

Abstract
BACKGROUND:
Neonatal epileptic seizures cause postictal dysregulation of cerebral blood flow. Hydrogen sulfide (H2S), a mediator with vasodilator and antioxidant properties, is produced in the brain by astrocyte cystathionine β-synthase (CBS). This study investigated whether H2S improves the cerebral vascular outcome of seizures.
METHODS:
Epileptic seizures were induced in newborn pigs using bicuculline. The effects of the CBS inhibitor aminooxyacetate (AOA) and the H2S donor NaHS on cerebral vascular outcome of seizures were examined in live pigs, cerebral endothelial cells, and cortical astrocytes.
RESULTS:
Brain H2S was elevated during seizures. AOA blocked H2S and reduced functional hyperemia in the epileptic brain. The endothelium- and astrocyte-dependent vasodilation of pial arterioles was impaired 48 h after seizures suggesting cerebral vascular dysfunction. Systemic NaHS elevated brain H2S and blocked reactive oxygen species in the epileptic brain and in primary endothelial cells and astrocytes during inflammatory and excitotoxic conditions. Postictal cerebrovascular dysfunction was exaggerated in H2S-inhibited pigs and minimized in NaHS-treated pigs.
CONCLUSIONS:
H2S elevation in the epileptic brain via activation of CBS contributes to functional hyperemia and exhibits cerebroprotective properties. The H2S donor NaHS enhances brain antioxidant defense and provides a therapeutic approach for preventing adverse cerebral vascular outcome of neonatal epileptic seizures.
INTRODUCTION
The gaseous mediator H2S is produced by the brain as a product of L-cysteine metabolism catalyzed by cystathionine β–synthase (CBS) and cystathionine γ–lyase (CSE).1–4 In the cerebral circulation, CBS is predominantly expressed in cortical astrocytes, whereas CSE is expressed mainly in cerebral vessels 1–6. The vasorelaxant effects of H2S have been demonstrated in cerebral and systemic circulations.2,9–11 Furthermore, H2S has been recently characterized as a component of the antioxidant defense mechanism.1,12,13 However, a paucity of information remains on the roles of H2S in the diseased brain.14–16 Remarkably, the vasodilator, antioxidant, and cytoprotective effects of H2S in the epileptic brain have not yet been investigated.
Neonatal epileptic seizures cause cerebral vascular injury leading to sustained dysregulation of cerebral blood flow (CBF) during the delayed postictal period.17–19 Seizures are associated with increased production of reactive oxygen species (ROS) in the brain.19 Oxidative stress is the major contributor to prolonged cerebral vascular dysfunction that involves both endothelial and astrocyte components of the neurovascular unit.17–19 Preservation of cerebral vascular functions (cerebroprotection) is an important component of neuroprotection, as the newborn brain is highly dependent on sustainable CBF regulation. Strengthening the antioxidant capacity of the neonatal brain may provide an effective approach for ameliorating the detrimental effects of epileptic seizures on cerebral vascular functions.
The present study addresses the hypothesis that H2S contributes to functional hyperemia and exhibits antioxidant and cerebroprotective properties in the epileptic brain. To address this hypothesis, we used a bicuculline-induced epileptic seizure model in newborn pigs as a a translationally relevant large animal model of neonatal cerebral vascular disease. We applied the closed cranial window technique to examine the dynamics and the contribution of H2S enzymatically produced in the brain to the delayed cerebral vascular outcome of seizures. We also used the H2S donor NaHS as a potential approach that may prevent the deteriorating effects of seizures in the neonatal cerebral circulation. In addition, we used primary cultures of cerebral microvascular endothelial cells and astrocytes from the newborn pig brain as an in vitro model of neurovascular injury to investigate the mechanism of H2S-mediated cerebroprotection. Overall, we collected in vivo and in vitro evidence that H2S protects the neonatal epileptic brain via multiple mechanisms that involve its vasodilator and antioxidant properties.
METHODS
Animals
Newborn pigs (1–5 days old, 1.5–3.0 kg, either sex) were purchased from a commercial breeder. The University of Tennessee Health Science Center (UTHSC) Animal Care and Use Committee (IACUC) reviewed and approved all procedures involving animals in compliance with National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals. All experiments were conducted according to the ARRIVE guidelines 2.0 for animal research.
Model of neonatal epileptic seizures
The GABAA receptor blocker bicuculline induces glutamatergic seizures by disrupting the normal balance between inhibitory and excitatory neurotransmitters. Pigs were initially anesthetized with ketamine-xylazine (33:2 mg/kg, i.m.) and maintained by administration of α-chloralose (50 mg/kg iv), as we described previously.17–20 Pigs were intubated, ventilated with room air, and instrumented to monitor systemic parameters and blood gases. The body temperature was maintained at 37–38°C by a servo-controlled heating pad. Pigs were paralyzed by pancuronium bromide (0.2 mg/kg, i.v.).19 Bicuculline (3 mg/kg, i.p.) produced a burst of epileptiform discharges and cerebral hyperemia that lasted for ~2 h.20 After recovery from seizures and anesthesia, pigs were extubated and transferred to the animal care facility for full recovery for 48 h. No major complications were noted during the postictal period.
Intravital microscopy via closed cranial windows
Closed cranial windows were surgically installed in anesthetized and ventilated pigs for measurements of pial arterioles and for collection of periarachnoid cerebrospinal fluid (pCSF) from the cortical surface as we described previously.6, 17–19 The space under the window (500 μl) was filled with artificial cerebrospinal fluid (aCSF) that contained (in mM): 3.0 KCl, 1.5 MgCl2, 1.5 CaCl2, 132 NaCl, 6.6 urea, 3.7 dextrose, and 24.6 NaHCO3 equilibrated with 6% CO2-6% O2-88% N2 to pH 7.3–7.35 at 37°C. Pial arteriolar diameter was measured using a digital video micrometer connected to a Wild Heerbrugg M3B Type-S intravital microscope. For intravital microscopy, three to four pial arterioles (30–100 μm) in each pig were selected for observation. pCSF (500 μl) was sampled from under the window in 10-min intervals before and during the 2-h ictal period for further detection of H2S production.
Cerebral vascular functions
Pial arterioles are major resistance arterioles that play a key role in regulation of CBF. Cerebral vascular functions were tested using the closed cranial window technique in control and postictal pigs by examining the responses of pial arterioles to: 1) the endothelium-dependent vasodilator, bradykinin (10−6 M); 2) the endothelium- and astrocyte-dependent vasodilator glutamate (10−4 M); 3) the astrocyte-dependent vasodilator adenosine diphosphate (ADP, 10−4 M); and 4) vascular smooth muscle-targeting vasodilator sodium nitroprusside (SNP). All compounds were topically applied under the cranial window at concentrations that produce submaximal vasodilation, as established in our previous publications.17,18
Primary cultures of cerebral microvascular endothelial cells and astrocytes
Cerebral microvessels (60–300 μm) and vessel-free parenchyma were collected by differential filtration of the brain cortex homogenates through nylon mesh filters as described.19,22,23 Cerebral microvascular endothelial cells (CMVEC) were dislodged from cerebral microvessels by treatment with collagenase-dispase and purified by the Percoll density gradient centrifugation.23 CMVEC were plated on Matrigel-coated surfaces and cultured in DMEM supplemented with 20% fetal bovine serum (FBS), endothelial cell growth supplement (ECGS, 30 μg/ml), heparin (1 U/ml), and antibiotic/antimycotic mixture for 5–6 d. 22 Cortical astrocytes from the brain cortex parenchyma were grown in the astrocyte-supporting DMEM supplemented with 20% FBS, epidermal growth factor (EGF, 10 ng/ml), and antibiotic/antimycotic mixture for 10–14 days.22 All experiments were performed using confluent quiescent cells.
ROS production
To detect ROS production, we used dihydroethidium (DHE), the blood-brain-barrier-permeable oxidant-sensitive probe, as we described previously.19,22,23 Oxidation of DHE produces the superoxide-specific product (2-hydroxyethidium) and a non-specific oxidation product (ethidium) which have overlapping fluorescence spectra and, if necessary can be separated using liquid chromatography–mass spectrometry (LC–MS).24 However, even without separation of these spectra, DHE fluorescence can be used as a general indicator of overall ROS production in tissues and cells.24 To measure ROS in brain tissue, intact and epileptic newborn pigs were injected with DHE (1.5 mg/kg, i.v.) as we described previously.19 Cerebral microvessels and astrocyte-enriched parenchyma were isolated from the brain cortex 1 h after DHE administration. The fluorescent products of DHE oxidation in the tissue lysates were detected by spectrofluorimetry. To detect ROS in endothelial cells and astrocytes, cells grown on 12-well Costar plates were exposed to seizure-related inflammatory and excitotoxic pro-oxidants TNF-α (30 ng/ml) or glutamate (2 mM) in the presence or absence of NaHS (50 μM) or the superoxide scavenger Tiron (1 mM) for 60 min at 37°C. DHE (20 μM) was added to the culture medium for the last 20 min of the incubation period.19 DHE fluorescence (excitation/emission, 485/590 nm) in fresh brain tissue and in cultured cells was measured by a Synergy HT microplate reader (BioTek Instruments; Winooski, VT) and normalized to the total amount of protein in the samples.
Apoptosis detection
DNA fragmentation, a key event in apoptosis, was detected by formation of cytoplasmic histone-complexed DNA fragments (mono- and oligonucleosomes).23 CMVEC or astrocytes were treated with TNF-α (30 ng/ml) or glutamate (2 mM) in the presence or absence of NaHS (20 μM) or Tiron (1 mM) for 3–5 h. Apoptotic DNA fragments were detected using a photometric enzyme immunoassay (ELISA PLUS, Roche Applied Science; Indianapolis, IN) and quantified on a Synergy HT multi-mode microplate reader (BioTek Instruments, Winooski, VT).
H2S detection
H2S levels in pCSF were measured using a H2S-selective electrode (Lazar Research Laboratories) on a Jenko Model 6230 microcomputer-based pH/mV/Temp meter (Jenko Electronics, LTD), as described elsewhere.21 pCSF samples were collected from the cranial windows in 500-μL tubes preloaded with NaOH (final concentration, 40 mM) to keep samples in alkaline pH for measurements of all H2S-derived species in solution. Sodium sulfide (Na2S; 0.5–10 μM) was used for a calibration curve as recommended by the manufacturer. The H2S detection limit was 0.4 ± 0.2 μM.
Materials
Bicuculline was purchased from Tocris (Minneapolis, MN); pancuronium bromide from Astra Pharmaceutical Products (Westborough, MA); NaHS from Cayman Chemical (Ann Arbor, MI); dihydroethidium from Life Technologies (Thermo Fisher Scientific, Waltham, MA); and cell culture reagents were from GE Healthcare Life Sciences (Pittsburg, PA). All other reagents were purchased from Sigma (St. Louis, MO).
Statistical analysis.
Data are presented as mean ± SD of absolute values or percentage of control. Data were analyzed by Student’s t-test and analysis of variance (ANOVA) for independent measurements. P values of < 0.05 were considered to be statistically significant.
RESULTS
Seizures elevate brain H2S production and produce cerebral hyperemia via CBS activation
Production of H2S in live brain was evaluated by measuring H2S levels in pCSF samples collected at 10 m intervals before and during a 2 h ictal period. The baseline level of H2S level in pCSF was 0.9 ± 0.3 μM (N = 6). These values confirm our previous measurements using H2S-selective electrode and gas chromatography-mass spectrometry (GCMS).6 Seizures caused a rapid elevation in H2S that was sustained for the duration of the ictal period (up to 2h) (Fig. 1A). The maximal elevation of H2S (3-fold) coincided with a maximal dilator response of the pial arterioles (60–80% above the baseline) during the initial 10–40 m ictal period (Fig. 1B).
Figure 1.
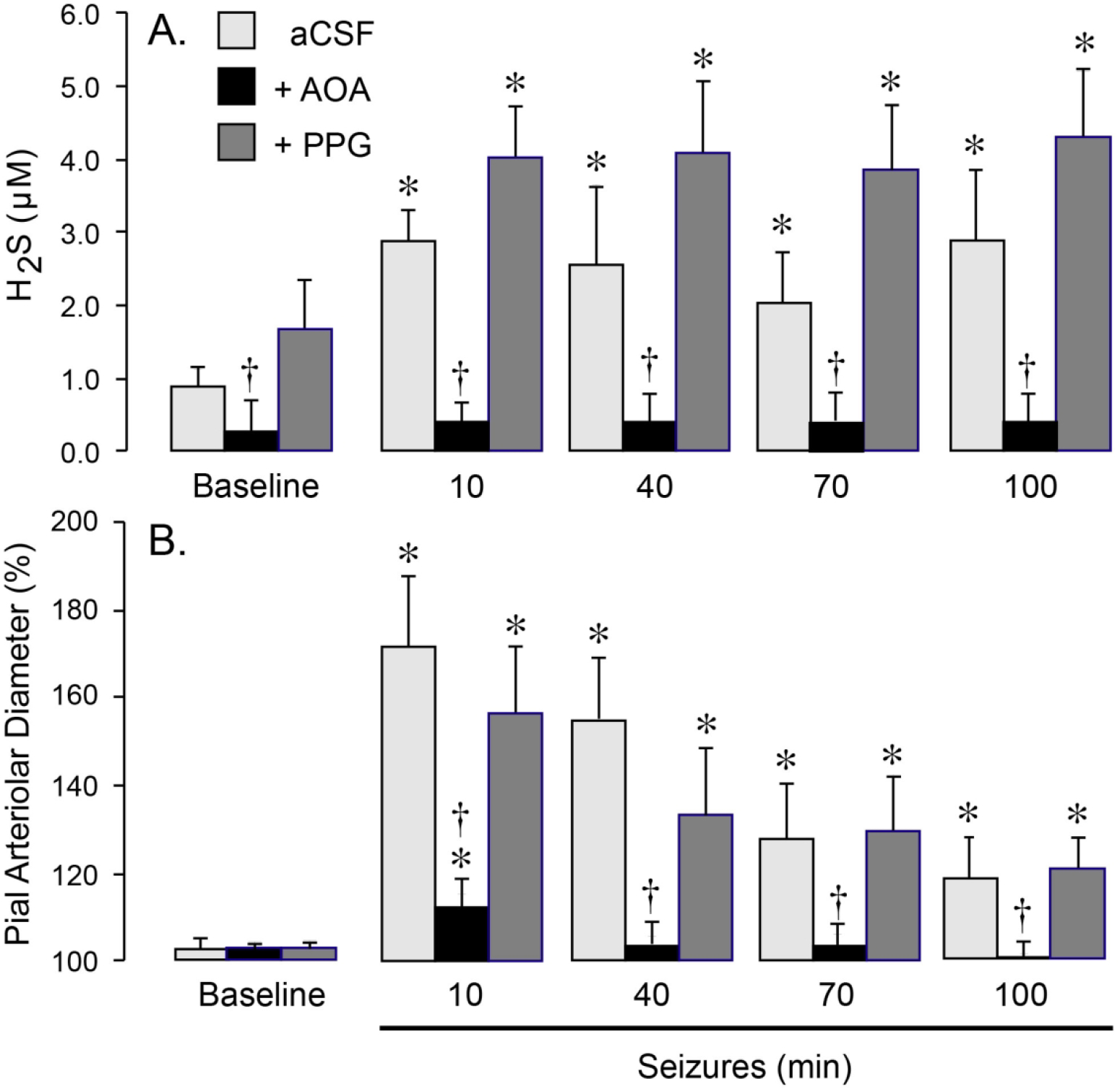
CBS contributes to brain H2S production (A) and pial arteriolar dilation (B) during seizures. Seizures were induced by bicuculline (3 mg/kg, i.p.) in newborn pigs, that were untreated (aCSF) or treated with selective inhibitors of CBS (AOA, 5 mM) or CSE (PPG, 5 mM) that were topically applied to the cortical surface. A: H2S concentrations in periarachnoid cerebrospinal fluid (pCSF) collected from the closed cranial windows in 10-m intervals before (Baseline) and 10–100 m after bicuculline administration. B: Dilator responses of pial arterioles to seizures 10–100 m after bicuculline administration. N = 6 pigs per group. Values are means ± SD. *P < 0.05, compared with the corresponding baseline values. †P < 0.05, compared with the corresponding values in untreated pigs.
To investigate the relative contributions of CBS and CSE to H2S generation, we used selective inhibitors of these enzymes. The CBS inhibitor aminooxyacetate (AOA, 5 mM), or the CSE inhibitor DL-propargyl glycin (PPG, 5 mM) was applied directly to the brain surface under the cranial window for the duration of the experiment (100 m). The concentrations of the inhibitors were selected based on preliminary and published data.6,25 Seizures are accompanied by tachycardia, which is a sensitive systemic indicator of epileptiform activity.17,26,27 Neither AOA or PPG altered the dynamics of tachycardia and mean arterial blood pressure (MABP; Table 1), suggesting that these inhibitors had no effects on bicuculline-evoked epileptiform neuronal discharges.
Table 1.
Systemic circulatory parameters during seizures in newborn pigs.
| Experiment | Time after Bicuculline (min) | MAPB, mm Hg | Heart Rate beats/min | Body T° (C) |
|---|---|---|---|---|
|
| ||||
| Control (aCSF) | 0 | 63 ± 8 | 151 ± 37 | 37.5 ± 0.4 |
| 10–20 | 68 ± 9 | 235 ± 30* | 37.8 ± 0.3 | |
| 30–50 | 65 ±7 | 218 ± 51* | 38.2 ± 0.9 | |
| 60–90 | 65 ±7 | 210 ± 31* | 38.2 ± 0.9 | |
| 100–120 | 60 ±8 | 186 ± 45 | 37.9 ± 0.5 | |
|
| ||||
| +AOA | 0 | 58 ± 6 | 168 ± 45 | 37.8 ± 0.6 |
| 10–20 | 59 ± 9 | 253 ± 3* | 37.9 ± 0.3 | |
| 30–50 | 60 ±8 | 248 ± 13* | 38.2 ± 0.9 | |
| 60–90 | 62 ±8 | 220 ± 11* | 38.2 ± 0.9 | |
| 100–120 | 58 ±9 | 210 ± 18 | 38.1 ± 0.5 | |
|
| ||||
| +PPG | 0 | 63 ± 5 | 165 ± 29 | 37.1 ± 0.6 |
| 10–20 | 75 ± 8 | 251 ± 6* | 37.5 ± 0.4 | |
| 30–50 | 69 ±7 | 258 ± 9* | 37.6 ± 0.4 | |
| 60–90 | 63 ±16 | 250 ± 11* | 37.5 ± 0.3 | |
| 100–120 | 62 ±11 | 220 ± 18* | 38.5 ± 0.1 | |
Values are means ± SD. N = 4 pigs in each group. Seizures were induced by bicuculline (3 mg/kg ip). aCSF (Control), the CBS inhibitor AOA (5 mM), or the CSE inhibitor PPG (5 mM) were placed to the brain surface under the cranial window 20 min before bicuculline and kept constant during the experiment.
P< 0.05 compared to the baseline values.
AOA reduced the baseline H2S and completely prevented H2S elevation during the ictal period (Fig. 1A). Furthermore, AOA blocked the dilator response of pial arterioles to seizures (Fig. 1B). These data suggest that CBS is the main contributor to brain H2S production and cerebral hyperemia during the ictal state. In contrast, in the presence of PPG, the cerebral vasodilator response to seizures remained unaltered (Fig. 1B), and we even observed a tendency to higher H2S levels during the basal and ictal states (P = 0.08) (Fig. 1A). These data provide evidence that an increased production of H2S in the brain via CBS activation is required for the functional cerebral hyperemia response to neuronal activation.
CBS-derived H2S attenuates adverse cerebrovascular outcome of epileptic seizures
To investigate whether endogenously produced H2S contributed to long term cerebral vascular outcome of seizures, we compared postictal cerebral vascular responses in newborn pigs with native or pharmacologically inhibited production of H2S in the brain. In 48 h postictal pigs with native levels of H2S production, the responses of pial arterioles to endothelium- and astrocyte-dependent dilators bradykinin, glutamate, and ADP were reduced by 50–60% (Fig. 2). In 48 h postictal pigs treated with the CBS inhibitor AOA (5 mg/kg, i.p.) before seizures, the responses of pial arterioles to bradykinin, glutamate and ADP were completely blocked (Fig. 2). In contrast, the CSE inhibitor PPG (5 mg/kg, i.p.) administered before seizures did not aggravate the 48h postictal cerebral vascular dysfunction (Fig. 2). These findings suggest that CBS-derived H2S lessens cerebral vascular dysfunction caused by seizures.
Figure 2.
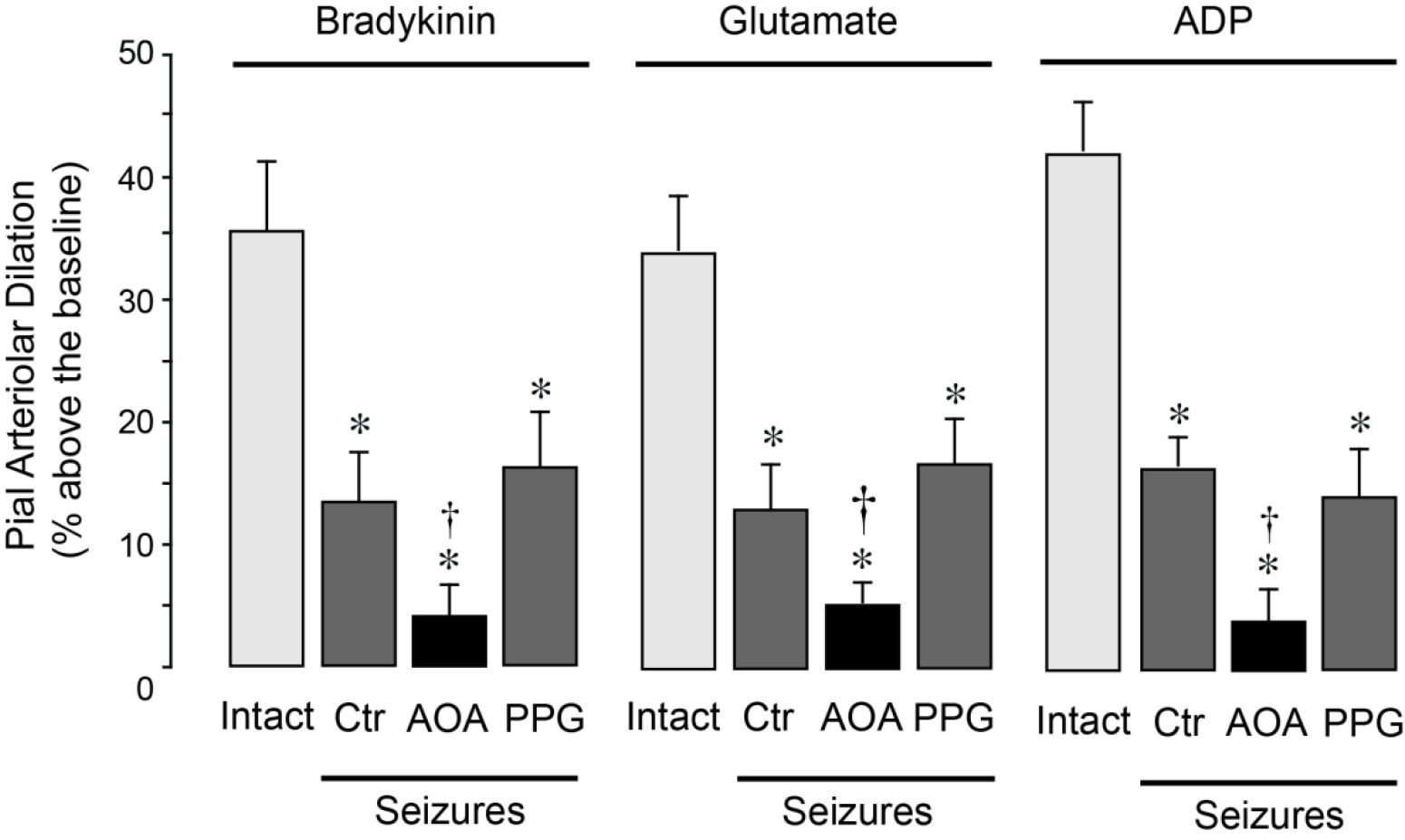
CBS inhibition aggravates long-term impairment of postictal cerebral vasodilator functions. Seizures were induced by bicuculline (3 mg/kg, i.p.) The responses of pial arterioles to topical endothelium- and astrocyte-dependent vasodilators bradykinin (BK, 10−6 M), glutamate (Glu, 10−4 M), and ADP (10−4 M) were evaluated in intact pigs and in the 48 h postictal pigs (Seizures) that were untreated (Ctrl) or treated with either the CBS inhibitor AOA (5 mg/kg, i.p.) or the CSE inhibitor PPG (5 mg/kg, i.p.) (N = 5 per group). Values are means ± SD. *P < 0.05 compared with the baseline (Base). †P < 0.05 compared with the corresponding responses in the intact group.
Systemic administration of NaHS elevates H2S in the brain and dilates pial arterioles
We investigated whether systemic administration of NaHS could deliver H2S to the brain during physiological conditions. NaHS (3 mg/kg) was aseptically administered to pigs either parenterally (i.p.) or enterally via an orogastric tube. Systemic parameters remained at physiological levels, and no changes were recorded during the first 80 m period after the NaHS administration (MABP, 60 ± 3 mm Hg; HR, 120 ± 5 bpm; core temperature, 37.5 ± 0.1° C). The pCSF samples were collected at 10 m intervals for H2S measurements. NaHS treatment caused a rapid and sustained increase in H2S levels (up to 3- to 4-fold) during the first 10–80 m after parenteral or enteral administration (Fig. 3A). Furthermore, NaHS administered via parenteral or enteral routes produced an immediate dilation of pial arterioles (10–25% above the baseline) that was maintained during the 80 m period (Fig. 3B). These findings suggest that systemic NaHS can be used as a pharmacological tool for delivery of H2S to the brain.
Figure 3.
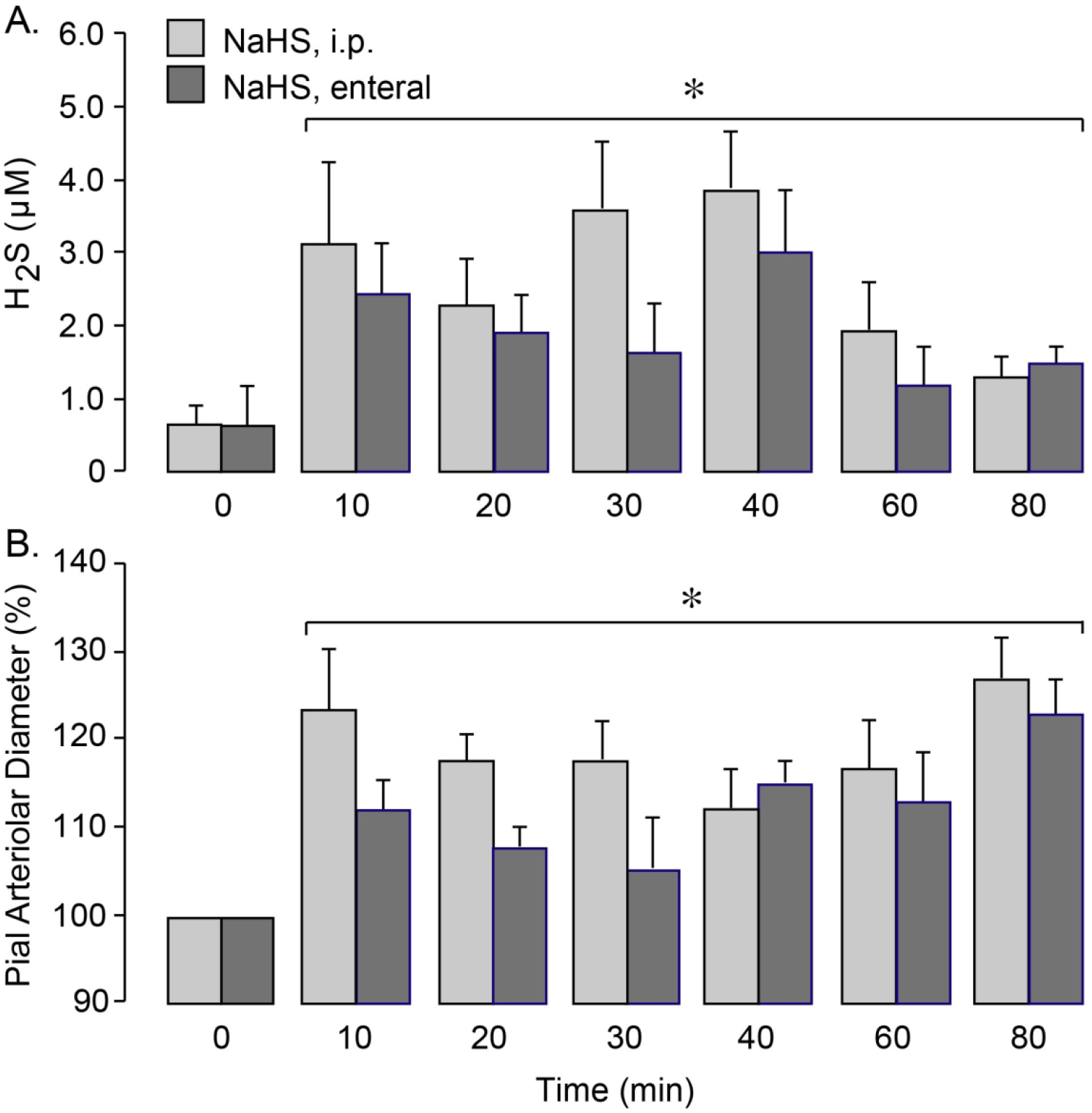
Systemic administration of NaHS increases H2S in the brain (A) and dilates pial arterioles (B). NaHS (3 mg/kg) was administered parenterally (i.p.) or enterally (orogastric tube). A: H2S levels in periarachnoid cerebrospinal fluid (pCSF) collected before (0 m) and 10–80 m after NaHS administration. B: Vasodilator responses of pial arterioles to NaHS administration. N = 6 pigs per group. Values are means ± SD. *P < 0.05, compared with the baseline values.
Antioxidant effects of NaHS in the epileptic brain
Our previous studies demonstrated that epileptic seizures increased production of ROS in the brain, as detected by DHE fluorescence.19 We determined the effects of systemic NaHS on brain oxidative stress induced by epileptic seizures. To this end, we measured DHE fluorescence in cerebral microvessels and astrocytes freshly isolated from the brain cortex in intact and epileptic pigs, untreated (seizure control) or pretreated with NaHS (3 mg/kg, i.p.) or Tiron (2g/kg, i.v.) (N=4 pigs per group) (Fig. 4A). A significant increase in DHE fluorescence indicative of ROS elevation was observed in cerebral vessels (~3-fold) and in cortical astrocytes (~1.5-fold) freshly isolated from the epileptic brain. In pigs pretreated with the superoxide scavenger Tiron (2 g/kg, i.v.) before bicuculline administration, the surge of DHE fluorescence in cerebral vessels and the brain parenchyma was largely prevented, suggesting that the superoxide anion is the major contributor to ROS elevation in the epileptic brain. Systemic NaHS (3 mg/kg, i.p.) also greatly reduced ROS elevation in both cerebral vessels and astrocyte-enriched brain cortex parenchyma (Fig. 4A), indicating strong antioxidant capacity of NaHS in the epileptic brain.
Figure 4.
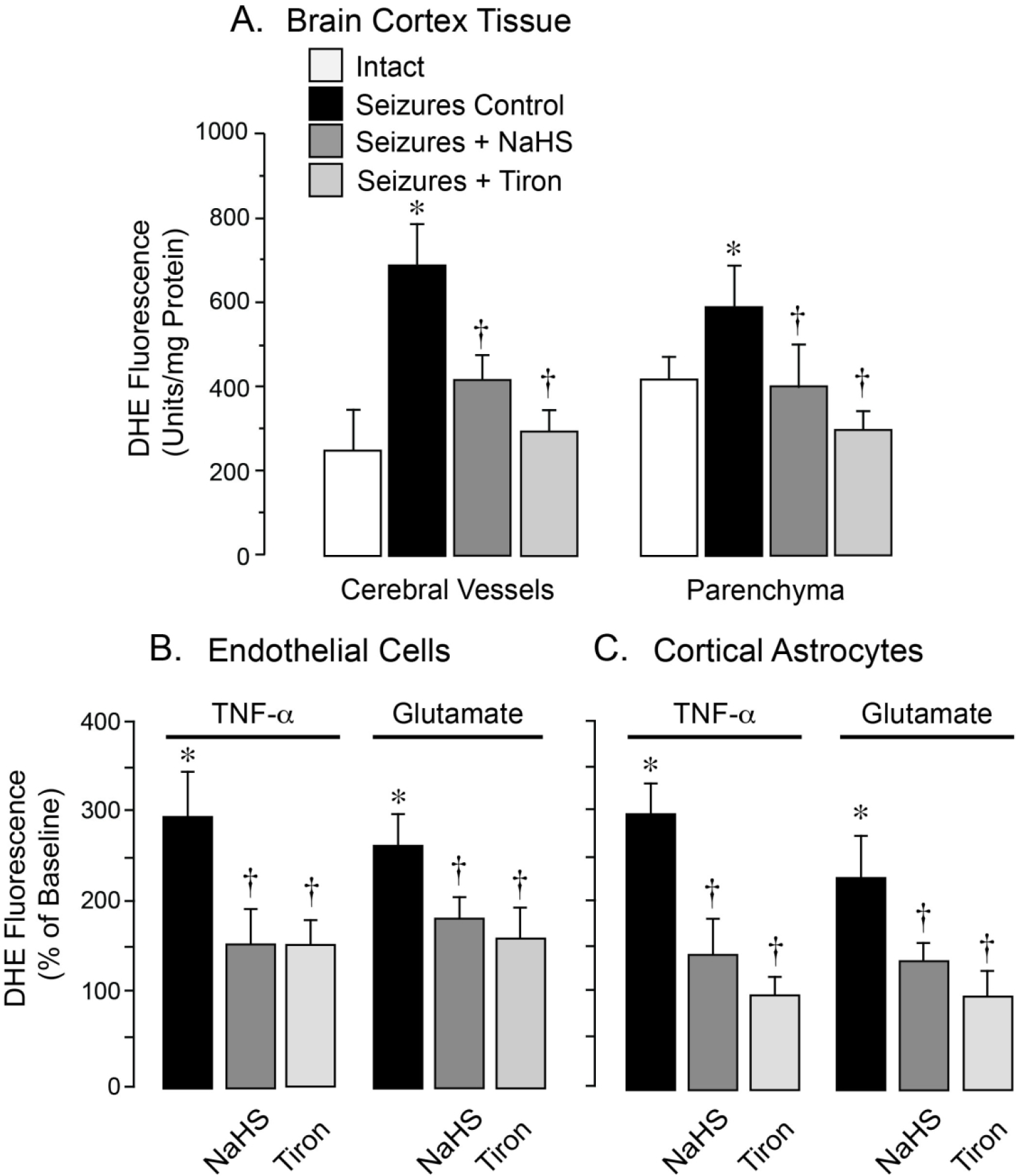
Antioxidant effects of NaHS in the epileptic brain (A) and in cultured neurovascular cells exposed to seizure-related pro-oxidants (B, C). ROS production was evaluated by the fluorescent products of DHE oxidation. A: Seizures were induced by bicuculline (3 mg/kg, i.p.) in untreated newborn pigs (Seizures Control) and in pigs pretreated with NaHS (3 mg/kg, i.p.) or the superoxide scavenger Tiron (2 g/kg, i.v.) Cerebral vessels and astrocyte-enriched brain cortex parenchyma were isolated 1 h after bicuculline administration (N = 4 animals per group). B, C: Primary cerebral microvascular endothelial cells (B) and cortical astrocytes (C) were treated for 1 h with pro-oxidants TNF-α (30 ng/ml) or glutamate (2 mM) in the absence or presence of NaHS (20 μM) or Tiron (1 mM). Values are means ± SD. *P < 0.05 compared with intact values. †P < 0.05 compared with seizure control values (A) or with TNF-α or glutamate alone (B, C).
Antioxidant effects of NaHS in primary neurovascular cells exposed to seizure-related pro-inflammatory and excitotoxic mediators
Cultured primary cerebral microvascular endothelial cells (CMVEC) and cortical astrocytes responded to 1 h treatment with seizure-related pro-oxidants TNF-α (30 ng/ml) and glutamate (2 mM) with a 2–3- fold surge in DHE fluorescence (Figs. 4B, 4C). Tiron (1 mM) largely inhibited both DHE responses providing evidence that superoxide is the major component of ROS species evoked in neurovascular cells (Figs. 4B, 4C). NaHS (20 μM) was as efficient as Tiron in reducing the ROS surge in CMVEC and astrocytes in response to these seizure-related pro-inflammatory and excitotoxic mediators, indicating strong antioxidant potential of the H2S donor molecule in neurovascular cells.
NaHS attenuates postictal cerebral vascular dysfunction caused by epileptic seizures
We investigated the effects of NaHS on long-term cerebral vascular outcome of seizures (Figs. 5A–5D). NaHS (3 mg/kg) was administered parenterally (i.p.) or enterally (orogastric tube) using preventive or therapeutic treatment protocols. In these experiments, cerebral vascular responses to endothelium/astrocyte–dependent and–independent vasodilators were evaluated in intact group (no seizures) and in five 48 h postictal groups of animals: 1) seizure control group (saline, 5 ml, i.p.); 2) preventive parenteral NaHS seizure group (NaHS administered i.p. 20 m before bicuculline), 3) preventive enteral NaHS seizure group (NaHS administered enterally 20 m before bicuculline), 4) therapeutic parenteral NaHS seizure group (NaHS administered i.p. 20 m after bicuculline), and 5) therapeutic enteral NaHS seizure group (NaHS administered enterally 20 m after bicuculline).
Figure 5.
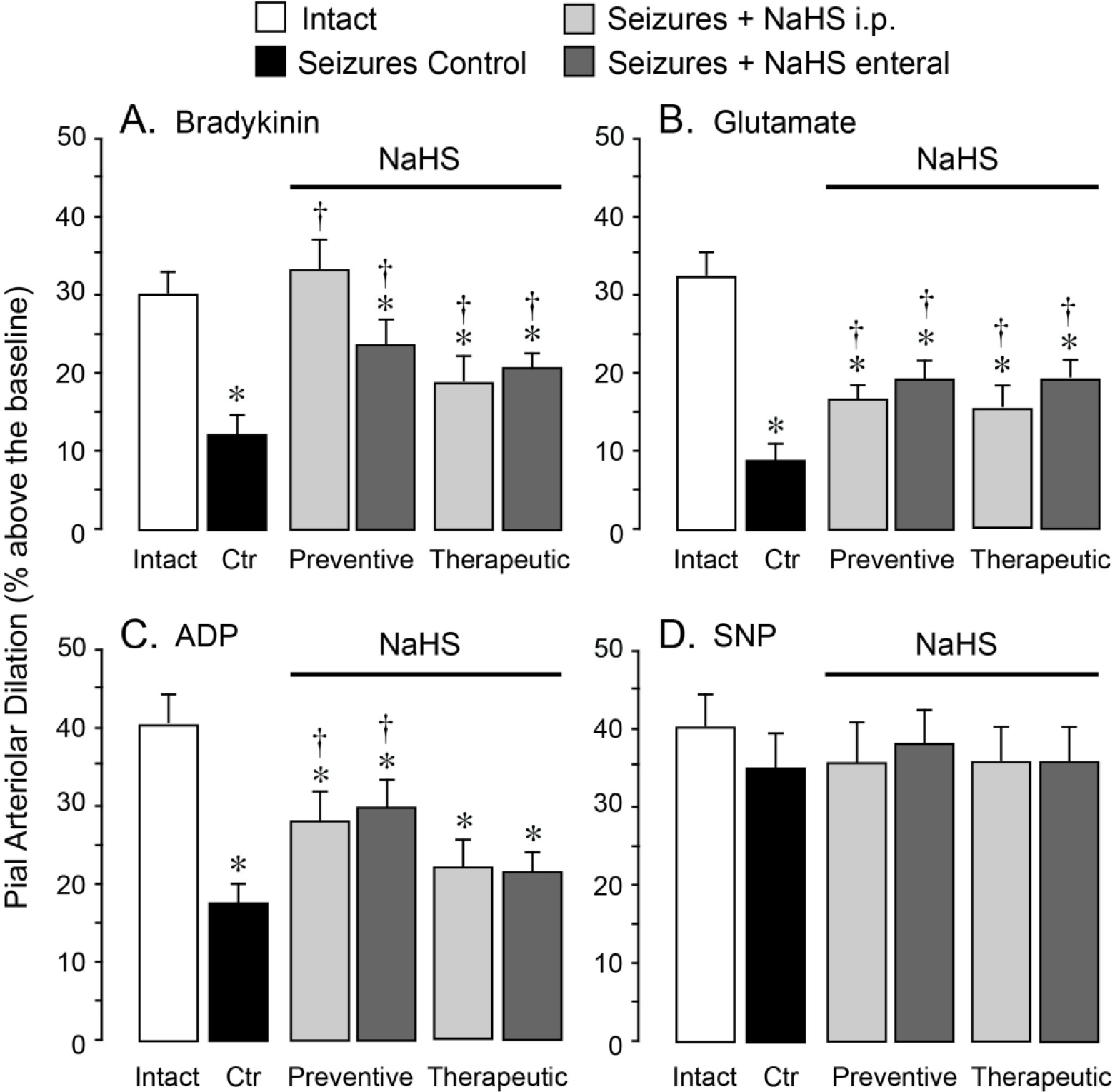
NaHS improves long-term cerebral vascular outcome of neonatal seizures. Seizures were induced by bicuculline (3 mg/kg, i.p.) in untreated newborn pigs (seizure control, Ctrl) and in pigs treated with NaHS. NaHS (3 mg/kg) was administered parenterally (i.p.) or enterally (orogastric tube) 20 m before (preventive protocol) or 20 m after (therapeutic protocol) bicuculline administration. The responses of pial arterioles to topical endothelium- and astrocyte-dependent vasodilators A, bradykinin (10−6 M), B, glutamate (10−4 M), or C, ADP (10−4 M), and to D, the smooth muscle-dependent vasodilator sodium nitroprusside (SNP, 10−6 M) were tested in the intact group (N = 10) and in the five 48 h postictal groups, either untreated (Seizure Control) or treated with NaHS (N = 5 per group). Values are means ± SD. *P < 0.05 compared with the corresponding responses in the intact group. †P < 0.05 compared with the corresponding responses in the seizure control group.
Postictal cerebral vascular responses to bradykinin, glutamate, and ADP were greatly reduced 48 h after seizures as compared to the intact group (Figs. 5A–5C). NaHS (3 mg/kg) administered parenterally or enterally 20 m before or after bicuculline greatly improved postictal cerebral vascular responses to these endothelium- and astrocyte-dependent vasodilators (Figs. 5A–5C). Postictal responses to endothelium- and astrocyte-independent vasodilator SNP remained intact, as in previous publications,18 and NaHS had no effect on these responses (Fig. 5D). Overall, parenterally or enterally administered NaHS exhibited remarkable long-term cerebroprotective effects when used in either preventive or therapeutic treatment protocols.
NaHS prevents apoptosis caused by oxidative stress in primary neurovascular cells
Cultured primary CMVEC and cortical astrocytes exposed to pro-inflammatory and excitotoxic mediators TNF-α and glutamate provide an appropriate in vitro model of seizure-related neurovascular oxidative stress (Fig. 4). We determined whether seizure-related oxidative stress leads to neurovascular cell death by apoptosis. CMVEC and astrocytes responded to a 3–5 h exposure to pro-oxidants TNF-α (30 ng/ml) or glutamate (2 mM) by DNA fragmentation, a key event in apoptosis (Figs. 6A, 6B). The superoxide scavenger Tiron (1mM) prevented DNA fragmentation in CMVEC and astrocytes (Figs. 6A, 6B), thus providing evidence that oxidative stress is the main cause of apoptosis in neurovascular cells. Treatment with NaHS (20 μM) also greatly reduced or fully prevented apoptosis of CMVEC and astrocytes exposed to TNF-α and glutamate (Figs. 6A, 6B). These data demonstrate that NaHS exhibits strong anti-apoptotic properties in the model of neurovascular injury by oxidative stress.
Figure 6.
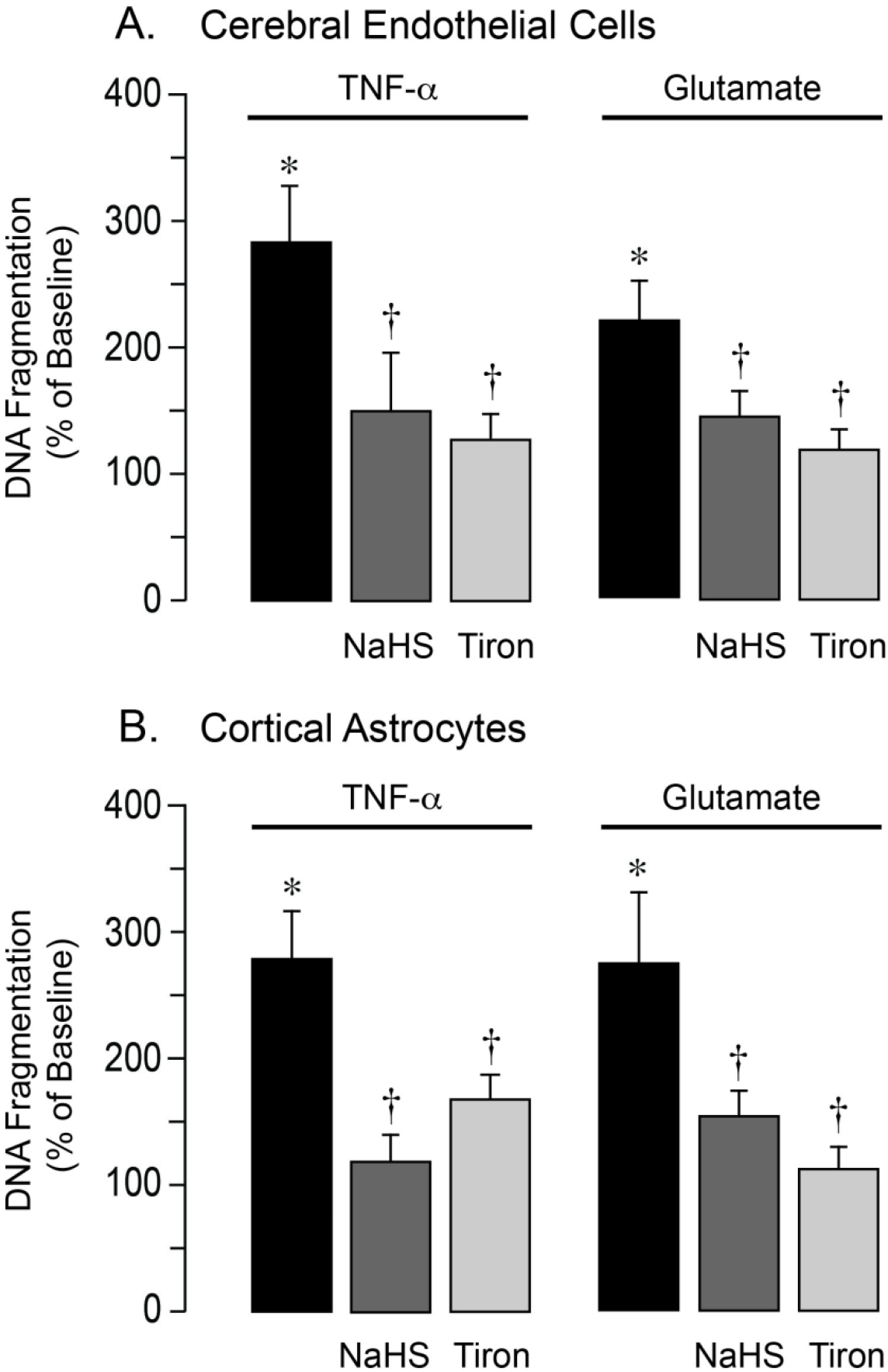
NaHS prevents apoptosis caused by oxidative stress in cerebral microvascular endothelial cells and cortical astrocytes. Cultured primary cerebral microvascular endothelial cells (A) and cortical astrocytes (B) from newborn pigs were treated with TNF-α (30 ng/ml) or glutamate (2 mM) for 3–5 h in the absence or presence of NaHS (20 μM) or the superoxide scavenger Tiron (1 mM). DNA fragmentation, a key event of apoptosis, was detected by ELISA. Data represent the average of 5 independent experiments. Values are means ± SD. *P < 0.05 compared with the baseline value. †P < 0.05 compared with TNF-α or glutamate alone.
DISCUSSION
We report our novel in vivo and in vitro findings that the gaseous messenger H2S exhibits antioxidant and cytoprotective actions in the epileptic brain. This conclusion is supported by the following observations: 1) during epileptic seizures, production of H2S in the brain is increased via activation of CBS that is localized mainly in astrocytes; 2) CBS-derived H2S is required for the functional hyperemia response to epileptiform discharges; 3) endogenous H2S reduces oxidative stress in the epileptic brain and mitigates postictal cerebral vascular dysfunction; 4) systemic administration of NaHS provides a pharmacological tool to increase brain H2S; 5) NaHS reduces brain oxidative stress and prevents cerebral vascular dysfunction caused by epileptic seizures, and 6) NaHS exhibits strong antioxidant and antiapoptotic effects in the cell model of neurovascular injury during excitotoxic and inflammatory conditions.
Cerebral circulatory disorders in newborns are the leading cause of neurological disabilities that represent serious healthcare problems in both cost and quality of life in survivors. An adequate blood supply to the neonatal brain is particularly important because of the rapid development of neurons and high susceptibility of the brain to inflammation. Using a clinically relevant newborn pig model, our studies demonstrated that epileptic seizures exert long-term debilitating effects on cerebral vascular functions in newborns that involve endothelial and astrocyte components of the neurovascular unit.17,18 Clinical studies also support the occurrence of cerebral vascular injury caused by epileptic seizures.28–30
Brain oxidative stress has been recognized as the major cause of neuronal and cerebral vascular injury caused by epileptic seizures.19,31,32 Our research focuses on endogenous antioxidant mechanisms in the neonatal brain. H2S enzymatically produced by the brain has emerged as a vasoactive gaseous mediator with vasodilator and antioxidant properties.1,2,13,33,34 The role of H2S in epilepsy remains elusive. Here we provided novel evidence that endogenous CBS-produced H2S is involved in the cerebral hyperemia response to epileptic seizures. Epileptic seizures increase pial arteriolar diameter indicative of CBF elevation concomitant with the epileptiform discharges.20 We report here that the functional hyperemia response was accompanied by immediate and sustained elevation in H2S production by the brain. H2S elevation occurred in a CBS-dependent manner, as suggested by its sensitivity to the CBS inhibitor AOA but not to the CSE inhibitor PPG. Furthermore, AOA selectively blocked bicuculline-induced vasodilation of pial arterioles, thus providing evidence that astrocyte CBS-dependent H2S elevation is required for the functional hyperemia response to seizures. Notably, the inhibitors of CBS or CSE did not affect the tachycardia response to seizures, suggesting that neuronal activation does not involve an H2S-dependent mechanism. Overall, these data demonstrated that the functional hyperemia response to epileptiform neuronal discharges involves astrocyte influences via CBS/H2S activation.
We presented novel evidence that astrocyte CBS-produced H2S contributes to antioxidant and cerebroprotective defense mechanism in the epileptic brain. Neonatal epileptic seizures produce sustained postictal cerebral vascular dysfunction related to the inability of resistance cerebral arterioles to adequately respond to endothelium- and astrocyte-mediated vasodilator stimuli, including bradykinin, ADP and glutamate. In pigs with AOA-inhibited CBS activity, pial arterioles were almost non-responsive to ADP and glutamate during the delayed postictal period (48 h after seizures), while the CSE inhibitor PPG did not aggravate these responses. These data suggested that activation of H2S production by astrocytic CBS during epileptic seizures exhibited a long-term cerebroprotective role by preserving CBF regulation during the delayed postictal period.
The use of pharmacological H2S donors provides an attractive approach to strengthening the antioxidant potential of H2S in the brain. We provide evidence that systemically administered NaHS delivered H2S to both normal and epileptic brain and exhibited cerebral vasodilator effects. Given that NaHS effectively elevated the H2S pool in the brain, we sought to define its potential therapeutic effects in improving postictal cerebral vascular functions. We show for the first time that systemic administration of NaHS resulted in acute antioxidant effects in the epileptic brain and improved long-term postictal cerebrovascular functions. Cerebroprotective effects of NaHS were observed after both parenteral and enteral routes of drug administration to epileptic pigs. Of clinical importance, NaHS administered during the advanced ictal state also exhibited potent cerebroprotective effects, thus providing the therapeutic opportunity for prevention of postictal cerebral vascular dysfunction. These novel findings demonstrate that systemic administration of NaHS represents a convenient therapeutic approach for improving the cerebral vascular outcome of neonatal epilepsy.
Overall, our present study fills the gap in knowledge on antioxidant and cytoprotective roles of H2S in cerebral circulation. These results uncover the importance of CBS-expressing astrocytes in the antioxidant defense mechanism in the epileptic brain. We demonstrated, for the first time, that astrocyte CBS-produced H2S has vasodilator, antioxidant, and cytoprotective effects in the epileptic brain. Importantly, these findings in a large animal model provide translationally relevant evidence of cerebroprotective effects of H2S donors in preventing long-term impairment of cerebral vascular functions and CBF dysregulation caused by epileptic seizures.
IMPACT:
Epileptic seizures in neonates lead to prolonged postictal cerebral vascular dysregulation.
The role of hydrogen sulfide (H2S), a mediator with vasodilator and antioxidant properties, in the epileptic brain has been explored.
Astrocytes are major sites of enzymatic H2S production in the epileptic brain.
Postictal cerebral vascular dysfunction is exaggerated when astrocyte H2S production is pharmacologically inhibited during seizures.
Postictal cerebral vascular dysfunction is minimized when the brain H2S is elevated by systemic administration of NaHS during seizures.
NaHS provides a therapeutic approach for improving cerebrovascular outcome of epileptic seizures via a mechanism that involves the antioxidant potential of H2S.
FUNDING
The works were supported by awards NS101717 (to H.P.) and NS105655 (to H.P.) from the National Institutes of Health.
Footnotes
COMPETING INTERESTS
The authors declare no competing interests.
CONSENT STATEMENT
Patient consent was not required.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study. All data generated or analyzed during this study are included in this published article.
REFERENCES
- 1.Kimura H Hydrogen sulfide (H2S) and polysulfide (H2Sn) signaling: the first 25 years. Biomolecules 11, 896 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Szabo C A timeline of hydrogen sulfide (H2S) research: From environmental toxin to biological mediator. Biochem. Pharmacol. 149, 5–19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Enokido Y, et al. Cystathionine beta-synthase, a key enzyme for homocysteine metabolism, is preferentially expressed in the radial glia/astrocyte lineage of developing mouse CNS. FASEB J. 19, 1854–1856 (2005). [DOI] [PubMed] [Google Scholar]
- 4.Zuhra K, Augsburger F, Majtan T & Szabo C Cystathionine-β-Synthase: Molecular Regulation and Pharmacological Inhibition. Biomolecules 10, 697 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lechpammer M et al. Upregulation of cystathionine β-synthase and p70S6K/S6 in neonatal hypoxic ischemic brain injury. Brain Pathol. 27, 449–458 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leffler CW et al. Hydrogen sulfide and cerebral microvascular tone in newborn pigs. Am. J. Physiol. Heart Circ. Physiol. 300, H440–H456 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liang GH et al. Hydrogen sulfide dilates cerebral arterioles by activating smooth muscle cell plasma membrane KATP channels. Am. J. Physiol. Heart Circ. Physiol. 300, H2088–H2095 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liang GH, Xi Q, Leffler CW & Jaggar JH Hydrogen sulfide activates Ca2+ sparks to induce cerebral arteriole dilatation. J. Physiol. 590, 2709–2720 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang R Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol. Rev. 92, 791–896 (2012). [DOI] [PubMed] [Google Scholar]
- 10.Yang G et al. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science 322, 587–590 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao W & Wang. R. H2S-induced vasorelaxation and underlying cellular and molecular mechanisms. Am. J. Physiol. Heart Circ. Physiol. 283, H474–H480 (2002). [DOI] [PubMed] [Google Scholar]
- 12.Calvert JW, Coetzee WA & Lefer DJ Novel insights into hydrogen sulfide-mediated cytoprotection. Antioxid. Redox Signal. 12: 1203–1217 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kabil O, Motl N & Banerjee R H2S and its role in redox signaling. Biochim. Biophys. Acta 1844, 1355–1366 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bai C & Zhao C Sodium hydrosulfide post-conditioning protects hippocampal CA1 neurons from neuronal cell injury in the rat model of transient global cerebral ischemia through activation of extracellular-regulated kinases signaling. Curr. Neurovasc. Res. 16, 156–165 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Jia J, Li J & Cheng J H2S-based therapies for ischaemic stroke: opportunities and challenges. Stroke Vasc. Neurol. 4, 63–66 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kamat PK et al. Hydrogen sulfide attenuates neurodegeneration and neurovascular dysfunction induced by intracerebral-administered homocysteine in mice. Neuroscience 252: 302–319 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harsono M et al. Selective head cooling during neonatal seizures prevents postictal cerebral vascular dysfunction without reducing epileptiform activity. Am. J. Physiol. Heart Circ. Physiol. 311, H1202–H1213 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu D, Pourcyrous M, Fedinec AL, Leffler CW & Parfenova H Preventing harmful effects of epileptic seizures on cerebrovascular functions in newborn pigs: Does sex matter? Ped. Res. 82, 881–887 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parfenova H, Leffler CW, Basuroy S, Liu J & Fedinec AL Antioxidant roles of heme oxygenase, carbon monoxide, and bilirubin in cerebral circulation during seizures. J.Cereb. Blood Flow Metab. 32, 1024–1034 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parfenova H, Daley ML, Carratu P & Leffler CW Heme oxygenase inhibition reduces neuronal activation evoked by bicuculline in newborn pigs. Brain Res. 1014, 87–96 (2004). [DOI] [PubMed] [Google Scholar]
- 21.Parfenova H, Liu J, Hoover DT & Fedinec AL Vasodilator effects of sulforaphane in cerebral circulation: A critical role of endogenously produced hydrogen sulfide and arteriolar smooth muscle KATP and BK channels in the brain. J. Cereb. Blood Flow Metab. 40, 1987–1996 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu J, Chandaka GK, Zhang R & Parfenova H Acute antioxidant and cytoprotective effects of sulforaphane in brain endothelial cells and astrocytes during inflammation and excitotoxicity. Pharmacol. Res. Perspect. 8, e00630 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Basuroy S, Tcheranova D, Bhattacharya S, Leffler CW & Parfenova H Nox4 NADPH oxidase–derived reactive oxygen species, via endogenous carbon monoxide, promote survival of brain endothelial cells during TNF-a-induced apoptosis. Am. J. Physiol. Cell Physiol. 300, C256–C265 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murphy MP et al. Guidelines for measuring reactive oxygen species and oxidative damage in cells and in vivo. Nat Metab 4:651–662 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Patel S et al. H2S mediates the vasodilator effect of endothelin-1 in the cerebral circulation. Am. J. Physiol. Heart Circ. Physiol. 315, H1759–H1764 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Elmpt WJ, Nijsen TM, Griep PA & Arends JB A model of heart rate changes to detect seizures in severe epilepsy. Seizure 15, 366–375 (2006). [DOI] [PubMed] [Google Scholar]
- 27.Greene BR et al. Heart and respiration rate changes in the neonate during electroencephalographic seizure. Med. Biol. Eng. Comput. 44, 27–34 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Shao LR & Stafstrom CE Pediatric Epileptic Encephalopathies: Pathophysiology and Animal Models. Semin. Pediatr. Neurol. 23, 98–107 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Abend NS & Wusthoff CJ Neonatal seizures and status epilepticus. J. Clin. Neurophysiol. 29, 441–448 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chapman KE, Raol YH & Brooks-Kayal A Neonatal seizures: controversies and challenges in translating new therapies from the lab to the isolette. Eur. J. Neurosci. 35, 1857–1865 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lombroso CT Neonatal seizures: gaps between the laboratory and the clinic. Epilepsia 48 S2: 83–106 (2007). [DOI] [PubMed] [Google Scholar]
- 32.Pestana RR, Kinjo ER, Hernandes MS & Britto LR Reactive oxygen species generated by NADPH oxidase are involved in neurodegeneration in the pilocarpine model of temporal lobe epilepsy. Neurosci. Lett. 484, 187–191 (2010). [DOI] [PubMed] [Google Scholar]
- 33.Waldbaum S & Patel M Mitochondria, oxidative stress, and temporal lobe epilepsy. Epilepsy Res. 88, 23–45 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nam B et al. In vivo detection of hydrogen sulfide in the brain of live mouse: application in neuroinflammation models. Eur. J. Nucl. Med. Mol. Imaging 49, 4073–4087 (2022). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study. All data generated or analyzed during this study are included in this published article.