

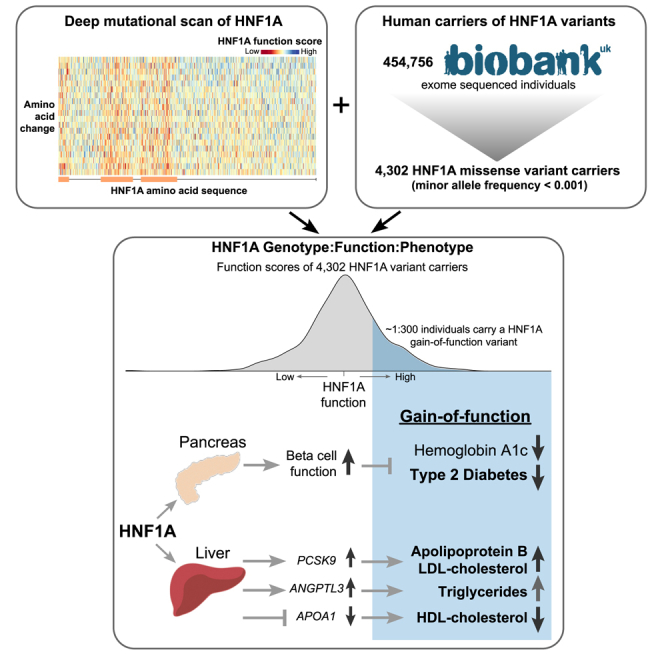
Summary
Loss-of-function mutations in hepatocyte nuclear factor 1A (HNF1A) are known to cause rare forms of diabetes and alter hepatic physiology through unclear mechanisms. In the general population, 1:100 individuals carry a rare, protein-coding HNF1A variant, most of unknown functional consequence. To characterize the full allelic series, we performed deep mutational scanning of 11,970 protein-coding HNF1A variants in human hepatocytes and clinical correlation with 553,246 exome-sequenced individuals. Surprisingly, we found that ∼1:5 rare protein-coding HNF1A variants in the general population cause molecular gain of function (GOF), increasing the transcriptional activity of HNF1A by up to 50% and conferring protection from type 2 diabetes (odds ratio [OR] = 0.77, p = 0.007). Increased hepatic expression of HNF1A promoted a pro-atherogenic serum profile mediated in part by enhanced transcription of risk genes including ANGPTL3 and PCSK9. In summary, ∼1:300 individuals carry a GOF variant in HNF1A that protects carriers from diabetes but enhances hepatic secretion of atherogenic lipoproteins.
Keywords: HNF1A, PCSK9, ANGPTL3, deep mutational scan, saturation mutagenesis, UK Biobank, gain of function, diabetes, inflammation, atherosclerosis, coronary artery disease
Graphical abstract
Highlights
-
•
Deep mutational scan of HNF1A characterized the function of 11,970 HNF1A variants
-
•
Gain-of-function HNF1A variants identified that increase transcriptional activity
-
•
1 in 300 rare HNF1A missense variants in humans confer gain of function
-
•
Human carriers of HNF1A gain-of-function variants are protected from T2D
DeForest et al. analyzed protein-coding variants in the gene HNF1A, which is associated with diabetes and hepatic physiology. They found that 1 in 5 rare protein-coding HNF1A variants in the general population actually increase HNF1A activity, leading to a lower risk of type 2 diabetes but elevated atherogenic lipoproteins.
Introduction
Hepatocyte nuclear factor 1 alpha (HNF1A) is a lineage-determining transcription factor expressed in several tissues including the liver and pancreas.1 Rare, complete loss-of-function (LOF) mutations in HNF1A have been shown to cause an autosomal dominant monogenic form of diabetes (MODY3)2,3 through deficiencies in pancreatic insulin secretion via a haploinsufficient genetic mechanism.4 Patients with MODY3 have been observed to carry altered levels of serum lipoproteins5 and lower levels of the inflammatory marker high-sensitivity C-reactive protein (hsCRP)6 than matched patients with type 2 diabetes (T2D), suggesting a potential liver-mediated functional role for HNF1A in metabolic disease pathogenesis. However, diabetes itself, through underlying insulin resistance or elevated blood glucose, can dysregulate serum lipids7 and systemic inflammation,8 and thus these observations in diabetic individuals with MODY3 cannot evidentiate HNF1A as a direct hepatic regulator of serum lipids or inflammation. Murine LOF studies have implicated HNF1A in bile acid secretion and cholesterol metabolism but are also confounded by a diabetic phenotype.9 Population-based genetic association studies have corroborated associations between HNF1A and T2D,10 serum lipoproteins,11 and hsCRP12 but do not provide insight into mechanism, as most of the associated variants (SNPs) are either non-coding or have minimal experimentally observable consequences on protein function.13 Finally, common HNF1A SNPs have been associated with coronary artery disease (CAD) risk,14,15 but these SNPs are also associated with T2D and serum lipids, both of which are known, independent CAD mediators.16,17 Thus, the relationship between HNF1A function, these risk factors, and the specific mechanisms of disease pathogenesis remains unclear.
Characterization of an allelic series of functional variants for the hepatic functions of HNF1A with clinical correlation would enable a mechanistic understanding of the complex role for HNF1A in driving metabolic disease in multiple tissues including the pancreas (via diabetes) and liver (serum lipids) with their respective directions of effect. In the past few years, dozens of novel protein-coding alleles in HNF1A have been identified from population-based exome sequencing.18 These variants are individually rare (minor allele frequency [MAF] < 0.001) and are mostly of unknown functional consequence but, in aggregate, are carried by over 1% of the population. The much greater allelic diversity of rare variants presents an opportunity to dissect the role of HNF1A in metabolic disease, if these variants could be functionally annotated at scale.
To address this gap in knowledge, we examined the full allelic series of protein-coding variants in HNF1A using deep mutational scanning, a high-throughput approach that has been successfully utilized to characterize protein-coding variants in clinically important genes.19,20,21 All possible single amino acid substitutions in HNF1A were tested for transcriptional activity in human hepatocytes. These comprehensive data were intersected with carriers of rare protein-coding variants (MAF < 0.001) identified from among 553,246 sequenced individuals in order to relate variant to function to phenotype for metabolic disease-associated factors and disease outcomes including T2D and CAD. We discover gain-of-function (GOF) variants that, while conferring protection from T2D, promote pro-inflammatory/thrombogenic gene expression and an atherogenic lipid profile through liver specific enhancement of HNF1A target genes.
Results
Identification of GOF substitutions from comprehensive functional testing of 11,970 HNF1A amino acid variants in human hepatocytes
To comprehensively assess the hepatic function of protein-coding variants in HNF1A, we synthesized a cDNA library comprised of all possible single amino acid permutations of HNF1A (630 amino acid protein × 19 amino acid changes = 11,970; Table S1) such that each transgene encoded a single amino acid substitution. The library was introduced into a human hepatocyte cell line previously engineered to lack endogenous HNF1A and at a dilution maximizing the number of cells receiving only a single copy of HNF1A transgene. The HNF1A transgenes were induced for 6 days via a doxycycline-inducible promoter, and cells were sorted via fluorescence-activated cell sorting (FACS) based on their protein expression level of TM4SF4, a cell surface protein shown to be a direct transcriptional target of HNF1A in pancreas and liver22,23 (Figure 1A). The population of cells bearing HNF1A transgenes was partitioned into two bins separated by a 10-fold expression intensity difference in TM4SF4: TM4SF4low and TM4SF4high (Figures 1A and S1). To recover and quantify the HNF1A variants in each TM4SF4 expression bin, HNF1A transgenes from the TM4SF4low and TM4SF4high cells were sequenced by targeted next-generation sequencing. Raw function scores were generated for each amino acid substitution at each site in HNF1A by determining the frequency of appearance of each variant in the TM4SF4low and TM4SF4high bins as we and others have previously done.20,24 Over 99% of the originally designed variants were recovered in sufficient quantity from the pooled screen to be assigned a function score (Figure S2; Table S2).
Figure 1.

Identification of GOF variants from comprehensive functional testing of 11,970 HNF1A variants in human hepatocytes
(A) A library of 11,970 HNF1A constructs was synthesized, with each construct encoding a single amino acid substitution. The construct library was introduced into HUH7 hepatocytes (deleted for endogenous HNF1A) at a dilution of one construct per cell. The resulting polyclonal population of HUH7 hepatocytes was separated via FACS according to the expression of the known HNF1A transcriptional target TM4SF4 and sorted into low (−) and high (+) bins of HNF1A activity. Activity cutoffs were established through flow cytometry experiments of HNF1A KO cells (dashed red line) and WT cells (dashed green line). Each bin of cells was sequenced at the transgenic HNF1A locus to identify and tabulate the introduced variants. Each HNF1A variant was assigned a function score based on its abundance in the low and high TM4SF4 expression bins.
(B) Heatmap of 11,970 HNF1A variant function scores, arranged according to the primary amino acid sequence (rows). Function scores lower than WT are shaded red, and function scores greater than WT are shaded blue. Function scores averaged (mean) at each amino acid position are plotted to the right, showing the level of tolerance for any amino acid substitution away from WT at each position.
(C) Mutation tolerance scores as described in (B) overlaid on the crystalized protein structure of HNF1A DNA-binding domain (PF04814). Positions intolerant of amino acid changes (i.e., lower function scores) are shaded red. Helices that make direct contacts with the DNA are the most intolerant of mutations.
(D) (Left panel) HNF1A function scores ranked for all 11,970 amino acid variants tested, and ClinVar-annotated pathogenic variants (n = 29) are highlighted in red. (Right panel) Function bins correspond to variants with function scores above (GOF), within (neutral), or below (LOF) ± 1 Z-score of the synonymous distribution, shown with the total number of variants per bin. Overlaid are the function score distributions of the 613 synonymous HNF1A variants tested (purple) and the 29 ClinVar pathogenic variants (red).
The raw function scores for all protein-coding HNF1A variants were compared with the known sequence:function relationships of HNF1A. First, the function scores were averaged (mean) at each of the 631 amino acid positions along HNF1A to obtain a “mutation tolerance” score representing the effect on function of substituting any non-wild-type (WT) amino acid at that position, i.e., low “mutation tolerance” represents a functionally important residue (Figure 1B). Several clusters of amino acids along the primary sequence exhibiting low mutation tolerance scores were observed and colocalized with known domains critical for HNF1A molecular function including the dimerization domain and the DNA-binding domain.25,26 The mutation tolerance scores were also overlaid onto the HNF1A DNA-binding domain crystal structure to evaluate the relationship between function and higher-order structure (Figure 1C). Within the extent of the available crystal structure, the amino acid residues comprising the alpha helices in the DNA-binding domain that directly contact the DNA double helix exhibit the lowest mutation tolerance scores, consistent with a high degree of functional conservation (Figure 1C).
Taking a conservative approach to minimize the effect of extreme values, the raw individual variant scores were added to the mutation tolerance score at that amino acid position (Figure 1B) and rescaled to the mean and standard deviation (SD) of the distribution of synonymous variants to provide an intuitive interpretation to the function scores (Figure 1D). Of the 11,970 variants tested, 2,190 fell into the putative LOF category (score < −1 SD), 6,952 fell into the neutral category (−1 SD ≤ score ≤ 1 SD), and, surprisingly, 2,740 fell into a putative GOF (score > 1 SD) category, indicating that those amino acid substitutions increased the transcriptional activity of HNF1A over WT in our assay, a finding not previously reported in over two decades of HNF1A functional variant characterization.2 As expected, variants categorized as putative LOF by our experiments occurred at codons that were more evolutionarily conserved in mammalian genomic sequences27 than those classified as neutral (p < 10−6 Wilcoxon rank-sum test; Figure S3). Variants categorized as putative GOF, however, could not be distinguished from neutral variants on the basis of evolutionary conservation, suggesting that they would be poorly annotated by computational prediction algorithms leveraging conservation metrics.28,29 To further benchmark our experiments, we extracted the function scores of pathogenic human HNF1A variants from ClinVar,30 a curated database of clinically significant human genetic variants (Figure 1D; Table S3), finding that the majority, but not all (n = 21/29), would be classified as LOF by our data. Thus, to explicitly quantify on a per-variant basis the confidence of LOF/GOF, a posterior error probability31 (PEP) was computed for every variant based on where its function score fell in the distribution of function scores for synonymous vs. non-synonymous variants (Figure 1D; Table S2).
GOF HNF1A variants are carried in the general population and increase transcriptional activity in multiple cellular contexts
To evaluate the human relevance of our deep mutational scan and assess if these putative GOF variants are present in people, we intersected our comprehensive dataset of HNF1A function scores with HNF1A protein-coding variants in the UK Biobank, a population-based cohort containing 454,756 exome-sequenced individuals.32,33 From the exome sequences, we extracted all HNF1A variants and identified 4,302 individuals who carried a protein-coding variant with a MAF less than 0.001 (∼1:100 individuals; Table S4). Of the 444 unique protein-coding variants identified, 335 (75%) were absent from ClinVar.30 The distribution of HNF1A function scores in the UK Biobank peaked at the same level as the distribution of synonymous variants (Figure 2A), consistent with prior observations that most protein-coding variants are not functional.34 The distribution of function scores in the UK Biobank also notably lacked the lowest-scoring pathogenic/MODY3 variants, as would be expected for a generally healthy population-based cohort32 and with the estimated UK prevalence of MODY3 being ∼5 per 100,000.35 We did observe a 22% prevalence of diagnosed diabetes among carriers of HNF1A frameshifting mutations (n = 62 carriers), which exceeded the 8% prevalence of diabetes in the general UK Biobank cohort. The presence of LOF HNF1A variants in population-based cohorts has been described previously,36 but remarkably, carriers of GOF variants have not previously been described in the many studies functionally characterizing human genetic variants in HNF1A.
Figure 2.

GOF HNF1A variants are carried in the general population and increase transcriptional activity in multiple cellular contexts
(A) (Left panel) Selected human HNF1A variants with function scores greater than WT highlighted in blue among the full distribution of all 11,970 amino acid variants tested. The shaded purple box represents ± 1 Z-score of the distribution of 613 synonymous HNF1A variants tested. (Right panel) The distributions of function scores of rare protein-coding HNF1A variants (n = 444) identified from 454,756 sequenced individuals in the UK Biobank (UKB) and of the 613 synonymous variants. An upper tail of function-increasing variants in UKB individuals is highlighted in blue (n = 94 unique variants in 1,469 individuals).
(B) Location of top-scoring variants selected for validation along the HNF1A protein primary amino acid sequence with number of human variant carriers identified in 454,756 exome sequences from the UKB.
(C) Putative GOF variants were individually recreated and tested for transcriptional activation using luciferase reporters in HeLa cells (which lack endogenous HNF1A activity) on the rat albumin promoter. Activity measurements are shown as percentages of WT HNF1A activity ± SEM; n = 3. Basal promoter activity in cells is measured by transfection of vectors lacking the HNF1A transgene (empty) (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, linear mixed model).
(D) The same variants described in (B) were tested for transcriptional activity in mouse insulinoma MIN6 cells on the HNF4A-P2 promoter.
(E) GOF HNF1A variant constructs were transfected into HNF1A-deleted HUH7 hepatocytes, and ANGPTL3 levels were measured by ELISA 48 h post-confluency (n = 3 per variant). Transfection with HNF1A p.L348F, p.T196A, and p.T515M increases ANGPTL3 secretion compared with WT (∗∗∗p < 0.0005,∗∗p < 0.005, ∗p < 0.05, linear mixed model).
To confirm and further characterize these putative GOF HNF1A variants carried by humans in the general population, we selected a series of high-scoring GOF variants that spanned across the protein domains of HNF1A (Figure 2A). These seven variants ranged in frequency from 3 carriers (p.L348F) to 421 carriers (p.T196A) in the 454,756 sampled exomes (Figure 2B). Each variant was recreated, transfected, and assessed for transcriptional activity using luciferase reporter assays as we37 and others38 have previously performed in two separate experimental contexts: (1) HeLa cells using the rat albumin promoter (Figure 2C) and (2) MIN6 insulinoma cells using the HNF4A-P2 promoter (Figure 2D). In HeLa cells, all selected variants showed significant increases in transcriptional activity over WT (ranging from 30% to 70%), whereas in MIN6 cells, the measured increase in transactivation was attenuated with only three of the tested variants exceeding the nominal threshold of significance. Notably, the background activity of HNF1A in HeLa cells was nil, whereas in MIN6 cells, it approached 50% of WT. Given that GOF was observed in two independent HNF1A-free cellular contexts (i.e., HeLa and HUH7 HNF1A-null cells), the attenuation in signal in the MIN6 cell system likely reflected decreased sensitivity in measuring increased HNF1A transcription in the setting of pre-existing high levels of endogenous HNF1A activity.
To assess the functional consequence of GOF HNF1A in hepatocytes, we developed a secretion assay for ANGPTL3, an emerging cardiovascular disease risk factor39 that encodes a hepatically secreted regulator of serum lipids40 whose gene is bound by HNF1A41 (Figure S4). After confirming that HNF1A functionally regulates production of ANGPTL3 from cultured hepatocytes (Figure S4B), we evaluated ANGPTL3 secretion in HNF1A-null hepatocytes and performed reconstitution experiments with WT and the identified GOF HNF1A variants. The p.L348F, p.T196A, and p.T515M variants, but not the p.P291Q variant, increased ANGPTL3 secretion when transfected into HNF1A-null HUH7 hepatocytes compared with WT (Figure 2E). Taken together, these cell culture experiments demonstrate that GOF HNF1A variants increase transcriptional activity in both pancreatic and hepatic cellular contexts.
HNF1A GOF variant carriers in the population are protected from T2D but show elevated levels of serum atherogenic risk factors
We examined the relationship between HNF1A function and T2D in carriers of rare protein-coding HNF1A variants ascertained from the UK Biobank and found an inverse association between HNF1A function and T2D risk in the full multi-ancestry cohort (odds ratio [OR] = 0.59 per SD increase in HNF1A function, 95% confidence interval [CI] = [0.48–0.72], p = 6 × 10−7, n = 4,341, logistic mixed-effects regression) (Figure 3A; Table S5). A similar association was obtained when restricting to European ancestry HNF1A rare variant carriers (OR = 0.56 per SD increase in HNF1A function, 95% CI = [0.43–0.73], p = 1.2 × 10−5, n = 3,095, logistic mixed-effects regression). These analyses included the 421 carriers of the p.T196A variant (Figure 2B). Given the previously known associations between LOF variants in HNF1A and diabetes,2,36,42 we performed a categorical analysis binning the HNF1A variants by function (GOF, neutral, LOF) and separately examined their association with T2D risk in the UK Biobank and in an independent, multi-ancestry T2D case:control cohort (AMP-T2D43; 20,791 cases, 24,440 controls) to disentangle the effect of GOF variants from the known relationship of LOF variants and increased risk of T2D (Figure 3B). In the meta-analysis, LOF variant carriers compared with neutral variant carriers had an increased risk of T2D as expected from prior studies (OR = 1.9, 95% CI = [1.41–2.63], p = 4.1 × 10−5, logistic regression). Remarkably, GOF variant carriers were protected from T2D (UK Biobank GOF carriers: n = 1,469, OR = 0.75, 95% CI = [0.59–0.95], p = 0.017, logistic regression), a signal that was strengthened when analyzed in combination with the AMP-T2D cohort (OR = 0.76, 95% CI = [0.62–0.93], p = 0.007, logistic regression; Figure 3B), evidentiating that genetically determined increases in HNF1A function have clinical consequences in humans. The association signal for GOF variants conferring protection from T2D was consistent over progressively increasing function score thresholds, although at the most stringent thresholds, we observed a loss of statistical power due to a steep decrease in the number of GOF variant carriers (Figure S5; Table S6). Furthermore, correction for independent common protein-coding HNF1A variants I27L (rs1169288) and A98V (rs1800574), which have been associated with increased risk of T2D,44 had minimal impact on these associations (Table S6).
Figure 3.

HNF1A GOF variant carriers in the population are protected from T2D but show elevated levels of serum atherogenic risk factors
Shape and fill indicate the ancestry and T2D status, respectively, of individuals included in each analysis. All phenotypes are adjusted for age, age2, sex, and the first 10 principal components of ancestry. T2D, type 2 diabetes; SD, standard deviation; LOF, loss of function; GOF, gain of function; CRP, C-reactive protein; LDL, low-density lipoprotein cholesterol; ApoB, apolipoprotein B; GGT, gamma-glutamyl transferase; TC, total cholesterol; TG, triglycerides; ApoA-I, apolipoprotein A-I; HDL, high-density lipoprotein cholesterol; HbA1c, glycated hemoglobin.
(A) Association of HNF1A function score and T2D risk using logistic regression in the UKB HNF1A rare protein-coding variant carriers identified among the 454,756 exome-sequenced individuals. Odds ratios and the 95% confidence interval are shown.
(B) Association of LOF and GOF HNF1A variants with T2D in the UKB and AMP-T2D (n = 20,791 cases, 24,440 controls) using logistic regression. Odds ratios and the 95% confidence interval are shown, and fixed effects inverse-variance meta-analysis was used to combine the results.
(C) Association of functional HNF1A variants with cardiovascular disease risk factors in the UKB rare variant carriers (European n = 3,089, multi-ancestry n = 4,302, linear mixed-effects regression). Effect size and the 95% confidence interval are shown for each phenotype.
(D) Association of liver-specific predicted HNF1A gene expression with disease risk factors tested in (C) among all UKB European genotyped individuals (n = 407,227, filled squares) and non-T2D European genotyped individuals (n = 376,043, open squares).
Given that individuals who carry pathogenic/MODY3 variants demonstrate alterations in hepatically secreted factors including hsCRP and lipoprotein profiles, we quantitatively examined the available biomarkers including hsCRP,17 lipoprotein levels,45 liver enzymes,46 and glycemic traits47 in HNF1A rare variant carriers in the UK Biobank by regressing the normalized values for each measurement against the HNF1A function scores of the variant carriers (Figure 3C). A positive relationship was identified between HNF1A function and serum hsCRP, gamma glutamyltransferase (GGT), serum low-density lipoprotein (LDL) cholesterol, and apolipoprotein B (ApoB), while an inverse relationship was identified between HNF1A function and serum high-density lipoprotein (HDL) cholesterol and ApoA-I. After excluding carriers with diagnosed diabetes, we examined the relationship between HNF1A function and glycemia, finding an inverse relationship between HNF1A function and glycosylated hemoglobin (HbA1c; Figure 3C). These findings were robust to correction for multiple hypothesis testing (Table S5) and suggest that genetic variants that increase HNF1A protein function increase serum hsCRP and GGT and promote an atherogenic lipoprotein profile by raising serum ApoB/LDL cholesterol levels and lowering ApoA-I/HDL cholesterol. However, in concordance with conferring protection from T2D, HNF1A function-increasing variants also decrease blood glucose in non-diabetic individuals.
In light of these observed pleiotropic and opposing effects of GOF in HNF1A on T2D and inflammatory/atherogenic factors, we examined the relationship between GOF HNF1A variants and CAD events in the UK Biobank and a cohort of individuals exome sequenced for early-onset myocardial infarction (MIGen48; 24,337 cases, 28,922 controls). We observed no decrease in CAD risk in GOF HNF1A variant carriers (OR = 1.15, 95% CI = [0.93–1.43], p = 0.21, logistic regression), suggesting that either the observed increase in hepatic pro-atherogenic factors is counterbalanced by the protection from T2D or that the analysis lacked statistical power due to the limited number of CAD cases available. Notably, our combined CAD cohort analysis had over 90% power to detect a decrease in risk of similar magnitude (∼25%) as observed with T2D and GOF variant carriers at the nominal type 1 error rate (α = 0.05), but a smaller effect size may not have risen to the level of statistical significance (Table S5).
To evaluate if these findings were the result of perturbed HNF1A activity in the liver, we turned to liver-specific HNF1A gene expression, reasoning that increasing the levels of intra-cellular HNF1A would produce a similar clinical effect as increasing intrinsic HNF1A protein function. To quantify the heritable component of HNF1A gene expression, we selected common, non-coding SNPs identified in European liver samples from the Genotype-Tissue Expression (GTEx) Project49,50 as expression quantitative trait loci (eQTLs) for HNF1A and combined them to produce a liver HNF1A gene expression score. After removing the carriers of rare protein-coding HNF1A variants shown in Figures 3A–3C, we assigned liver-specific HNF1A expression scores to the remaining 407,227 European individuals using established methods (Functional Summary-based Imputation [FUSION]).51 These liver-specific HNF1A expression scores were then regressed against the same clinical phenotypes as the rare, protein-coding variant analysis (Figure 3C). This method confirmed that increased HNF1A gene expression encoded by liver eQTLs conferred increased levels of hsCRP (p < 10−46, linear model; Figure 3D) as would be expected given the known biology of CRP being synthesized and secreted by hepatocytes.52 Similarly, serum GGT and LDL cholesterol were associated with increased liver-specific HNF1A expression scores, whereas HDL and ApoA-I showed a nominal inverse association. Notably, glycemia measured by HbA1c (p = 0.09, linear model) did not associate with liver-specific HNF1A expression as would be expected from the known pancreatic role of HNF1A in regulating blood glucose.53
To evaluate if these associations were being driven by or distinct from T2D, we repeated the above analyses excluding all 41,923 individuals (including 539 carriers of rare-protein coding variants) with diabetes from the UK Biobank population and recomputed the regression (Figures 3C and 3D; Table S5) of clinical measurements against HNF1A function/expression. With regard to liver-specific HNF1A gene expression (n = 376,043 samples without T2D), the analyses were well powered and showed similar effect size, directionality, and significance of all quantitative trait associations, suggesting a causal relationship with HNF1A that is independent of T2D (Figure 3D). Effect sizes in the rare, protein-coding variant analysis also remained directionally consistent, but several associations no longer passed the nominal threshold of statistical significance, likely due to a loss of power (n = 3,802 rare-protein coding variant carriers without T2D; Table S5). In summary, our data indicate that increased hepatic function/expression of HNF1A increases serum hsCRP, GGT, and LDL while decreasing HDL/ApoA-I independently of its effects on T2D and that increased HNF1A function decreases blood glucose but not through the liver.
HNF1A transcriptionally regulates complement activation, coagulation, and pro-atherogenic gene expression programs in hepatocytes
To understand the molecular mechanisms by which functional genetic variants in HNF1A regulate serum lipids, we deleted HNF1A in human hepatocytes using CRISPR-Cas9 and transcriptionally profiled the resulting cells using mRNA sequencing (Figure 4A; Table S7). Experiments were carried out in both HUH7 and Hep3B cell lines to mitigate confounding cell line artifacts. Many of the top differentially expressed genes are known transcriptional targets of HNF1A including FABP1,41 FGB,54 and TM4SF4.23 To examine which molecular pathways were perturbed by the loss of HNF1A, we performed gene set enrichment analysis (GSEA)55,56 on the full list of differentially expressed genes in WT and HNF1A knockout (KO) cells (Figures 4B and S6; Table S8). The GSEA showed that HNF1A-null cells downregulated multiple lipid metabolism pathways including “fatty acid metabolism” (MsigDB: M5935), “cholesterol homeostasis” (MsigDB: M5892), and “bile acid metabolism” (MsigDB: 5948), consistent with previous findings from murine Hnf1a LOF models.9 Additionally, pathways for “coagulation” (MsigDB: M5946) and “complement” (MsigDB: M5921) were also significantly downregulated in the absence of HNF1A, and a more refined examination of subpathways underlying these further showed a downregulation in “platelet activation” (MsigDB: M10857) and “wound healing” (MsigDB: M12074).
Figure 4.

HNF1A transcriptionally regulates complement activation, coagulation, and pro-atherogenic gene expression programs in hepatocytes
(A) Differentially expressed genes between WT and HNF1A KO HUH7 and Hep3B cell lines quantified by mRNA sequencing (n = 3 per group). The dotted red line indicates an adjusted p value threshold of 0.05, with genes falling above the line meeting a global significance threshold for differential expression.
(B) Gene set enrichment analysis (GSEA) was performed on mRNA sequencing data shown in (A) to identify pathways altered by deletion of HNF1A (HNF1A KO vs. WT). Significant genesets in at least one cell line are shown. The effect of GOF in HNF1A on these pathways was examined by complementing HNF1A KO HUH7 cells with WT HNF1A and the GOF p.T196A variant (n = 3 per group), followed by mRNA sequencing, differential expression, and GSEA (HNF1A p.T196A vs. WT). Downregulated pathways (negative normalized enrichment scores) are shaded yellow, and upregulated pathways are shaded blue. Size represents the -log10(p value), and significantly enriched pathways (p < 0.05) are outlined in red.
(C) HNF1A was reintroduced via doxycycline-inducible transgenes into HNF1A KO hepatocyte cell lines and global gene expression was measured (TPM, transcripts per million; n = 3 per group). In a dose-dependent fashion, HNF1A regulates genes involved in oxidative stress and lipid regulation pathways. Significance levels from regressing TPM on HNF1A levels are shown for each gene and cell type if significant (∗∗∗p < 10−6, ∗∗p < 10−3, ∗p < 0.05, linear model). GGT1, gamma glutamyltransferase 1; PCSK9, proprotein convertase subtilisin/kexin type 9; APOA1, apolipoprotein A1; APOB, apolipoprotein B.
To evaluate global transcriptional changes from HNF1A GOF variants, we assessed the global transcriptional effects of expressing HNF1A WT and the most commonly found human GOF variant HNF1A p.T196A (Figure 2B). We performed short-term complementation experiments, expressing HNF1A WT and p.T196A in HNF1A-null HUH7 cells for 18 h from in-vitro-transcribed mRNA at levels similar to endogenous HNF1A in WT HUH7 cells (Figure S7). GSEA of genes differentially expressed between the HNF1A WT- and p.T196A variant-complemented cells (Table S9) showed that relative to WT, the p.T196A variant more strongly upregulated several of the lipid metabolism and coagulation/complement activation pathways that were downregulated in HNF1A KO cells (Figure 4B; Table S8), supporting a molecular mechanism of GOF that spans multiple transcriptional pathways.
To validate specific effector genes that could explain the observed inflammatory, oxidative stress, and atherogenic serum lipoprotein gene expression profiles in HNF1A variant carriers, we performed independent experiments evaluating HNF1A:effector gene dose responsiveness. Doxycycline-inducible HNF1A transgenes were introduced into HNF1A-null cells and exposed to varying doses of doxycycline to evaluate HNF1A:gene dose-response curves, quantified by regressing gene transcripts per million (TPM) onto HNF1A dose (Figure 4C). The most straightforward example was for serum GGT, a marker of oxidative stress,57 which is hepatically secreted and positively regulated by HNF1A (GGT1 p < 10−3, linear model, HUH7; Figure 4C) and whose serum values are positively correlated with genetically mediated increases in HNF1A function/liver-specific gene expression (Figures 3C and 3D). Effector genes that could account for the serum lipoprotein profile associated with GOF in HNF1A and showed dose response included the LDL receptor recycling regulator proprotein convertase subtilisin/kexin type 958 (PCSK9 p < 10−6, linear model, HUH7) and ApoA1, the major lipoprotein component of HDL (APOA1 p < 10−6, linear model, Hep3B). Conversely, a negative HNF1A:gene expression dose response was observed for ApoB, the major lipoprotein component of LDL (APOB p < 10−3, linear model; HUH7), suggesting that the positive correlation observed between serum ApoB levels with genetically mediated increases in HNF1A function/expression (Figures 3C and 3D) is not a hepatocyte cell-autonomous effect of HNF1A.
To corroborate direct transcriptional regulation of these genes by HNF1A, we identified HNF1A consensus binding motifs within a 5 kb genomic window of the transcription start sites (TSSs) and overlaid these with HNF1A ChIP-seq data from HepG2 cells made available through the ENCODE project.59 The PCSK9 and GGT1 loci showed ChIP-seq peaks coincident with high-scoring HNF1A consensus motifs near the TSS (Table S10), strongly supporting direct transcriptional regulation. The APOA1 and APOB loci showed more modest evidence of direct regulation having only ChIP-seq peaks near the TSS but lacking HNF1A consensus motifs.
Discussion
In this study, we combine massively parallel functional characterization and saturation mutagenesis with genetic and clinical data from almost 600,000 individuals to evaluate a full allelic series of protein-coding variants in HNF1A and their impact on human health. By correlating variant, cellular function, and clinical phenotype in thousands of individuals, we find that the general human population contains carriers of GOF variants that increase the transcriptional activity of HNF1A by 30%–50%. These GOF variants have both beneficial and pathological clinical consequences in the people who carry them through pleiotropic actions in the pancreas and liver, respectively. While GOF variants confer a ∼30% decrease in T2D risk they, likely through liver-specific transcriptional effects, promote an atherogenic lipid profile (increased serum ApoB/LDL and decreased ApoA-I/HDL). Lastly, we demonstrate that the hepatic transcriptional regulation of key effector genes by HNF1A, including APOA1, ANGPTL3, and PCSK9, mediates at least part of the pro-atherogenic lipid profile observed in GOF variant carriers.
Here, we present a description of function-increasing genetic variants in HNF1A and their presence in the human population, an unexpected finding given that thousands of individuals have undergone HNF1A gene sequencing and dozens of protein-coding variants have been functionally characterized over the past two decades.60,61 We note that in contrast to LOF variants, GOF HNF1A variants showed no evidence of being under evolutionary constraint (Figure S3) and thus would escape discovery by computational prediction programs28,29 that rely on evolutionary conservation, highlighting the importance of taking an experimental approach to pathogenicity prediction at clinically important genes. In retrospect, some variants we have demonstrated as GOF have been previously identified and experimentally tested showing transactivation signals greater than WT38 but due to inconsistent results across different experiments or labs were labeled WT-like. For example, the p.T196A variant showed activity greater than WT in transformed COS-7 monkey fibroblasts but not in mouse insulinoma cells,38 whereas in a recent study,62 p.P291Q and p.T196A showed increased transcriptional activity over WT in both HeLa and rat insulinoma cells but at only one of two expert labs testing the same variants. In our own experiments, we were able to most clearly resolve variants with function greater than WT in cell systems without endogenous HNF1A (HUH7 KO and HeLa cells; Figure 2C) using the standard transactivation assays, but the signal was more challenging to identify in MIN6 cells with a high level of basal HNF1A activity (Figure 2D). The variable function of GOF variants in MIN6 cells could also be attributed to differences in HNF1A function in human hepatocytes (in which GOF was called) vs. the mouse beta cells from which MIN6 are derived. Moreover, such GOF variants, by virtue of conferring protection from diabetes, were less likely to be identified in previous studies that selected diabetic individuals for sequencing. By functionally characterizing all possible missense variants in a single experiment, our study revealed the full spectrum of possible functional variation including both loss and gain. We further benefited from ascertainment of HNF1A variants in the UK Biobank, a general population-based cohort not enriched for diabetes and therefore not depleted of GOF HNF1A variant carriers. In the context of another recent study that identified GOF variant carriers in MC4R in the UK Biobank,63 our study highlights the potential for discovering novel genetic mechanisms of disease by conducting large scale functional variant characterization and clinical correlation in population-based cohorts.
Through the identification of a full allelic series of HNF1A variants, our study advances previously reported genetic associations between HNF1A, clinical biomarkers and T2D42 by providing tissue-specific, mechanistic insights and unexpected directional relationships. In the general population our data support that GOF in HNF1A reduces T2D risk and glycemia through enhanced beta cell function, which complements the mechanisms by which LOF HNF1A variants are known to cause T2D/MODY3.3,36 In the liver, however, we find that GOF in HNF1A is metabolically pathogenic, causing increases in hepatically derived lipoprotein risk factors, which have been shown causal for CAD as well as increases in correlative, if not directly causal, factors such as hsCRP and GGT.16,17,46,64,65 Despite a significant decrease in diabetes risk in GOF variant carriers, we found no signal for protection from CAD risk in GOF variant carriers and propose that the increase in hepatic pro-atherogenic factors is counterbalanced by the decrease in T2D risk (Figure 5). Furthermore, the identified genetic associations between GOF in HNF1A, T2D, and atherogenic lipoproteins remained robust upon correction for common HNF1A protein-coding variants as well as the heritable component of hepatic HNF1A gene expression (Table S6). An important medical implication of these findings is that HNF1A “replacement” gene therapy for patients with HNF1A MODY, which is technically feasible through mRNA therapeutics,66 should be approached with caution, as hepatic increases in HNF1A would be clinically undesirable.
Figure 5.

Graphical summary of proposed mechanism through which GOF in HNF1A decreases diabetes risk through pancreas-specific mechanisms but increases the hepatic secretion of atherogenic risk factors
Carriers of GOF variants in HNF1A have reduced T2D risk and HbA1c through enhanced beta cell function but have increases in CRP and GGT and present a pro-atherogenic profile (suppression of ApoA-I/HDL levels and increased hepatic secretion of ANGPTL3 and ApoB/LDL levels through increased transcription of PCSK9). Red arrows indicate metabolically harmful outcomes, and blue arrows represent metabolically beneficial outcomes. GOF, gain of function; CRP, C-reactive protein; GGT, gamma-glutamyl transferase; ApoA-I, apolipoprotein A-I; HDL, high-density lipoprotein cholesterol; ANGPTL3, angiopoietin like 3; TG, triglycerides; PCSK9, proprotein convertase subtilisin/kexin type 9; ApoB, apolipoprotein B; LDL, low-density lipoprotein cholesterol; HbA1c, glycated hemoglobin.
On the other hand, liver-specific inhibition of HNF1A could be therapeutically attractive. At the molecular level, our data suggest that the suppression of ApoA-I/HDL levels by HNF1A is likely cell autonomous, as it occurred in cultured hepatocytes. Similarly, we find that HNF1A and GOF-causing variants increase hepatocyte secretion of ANGPTL3 (Figure 2E), an emerging cardiovascular risk factor that inhibits endothelial lipase resulting in decreased clearance of triglyceride-rich lipoproteins.67 Although we did not find significantly increased serum triglyceride levels in HNF1A GOF variant carriers (Figure 3C), ANGPTL3 has been proposed to promote atherosclerosis independent of triglycerides by increasing inflammation and angiogenesis and inhibiting reverse cholesterol transport.68 Additionally, we propose that the increase in serum LDL cholesterol in carriers of rare HNF1A GOF variants is at least partially mediated by a direct transcriptional activation by HNF1A of PCSK9 (Figures 4 and 5), a hepatic protease that regulates hepatic uptake of LDL69,70 and is a pharmacological target for lipid-lowering therapies.71,72,73 The lack of an HNF1A motif at the APOB promoter as well as the lack of positive HNF1A-APOB dose:response (Figure 4C) in cultured hepatocytes suggests the increase in serum ApoB levels from genetically mediated increase in HNF1A function/expression is indirect and is driven predominantly by increased serum LDL persistence from reduced LDLR recycling that would be caused by increasing PCSK9 transcription74 (Figure 5). Concordantly, prior studies have found that liver-specific HNF1A inhibition decreases PCSK9 levels, increases LDLR persistence, and results in decreased serum total and LDL cholesterol.75,76 Intriguingly a recent phase 1 trial of a non-statin lipid-lowering compound was demonstrated to prevent the dimerization and transactivation of HNF1A at its transcriptional targets, ANGPTL3 and PCSK9, resulting in lowered serum triglycerides and LDL cholesterol, respectively.77 Our study predicts that GOF variant carriers in HNF1A would be highly responsive to this novel lipid-lowering compound. Furthermore, by virtue of having increased PCSK9 expression, HNF1A GOF carriers would also define a subpopulation that may respond to PCSK9 inhibitors more favorably for LDL reduction than conventional statin medications, which are known to increase levels of PCSK9.78
In summary, we find that ∼1:300 (Table S4) individuals in the general population carry GOF variants in HNF1A that are not identifiable by computational prediction tools and pleiotropically decrease diabetes risk while increasing hepatic secretion of atherogenic lipids. In an era of rapidly deployable gene therapy,79 understanding the clinical consequences of increasing or decreasing gene function in humans bottlenecks therapeutic development. As biobanks continue to grow in size and phenotypic complexity, this study demonstrates the utility of comprehensive functional characterization to distill novel mechanisms of disease and identify adverse effects prior to undertaking drug development and putting clinical trial participants at risk.
Limitations of the study
Limitations of our study include the precision of HNF1A function scores generated in our massively parallel assay and the cell models utilized to elucidate molecular mechanisms. Specifically, the use of TM4SF4 as the readout for HNF1A function would not capture the complexity of HNF1A transactivation functions across all of its targets. While our observation of a class of amino acid substitutions that increase the transcriptional activity of HNF1A (Figures 1D and 2A) is strongly supported by experimental validation in multiple independent experiments (Figures 2C–2E and 4B), the precise level of increase in function conferred by any particular variant contains biological (different cellular contexts) and experimental (performed in different labs) uncertainties that limit their interpretation. For example, the top-scoring GOF variant p.P291Q (Figure 2A) replicated in HeLa cells (Figure 2C) but did not show increase over WT in MIN6 cells (Figure 2D) or ANGPTL3 secretion (Figure 2E) assays. Conversely, p.T196A was the lowest scoring of the selected GOF variants but showed higher function than other variants in subsequent assays. Thus, we would caution against utilizing these function scores directly to provide an individualized risk estimate to a person carrying a protein-coding HNF1A variant. If used as a standalone classifier for GOF/LOF, our function scores would meet the 90% probability standard for clinical variant interpretation80 for only 43 variants (Table S2). Additionally, due to the design of our deep mutational scan, effects of splice variants are not detectable, and functional interpretation of such variants should be approached with caution. Furthermore, our mechanistic evaluation in multiple human hepatocyte cell lines could miss important regulatory mechanisms due to the limitations of in vitro cell culture models. For example, our cellular data did not identify direct regulation of CRP by HNF1A as it is poorly expressed and lacks evidence for binding in the publicly available HNF1A ChIP sequencing (ChIP-seq) data59 (Table S10), but previous investigators have reported HNF1A binding at the CRP promoter,81,82,83 supporting a direct regulation.
Consortia
The members of Alnylam Human Genetics are Aimee M. Deaton, Rachel A. Hoffing, Aaron M. Holleman, Lynne Krohn, Philip LoGerfo, Paul Nioi, Mollie E. Plekan, and Lucas D. Ward.
The members of AMP-T2D are Gonçalo R. Abecasis, Carlos A. Aguilar-Salinas, David M. Altshuler, Gil Atzmon, Francisco Barajas-Olmos, Aris Baras, Nir Barzilai, Graeme I. Bell, Thomas W. Blackwell, John Blangero, Michael Boehnke, Eric Boerwinkle, Lori L. Bonnycastle, Erwin P. Bottinger, Donald W. Bowden, Jennifer A. Brody, Brian Burke, Noël P. Burtt, David J. Carey, Lizz Caulkins, Federico Centeno-Cruz, John C. Chambers, Juliana Chan, Edmund Chan, Ling Chen, Siying Chen, Ching-Yu Cheng, Francis S. Collins, Cecilia Contreras-Cubas, Adolfo Correa, Maria Cortes, Nancy J. Cox, Emilio Córdova, Dana Dabelea, Paul S. de Vries, Ralph A. DeFronzo, Frederick E. Dewey, Lawrence Dolan, Kimberly L. Drews, Ravindranath Duggirala, Josée Dupuis, Maria Elena Gonzalez, Amanda Elliott, Maria Eugenia Garay-Sevilla, Jason Flannick, Jose C. Florez, James S. Floyd, Philippe Frossard, Christian Fuchsberger, Stacey B. Gabriel, Humberto García-Ortiz, Christian Gieger, Benjamin Glaser, Clicerio Gonzalez, Niels Grarup, Leif Groop, Myron Gross, Christopher A. Haiman, Sohee Han, Craig L. Hanis, Torben Hansen, Nancy L. Heard-Costa, Susan R. Heckbert, Brian E. Henderson, Soo Heon Kwak, Anne U. Jackson, Young Jin Kim, Marit E. Jørgensen, Megan Kelsey, Bong-Jo Kim, Ryan Koesterer, Heikki A. Koistinen, Jaspal S. Kooner, Johanna Kuusisto, Markku Laakso, Leslie A. Lange, Joseph B. Leader, Juyoung Lee, Jong-Young Lee, Donna M. Lehman, H. Lester Kirchner, Allan Linneberg, Jianjun Liu, Ching-Ti Liu, Ruth J.F. Loos, Valeriya Lyssenko, Ronald C.W. Ma, Anubha Mahajan, Alisa K. Manning, Juan Manuel Malacara-Hernandez, Anthony Marcketta, Angélica Martínez-Hernández, Karen Matsuo, Elizabeth Mayer-Davis, James B. Meigs, Thomas Meitinger, Elvia Mendoza-Caamal, Josep M. Mercader, Hyun Min Kang, Karen L. Mohlke, Andrew P. Morris, Andrew D. Morris, Alanna C. Morrison, Anne Ndungu, Maggie C.Y. Ng, Peter Nilsson, Christopher J. O’Donnell, Colm O’Dushlaine, Lorena Orozco, Colin N.A. Palmer, James S. Pankow, Anthony J. Payne, Oluf Pedersen, Catherine Pihoker, Wendy S. Post, Michael Preuss, Bruce M. Psaty, Asif Rasheed, Alexander P. Reiner, Cristina Revilla-Monsalve, Stephen S. Rich, Neil R. Robertson, Jerome I. Rotter, Danish Saleheen, Nicola Santoro, Claudia Schurmann, Laura J. Scott, Mark Seielstad, Yoon Shin Cho, E. Shyong Tai, Xueling Sim, Robert Sladek, Kerrin S. Small, Xavier Soberón, Kyong Soo Park, Timothy D. Spector, Konstantin Strauch, Heather M. Stringham, Tim M. Strom, Claudia H.T. Tam, Tanya M. Teslovich, Farook Thameem, Brian Tomlinson, Jason M. Torres, Russell P. Tracy, Tiinamaija Tuomi, Jaakko Tuomilehto, Teresa Tusié-Luna, Miriam S. Udler, Rob M. van Dam, Ramachandran S. Vasan, Marijana Vujkovic, Shuai Wang, Ryan P. Welch, Jennifer Wessel, N. William Rayner, James G. Wilson, Daniel R. Witte, Tien-Yin Wong, Wing Yee So, Mi Yeong Hwang, Yik Ying Teo, and Philip Zeitler.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Human TM4SF4 APC-conjugated Antibody | R&D systems | Cat#FAB7998A |
| Critical commercial assays | ||
| QIAamp DNA Mini Kit | Qiagen | Cat#51304 |
| Nextera XT DNA Library Prep | Illumina | Cat#FC-131-1096 |
| Dual Luciferase Assay System | Promega | Cat#E1500 |
| ANGPTL3 ELISA | R&D Systems | Cat#DANL30 |
| Deposited data | ||
| HNF1A function scores | This paper (Table S2) | https://medschool.ucsd.edu/som/medicine/divisions/endocrinology/research/labs/majithia/data/Pages/default.aspx |
| Experimental models: Cell lines | ||
| HUH7 | JCRB | JCRB0403 |
| Hep3B | ATCC | HB-8064 |
| Oligonucleotides | ||
| HNF1A KO sgRNA (CTGTCCCAACACCTCAACAA) | This paper | N/A |
| Primers and sequences for functional testing, see Table S11 | This paper | N/A |
| Software and algorithms | ||
| ORFcall | Majithia et al.20 | https://github.com/tedsharpe/ORFCall |
| R statistical software v3.5.1 | N/A | http://www.R-project.org/ |
| lme4 v1.1-17, lmerTest v3.1-2 | N/A | https://github.com/lme4/lme4 |
| SnpEff v4.3 | Cingolani et al.84 | https://pcingola.github.io/SnpEff/se_introduction/ |
| Kallisto v0.44.0 | Bray et al.85 | https://pachterlab.github.io/kallisto/ |
| DESeq2 v1.22.1 | Love et al.86 | http://www.bioconductor.org/packages/release/bioc/html/DESeq2.html |
| fGSEA v1.8.0 | Subramanian et al.55; Mootha et al.56 | https://doi.org/10.18129/B9.bioc.fgsea |
Resource availability
Lead contact
Further information and requests should be directed to and will be fulfilled by the lead contact, Amit Majithia (amajithia@ucsd.edu).
Materials availability
This study did not generate new unique reagents.
Experimental model and study participant details
Cell line generation
HUH7 and Hep3B cells were cultured in DMEM (Life Technologies #11995065) supplemented with 10% FBS (Sigma #F2442) and L-Glutamine and antibiotics (Omega PG-30). Polyclonal HNF1A-null cells were generated by introducing the endonuclease Cas9 and sgRNA (CTGTCCCAACACCTCAACAA) targeting exon 2 of human HNF1A into cells by lentiviral transduction as we have previously described.20
UK Biobank cohort
The UK Biobank is a prospective cohort study with genotypic and phenotypic data that enrolled approximately 500,000 individuals aged 40–69 from across the United Kingdom.32 The UK Biobank study was approved by the Research Ethics Committee and informed consent was obtained from all participants. Analysis of UK Biobank data was conducted under application numbers 51436 and 26041.
Method details
Synthesis of all possible HNF1A amino acid variants
Human HNF1A cDNA (CCDS9209.1) was recoded by selecting alternative codons to eliminate the CRISPR/CAS9 binding site and optimize expression (Table S11). This cDNA corresponds to the longest amino acid coding sequence of HNF1A expressed in the liver and also the most abundant.50 Constructs encoding all 19 alternative amino acids at each position were synthesized (Twist Bioscience, Table S1). An additional 613 constructs encoding synonymous variants spread across the coding sequence were also synthesized and included. The HNF1A construct library described above was cloned into a lenti-viral expression vector by simple restriction cloning, sequence verified for quality and completeness (Figure S2), and transfected into a packaging cell line to produce pooled lenti-virus.20
Pooled experimental assay of 11,970 HNF1A variants
The lentiviral library of HNF1A amino acid variants was introduced into HUH7 HNF1A-null cells under a doxycycline responsive promoter to enable controlled protein expression.87 To minimize doubly infected cells, 1.2 × 107 viral particles were combined with 4 × 107 cells after which uninfected cells were eliminated by selection with 4 μg/mL puromycin. At least 107 cells were infected to ensure that each HNF1A variant was independently represented in 1000 cells. The resulting polyclonal population of HUH7 hepatocytes (each cell containing a different HNF1A variant) was stimulated with doxycycline (5 μg/mL) for 6 days and then immuno-stained for TM4SF4 (R&D systems FAB7998A). Stained cells were sorted using FACS (FACS Aria II and BD Influx) into two equal bins: high expression and low expression bins based on TM4SF4 expression with a 10-fold expression intensity difference between each bin (Figure 1A). Control experiments with HNF1A-null and WT cells were performed to identify the TM4SF4 expression distributions of cells with and without HNF1A (Figure 1A dashed red and dashed green lines and Figure S1). These distributions helped to establish TM4SF4 expression cutoffs for the FACS sorting. Three independent sorting experiments were performed, sorting ∼500,000 cells per bin per replicate. To re-identify and quantitate the HNF1A variants in the TM4SF4 ‘high’ and ‘low’ bins, genomic DNA (gDNA) was extracted (Qiagen #51304) from each sample of 500,000 cells and the integrated proviral HNF1A transgenes recovered by PCR. Each sample of purified gDNA was split into 14 separate 100 μL PCR reactions (∼400ng gDNA per reaction) and amplified using Herculase II (Agilent #600675) according to the manufacturer protocol using a Tm of 57°C (see Table S11 for primer sequences). All 14 PCR reactions per sample were pooled together, and the transgenes were purified (Qiagen #28104) and prepared for sequencing (Illumina Nextera #FC-131-1096) according to the manufacturer’s protocol. The six samples were pooled and sequenced to obtain approximately 350 million paired-end 100-base reads (Illumina NovaSeq) resulting in a 12x base coverage assuming a target size of 6 × 109 bp (2000 bp transgene x 12,000 variants x 250 observations per variant). Raw sequencing reads were aligned to the reference HNF1A recoded cDNA sequence (Table S11) using ORFCall (https://github.com/tedsharpe/ORFCall)20 and the number of occurrences of each amino acid at each position along the coding region were counted and tabulated.
Calculation of function scores
We constructed a likelihood function based on the log odds of an amino acid variant being in the fraction with high or low TM4SF4. The log odds for each amino acid variant was estimated by maximizing a likelihood function based on the observed counts of each amino acid variant in the fractions with high and low TM4SF4 normalized to the total read depth at that amino acid position. Data were combined across experimental replicates. To avoid spuriously high or low log-odds estimates for any given variant, we constrained the log-odds estimate with a Gaussian prior whose parameters were estimated from data combined across all variants. These procedures were analogous to those we have previously described.20 Variants which had total counts across all replicates and bins less than the known low count variants from the original library design were not assigned a function score (125 variants). A mutation tolerance score was calculated for each of the 631 amino acid positions along HNF1A by taking the mean of all assigned function scores at that position.
To calculate the posterior error probability (PEP) that any particular variant was not neutral, we computed the probability that it belonged to the synonymous function score distribution instead of the nonsynonymous distribution using the formula PEP_nonWT = Psynonymous/(Psynonymous + Pnonsynonymous).31 The probability density functions for Psynonymous and Pnonsynonymous were generated by linearly interpolating the distribution of function scores (“combined_function_score” Table S2) for synonymous and nonsynonymous variants using an interpolation function as instantiated in R v3.5.1.
Genetic data
For all exome sequenced cohorts, variants within the genomic coordinates of HNF1A (chr12:120,977,683-121,002,512, GRCh38) were extracted, and variant annotation was performed using SnpEff v4.3.84 Nomenclature used for missense variants is for the canonical HNF1A transcript ENST00000257555.10; protein ENSP00000257555.4. Disruptive variants (i.e. splice region variants, stop gained variants, or frameshift variants) were assigned the function score equal to the lowest known pathogenic variant functional score, homozygous carriers of rare HNF1A protein-coding variants (n = 5) were removed from all analyses, and compound heterozygotes (n = 39) were included twice as single variant carriers.
Metabolic biomarkers and clinical phenotypes
Clinical measurements from all 502,489 UK Biobank participants were log transformed, if not normally distributed (Table S5) and residualized adjusting for age, age2 and sex, then normalized using a rank based inverse normal transformation.88 Diabetes mellitus designation was based on a compilation of self-report or ICD-10 codes of E11 in hospitalization or primary care records.89,90
Functional validation of GOF HNF1A variants
Each GOF variant selected for validation (Figure 2A) was recreated as a transgene based on the native sequence of the cDNA (CCDS9209.1) with the exact human carrier nucleotide change observed in the UK Biobank (Table S11). The constructed variants were tested for transactivation potential in HeLa cells as we have previously published37 using the rat albumin promoter. All transactivation experiments included three parallels performed on three independent experimental days (nine readings in total).
Transcriptional activity of the HNF1A GOF variants was also tested in MIN6 cells using the HNF4aP2 promoter.38 Cells were co-transfected with a DNA mixture consisting of 10 ng of pGL-Luc and pGL4.75, and 10 ng of variant or WT HNF1A. The level of the transcription factor activity was evaluated by measuring firefly luciferase activity relative to renilla luciferase activity using the Dual Luciferase Assay System (Promega #E1500), as described by the manufacturer.
Assessment of ANGPTL3 levels in cultured hepatocytes
HNF1A constructs (WT, p.T260M, and p.P447L) were introduced into WT and HNF1A KO human HUH7 cells by lentiviral induction under a doxycycline responsive promoter as above. Cells were stimulated with doxycycline (0, 0.5, 1, 5 μg/mL) for 5 days. Following doxycycline incubation, the spent media was collected for ANGPTL3 ELISA analysis (R&D Systems #DANL 30) and the cells for protein quantification (RIPA buffer + protease inhibitor) according to the manufacturer’s protocol. GOF variant constructs were transfected into HUH7 hepatocytes using TransIT-LT1 transfection reagent (Mirus Bio), puromycin selection was performed to remove untransfected cells, and cells were incubated in fresh media for 24 h before harvest for ELISA and protein quantification as described above.
Functional genomic analysis of HNF1A deleted hepatocytes and complementation with GOF variants
HNF1A-null HUH7 and Hep3b cells were generated as described above, and WT HNF1A was reintroduced using a doxycycline-inducible promoter (0, 2, 5 μg/mL) in triplicate. mRNA was extracted (Qiagen #74104) and sequenced (Illumina TruSeq) according to the manufacturers’ protocol to a depth of at least 30 million reads per sample. Raw reads were aligned using Kallisto (v0.44.0)85 with default parameters to generate gene counts per cell. DESeq2 (R v3.5.1, package v1.22.1)86 was used to perform differential expression analysis with effect sizes and p values obtained from the Wald test as instantiated within DESeq2. Gene set enrichment analysis (GSEA)55,56 was conducted using the fgsea package with 10000 permutations (package v1.8.091) utilizing Hallmark gene sets92 and Gene Ontology biological process gene sets93,94 related to significantly enriched Hallmark gene sets (cholesterol homeostasis, fatty acid metabolism, bile acid metabolism, coagulation, and complement activation). Significance was determined by a two-sided enrichment p value with Benjamini–Hochberg correction for multiple hypotheses as instantiated within the fgsea software package.
Complementation of HNF1A-null HUH7 cells with mRNA encoding WT HNF1A or the GOF p.T196A variant was essentially performed as described before,95 with modifications. Briefly, cDNA constructs were amplified from the expression plasmids described above with primers that introduce a T7 promoter and AG as the first two transcribed nucleotides and that add a 179-nt-long poly(A) tail to the 3′ end. PCR products were phenol/chloroform-extracted and in vitro-transcribed with T7 RNA Polymerase HiScribe Mix (NEB E2040S), CleanCap Reagent AG (Tri-Link N-7113) and 5-methoxyuridine triphosphate instead of UTP for 2 h at 37°C to generate capped, polyadenylated mRNA. IVT mRNA was precipitated with 2.5 M LiCl final, washed twice with 80% EtOH and dissolved in water. For each construct, 106 HNF1A-null HUH7 cells were electroporated with 2 μg mRNA in 100 μL Buffer R with four 10-millisecond pulses at 1230 V using a Neon Transfection System (ThermoFisher MPK10025). Cells were cultured for 18 h in 5 mL medium in 60 mm plates, and mRNA was extracted (Zymo R1050), sequenced (Illumina Stranded mRNA), and gene expression analyzed as above. Expression of HNF1A was verified by Western blot, and EGFP mRNA was used as electroporation control. Differential expression analysis between HNF1A-null HUH7 complemented with EGFP and WT HNF1A showed a profile of differentially expressed genes similar to HNF1A-null vs. HNF1A WT cells (top genes in both experiments included FGB, FABP1, PCSK9, and ANGPTL3; Tables S7 and S9).
HNF1A binding motifs were scored using JASPAR96 consensus matrix sequence matches 5000 bp before and after the genomic coordinates of the transcriptional start site for each gene of interest. HNF1A HepG2 ChIP-seq bigWig signal p value and IDR ranked peaks result files from both replicates were downloaded from ENCODE experiment ENCSR633HRJ.97
Quantification and statistical analysis
Genotype-function-phenotype association analyses
To examine the association between the full continuum of HNF1A function score and diabetes status in the UK Biobank, we performed logistic mixed effects regression (lme4 v1.1-17, lmerTest v3.1-2) with variant identity as a random effect in order to account for multiple carriers of the same variant, adjusting for age, age,2 sex, and the first 10 genetic principal components of ancestry (computed by UK Biobank for multi-ancestry analyses (Field 22009) and computed for European ancestry participants as previously described89). To account for related individuals, in the full multi-ancestry analysis we randomly removed one participant if a pair of them had a genetic relationship equal or closer than a second-degree relative, and in the European analysis, related individuals were identified and excluded as per Deaton et al.98 To examine the association between functional categories (i.e. GOF/LOF) and diabetes or CAD status in the UK Biobank, we performed logistic regression (glm function, R stats v3.5.1) adjusting for covariates previously mentioned. To test the effect of categorization threshold on T2D, GOF/LOF variants were re-categorized based on increasingly stringent cutoffs defined according to the spread of synonymous variant distributions (+/− 1SD, 1.25, 1.5, 1.75, 2) and the effect on T2D risk in HNF1A protein-coding variant carriers in the UK Biobank was recomputed for each recategorization threshold as above. To assess the effect of common HNF1A protein-coding variation on the identified associations with T2D, we first identified all independent HNF1A protein coding SNPs with MAF >0.01. Two such SNPs were identified, p.I27L (rs1169288) and p.A98V (rs1800574), and incorporated as a covariates in the regressions of T2D on HNF1A function score/categorized HNF1A variants.
Analysis in the AMP-T2D-GENES study was conducted as previously described.43 Briefly, we conducted two burden tests -- one including gain of function variants and one including loss of function variants -- using the EPACTS software package, regressing sample phenotype on genotype dosage. We conducted each test across all unrelated individuals pooled together and included principal components of ancestry covariates (computed from common variants) as well as covariates for sample cohort of origin and sequencing technology. The effect size and significance of the burden test was evaluated using the two-sided Firth logistic regression test. Meta-analysis for the UK Biobank and AMP-T2D cohorts was conducted using fixed-effect inverse-variance meta-analysis as implemented by METAL.99
To test the association of rare HNF1A variants in the MIGen ExSeq dataset with the risk of coronary artery disease, a Firth logistic regression was applied with sex and top 10 principal components of ancestry as covariates and cleaned to second-degree of relationship. The Firth logistic regression is a test robust to association testing in the context of low carrier counts or case-control imbalance.100 For each person, the genetic score of the qualified rare variant carriers was coded as 1, and 0 otherwise. Meta-analysis for the UK Biobank and MIGen ExSeq cohorts was conducted using fixed-effect inverse-variance meta-analysis as implemented by METAL.99 Power calculations were performed using the Purcell et al.101 genetic power calculator with the following parameters: high risk/marker allele frequency = cumulative GOF allele frequency = 0.002, a 5.4% prevalence of CAD in the UK Biobank, genotype relative risk = GOF OR for T2D = 0.75, total number of CAD cases in MIGEN and UK Biobank = 49,666, D’ = 1, and control:cases ratio = 9.44. Power is reported for α = 0.05.
Quantitative phenotype associations with function scores were performed within the UK Biobank using the normalized clinical measurements described above and a linear mixed model with function score as a fixed effect, adjusting for 10 genetic principal components of ancestry as described above, and with variant identity as a random effect in order to account for multiple carriers of the same variant (lme4 v1.1-17, lmerTest v3.1-2).
To quantify liver specific HNF1A gene expression, the Functional Summary-based Imputation (FUSION) framework for generating individual-level predicted gene expression51 was applied to UK Biobank genotyped individuals. First, to ensure that this analysis was independent of our analysis of protein-coding variants, carriers of rare protein-coding HNF1A variants (n = 2,203) in the UK Biobank were excluded. Next, all HNF1A (ENSG00000135100) expression reference weights computed in European liver RNA-sequenced samples from the Genotype-Tissue Expression (GTEx) Project v8 were extracted from the FUSION archive (http://gusevlab.org/projects/fusion/#gtex-v8-multi-tissue-expression) (n = 466 SNPs with precomputed expression weights, Table S12). These expression weights and their corresponding imputed genotypes in the UK Biobank European individuals (n = 407,227) were combined into a predicted gene expression score for each individual by computing a linear combination across the HNF1A SNPs expression weights and genotypes, then dividing by the number of non-missing SNPs as implemented in the Plink score utility.102 Phenotypes were normalized as described above and regressed onto predicted HNF1A liver expression scores using a generalized linear model (glm function, R stats v3.5.1) adjusting for 10 genetic principal components of ancestry. Regressions additionally included as a covariate individual level genotype of the common HNF1A variant rs1169288 (encoding p.I27L) which has previously been associated with metabolic phenotypes15,103 and was significantly associated with the predicted HNF1A liver expression score (p < 2 × 10−16, linear model). To assess potential confounding of the HNF1A liver gene expression score with rare-variant phenotypic associations, the models for T2D and quantitative trait regression on HNF1A function score were adjusted for each individual’s computed HNF1A liver gene expression. This analysis was performed in the subset of UK Biobank European participants for whom liver eQTLs are available.
P-values from the regression of categorical phenotypes against HNF1A category were adjusted for multiple comparisons using Bonferroni correction for two categories (LOF and GOF), and all p values from the regression of quantitative traits against HNF1A function score/liver expression score were adjusted using Bonferroni correction for six phenotype groups (C-reactive protein, LDL/Apolipoprotein B, Cholesterol/Triglycerides, HDL/Apolipoprotein A, Gamma glutamyltransferase, and HbA1c).
Acknowledgments
We would like to thank Joseph Witztum and Vikas Bansal for helpful discussions. We are grateful to Xiaolan Zhang and Kyle Sanchez for assistance with preparing the HNF1A variant library, cellular screening/FACS, and production of HNF1A-null hepatocyte cell lines. We would also like to thank Twist Bioscience for constructing the HNF1A variant library. Henrietta Lacks and the HeLa cell line that was established from her tumor cells without her knowledge or consent in 1951 have made significant contributions to scientific progress and advances in human health. We are grateful to Henrietta Lacks, now deceased, and to her surviving family members for their contributions to biomedical research. This work was supported by grants from the National Institute of Diabetes and Digestive and Kidney Diseases (1R03DK113328 and 1R01DK123422 to A.R.M. and 1R01DK125490 to J.F. and A.R.M.), a UCSD/UCLA Pilot and Feasibility grant (P30 DK063491 to A.R.M.), and a Ruth L. Kirschstein Institutional National Research Service Award T32 GM008666 from the National Institute of General Medical Sciences (to N.D.). S.Heinz. received support from NIDDK grants P30DK063491 and P30DK120515 and NIGMS R01GM129523. G.M.P. is supported by NHLBI grants R01HL142711 and R01HL127564. This publication includes data generated at the UC San Diego IGM Genomics Center utilizing an Illumina NovaSeq 6000 that was purchased with funding from a National Institutes of Health SIG grant (S10 OD026929).
Author contributions
N.D. and A.R.M. designed the study. N.D., B.K., S. Hu, R.I., L.K., M.W., J.G., X.D., C.D.A.S., and A.M.D. generated/acquired and analyzed the data. N.D., B.K., S. Hu, R.I., M.W., R.A., J.F., G.M.P., S. Heinz, P.L.S.M.G., A.M.D., A.V.K., J.O., V.R., and A.R.M. were involved in data interpretation. N.D. and A.R.M. drafted the manuscript. N.D., B.K., R.I., L.K., M.W., X.D., V.M., G.M.P., P.L.S.M.G., A.M.D., A.V.K., J.O., V.R., S. Heinz, and A.R.M. were involved in critical manuscript revision.
Declaration of interests
A.M.D. and L.K. are employees and shareholders of Alnylam Pharmaceuticals.
Published: May 30, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xgen.2023.100339.
Supplemental information
Data and code availability
-
•
Function scores for HNF1A missense variants found in Table S2 can be queried interactively via our custom data portal found at https://medschool.ucsd.edu/som/medicine/divisions/endocrinology/research/labs/majithia/data/Pages/default.aspx. All UK Biobank data used in this study are accessible through application to UK Biobank.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Lau H.H., Ng N.H.J., Loo L.S.W., Jasmen J.B., Teo A.K.K. The molecular functions of hepatocyte nuclear factors - in and beyond the liver. J. Hepatol. 2018;68:1033–1048. doi: 10.1016/j.jhep.2017.11.026. [DOI] [PubMed] [Google Scholar]
- 2.Fajans S.S., Bell G.I. MODY: history, genetics, pathophysiology, and clinical decision making. Diabetes Care. 2011;34:1878–1884. doi: 10.2337/dc11-0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yamagata K., Oda N., Kaisaki P.J., Menzel S., Furuta H., Vaxillaire M., Southam L., Cox R.D., Lathrop G.M., Boriraj V.V., et al. Mutations in the hepatocyte nuclear factor-1alpha gene in maturity-onset diabetes of the young (MODY3) Nature. 1996;384:455–458. doi: 10.1038/384455a0. [DOI] [PubMed] [Google Scholar]
- 4.Lehto M., Tuomi T., Mahtani M.M., Widén E., Forsblom C., Sarelin L., Gullström M., Isomaa B., Lehtovirta M., Hyrkkö A., et al. Characterization of the MODY3 phenotype. Early-onset diabetes caused by an insulin secretion defect. J. Clin. Invest. 1997;99:582–591. doi: 10.1172/JCI119199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McDonald T.J., McEneny J., Pearson E.R., Thanabalasingham G., Szopa M., Shields B.M., Ellard S., Owen K.R., Malecki M.T., Hattersley A.T., et al. Lipoprotein composition in HNF1A-MODY: differentiating between HNF1A-MODY and type 2 diabetes. Clin. Chim. Acta. 2012;413:927–932. doi: 10.1016/j.cca.2012.02.005. [DOI] [PubMed] [Google Scholar]
- 6.McDonald T.J., Shields B.M., Lawry J., Owen K.R., Gloyn A.L., Ellard S., Hattersley A.T. High-sensitivity CRP discriminates HNF1A-MODY from other subtypes of diabetes. Diabetes Care. 2011;34:1860–1862. doi: 10.2337/dc11-0323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goldberg I.J. Clinical review 124: diabetic dyslipidemia: causes and consequences. J. Clin. Endocrinol. Metab. 2001;86:965–971. doi: 10.1210/jcem.86.3.7304. [DOI] [PubMed] [Google Scholar]
- 8.King D.E., Mainous A.G., 3rd, Buchanan T.A., Pearson W.S. C-reactive protein and glycemic control in adults with diabetes. Diabetes Care. 2003;26:1535–1539. doi: 10.2337/diacare.26.5.1535. [DOI] [PubMed] [Google Scholar]
- 9.Shih D.Q., Bussen M., Sehayek E., Ananthanarayanan M., Shneider B.L., Suchy F.J., Shefer S., Bollileni J.S., Gonzalez F.J., Breslow J.L., et al. Hepatocyte nuclear factor-1α is an essential regulator of bile acid and plasma cholesterol metabolism. Nat. Genet. 2001;27:375–382. doi: 10.1038/86871. [DOI] [PubMed] [Google Scholar]
- 10.Voight B.F., Scott L.J., Steinthorsdottir V., Morris A.P., Dina C., Welch R.P., Zeggini E., Huth C., Aulchenko Y.S., Thorleifsson G., et al. Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat. Genet. 2010;42:579–589. doi: 10.1038/ng.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Teslovich T.M., Musunuru K., Smith A.V., Edmondson A.C., Stylianou I.M., Koseki M., Pirruccello J.P., Ripatti S., Chasman D.I., Willer C.J., et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466:707–713. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu Y., McDade T.W., Kuzawa C.W., Borja J., Li Y., Adair L.S., Mohlke K.L., Lange L.A. Genome-wide association with C-reactive protein levels in CLHNS: evidence for the CRP and HNF1A loci and their interaction with exposure to a pathogenic environment. Inflammation. 2012;35:574–583. doi: 10.1007/s10753-011-9348-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Giuffrida F.M.A., Furuzawa G.K., Kasamatsu T.S., Oliveira M.M., Reis A.F., Dib S.A. HNF1A gene polymorphisms and cardiovascular risk factors in individuals with late-onset autosomal dominant diabetes: a cross-sectional study. Cardiovasc. Diabetol. 2009;8:28. doi: 10.1186/1475-2840-8-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Erdmann J., Grosshennig A., Braund P.S., König I.R., Hengstenberg C., Hall A.S., Linsel-Nitschke P., Kathiresan S., Wright B., Trégouët D.A., et al. New susceptibility locus for coronary artery disease on chromosome 3q22.3. Nat. Genet. 2009;41:280–282. doi: 10.1038/ng.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reiner A.P., Gross M.D., Carlson C.S., Bielinski S.J., Lange L.A., Fornage M., Jenny N.S., Walston J., Tracy R.P., Williams O.D., et al. Common coding variants of the HNF1A gene are associated with multiple cardiovascular risk phenotypes in community-based samples of younger and older European-American adults: the Coronary Artery Risk Development in Young Adults Study and the Cardiovascular Health Study. Circ. Cardiovasc. Genet. 2009;2:244–254. doi: 10.1161/CIRCGENETICS.108.839506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reiner Ž. Hypertriglyceridaemia and risk of coronary artery disease. Nat. Rev. Cardiol. 2017;14:401–411. doi: 10.1038/nrcardio.2017.31. [DOI] [PubMed] [Google Scholar]
- 17.Lagrand W.K., Visser C.A., Hermens W.T., Niessen H.W., Verheugt F.W., Wolbink G.J., Hack C.E. C-reactive protein as a cardiovascular risk factor: more than an epiphenomenon? Circulation. 1999;100:96–102. doi: 10.1161/01.cir.100.1.96. [DOI] [PubMed] [Google Scholar]
- 18.Flannick J., Beer N.L., Bick A.G., Agarwala V., Molnes J., Gupta N., Burtt N.P., Florez J.C., Meigs J.B., Taylor H., et al. Assessing the phenotypic effects in the general population of rare variants in genes for a dominant Mendelian form of diabetes. Nat. Genet. 2013;45:1380–1385. doi: 10.1038/ng.2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fowler D.M., Fields S. Deep mutational scanning: a new style of protein science. Nat. Methods. 2014;11:801–807. doi: 10.1038/nmeth.3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Majithia A.R., Tsuda B., Agostini M., Gnanapradeepan K., Rice R., Peloso G., Patel K.A., Zhang X., Broekema M.F., Patterson N., et al. Prospective functional classification of all possible missense variants in PPARG. Nat. Genet. 2016;48:1570–1575. doi: 10.1038/ng.3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Findlay G.M., Daza R.M., Martin B., Zhang M.D., Leith A.P., Gasperini M., Janizek J.D., Huang X., Starita L.M., Shendure J. Accurate classification of BRCA1 variants with saturation genome editing. Nature. 2018;562:217–222. doi: 10.1038/s41586-018-0461-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abel E.V., Goto M., Magnuson B., Abraham S., Ramanathan N., Hotaling E., Alaniz A.A., Kumar-Sinha C., Dziubinski M.L., Urs S., et al. HNF1A is a novel oncogene that regulates human pancreatic cancer stem cell properties. Elife. 2018;7:e33947. doi: 10.7554/eLife.33947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Servitja J.-M., Pignatelli M., Maestro M.A., Cardalda C., Boj S.F., Lozano J., Blanco E., Lafuente A., McCarthy M.I., Sumoy L., et al. Hnf1alpha (MODY3) controls tissue-specific transcriptional programs and exerts opposed effects on cell growth in pancreatic islets and liver. Mol. Cell Biol. 2009;29:2945–2959. doi: 10.1128/MCB.01389-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Giacomelli A.O., Yang X., Lintner R.E., McFarland J.M., Duby M., Kim J., Howard T.P., Takeda D.Y., Ly S.H., Kim E., et al. Mutational processes shape the landscape of TP53 mutations in human cancer. Nat. Genet. 2018;50:1381–1387. doi: 10.1038/s41588-018-0204-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chi Y.-I., Frantz J.D., Oh B.-C., Hansen L., Dhe-Paganon S., Shoelson S.E. Diabetes mutations delineate an atypical POU domain in HNF-1α. Mol. Cell. 2002;10:1129–1137. doi: 10.1016/s1097-2765(02)00704-9. [DOI] [PubMed] [Google Scholar]
- 26.Rose R.B., Endrizzi J.A., Cronk J.D., Holton J., Alber T. High-resolution structure of the HNF-1α dimerization domain. Biochemistry. 2001;40:3242. doi: 10.1021/bi0151263. [DOI] [PubMed] [Google Scholar]
- 27.Cooper G.M., Stone E.A., Asimenos G., NISC Comparative Sequencing Program. Green E.D., Batzoglou S., Sidow A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005;15:901–913. doi: 10.1101/gr.3577405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Adzhubei I., Jordan D.M., Sunyaev S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013;Chapter 7:Unit7.20. doi: 10.1002/0471142905.hg0720s76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rentzsch P., Witten D., Cooper G.M., Shendure J., Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47:D886–D894. doi: 10.1093/nar/gky1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Landrum M.J., Lee J.M., Benson M., Brown G.R., Chao C., Chitipiralla S., Gu B., Hart J., Hoffman D., Jang W., et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46:D1062–D1067. doi: 10.1093/nar/gkx1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Käll L., Storey J.D., MacCoss M.J., Noble W.S. Posterior error probabilities and false discovery rates: two sides of the same coin. J. Proteome Res. 2008;7:40–44. doi: 10.1021/pr700739d. [DOI] [PubMed] [Google Scholar]
- 32.Bycroft C., Freeman C., Petkova D., Band G., Elliott L.T., Sharp K., Motyer A., Vukcevic D., Delaneau O., O’Connell J., et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562:203–209. doi: 10.1038/s41586-018-0579-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Backman J.D., Li A.H., Marcketta A., Sun D., Mbatchou J., Kessler M.D., Benner C., Liu D., Locke A.E., Balasubramanian S., et al. Exome sequencing and analysis of 454,787 UK Biobank participants. Nature. 2021;599:628–634. doi: 10.1038/s41586-021-04103-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tennessen J.A., Bigham A.W., O’Connor T.D., Fu W., Kenny E.E., Gravel S., McGee S., Do R., Liu X., Jun G., et al. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337:64–69. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shields B.M., Hicks S., Shepherd M.H., Colclough K., Hattersley A.T., Ellard S. Maturity-onset diabetes of the young (MODY): how many cases are we missing? Diabetologia. 2010;53:2504–2508. doi: 10.1007/s00125-010-1799-4. [DOI] [PubMed] [Google Scholar]
- 36.Najmi L.A., Aukrust I., Flannick J., Molnes J., Burtt N., Molven A., Groop L., Altshuler D., Johansson S., Bjørkhaug L., et al. Functional investigations of HNF1A identify rare variants as risk factors for type 2 diabetes in the general population. Diabetes. 2017;66:335–346. doi: 10.2337/db16-0460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Balamurugan K., Bjørkhaug L., Mahajan S., Kanthimathi S., Njølstad P.R., Srinivasan N., Mohan V., Radha V. Structure-function studies of HNF1A (MODY3) gene mutations in South Indian patients with monogenic diabetes. Clin. Genet. 2016;90:486–495. doi: 10.1111/cge.12757. [DOI] [PubMed] [Google Scholar]
- 38.Galán M., García-Herrero C.M., Azriel S., Gargallo M., Durán M., Gorgojo J.-J., Andía V.M., Navas M.-A. Differential effects of HNF-1α mutations associated with familial young-onset diabetes on target gene regulation. Mol. Med. 2011;17:256–265. doi: 10.2119/molmed.2010.00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dewey F.E., Gusarova V., Dunbar R.L., O’Dushlaine C., Schurmann C., Gottesman O., McCarthy S., Van Hout C.V., Bruse S., Dansky H.M., et al. Genetic and pharmacologic inactivation of ANGPTL3 and cardiovascular disease. N. Engl. J. Med. 2017;377:211–221. doi: 10.1056/NEJMoa1612790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Musunuru K., Pirruccello J.P., Do R., Peloso G.M., Guiducci C., Sougnez C., Garimella K.V., Fisher S., Abreu J., Barry A.J., et al. Exome sequencing, ANGPTL3 mutations, and familial combined hypolipidemia. N. Engl. J. Med. 2010;363:2220–2227. doi: 10.1056/NEJMoa1002926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Odom D.T., Zizlsperger N., Gordon D.B., Bell G.W., Rinaldi N.J., Murray H.L., Volkert T.L., Schreiber J., Rolfe P.A., Gifford D.K., et al. Control of pancreas and liver gene expression by HNF transcription factors. Science. 2004;303:1378–1381. doi: 10.1126/science.1089769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.SIGMA Type 2 Diabetes Consortium. Estrada K., Aukrust I., Bjørkhaug L., Burtt N.P., Mercader J.M., García-Ortiz H., Huerta-Chagoya A., Moreno-Macías H., Walford G., et al. Association of a low-frequency variant in HNF1A with type 2 diabetes in a Latino population. JAMA. 2014;311:2305–2314. doi: 10.1001/jama.2014.6511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Flannick J., Mercader J.M., Fuchsberger C., Udler M.S., Mahajan A., Wessel J., Teslovich T.M., Caulkins L., Koesterer R., Barajas-Olmos F., et al. Exome sequencing of 20,791 cases of type 2 diabetes and 24,440 controls. Nature. 2019;570:71–76. doi: 10.1038/s41586-019-1231-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gaulton K.J., Ferreira T., Lee Y., Raimondo A., Mägi R., Reschen M.E., Mahajan A., Locke A., Rayner N.W., Robertson N., et al. Genetic fine mapping and genomic annotation defines causal mechanisms at type 2 diabetes susceptibility loci. Nat. Genet. 2015;47:1415–1425. doi: 10.1038/ng.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Prospective Studies Collaboration. Lewington S., Whitlock G., Clarke R., Sherliker P., Emberson J., Halsey J., Qizilbash N., Peto R., Collins R. Blood cholesterol and vascular mortality by age, sex, and blood pressure: a meta-analysis of individual data from 61 prospective studies with 55,000 vascular deaths. Lancet. 2007;370:1829–1839. doi: 10.1016/S0140-6736(07)61778-4. [DOI] [PubMed] [Google Scholar]
- 46.Targher G., Byrne C.D. Circulating markers of liver function and cardiovascular disease risk. Arterioscler. Thromb. Vasc. Biol. 2015;35:2290–2296. doi: 10.1161/ATVBAHA.115.305235. [DOI] [PubMed] [Google Scholar]
- 47.Nielson C., Lange T., Hadjokas N. Blood glucose and coronary artery disease in nondiabetic patients. Diabetes Care. 2006;29:998–1001. doi: 10.2337/diacare.295998. [DOI] [PubMed] [Google Scholar]
- 48.Myocardial Infarction Genetics Consortium Investigators. Stitziel N.O., Won H.-H., Morrison A.C., Peloso G.M., Do R., Lange L.A., Fontanillas P., Gupta N., Duga S., et al. Inactivating mutations in NPC1L1 and protection from coronary heart disease. N. Engl. J. Med. 2014;371:2072–2082. doi: 10.1056/NEJMoa1405386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.GTEx Consortium Genetic effects on gene expression across human tissues (2017) Nature. 2017;550:204–213. doi: 10.1038/nature24277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.GTEx Consortium The genotype-tissue expression (GTEx) project. Nat. Genet. 2013;45:580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gusev A., Ko A., Shi H., Bhatia G., Chung W., Penninx B.W.J.H., Jansen R., de Geus E.J.C., Boomsma D.I., Wright F.A., et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet. 2016;48:245–252. doi: 10.1038/ng.3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pepys M.B., Hirschfield G.M. C-reactive protein: a critical update. J. Clin. Invest. 2003;111:1805–1812. doi: 10.1172/JCI18921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yamagata K. Roles of HNF1α and HNF4α in pancreatic β-cells: lessons from a monogenic form of diabetes (MODY) Vitam. Horm. 2014;95:407–423. doi: 10.1016/B978-0-12-800174-5.00016-8. [DOI] [PubMed] [Google Scholar]
- 54.Dalmon J., Laurent M., Courtois G. The human beta fibrinogen promoter contains a hepatocyte nuclear factor 1-dependent interleukin-6-responsive element. Mol. Cell Biol. 1993;13:1183–1193. doi: 10.1128/mcb.13.2.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mootha V.K., Lindgren C.M., Eriksson K.-F., Subramanian A., Sihag S., Lehar J., Puigserver P., Carlsson E., Ridderstråle M., Laurila E., et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 57.Lee D.-H., Blomhoff R., Jacobs D.R., Jr. Is serum gamma glutamyltransferase a marker of oxidative stress? Free Radic. Res. 2004;38:535–539. doi: 10.1080/10715760410001694026. [DOI] [PubMed] [Google Scholar]
- 58.Kathiresan S., Srivastava D. Genetics of human cardiovascular disease. Cell. 2012;148:1242–1257. doi: 10.1016/j.cell.2012.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Partridge E.C., Chhetri S.B., Prokop J.W., Ramaker R.C., Jansen C.S., Goh S.-T., Mackiewicz M., Newberry K.M., Brandsmeier L.A., Meadows S.K., et al. Occupancy maps of 208 chromatin-associated proteins in one human cell type. Nature. 2020;583:720–728. doi: 10.1038/s41586-020-2023-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bellanné-Chantelot C., Carette C., Riveline J.-P., Valéro R., Gautier J.-F., Larger E., Reznik Y., Ducluzeau P.-H., Sola A., Hartemann-Heurtier A., et al. The type and the position of HNF1A mutation modulate age at diagnosis of diabetes in patients with maturity-onset diabetes of the young (MODY)-3. Diabetes. 2008;57:503–508. doi: 10.2337/db07-0859. [DOI] [PubMed] [Google Scholar]
- 61.Ellard S. Hepatocyte nuclear factor 1 alpha (HNF-1α) mutations in maturity-onset diabetes of the young. Hum. Mutat. 2000;16:377–385. doi: 10.1002/1098-1004(200011)16:5<377::AID-HUMU1>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 62.Althari S., Najmi L.A., Bennett A.J., Aukrust I., Rundle J.K., Colclough K., Molnes J., Kaci A., Nawaz S., van der Lugt T., et al. Unsupervised clustering of missense variants in HNF1A using multidimensional functional data aids clinical interpretation. Am. J. Hum. Genet. 2020;107:670–682. doi: 10.1016/j.ajhg.2020.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lotta L.A., Mokrosiński J., Mendes de Oliveira E., Li C., Sharp S.J., Luan J. ’an, Brouwers B., Ayinampudi V., Bowker N., Kerrison N., et al. Human gain-of-function MC4R variants show signaling bias and protect against obesity. Cell. 2019;177:597–607.e9. doi: 10.1016/j.cell.2019.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Toth P.P. High-density lipoprotein and cardiovascular risk. Circulation. 2004;109:1809–1812. doi: 10.1161/01.CIR.0000126889.97626.B8. [DOI] [PubMed] [Google Scholar]
- 65.Kannel W.B., Anderson K., Wilson P.W. White blood cell count and cardiovascular disease. Insights from the Framingham Study. JAMA. 1992;267:1253–1256. [PubMed] [Google Scholar]
- 66.Sahin U., Karikó K., Türeci Ö. mRNA-based therapeutics—developing a new class of drugs. Nat. Rev. Drug Discov. 2014;13:759–780. doi: 10.1038/nrd4278. [DOI] [PubMed] [Google Scholar]
- 67.Kersten S. 2017. Angiopoietin-like 3 in Lipoprotein Metabolism. [DOI] [PubMed] [Google Scholar]
- 68.Lupo M.G., Ferri N. Angiopoietin-like 3 (ANGPTL3) and atherosclerosis: lipid and non-lipid related effects. J. Cardiovasc. Dev. Dis. 2018;5:39. doi: 10.3390/jcdd5030039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Horton J.D., Cohen J.C., Hobbs H.H. Molecular biology of PCSK9: its role in LDL metabolism. Trends Biochem. Sci. 2007;32:71–77. doi: 10.1016/j.tibs.2006.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sun H., Samarghandi A., Zhang N., Yao Z., Xiong M., Teng B.-B. Proprotein convertase subtilisin/kexin type 9 interacts with apolipoprotein B and prevents its intracellular degradation, irrespective of the low-density lipoprotein receptor. Arterioscler. Thromb. Vasc. Biol. 2012;32:1585–1595. doi: 10.1161/ATVBAHA.112.250043. [DOI] [PubMed] [Google Scholar]
- 71.Graham M.J., Lemonidis K.M., Whipple C.P., Subramaniam A., Monia B.P., Crooke S.T., Crooke R.M. Antisense inhibition of proprotein convertase subtilisin/kexin type 9 reduces serum LDL in hyperlipidemic mice. J. Lipid Res. 2007;48:763–767. doi: 10.1194/jlr.C600025-JLR200. [DOI] [PubMed] [Google Scholar]
- 72.Frank-Kamenetsky M., Grefhorst A., Anderson N.N., Racie T.S., Bramlage B., Akinc A., Butler D., Charisse K., Dorkin R., Fan Y., et al. Therapeutic RNAi targeting PCSK9 acutely lowers plasma cholesterol in rodents and LDL cholesterol in nonhuman primates. Proc. Natl. Acad. Sci. USA. 2008;105:11915–11920. doi: 10.1073/pnas.0805434105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Duff C.J., Scott M.J., Kirby I.T., Hutchinson S.E., Martin S.L., Hooper N.M. Antibody-mediated disruption of the interaction between PCSK9 and the low-density lipoprotein receptor. Biochem. J. 2009;419:577–584. doi: 10.1042/BJ20082407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kotowski I.K., Pertsemlidis A., Luke A., Cooper R.S., Vega G.L., Cohen J.C., Hobbs H.H. A spectrum of PCSK9 alleles contributes to plasma levels of low-density lipoprotein cholesterol. Am. J. Hum. Genet. 2006;78:410–422. doi: 10.1086/500615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shende V.R., Wu M., Singh A.B., Dong B., Kan C.F.K., Liu J. Reduction of circulating PCSK9 and LDL-C levels by liver-specific knockdown of HNF1α in normolipidemic mice. J. Lipid Res. 2015;56:801–809. doi: 10.1194/jlr.M052969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dong B., Singh A.B., Shende V.R., Liu J. Hepatic HNF1 transcription factors control the induction of PCSK9 mediated by rosuvastatin in normolipidemic hamsters. Int. J. Mol. Med. 2017;39:749–756. doi: 10.3892/ijmm.2017.2879. [DOI] [PubMed] [Google Scholar]
- 77.Wang J., Zhao J., Yan C., Xi C., Wu C., Zhao J., Li F., Ding Y., Zhang R., Qi S., et al. Identification and evaluation of a lipid-lowering small compound in preclinical models and in a Phase I trial. Cell Metab. 2022;34:667–680.e6. doi: 10.1016/j.cmet.2022.03.006. [DOI] [PubMed] [Google Scholar]
- 78.Dubuc G., Chamberland A., Wassef H., Davignon J., Seidah N.G., Bernier L., Prat A. Statins upregulate PCSK9, the gene encoding the proprotein convertase neural apoptosis-regulated convertase-1 implicated in familial hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol. 2004;24:1454–1459. doi: 10.1161/01.ATV.0000134621.14315.43. [DOI] [PubMed] [Google Scholar]
- 79.Kim J., Hu C., Moufawad El Achkar C., Black L.E., Douville J., Larson A., Pendergast M.K., Goldkind S.F., Lee E.A., Kuniholm A., et al. Patient-customized oligonucleotide therapy for a rare genetic disease. N. Engl. J. Med. 2019;381:1644–1652. doi: 10.1056/NEJMoa1813279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Richards S., Aziz N., Bale S., Bick D., Das S., Gastier-Foster J., Grody W.W., Hegde M., Lyon E., Spector E., et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nishikawa T., Hagihara K., Serada S., Isobe T., Matsumura A., Song J., Tanaka T., Kawase I., Naka T., Yoshizaki K. Transcriptional complex formation of c-Fos, STAT3, and hepatocyte NF-1 alpha is essential for cytokine-driven C-reactive protein gene expression. J. Immunol. 2008;180:3492–3501. doi: 10.4049/jimmunol.180.5.3492. [DOI] [PubMed] [Google Scholar]
- 82.Toniatti C., Demartis A., Monaci P., Nicosia A., Ciliberto G. Synergistic trans-activation of the human C-reactive protein promoter by transcription factor HNF-1 binding at two distinct sites. EMBO J. 1990;9:4467–4475. doi: 10.1002/j.1460-2075.1990.tb07897.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li S.P., Goldman N.D. Regulation of human C-reactive protein gene expression by two synergistic IL-6 responsive elements. Biochemistry. 1996;35:9060–9068. doi: 10.1021/bi953033d. [DOI] [PubMed] [Google Scholar]
- 84.Cingolani P., Platts A., Wang L.L., Coon M., Nguyen T., Wang L., Land S.J., Lu X., Ruden D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bray N.L., Pimentel H., Melsted P., Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016;34:525–527. doi: 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- 86.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:1550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Majithia A.R., Flannick J., Shahinian P., Guo M., Bray M.-A., Fontanillas P., Gabriel S.B., et al. GoT2D Consortium. NHGRI JHS/FHS Allelic Spectrum Project. SIGMA T2D Consortium Rare variants in PPARG with decreased activity in adipocyte differentiation are associated with increased risk of type 2 diabetes. Proc. Natl. Acad. Sci. USA. 2014;111:13127–13132. doi: 10.1073/pnas.1410428111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Klarin D., Damrauer S.M., Cho K., Sun Y.V., Teslovich T.M., Honerlaw J., Gagnon D.R., DuVall S.L., Li J., Peloso G.M., et al. Genetics of blood lipids among ∼300,000 multi-ethnic participants of the Million Veteran Program. Nat. Genet. 2018;50:1514–1523. doi: 10.1038/s41588-018-0222-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Khera A.V., Chaffin M., Aragam K.G., Haas M.E., Roselli C., Choi S.H., Natarajan P., Lander E.S., Lubitz S.A., Ellinor P.T., et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 2018;50:1219–1224. doi: 10.1038/s41588-018-0183-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tan X., Benedict C. Increased risk of myocardial infarction among patients with type 2 diabetes who carry the common rs10830963 variant in the MTNR1B gene. Diabetes Care. 2020;43:2289–2292. doi: 10.2337/dc20-0507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Korotkevich G., Sukhov V., Sergushichev A. Fast gene set enrichment analysis. Cold Spring Harb. Lab. 2019:060012. doi: 10.1101/060012. [DOI] [Google Scholar]
- 92.Liberzon A., Birger C., Thorvaldsdóttir H., Ghandi M., Mesirov J.P., Tamayo P. The molecular signatures database Hallmark gene set collection. Cell Syst. 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gene Ontology Consortium The Gene Ontology resource: enriching a GOld mine. Nucleic Acids Res. 2021;49:D325–D334. doi: 10.1093/nar/gkaa1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Heinz S., Texari L., Hayes M.G.B., Urbanowski M., Chang M.W., Givarkes N., Rialdi A., White K.M., Albrecht R.A., Pache L., et al. Transcription elongation can affect genome 3D structure. Cell. 2018;174:1522–1536.e22. doi: 10.1016/j.cell.2018.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Fornes O., Castro-Mondragon J.A., Khan A., van der Lee R., Zhang X., Richmond P.A., Modi B.P., Correard S., Gheorghe M., Baranašić D., et al. JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2020;48:D87–D92. doi: 10.1093/nar/gkz1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Deaton A.M., Parker M.M., Ward L.D., Flynn-Carroll A.O., BonDurant L., Hinkle G., Akbari P., Lotta L.A., Baras A., Nioi P., et al. Gene-level analysis of rare variants in 379,066 whole exome sequences identifies an association of GIGYF1 loss of function with type 2 diabetes. Sci. Rep. 2021;11:21565. doi: 10.1038/s41598-021-99091-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Willer C.J., Li Y., Abecasis G.R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–2191. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Dey R., Schmidt E.M., Abecasis G.R., Lee S. A fast and accurate algorithm to test for binary phenotypes and its application to PheWAS. Am. J. Hum. Genet. 2017;101:37–49. doi: 10.1016/j.ajhg.2017.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Purcell S., Cherny S.S., Sham P.C. Genetic Power Calculator: design of linkage and association genetic mapping studies of complex traits. Bioinformatics. 2003;19:149–150. doi: 10.1093/bioinformatics/19.1.149. [DOI] [PubMed] [Google Scholar]
- 102.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A.R., Bender D., Maller J., Sklar P., de Bakker P.I.W., Daly M.J., et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Reiner A.P., Barber M.J., Guan Y., Ridker P.M., Lange L.A., Chasman D.I., Walston J.D., Cooper G.M., Jenny N.S., Rieder M.J., et al. Polymorphisms of the HNF1A gene encoding hepatocyte nuclear factor-1α are associated with C-reactive protein. Am. J. Hum. Genet. 2008;82:1193–1201. doi: 10.1016/j.ajhg.2008.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Function scores for HNF1A missense variants found in Table S2 can be queried interactively via our custom data portal found at https://medschool.ucsd.edu/som/medicine/divisions/endocrinology/research/labs/majithia/data/Pages/default.aspx. All UK Biobank data used in this study are accessible through application to UK Biobank.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.