

Abstract
The pathological aggregation of α-synuclein (αS) into amyloid fibrils is the hallmark of Parkinson’s disease (PD). The self-assembly and membrane interactions of αS are mainly governed by the seven imperfect 11-residue repeats of the XKTKEGVXXXX motif around residues 1–95. However, the particular role of each repeat in αS fibrillization remains unclear. To answer this question, we studied the aggregation dynamics of each repeat with up to ten peptides in silico by conducting multiple independent micro-second atomistic discrete molecular dynamics (DMD) simulations. Our simulations revealed that only repeats R3 and R6 readily self-assembled into β-sheet-rich oligomers, while the other repeats maintained as unstructured monomers with weak self-assembly and β-sheet propensities. The self-assembly process of R3 featured frequent conformational changes with β-sheet formation mainly in the non-conserved hydrophobic tail, whereas R6 spontaneously self-assembled into extended and stable cross-β structures. These results of seven repeats are consistent with their structures and organization in recently solved αS fibrils. As the primary amyloidogenic core, R6 were buried inside the central cross-β core of all αS fibrils, attracting the hydrophobic tails of adjacent R4, R5, and R7 repeats forming β-sheets around R6 in the core. Further away from R6 in the sequence but with a moderate amyloid aggregation propensity, the R3 tail could serve as a secondary amyloidogenic core and form independent β-sheets in the fibril. Overall, our results demonstrate the critical role of R3 and R6 repeats in αS amyloid aggregation and suggest their potential as targets for the peptide-based and small molecule amyloid inhibitors.
Graphical Abstract
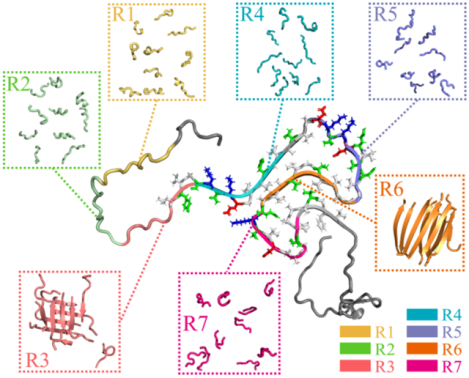
Introduction
Misfolding and pathological aggregation of α-synuclein (αS) into amyloid fibrils are associated with several neurodegenerative diseases, including Parkinson’s disease (PD), multiple system atrophy, and dementia with Lewy bodies.1, 2 Similar to other amyloid disease-related peptides (e.g., amyloid-β, islet amyloid polypeptide, and tau), the fibrillization of αS features a common sigmoidal curve with three phases, corresponding to the nucleation of monomers into oligomers and proto-fibrils, as well as the rapid elongation and saturation of proto-fibrils into mature fibrils.3, 4 The monomers of αS are generally disordered in solution and assume partial helices in the presence of lipid membranes.5, 6 The experimentally determined patient-derived αS fibrils feature a common cross-β core with closely packed parallel in-register β-sheets and the composite β-strands aligned perpendicularly to the fibril axis.4, 7, 8 Soluble oligomers of αS formed during the early amyloid aggregation stage are more toxic than the mature fibrils9, but their structures are still elusive. Prior studies have demonstrated that inhibiting αS aggregation can effectively prevent its cytotoxicity, making the development of anti-amyloid inhibitors a promising therapeutic strategy against PD.10–12 Therefore, characterizing the early stages of αS aggregation and elucidating the molecular-level aggregation mechanism are crucial for understanding the pathogenesis of αS aggregation-related diseases such as PD, as well as for designing therapeutic amyloid inhibitors.
The sequence of full-length αS comprises three distinct domains: one positively charged N-terminus domain of residues 1–60, which adopts an α-helix upon binding to negatively charged membranes13; one hydrophobic central domain of residues 61–95 known as non-amyloid component (NAC), which is necessary and sufficient for the aggregation of αS8, 14; and one strongly acidic and negatively charged C-terminal domain, which is very flexible in the monomers, oligomers, and fibrils of αS7. Monomers of αS are natively disordered with transient long-range intradomain interactions, resulting into a smaller radius of gyration than expected for a fully unfolded 140-residue protein.15 Prior experimental and computational studies have shown that the long-range interactions drive αS monomers to form some metastable compact globular conformations.3, 16, 17 Apart from the NAC domain, a portion of the N-terminal domain also participates in forming the cross-β core of αS fibrils.7 In the fibrillar form, the αS adopts an in-register parallel β-sheet conformation with the β-sheet residues spanning 38–95, while the rest of the terminal residues likely assume unstructured conformations.7, 18–20 The truncation of the C-terminus enhances the aggregation rate of αS, suggesting that the presence of the C-terminal tail might protect against amyloid aggregation.17, 21
In the N-terminal and NAC domains important for αS aggregation, there are seven imperfect 11-residue repeats with a core sequence motif of KTKTGV (also known as the XKTKEGVXXXX motif, all repeat sequences listed in Table 1) are present, postulated to be responsible for membrane binding and amyloid aggregation of αS.4, 22 Deletion of the first two repeats of residues 9–30 enhances the aggregation rate of αS22. The aggregation tendency and cytotoxicity of αS would be significantly suppressed when the segment of residues 36–42 from repeat 3 or 45–57 from repeat 4 are deleted.4 Prior experimental study has shown that residues 47–56 preceding the NAC domain, known as preNAC, play an important role in the aggregation of αS.8 Six out of seven αS familial single-point mutations associated with early-onset PD including E30P, E46K, H50Q, G51D, A53T/E/V are located within the 4th repeat and lead to distinct aggregation tendencies and cytotoxicity.23, 24 Given the important roles of these repeats in αS amyloid aggregation, it is necessary to characterize the propensity of each individual repeat to self-assemble and form β-sheet rich aggregates.
Table 1.
The details of molecule systems in our DMD simulations, including the amino acid sequence of each αS repeat (System), the number of residues per chain (Residues per peptide), the number of simulated peptides (Number peptide), the corresponding dimensions of the cubic simulation box (Box size), the length of each DMD simulations (Time), the number of independent DMD simulations performed (DMD run), and the accumulative total simulation time (Total time).
| System | Number peptide | Box size (nm) | Time (μs) | DMD run | Total time (μs) | ||
|---|---|---|---|---|---|---|---|
| Repeat | Sequence | Residues per peptide | |||||
| R1 | 9SKAKEGVVAAA19 | 11 | 10 | 9.0 | 1.0 | 30 | 30.0 |
| R2 | 20EKTKQGVAEAA30 | 11 | 10 | 9.0 | 1.0 | 30 | 30.0 |
| R3 | 31GKTKEGVLYVG41 | 11 | 10 | 9.0 | 1.0 | 30 | 30.0 |
| R4 | 42SKTKEGVVHGV52 | 11 | 10 | 9.0 | 1.0 | 30 | 30.0 |
| R5 | 57EKTKEQVTNVG67 | 11 | 10 | 9.0 | 1.0 | 30 | 30.0 |
| R6 | 68GAVVTGVTAVA78 | 11 | 10 | 9.0 | 1.0 | 30 | 30.0 |
| R7 | 79QKTVEGAGSIA89 | 11 | 10 | 9.0 | 1.0 | 30 | 30.0 |
In this study, we systematically examined the self-assembly dynamics of each imperfect repeat of αS through a large number of long-timescale atomistic discrete molecular dynamics (DMD) simulations. DMD is a rapid and predictive molecular dynamics algorithm widely used to study protein folding and amyloid aggregation.17, 25–28 For each repeat, we conducted 30 independent aggregation simulations of ten peptides with each simulation lasting 1000 ns. Our simulations revealed that only R3 and R6 readily self-assembled into β-sheet-rich oligomers with distinct conformational dynamics. R3 oligomers were metastable with frequent dissociation/reassociation, and β-sheets were primarily formed by hydrophobic non-conserved tails. In contrast, R6 repeats readily formed extended stable β-sheets, with all residues participating in β-sheet formation, consistent with prior computational and experimental studies8, 14. The other repeats mainly assumed isolated and unstructured monomers. Our results of all seven repeats are consistent with their structures and organization in recently solved αS fibrils.7, 18–20 Specifically, the weak β-sheet and aggregation tendencies of R1 and R2 render them structurally-unresolved, and thus, the experimentally determined αS fibril starts from the hydrophobic tail of R3 until the end of the NAC region7, 18–20. With the highest amyloid aggregation propensity, R6 repeats were buried inside the central cross-β core in the αS fibril.18–20 Although the isolated R4, R5, and R7 repeats around R6 displayed weak self-assembly tendencies for forming β-sheet-rich aggregates, the fibrillization of the R6 repeat could facilitate their hydrophobic tails adopting β-sheets. Further away from R6 in the sequence but with a moderate amyloid aggregation propensity, R3 tail could form serve a secondary amyloidogenic core and form β-sheet in the fibril. Therefore, R3 hydrophobic tail and R6 might serve as the targets in designing anti-amyloid inhibitors10, 11, 29, or, even as peptide-based competitive inhibitors30–32.
Materials and Methods
Molecular Systems.
The amino acid sequences of the seven imperfect repeats of α-synuclein were listed in Table 1. The initial structure of each repeat was based on the full-length α-synuclein monomer (PDB: 1XQ8), which was determined under micelle-bound conditions33. To achieve sufficient conformational sampling, we performed thirty independent DMD simulations for each repeat, using different initial states (coordinates, orientations, and velocities). Each DMD trajectory lasted 1.0 μs to ensure that equilibrium was attained. In the starting configuration of each independent simulation, ten peptides were randomly placed into a 9.0 nm cubic simulation box, with a minimum inter-peptide distance of no less than 1.5 nm. Further details of all the simulations could be found in Table 1.
Details of DMD Simulations.
All the simulations were performed in the canonical NVT ensemble using implicit-solvent united-atom discrete molecular dynamics (DMD) simulations at 300K with the Medusa force field, which has been thoroughly benchmarked for its ability to accurately predict protein stability change upon mutation and protein-ligand binding affinity34, 35. In the Medusa force field, bonded interactions, including covalent bonds, bond angles, and dihedrals, as well as non-bonded interactions such as van der Waals and electrostatic terms, are used to describe biomolecules.36 The VDW parameters of the Medusa force field are based on the CHARMM force field.37 Water is implicitly modeled using the EEF1 implicit solvation model developed by Lazaridis and Karplus.38 Hydrogen bond interactions are explicitly modeled using a reaction-like approach.39 The screened electrostatic interactions are modeled using the Debye–Hückel approximation with the Debye length set to 10 Å. The temperature is maintained by the Anderson thermostat.40 The units of mass, time, length, and energy used in our DMD simulations are 1 Da, ~50 fs, 1 Å, and 1 kcal/mol, respectively. By approximating interaction potentials with step functions, the sampling efficiency of DMD is significantly enhanced without frequent calculations of forces and accelerations (e.g., every ~1–2 fs) in traditional MD simulations. Implicit-solvent methods reduce the number of particles in atomistic simulations by approximating the solvent as a continuum, resulting in faster sampling of the conformational space.41, 42 The predictive power of DMD simulation with the Medusa force field and EEF1 implicit solvation model has been demonstrated in our previous studies, including the capture of native states with both secondary and tertiary structures in ab initio protein folding simulations36, 43, and the demonstration of fully consistent computationally-derived conformational ensembles with single-molecule FRET measurements in the conformational dynamics of multi-domain proteins44, 45. In addition, the accuracy of Medusa force field with EEF1 implicit solvation model in the aggregation of functional amyloid peptides of suckerin46 and pathological amyloid polypeptides of hIAPP26 has been benchmarked by the standard MD simulations with an explicit solvent model in our prior studies. With significantly enhanced sampling efficiency, the DMD algorithm has been widely used to study protein folding and amyloid aggregation both by our group17, 25, 26 and by others27, 28, 47.
Analysis methods.
The secondary structure was determined by using the DSSP (dictionary secondary structure of the protein) method.48 A contact was considered to have occurred when the minimum distance between heavy atoms of two nonconsecutive residues was within 0.55 nm.49 Two peptides were considered to belong to the same oligomer if they were inter-connected by at least one intermolecular contact.50 The total number of peptides within an oligomer was defined as the oligomer size. A hydrogen bond was considered if the distance between the backbone N and O atoms was ≤3.5 Å and the angle of N–H•••O ≥ 120°.51 Two peptides were considered to form a β-sheet if at least two consecutive residues from each β-strand conformation were inter-connected by more than two backbone hydrogen bonds. A β-sheet oligomer was defined as multiple β-sheets inter-connected by at least one heavy atom contact pair. The total number of β-strands in a β-sheet oligomer and a β-sheet layer was defined as the β-sheet oligomer size and the β-sheet size, respectively. The β-strand length referred to the number of consecutive residues that adopt a β-sheet structure in each peptide. To construct a two-dimensional (2D) free energy surface, the probability of a conformational state having a certain parameter value of x and y, denoted as P(x, y), was calculated and then used to compute the free energy surface as −RTln P(x, y).
Results and discussion
For each of the seven imperfect repeats, thirty independent micro-second aggregation simulations of ten peptides were performed to ensure sufficient sampling (Methods). The convergence of simulations was examined by the time evolution of several structural and energetic parameters, including the radius gyration (Rg), inter-peptide hydrogen bonds and contacts, and the probabilities of β-sheet and unstructured (random coil and bend, third column) formations. As illustrated by a typical trajectory of each molecular system (Figure S1, randomly selected out of 30 independent simulation trajectories), all systems reached the steady states in the last 400 ns for the above monitored parameters. The ensemble-averaged time evolution of these parameters over 30 independent simulations also showed no many changes during the last 400 ns, indicating that all the molecular systems were equilibrated (Figure S2). As an intrinsically disordered protein, αS is highly dynamic and undergo constant conformational changes in solution5, 6. Compared to the micelle-bound form, the cytosolic form is expected to be less helical and have more coils5, 6. There are no solution structures available in the PDB for the cytosolic form of αS17. Here, we only used the micelle-bound PDB structure of αS as the starting conformation for each repeat. These initially helical fragments rapidly converted to coils in our simulations (Figures S1–2), suggesting that our results were independent of the initial structures. The aggregation convergence of each repeat was assessed by analyzing the time evolution of the largest oligomer size in each of the 30 independent DMD trajectories, as well as the ensemble-average mass-weighted oligomer size (Figure S3). The significant differences in the largest oligomer sizes among the independent trajectories for each repeat indicated efficient sampling was achieved. Moreover, there were no significant fluctuations in the average oligomer size during last 400 ns, suggesting that all simulations were reasonably converged. Therefore, all the last 400 ns of 30 independent DMD trajectories were used for the equilibrium analysis of each repeat.
R3 and R6 repeats of αS spontaneously self-assemble into β-sheet-rich aggregates, while the other repeats showed weaker aggregation and β-sheet propensities.
We first analyzed the self-assembly dynamics of each αS repeat by examining the time evolution of the largest oligomer size, the total number of inter-peptide hydrogen bonds and contacts, and the content of unstructured (i.e., random coil and bend) and β-sheet conformations (Figure 1). The analysis revealed that the R1, R2, R4, R5, and R7 repeats had similar self-assembly dynamics by which the peptides could only transiently aggregate into small-size oligomers. The oligomer sizes were mostly less than 5, which was much smaller than the system size of 10. The frequent fluctuation of the largest oligomer size and the total number of inter-peptide hydrogen bonds and contacts suggested the aggregates of R1, R2, R4, R5, and R7 repeats were very dynamic and easily dissociated into isolated monomers. Time evolution of the secondary structure content showed that these repeats predominantly adopted unstructured conformations (> 70%) with little β-sheet structures (< 5%). Representative snapshots along the trajectories also demonstrated that they indeed mostly stayed as monomers adopting random coil and bend structures and had only a few small-size oligomers. In contrast, R3 and R6 repeats readily self-assembled into large β-sheet rich oligomers, but featured distinct dynamics. For instance, the largest oligomer size of R3 fluctuated around 6–10 after reaching equilibration, displaying frequent association and dissociation (Figure 1c). R6, on the other hand, formed a stable decamer with minimal fluctuations in the oligomer size (Figure 1f). Furthermore, R6 aggregates had a higher number of inter-peptide hydrogen bonds and contacts, as well as a higher β-sheet content compared to R3 aggregates. Representative snapshots revealed that R6 decamers featured highly ordered β-sheets. Conversely, R3 formed partial β-sheets in the oligomers, and these β-sheet oligomers were in equilibrium with unstructured monomers.
Figure 1. Self-assembly dynamics of αS repeats.
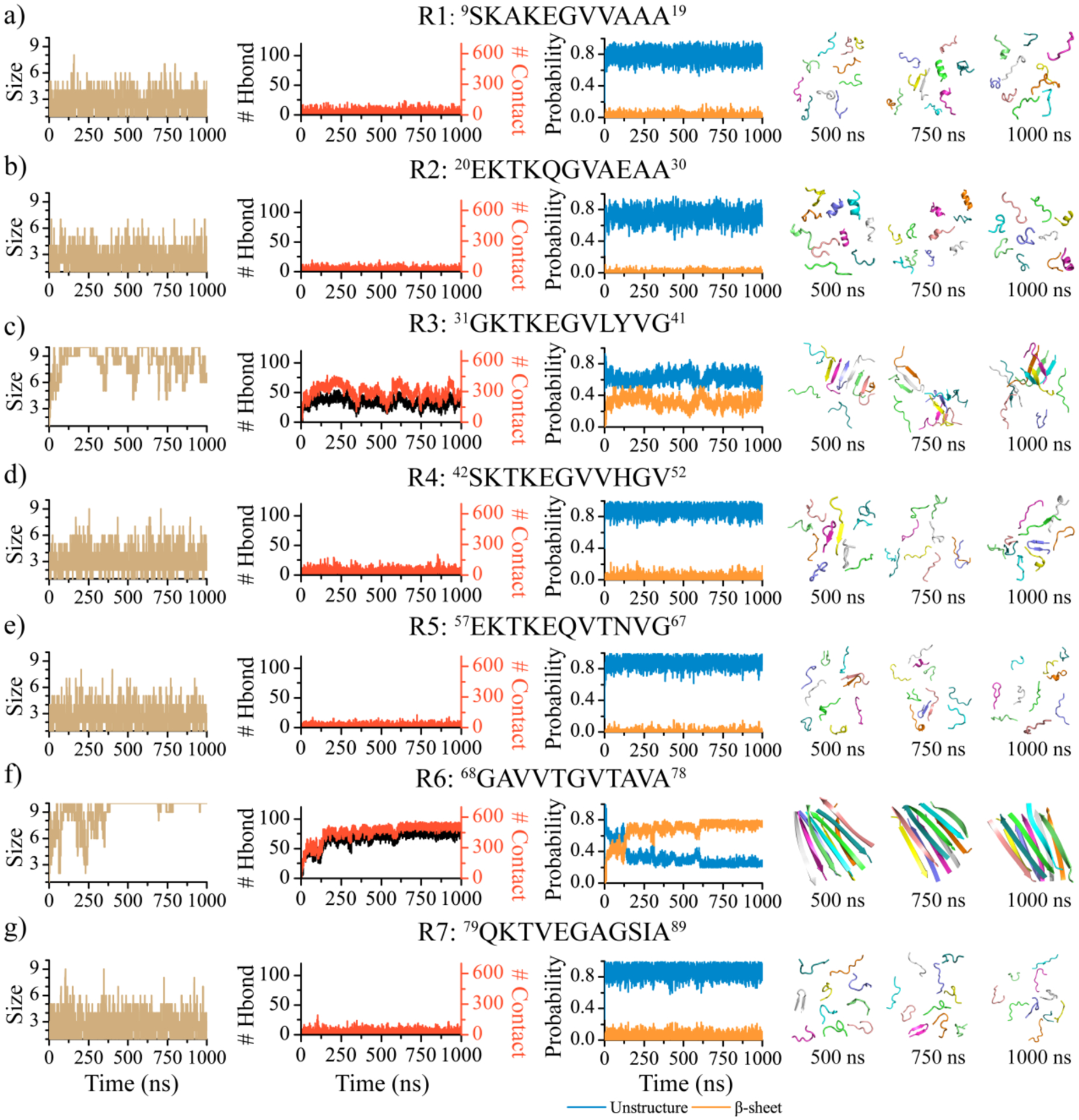
One representative trajectory is randomly selected from 30 independent DMD simulations to illustrate the self-assembly dynamics of each αS repeat: R1 a), R2 b), R3 c), R4 d), R5e), R6 f), and R7 g). The first column displays the time evolution of the largest oligomer size. The second column shows the time evolution of the total number of inter-molecular hydrogen bonds and contacts. The third column presents the averaged contents of the unstructured and β-sheet conformations as a function of simulation time. Snapshots along the simulation trajectory at 500, 750, and 1000 ns are presented to the right.
In order to explore the intricate details of the self-assembly dynamics of each αS repeat, we calculated the potential of mean force (PMF, i.e., the effective aggregation free energy landscape) as a function of the oligomer size and the average number of residues adopted β-sheet structures per repeat within the aggregates (Figure 2). All the 1000 ns DMD trajectories from independent simulations were included in the PMF analysis in order to capture the whole self-assembly process. To demonstrate the self-assembly dynamics on the free energy landscape, we also showed for each repeat a randomly selected trajectory the corresponding time evolution of the sizes of the largest oligomer, the largest β-sheet oligomer, the largest β-sheet, and the mass-weighted average β-sheet (Figure 2). As depicted in Figure 2, representative aggregates of R1, R2, R4, R5, and R7 that corresponded to free energy basins were comprised of only a small number of peptides. Interestingly, larger oligomers tended to have lower β-sheet contents. Hence, R1, R2, R4, R5, and R7 repeats of αS had low propensity to spontaneously self-assemble into β-sheet rich aggregates.
Figure 2. Self-assembly free energy landscape and aggregation conformational dynamics analysis for αS repeats.
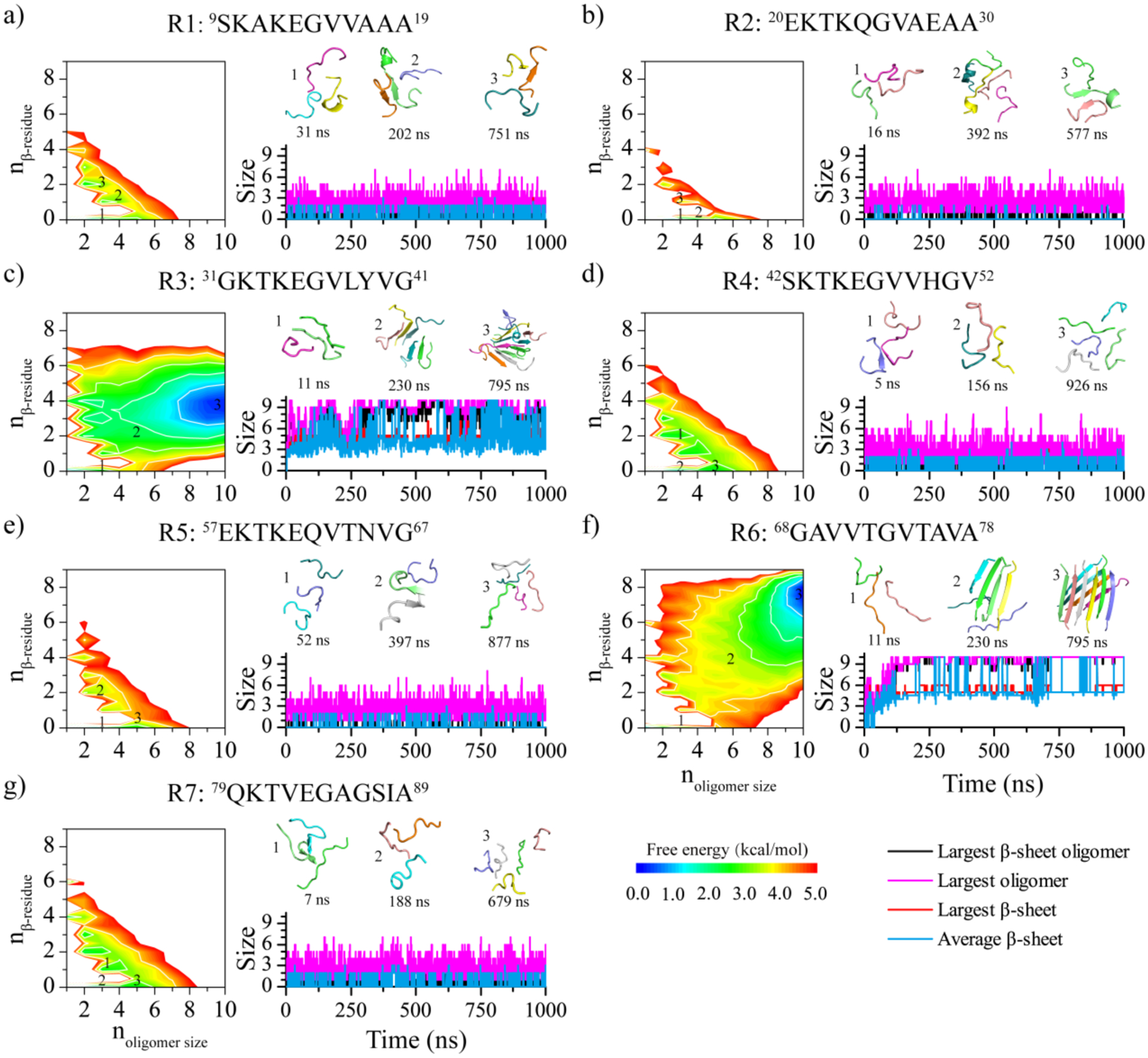
The aggregation free energy landscapes for the seven αS repeats a-g) are presented on the left as a function of oligomer size (noligomer size) and the average number of residues per chain adopting the β-sheet conformation (nβ-residue). The whole 1000 ns simulation data from 30 independent DMD trajectories are included to capture all self-assembly conformations during the aggregation process. One trajectory, randomly selected from the 30 independent DMD trajectories, is presented on the right to illustrate the self-assembly conformational dynamics of the corresponding repeat. The self-assembly dynamics are monitored by the time evolution of the largest oligomer size, largest β-sheet oligomer size, largest β-sheet, and average β-sheet. The snapshots along with the assemblies are shown in the inset on the right, and their corresponding states in the free energy landscape are labeled.
The aggregation free energy landscapes of R3 and R6 displayed distinctive features, although they both had large β-sheet rich oligomers as the lowest energy basin (Figure 2c,f). Compared to R6, the lowest free energy basin of R3 was wider in terms of oligomer sizes and had smaller β-sheet content. Specifically, the lowest free energy oligomers of R3 had the oligomers size ~7–10 and only ~3–5 residues adopting β-sheet conformation per chain (e.g., labeled 3 in Figure 2c). In contrast, the lowest free energy basin of R6 corresponded to oligomers with the size ~9–10 and ~7–8 residues per peptide adopting β-sheet structures (labeled 3 in Figure 2f). The analysis suggested that R6 formed more stable oligomers with higher β-sheet contents than R3.
Previous experiments have shown that deleting the R1 and R2 enhanced αS aggregation, while adding two more R1 and R2 inhibited αS fibrillization22. Deleting the seven-residue sequence of 37VLYVGSK43 from R3 abolished the aggregation of αS within 100 hours, while cutting residues 48VVHGVAT54 around the R4 only delayed αS fibrillization4. ThT fluorescence assays demonstrated that the aggregation kinetics of αS36–140 were much faster than those of αS1–140, αS14–140, and αS41–14052. The fibrillization of α-synuclein was significantly inhibited when some residues from R6 were deleted53. The β-synuclein (βS) had 90% homology to αS showed a dramatically weak aggregation due to the lack of the R6 repeat.54 In addition, prior experimental measurements have demonstrated that pure R6 repeat, also known as NACore, can efficiently aggregate into in-register cross-β fibrils without any assistance from the other repeats8. All of these results suggest that R3 and R6 played more important roles in driving the aggregation of αS than the other repeats, consistent with our simulation results that only R3 and R6 were capable of forming β-sheet-rich oligomers. Moreover, the aggregation tendency and β-sheet propensity of R6 were much stronger than those of R3. The observed difference in the self-assembly dynamics between R6 and R3 repeats can be attributed to the higher hydrophobicity of R6. Specifically, the head sequence of R3 shares the same polar and charged XKTKEGV motif with R1, R2, R4, R5, and R7 repeats. But the head sequence of R6, 68GAVVTG73, is highly hydrophobic (as illustrated by the sequence alignment in Figure S4). The primary distinction between R3 and other repeats including R1, R2, R4, R5, and R7 is the relatively stronger hydrophobicity of the non-conserved tails, because the hydrophobicity of leucine, valine, and isoleucine is much stronger than alanine or glycine55, 56. The non-conserved tail of R3 has two strong hydrophobic residues of leucine and valine. R4 also contains two valine residues, but they are separated by a polar histidine, while R3 has more hydrophobic tyrosine instead.
R6 formed extended β-sheets in the aggregates, whereas the R3 aggregates had the non-conserved hydrophobic tails forming partial β-sheets.
Next, we conducted a conformational analysis of the equilibrium aggregates of each repeat to study the conformational characteristics of αS repeat self-assemblies. The probability distributions of the mass-weighted oligomer size, β-sheet oligomer size, and β-sheet size were calculated for each molecular system (Figure 3a–c). The analysis also revealed that the R1, R2, R4, R5, and R7 repeats exhibited a strong preference of forming isolated monomers (greater than 50%) and dimers (more than 20%), with the observation of oligomers larger than 4 very rare (Figure 3a). For these repeats, the populations of β-sheet oligomers larger than two were too weak to be observed (Figure 3b). Conversely, R3 and R6 predominantly formed oligomers larger than 8, with small-size oligomers being extremely rare. The size probability distributions of R3 and R6 β-sheet oligomers were similar to their corresponding oligomer size distributions, indicating that all peptides in their aggregations formed β-sheets. The β-sheet size distribution within the aggregates of R3 and R6 ranged ~2–10, with the peaks around ~4–6, suggesting that their β-sheet oligomers were often composed of more than one β-sheets.
Figure 3. Equilibrium conformational and structural analyses of the αS repeat assemblies.
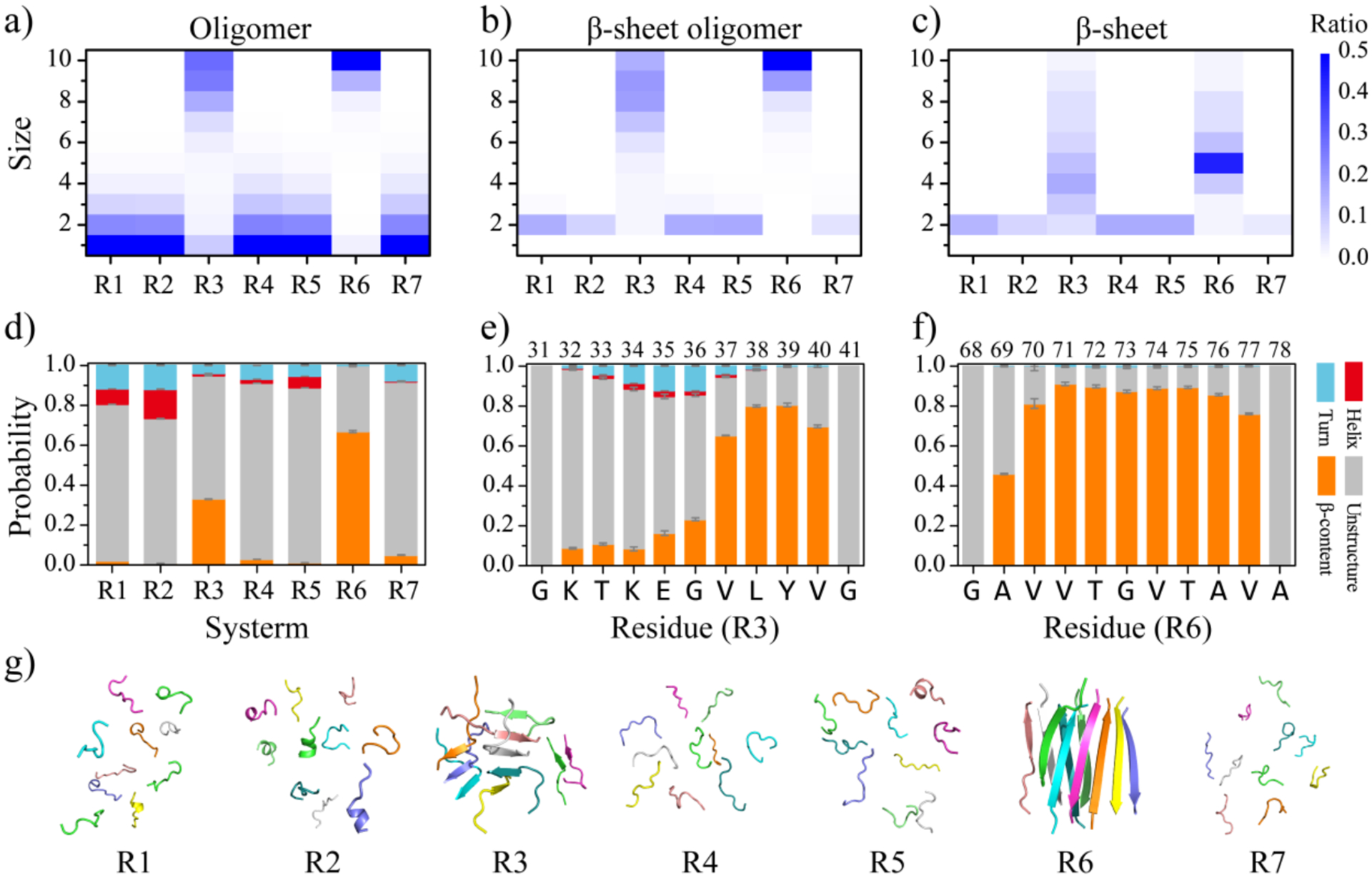
The probability distribution of the oligomer size a), β-sheet oligomer size b), and β-sheet size c) for the self-assemblies formed by each αS repeat are presented. To avoid bias from the initial state, only the conformations of the last 400 ns from all independent simulations are used for the conformational analysis. The average secondary structure contents in terms of unstructured, β-sheet, helix, and turn formations for the aggregates of each αS repeat are shown d). The propensities of each residue to adopt the coil and bend, β-sheet, helix, and turn for the R3 e) and R6 f) repeats are presented. The final snapshot for each repeat, randomly selected from 30 independent DMD trajectories, is also shown g).
The analysis of the secondary structure content revealed that the R1, R2, R4, R5, and R7 repeats mainly assumed unstructured conformations, such as random coil and bend, with a high propensity ranging ~72.3%−88.1% (Figure 3d). These repeats had low β-sheet contents, which were less than 3.0% (Figure 3d). In addition, we also observed helical formations in the self-assemblies of R1, R2, and R5 repeats, accounting for 7.5%, 14.7%, and 5.5%, respectively (Figures 3d&S5). The average β-sheet propensity of R3 and R6 aggregates was ~32.5% and ~66.8%, respectively. Further analysis of the secondary structure propensity of each residue revealed that the β-sheet structures of R3 were mainly formed by residues V37, L38, Y39, and V40 around the non-conserved hydrophobic tail, with a propensity greater than 60% (Figure 3e). In contrast, all residues from R6 repeats displayed strong β-sheet propensity, with residues V70-V77 having a β-sheet propensity greater than 70% (Figure 3f). Another recent MD simulation study has indicated that the β-sheet stability of residues V37-V40 within R3 and V70-V77 from R6 in preformed αS dimeric fibrils was significantly greater than that of other regions57. The final representative snapshots, randomly selected from 30 independent DMD trajectories, confirmed that only R3 and R6 could self-assemble into β-sheet-rich oligomers, while the other repeats mainly adopted isolated monomeric states with the population of oligomeric aggregates very rare (Figure 3g).
The probability distribution of β-strand lengths was calculated for the self-assemblies of R3 and R6 in Figure 4a. The β-strand length denotes the number of the consecutive residues that adopt β-sheet structures. R3 repeats predominantly comprised short β-strands with ~4 residues in length, while the β-strands within the R6 self-assemblies were mostly ~8–9 residues long, owing to the majority of residues assuming β-sheet conformations. Prior computational and experimental studies also revealed that the R6 repeats mainly formed extended cross-β structures.8, 14 As a result, the end-to-end distances of R6 were significantly larger than that of R3 in the aggregates (Figure 4b). The analysis of residue-pairwise contact frequency maps revealed that the interactions among hydrophobic and aromatic residues drove the segment of 37VLYV40 from R3 repeats to form both parallel and anti-parallel β-sheets (Figure 4c). Compared to the interactions among hydrophobic residues, the residue-pairwise frequencies involved by the charged and polar N-terminal residues were relatively weak (e.g., the cation-pi58 interactions of K32-Y39 and K34-Y39). Therefore, the root-mean-square fluctuation (RMSF) per residue within the self-assemblies of R3 indicated that the residues from the hydrophilic head were much more dynamic (Figure S7a). In contrast, the N-terminal head 68GAVVTGV74 of R6 was much more hydrophobic than the polar and charged conserved XKTKEGV motif. The RMSF analysis revealed that the average fluctuation of residues from R6 was much smaller than R3 (Figure S7b). The diagonal pattern observed in the main-chain contact frequencies and the corresponding representative snapshot suggested that all R6 residues participated in forming both parallel and anti-parallel β-sheets (Figure 4d), thereby stabilizing the interactions among the hydrophobic residues (Figure S6). The X-ray determined fibril structure of R6 showed parallel R6 repeats within the β-sheet layer but anti-parallel between β-layers8, suggesting that side-chain interactions may drive the formation of either parallel or anti-parallel β-sheets in R6 aggregates due to the sharing of similar residue-pairwise contact patterns. Overall, our results demonstrated that R6 repeats predominantly aggregated into extended β-sheet oligomers, whereas R3 repeats only aggregated partial β-sheets around the hydrophobic tail within their self-assemblies.
Figure 4. The analysis of β-sheet structures in the self-assemblies of R3 and R6 repeats.
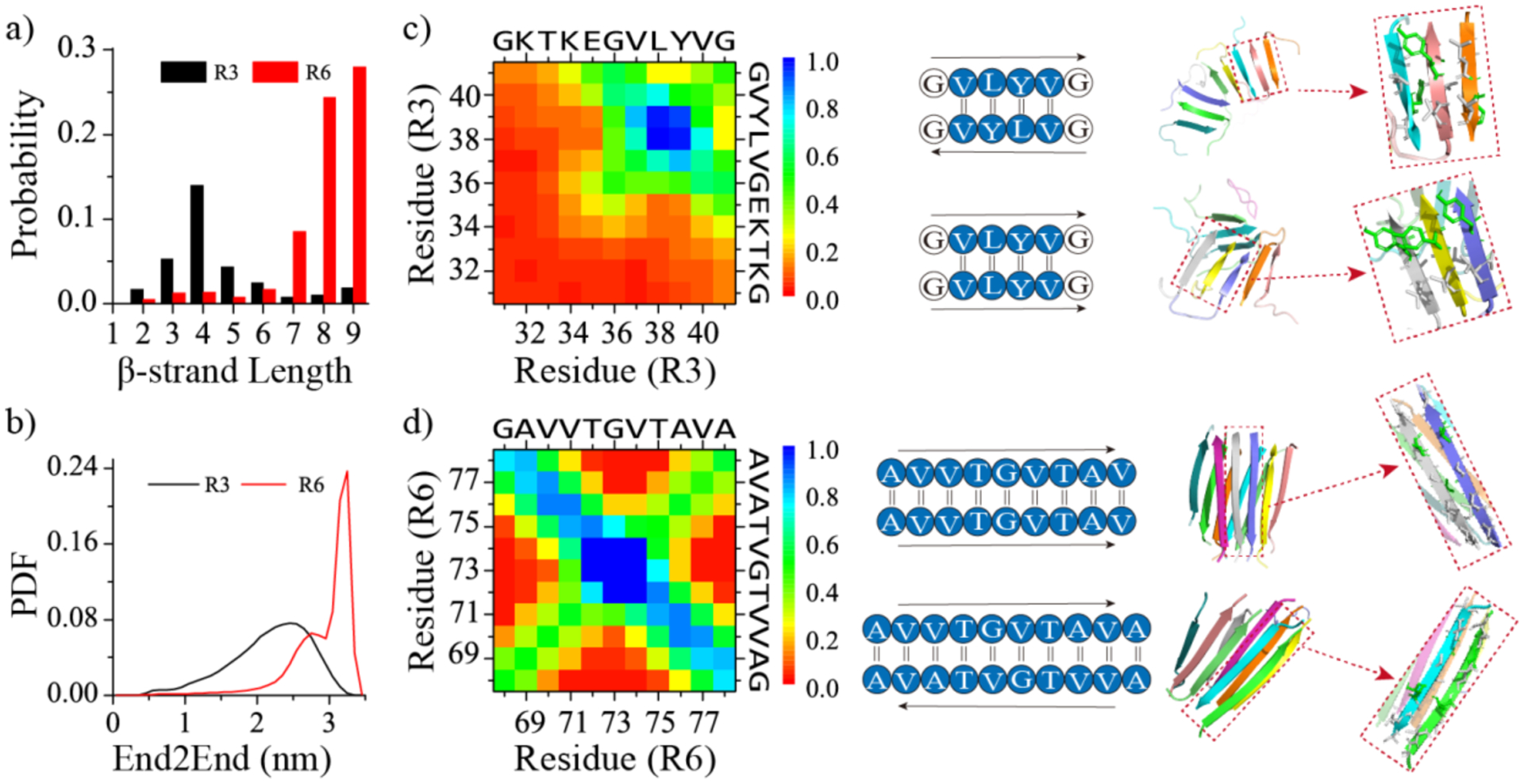
Probability distribution of β-strand length for each peptide within the self-assemblies of R3 and R6 repeats a). Probability distribution of end-to-end (End2End) distances for each R3 and R6 repeat within their aggregates b). Residue-pairwise intermolecular contact frequency maps between main-chain atoms within the self-assemblies of R3 c) and d). Representative parallel and anti-parallel β-sheet contact motifs are shown on the right. Only the last 400 ns of each 1000 ns independent simulation are used for the analysis.
The computationally derived aggregation and β-sheet propensities of αS repeats are consistent with their structures and organizations in experimentally solved αS fibrils.
Based on our systematic DMD simulations, only R3 and R6 repeats were capable of spontaneously self-assembling into β-sheet-rich oligomers. The fibril-like aggregates of R6 were more stable, while the R3 oligomers were dynamic. Hence, R6 can be regarded as the primary amyloidogenic core, and R3 as the secondary core. The computationally derived self-assembly and β-sheet propensities of αS repeats are consistent with their structures and organizations in recently solved αS fibrils,7, 18–20 including structures with PDB ID 2N0A18, 6H6B19, and 8H0320 (Figure 5a–c). The experimentally resolved structures of available αS fibrils usually start from the hydrophobic tail of the R3 and extended to the end of the NAC region7, 18–20, suggesting that R1 and R2 are likely unstructured in the fibrils and that R1 and R2 have low propensity to self-assembly in to β-sheet conformations. These observations along with prior experimental measurements where the presence of two more additional R1 and R2 repeats inhibited αS aggergation22 are consistent with our simulation results that the R1 and R2 repeats mainly assumed unstructured isolated monomers in silico. As the primary amyloidogenic core with the strongest aggregation propensity, R6 repeats were buried inside as the central cross-β core of all available αS fibrils. The hydrophobic tails of the R4, R5, and R7 repeats were packed around the hydrophobic interfaces of R6 β-sheets (Figure 5a–c). With 90% homologs to αS but lacking R6 repeat, the aggregation tendency of βS was significantly weaked54, 59, supporting the crucial role of R6 in driving the fibrillization of αS. This observation is also consistent with our simulation results that R4, R5, and R7 repeats alone mainly adopted isolated monomers rather than the β-sheet-rich oligomers in silico. Our simulation results along the available fibril structures suggested that the fibrillization of the R6 repeat could facilitate hydrophobic tails of adjacent R4, R5, and R7 repeats adopting β-sheet structures. As the secondary amyloidogenic core of αS, the R3 tail could form β-sheet in the peripherals of cross-β core, either pack against the R6 sheet (8H03, Figure 5c) or form an initial β-sheet independently in αS fibrils18, 19 (2N0A, 6H6B, Figure 5a,b). Deletion of the segment of 36GVLYVGS42 of the R3 hydrophobic tail suppressed αS from forming amyloid fibrils further confirming the important role of R3 in αS aggregation4. Together, R3 and R6 repeats play crucial roles in driving amyloid aggregation of αS and also determining the fibril morphology in terms of the structural organization or “fold” of αS.
Figure 5. The structures and organization of αS repeats in experimentally determined αS fibrils.
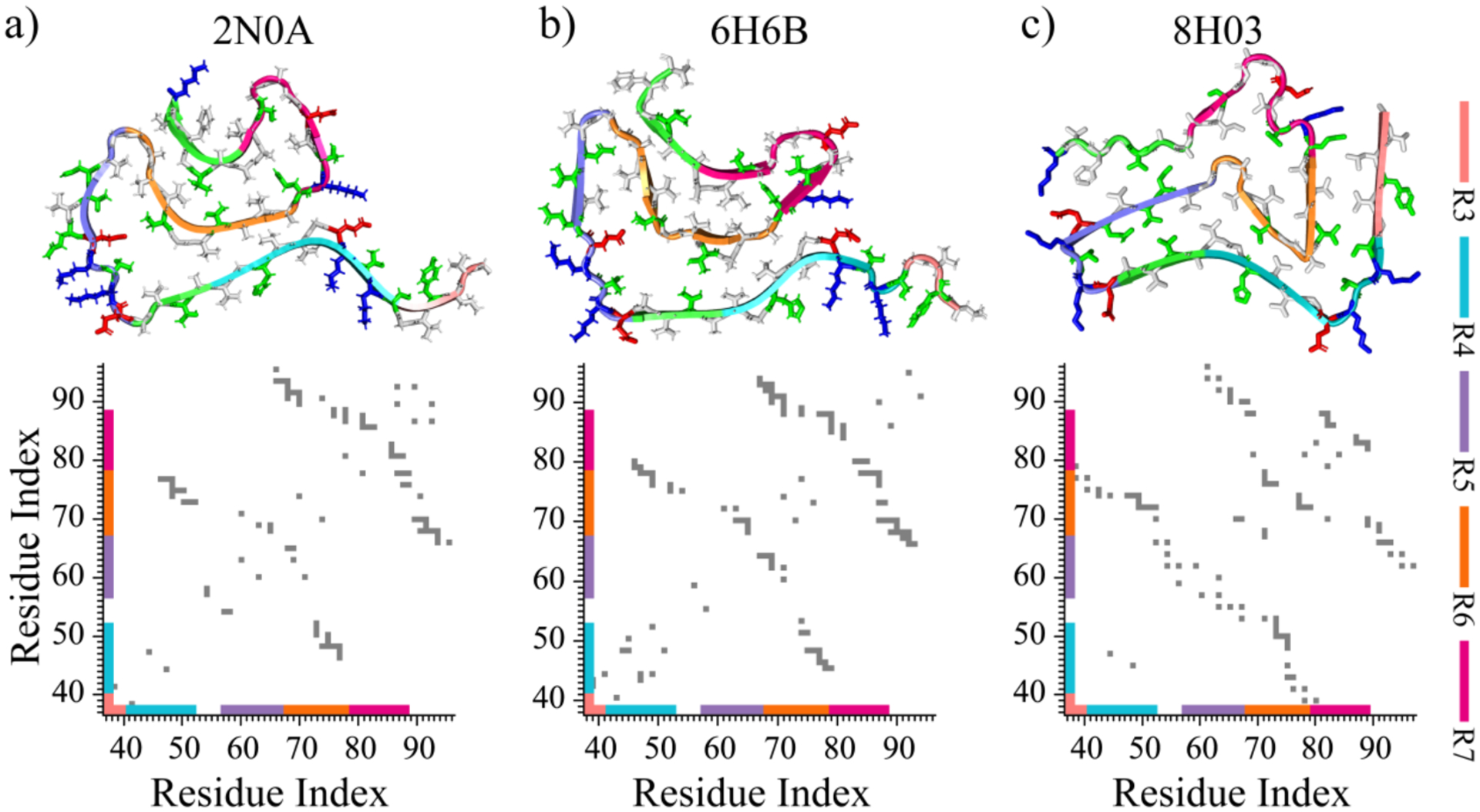
Three representative models of experimentally determined αS fibril structures are presented at the top, with their corresponding intra-peptide contact frequency maps illustrated at the bottom a-c). Each repeat of αS is colored differently in the cartoon for clarity. The side chains of hydrophobic, hydrophilic, negatively charged, and positively charged residues are colored white, green, red, and blue, respectively.
Conclusions
In this study, we investigated the self-assembly dynamics of the seven imperfect 11-residue repeats with the XKTKEGVXXXX motif, known to be responsible for membrane binding and amyloid aggregation of αS4,22. For each αS repeat, we performed multiple independent atomistic microsecond-long DMD aggregation simulations using an implicit-solvent approach, with up to 10 peptides simulated. Our simulation results revealed that only the R3 and R6 repeats could readily self-assemble into β-sheet-rich oligomers, while the other repeats – R1, R2, R4, R5, and R7 – had weak aggregation tendencies and mainly existed as unstructured monomers. The β-sheet-rich oligomers formed by the R3 were dynamic and underwent frequently dissociation/reassociation transitions. The β-sheets were formed by the non-conserved hydrophobic tail of R3. In contrast, the R6 repeats readily formed extended stable β-sheets, with nearly all residues participating in β-sheet formation. The aggregation kinetics of each αS repeats in our simulation may be faster than in standard explicit MD simulations, as it is known that implicit-solvent methods enhance conformational sampling by reducing solvent viscosity rather than altering the free energy landscape41, 42. Aggregation of αS in the cellular membrane environment occurs more rapidly than in a solution.60 Understanding the effects of the lipid membrane on αS is crucial and requires further investigation in future studies. The computationally derived aggregation and β-sheet propensities of αS repeats are consistent with their structures and organizations in experimentally resolved αS fibrils7, 18–20. For instance, the weak β-sheet and aggregation tendencies of R1 and R2 render them structurally-unresolved in the experimentally determined αS fibrils.7, 18–20 With the highest amyloid aggregation propensity, R6 repeats were buried inside as the central cross-β core, facilitating the hydrophobic tails of adjacent R4, R5, and R7 adopting β-sheets in the αS fibril7, 18–20. Far away from R6 in the sequence but with a moderate amyloid aggregation propensity, the R3 could also form cross-β independently at the beginner of the αS fibril18, 19. In summary, our findings suggest that the self-assembly of R6 and the R3 tail drives the aggregation of αS. Therefore, R6 and R3 hydrophobic tail – the primary and secondary amyloidogenic cores of αS – might serve as the targets in designing anti-amyloid inhibitors10, 11, 29, or, even as used for peptide-based competitive inhibitors30–32.
Supplementary Material
Acknowledgments.
This work was supported in part by the National Natural Science Foundation of China (Grant No. 11904189 and 82171527), Ningbo Top Medical and Health Research Program (No.2022020304), and NIH (R35GM145409). Computer simulations were supported by the multi-scale computational modeling core of NIH P20GM121342. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NSFC and NIH.
Footnotes
Supporting Information: The Supporting Information is available free of charge at the website:
The convergence assessments for the simulations of each repeat (Figure S1–S3); the sequence alignment of αS repeats(Figure S4); the secondary structure analysis for each residue in self-assemblies of R1, R2, R4, R5, and R7 (Figure S5); the residue-pairwise contact frequency analysis within the self-assemblies of R3 and R6 repeats (Figure S6); the root-mean-square fluctuation (RMSF) analysis for the self-assemblies of R3 and R6 (Figure S7). (PDF).
The authors declare no competing financial interest.
Data and Software Availability.
DMD simulation engine is available at Molecules In Action, LLC.
(www.moleculesinaction.com). Initial conformations, input parameter and topology files for DMD simulation, and representative DMD output trajectories for each system are available (https://doi.org/10.5281/zenodo.7787407).
References
- 1.Fanning S; Selkoe D; Dettmer U, Parkinson’s disease: proteinopathy or lipidopathy? NPJ Parkinsons Dis 2020, 6, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ke PC; Sani MA; Ding F; Kakinen A; Javed I; Separovic F; Davis TP; Mezzenga R, Implications of peptide assemblies in amyloid diseases. Chem Soc Rev 2017, 46, 6492–6531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen J; Zaer S; Drori P; Zamel J; Joron K; Kalisman N; Lerner E; Dokholyan NV, The structural heterogeneity of alpha-synuclein is governed by several distinct subpopulations with interconversion times slower than milliseconds. Structure 2021, 29, 1048–1064 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Doherty CPA; Ulamec SM; Maya-Martinez R; Good SC; Makepeace J; Khan GN; van Oosten-Hawle P; Radford SE; Brockwell DJ, A short motif in the N-terminal region of alpha-synuclein is critical for both aggregation and function. Nat Struct Mol Biol 2020, 27, 249–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bartels T; Choi JG; Selkoe DJ, alpha-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 2011, 477, 107–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iyer A; Claessens M, Disruptive membrane interactions of alpha-synuclein aggregates. Biochim Biophys Acta Proteins Proteom 2019, 1867, 468–482. [DOI] [PubMed] [Google Scholar]
- 7.Guerrero-Ferreira R; Kovacik L; Ni D; Stahlberg H, New insights on the structure of alpha-synuclein fibrils using cryo-electron microscopy. Curr Opin Neurobiol 2020, 61, 89–95. [DOI] [PubMed] [Google Scholar]
- 8.Rodriguez JA; Ivanova MI; Sawaya MR; Cascio D; Reyes FE; Shi D; Sangwan S; Guenther EL; Johnson LM; Zhang M; Jiang L; Arbing MA; Nannenga BL; Hattne J; Whitelegge J; Brewster AS; Messerschmidt M; Boutet S; Sauter NK; Gonen T; Eisenberg DS, Structure of the toxic core of alpha-synuclein from invisible crystals. Nature 2015, 525, 486–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Celej MS; Sarroukh R; Goormaghtigh E; Fidelio GD; Ruysschaert JM; Raussens V, Toxic prefibrillar alpha-synuclein amyloid oligomers adopt a distinctive antiparallel beta-sheet structure. Biochem J 2012, 443, 719–726. [DOI] [PubMed] [Google Scholar]
- 10.Santos J; Gracia P; Navarro S; Pena-Diaz S; Pujols J; Cremades N; Pallares I; Ventura S, alpha-Helical peptidic scaffolds to target alpha-synuclein toxic species with nanomolar affinity. Nat Commun 2021, 12, 3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Santos J; Cuellar J; Pallarès I; Byrd EJ; Lends A; Moro F; Abdul-Shukkoor MB; Pujols J; Velasco-Carneros L; Sobott F; Otzen DE; Calabrese AN; Muga A; Pedersen JS; Loquet A; Valpuesta JM; Radford SE; Ventura S, The structural architecture of an α-synuclein toxic oligomer. bioRxiv 2023, 2023.02.10.527650. [Google Scholar]
- 12.Scheidt T; Carozza JA; Kolbe CC; Aprile FA; Tkachenko O; Bellaiche MMJ; Meisl G; Peter QAE; Herling TW; Ness S; Castellana-Cruz M; Benesch JLP; Vendruscolo M; Dobson CM; Arosio P; Knowles TPJ, The binding of the small heat-shock protein alphaB-crystallin to fibrils of alpha-synuclein is driven by entropic forces. Proc Natl Acad Sci U S A 2021, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bodner CR; Dobson CM; Bax A, Multiple tight phospholipid-binding modes of alpha-synuclein revealed by solution NMR spectroscopy. J Mol Biol 2009, 390, 775–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun Y; Kakinen A; Zhang C; Yang Y; Faridi A; Davis TP; Cao W; Ke PC; Ding F, Amphiphilic surface chemistry of fullerenols is necessary for inhibiting the amyloid aggregation of alpha-synuclein NACore. Nanoscale 2019, 11, 11933–11945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Theillet FX; Binolfi A; Bekei B; Martorana A; Rose HM; Stuiver M; Verzini S; Lorenz D; van Rossum M; Goldfarb D; Selenko P, Structural disorder of monomeric alpha-synuclein persists in mammalian cells. Nature 2016, 530, 45–50. [DOI] [PubMed] [Google Scholar]
- 16.Brodie NI; Popov KI; Petrotchenko EV; Dokholyan NV; Borchers CH, Conformational ensemble of native alpha-synuclein in solution as determined by short-distance crosslinking constraint-guided discrete molecular dynamics simulations. PLoS Comput Biol 2019, 15, e1006859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Y; Wang Y; Liu Y; Wei G; Ding F; Sun Y, Molecular insights into the misfolding and dimerization dynamics of the full-length alpha-synuclein from atomistic discrete molecular dynamics simulations. ACS Chem Neurosci 2022, 13, 3126–3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tuttle MD; Comellas G; Nieuwkoop AJ; Covell DJ; Berthold DA; Kloepper KD; Courtney JM; Kim JK; Barclay AM; Kendall A; Wan W; Stubbs G; Schwieters CD; Lee VM; George JM; Rienstra CM, Solid-state NMR structure of a pathogenic fibril of full-length human alpha-synuclein. Nat Struct Mol Biol 2016, 23, 409–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guerrero-Ferreira R; Taylor NM; Mona D; Ringler P; Lauer ME; Riek R; Britschgi M; Stahlberg H, Cryo-EM structure of alpha-synuclein fibrils. Elife 2018, 7, e36402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fan Y; Sun Y; Yu W; Tao Y; Xia W; Liu Y; Zhao Q; Tang Y; Sun Y; Liu F; Cao Q; Wu J; Liu C; Wang J; Li D, Conformational change of alpha-synuclein fibrils in cerebrospinal fluid from different clinical phases of Parkinson’s disease. Structure 2023, 31, 78–87 e5. [DOI] [PubMed] [Google Scholar]
- 21.Farzadfard A; Pedersen JN; Meisl G; Somavarapu AK; Alam P; Goksoyr L; Nielsen MA; Sander AF; Knowles TPJ; Pedersen JS; Otzen DE, The C-terminal tail of alpha-synuclein protects against aggregate replication but is critical for oligomerization. Commun Biol 2022, 5, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kessler JC; Rochet JC; Lansbury PT Jr., The N-terminal repeat domain of alpha-synuclein inhibits beta-sheet and amyloid fibril formation. Biochemistry 2003, 42, 672–678. [DOI] [PubMed] [Google Scholar]
- 23.Pancoe SX; Wang YJ; Shimogawa M; Perez RM; Giannakoulias S; Petersson EJ, Effects of mutations and post-translational modifications on alpha-synuclein in vitro aggregation. J Mol Biol 2022, 434, 167859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stephens AD; Zacharopoulou M; Moons R; Fusco G; Seetaloo N; Chiki A; Woodhams PJ; Mela I; Lashuel HA; Phillips JJ; De Simone A; Sobott F; Schierle GSK, Extent of N-terminus exposure of monomeric alpha-synuclein determines its aggregation propensity. Nat Commun 2020, 11, 2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu YY; Wang Y; Zhang Y; Zou Y; Wei GH; Ding F; Sun YX, Structural perturbation of monomers determines the amyloid aggregation propensity of calcitonin variants. Journal of Chemical Information and Modeling 2022, 63, 308–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Y; Liu YY; Zhang Y; Wei GH; Ding F; Sun YX, Molecular insights into the oligomerization dynamics and conformations of amyloidogenic and non-amyloidogenic amylin from discrete molecular dynamics simulations. Phys Chem Chem Phys 2022, 24, 21773–21785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Emperador A; Orozco M, Discrete molecular dynamics approach to the study of disordered and aggregating proteins. J Chem Theory Comput 2017, 13, 1454–1461. [DOI] [PubMed] [Google Scholar]
- 28.Xiao X; Wang Y; Seroski DT; Wong KM; Liu R; Paravastu AK; Hudalla GA; Hall CK, De novo design of peptides that coassemble into beta sheet-based nanofibrils. Sci Adv 2021, 7, eabf7668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Horsley JR; Jovcevski B; Wegener KL; Yu J; Pukala TL; Abell AD, Rationally designed peptide-based inhibitor of Abeta42 fibril formation and toxicity: a potential therapeutic strategy for Alzheimer’s disease. Biochem J 2020, 477, 2039–2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun Y; Ding F, alphaB-crystallin chaperone inhibits abeta aggregation by capping the beta-sheet-rich oligomers and fibrils. J Phys Chem B 2020, 124, 10138–10146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hochberg GK; Ecroyd H; Liu C; Cox D; Cascio D; Sawaya MR; Collier MP; Stroud J; Carver JA; Baldwin AJ; Robinson CV; Eisenberg DS; Benesch JL; Laganowsky A, The structured core domain of alphaB-crystallin can prevent amyloid fibrillation and associated toxicity. Proc Natl Acad Sci U S A 2014, 111, E1562–E1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saini RK; Goyal D; Goyal B, Targeting human islet amyloid polypeptide aggregation and toxicity in type 2 diabetes: an overview of peptide-based inhibitors. Chem Res Toxicol 2020, 33, 2719–2738. [DOI] [PubMed] [Google Scholar]
- 33.Ulmer TS; Bax A; Cole NB; Nussbaum RL, Structure and dynamics of micelle-bound human alpha-synuclein. J Biol Chem 2005, 280, 9595–9603. [DOI] [PubMed] [Google Scholar]
- 34.Yin S; Ding F; Dokholyan NV, Eris: an automated estimator of protein stability. Nat Methods 2007, 4, 466–467. [DOI] [PubMed] [Google Scholar]
- 35.Yin S; Biedermannova L; Vondrasek J; Dokholyan NV, MedusaScore: an accurate force field-based scoring function for virtual drug screening. J Chem Inf Model 2008, 48, 1656–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shirvanyants D; Ding F; Tsao D; Ramachandran S; Dokholyan NV, Discrete molecular dynamics: an efficient and versatile simulation method for fine protein characterization. J Phys Chem B 2012, 116, 8375–8382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brooks BR; Bruccoleri RE; Olafson BD; States DJ; Swaminathan S; Karplus M, Charmm - a program for macromolecular energy, minimization, and dynamics calculations. J Comput Chem 1983, 4, 187–217. [Google Scholar]
- 38.Lazaridis T; Karplus M, Effective energy function for proteins in solution. Proteins 1999, 35, 133–152. [DOI] [PubMed] [Google Scholar]
- 39.Ding F; Borreguero JM; Buldyrey SV; Stanley HE; Dokholyan NV, Mechanism for the alpha-helix to beta-hairpin transition. Proteins 2003, 53, 220–228. [DOI] [PubMed] [Google Scholar]
- 40.Andersen HC, Molecular-dynamics simulations at constant pressure and-or temperature. J Chem Phys 1980, 72, 2384–2393. [Google Scholar]
- 41.Shen My MY; Freed KF, Long time dynamics of Met-enkephalin: comparison of explicit and implicit solvent models. Biophys J 2002, 82, 1791–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Anandakrishnan R; Drozdetski A; Walker RC; Onufriev AV, Speed of conformational change: comparing explicit and implicit solvent molecular dynamics simulations. Biophys J 2015, 108, 1153–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ding F; Tsao D; Nie H; Dokholyan NV, Ab initio folding of proteins with all-atom discrete molecular dynamics. Structure 2008, 16, 1010–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yanez Orozco IS; Mindlin FA; Ma J; Wang B; Levesque B; Spencer M; Rezaei Adariani S; Hamilton G; Ding F; Bowen ME; Sanabria H, Identifying weak interdomain interactions that stabilize the supertertiary structure of the N-terminal tandem PDZ domains of PSD-95. Nat Commun 2018, 9, 3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hamilton GL; Saikia N; Basak S; Welcome FS; Wu F; Kubiak J; Zhang C; Hao Y; Seidel CAM; Ding F; Sanabria H; Bowen ME, Fuzzy supertertiary interactions within PSD-95 enable ligand binding. Elife 2022, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu Y; Wang Y; Tong C; Wei G; Ding F; Sun Y, Molecular insights into the self-assembly of block copolymer suckerin polypeptides into nanoconfined beta-sheets. Small 2022, 18, e2202642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brodie NI; Popov KI; Petrotchenko EV; Dokholyan NV; Borchers CH, Solving protein structures using short-distance cross-linking constraints as a guide for discrete molecular dynamics simulations. Sci Adv 2017, 3, e1700479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kabsch W; Sander C, Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–637. [DOI] [PubMed] [Google Scholar]
- 49.Sun Y; Kakinen A; Xing Y; Faridi P; Nandakumar A; Purcell AW; Davis TP; Ke PC; Ding F, Amyloid self-assembly of hIAPP8–20 via the accumulation of helical oligomers, alpha-helix to beta-sheet transition, and formation of beta-barrel intermediates. Small 2019, 15, e1805166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sun Y; Ge X; Xing Y; Wang B; Ding F, Beta-barrel oligomers as common intermediates of peptides self-assembling into cross-beta aggregates. Sci Rep 2018, 8, 10353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang Y; Liu Y; Zhao W; Sun Y, Hydroxylated single-walled carbon nanotube inhibits beta2m(21)(−)(31) fibrillization and disrupts pre-formed proto-fibrils. Int J Biol Macromol 2021, 193, 1–7. [DOI] [PubMed] [Google Scholar]
- 52.McGlinchey RP; Ni X; Shadish JA; Jiang J; Lee JC, The N terminus of alpha-synuclein dictates fibril formation. Proc Natl Acad Sci U S A 2021, 118, e2023487118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Giasson BI; Murray IV; Trojanowski JQ; Lee VM, A hydrophobic stretch of 12 amino acid residues in the middle of alpha-synuclein is essential for filament assembly. J Biol Chem 2001, 276, 2380–2386. [DOI] [PubMed] [Google Scholar]
- 54.Yang X; Williams JK; Yan R; Mouradian MM; Baum J, Increased dynamics of alpha-synuclein fibrils by beta-synuclein leads to reduced seeding and cytotoxicity. Sci Rep 2019, 9, 17579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ide M; Maeda Y; Kitano H, Effect of hydrophobicity of amino acids on the structure of water. J Phys Chem B 1997, 101, 7022–7026. [Google Scholar]
- 56.Radzicka A; Wolfenden R, Comparing the polarities of the amino-acids - side-chain distribution coefficients between the vapor-phase, cyclohexane, 1-octanol, and neutral aqueous-solution. Biochemistry 1988, 27, 1664–1670. [Google Scholar]
- 57.Lan-Mark S; Miller Y, Insights into the interactions that trigger the primary nucleation of polymorphic alpha-synuclein dimers. ACS Chem Neurosci 2022, 13, 370–378. [DOI] [PubMed] [Google Scholar]
- 58.Infield DT; Rasouli A; Galles GD; Chipot C; Tajkhorshid E; Ahern CA, Cation-pi interactions and their functional roles in membrane proteins. J Mol Biol 2021, 433, 167035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jain MK; Singh P; Roy S; Bhat R, Comparative analysis of the conformation, aggregation, interaction, and fibril morphologies of human alpha-, beta-, and gamma-synuclein proteins. Biochemistry 2018, 57, 3830–3848. [DOI] [PubMed] [Google Scholar]
- 60.Fakhree MAA; Nolten IS; Blum C; Claessens MMAE, Different conformational subensembles of the intrinsically disordered protein alpha-synuclein in cells. J Phys Chem Lett 2018, 9, 1249–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.