

Summary
Genome-wide association studies (GWASs) have uncovered numerous trait-associated loci across the human genome, most of which are located in noncoding regions, making interpretation difficult. Moreover, causal variants are hard to statistically fine-map at many loci because of widespread linkage disequilibrium. To address this challenge, we present a strategy utilizing transcription factor (TF) binding quantitative trait loci (bQTLs) for colocalization analysis to identify trait associations likely mediated by TF occupancy variation and to pinpoint likely causal variants using motif scores. We applied this approach to PU.1 bQTLs in lymphoblastoid cell lines and blood cell trait GWAS data. Colocalization analysis revealed 69 blood cell trait GWAS loci putatively driven by PU.1 occupancy variation. We nominate PU.1 motif-altering variants as the likely shared causal variants at 51 loci. Such integration of TF bQTL data with other GWAS data may reveal transcriptional regulatory mechanisms and causal noncoding variants underlying additional complex traits.
Keywords: genome-wide association study, transcription factor binding quantitative trait locus, TF bQTL, colocalization, variant-to-function, fine-mapping, noncoding variants, PU.1, blood cell traits, transcription factors, ChIP-seq
Graphical abstract
Highlights
-
•
69 PU.1 binding QTLs colocalize with blood cell trait associations
-
•
PU.1 motif-altering variants are likely causal at 51 colocalized loci
-
•
Variants affect chromatin accessibility, histone marks, and gene expression levels
-
•
TF-centered strategy pinpoints likely causal variants and mechanisms at GWAS loci
Identifying the causal variants and mechanisms of noncoding genomic loci associated with traits is challenging. Jeong and Bulyk present a computational strategy to utilize population-level data on transcription factor (TF) occupancy to pinpoint trait-associated loci that are likely driven by variants altering the TF’s binding site motif and binding levels.
Introduction
A recurring challenge in genome-wide association studies (GWASs) is the difficulty of identifying causal variants as well as formulating corresponding variant-to-function (V2F) hypotheses.1 Pinpointing causal variants is important because it guides subsequent validation experiments2,3,4 and development of potential therapies.5 More precise identification of causal variants (e.g., fine-mapping) also leads to better genetic risk predictions across various traits and diseases.6,7 However, widespread linkage disequilibrium (LD) typically prevents effective statistical fine-mapping, especially for common variants.1,8 Moreover, most of the genome-wide significant loci are noncoding and likely have regulatory functions.9,10 Variants predicted to affect transcription factor (TF) binding across the genome explain a large proportion of genetic associations with traits (i.e., heritability enrichment).11,12 In practice, noncoding variants are much harder to interpret than coding variants because predicting the effects of noncoding variants on TF binding in vivo is challenging. Some commonly used approaches to predict affected TFs include searching for overlapping TF chromatin immunoprecipitation sequencing (ChIP-seq) peaks13,14 and TF binding site motifs.8,15 However, such approaches lack evidence specifically demonstrating the variants’ effects on in vivo TF binding. Furthermore, many TFs within a TF family recognize very similar motifs16 while also binding to distinct genomic loci,17 adding to the challenge of pinpointing the causal TF. Therefore, an approach to effectively pinpoint regulatory variants and their effects on in vivo TF binding at individual GWAS loci is essential.
An effective method to capture the genetic effects on in vivo TF binding is TF binding quantitative trait loci (bQTLs)18,19,20 (i.e., genomic loci where the TF occupancy level, as measured by ChIP-seq, is significantly associated with a genetic variant). An earlier study attempted to link specific TF bQTLs to individual GWAS loci simply based on a single variant’s association signal,19 but this method is prone to false positive findings because the two associations could be driven by distinct variants merely in LD with each other.21 Instead, we aimed to link TF bQTLs and GWAS loci by applying colocalization analysis,21,22,23,24 which is a widely accepted statistical approach to specifically test the hypothesis that genetic signals are shared between a pair of traits (e.g., TF binding and GWAS trait). Significant colocalization suggests that a genetic variant affects TF binding as well as the studied downstream trait.25
A key benefit of TF bQTL colocalization lies in the observation that TF binding variation is often driven by variants altering the motif of the corresponding TF at its binding site.26,27 With a TF motif model, such as gapped k-mer support vector machine (gkm-SVM),28,29 we can recognize a variant that overlaps the TF’s binding motif and changes its predicted affinity in the direction concordant with the changes in TF binding level. This is advantageous because we can pinpoint such a motif-altering variant at the binding site even when association statistics alone cannot readily identify the likely causal variant because of LD. In other words, if some TF’s bQTL significantly colocalizes with a GWAS signal, and that TF’s motif-altering variant is among the top associated variants, then that variant is likely to explain the GWAS association. We contrast this with expression QTL (eQTL) or methylation QTL (mQTL) colocalization,30,31 where the TF is unknown. By pinpointing the candidate causal TF in a GWAS locus through TF bQTL colocalization, we can also prioritize the corresponding TF motif-altering variants as the likely causal regulatory variants underlying TF binding variation and the GWAS traits.
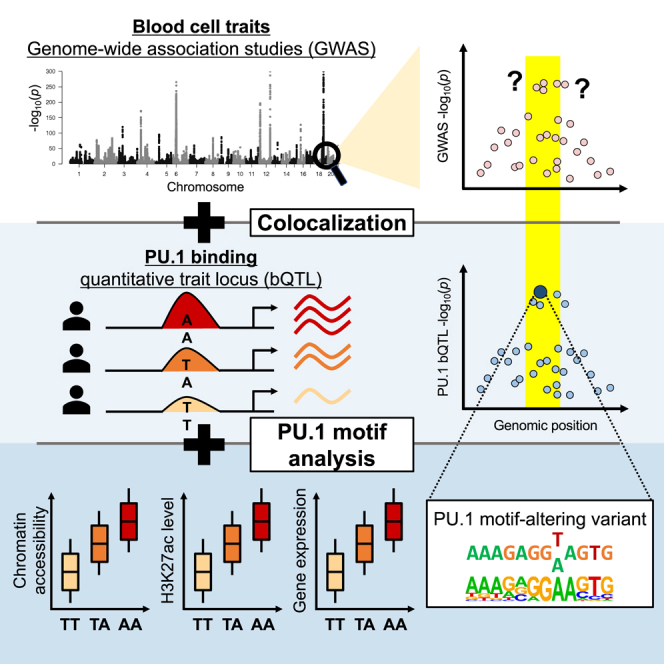
Hence, we present a strategy (1) to analyze colocalization of TF bQTLs at GWAS loci to highlight TF binding sites that potentially mediate the GWAS associations25 and (2) to utilize TF motif models to nominate variants altering the corresponding TF motifs at those binding sites as likely shared causal variants underlying both phenotypes (Figure 1A). By performing TF bQTL colocalization analysis with GWAS data to fine-map putative causal variants that affect in vivo TF binding within individual GWAS loci, we aim to add to the understanding of the direct molecular consequences of trait-associated genetic variation.
Figure 1.

Relevance of PU.1 bQTLs in LCLs to blood cell trait associations
(A) Left: blood cell trait-associated loci may have overlapping PU.1 bQTLs and, potentially, expression QTL (eQTL) associations. Right: significant colocalization suggests that the causal variants are shared. If there is a PU.1 motif-altering variant at a colocalized PU.1 bQTL, then the variant is likely to be the shared causal variant. exp, expression.
(B) Comparison of changes in motif score (Δ gkm-SVM) and estimated bQTL effect sizes at PU.1 motif-altering variants within the 200-bp PU.1 ChIP-seq peaks. The color represents the –log10(p) of PU.1 bQTL association (linear regression). The insets show examples of variants’ effects on PU.1 gkm-SVM score and their nucleotide change within a PU.1 motif. At the variant position, the top and bottom bases are reference and variant alleles, respectively.
(C) Number of significant PU.1 bQTLs with PU.1 motif-altering variants at each region within the 200-bp PU.1 ChIP-seq peaks. ∗∗∗: p < 2.2 × 10−16 (Fisher’s exact test).
See also Figures S1 and S2 and Tables S1 and S2.
We carried out this strategy with blood cell trait GWAS32 and bQTL data for the hematopoietic master regulator PU.1 from lymphoblastoid cell lines (LCLs),18,26 which are immortalized B cell lines. PU.1 bQTLs in neutrophils have been found previously to colocalize with immune disease susceptibility loci but have not been used to fine-map the causal variants.25 Blood cell traits (e.g., lymphocyte counts, hemoglobin concentrations) are indicators of various diseases; for instance, individuals with low lymphocyte counts are more susceptible to infections, including severe coronavirus disease 2019 (COVID-19).33,34,35 PU.1 has a role in specifying myeloid and lymphoid lineages during hematopoiesis,36,37 and SPI1, the gene encoding PU.1, is expressed throughout progenitor cell types38 (Figure S1). A recent fine-mapping analysis of blood cell trait GWASs reported that PU.1 was the TF with the highest number of fine-mapped noncoding variants altering its DNA binding site motif,15 suggesting that PU.1 motif-altering variants might drive many blood cell trait association signals.
To identify blood cell trait associations that may be driven by a variant altering PU.1 binding, we analyzed publicly available PU.1 ChIP-seq data from LCLs across 49 individuals18,26 and identified 1,497 PU.1 bQTLs. PU.1 bQTLs colocalized with at least one blood cell trait association at 69 loci; for 51 of these loci, we identified PU.1 motif-altering variants as the likely causal variants. Our approach allowed us to overcome the limitations of statistical fine-mapping in resolving these GWAS signals to single causal variants. Most of those PU.1 motif-altering variants were also associated with other regulatory phenotypes, such as chromatin accessibility and histone mark levels, in LCLs. By also incorporating transcriptome data for LCLs, we identified several putative causal genes for traits, including lymphocyte and monocyte counts. Our results illustrate the utility of TF bQTL datasets for fine-mapping trait-associated noncoding loci and in generating mechanistic V2F models of gene dysregulation for traits of biomedical importance.
Results
PU.1 motif-altering variants are likely causal for PU.1 bQTL associations
First, we reanalyzed available PU.1 ChIP-seq data for LCLs from 49 individuals.18,26 These individuals are all of European ancestry, and their genotypes are available through the 1000 Genomes Project39 (Table S1). After peak calling and normalization of the PU.1 ChIP-seq read counts, we tested for significant genetic associations with common variants (minor-allele frequency [MAF] > 0.05) within 100 kb of each ChIP-seq peak. In total, we identified 1,497 significant PU.1 bQTLs (false discovery rate [FDR] < 5%).
We next inspected the contribution of PU.1 motif-altering variants to PU.1 bQTLs. First, we verified that PU.1-occupied regions were enriched for a match to the PU.1 binding site motif, identified by a position weight matrix (PWM), near the center of the ChIP-seq peaks (Figure S2A). This suggests that most of these sites are bound directly by PU.1. Next, we evaluated whether PU.1 motif-altering variants affect PU.1 binding by training a motif score model gkm-SVM to learn gapped k-mers that are overrepresented in PU.1-occupied sequences. PU.1 can bind DNA as a monomer and as a heterodimer with either interferon regulatory factor 4 (IRF4) or IRF8,40 and the model correctly captured PU.1 and PU.1:IRF composite motifs (Figure S2B). Changes in gkm-SVM scores predict effects of variants on TF binding better than PWMs,41 which imprecisely assume each nucleotide to affect binding independently. Consistent with our expectations, the predicted change in gkm-SVM scores for single-nucleotide polymorphisms (SNPs) within PU.1 motifs was significantly correlated with estimated PU.1 bQTL effect sizes (Pearson r = 0.80, p = 3.6 × 10−310; Figure 1B; Table S2). This strong positive correlation supports the model that PU.1 motif-altering variants, if present, are likely causal for those PU.1 bQTLs. Furthermore, significant PU.1 bQTLs with a motif-altering variant (determined by gkm-SVM) showed that such variants are more concentrated toward the peak centers compared with PU.1 bQTLs without one (two-sided Fisher’s exact test, p = 3.1 × 10−18; Figure 1C), consistent with the expectation that PU.1 motif-altering variants directly affect PU.1 occupancy. Hence, we considered that PU.1 bQTLs colocalized with blood cell trait association would likely be driven by PU.1 motif-altering variants, if present (Figure 1A).
PU.1 binding sites and PU.1 bQTLs in LCLs are enriched for blood cell trait association
To verify the relevance of these PU.1 bQTLs for investigations of blood cell traits, we evaluated whether the PU.1 bQTLs are more likely to be significantly associated with blood cell traits than expected by chance. We analyzed GWAS data for 28 blood cell traits from the UK Biobank32 (Table S3). As a background expectation, we constructed 250 sets of null variants matched with PU.1 bQTL lead variants for allele frequency, number of tagging variants (LD r2 > 0.5), and distance to the closest transcription start site (TSS). The significant PU.1 bQTLs were more likely to tag lead variants associated (i.e., p < 5 × 10−8) with myeloid lineage traits (e.g., monocyte and neutrophil count) and lymphoid lineage traits (e.g., lymphocyte count) than the sets of null variants (adjusted empirical p < 0.05) (Figures 2A and S2C). This is consistent with the known role of PU.1 in myeloid and lymphoid differentiation.36,37 In contrast, PU.1 bQTLs were not enriched for other traits like type 2 diabetes or height (Figures 2A and S2D).
Figure 2.

Colocalization of blood cell trait GWAS and PU.1 bQTLs
(A) Enrichment of PU.1 bQTLs for associations with specific blood cell traits and control traits (i.e., height and type 2 diabetes). Traits with empirical adjusted p < 0.05 (above the dashed line) and control traits are labeled. Lym, lymphocyte; Neut, neutrophil; Mono, monocyte. Abbreviations of blood cell traits are further described in Table S3.
(B) Colocalization results from JLIM and Coloc. Each point is a PU.1 bQTL-trait pair. The number shown in each quadrant is the number of points within the significance category. Dashed lines indicate the respective significance thresholds (JLIM, p < 0.01172 [FDR 5%]; Coloc, PP[colocalized] > 0.5).
(C) The types of putative causal variants at colocalized PU.1 bQTLs that alter PU.1 motifs or the copy number of the PU.1 occupancy site. SNPs, indels, and multivariants alter PU.1 motifs. CNV, copy number variation altering the copy number of PU.1 binding sites; Multi, multiple variants in perfect LD (r2 = 1) within a PU.1 motif sequence; Unk (unknown), No variant-altering PU.1 motif sequence or its copy number.
(D) Number of PU.1 motif-altering SNPs at each nucleotide position at colocalized PU.1 binding sites. Motif logos are from the Homer42 database.
(E) Blood cell trait GWAS credible set size at loci with colocalized PU.1 bQTLs and a PU.1 motif-altering variant. Only 25 loci with fine-mapping result in Vuckovic et al.8 are represented.
See also Figures S3 and S4; Tables S3, S4, S5, S6, S7, S8, and S9; and Note S1.
PU.1 bQTL colocalization with blood cell trait associations
To identify candidate loci to test for potential colocalization of PU.1 bQTL and blood cell trait associations, we filtered all significant PU.1 bQTLs for loci with at least one blood cell trait association at p < 10−6. We reasoned that suggestive loci with p < 10−6 that colocalize with PU.1 bQTLs could be weaker, but likely functional, associations. This resulted in a total of 1,621 such PU.1 bQTL-trait pairs, comprising 367 unique loci. We then applied two distinct colocalization methods, joint likelihood mapping (JLIM)23 and Coloc,22 to test for robust colocalization (Table S4). JLIM is a frequentist method testing the significance of the shared association by a permutation p value, while Coloc is a Bayesian method estimating the posterior probability of colocalization. Each method can exhibit different performance depending on the LD structure of the loci;23 therefore, we reasoned that requiring significant colocalization by both methods would enrich true positive cases. We used a significance threshold of p < 0.01172 (FDR < 5%) for JLIM and posterior probability of colocalization (PP[colocalization]) > 0.5 for Coloc.
The statistically significant colocalization of PU.1 bQTL-trait pairs identified by JLIM and Coloc was overall consistent (Pearson r = 0.73, p = 6.8 × 10−270; Figure 2B). We identified a total of 190 (11.7%) PU.1-trait pairs, spanning 69 unique loci, that were significant by both methods. We also found 1,196 (73.8%) cases where a variant that was significant for PU.1 bQTL and blood cell traits did not exhibit significant colocalization by either JLIM or Coloc. This highlights the importance of performing colocalization analysis to distinguish loci with statistical evidence of shared causal variants from those where the variants associated with each trait are merely in LD with each other.21 The remaining 235 (14.5%) pairs showed discordant results between the two methods, which could potentially stem from lack of statistical power due to weak association signals or many variants showing high LD with the lead variant (Figure S3; Note S1). This discrepancy justifies the rationale of applying both methods to identify high-confidence colocalization.
Most (56 of 69) loci showing high-confidence colocalization had some biologically plausible putative causal variants that directly affect PU.1 binding sequences (Figures 2C and S4A; Table S5). 43 (62.3%) loci had a SNP altering a PU.1 motif, while 7 (10.1%) had a short insertion or deletion (indel) variant. In addition, there was one locus where two adjacent SNPs were in perfect LD (r2 = 1) and altered a single PU.1 motif sequence (Figure S4A; Table S6). These SNPs and short indels showed a balance of gained and lost PU.1 binding (two-sided binomial test, p = 0.67), and changes in gkm-SVM motif scores were highly correlated with the estimated PU.1 bQTL effect sizes (Pearson r = 0.89, p = 5.2 × 10−18; Figure S4B). The PU.1 motif-altering SNPs at colocalized loci were distributed within the PU.1 or PU.1:IRF motif, with the highest frequencies at the core “GGAAG” positions (Figure 2D; Table S7). There were also 5 loci with large deletions that completely removed the PU.1 binding site, which we were able to uncover because the 1000 Genomes Project (1KGP)39 genotypes included structural variants (Figure S4C). Whether the deletions are true causal variants will need to be tested experimentally in future studies. From here on, “PU.1 motif-altering variants” refers to the 51 variants that are not structural variants.
To evaluate the benefits of our approach in pinpointing the putative causal variant and TF, we retrieved fine-mapping results for 25 colocalized loci with a PU.1 motif-altering variant (i.e., SNP or indel) from a recent blood cell trait GWAS study8 (Note S2). 19 of these 25 (76%) loci had more than 10 variants in the 95% credible set (i.e., minimal set of variants that have 95% posterior probability of containing the causal variant), none of which was fine-mapped to a single variant (Figure 2E; Table S8). Without TF bQTL colocalization, existing approaches to narrow down candidate variants and hypothesize the causal TF typically include filtering for variants in accessible chromatin and scanning for any TF motif alterations.15 When we applied such an approach to the 25 PU.1 bQTL colocalized loci, it still led to multiple candidate variants (on average, 4.9 variants per locus), corresponding to numerous LCL-expressed (transcripts per million [TPM] > 1) TFs with motif alterations (on average, 13.8 unique TFs with a motif alteration per SNP; Table S9). In contrast, despite the difficulty in fine-mapping due to LD structure, we were able to pinpoint single putative causal variants in these loci using a specific TF’s (i.e., PU.1) motif information. Because PU.1 bQTL colocalization nominates PU.1 as the causal TF in those blood cell trait GWAS loci, it also narrows down the search for putative causal variants to PU.1 motif-altering variants.
Across the blood cell traits, those related to white blood cells (e.g., white blood cell count, lymphocyte count, neutrophil count) showed a higher proportion of the tested loci showing colocalization than red blood cell or platelet traits (Figure 3A). This relative enrichment is similar to that of tagging variants observed in Figure 1B. Some loci showed association with multiple blood cell traits; those traits were mostly closely related, like neutrophil count and neutrophil percentage (Figures 3B and 3C).
Figure 3.

Distribution of colocalized loci across the genome
(A) Proportion of tested loci with significant colocalization. The colors represent the trait groups. The blood cell traits highlighted in yellow correspond to white blood cell traits. Baso, basophil; Eosino, eosinophil; WBC, white blood cell; Hb conc, hemoglobin concentration; Ht, hematocrit; MCH, mean corpuscular hemoglobin; MCV, mean corpuscular volume; MSCV, mean sphered corpuscular volume; RBC, red blood cell; dist, distribution; HLSR, high-light-scatter reticulocyte; Imm ret frac, immature reticulocyte fraction; Ret, reticulocyte; MPV, mean platelet volume; Plt, platelet. Abbreviations of blood cell traits are further described in Table S3.
(B) Fuji plot depicting the genomic distribution of blood cell trait-associated loci that show high-confidence colocalization with PU.1 bQTLs. Tracks are colored by trait group as in (A).
(C) Number of traits with which each PU.1 bQTL colocalizes. The panel is at the center. Bars representing each trait are stacked at each locus.
Most PU.1 bQTLs alter chromatin activity, and some affect gene expression
Additional regulatory phenotype data allowed us to derive specific hypotheses about gene-regulatory mechanisms that are perturbed by the variants. First, we reanalyzed assay for transposase-accessible chromatin using sequencing (ATAC-seq)43 and histone mark ChIP-seq data for LCLs44 to generate QTL statistics for chromatin accessibility and active histone mark (histone H3 lysine 27 acetylation [H3K27ac] and histone H3 lysine 4 monomethylation [H3K4me1]) levels. These chromatin phenotypes can indicate regulatory regions in the genome,45,46 and a direct consequence of PU.1 binding alteration is likely to be in the cognate chromatin region. In fact, the majority (>60%) of the colocalized PU.1 binding sites with PU.1 motif-altering variants showed overlap with each of the chromatin phenotypes (Figure 4A). The presence of PU.1 binding sites that are not accessible is consistent with earlier observations.47 Moreover, there were significant QTL signals (FDR < 5%) that were in LD (r2 > 0.8) with the PU.1 motif-altering variants in more than 70% of the overlapping peaks. 20 of the PU.1 bQTLs showed chromatin accessibility QTLs (caQTLs), H3K27ac histone QTLs (hQTLs), and H3K4me1 hQTLs, while 16 showed at least one of the three (Figure 4B; Tables S10, S11, and S12). These effects all showed concordant directions as the effects of PU.1 motif-altering variants on PU.1 binding (Figure 4C). Thus, most PU.1 motif-altering variants are supported by measured chromatin effects. The rest of the variants may show chromatin effects in a different cellular context.
Figure 4.

Regulatory effects of the colocalized PU.1 motif-altering variants
(A) Number of colocalized PU.1 motif-altering variants that overlap ATAC-seq or histone mark (H3K27ac or H3K4me1) ChIP-seq peaks and that are in LD (r2 > 0.8) with those regulatory QTLs.
(B) Upset plot showing the number of colocalized PU.1 motif-altering variants that are in LD (r2 > 0.8) with different sets of regulatory QTLs. caQTL, chromatin accessibility QTL; hQTL, histone QTL.
(C) Comparison of PU.1 bQTL effects (i.e., regression effect size) with other regulatory QTL effects. Each point corresponds to a PU.1 motif-altering variant. The colors match those in (A). The error bars represent standard errors. Pearson correlation coefficient is calculated only for those points showing significant regulatory QTLs.
(D) Comparison of PU.1 bQTL effects and PU.1 ChIP-seq allelic imbalance effect (i.e., log2[allelic fold change] estimated from weighted linear regression). The effect is with respect to the alternate alleles. The error bars represent standard errors.
(E) Comparison of PU.1 bQTL effects with eQTL effects. Each point corresponds to a PU.1 motif-altering variant. For rs3808619, which had multiple eQTL signals, only the value for the closest gene, ZC2HC1A, is shown. The error bars represent standard errors.
See also Tables S10, S11, S12, S13, and S14.
To further corroborate the variants’ effect on PU.1 binding, we estimated their ChIP-seq allelic imbalance effects in heterozygous individuals. A PU.1 motif-altering variant that is causally associated with PU.1 binding would exhibit allelic imbalance signals that are consistent with estimated bQTL effects.48,49 For all 44 PU.1 motif-altering SNPs, the estimated allelic imbalance effects showed directions and magnitudes that are concordant with those of the PU.1 bQTL estimates (Pearson r = 0.94, p = 4.2 × 10−43; Figure 4D; Table S13).
We next searched to see whether PU.1 motif-altering variants that colocalized with blood cell trait association signals were in LD (r2 > 0.8) with eQTLs in LCLs. Interestingly, just 9 PU.1 motif-altering variants were in LD with eQTL lead variants, and one was in LD with a secondary eQTL signal (i.e., a weaker signal independent of the strongest, primary eQTL; Table S14). Nine of 10 of these variants showed the same effect directions for PU.1 bQTLs and eQTLs (Figure 4E). The remaining colocalized PU.1 motif-altering variants might drive eQTL signals under other experimental conditions and/or cell types. Among the examples with an eQTL signal in LCLs, we selected 3 loci to describe further. We show one example where a PU.1 motif-altering SNP (rs12517864) represents a secondary eQTL for ZNF608 in LCLs, and only this secondary signal colocalizes with lymphocyte count association. An eQTL-centric analysis in LCLs would have missed this locus without accounting for multiple independent signals, highlighting the power of the use of TF bQTL data in colocalization analysis with GWAS data. Two other examples show reporter assay results corroborating the regulatory effects of PU.1 motif-altering variants identified in colocalized loci.
bQTL colocalization reveals a putative causal variant that is not the primary eQTL
Causal genes at a trait-associated locus frequently have been identified using eQTL data for nearby genes.50,51 However, eQTLs can often have multiple independent signals,51 and these signals detected in any one cell type may not all be associated with a GWAS trait, such as when the regulatory effects manifest themselves only in certain cellular contexts. This complicates colocalization analyses that often assume a single shared causal variant at a locus.22,23 In contrast, TF bQTLs capture regulatory effects of individual regulatory elements. Therefore, TF bQTL colocalization analysis can isolate the effects of variants on specific regulatory elements, lowering the probability of multiple causal variants compared with that of eQTLs.
For example, the ZNF608 locus shows significant colocalization of PU.1 bQTLs and lymphocyte count association (JLIM p = 2.0 × 10−5 and Coloc PP[colocalization] = 0.78; Figures 5A and S5A; Table S4). As expected, the top association signal for PU.1 binding and lymphocyte count align (Figure 5A). The exact molecular function of ZNF608 remains unclear. Nonetheless, a study of follicular lymphoma (FL), a type of cancer in which B lymphocytes divide uncontrollably, found ZNF608 to be among the 39 genes significantly enriched for missense or predicted-loss-of-function (pLOF) somatic mutations in FL patients.52 This finding suggests that the gene may play a role in B lymphocyte development. The associated PU.1 binding site is located about 257 kb upstream of the ZNF608 promoter, and the SNP rs12517864, which increases the PU.1 binding motif score (0.68→2.69), is located near the center of the PU.1 occupancy site (Figure 5B).
Figure 5.

PU.1 motif alteration pinpoints a lymphocyte-count-associated variant that is a secondary ZNF608 eQTL variant
(A) PU.1 bQTL and lymphocyte count association signals. The PU.1 motif-altering variant rs12517864 is shown as a purple diamond, and the ZNF608 eQTL lead variant rs2028854 is shown as a yellow diamond. Vertical dashed lines mark the position of these two variants. Points are colored by LD r2 with respect to rs12517864.
(B) The effect of rs2028854 on the sequence with respect to the PU.1 binding motif.
(C) ZNF608 locus genome tracks of PU.1 ChIP-seq, ATAC-seq, and H3K4me1 and H3K27ac ChIP-seq assayed in GM12878.
(D) Boxplots of the effect of rs12517864 dosage on various molecular phenotypes shown in (C), using the same colors. For PU.1 ChIP-seq data, there were no individuals with a homozygous alternate allele (AA). All data points are superimposed over the boxplots.
(E) Gene track showing ZNF608 and the two variants. The weights of the red curves indicate the capture Hi-C analysis of genomic organization (CHiCAGO) scores calculated by Javierre et al.,53 representing physical interaction.
(F) Top: primary ZNF608 eQTL signals in LCLs. LD r2 is calculated with respect to rs2028854, the lead variant. Bottom: ZNF608 eQTL signals in LCLs conditioned on the rs2028854 dosage. Points are colored as in (A).
(G) Fine-mapping result of ZNF608 eQTL signals in LCLs, using SuSiE.54 Points are colored by the credible set to which they belong. PIP, posterior inclusion probability.
(H) Boxplots of ZNF608 expression levels (count per million [CPM]) through lymphocyte differentiation and across various lymphocyte types. All data points are superimposed over the boxplot. HSC, hematopoietic stem cell; MPP, multipotent progenitor; LMPP, lymphoid-primed multipotent progenitor; CLP, common lymphoid progenitor; B, B cell; CD4T, CD4+ T cell; CD8T, CD8+ T cell; NK, natural killer.
(I) ZNF608 eQTL association signals in naive B cells (Database of Immune Cell Expression, Expression Quantitative Trait Loci and Epigenomics [DICE]55). Points are colored as in (A).
See also Figure S5.
Multiple lines of evidence support the regulatory effect of rs12517864. Based on our reanalysis of ATAC-seq43 and histone mark ChIP-seq data for LCLs,44 we found that rs12517864 is significantly associated with each of these molecular phenotypes that overlap the PU.1 binding site (Figures 5C and 5D). This observation suggests that the variant, if causal, likely affects gene regulation. Consistent with the observation that PU.1 is generally an activator,56,57 increased PU.1 binding was associated with increased chromatin accessibility and active histone marks—H3K27ac and H3K4me1 (p = 1.9 × 10−24, 9.0 × 10−20, and 1.4 × 10−10, respectively; Tables S10, S11, and S12). Furthermore, the variant falls within a fragment that physically interacts only with the ZNF608 promoter in primary B cells according to promoter-capture Hi-C (PCHi-C) data,53 supporting the model that rs12517864 directly regulates ZNF608 (Figure 5E).
Surprisingly, initial inspection of ZNF608 eQTL signals in LCLs58 seemed contradictory because the lead variant for this eQTL (rs2028854) is located elsewhere, 200 kb upstream of the ZNF608 promoter, and is not strongly associated with lymphocyte count32 (p = 0.04; Figures 5E and 5F). We therefore examined the possibility of multiple independent ZNF608 eQTL signals in LCLs by performing a conditional analysis on the lead variant as well as fine-mapping using the “sum of single effects” (SuSiE) model,54 which can detect multiple signals. When conditioned on the lead eQTL SNP rs2028854, association of rs12517864 to ZNF608 expression became much stronger (p = 2.03 × 10−7), with a positive effect direction (Figure 5F; Table S14). The effect direction is consistent with the increased chromatin activity of the enhancer by rs12517864 (Figure 5D). Moreover, the fine-mapping analysis identified two independent credible sets for ZNF608 eQTL signal, one of which contained rs12517864 as the variant with the highest posterior inclusion probability (PIP = 0.07), demonstrating that this variant is likely to be causally associated with ZNF608 expression level (Figure 5G).
Because only one of the two independent ZNF608 eQTL signals in LCLs is associated with lymphocyte count, we hypothesized that even though both SNPs are significant eQTLs in LCLs, only rs12517864 (i.e., the secondary eQTL signal), and not rs2028854 (i.e., the primary eQTL signal), modulates ZNF608 expression in the causal cell type. Analysis of RNA-seq data for various blood cells38 revealed that ZNF608 is highly expressed in common lymphoid progenitors and B cells (Figure 5H). Inspection of eQTL data for B cells in the eQTL Catalogue55,59 showed that only rs12517864, and not rs2028854 (p = 0.25), is significantly associated with increased ZNF608 expression (p = 4.39 × 10−5; Figure 5I). Although we cannot unambiguously conclude that B cells are the causal cell type, rs12517864 is likely the only variant that increases lymphocyte count through increased ZNF608 expression (Figure S5B).
Because the ZNF608 locus demonstrates an interesting genetic architecture, we searched for additional such examples. Based on conditional eQTL analysis, the ZNF608 locus was the only example with a PU.1 motif-altering variant representing a secondary eQTL signal. We also applied statistical fine-mapping54 on eQTLs at PU.1 bQTL colocalized loci to look for GWAS loci with only one of multiple eQTL signals colocalizing. However, the ZNF608 locus was the only example with a gene with more than one independent eQTL signal at blood cell trait GWAS loci that colocalized with PU.1 bQTL signals.
Blood cell trait-associated PU.1 motif-altering variants show regulatory effects in reporter assays
To verify that the nominated PU.1 motif-altering variants are indeed regulatory variants, we inspected massively parallel reporter assay (MPRA) study data,60,61 which measured the regulatory effects of two such variants. rs5827412, a PU.1 motif-altering short deletion in the LRRC25 locus, was associated with a lower monocyte percentage (p = 1.3 × 10−96) and lowered reporter activity61 (two-sided t test, p = 6.9 × 10−5). rs3808619, a PU.1 motif-altering SNP at the promoter of ZC2HC1A, was associated with a lower lymphocyte count (p = 2.3 × 10−98) and increased reporter activity61 (two-sided t test, p = 0.006).
LRRC25, also called monocyte and plasmacytoid-activated protein (MAPA), is a gene necessary for differentiation of granulocytes, which share lineages with monocytes.62 At this locus, we found that the PU.1 bQTL signal showed significant colocalization with monocyte count and percentage, neutrophil count and percentage, and white blood cell count association signals8,32 (JLIM p = 5 × 10−5, 4 × 10−5, 1 × 10−5, 1 × 10−5, and 1 × 10−5, respectively, and Coloc PP[colocalization] = 0.99, 0.99, 0.99, 0.99, and 0.98, respectively; Figures 6A and S6A; Table S4). As the association Z scores show, variants significantly associated with lower PU.1 binding are also associated with a lower monocyte percentage, consistent with colocalization (Figure 6A). In contrast, the direction of effect is reversed for neutrophil count and percentage and white blood cell count (Figure S6A). The corresponding PU.1 binding site contains a short deletion rs5827412 that lowers the PU.1 motif score and is associated with reduced PU.1 binding as well as chromatin accessibility, active histone mark levels, and LRRC25 expression43,44,58 (Figures 6B and S6B), which is expected from PU.1’s likely role as an activator.56,57 Consistent with reduced PU.1 binding, rs5827412 significantly reduced regulatory activity in a reporter assay61 (two-sided t test, p = 6.9 × 10−5; Figure 6C, left bar); data from another study suggested concordant direction of effect despite not being statistically significant60 (negative binomial regression, p = 0.26; Figure 6C, right bar). Next, we analyzed available ATAC-seq data from SPI1, the gene encoding PU.1, knockout pro-B cell lines (RS4;11) to verify whether PU.1 is likely to be the trans factor for the regulatory variant.63 We found that, across triplicates for each genotype, SPI1 knockout resulted in significantly reduced chromatin accessibility at sites of PU.1 occupancy genome wide64 (chi-square test, p < 1 × 10−300; Figure S6C). Indeed, the activity of the regulatory element that contains rs5827412 is likely dependent on PU.1 binding because SPI1 knockout cell lines showed reduced chromatin accessibility at this region (DESeq2-adjusted p = 8.73 × 10−5; Figure 6D). RNA sequencing (RNA-seq) data for 13 blood cell types38 indicate that LRRC25 is specifically expressed in monocytes at a much higher level than in other blood cell types and is sharply upregulated as progenitor cells differentiate to monocytes (Figures 6E and S6D). Consistent with the variant’s strongest effect on monocyte percentage (p = 1.3 × 10−96) and monocyte-specific expression of LRRC25, we found that rs5827412 is also significantly associated with reduced LRRC25 expression in monocytes65 (p = 3.78 × 10−22; Figure 6F) and is in a regulatory element that is accessible throughout monocyte differentiation (Figure 6G). Altogether, our results provide strong support for rs5827412 reducing LRRC25 gene expression levels in monocytes and decreasing monocyte percentage while increasing neutrophil percentage.
Figure 6.

PU.1 motif-altering deletion rs5827412 at the LRRC25 locus associated with lower monocyte counts
(A) Association Z scores of variants in the locus with PU.1 binding and monocyte percentage. The sign of the Z score is the effect direction of the AA of each variant. The points are colored by LD r2 with respect to rs5827412 (purple diamond).
(B) The effect of rs5827412 on the PU.1 motif. Dashes indicate gaps in the alignment, reflecting the short deletion.
(C) Negative allelic skew (i.e., reduced reporter activity) by rs5827412 in log2 fold change. Error bars indicate 95% confidence intervals. ∗: adjusted p < 0.05.
(D) A boxplot showing PU.1-dependent reduction in chromatin accessibility levels (CPM) at the regulatory element surrounding rs5827412 in control pro-B cell lines (SPI1+/+) and counterparts with SPI1 knocked out (SPI1−/−). Regions highlighted in yellow marks the accessible region corresponding to the boxplot. All data points are superimposed over the boxplot. n = 3 for each condition. ∗: DESeq2-adjusted p < 0.05.
(E) A boxplot showing LRRC25 expression levels (CPM) through monocyte differentiation. All data points are superimposed over the boxplot. CMP, common myeloid progenitor; GMP, granulocyte-macrophage progenitor.
(F) Mono LRRC25 eQTL association. Downward and upward triangles indicate the direction of effect (down- and upregulation, respectively) for variants with p < 1 × 10−3. A purple triangle and dashed line mark rs5827412.
(G) LRRC25 locus ATAC-seq tracks as fold enrichment over average (range, 0–40) for various blood cell types through monocyte differentiation. A purple diamond and dashed line mark rs5827412.
In the ZC2HC1A locus, which is primarily associated with lymphocyte count and percentage32 (p = 1.9 × 10−84 and 6.3 × 10−58, respectively), a PU.1 motif-altering SNP, rs3808619, is among more than 40 tightly linked (LD r2 ≈ 1) variants (Figures 7A, 7B, S7A and S7B; Table S4). Currently, ZC2HC1A is a functionally uncharacterized gene. Based on a UK Biobank fine-mapping study,66 44 variants comprise the 95% credible set at this locus, and none has a PIP greater than 0.1 (Figure 7C). From statistical fine-mapping alone, one would not be able to pinpoint the causal variant. However, we found that rs3808619 is the only PU.1 motif-altering variant found within the associated PU.1 binding site at the ZC2HC1A promoter. rs3808619 increases the strength of a PU.1 motif, resulting in a higher-affinity DNA binding site (Figure 7A). Of multiple variants in this locus that were in high LD with rs3808619 and were tested for reporter activity (59 variants in Abell et al.60 and 30 variants in Tewhey et al.61), only rs3808619 showed significantly increased reporter activity (negative binomial regression p = 5.7 × 10−5 and two-sided t test p = 0.006, respectively) that is concordant in direction with that of the variant’s associations with elevated chromatin accessibility, active histone mark levels, and ZC2HC1A expression in LCLs43,44,58 (Figures 7D–7F). Finally, similar to the previous example, we detected significantly reduced chromatin accessibility levels at the ZC2HC1A promoter in SPI1 knockout cell lines63 (DESeq2-adjusted p = 1.76 × 10−13), supporting the likely role of PU.1 at this promoter (Figure 7G). rs3808619 is also associated with multiple sclerosis67 (p = 1.1 × 10−9; Figures S7C and S7D), suggesting that it plays a multifactorial role in immune-mediated diseases. Our results suggest that a direct consequence of rs3808619, which is associated with a lower lymphocyte count, is likely ZC2HC1A upregulation (Note S3).
Figure 7.

ZC2HC1A locus: PU.1 motif alteration highlights a regulatory variant among those in high LD
(A) The effect of rs3808619 on the PU.1 composite motif.
(B) PU.1 bQTL and lymphocyte count association signal at the ZC2HC1A locus. PU.1 motif-altering variant rs3808619 is marked with a purple diamond and a dashed line.
(C) PIP of variants in the 95% credible set of lymphocyte count association at the ZC2HC1A locus. rs3808619 is marked as in (B).
(D) ZC2HC1A locus genome tracks of PU.1 ChIP-seq, ATAC-seq, and H3K4me1, histone H3 lysine 4 trimethylation (H3K4me3), and H3K27ac ChIP-seq assayed in GM12878. rs3808619 is marked as in (B). The highlighted regions correspond to molecular phenotypes with QTL associations in (E).
(E) The effect of rs3808619 dosage on various molecular phenotypes shown in (D). All data points are superimposed over the boxplot.
(F) Regulatory effects of rs3808619 and 58 tagging variants in a reporter assay. MPRA allelic effect corresponds to log2 fold change of regulatory activity of the oligo sequence with the AA over that with the reference allele. The inset shows the allelic skew estimates with error bars depicting the 95% confidence intervals from Abell et al.60 and Tewhey et al.61 ∗: adjusted p < 0.05.
(G) PU.1-dependent reduction in chromatin accessibility levels (CPM) at the regulatory element surrounding rs3808619 in control pro-B cell lines (SPI1+/+) and counterparts with SPI1 knocked out (SPI1−/−). n = 3 for each condition. ∗: DESeq2 adjusted p < 0.05. The panel is formatted as in Figure 6D.
Discussion
Our results with PU.1 binding and blood cell trait GWAS data demonstrate the utility of TF bQTL data in identifying which of many variants in LD are the likely causal regulatory variants underlying GWAS trait associations. If a TF bQTL signal shows significant colocalization with a GWAS signal, and if there is a motif-altering variant for that TF in the binding site, then that variant is likely to be the causal variant for both associations. Incorporating PU.1 bQTLs in our colocalization analysis conferred two key advantages: (1) identification of trait-associated regulatory elements, in which PU.1 binding is altered, and (2) identification of putatively causal PU.1 motif-altering variants. Together, they highlight a likely transcriptional regulatory mechanism underlying the trait association. In contrast, alternative approaches to narrow down variants in accessible chromatin and search for altered motifs often do not show the same level of precision. Moreover, eQTL colocalization cannot assist fine-mapping in this way because there is no prior expectation that a specific noncoding region regulates the associated gene and that a regulatory variant alters a certain TF binding site motif. TF bQTLs offer a unique opportunity in this aspect.
For instance, in the ZNF608 locus, pinpointing the putative causal variant and associated regulatory element would have been difficult without PU.1 bQTLs. The lead eQTL signal for ZNF608 in LCLs did not colocalize with the lymphocyte count association (Figure 5). Such a situation may partially explain the observation that many significant eQTL signals failed to colocalize with the GWAS associations using existing colocalization methods.23 However, this locus was the only such example in our study. Nevertheless, this example motivates applying TF bQTL colocalization to isolate independent eQTL signals and generating eQTL data in trait-relevant cell types.68 Moreover, applying colocalization methods that allow multiple causal variants to eQTLs69 would be useful when accurate LD matrices or individual genotypes are available for both traits, which is often not the case for GWAS data.
Despite finding several examples of PU.1 motif-altering variants driving a change in gene expression level, only 10 of 51 such loci showed eQTL signals in LCLs. This observation is not unlike reports showing that, although GWAS loci are enriched in eQTL signals,70 only a small subset of GWAS loci shows colocalization with eQTLs.23,30,71 Nevertheless, most PU.1 motif-altering variants that colocalized with blood cell trait associations showed effects on allelic imbalance in PU.1 ChIP-seq, on chromatin accessibility, and on histone marks (Figure 4). These variants are likely to be true functional regulatory variants, so it is mysterious that eQTL effects are not detected in the same cell type (i.e., LCLs). A possible explanation is that, even though the variants alter PU.1 binding in LCLs, their effects on gene expression are manifested in a different cell type, such as progenitor cell types during hematopoiesis, or under particular environmental conditions. Uncovering other possible reasons for the lack of eQTL signals at those loci is crucial for understanding how the different layers of gene regulation affect complex traits.
A prior study that performed colocalization analysis of neutrophils' PU.1 bQTLs and immune disease GWASs found that a minority (<50%) of colocalized variants altered PU.1 motifs.25 In contrast, we found that the majority (87%) of the colocalized blood cell trait GWAS loci had a variant that altered a PU.1 motif (Figure 2C). This is an enrichment over just 34% of all LCLs’ PU.1 bQTLs, colocalized or not, harboring a PU.1 motif-altering variant (Figure 1C). The increased proportion of PU.1 motif-altering variants present in this study may be due to PU.1’s central role in blood cell traits36 and highlights the increased likelihood that PU.1 binding is mediating the genetic effects on blood cell traits.
We observed that only a minority of the tested GWAS loci (69 of 367) showed significant colocalization. This is not surprising because we selected candidate loci solely based on the marginal association with PU.1 binding and blood cell traits23 without filtering for high LD between the two lead variants23 to “cast a wide net” for discovery. This observation is a testament to the importance of performing colocalization analysis to distinguish loci with a single causal variant for the two phenotypes (here, PU.1 binding and a particular blood cell trait) from those with distinct tagging variants responsible for the individual phenotypes. Furthermore, even though PU.1 bQTLs were enriched for blood cell trait association (Figure 2A), they explained only a subset of all associated loci, likely indicating that other TFs are mediating genetic effects at other associated loci.
We offer guidelines for broad application of colocalization analysis with TF bQTLs. First, high-quality ChIP-grade antibodies72 or, alternatively, cell lines in which the TF has been epitope tagged are essential. Second, TFs for bQTL analysis, as well as the cell type for the ChIP experiments, must be selected to be relevant to the trait or disease of interest. The feasibility of our analysis relied on the importance of PU.1, a known hematopoietic master regulator, and LCLs, a model of mature B cells, for specific blood cell traits, such as lymphocyte count and monocyte count. Because generating TF ChIP-seq data across multiple genotyped samples can be cumbersome, selecting the trait-relevant TF and cell type is critical. Future studies will need to validate the regulatory functions of the variants in the relevant primary cell types.
Future studies could use TF bQTL data in colocalization analysis to elucidate the ever-increasing number of trait-associated loci.1 When TFs important for a trait are known, TF bQTLs identified in the relevant cell type(s) could mediate a subset of trait associations, shedding light on putative causal variants as well as the pathogenic mechanisms. Such colocalization analysis with TF bQTL data uniquely provides a path to pinpointing causal regulatory elements and variants and, thus, a smaller set of mechanistic hypotheses to test experimentally to verify the underlying causes of the disease.
Limitations of the study
The power of statistical tests, including QTL analysis (i.e., linear regression) and colocalization analysis, depends on the sample size of the data. In this proof-of-concept study, in which we analyzed PU.1 ChIP-seq data from 49 samples, we detected 1,497 significant PU.1 bQTLs and 69 robustly colocalized loci across blood cell traits. However, we anticipate that a larger sample size could increase the power to detect more loci with weaker but significant bQTL and colocalization signals. Moreover, colocalization and genetic association are not, in themselves, tests of causality. We incorporated colocalization and PU.1 motif analyses to identify strong candidates for causal variants and their molecular mechanisms at blood cell trait-associated loci. For two examples, we were able to show that MPRA studies measured the significant regulatory effects of the identified variants in an episomal context (Figures 6 and 7). However, whether the associated regulatory effects of these variants cause downstream changes in blood cell traits needs to be validated with a genetic perturbation experiment that models blood cell traits.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| LCL PU.1 and histone mark ChIP-seq data | Waszak et al.,18 Delaneau et al.44 | EMBL-EBI: E-MTAB-3657, EMBL-EBI: E-MTAB-1884 |
| GM12878 control ChIP-seq | Dunham et al.73 | ENCODE: ENCFF032WUR, ENCFF426WJH, ENCFF508HCX, ENCFF537DAJ, ENCFF812HUT, ENCFF837IOW, ENCFF849LYY, ENCFF892TNJ |
| LCL ATAC-seq data | Kumasaka et al.43 | ENA: ERP110508 |
| Processed LCL RNA-seq data | Lappalainen et al.58 | EMBL-EBI: E-GEUV-1 |
| Human reference genome NCBI build 37, GRCh37 | Genome Reference Consortium | http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/ |
| 1000 Genomes Project Phase 3 data | Auton et al.39 | ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20130502 |
| 1000 Genomes Project High-coverage data | Byrska-Bishop et al.74 | http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/1000G_2504_high_coverage/working/20220422_3202_phased_SNV_INDEL_SV/ |
| UK Biobank blood cell trait GWAS summary statistics | Canela-Xandri et al.,32 Vuckovic et al.8 | http://geneatlas.roslin.ed.ac.uk/, ftp://ftp.sanger.ac.uk/pub/project/humgen/summary_statistics/UKBB_blood_cell_traits |
| Type 2 diabetes GWAS lead SNPs | Mahajan et al.13 | https://www.nature.com/articles/s41588-018-0241-6 |
| Height GWAS lead SNPs | Wood et al.75 | https://www.nature.com/articles/ng.3097 |
| ATAC-seq data from blood cell types | Corces et al.38 | GEO: GSE74912 |
| RNA-seq data from blood cell types | Corces et al.38 | GEO: GSE74246 |
| Monocyte eQTL data | Chen et al.65 | http://blueprint-dev.bioinfo.cnio.es/WP10/qtls |
| Naive B cell eQTL data | Schmiedel et al.55 | ftp://ftp.ebi.ac.uk/pub/databases/spot/eQTL/sumstats/Schmiedel_2018/ge/Schmiedel_2018_ge_monocyte.all.tsv.gz |
| Blood cell traits' GWAS fine-mapping data | Vuckovic et al.,8 Kanai et al.66 | https://github.com/bloodcellgwas/manuscript_code/tree/master/data/finemap_bedfiles/ukbb_v2, https://www.finucanelab.org/data |
| LCL massively parallel reporter assay (MPRA) data | Tewhey et al.,61 Abell et al.60 | https://www.sciencedirect.com/science/article/pii/S0092867416304214, https://www.science.org/doi/10.1126/science.abj5117 |
| ATAC-seq data from SPI1 knockout study | Le Coz et al.63 | EMBL-EBI: E-MTAB-8676 |
| RS4; 11 PU.1 ChIP-seq data | Wu et al.64 | GEO: GSE71616 |
| Software and algorithms | ||
| Bowtie2 | Langmead et al.76 | https://bowtie-bio.sourceforge.net/bowtie2 |
| WASP | Van de Geijn et al.77 | https://github.com/bmvdgeijn/WASP |
| GSNAP | Wu et al.78 | http://research-pub.gene.com/gmap/ |
| MACS2 | Zhang et al.79 | https://pypi.org/project/MACS2/ |
| FeatureCounts | Liao et al.80 | https://subread.sourceforge.net/featureCounts.html |
| Bedtools | Quinlan et al.81 | https://bedtools.readthedocs.io |
| PWMScan | Ambrosini et al.82 | https://ccg.epfl.ch/pwmtools/pwmscan.php |
| LS-GKM (gkm-SVM) | Lee et al.83 | https://github.com/Dongwon-Lee/lsgkm |
| Michigan Imputation Server | Das et al.84 | https://imputationserver.sph.umich.edu |
| BCFtools | Danecek et al.85 | https://github.com/samtools/bcftools |
| PLINK v1.9 | Chang et al.86 | https://www.cog-genomics.org/plink/1.9 |
| PEER | Stegle et al.87 | https://github.com/PMBio/peer |
| QTLtools | Delaneau et al.88 | https://qtltools.github.io/qtltools/ |
| SNPsnap | Pers et al.89 | https://data.broadinstitute.org/mpg/snpsnap |
| JLIM 2.0 | Chun et al.90 | https://github.com/cotsapaslab/jlim |
| Coloc | Giambartolomei et al.22 | https://github.com/chr1swallace/coloc |
| Custom scripts to perform colocalization analyses | This paper | https://doi.org/10.5281/zenodo.7837982 |
| Fujiplot | Kanai et al.91 | https://github.com/mkanai/fujiplot |
| ComplexUpset | Lex et al.92 | https://github.com/krassowski/complex-upset |
| RASQUAL | Kumasaka et al.93 | https://github.com/natsuhiko/rasqual |
| MotifbreakR | Coetzee et al.94 | https://github.com/Simon-Coetzee/motifBreakR |
| LocusCompareR | Liu et al.21 | https://github.com/boxiangliu/locuscomparer |
| SusieR | Wang et al.54 | https://github.com/stephenslab/susieR |
| DESeq2 | Love et al.95 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Integrative Genomics Viewer | Robinson et al.96 | https://software.broadinstitute.org/software/igv/ |
| Custom scripts to generate figures | This paper | https://doi.org/10.5281/zenodo.7837894 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Martha L. Bulyk (mlbulyk@genetics.med.harvard.edu).
Materials availability
This study did not generate new unique reagents.
Method details
PU.1 ChIP-seq data processing
We downloaded PU.1 ChIP-seq fastq files from EMBL-EBI ArrayExpress under accession “ArrayExpress: E-MTAB-3657”18 (n = 45) and “ArrayExpress: E-MTAB-1884”26 (n = 4). The list of samples is provided in Table S1. We mapped the reads to the hg19 reference genome supplemented with the Epstein-Barr virus (EBV) genome using Bowtie 2.76 In order to eliminate reference allele bias in read mapping, we applied WASP77 to filter reads that mapped to a different position when variants were added, and used GSNAP,78 which is an SNP-tolerant read alignment method, to remap filtered out reads.
PU.1 ChIP-seq peaks were called using model-based analysis of ChIP-seq version 2 (MACS2).79 For equal representation, we subsampled 5 million reads from each sample and performed peak calling on the aggregate alignment file. To account for the size of the merged read set, we downloaded 8 available control ChIP-seq samples in GM12878 from ENCODE73 (File IDs: ENCFF032WUR, ENCFF426WJH, ENCFF508HCX, ENCFF537DAJ, ENCFF812HUT, ENCFF837IOW, ENCFF849LYY, ENCFF892TNJ) for peak calling. To define 200-bp sequences occupied by PU.1, we took the summits and extended them by 100 bp in each direction. In total, there were 78,720 peaks.
PU.1 binding quantitative trait loci
First, we quantified the PU.1 binding levels at identified occupancy sites. We counted the number of reads overlapping each 200-bp peak using featureCounts.80 For each sample, the read counts were normalized for library size using trimmed mean of M-values97 so that the values are comparable across the samples. Then, the phenotype values were further normalized to follow a standard normal distribution across the samples, using quantile normalization, similar to the GTEx protocol.51 Finally, in order to eliminate the effect of variables, such as batch, gender, and ancestry, we used PEER87 to residualize the phenotype values, correcting for batch (i.e., which publication), sex, and 3 genotype principal components, as well as 10 PEER factors.
Second, we obtained the genotypes of the LCL samples from the 1000 Genomes Project data.39 4 out of 49 samples only had microarray genotype data from Illumina Omni2.5 chips, and these genotypes were phased and imputed using the European samples of the 1000 Genomes project phase 3 data39 on the Michigan Imputation Server.84 Genotypes of all samples were converted to biallelic form and aggregated. Afterward, variants with minor allele frequency less than 5% were removed from the PU.1 binding quantitative trait loci analysis.
Finally, we tested for genetic associations to PU.1 binding levels using the phenotype matrix and the genotype data. We utilized QTLtools88 to approximate linear regression efficiently while also correcting for multiple hypotheses tested with permutations and false discovery rate estimation. For each PU.1 occupancy site, variants within 100 kb were included in the QTL analysis. In the end, there were 1,497 significant PU.1 bQTLs at FDR <5%.
UK Biobank blood cell trait GWAS summary statistics
We downloaded 28 blood cell trait GWAS summary statistics from UK Biobank32 for the colocalization analysis. The authors performed a linear mixed model-based regression analysis on 452,264 White British individuals using rank-normalized phenotypes. The 28 blood cell traits are listed in Table S3. One limitation of these summary statistics is that the authors used the Haplotype Reference Consortium imputation panel, which only included SNPs by design, for imputation98 (Note S2). Thus, short deletions like rs5827412 were missing in these summary statistics. For Figure 6, we verified that the variant is associated with decreased monocyte percentage and increased neutrophil percentage in summary statistics from another analysis of the UK Biobank data,8 and utilized these data for visualization.
Fold enrichment of GWAS signal in PU.1 bQTLs
We first generated 250 sets of null variants matched with the significant PU.1 bQTL lead variants for allele frequency, number of tagging SNPs (LD r2 > 0.5), and distance to the closest transcription start site (TSS), using SNPsnap.89 250 sets of null variants were successfully generated for 1,292 of the PU.1 bQTL lead variants, so we restricted the downstream analysis within them. Using the distribution of number of variants tagging (r2 > 0.8) trait-associated lead variants as the background, we computed the fold enrichment of the number of PU.1 bQTLs tagging those variants. The empirical p values are derived for each blood cell trait by counting how many sets had SNPs tagging (r2 > 0.8) trait-associated variants more than or equal to the number of PU.1 bQTLs tagging them and dividing by 251. The p values were adjusted using qvalue package in R. For non-blood traits, lead SNPs from GWAS of type 2 diabetes13 and height75 were used.
Position weight matrix and gkm-SVM PU.1 motif models
To initially scan for the position of PU.1 motif sequences within occupancy sites, we used PWMScan.82 With a PU.1 (SPI1) motif position weight matrix (PWM) selected within the tool (CISBP: M6119_1) we scanned for the motif (p < 10−5) within PU.1 occupancy sites, which resulted in a total of 30,812 instances. To determine the relative location of PU.1 motifs within the PU.1 occupancy sites, we subtracted the start or end position of the motif from the center position of the 200-bp PU.1 peak, depending on the strand (Figure S2A).
Afterward, we trained a PU.1 motif model using gkm-SVM, as a more sophisticated counterpart to PWM. We used the 200-bp sequences detected to be PU.1 occupancy sites for positive sequences in the training set. We left out PU.1 occupancy sites with a variant overlapping PU.1 motifs identified using PWMs (i.e., one of the alleles with log likelihood score >8) from the training set so that the model effectively captures the motif sequences and excludes potentially causal PU.1 bQTLs. We generated negative sequences using the ‘genNullSeqs’ function in the gkmSVM R package. Then, we trained the model using default parameters with LS-GKM,83 which is a faster implementation from the developers. Throughout the study, we defined PU.1 motif-altering variants as those where one of the alleles shows a gkm-SVM score greater than 0 for a 30-bp sequence centered at the variant, and the variant induces a non-zero change.
Colocalization analysis using JLIM and Coloc
We selected 1,621 PU.1-trait pairs at loci where the significant PU.1 bQTLs also show at least one blood cell trait association at p < 10−6 to perform colocalization. For JLIM,23,90 we used the default parameters. p values were derived by permuting the PU.1 binding level matrix. For Coloc,22 we used the prior parameters p1 = 10−4, p2 = 10−4, and p12 = 10−6, which is more conservative than the default, and ran Coloc on the summary statistics. For both analyses, we considered variants within a 200-kb window around the GWAS lead variant. We used a significance threshold of p < 0.01172 (FDR <5%) for JLIM and posterior probability of colocalization (PP(Colocalization)) > 0.5. The FDR cutoff for JLIM was determined by the equation:
where pcutoff is the p value cutoff, N is the number of PU.1-trait loci tested, and PJLIM is the JLIM p value.
Chromatin accessibility, histone mark, and expression QTLs in LCLs
ATAC-seq43 (n = 100), histone mark ChIP-seq (n = 15813 and n = 234, respectively), and RNA-seq58 (n = 373) data were downloaded from European Nucleotide Archive ("ENA: ERP110508"), EMBL-EBI ArrayExpress ("ArrayExpress: E-MTAB-3657" and "ArrayExpress: E-GEUV-1"), respectively. ATAC-seq data were only available as bam files, so we used bamtofastq command from bedtools81 to extract reads. We processed ATAC-seq and histone mark ChIP-seq read data similarly to PU.1 ChIP-seq data (i.e., alignment, duplicate removal, peak calling, quantification, and then probabilistic estimation of expression residuals [PEER]87 normalization). The processed gene expression matrix derived from RNA-seq was downloaded directly.
We obtained the genotypes of the LCL samples from the 1000 Genomes Project data. We imputed 9 out of 100, 9 out of 160, and 15 out of 373 samples, respectively, from available microarray data to the 1000 Genomes Project phase 3 data39 on the Michigan Imputation Server.84 Common variants (MAF >5%) from the merged genotypes and the prepared phenotype matrices were used to test genetic associations to the corresponding molecular phenotypes with QTLtools.88
We counted the number of significant chromatin accessibility QTLs (caQTLs) and histone QTLs (hQTLs) that are in LD (r2 > 0.8) with PU.1 motif-altering variants. Since PU.1 binding alteration would affect chromatin that it binds, we considered only those ATAC-seq and ChIP-seq peaks that overlapped the corresponding PU.1 ChIP-seq peak. LD between the lead variants was determined using the genotypes of 373 European samples with gene expression data.
To count how many eQTL signals are in LD with colocalized PU.1 motif-altering variants, we searched not only for primary eQTL signals but also for secondary eQTL signals by conditioning on the primary lead variants. For fine-mapping the ZNF608 locus, as in Figure 5D, we applied SuSiE54 using default parameters and the genotype matrix of variants within 1 Mb of the gene’s transcriptional start site. This same fine-mapping approach was used to search for other examples of colocalized PU.1 motif-altering variants with multiple eQTL signals where only one colocalizes with the GWAS signal.
Searching for accessible variants in GWAS credible sets and their TF motif alterations
We first ascertained 25 PU.1 bQTL colocalized GWAS loci that had a credible set provided in a published blood cell trait GWAS fine-mapping study.8 Chromatin accessibility annotation was derived from the 100 LCL ATAC-seq samples mentioned above. We scanned the ascertained credible set variants for those in accessible chromatin using bedtools.81 Then, we searched for which TFs’ motifs were altered by these variants, using motifbreakR.94 We considered all 2,817 human TF motifs collected in the tool’s dataset “motifbreakR_motif”. The dataset includes multiple versions of some TFs’ motifs because the PWMs were collected from multiple sources. We used “filterp = TRUE” option with threshold of p = 5 × 10−4. The PWM scoring method was set to “ic”. Since there can be redundant occurrences of motif alterations for the same TF across the PWM databases, we considered the number of unique TFs. Lastly, to filter the motifs for those corresponding to TFs expressed in LCLs, we considered TFs with median gene expression TPM >1 across 373 LCL samples.58
QTL analysis for rs74267027 missing in 1000 Genomes phase 3 data
PU1_67321 (chr17:16,171,568-16,171,767) significantly colocalized with blood cell traits – lymphocyte percentage, neutrophil percentage, neutrophil count, and white blood cell count (JLIM p = 5 × 10−5, 5 × 10−5, 5 × 10−5, and 6 × 10−5, respectively, and Coloc PP(Colocalization) = 0.85, 0.85, 0.82, and 0.71, respectively). Initially with 1000 Genomes project phase 3 data,39 there was no PU.1 motif-altering variant. However, with closer inspection, a short deletion rs74267027 that alters a PU.1 binding motif at this site was present in the recently published high-coverage genotype data74 (Table S5). Therefore, we used the genotype information in the high-coverage genotype data to estimate its QTL effect for PU.1 binding, chromatin accessibility and histone mark levels.
PU.1 ChIP-seq allelic imbalance effects of PU.1 motif-altering variants
We analyzed PU.1 ChIP-seq data across 49 individuals to estimate the effect of prioritized variants on allele-specific PU.1 binding. First, we counted the number of PU.1 ChIP-seq reads containing the reference or the alternate allele using createASVCF.sh script from the robust allele-specific quantification and quality control (RASQUAL) package93. For 44 PU.1 motif-altering SNPs that colocalized with blood cell traits association, we identified heterozygous individuals and determined the log2 allelic fold change between the two haplotypes within each sample. In order to account for samples with no reads containing either allele, we added a pseudocount of 0.5 to both the denominator and the numerator. Consider an individual with the genotype “1|0”, where 0 and 1 are reference and alternate alleles, respectively. Haplotype 1 reads would contain the alternate allele, while haplotype 2 reads would contain the reference allele. Then,
Next, we followed the allelic imbalance model presented by Liang and colleagues49 to estimate the variant effect on allelic imbalance across individuals. The only difference from that model is the pseudocount of 0.5 that we added to the number of reads from each haplotype. Individuals with “1|0” and “0|1” genotypes will be encoded as “1” and “-1”, respectively. We performed weighted linear regression where the weights were
to estimate the variant’s effect on allelic imbalance. This weighting scheme effectively puts more weight on samples with a higher number of reads.
Chromatin accessibility and gene expression levels across blood cell types
ATAC-seq and RNA-seq data from multiple blood cell types throughout hematopoiesis were downloaded from GEO series GSE74912 and GSE74246, respectively.38 We aligned ATAC-seq read data to the hg19 reference genome, and merged data from each cell type for visualization. The genome tracks in Figure 6G were generated with fold enrichment over average genome coverage to account for library size differences. We downloaded the count matrix for RNA-seq and converted them to counts per million for comparison across cell types.
MPRA data analysis
We downloaded MPRA analysis tables from the two studies.60,61 We extracted statistics for rs5827412 and rs3808619, which were the only two putative causal PU.1 motif-altering variants at colocalized loci with MPRA data. For rs3808619, we also extracted the statistics for the other 29 and 58 variants tagging rs3808619 from Tewhey et al. and Abell et al., respectively. From Tewhey et al. data, we referred to the combined LCL analysis statistics, and from Abell et al. data, we referred to the allele effect statistics to measure the regulatory effects of variants.
Differential accessibility analysis in SPI1 knockout RS4; 11 lines
ATAC-seq data from wild type and SPI1 knockout RS4; 11 cell lines were downloaded from EMBL-EBI ArrayExpress under accession “ArrayExpress: E-MTAB-8676”.63 We aligned the reads using Bowtie276 and removed duplicate alignments using scripts from WASP.77 Then, we pooled the three replicates per genotype to call accessible regions using MACS279 with q < 0.05 cutoff, and the two sets of accessible regions were merged using bedtools.81 After counting the number of reads from each region using featureCount,80 we tested for differential accessibility using DESeq2.95 PU.1 ChIP-seq and input DNA data from unstimulated RS4; 11 cell lines were downloaded from GEO series GSE71616.64 After alignment using Bowtie276 and duplicate removal,77 we called peaks using MACS2.79 Accessible regions were stratified by whether they intersect identified PU.1 occupancy sites. The significance of observing reduced accessibility in SPI1 knockout lines was tested using a chi square test.
Quantification and statistical analysis
Details of the statistical analyses are described in the relevant sections of the method details or in the figure legends.
Acknowledgments
We thank all members of the Bulyk lab and members of the Raychaudhuri lab including, but not limited to, Soumya Raychaudhuri, Kazuyoshi Ishigaki, Saori Sakaue, Tiffany Amariuta, Yang Luo, and Samira Asgari for valuable feedback. We thank Vijay Sankaran, Shamil Sunyaev, and Alexander Gusev for helpful discussions throughout the work. We also thank Shubham Khetan, Shamil Sunyaev, and Soumya Raychaudhuri for critical reading of the manuscript. This work was funded by a grant from the Brigham and Women's Hospital's Fund to Sustain Research Excellence and NIH grant R01 HG010501.
Author contributions
R.J. and M.L.B. conceived and designed the research project. R.J. performed all analyses and prepared the figures. M.L.B. supervised the research. R.J. and M.L.B. wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: May 17, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xgen.2023.100327.
Supplemental information
GWAS statistics are based on Canela-Xandri et al. [S8].
GWAS statistics and fine-mapping results are from Vuckovic et al. [S7].
Only single nucleotide polymorphisms were included in the analysis. Each locus is defined by blood cell trait and a colocalized PU.1 bQTL.
Rank is based on conditional analysis.
Data and code availability
Code and processed data for performing colocalization analysis in this study are available at https://doi.org/10.5281/zenodo.7837982. Code and processed data for generating the figures are available at https://doi.org/10.5281/zenodo.7837894. All other data used in the analysis are publicly available and listed in the key resources table.
References
- 1.Claussnitzer M., Cho J.H., Collins R., Cox N.J., Dermitzakis E.T., Hurles M.E., Kathiresan S., Kenny E.E., Lindgren C.M., MacArthur D.G., et al. A brief history of human disease genetics. Nature. 2020;577:179–189. doi: 10.1038/s41586-019-1879-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Claussnitzer M., Dankel S.N., Kim K.-H., Quon G., Meuleman W., Haugen C., Glunk V., Sousa I.S., Beaudry J.L., Puviindran V., et al. FTO obesity variant circuitry and adipocyte browning in humans. N. Engl. J. Med. 2015;373:895–907. doi: 10.3389/fgene.2015.00318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nasser J., Bergman D.T., Fulco C.P., Guckelberger P., Doughty B.R., Patwardhan T.A., Jones T.R., Nguyen T.H., Ulirsch J.C., Lekschas F., et al. Genome-wide enhancer maps link risk variants to disease genes. Nature. 2021;593:238–243. doi: 10.1038/s41586-021-03446-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.International Common Disease Alliance International common disease alliance white paper v1.0. 2020. https://www.icda.bio
- 5.Visscher P.M., Wray N.R., Zhang Q., Sklar P., McCarthy M.I., Brown M.A., Yang J. 10 Years of GWAS discovery: biology, function, and translation. Am. J. Hum. Genet. 2017;101:5–22. doi: 10.1016/j.ajhg.2017.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Amariuta T., Ishigaki K., Sugishita H., Ohta T., Koido M., Dey K.K., Matsuda K., Murakami Y., Price A.L., Kawakami E., et al. Improving the trans-ancestry portability of polygenic risk scores by prioritizing variants in predicted cell-type-specific regulatory elements. Nat. Genet. 2020;52:1346–1354. doi: 10.1038/s41588-020-00740-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weissbrod O., Kanai M., Shi H., Gazal S., Peyrot W.J., Khera A.V., Okada Y., Biobank Japan Project. Martin A.R., Finucane H.K., Price A.L. Leveraging fine-mapping and multipopulation training data to improve cross-population polygenic risk scores. Nat. Genet. 2022;54:450–458. doi: 10.1038/s41588-022-01036-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vuckovic D., Bao E.L., Akbari P., Lareau C.A., Mousas A., Jiang T., Chen M.-H., Raffield L.M., Tardaguila M., Huffman J.E., et al. The polygenic and monogenic basis of blood traits and diseases. Cell. 2020;182:1214–1231.e11. doi: 10.1016/j.cell.2020.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maurano M.T., Humbert R., Rynes E., Thurman R.E., Haugen E., Wang H., Reynolds A.P., Sandstrom R., Qu H., Brody J., et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gusev A., Lee S.H., Trynka G., Finucane H., Vilhjálmsson B.J., Xu H., Zang C., Ripke S., Bulik-Sullivan B., Stahl E., et al. Partitioning heritability of regulatory and cell-type-specific variants across 11 common diseases. Am. J. Hum. Genet. 2014;95:535–552. doi: 10.1016/j.ajhg.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Amariuta T., Luo Y., Gazal S., Davenport E.E., van de Geijn B., Ishigaki K., Westra H.J., Teslovich N., Okada Y., Yamamoto K., et al. IMPACT: genomic annotation of cell-state-specific regulatory elements inferred from the epigenome of bound transcription factors. Am. J. Hum. Genet. 2019;104:879–895. doi: 10.1016/j.ajhg.2019.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van de Geijn B., Finucane H., Gazal S., Hormozdiari F., Amariuta T., Liu X., Gusev A., Loh P.R., Reshef Y., Kichaev G., et al. Annotations capturing cell type-specific TF binding explain a large fraction of disease heritability. Hum. Mol. Genet. 2020;29:1057–1067. doi: 10.1093/hmg/ddz226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mahajan A., Taliun D., Thurner M., Robertson N.R., Torres J.M., Rayner N.W., Payne A.J., Steinthorsdottir V., Scott R.A., Grarup N., et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat. Genet. 2018;50:1505–1513. doi: 10.1038/s41588-018-0241-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ramdas S., Judd J., Graham S.E., Kanoni S., Wang Y., Surakka I., Wenz B., Clarke S.L., Chesi A., Wells A., et al. A multi-layer functional genomic analysis to understand noncoding genetic variation in lipids. Am. J. Hum. Genet. 2022;109:1366–1387. doi: 10.1016/j.ajhg.2022.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ulirsch J.C., Lareau C.A., Bao E.L., Ludwig L.S., Guo M.H., Benner C., Satpathy A.T., Kartha V.K., Salem R.M., Hirschhorn J.N., et al. Interrogation of human hematopoiesis at single-cell and single-variant resolution. Nat. Genet. 2019;51:683–693. doi: 10.1038/s41588-019-0362-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lambert S.A., Jolma A., Campitelli L.F., Das P.K., Yin Y., Albu M., Chen X., Taipale J., Hughes T.R., Weirauch M.T. The human transcription factors. Cell. 2018;172:650–665. doi: 10.1016/j.cell.2018.01.029. [DOI] [PubMed] [Google Scholar]
- 17.Zhao B., Barrera L.A., Ersing I., Willox B., Schmidt S.C.S., Greenfeld H., Zhou H., Mollo S.B., Shi T.T., Takasaki K., et al. The NF-κB genomic landscape in lymphoblastoid B cells. Cell Rep. 2014;8:1595–1606. doi: 10.1016/j.celrep.2014.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Waszak S.M., Delaneau O., Gschwind A.R., Kilpinen H., Raghav S.K., Witwicki R.M., Orioli A., Wiederkehr M., Panousis N.I., Yurovsky A., et al. Population variation and genetic control of modular chromatin architecture in humans. Cell. 2015;162:1039–1050. doi: 10.1016/j.cell.2015.08.001. [DOI] [PubMed] [Google Scholar]
- 19.Tehranchi A.K., Myrthil M., Martin T., Hie B.L., Golan D., Fraser H.B. Pooled ChIP-seq links variation in transcription factor binding to complex disease risk. Cell. 2016;165:730–741. doi: 10.1016/j.cell.2016.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ding Z., Ni Y., Timmer S.W., Lee B.K., Battenhouse A., Louzada S., Yang F., Dunham I., Crawford G.E., Lieb J.D., et al. Quantitative genetics of CTCF binding reveal local sequence effects and different modes of X-Chromosome association. PLoS Genet. 2014;10:e1004798. doi: 10.1371/journal.pgen.1004798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu B., Gloudemans M.J., Rao A.S., Ingelsson E., Montgomery S.B. Abundant associations with gene expression complicate GWAS follow-up. Nat. Genet. 2019;51:768–769. doi: 10.1038/s41588-019-0404-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Giambartolomei C., Vukcevic D., Schadt E.E., Franke L., Hingorani A.D., Wallace C., Plagnol V. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 2014;10:e1004383. doi: 10.1371/journal.pgen.1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chun S., Casparino A., Patsopoulos N.A., Croteau-Chonka D.C., Raby B.A., De Jager P.L., Sunyaev S.R., Cotsapas C. Limited statistical evidence for shared genetic effects of eQTLs and autoimmune-disease-associated loci in three major immune-cell types. Nat. Genet. 2017;49:600–605. doi: 10.1038/ng.3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hukku A., Pividori M., Luca F., Pique-Regi R., Im H.K., Wen X. Probabilistic colocalization of genetic variants from complex and molecular traits: promise and limitations. Am. J. Hum. Genet. 2021;108:25–35. doi: 10.1101/2020.07.01.182097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Watt S., Vasquez L., Walter K., Mann A.L., Kundu K., Chen L., Sims Y., Ecker S., Burden F., Farrow S., et al. Genetic perturbation of PU.1 binding and chromatin looping at neutrophil enhancers associates with autoimmune disease. Nat. Commun. 2021;12:2298. doi: 10.1038/s41467-021-22548-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kilpinen H., Waszak S.M., Gschwind A.R., Raghav S.K., Witwicki R.M., Orioli A., Migliavacca E., Wiederkehr M., Gutierrez-Arcelus M., Panousis N.I., et al. Coordinated effects of sequence variation on DNA binding, chromatin structure, and transcription. Science. 2013;342:744–747. doi: 10.1126/science.1242463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deplancke B., Alpern D., Gardeux V. The genetics of transcription factor DNA binding variation. Cell. 2016;166:538–554. doi: 10.1016/j.cell.2016.07.012. [DOI] [PubMed] [Google Scholar]
- 28.Ghandi M., Lee D., Mohammad-Noori M., Beer M.A. Enhanced regulatory sequence prediction using gapped k-mer features. PLoS Comput. Biol. 2014;10:e1003711. doi: 10.1371/journal.pcbi.1003711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee D., Gorkin D.U., Baker M., Strober B.J., Asoni A.L., McCallion A.S., Beer M.A. A method to predict the impact of regulatory variants from DNA sequence. Nat. Genet. 2015;47:955–961. doi: 10.1038/ng.3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barbeira A.N., Bonazzola R., Gamazon E.R., Liang Y., Park Y., Kim-Hellmuth S., Wang G., Jiang Z., Zhou D., Hormozdiari F., et al. Exploiting the GTEx resources to decipher the mechanisms at GWAS loci. Genome Biol. 2021;22:49. doi: 10.1186/s13059-020-02252-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oliva M., Demanelis K., Lu Y., Chernoff M., Jasmine F., Ahsan H., Kibriya M.G., Chen L.S., Pierce B.L. DNA methylation QTL mapping across diverse human tissues provides molecular links between genetic variation and complex traits. Nat. Genet. 2023;55:112–122. doi: 10.1038/s41588-022-01248-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Canela-Xandri O., Rawlik K., Tenesa A. An atlas of genetic associations in UK Biobank. Nat. Genet. 2018;50:1593–1599. doi: 10.1038/s41588-018-0248-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guan W.-J., Ni Z.-Y., Hu Y., Liang W.-H., Ou C.-Q., He J.-X., Liu L., Shan H., Lei C.-L., Hui D.S.C., et al. Clinical characteristics of coronavirus disease 2019 in China. N. Engl. J. Med. 2020;382:1708–1720. doi: 10.1056/NEJMoa2002032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Terpos E., Ntanasis-Stathopoulos I., Elalamy I., Kastritis E., Sergentanis T.N., Politou M., Psaltopoulou T., Gerotziafas G., Dimopoulos M.A. Hematological findings and complications of COVID-19. Am. J. Hematol. 2020;95:834–847. doi: 10.1002/ajh.25829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang S., Sheng Y., Tu J., Zhang L. Association between peripheral lymphocyte count and the mortality risk of COVID-19 inpatients. BMC Pulm. Med. 2021;21:55. doi: 10.1186/s12890-021-01422-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fisher R.C., Scott E.W. Role of PU.1 in hematopoiesis. Stem Cell. 1998;16:25–37. doi: 10.1002/stem.160025. [DOI] [PubMed] [Google Scholar]
- 37.Rothenberg E.V., Hosokawa H., Ungerbäck J. Mechanisms of action of hematopoietic transcription factor PU.1 in initiation of T-cell development. Front. Immunol. 2019;10:228. doi: 10.3389/fimmu.2019.00228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Corces M.R., Buenrostro J.D., Wu B., Greenside P.G., Chan S.M., Koenig J.L., Snyder M.P., Pritchard J.K., Kundaje A., Greenleaf W.J., et al. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat. Genet. 2016;48:1193–1203. doi: 10.1038/ng.3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.1000 Genomes Project Consortium. Auton A., Brooks L.D., Durbin R.M., Garrison E.P., Kang H.M., Korbel J.O., Marchini J.L., McCarthy S., McVean G.A., Abecasis G.R. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Escalante C.R., Brass A.L., Pongubala J.M.R., Shatova E., Shen L., Singh H., Aggarwal A.K. Crystal structure of PU.1/IRF-4/DNA ternary complex. Mol. Cell. 2002;10:1097–1105. doi: 10.1016/S1097-2765(02)00703-7. [DOI] [PubMed] [Google Scholar]
- 41.Yan J., Qiu Y., Ribeiro Dos Santos A.M., Yin Y., Li Y.E., Vinckier N., Nariai N., Benaglio P., Raman A., Li X., et al. Systematic analysis of binding of transcription factors to noncoding variants. Nature. 2021;591:147–151. doi: 10.1038/s41586-021-03211-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kumasaka N., Knights A.J., Gaffney D.J. High-resolution genetic mapping of putative causal interactions between regions of open chromatin. Nat. Genet. 2019;51:128–137. doi: 10.1038/s41588-018-0278-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Delaneau O., Zazhytska M., Borel C., Giannuzzi G., Rey G., Howald C., Kumar S., Ongen H., Popadin K., Marbach D., et al. Chromatin three-dimensional interactions mediate genetic effects on gene expression. Science. 2019;364:eaat8266. doi: 10.1126/science.aat8266. [DOI] [PubMed] [Google Scholar]
- 45.Klemm S.L., Shipony Z., Greenleaf W.J. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 2019;20:207–220. doi: 10.1038/s41576-018-0089-8. [DOI] [PubMed] [Google Scholar]
- 46.Heintzman N.D., Hon G.C., Hawkins R.D., Kheradpour P., Stark A., Harp L.F., Ye Z., Lee L.K., Stuart R.K., Ching C.W., et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459:108–112. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pham T.H., Minderjahn J., Schmidl C., Hoffmeister H., Schmidhofer S., Chen W., Längst G., Benner C., Rehli M. Mechanisms of in vivo binding site selection of the hematopoietic master transcription factor PU.1. Nucleic Acids Res. 2013;41:6391–6402. doi: 10.1093/nar/gkt355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mohammadi P., Castel S.E., Brown A.A., Lappalainen T. Quantifying the regulatory effect size of cis-acting genetic variation using allelic fold change. Genome Res. 2017;27:1872–1884. doi: 10.1101/gr.216747.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liang Y., Aguet F., Barbeira A.N., Ardlie K., Im H.K. A scalable unified framework of total and allele-specific counts for cis-QTL, fine-mapping, and prediction. Nat. Commun. 2021;12:1424. doi: 10.1038/s41467-021-21592-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hormozdiari F., van de Bunt M., Segrè A.V., Li X., Joo J.W.J., Bilow M., Sul J.H., Sankararaman S., Pasaniuc B., Eskin E. Colocalization of GWAS and eQTL signals detects target genes. Am. J. Hum. Genet. 2016;99:1245–1260. doi: 10.1016/j.ajhg.2016.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.GTEx Consortium The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 2020;369:1318–1330. doi: 10.1126/science.aaz1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Krysiak K., Gomez F., White B.S., Matlock M., Miller C.A., Trani L., Fronick C.C., Fulton R.S., Kreisel F., Cashen A.F., et al. Recurrent somatic mutations affecting B-cell receptor signaling pathway genes in follicular lymphoma. Blood. 2017;129:473–483. doi: 10.1182/blood-2016-07-729954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Javierre B.M., Burren O.S., Wilder S.P., Kreuzhuber R., Várnai C., Sewitz S., Cairns J., Wingett S.W., Várnai C., Thiecke M.J., et al. Lineage-specific genome architecture links enhancers and non-coding disease variants to target gene promoters. Cell. 2016;167:1369–1384.e19. doi: 10.1016/j.cell.2016.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang G., Sarkar A., Carbonetto P., Stephens M. A simple new approach to variable selection in regression, with application to genetic fine mapping. J. R. Stat. Soc. Ser. B Stat. Methodol. 2020;82:1273–1300. doi: 10.1111/rssb.12388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schmiedel B.J., Singh D., Madrigal A., Valdovino-Gonzalez A.G., White B.M., Zapardiel-Gonzalo J., Ha B., Altay G., Greenbaum J.A., McVicker G., et al. Impact of genetic polymorphisms on human immune cell gene expression. Cell. 2018;175:1701–1715.e16. doi: 10.1016/j.cell.2018.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Natoli G., Ghisletti S., Barozzi I. The genomic landscapes of inflammation. Genes Dev. 2011;25:101–106. doi: 10.1101/gad.2018811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Minderjahn J., Schmidt A., Fuchs A., Schill R., Raithel J., Babina M., Schmidl C., Gebhard C., Schmidhofer S., Mendes K., et al. Mechanisms governing the pioneering and redistribution capabilities of the non-classical pioneer PU.1. Nat. Commun. 2020;11:402. doi: 10.1038/s41467-019-13960-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lappalainen T., Sammeth M., Friedländer M.R., ’T Hoen P.A.C., Monlong J., Rivas M.A., Gonzàlez-Porta M., Kurbatova N., Griebel T., Ferreira P.G., et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature. 2013;501:506–511. doi: 10.1038/nature12531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kerimov N., Hayhurst J.D., Peikova K., Manning J.R., Walter P., Kolberg L., Samoviča M., Sakthivel M.P., Kuzmin I., Trevanion S.J., et al. A compendium of uniformly processed human gene expression and splicing quantitative trait loci. Nat. Genet. 2021;53:1290–1299. doi: 10.1038/s41588-021-00924-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Abell N.S., DeGorter M.K., Gloudemans M.J., Greenwald E., Smith K.S., He Z., Montgomery S.B. Multiple causal variants underlie genetic associations in humans. Science. 2022;375:1247–1254. doi: 10.1126/science.abj5117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tewhey R., Kotliar D., Park D.S., Liu B., Winnicki S., Reilly S.K., Andersen K.G., Mikkelsen T.S., Lander E.S., Schaffner S.F., Sabeti P.C. Direct identification of hundreds of expression-modulating variants using a multiplexed reporter assay. Cell. 2016;165:1519–1529. doi: 10.1016/j.cell.2016.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu W., Li T., Wang P., Liu W., Liu F., Mo X., Liu Z., Song Q., Lv P., Ruan G., Han W. LRRC25 plays a key role in all-trans retinoic acid-induced granulocytic differentiation as a novel potential leukocyte differentiation antigen. Protein Cell. 2018;9:785–798. doi: 10.1007/s13238-017-0421-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Le Coz C., Nguyen D.N., Su C., Nolan B.E., Albrecht A.V., Xhani S., Sun D., Demaree B., Pillarisetti P., Khanna C., et al. Constrained chromatin accessibility in PU.1-mutated agammaglobulinemia patients. J. Exp. Med. 2021;218:e20201750. doi: 10.1084/jem.20201750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wu J.N., Pinello L., Yissachar E., Wischhusen J.W., Yuan G.-C., Roberts C.W.M. Functionally distinct patterns of nucleosome remodeling at enhancers in glucocorticoid-treated acute lymphoblastic leukemia. Epigenet. Chromatin. 2015;8:53. doi: 10.1186/s13072-015-0046-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen L., Ge B., Casale F.P., Vasquez L., Kwan T., Garrido-Martín D., Watt S., Yan Y., Kundu K., Ecker S., et al. Genetic drivers of epigenetic and transcriptional variation in human immune cells. Cell. 2016;167:1398–1414.e24. doi: 10.1016/j.cell.2016.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kanai M., Ulirsch J.C., Karjalainen J., Kurki M., Karczewski K.J., Fauman E., Wang Q.S., Jacobs H., Aguet F., Ardlie K.G., et al. Insights from complex trait fine-mapping across diverse populations. medRxiv. 2021 doi: 10.1101/2021.09.03.21262975. Preprint at. [DOI] [Google Scholar]
- 67.International Multiple Sclerosis Genetics Consortium IMSGC. Beecham A.H., Patsopoulos N.A., Xifara D.K., Davis M.F., Kemppinen A., Cotsapas C., Shah T.S., Spencer C., Booth D., et al. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat. Genet. 2013;45:1353–1360. doi: 10.1038/ng.2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Umans B.D., Battle A., Gilad Y. Where are the disease-associated eQTLs? Trends Genet. 2021;37:109–124. doi: 10.1016/j.tig.2020.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wallace C. A more accurate method for colocalisation analysis allowing for multiple causal variants. PLoS Genet. 2021;17:e1009440. doi: 10.1371/journal.pgen.1009440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nicolae D.L., Gamazon E., Zhang W., Duan S., Dolan M.E., Cox N.J. Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet. 2010;6:e1000888. doi: 10.1371/journal.pgen.1000888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Connally N.J., Nazeen S., Lee D., Shi H., Stamatoyannopoulos J., Chun S., Cotsapas C., Cassa C.A., Sunyaev S.R. The missing link between genetic association and regulatory function. Elife. 2022;11:e74970. doi: 10.7554/eLife.74970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Baker M. Reproducibility crisis: blame it on the antibodies. Nature. 2015;521:274–276. doi: 10.1038/521274a. [DOI] [PubMed] [Google Scholar]
- 73.ENCODE Project Consortium. Kundaje A., Aldred S.F., Collins P.J., Davis C.A., Doyle F., Epstein C.B., Frietze S., Harrow J., Kaul R., et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Byrska-Bishop M., Evani U.S., Zhao X., Basile A.O., Abel H.J., Regier A.A., Corvelo A., Clarke W.E., Musunuri R., Nagulapalli K., et al. High-coverage whole-genome sequencing of the expanded 1000 Genomes Project cohort including 602 trios. Cell. 2022;185:3426–3440.e19. doi: 10.1016/j.cell.2022.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wood A.R., Esko T., Yang J., Vedantam S., Pers T.H., Gustafsson S., Chu A.Y., Estrada K., Luan J., Kutalik Z., et al. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat. Genet. 2014;46:1173–1186. doi: 10.1038/ng.3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Van De Geijn B., Mcvicker G., Gilad Y., Pritchard J.K. WASP: allele-specific software for robust molecular quantitative trait locus discovery. Nat. Methods. 2015;12:1061–1063. doi: 10.1038/nmeth.3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wu T.D., Nacu S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics. 2010;26:873–881. doi: 10.1093/bioinformatics/btq057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., Liu X.S. Model-based analysis of ChIP-seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liao Y., Smyth G.K., Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 81.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ambrosini G., Groux R., Bucher P. PWMScan: a fast tool for scanning entire genomes with a position-specific weight matrix. Bioinformatics. 2018;34:2483–2484. doi: 10.1093/bioinformatics/bty127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lee D. LS-GKM: a new gkm-SVM for large-scale datasets. Bioinformatics. 2016;32:2196–2198. doi: 10.1093/bioinformatics/btw142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Das S., Forer L., Schönherr S., Sidore C., Locke A.E., Kwong A., Vrieze S.I., Chew E.Y., Levy S., McGue M., et al. Next-generation genotype imputation service and methods. Nat. Genet. 2016;48:1284–1287. doi: 10.1038/ng.3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Danecek P., Bonfield J.K., Liddle J., Marshall J., Ohan V., Pollard M.O., Whitwham A., Keane T., McCarthy S.A., Davies R.M., et al. Twelve years of SAMtools and BCFtools. GigaScience. 2021;10:giab008. doi: 10.1093/gigascience/giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chang C.C., Chow C.C., Tellier L.C., Vattikuti S., Purcell S.M., Lee J.J. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience. 2015;4:7. doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Stegle O., Parts L., Piipari M., Winn J., Durbin R. Using probabilistic estimation of expression residuals (PEER) to obtain increased power and interpretability of gene expression analyses. Nat. Protoc. 2012;7:500–507. doi: 10.1038/nprot.2011.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Delaneau O., Ongen H., Brown A.A., Fort A., Panousis N.I., Dermitzakis E.T. A complete tool set for molecular QTL discovery and analysis. Nat. Commun. 2017;8:15452. doi: 10.1038/ncomms15452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pers T.H., Timshel P., Hirschhorn J.N. SNPsnap: a Web-based tool for identification and annotation of matched SNPs. Bioinformatics. 2015;31:418–420. doi: 10.1093/bioinformatics/btu655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chun S., Akle S., Teodosiadis A., Cade B.E., Wang H., Sofer T., Evans D.S., Stone K.L., Gharib S.A., Mukherjee S., et al. Leveraging pleiotropy to discover and interpret GWAS results for sleep-associated traits. PLoS Genet. 2022;18:e1010557. doi: 10.1371/journal.pgen.1010557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kanai M., Akiyama M., Takahashi A., Matoba N., Momozawa Y., Ikeda M., Iwata N., Ikegawa S., Hirata M., Matsuda K., et al. Genetic analysis of quantitative traits in the Japanese population links cell types to complex human diseases. Nat. Genet. 2018;50:390–400. doi: 10.1038/s41588-018-0047-6. [DOI] [PubMed] [Google Scholar]
- 92.Lex A., Gehlenborg N., Strobelt H., Vuillemot R., Pfister H. UpSet: visualization of intersecting sets. IEEE Trans. Vis. Comput. Graph. 2014;20:1983–1992. doi: 10.1109/TVCG.2014.2346248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kumasaka N., Knights A.J., Gaffney D.J. Fine-mapping cellular QTLs with RASQUAL and ATAC-seq. Nat. Genet. 2016;48:206–213. doi: 10.1038/ng.3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Coetzee S.G., Coetzee G.A., Hazelett D.J. motifbreakR: an R/Bioconductor package for predicting variant effects at transcription factor binding sites. Bioinformatics. 2015;31:3847–3849. doi: 10.1093/bioinformatics/btv470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Robinson J.T., Thorvaldsdóttir H., Winckler W., Guttman M., Lander E.S., Getz G., Mesirov J.P. Integrative genomics viewer. Nat. Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Robinson M.D., Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11:R25. doi: 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.McCarthy S., Das S., Kretzschmar W., Delaneau O., Wood A.R., Teumer A., Kang H.M., Fuchsberger C., Danecek P., Sharp K., et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet. 2016;48:1279–1283. doi: 10.1038/ng.3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
GWAS statistics are based on Canela-Xandri et al. [S8].
GWAS statistics and fine-mapping results are from Vuckovic et al. [S7].
Only single nucleotide polymorphisms were included in the analysis. Each locus is defined by blood cell trait and a colocalized PU.1 bQTL.
Rank is based on conditional analysis.
Data Availability Statement
Code and processed data for performing colocalization analysis in this study are available at https://doi.org/10.5281/zenodo.7837982. Code and processed data for generating the figures are available at https://doi.org/10.5281/zenodo.7837894. All other data used in the analysis are publicly available and listed in the key resources table.