synuclein in Parkinson’s disease (PD)5. Despite differences in primary, secondary and tertiary structures of these amyloidogenic proteins (e.g., Aβ, Tau, α-synuclein, and human amylin), mounting experimental studies have established that the amyloid fibrils share similar cross-β core structures with in-registered β-strands within each proto-filament aligned perpendicular to the fibril axis1, 6, 7. In addition, all the amyloid disease-related peptides feature a common nucleation–growth self-assembly kinetics, where monomers first nucleate into soluble oligomers and β-sheet-rich aggregates before their rapid elongation into proto-fibrils and saturation of mature fibrils8–10. Increasing evidence reveals that soluble low-molecular-weight oligomers formed during the early aggregation stage are much more cytotoxic than the mature fibrils9, 11, 12. Prior numerous studies have demonstrated that inhibiting the fibrillization of the amyloid peptide could effectively mitigate aggregation-mediated cytotoxicity13–16. Therefore, the inhibition of pathological fibrillization of amyloid proteins is considered as a promising strategy for the future cure of amyloid diseases.
To mitigate the cytotoxicity of amyloidosis, the modulation of amyloid aggregation by naturally-occurring small-molecules (e.g., EGCG15–17, dopamine18–20, and resveratrol21, 22), nanoparticles (e.g., graphene oxide quantum dot23–26, fullerene derivative14, 27–29, MoS230, 31), and peptides or proteins (e.g., αB-crystallin protein32–34, antimicrobial α-defensins35) has been widely studied in vitro, in vivo, and in silico. For example, the presence of these amyloid inhibitors15–26 could not only inhibit amyloid fibrillization but also suppress the cytotoxicity of amyloid peptides. Although there have been reports of various compounds, including polyphenols14–17, 36, 37 and inorganic nanoparticles23–26, 30, 31 effectively inhibiting the pathological aggregation of amyloid proteins (e.g. Aβ, hIAPP, and tau), their potential side effects and pharmacological efficacy are still unknown38. The endogenous proteins32–34, 39, featuring amyloid-inhibiting effects against pathological aggregation of amyloid proteins have attracted broad interest due to their intrinsic biocompatibility, biological origin, and low immunogenicity. For example, αB-crystallin, widely expressed in the human body (including the brain, retina, and eye lens), could effectively prevent fibrillization and reduce cytotoxicity of multiple amyloid proteins, including α-synuclein, tau, and Aβ32–34. A recent experimental study has shown that the SEVI-fragments (semen-derived enhancer of viral infection) of prostatic acid phosphatase (PAP248–286) could completely inhibit Aβ aggregation at substoichiometric concentrations, prevent the growth of preformed Aβ fibrils, and reduce Aβ-induced cell toxicity39. Various methods for disrupting mature Aβ fibril structures have been reported, including ultrasound wave irradiation40, 41 and infrared laser irradiation42, 43, which have been evaluated through both experimental measurements40, 42 and computational simulations41, 43. Interestingly, it has been found that the presence of SEVI alone is sufficient to destroy Aβ fibril structures39. However, the inhibition mechanism of SEVI against amyloid aggregation of Aβ remains to be fully established. A better understanding of the inhibition mechanism at the molecular level will be helpful for the design of future anti-amyloidosis peptide inhibitors against AD, as well as other amyloid diseases.
Aβ peptides, major constituent of senile plaques in AD, are cleaved off from the amyloid precursor protein by β- and γ-secretases4, 44. Aβ40 and Aβ42 are the two most abundant isoforms in senile plaques, and Aβ42 features higher aggregation propensity and cytotoxicity45. Aβ monomers mainly adopt random coil and partial β-sheet structures in solution and readily aggregate into β-sheet dominated fibril structures in vitro8. SEVI is a representative amyloidogenic fragment derived from peptides of prostatic acid phosphatase (also known as PAP248–286) and is naturally present in human semen46. Similar to Aβ, the self-assembly kinetics of SEVI forming cross-β fibrillar structure also follows the nucleation-dependent elongation mechanism47. The self-assembly of SEVI is strongly sensitive to the factors that affect electrostatic interactions47, 48. Because the SEVI contains many cationic residues, it leads to strong charge repulsion between the monomers. Thus, fibrillization of SEVI requires extensive time and agitation46. The aggregation of SEVI only occurs at neutral pH and high salt concentrations (above ~100 mM), but not in the absence of salt or at acidic pH49. Decreasing the concentration of Zn2+ would result in SEVI fibrils dissociation, indicating the physiologic concentrations of zinc in semen protect the stability of SEVI fibrils50. Interestingly, cross-interaction between Aβ42 and SEVI completely inhibits pathological aggregation and elongation of preformed fibrils of Aβ4239.
To investigate the inhibition mechanism of SEVI against Aβ fibrillization, we systematically studied the interactions between Aβ42 and SEVI by applying multiple long-timescale discrete molecular dynamics (DMD) simulations51, 52. DMD is a rapid and predictive molecular dynamics algorithm widely used to study protein folding and misfolding by both our group53, 54 and others55–57. Our results revealed that SEVI was intrinsically disordered with dynamic formation of helixes and had a significantly weaker self-aggregation tendency compared to Aβ42. Simulations of one SEVI mixed with an Aβ42 showed the two peptides preferred to bind each other and the cross-interaction hot-spots corresponded to residues 14–22&31–39 in SEVI and 10–21&30–41 in Aβ42 (the well-known amyloidogenic core regions8). Co-aggregation simulations of two SEVI and two Aβ42 peptides showed that SEVI preferred interacting with Aβ42 rather than the SEVI themselves. In their hetero-aggregates, Aβ42 mostly formed β-sheets buried inside with β-strand edges capped by SEVI in the outer layer. In simulations of SEVI mixed with preformed Aβ42 proto-fibril, SEVI was observed to both capping to the β-sheet elongation ends and binding to the lateral fibril surfaces. With the highly charged SEVI occupying the elongation surfaces of Aβ monomers, oligomers, and proto-fibrils, their growth to higher molecular weight oligomers, nucleation of proto-fibrils, and rapid fibril elongation should be prevented correspondingly. The binding of SEVI to the Aβ42 fibril lateral surface could also inhibit secondary nucleation58. Together, this study reveals a complete picture of the inhibitory mechanism of Aβ aggregation by the endogenous SEVI protein, providing theoretical insights into the development of novel therapeutic strategies against AD.
Methods and materials
Molecular systems.
The amino acid sequences and initial structures of SEVI (PDB: 2l3h59) and Aβ42 (PDB: 1z0q60) used in our simulations are shown in Figure S1. To investigate the SEVI effects on the amyloid aggregation of Aβ42, multiple molecular systems (summarized in Table S1) were set up for simulations, including one peptide of SEVI and Aβ42 monomers, two peptides of either two SEVI, two Aβ42, or one SEVI mixed with one Aβ42, and four peptides of two SEVI mixed with two Aβ42. For each system, fifty independent DMD simulations were performed, starting with different initial configurations (i.e., coordinates and velocities in multiple-peptide simulations and velocities in the monomeric simulations). Each independent simulation lasted 600 ns in the one-peptide system and 1200 ns in the multiple-peptide simulations. The peptides were initially randomly placed in a cubic simulation box (the corresponding box size of each system was summarized in Table S1) with different orientations and a minimum inter-molecular atomic distance of 1.5 nm. In addition, interactions of SEVI monomer and Aβ42 fibril were also investigated by the simulation of one SEVI mixed with a pre-formed Aβ42 fibril composed of 20 peptides (PDB: 5oqv7, Figure S1). Fifty independent DMD simulations, with each during the time up to 600 ns, were also performed starting from different initial coordinates and velocities. Initially, the SEVI monomer and Aβ42 fibrils were randomly placed in a 12 nm cubic simulation box with a minimum inter-molecular atomic distance of 1.5 nm. The Aβ42 fibril was set static to reduce the computational cost.
Discrete molecular dynamics (DMD) simulations.
All simulations were performed utilizing the atomic DMD with implicit solvent at 300 K. DMD is a rapid and predictive molecular dynamics (MD) algorithm, in which optimized stepwise functions modeled the continuous potential functions in traditional MD51. The step function potentials were adapted from the Medusa force field, which has been well benchmarked for the accurate prediction of protein stability change upon mutation, protein-ligand binding affinity61, 62, as well as ab initio protein folding52. Similar to most traditional MD force fields, both bonded interactions (i.e., covalent bonds, bond angles, and dihedrals) and non-bonded interactions (i.e., van der Waals, solvation, hydrogen bond, and electrostatic terms) were considered in the Medusa force field. Solvation energy was calculated by the effective energy function proposed by Lazaridis and Karplus63. The hydrogen bond was explicitly modeled by a reaction-like algorithm52. The screened electrostatic interactions between charged atoms were computed by the Debye–Hückel approximation with the Debye length assigned of ~10 Å at the physiological condition. DMD software is available to academic researchers via the Molecules In Action, LLC (www.moleculesinaction.com). The units of mass, time, length, and energy used in our simulations were 1 Da, ~50 fs, 1 Å, and 1 kcal/mol, respectively. With a rapid computational speed and enhanced sampling efficiency, DMD has been widely used to study protein folding and aggregation both by our group and others53–57.
Analysis methods.
The secondary structure was calculated using the DSSP (Define Secondary Structure of Protein) method64. A hydrogen bond was considered to be formed once the N···O distance was less than 3.5 Å and the N–H···O angle was larger than 150°65. A pairwise residue contact was defined when the distance between the heavy atoms from two non-sequential sidechain/main chains was within 0.65 nm. Cluster analysis was performed using the Daura algorithm and a backbone atoms deviation cutoff of 0.55 nm66. A two-dimensional (2D) free energy (also known as the potential mean force) surface was constructed using −RT ln P(x, y), where P(x, y) is the probability of a conformation having a certain parameter value of x and y. The radial distribution function g(r) of Cα atom of each peptide corresponding to the complex center was calculated by the following equation g(r) = Nr,r+dr/(4πr2dr), where Nr,r+dr is the number of atoms within distances of r and r+dr away from the center of the complex14.
Results and discussion
Conformational ensembles of SEVI monomers were populated with transiently formed helices, and Aβ42 monomers with dynamic β-sheets.
The conformational dynamics of SEVI and Aβ42 monomers were investigated by fifty independent 600-ns DMD simulations. The conformational sampling efficiency and equilibrium assessments were examined by the structural parameters of the radius gyration (Rg), the number of backbone hydrogen bonds and heavy atom contacts, and the secondary structure content as a function of simulation time (Figures S2&S3). The significant fluctuations without long-terms trends indicated that our long-timescale DMD simulation was not trapped and that sufficient sampling was achieved. Only the last 300 ns simulation data from each independent simulation trajectory was used for the conformational analysis to avoid potential biases from the initial structures.
Time evolution of the secondary structure per residue suggested the SEVI monomer dynamically adopted helical conformations (Figures 1a&S2). Transient β-sheets were also observed in SEVI monomers but were very rare (Figures 1a&S2). The average content of each secondary structure showed that monomeric SEVI was predominantly populated with unstructured and helical conformations with a probability of ~46.8% and ~39.2% (Figure 1b), respectively. The β-sheet content was only ~4.2% (Figure 1b). The result is consistent with prior NMR and CD characterizations, which suggested that the monomeric peptide was dynamic with partial helical conformations in aqueous solution67, 68. The helices of SEVI monomers were mainly formed by residues 4–11 and 18–26, with an average probability up to ~60.0% (Figure 1c). C-terminal residues 27–39 were predominantly unstructured (> 50%). A weak helical tendency of ~30% around residues 33–36 was observed (Figure 1c). The high intra-chain residue-pairwise contact frequencies along the diagonal around residues 4–11, 18–26, and 33–36 were indicative of a high helical propensity in these regions (Figure S4a). Helices in these regions were also found in SEVI monomeric structures under membrane mimic environments determined by the NMR spectroscopy59, 67, 68. Relatively weak intra-chain interactions between residues 15–21 and 32–38 (with contact frequencies less than 0.2) with a contact pattern perpendicular to the diagonal reflected the observation that monomeric SEVI may form a transient β-hairpin structure (Figures 1a&S4a). Prior experimental studies also suggested that only the central and C-terminal regions of SEVI participated in forming β-sheet aggregates69. The conformational free energy landscape estimated by the potential mean force (PMF) along with the probability distribution of overall helix and β-sheet contents demonstrated that monomeric SEVI was much more favorable to form helixes than β-sheets (Figures 1d&S5a). Using clustering analysis of conformational ensembles, the centroid structures of the top 4 most populated SEVI monomer conformations were indeed populated with helices (Figure 1e). SEVI monomer mainly adopted unstructured conformations with partial helixes agreed with prior experimental measurements59, 68.
Figure 1. Conformational dynamics analysis of SEVI and Aβ monomer.
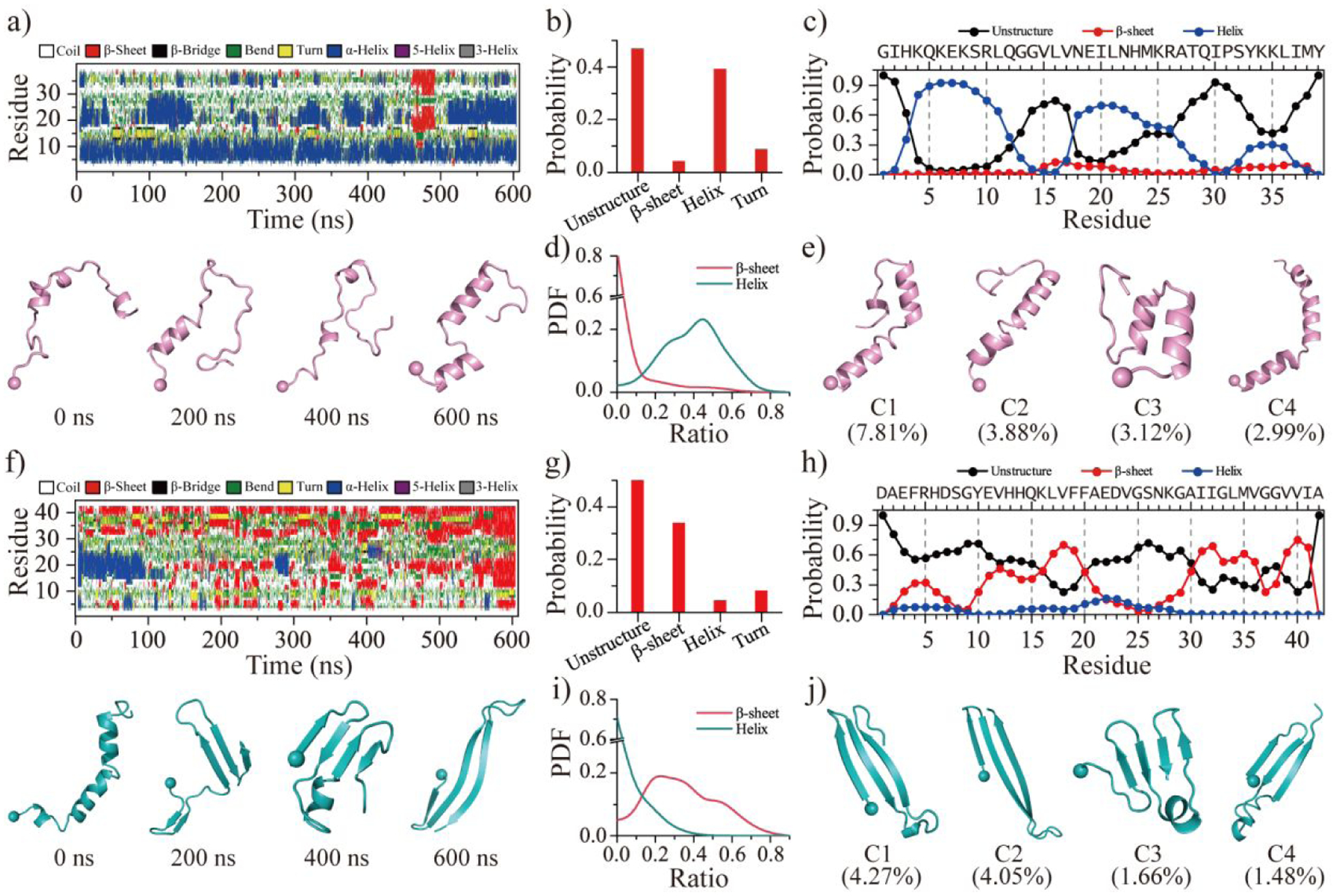
The time evolution of the secondary structure for each residue from SEVI a) and Aβ f). The snapshots of each monomer along the simulation trajectory are also presented every 200 ns. The average secondary structure contents of unstructured (coil and bend), β-sheet, helix, and turn conformations for SEVI b) and Aβ g) monomer during the last 300 ns DMD simulations. The propensity of each residue from SEVI c) and Aβ h) adopted the unstructured (coil and bend), β-sheet, and helix during the last 300 ns. The probability distribution as a function of the secondary structure contents of β-sheet and helix for each SEVI d) and Aβ i) monomer. Representative monomeric conformations of the top four most-populated clusters of SEVI e) and Aβ j). The N-terminal Cα atom is highlighted as a bead. Monomers of SEVI and Aβ are colored pink and cyan, respectively.
In contrast, monomeric simulation of Aβ42 showed that the peptide formed dynamic β-sheets instead of helices (Figures 1f&S3), consistent with FRET and CD spectra experiments70, 71. The main secondary structures of Aβ42 monomer were unstructured coil and β-sheet with a probability of ~52.2% and ~33.9%, respectively (Figure 1g). Helical formations were only ~4.3% formed by residues 20–25 (Figure 1h). Residues 10–21 and 31–41 of Aβ42 displayed strong tendency of forming β-sheets with an average propensity over 50% (Figure 1h), as revealed by prior experimental and simulation studies3, 71, 72. In addition, the N-terminal residues 3–7 also displayed weak β-sheet propensities (~20–25%) (Figure 1h). The intra-chain contact frequency map of Aβ42 featured three contact patterns perpendicular to the diagonal, including residues 2–8 vs 11–17, 11–22 vs 30–41, and 31–34 vs 38–41 correspond to multiple strand-turn-strand motifs (Figure S4b). Another recent study using Hamiltonian replica-permutation molecular dynamics simulations73, 74 for Aβ40 and Aβ42 also found similar β-hairpin motifs75. Interestingly, these β-strands were also present as the cross-β cores in many experimentally-determined Aβ amyloid fibrils7, 76, 77. Residues 1–14 had the highest coil propensity (> 60%) comparing to the rest of the sequence. Due to the highly conformational flexibility, the N-terminal residues 1–10 were found missing in most fibril models77. The conformational free energy landscape, the probability distribution of overall helix and β-sheet contents, and the top 4 most populated conformations of Aβ42 monomer further confirmed that dynamical β-sheets were much more abundant than helices (Figures 1i&1j&S5b). Overall, our simulation showed that both SEVI and Aβ42 were indeed intrinsically disordered with transiently formed residual structures. SEVI monomers preferred to form transient helices, while Aβ42 monomers formed β-sheets.
Contrasting to Aβ42 that readily aggregated into β-sheet oligomers, SEVI displayed a significantly weak self-aggregation propensity but preferred to bind Aβ42.
To investigate the effects of SEVI on the aggregation of Aβ42, dimerization simulation of two SEVI, two Aβ42, and one SEVI mixed with one Aβ42 were performed. For each molecular system, we performed fifty independent DMD trajectories with each simulation lasting 1200 ns. The time evolution of the radius gyration, the number of hydrogen bonds and contacts, and the content of each secondary structure suggested that all the simulations were well equilibrium and reached their steady states in the last 600 ns (Figures S6–S8).
Dimerization dynamics of SEVI featured frequent fluctuations in the inter-peptide backbone hydrogen bonds and atomic contacts with frequent sampling of unbound states, suggesting dimers of SEVI were unstable and easily dissociated into monomers (Figures 2a&S6). The time evolution of the secondary structure per residue demonstrated that the SEVI conformations were populated more with helixes than β-sheets. The conformational free energy landscape was projected in terms of the radius gyration and the number of inter-peptide backbone hydrogen bonds stabilizing inter-peptide β-sheets using the last 600 ns of all fifty independent simulations (Figure 2b). There were three energy basins centered around (1.5, 0), (1.6, 7), and (1.6, 20), respectively. SEVI dimers with fewer inter-peptide hydrogen displayed lower free energy values, indicating that the dimerization tendency of SEVI forming β-sheet-rich aggregates was relatively weak (snapshots 1–3 in Figure 2b). Because SEVI contains many cationic residues, this leads to strong charge repulsion between the monomers when they aggregate. Differently, two Aβ42 peptides readily aggregated into a stable dimer stabilized a large number of inter-chain hydrogen bonds and contacts (Figures 2c&S7). Aβ42 dimers with ~9–15 inter-chain hydrogen bonds were the most populated conformational state, corresponding to the lowest free energy basin (Figure 2d) and indicating that Aβ42 had a strong aggregation tendency of forming β-sheet-rich aggregates. The dynamic association and dissociation observed in the homo-dimerization of SEVI disappeared in the hetero-dimerization of SEVI mixed with Aβ42, in which SEVI readily bound to Aβ42 and formed a stable β-sheets-rich hetero-dimer stabilized inter-peptide hydrogen bonds (Figures 2e&S8). SEVI-Aβ42 hetero-dimers had significantly more inter-peptide hydrogen bonds than SEVI homo-dimer as revealed by comparison of states in the free energy landscapes (Figures 2b&f), indicating the SEVI-Aβ42 cross-interaction was stronger than the self-association of SEVI.
Figure 2. Homo/hetero-dimerization dynamics and free energy landscape of SEVI and Aβ.
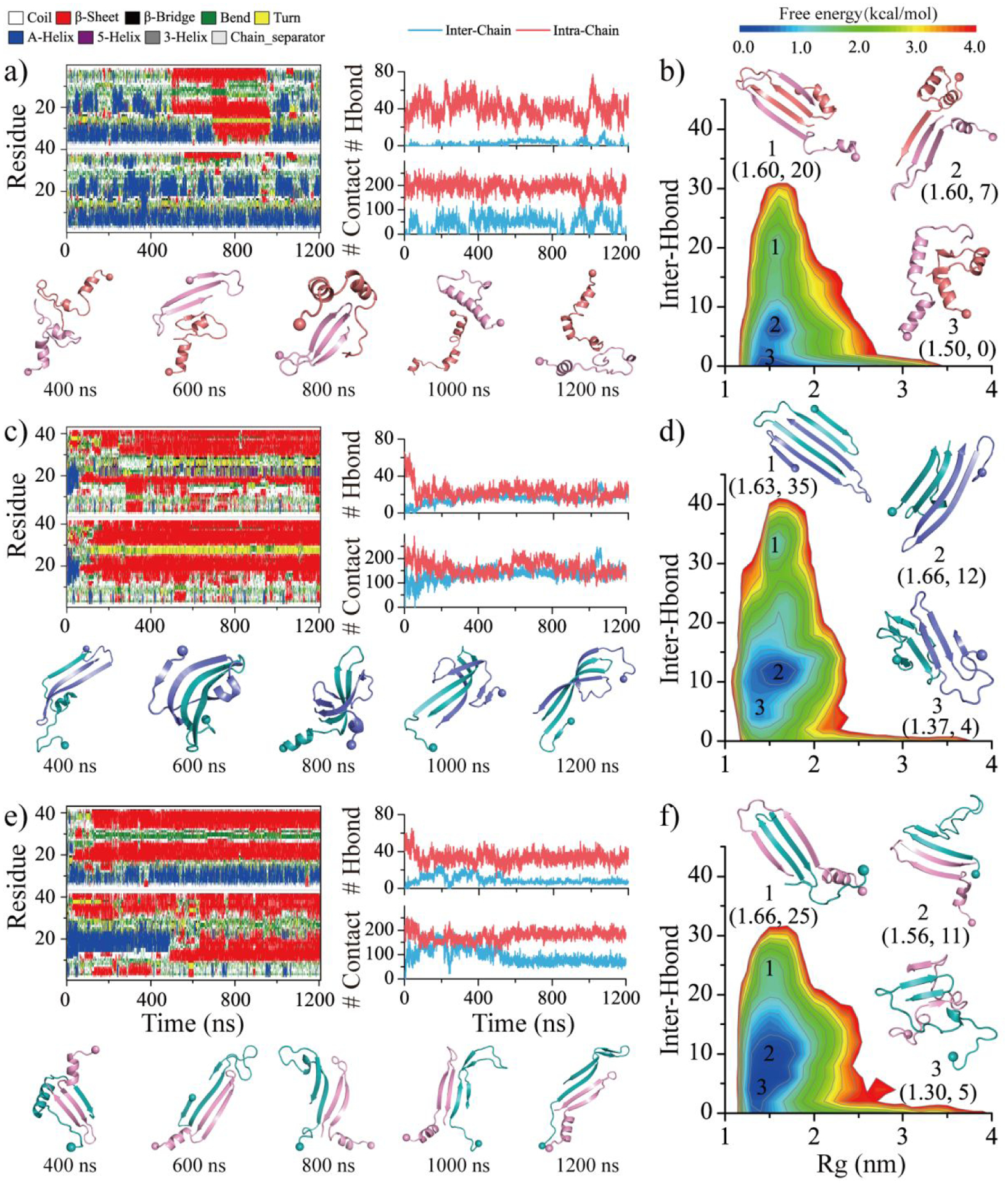
The time evolution of the secondary structure for each residue (first column) and the number of backbone hydrogen bonds and heavy contacts (second column) for the simulations of two SEVI a), two Aβ c), and one SEVI along with one Aβ e) peptides. According to the simulation time, the snapshots are presented every 400 ns. The conformational free energy landscape as a function of the radius gyration (Rg) and the total number of inter-chain backbone hydrogen bonds in self-assemblies of SEVI homo-dimer b), Aβ homo-dimer d), and SEVI-Aβ hetero-dimer f). Three representative structures labeled in the PMFs are also shown as insets. Only the last 600 ns simulation data from each independent DMD trajectory is used for the conformational free energy landscape analysis. For clarity, the SEVI peptides are colored red and pink, and the Aβ are colored blue and cyan.
SEVI bound to Aβ42 via β-sheet pairing and the binding enhanced β-sheet propensity of SEVI.
Compared to SEVI monomers, the homo-dimerization of SEVI enhanced the β-sheet content along with a small decrease in the helical content (Figure 3a). But the helical structure (~29.9%) was still much higher than β-sheet (~19.5%) in the homo-dimerization simulations. The presence of Aβ42 significantly increased the β-sheet propensity of SEVI during their hetero-dimerization, in which the β-sheet and helix content of SEVI was 32.2% and 24.3% (Figure 3a). Monomeric SEVI featured three helical regions around residues 4–11 (~83.5%), 18–26 (~59.4%), and 33–36 (~28.6%). The average helical propensity of SEVI residues 18–26 and 33–36 significantly decreased upon forming both SEVI homo-dimer and SEVI-Aβ42 hetero-dimer (Figure 3b). But residues 4–11 still stayed in helical conformation in both SEVI-SEVI homo-dimer and SEVI-Aβ42 hetero-dimer, consistent with prior experimental measurements69. Specifically, N-terminal residues 1–14 of aggregated SEVI were unprotected from hydrogen-deuterium exchange (HDX) and susceptible to proteolytic cleavage, suggesting that the region didn’t form β-sheet core in aggregation69. The β-sheets were mostly formed by SEVI residues 16–25 and 31–39 with an averaged propensity of ~34.0% in the SEVI-SEVI homo-dimer and ~52.6% in the SEVI-Aβ42 hetero-dimer. Interestingly, previous experimental assays also suggested that residues 13–18 and 33–39 were involved in forming the β-sheet core of SEVI fibrils69, 78. The β-sheet content of Aβ42 in the isolated monomer, ~33.9%, was increased to 54.8% in the Aβ42-Aβ42 homo-dimer and 49.5% in the SEVI-Aβ42 hetero-dimer (Figure 3c). The secondary structure propensity of each residue revealed that the β-sheet propensity around Aβ42 residues 11–22 and 31–41 was enhanced to ~80.0% in the Aβ42 homo-dimer and SEVI-Aβ42 hetero-dimer (Figure 3d). Overall, the cross-interactions between SEVI and Aβ42 drove them aggregated into β-sheet-rich structures, which enhanced the β-sheet content of SEVI.
Figure 3. Conformal analysis for the homodimers and hetero-dimers of SEVI and Aβ.
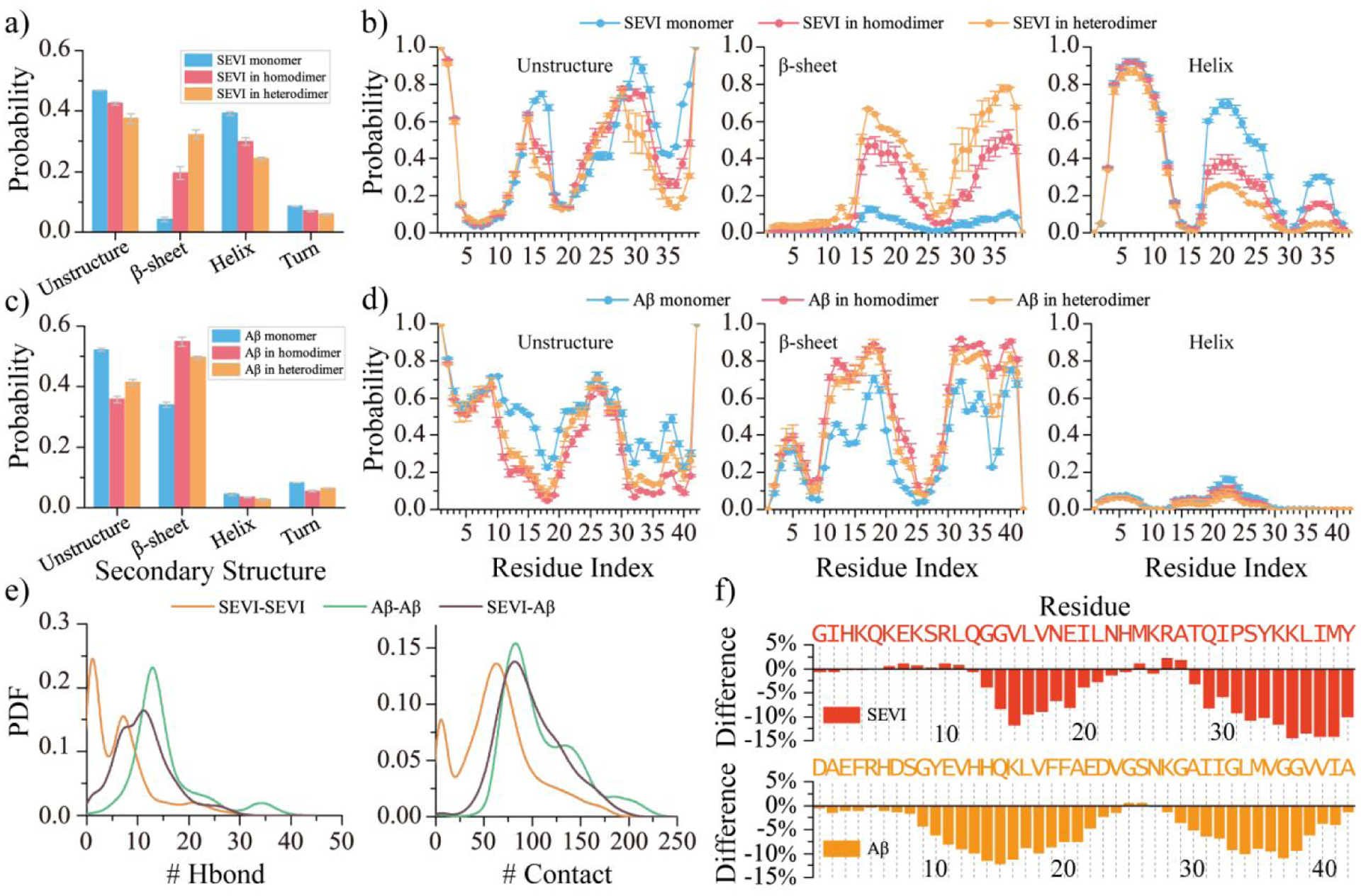
The average secondary content of SEVI a) and Aβ c) peptide in monomer, homo-dimer, and hetero-dimer. Probability of each residue from SEVI b) and Aβ d) adopting unstructured, β-sheet, and helix formations in monomer, homodimer, and heterodimer. The probability distribution of inter-peptide backbone hydrogen bonds and contacts in the SEVI and Aβ homo-dimer and hetero-dimer e). The change ratio of accessible surface area per residue of SEVI (upper) and Aβ (bottom) in the hetero-dimer compared to in the SEVI and Aβ isolated monomer f).
The intra-peptide residue-pairwise contact frequency map of SEVI homo-dimer (Figure S9a) features two helical patterns (~50%) along the diagonal around residues 3–15 and 18–27 (~50%) and a weak β-hairpin contact pattern (~25%) with β-strand formed by residues 14–22 and 31–39. Inter-peptide β-sheet contact patterns with relatively weak frequencies (less than 15%) among residues 16–25 and 29–38 were driven by the hydrophobic interactions (Figure S9a). N-terminal residues 1–13 did not participate in forming inter-peptide β-sheets in agreement with prior HDX-MS measurements69. In Aβ42 homo-dimers, contact patterns of intra-chain β-hairpins and inter-chain β-sheets were widely observed (~30%), with the β-strands mainly formed by residues 10–21 and 30–41 (Figure S9b). Representative structured contact patterns and corresponding structures revealed that Aβ42 peptides formed inter-chain β-hairpins and also inter-chain β-sheets via pairing of β-sheet edges, similar to prior simulation studies3, 8, 27, 75. The contact frequency analysis of SEVI-Aβ42 hetero-dimers revealed that Aβ42 residues 10–41 predominantly adopted in β-hairpin structures, which then paired with SEVI residues 14–39 by forming inter-peptide β-strands (Figure 4). The cross-interaction hot-spot binding regions mostly include Aβ42 residues 10–21 and 30–41 and SEVI residues 14–22 and 31–39. The analysis of representative binding motifs revealed that inter-peptide β-sheets were stabilized by interactions among hydrophobic residues in the binding hot-spot regions (Figure 4). In addition to β-sheets, weak helical patterns among residues 18–27 of SEVI (~30%) were also observed in the SEVI-Aβ42 hetero-dimer. SEVI residues 3–15 were still predominantly adopting helices (~50%). The SEVI-Aβ42 hetero-dimer had more inter-chain backbone hydrogen bonds and contacts than the homo-dimer of SEVI but less than the Aβ42 homo-dimer (Figure 3e). Also as expected, SEVI-Aβ42 cross-interaction decreased the exposed surface areas of both SEVI and Aβ42 round their hot-spot binding regions due to their formation of the inter-chain β-sheets (Figure 3f). Prior experimental and computational studies have shown that the exposed β-hairpin edges could accelerate the intermolecular β-sheet formation8, 75, 79. Since the SEVI was highly positively charged with a very weak self-aggregation tendency, the capping of SEVI around the amyloidogenic region of Aβ42 may suppress the amyloid aggregation of Aβ42.
Figure 4. Residue-pairwise contact frequency of SEVI-Aβ hetero-dimer.
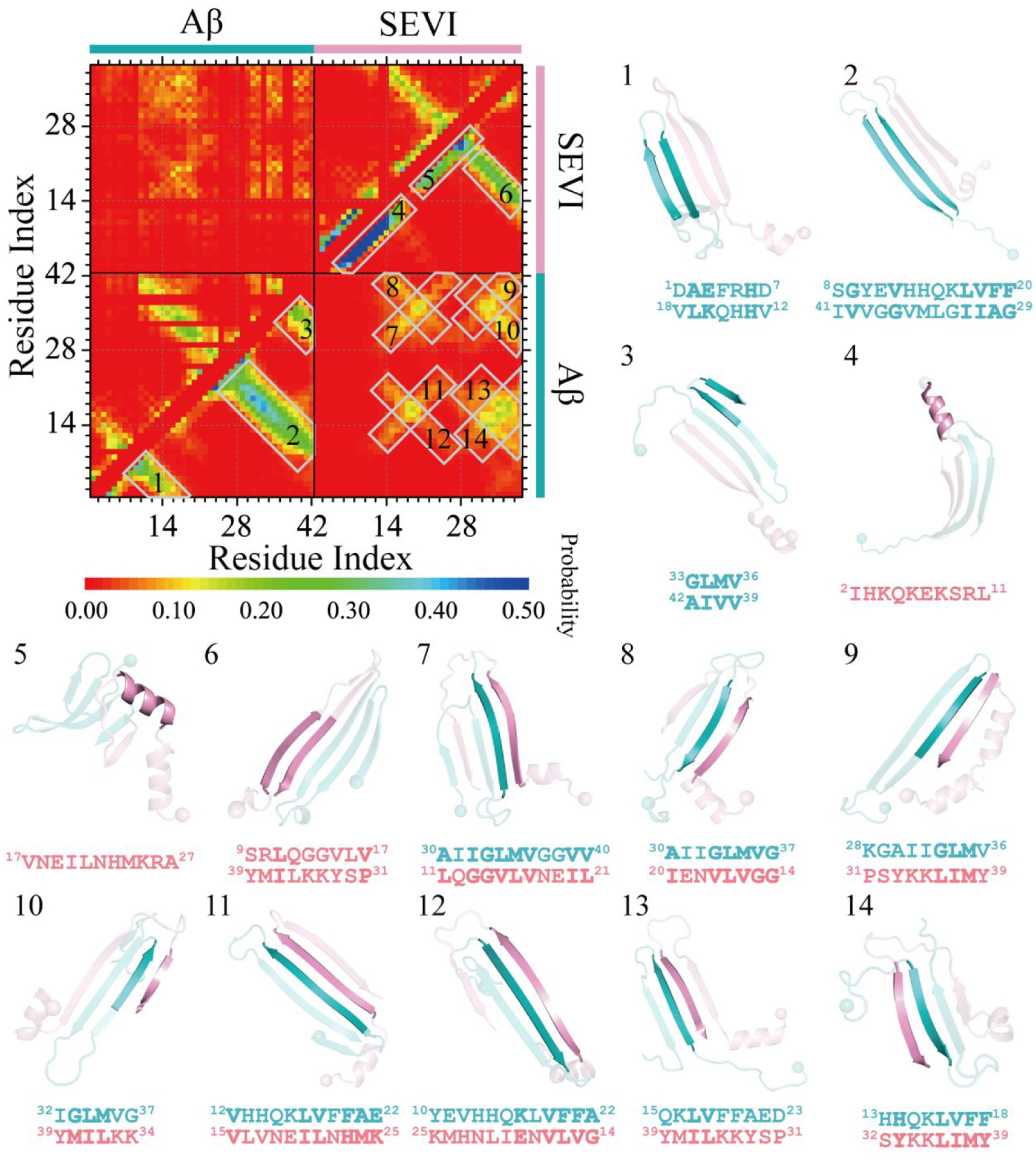
The residue-pairwise contact frequency maps are computed between main-chain atoms (lower diagonal) and side-chain atoms (upper diagonal) based on the last 600 ns trajectories of 50 independent DMD simulations after reaching the saturation state. The representative structured motifs with high contact frequency patterns, mostly corresponding to the helices or β-sheets labeled as 1–14 in the contact frequency map, are also presented.
Aβ42 formed β-sheets buried inside the SEVI-Aβ42 hetero-aggregates with the β-sheet edges capped by SEVI in the outer layer.
To investigate the effects of SEVI on the aggregation of Aβ42, we further investigated the co-aggregation of two SEVI mixed with two Aβ42. Fifty independent DMD trajectories started from different initial states, with each lasting 1200 ns to achieve sufficient conformational sampling. All simulations were well equilibrated and reached their stead states during the last 600 ns by examining the structural parameters of radius gyration, the total number of intra- and inter-chain backbone hydrogen bonds and contacts, and each secondary structure content as a function of simulation time (Figure S10).
The co-aggregation dynamics suggested that SEVI could cap the β-sheet edges of both Aβ42 monomer and dimer, forming a stable SEVI-Aβ42 hetero-aggregate (Figures S10&5a–b). For example, regardless of whether two Aβ42 formed inter-chain contacts and hydrogen bonds or not, the SEVI always interacted with Aβ42 and converted into β-sheets by forming SEVI-Aβ42 contacts and backbone hydrogen bonds (Figure 5a–b). The hetero-aggregates were stabilized by Aβ42-Aβ42 and SEVI-Aβ42 inter-peptide contacts and backbone hydrogen bonds but lacked SEVI-SEVI contacts and hydrogen bonds (Figure 5c). The hetero-aggregates were further analyzed by projected the conformational free energy landscapes onto different pairs of inter-molecular backbone hydrogen bonds, including these among SEVI (SEVI-SEVI), Aβ42 (Aβ42-Aβ42 ), or between SEVI and Aβ42 (SEVI-Aβ42) in Figure 5d&e. The hetero-oligomers featured low free energy with the number of SEVI-SEVI and SEVI-Aβ42 back bond hydrogen bonds ~0–5 and 30–76, respectively (Figure 5d). The hetero-tetramer had two distinct states in terms of the number of the Aβ42-Aβ42 hydrogen bonds (Figure 5c&e), 0–5 and 9–11, indicating that two Aβ42 peptides could be either separately (e.g., Figure 5b) or in contact with each other (e.g., Figure 5a). The radius distribution function of the Cα atom from each peptide demonstrated that the Aβ42 were buried inside while the SEVI peptides were exposed in the outer layer (Figure 5f). Overall, our analysis suggested that Aβ42 mainly formed β-sheets buried inside with β-sheet edges caped by the highly charged SEVI peptides. Since the SEVI exposed outer layer had a relatively weak aggregation tendency, which would prevent the further aggregation of additional Aβ42.
Figure 5. Co-aggregation dynamic and conformation analysis for two SEVI mixed with two Aβ peptides.
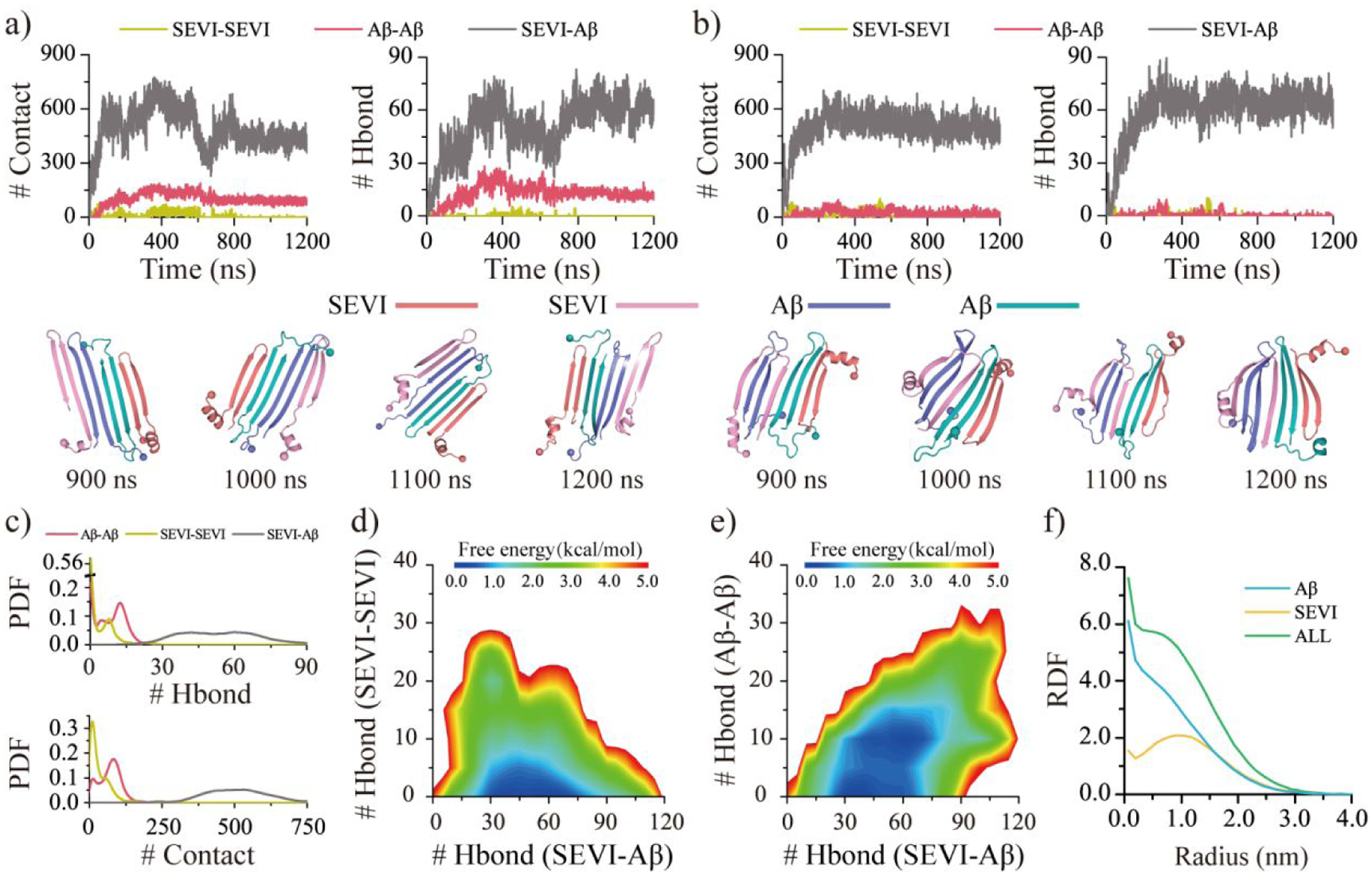
The co-aggregation dynamics are monitored by the time evolution of the number of the inter-peptide Aβ-Aβ, SVEI-SEVI, and SEVI-Aβ contacts and hydrogen bonds a&b). The snapshots during the last 300 ns are shown every 100 ns. Representative trajectories with two Aβ separately a) and jointly b) attached to the SEVI peptide are randomly selected from 50 independent DMD trajectories. The inter-peptide interactions are analyzed by the probability distribution of Aβ-Aβ, SVEI-SEVI, and SEVI-Aβ intermolecular contacts and hydrogen bonds c). The conformational free energy landscape as a function of the number of the inter-peptide SEVI-Aβ and SVEI-SEVI hydrogen bonds d) along with SEVI-Aβ and Aβ-Aβ hydrogen bonds e) in each co-aggregates. The radius distribution function (RDF) of Cα atoms from Aβ and SVEI corresponds to the geometry center of their hetero-aggregates f). Only the last 600 ns simulation data from each independent DMD trajectory is used for the conformational analysis. For clarity, the SEVI peptides are colored red and pink, and Aβ are colored blue and cyan.
The SEVI could bind to both the lateral and elongation surfaces of preformed Aβ42 proto-fibril.
Addition of the preformed fibrils could promote the fibrillization of amyloid peptides through rapid growth via monomer addition by binding to the fibril elongation surface and secondary nucleation by binding to the fibril lateral surface80. The interaction between SEVI monomer and a preformed Aβ42 fibril was therefore investigated. Fifty independent 600-ns DMD simulations were performed starting from different initial structures, in which the monomeric SEVI was randomly placed 1.5 nm away from a 20-peptide Aβ42 fibril. We kept the Aβ42 fibril structure static in our simulations to reduce computational costs, despite the knowledge that only one end of the fibril fluctuates81–83, due to the observation that capping of SEVI had little effect on β-sheet formation of Aβ42 in their hetero-aggregates (Figure S11). The binding dynamics were monitored by the time evolution of inter-peptide contacts and backbone hydrogen bonds between SEVI monomer and Aβ42 fibril along with the secondary structure of each SEVI residue (Figure 6a–c). SEVI mostly adopted helical conformation when it bound to the lateral surface of the Aβ42 fibril (Figure 6a&b). Once the SEVI diffused to the elongation end, SEVI readily converted into β-sheet structures and capped the fibril growth edge (Figure 6b&c). The potential mean force as a function of the number of intermolecular contacts and backbone hydrogen bonds between SEVI and Aβ42 fibril was calculated using the last 200 ns data from 50 independent DMD trajectories. There were two energy basins with the number of intermolecular contacts and backbone hydrogen bonds centered around (74, 0) and (101, 5) corresponding to the SEVI binding to the lateral and elongation surfaces of Aβ42 fibril (snapshots 1&2 in Figure 6d). SEVI conformations with most residues forming high β-sheet with the Aβ42 fibril at elongation edge were also observed (snapshots 3&4 in Figure 6d), which featured a high free energy due to the loss of entropy. Residue-pairwise contact frequency showed that the SEVI mainly anchored around the negatively charged residues E22 and D23 of Aβ42 stabilizing by the electrostatic attraction (Figures 6e&S12). Although further study is required to understand the effect of SEVI binding on the structural stability of Aβ42 fibril, our results indicated that SEVI binding buried the lateral and elongation surfaces of Aβ42 fibril, which may potentially suppress both secondary nucleation and fibril growth through monomer addition.
Figure 6. The binding dynamic analysis of SEVI monomer to Aβ fibril.
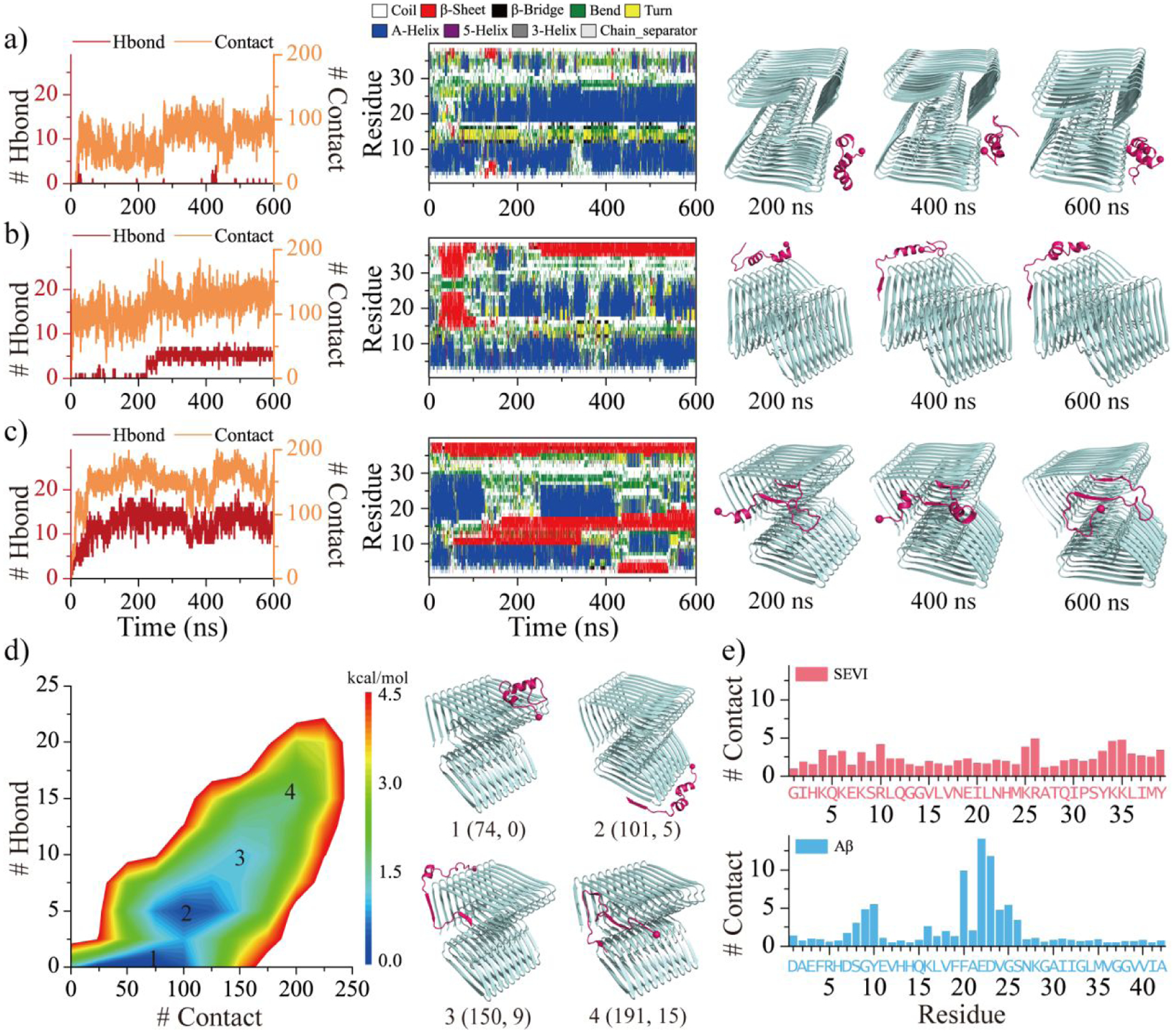
Interactions between SEVI monomer to Aβ fibril are monitored by the time evolution of the number of backbone hydrogen bonds and residue-pairwise contacts between SEVI and Aβ (left panel), and the secondary structure of each residue of SEVI monomer (middle panel) a-c). The corresponding snapshots are presented every 200 ns on the right. Three representative trajectories with the binding region mainly around lateral surface a), lateral mixed with elongation surfaces b), and elongation surface c) are selected from 50 independent DMD runs. The potential mean force as a function of the number of residue-pairwise contacts and backbone hydrogen bonds formed between SEVI and Aβ d). Four representative structures labeled 1–4 in the PMFs (1, 2, 3, 4) are also shown on the right. The average number of SEVI-Aβ contact per residue from SEVI (upper) and Aβ (bottom) e).
Conclusions
In this study, we systematically investigated the inhibition mechanism of SEVI against pathological aggregation of Aβ42 by applying multiple long-timescale atomistic DMD simulations with implicit solvent model. Our results revealed that monomers of both SEVI and Aβ42 were very dynamic and featured frequent conformational changes and transient formation of ordered secondary structures. SEVI monomers formed transient helices, but Aβ42 monomers formed dynamic β-sheets. The self-assembly propensity of SEVI was found to be very weak. The SEVI dimers were unstable, lacked β-sheets, and easily dissociated into helical or unstructured monomers. Aβ42 displayed a significant aggregation tendency. Two Aβ42 peptides readily self-assembled into stable β-sheet-rich oligomers. Compared to the SEVI homo-dimers, the SEVI-Aβ42 hetero-dimers were much more stable. The SEVI-Aβ42 cross-interaction enhanced the β-sheet content of SEVI by forming inter-molecular β-sheets with the amyloidogenic core regions of Aβ42. Co-aggregation simulation of two SEVI and two Aβ42 showed that SEVI preferred to interact with Aβ42 rather than the SEVI themselves. The Aβ42 peptides mainly formed β-sheets buried inside with the β-sheet edges capped by SEVI outside in the hetero-aggregates. Simulations of SEVI in the presence of a preformed Aβ42 proto-fibril demonstrated that SEVI could both cap the growth ends and bind the lateral surfaces of the Aβ42 proto-fibril. With the β-sheets edges of Aβ monomers, oligomers, and proto-fibrils occupied by the highly charged SEVI, the corresponding growth to higher molecular weight oligomers, nucleation of proto-fibrils, and rapid fibril elongation could be inhibited. It is worth noting that the capping strategy to prevent edge-to-edge aggregation has been observed not only with SEVI, but also with other amyloid inhibitors32, 33. For instance, αB-crystallin has been found to inhibit Aβ aggregation by capping the β-sheet elongation edge34, 84. Furthermore, a similar “negative design” approach that involves strategically positioning charged residues at the β-sheet edges has also been used in the design of amyloid inhibitors85, 86. Mechanistic insights obtained from our systematic computational studies may aid in the development of novel therapeutic strategies to modulate the pathological aggregation of amyloid protein in degenerative diseases.
Supplementary Material
Acknowledgments.
This work was supported in part by the National Natural Science Foundation of China (Grant No. 52007087, 11904189 and 82171527), NSF CBET-1553945, and NIH R35GM145409. Computer simulations were supported by the multi-scale computational modeling core of NIH P20GM121342. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NSFC, NIH, and NSF.
Footnotes
Supporting Information:
The Supporting Information is available free of charge on the website. The details of molecular systems in our DMD simulations (Table S1); the amino acid sequences and structure of SEVI and Aβ (Figure S1); the convergence assessments of each monomeric SEVI and Aβ simulation (Figures S2–S3);the residue-pairwise contact frequency of SEVI and Aβ monomer (Figure S4); the conformational free energy landscape of SEVI and Aβ monomers (Figure S5); the conformational sampling efficiency assessments and equilibrium analyses for the dimerization simulation of two Aβ, two SEVI, and one Aβ mixed with one SEVI (Figures S6–S8); the residue-pairwise contact frequency of SEVI and Aβ homo-dimer (Figure S9); the conformational sampling efficiency assessments and equilibrium analyses for the simulation of two SEVI mixed with two Aβ peptides (Figure S10); the effects of SEVI on the β-sheet structure of Aβ42 in their hetero-aggregates (Figure S11); the interaction of SEVI monomer and Aβ proto-fibril analysis (Figure S12) (PDF).
The authors declare no competing financial interest.
Data and Software Availability.
DMD simulation engine is available at Molecules In Action, LLC. (www.moleculesinaction.com). Initial conformations, input parameter and topology files for DMD simulation, and representative DMD output trajectories for each system are available (https://doi.org/10.5281/zenodo.7732322).
References.
- 1.Iadanza MG; Jackson MP; Hewitt EW; Ranson NA; Radford SE, A new era for understanding amyloid structures and disease. Nat Rev Mol Cell Biol 2018, 19, 755–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ke PC; Sani MA; Ding F; Kakinen A; Javed I; Separovic F; Davis TP; Mezzenga R, Implications of peptide assemblies in amyloid diseases. Chem Soc Rev 2017, 46, 6492–6531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nguyen PH; Ramamoorthy A; Sahoo BR; Zheng J; Faller P; Straub JE; Dominguez L; Shea JE; Dokholyan NV; De Simone A; Ma B; Nussinov R; Najafi S; Ngo ST; Loquet A; Chiricotto M; Ganguly P; McCarty J; Li MS; Hall C; Wang Y; Miller Y; Melchionna S; Habenstein B; Timr S; Chen J; Hnath B; Strodel B; Kayed R; Lesne S; Wei G; Sterpone F; Doig AJ; Derreumaux P, Amyloid Oligomers: A Joint Experimental/Computational Perspective on Alzheimer’s Disease, Parkinson’s Disease, Type II Diabetes, and Amyotrophic Lateral Sclerosis. Chem Rev 2021, 121, 2545–2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Busche MA; Hyman BT, Synergy between amyloid-beta and tau in Alzheimer’s disease. Nat Neurosci 2020, 23, 1183–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Henderson MX; Trojanowski JQ; Lee VM, alpha-Synuclein pathology in Parkinson’s disease and related alpha-synucleinopathies. Neurosci Lett 2019, 709, 134316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gallardo R; Ranson NA; Radford SE, Amyloid structures: much more than just a cross-beta fold. Curr Opin Struct Biol 2020, 60, 7–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gremer L; Scholzel D; Schenk C; Reinartz E; Labahn J; Ravelli RBG; Tusche M; Lopez-Iglesias C; Hoyer W; Heise H; Willbold D; Schroder GF, Fibril structure of amyloid-beta(1–42) by cryo-electron microscopy. Science 2017, 358, 116–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun Y; Kakinen A; Wan X; Moriarty N; Hunt CPJ; Li Y; Andrikopoulos N; Nandakumar A; Davis TP; Parish CL; Song Y; Ke PC; Ding F, Spontaneous Formation of beta-sheet Nano-barrels during the Early Aggregation of Alzheimer’s Amyloid Beta. Nano Today 2021, 38, 101125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun Y; Kakinen A; Xing Y; Pilkington EH; Davis TP; Ke PC; Ding F, Nucleation of beta-rich oligomers and beta-barrels in the early aggregation of human islet amyloid polypeptide. Biochim Biophys Acta Mol Basis Dis 2019, 1865, 434–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rane AR; Paithankar H; Hosur RV; Choudhary S, Modulation of alpha-synuclein fibrillation by plant metabolites, daidzein, fisetin and scopoletin under physiological conditions. Int J Biol Macromol 2021, 182, 1278–1291. [DOI] [PubMed] [Google Scholar]
- 11.Abedini A; Plesner A; Cao P; Ridgway Z; Zhang J; Tu LH; Middleton CT; Chao B; Sartori DJ; Meng F; Wang H; Wong AG; Zanni MT; Verchere CB; Raleigh DP; Schmidt AM, Time-resolved studies define the nature of toxic IAPP intermediates, providing insight for anti-amyloidosis therapeutics. Elife 2016, 5, e12977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Benilova I; Karran E; De Strooper B, The toxic Abeta oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat Neurosci 2012, 15, 349–357. [DOI] [PubMed] [Google Scholar]
- 13.Ke PC; Pilkington EH; Sun Y; Javed I; Kakinen A; Peng G; Ding F; Davis TP, Mitigation of Amyloidosis with Nanomaterials. Adv Mater 2020, 32, e1901690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun Y; Kakinen A; Zhang C; Yang Y; Faridi A; Davis TP; Cao W; Ke PC; Ding F, Amphiphilic surface chemistry of fullerenols is necessary for inhibiting the amyloid aggregation of alpha-synuclein NACore. Nanoscale 2019, 11, 11933–11945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bieschke J; Russ J; Friedrich RP; Ehrnhoefer DE; Wobst H; Neugebauer K; Wanker EE, EGCG remodels mature alpha-synuclein and amyloid-beta fibrils and reduces cellular toxicity. Proc Natl Acad Sci U S A 2010, 107, 7710–7715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ehrnhoefer DE; Bieschke J; Boeddrich A; Herbst M; Masino L; Lurz R; Engemann S; Pastore A; Wanker EE, EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nat Struct Mol Biol 2008, 15, 558–566. [DOI] [PubMed] [Google Scholar]
- 17.Mo Y; Lei J; Sun Y; Zhang Q; Wei G, Conformational Ensemble of hIAPP Dimer: Insight into the Molecular Mechanism by which a Green Tea Extract inhibits hIAPP Aggregation. Sci Rep 2016, 6, 33076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen Y; Li X; Zhan C; Lao Z; Li F; Dong X; Wei G, A Comprehensive Insight into the Mechanisms of Dopamine in Disrupting Abeta Protofibrils and Inhibiting Abeta Aggregation. ACS Chem Neurosci 2021, 12, 4007–4019. [DOI] [PubMed] [Google Scholar]
- 19.Li J; Zhu M; Manning-Bog AB; Di Monte DA; Fink AL, Dopamine and L-dopa disaggregate amyloid fibrils: implications for Parkinson’s and Alzheimer’s disease. FASEB J 2004, 18, 962–964. [DOI] [PubMed] [Google Scholar]
- 20.Ono K; Hasegawa K; Naiki H; Yamada M, Anti-Parkinsonian agents have anti-amyloidogenic activity for Alzheimer’s beta-amyloid fibrils in vitro. Neurochem Int 2006, 48, 275–285. [DOI] [PubMed] [Google Scholar]
- 21.Nedumpully-Govindan P; Kakinen A; Pilkington EH; Davis TP; Chun Ke P; Ding F, Stabilizing Off-pathway Oligomers by Polyphenol Nanoassemblies for IAPP Aggregation Inhibition. Sci Rep 2016, 6, 19463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mishra R; Sellin D; Radovan D; Gohlke A; Winter R, Inhibiting islet amyloid polypeptide fibril formation by the red wine compound resveratrol. Chembiochem 2009, 10, 445–449. [DOI] [PubMed] [Google Scholar]
- 23.Wang M; Sun Y; Cao X; Peng G; Javed I; Kakinen A; Davis TP; Lin S; Liu J; Ding F; Ke PC, Graphene quantum dots against human IAPP aggregation and toxicity in vivo. Nanoscale 2018, 10, 19995–20006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Faridi A; Sun Y; Mortimer M; Aranha RR; Nandakumar A; Li Y; Javed I; Kakinen A; Fan Q; Purcell AW; Davis TP; Ding F; Faridi P; Ke PC, Graphene quantum dots rescue protein dysregulation of pancreatic beta-cells exposed to human islet amyloid polypeptide. Nano Res 2019, 12, 2827–2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim D; Yoo JM; Hwang H; Lee J; Lee SH; Yun SP; Park MJ; Lee M; Choi S; Kwon SH; Lee S; Kwon SH; Kim S; Park YJ; Kinoshita M; Lee YH; Shin S; Paik SR; Lee SJ; Lee S; Hong BH; Ko HS, Graphene quantum dots prevent alpha-synucleinopathy in Parkinson’s disease. Nat Nanotechnol 2018, 13, 812–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ghaeidamini M; Bernson D; Sasanian N; Kumar R; Esbjorner EK, Graphene oxide sheets and quantum dots inhibit alpha-synuclein amyloid formation by different mechanisms. Nanoscale 2020, 12, 19450–19460. [DOI] [PubMed] [Google Scholar]
- 27.Sun Y; Qian Z; Wei G, The inhibitory mechanism of a fullerene derivative against amyloid-beta peptide aggregation: an atomistic simulation study. Phys Chem Chem Phys 2016, 18, 12582–12591. [DOI] [PubMed] [Google Scholar]
- 28.Bobylev AG; Kraevaya OA; Bobyleva LG; Khakina EA; Fadeev RS; Zhilenkov AV; Mishchenko DV; Penkov NV; Teplov IY; Yakupova EI; Vikhlyantsev IM; Troshin PA, Anti-amyloid activities of three different types of water-soluble fullerene derivatives. Colloids Surf B Biointerfaces 2019, 183, 110426. [DOI] [PubMed] [Google Scholar]
- 29.Ishida Y; Fujii T; Oka K; Takahashi D; Toshima K, Inhibition of amyloid beta aggregation and cytotoxicity by photodegradation using a designed fullerene derivative. Chem Asian J 2011, 6, 2312–2315. [DOI] [PubMed] [Google Scholar]
- 30.Sun LJ; Qu L; Yang R; Yin L; Zeng HJ, Cysteamine functionalized MoS2 quantum dots inhibit amyloid aggregation. Int J Biol Macromol 2019, 128, 870–876. [DOI] [PubMed] [Google Scholar]
- 31.Wang J; Liu L; Ge D; Zhang H; Feng Y; Zhang Y; Chen M; Dong M, Differential Modulating Effect of MoS2 on Amyloid Peptide Assemblies. Chemistry 2018, 24, 3397–3402. [DOI] [PubMed] [Google Scholar]
- 32.Hochberg GK; Ecroyd H; Liu C; Cox D; Cascio D; Sawaya MR; Collier MP; Stroud J; Carver JA; Baldwin AJ; Robinson CV; Eisenberg DS; Benesch JL; Laganowsky A, The structured core domain of alphaB-crystallin can prevent amyloid fibrillation and associated toxicity. Proc Natl Acad Sci U S A 2014, 111, E1562–E1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mainz A; Peschek J; Stavropoulou M; Back KC; Bardiaux B; Asami S; Prade E; Peters C; Weinkauf S; Buchner J; Reif B, The chaperone alphaB-crystallin uses different interfaces to capture an amorphous and an amyloid client. Nat Struct Mol Biol 2015, 22, 898–905. [DOI] [PubMed] [Google Scholar]
- 34.Sun Y; Ding F, alphaB-Crystallin Chaperone Inhibits Abeta Aggregation by Capping the beta-Sheet-Rich Oligomers and Fibrils. J Phys Chem B 2020, 124, 10138–10146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Y; Liu Y; Tang Y; Zhang D; He H; Wu J; Zheng J, Antimicrobial alpha-defensins as multi-target inhibitors against amyloid formation and microbial infection. Chem Sci 2021, 12, 9124–9139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ono K; Li L; Takamura Y; Yoshiike Y; Zhu LJ; Han F; Mao X; Ikeda T; Takasaki J; Nishijo H; Takashima A; Teplow DB; Zagorski MG; Yamada M, Phenolic Compounds Prevent Amyloid beta-Protein Oligomerization and Synaptic Dysfunction by Site-specific Binding. Journal of Biological Chemistry 2012, 287, 14631–14643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ngoc LLN; Itoh SG; Sompornpisut P; Okumura H, Replica-permutation molecular dynamics simulations of an amyloid-beta(16–22) peptide and polyphenols. Chem Phys Lett 2020, 758, 137913. [Google Scholar]
- 38.Doig AJ; del Castillo-Frias MP; Berthoumieu O; Tarus B; Nasica-Labouze J; Sterpone F; Nguyen PH; Hooper NM; Faller P; Derreumaux P, Why Is Research on Amyloid-beta Failing to Give New Drugs for Alzheimer’s Disease? Acs Chemical Neuroscience 2017, 8, 1435–1437. [DOI] [PubMed] [Google Scholar]
- 39.Tang Y; Zhang D; Zhang Y; Liu Y; Miller Y; Gong K; Zheng J, Cross-seeding between Abeta and SEVI indicates a pathogenic link and gender difference between alzheimer diseases and AIDS. Commun Biol 2022, 5, 417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chatani E; Lee YH; Yagi H; Yoshimura Y; Naiki H; Goto Y, Ultrasonication-dependent production and breakdown lead to minimum-sized amyloid fibrils. P Natl Acad Sci USA 2009, 106, 11119–11124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Okumura H; Itoh SG, Amyloid Fibril Disruption by Ultrasonic Cavitation: Nonequilibrium Molecular Dynamics Simulations. Journal of the American Chemical Society 2014, 136, 10549–10552. [DOI] [PubMed] [Google Scholar]
- 42.Kawasaki T; Yaji T; Ohta T; Tsukiyama K; Nakamura K, Dissociation of beta-Sheet Stacking of Amyloid beta Fibrils by Irradiation of Intense, Short-Pulsed Mid-infrared Laser. Cell Mol Neurobiol 2018, 38, 1039–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Okumura H; Itoh SG; Nakamura K; Kawasaki T, Role of Water Molecules and Helix Structure Stabilization in the Laser-Induced Disruption of Amyloid Fibrils Observed by Nonequilibrium Molecular Dynamics Simulations. J Phys Chem B 2021, 125, 4964–4976. [DOI] [PubMed] [Google Scholar]
- 44.Gouras GK; Olsson TT; Hansson O, beta-Amyloid peptides and amyloid plaques in Alzheimer’s disease. Neurotherapeutics 2015, 12, 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yan Y; Wang C, Abeta42 is more rigid than Abeta40 at the C terminus: implications for Abeta aggregation and toxicity. J Mol Biol 2006, 364, 853–862. [DOI] [PubMed] [Google Scholar]
- 46.Munch J; Rucker E; Standker L; Adermann K; Goffinet C; Schindler M; Wildum S; Chinnadurai R; Rajan D; Specht A; Gimenez-Gallego G; Sanchez PC; Fowler DM; Koulov A; Kelly JW; Mothes W; Grivel JC; Margolis L; Keppler OT; Forssmann WG; Kirchhoff F, Semen-derived amyloid fibrils drastically enhance HIV infection. Cell 2007, 131, 1059–1071. [DOI] [PubMed] [Google Scholar]
- 47.Olsen JS; DiMaio JT; Doran TM; Brown C; Nilsson BL; Dewhurst S, Seminal plasma accelerates semen-derived enhancer of viral infection (SEVI) fibril formation by the prostatic acid phosphatase (PAP248–286) peptide. J Biol Chem 2012, 287, 11842–11849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Morel B; Varela L; Azuaga AI; Conejero-Lara F, Environmental conditions affect the kinetics of nucleation of amyloid fibrils and determine their morphology. Biophys J 2010, 99, 3801–3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ye Z; French KC; Popova LA; Lednev IK; Lopez MM; Makhatadze GI, Mechanism of fibril formation by a 39-residue peptide (PAPf39) from human prostatic acidic phosphatase. Biochemistry 2009, 48, 11582–11591. [DOI] [PubMed] [Google Scholar]
- 50.Martellini JA; Cole AL; Svoboda P; Stuchlik O; Chen LM; Chai KX; Gangrade BK; Sorensen OE; Pohl J; Cole AM, HIV-1 enhancing effect of prostatic acid phosphatase peptides is reduced in human seminal plasma. PLoS One 2011, 6, e16285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shirvanyants D; Ding F; Tsao D; Ramachandran S; Dokholyan NV, Discrete molecular dynamics: an efficient and versatile simulation method for fine protein characterization. J Phys Chem B 2012, 116, 8375–8382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ding F; Tsao D; Nie H; Dokholyan NV, Ab initio folding of proteins with all-atom discrete molecular dynamics. Structure 2008, 16, 1010–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang Y; Wang Y; Liu Y; Wei G; Ding F; Sun Y, Molecular Insights into the Misfolding and Dimerization Dynamics of the Full-Length alpha-Synuclein from Atomistic Discrete Molecular Dynamics Simulations. ACS Chem Neurosci 2022, 13, 3126–3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang Y; Liu Y; Zhao W; Sun Y, Hydroxylated single-walled carbon nanotube inhibits beta2m(21)(−)(31) fibrillization and disrupts pre-formed proto-fibrils. Int J Biol Macromol 2021, 193, 1–7. [DOI] [PubMed] [Google Scholar]
- 55.Brodie NI; Popov KI; Petrotchenko EV; Dokholyan NV; Borchers CH, Solving protein structures using short-distance cross-linking constraints as a guide for discrete molecular dynamics simulations. Sci Adv 2017, 3, e1700479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bunce SJ; Wang Y; Stewart KL; Ashcroft AE; Radford SE; Hall CK; Wilson AJ, Molecular insights into the surface-catalyzed secondary nucleation of amyloid-beta(40) (Abeta(40)) by the peptide fragment Abeta(16–22). Sci Adv 2019, 5, eaav8216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Emperador A; Orozco M, Discrete Molecular Dynamics Approach to the Study of Disordered and Aggregating Proteins. J Chem Theory Comput 2017, 13, 1454–1461. [DOI] [PubMed] [Google Scholar]
- 58.Thacker D; Sanagavarapu K; Frohm B; Meisl G; Knowles TPJ; Linse S, The role of fibril structure and surface hydrophobicity in secondary nucleation of amyloid fibrils. Proc Natl Acad Sci U S A 2020, 117, 25272–25283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nanga RP; Brender JR; Vivekanandan S; Popovych N; Ramamoorthy A, NMR structure in a membrane environment reveals putative amyloidogenic regions of the SEVI precursor peptide PAP(248–286). J Am Chem Soc 2009, 131, 17972–17979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tomaselli S; Esposito V; Vangone P; van Nuland NA; Bonvin AM; Guerrini R; Tancredi T; Temussi PA; Picone D, The alpha-to-beta conformational transition of Alzheimer’s Abeta-(1–42) peptide in aqueous media is reversible: a step by step conformational analysis suggests the location of beta conformation seeding. Chembiochem 2006, 7, 257–267. [DOI] [PubMed] [Google Scholar]
- 61.Yin S; Biedermannova L; Vondrasek J; Dokholyan NV, MedusaScore: an accurate force field-based scoring function for virtual drug screening. J Chem Inf Model 2008, 48, 1656–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yin S; Ding F; Dokholyan NV, Eris: an automated estimator of protein stability. Nat Methods 2007, 4, 466–467. [DOI] [PubMed] [Google Scholar]
- 63.Lazaridis T; Karplus M, Effective energy functions for protein structure prediction. Curr Opin Struct Biol 2000, 10, 139–145. [DOI] [PubMed] [Google Scholar]
- 64.Kabsch W; Sander C, Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [DOI] [PubMed] [Google Scholar]
- 65.Wang Y; Liu Y; Zhang Y; Wei G; Ding F; Sun Y, Molecular insights into the oligomerization dynamics and conformations of amyloidogenic and non-amyloidogenic amylin from discrete molecular dynamics simulations. Phys Chem Chem Phys 2022, 24, 21773–21785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Daura X; Gademann K; Jaun B; Seebach D; van Gunsteren WF; Mark AE, Peptide folding: When simulation meets experiment. Angew Chem Int Edit 1999, 38, 236–240. [Google Scholar]
- 67.Brender JR; Hartman K; Gottler LM; Cavitt ME; Youngstrom DW; Ramamoorthy A, Helical conformation of the SEVI precursor peptide PAP248–286, a dramatic enhancer of HIV infectivity, promotes lipid aggregation and fusion. Biophys J 2009, 97, 2474–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brender JR; Nanga RP; Popovych N; Soong R; Macdonald PM; Ramamoorthy A, The amyloidogenic SEVI precursor, PAP248–286, is highly unfolded in solution despite an underlying helical tendency. Biochim Biophys Acta 2011, 1808, 1161–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.French KC; Makhatadze GI, Core sequence of PAPf39 amyloid fibrils and mechanism of pH-dependent fibril formation: the role of monomer conformation. Biochemistry 2012, 51, 10127–10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Meng F; Bellaiche MMJ; Kim JY; Zerze GH; Best RB; Chung HS, Highly Disordered Amyloid-beta Monomer Probed by Single-Molecule FRET and MD Simulation. Biophys J 2018, 114, 870–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ono K; Condron MM; Teplow DB, Structure-neurotoxicity relationships of amyloid beta-protein oligomers. Proc Natl Acad Sci U S A 2009, 106, 14745–14750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rosenman DJ; Connors CR; Chen W; Wang C; Garcia AE, Abeta monomers transiently sample oligomer and fibril-like configurations: ensemble characterization using a combined MD/NMR approach. J Mol Biol 2013, 425, 3338–3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Itoh SG; Okumura H, Replica-Permutation Method with the Suwa-Todo Algorithm beyond the Replica-Exchange Method. J Chem Theory Comput 2013, 9, 570–581. [DOI] [PubMed] [Google Scholar]
- 74.Itoh SG; Okumura H, Hamiltonian replica-permutation method and its applications to an alanine dipeptide and amyloid-beta(29–42) peptides. J Comput Chem 2013, 34, 2493–2497. [DOI] [PubMed] [Google Scholar]
- 75.Itoh SG; Yagi-Utsumi M; Kato K; Okumura H, Key Residue for Aggregation of Amyloid-beta Peptides. ACS Chem Neurosci 2022, 13, 3139–3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yang Y; Arseni D; Zhang W; Huang M; Lovestam S; Schweighauser M; Kotecha A; Murzin AG; Peak-Chew SY; Macdonald J; Lavenir I; Garringer HJ; Gelpi E; Newell KL; Kovacs GG; Vidal R; Ghetti B; Ryskeldi-Falcon B; Scheres SHW; Goedert M, Cryo-EM structures of amyloid-beta 42 filaments from human brains. Science 2022, 375, 167–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Paravastu AK; Leapman RD; Yau WM; Tycko R, Molecular structural basis for polymorphism in Alzheimer’s beta-amyloid fibrils. Proc Natl Acad Sci U S A 2008, 105, 18349–18354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sievers SA; Karanicolas J; Chang HW; Zhao A; Jiang L; Zirafi O; Stevens JT; Munch J; Baker D; Eisenberg D, Structure-based design of non-natural amino-acid inhibitors of amyloid fibril formation. Nature 2011, 475, 96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Abelein A; Abrahams JP; Danielsson J; Graslund A; Jarvet J; Luo J; Tiiman A; Warmlander SK, The hairpin conformation of the amyloid beta peptide is an important structural motif along the aggregation pathway. J Biol Inorg Chem 2014, 19, 623–34. [DOI] [PubMed] [Google Scholar]
- 80.Aprile FA; Sormanni P; Perni M; Arosio P; Linse S; Knowles TPJ; Dobson CM; Vendruscolo M, Selective targeting of primary and secondary nucleation pathways in Abeta42 aggregation using a rational antibody scanning method. Sci Adv 2017, 3, e1700488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ban T; Hamada D; Hasegawa K; Naiki H; Goto Y, Direct observation of amyloid fibril growth monitored by thioflavin T fluorescence. Journal of Biological Chemistry 2003, 278, 16462–16465. [DOI] [PubMed] [Google Scholar]
- 82.Ban T; Hoshino M; Takahashi S; Hamada D; Hasegawa K; Naiki H; Goto Y, Direct observation of A beta amyloid fibril growth and inhibition. Journal of Molecular Biology 2004, 344, 757–767. [DOI] [PubMed] [Google Scholar]
- 83.Okumura H; Itoh SG, Structural and fluctuational difference between two ends of Abeta amyloid fibril: MD simulations predict only one end has open conformations. Sci Rep 2016, 6, 38422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Xu Z; Gong Y; Zou Y; Wan J; Tang J; Zhan C; Wei G; Zhang Q, Dissecting the Inhibitory Mechanism of the alphaB-Crystallin Domain against Abeta(42) Aggregation and Its Effect on Abeta(42) Protofibrils: A Molecular Dynamics Simulation Study. ACS Chem Neurosci 2022, 13, 2842–2851. [DOI] [PubMed] [Google Scholar]
- 85.Tas K; Volta BD; Lindner C; El Bounkari O; Hille K; Tian Y; Puig-Bosch X; Ballmann M; Hornung S; Ortner M; Prem S; Meier L; Rammes G; Haslbeck M; Weber C; Megens RTA; Bernhagen J; Kapurniotu A, Designed peptides as nanomolar cross-amyloid inhibitors acting via supramolecular nanofiber co-assembly. Nat Commun 2022, 13, 5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cheng PN; Liu C; Zhao M; Eisenberg D; Nowick JS, Amyloid beta-sheet mimics that antagonize protein aggregation and reduce amyloid toxicity. Nat Chem 2012, 4, 927–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
DMD simulation engine is available at Molecules In Action, LLC. (www.moleculesinaction.com). Initial conformations, input parameter and topology files for DMD simulation, and representative DMD output trajectories for each system are available (https://doi.org/10.5281/zenodo.7732322).