

Abstract
We present a 10‐year follow‐up and describe our experience in managing a case of neonatal severe primary hyperparathyroidism (NSHPT) for the first time in Iran. Microcephaly, mental retardation, and epilepsy may be long time sequels of NSHPT. The brain MRI findings are compatible with an old hypoxic–ischemic event.
Keywords: epilepsy, mental retardation, microcephaly, neonatal severe primary hyperparathyroidism
1. INTRODUCTION
Neonatal severe primary hyperparathyroidism (NSHPT) is a rare, potentially life‐threatening autosomal recessive disease characterized by severe hyperparathyroidism, marked hypercalcemia, and metabolic bone disease. 1 Patients mainly present with poor feeding, failure to thrive, hypotonia, lethargy, polyuria, dehydration, respiratory distress, intestinal dysmotility, and skeletal demineralization during the first few weeks after birth. 2 , 3 , 4 , 5 , 6
If not promptly diagnosed and treated, NSHPT can be associated with high mortality or irreversible neurodevelopmental, renal, skeletal, or cardiologic complications. 2 Although successful medical management of NSHPT has been recently reported, 3 , 4 early parathyroidectomy followed by calcium supplementation and regular monitoring of serum calcium and PTH levels has been traditionally recommended as the definite therapy. 2 , 5 , 6 However, neuromotor abnormalities may persist even after successful treatment, which warrants the long‐term follow‐up of these patients. 1
Considering the importance of the long‐term outcome and prognosis of NSHPT, especially regarding neurological development and endocrine problems, we present a 10‐year follow‐up on a previously reported case of NSHPT who underwent total parathyroidectomy on the 11th day of life.
2. CASE PRESENTATION
A 10‐year‐old boy, previously diagnosed with NSHPT, who underwent a total parathyroidectomy on the 11th day of his life, was brought to our clinic for assessment of long‐term outcome. He was born full‐term to consanguineous parents and delivered by cesarean section. He was admitted on the 8th day of life due to poor feeding and hypotonia. He was diagnosed with NHSPT based on severe hypercalcemia (Ca = 35 mg/dL), marked hyperparathyroidism (PTH = 640 pmol/L) alongside with other findings consistent with NSHPT. The medical treatment, including intravenous normal saline, hydrocortisone, furosemide, calcitonin, and pamidronate, failed to stabilize serum calcium levels. The patient underwent then a total parathyroidectomy. The operation proved to be successful by a significant drop in serum calcium and PTH levels. Calcium and vitamin D supplements a few days after surgery were started. Although his NICU‐stay was prolonged due to Klebsiella septic episodes treated with antibiotics and IVIG, he had normal calcium level by the end of the forth postoperative week while he was on calcium supplementation. His neonatal presentation has been described in detail in another article. 7
Following hospital discharge, the patient was not brought for follow‐up visits. When he was 5 years old, he was admitted at another medical center due to new onset of seizures. Antiepileptic medication was initiated. According to the patient's mother, seizures recurred at the age of 7 (electroencephalography [EEG] showing multiple epileptic discharges in a normal background) and Carbamazepine was then prescribed. The child experienced another recurrence at the age of 9 and is still on Carbamazepine. Moreover, he has learning difficulties since school age and is currently attending a special school. According to his mother, the boy had normal serum calcium and PTH levels on occasions when laboratory investigations were performed. Unfortunately, previous laboratory data are not available. At the moment, he is taking no calcium/vitamin D supplements.
At the present visit, the patient's weight and height were 30 kg (50th percentile) and 132 cm (10th percentile), respectively, which is considered normal based on his age and his parents' weight and height. Patient's head circumference was 48 cm (>2 standard deviations [SDs] below the mean for age and sex) which is consistent with microcephaly. The rest of his physical examination was within normal limits. Additional investigations, including brain MRI, EEG, Wechsler Intelligence Scale for Children (WISC‐IV), hand and wrist radiographs, calcium, phosphorus, and PTH serum levels, were performed.
On EEG, multiple epileptic discharges in a normal background were observed, which is similar to the previous EEG performed at the age of 7. Brain MRI revealed periventricular white matter volume loss with extension to the perirolandic region, which probably is a sequel of hypoxic–ischemic damage (Figures 1, 2, 3). Based on the results of WISC‐IV, the full‐scale IQ of the patient was calculated to be 54, which is considered a mild intellectual disability. Patient's bone age was normal for his age based on hand and wrist radiographs. Laboratory investigations revealed normal serum calcium, phosphorus, and PTH levels.
FIGURE 1.

Axial‐flair view: Periventricular abnormal hyperintensities.
FIGURE 2.

Sagital‐T2: Thinning of corpus callosum.
FIGURE 3.
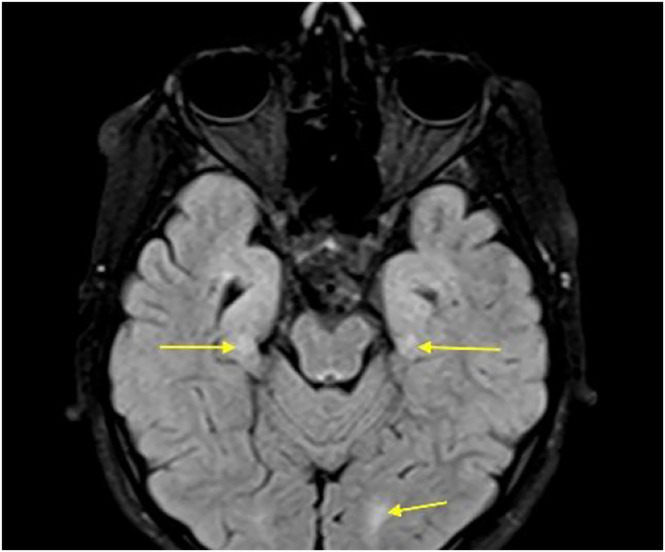
Axial‐flair: Bilateral perirolandic and occipital hyperintensities.
3. DISCUSSION AND CONCLUSION
Among hereditary causes of primary hyperparathyroidism, NSHPT is a highly uncommon autosomal recessive disorder, mostly produced by homozygous or compound heterozygous inactivating mutations in the gene encoding calcium sensing receptor (CaSR), a G‐protein‐coupled receptor expressed in the parathyroid glands, renal tubular cells, bones, and other organs, whose primary function is to maintain calcium homeostasis. Decreased sensitivity of the CaSR receptors to extracellular calcium results in PTH‐hyperproduction and consequently, severe hypercalcemia, 1 which can be potentially fatal or associated with severe neurodevelopmental impairment in untreated NSHPT patients. 8 , 9 On the contrary, early diagnosis and treatment of NSHPT usually lead to gradual improvement in growth and neurodevelopmental milestones. 3 , 4 , 6
Ten years after initial presentation, our patient has mild intellectual disability, microcephaly and he is under treatment for epilepsy. According to his mother, the levels of serum calcium and PTH on occasional laboratory tests performed at earlier age were normal. Although not common, neuromotor retardation may persist even in otherwise successfully treated NSHPT patients, 1 Savas‐Erdeve et al. reported mild mental retardation in a 15‐year‐old girl with a history of NSHPT, in whom normocalcemia was maintained by total parathyroidectomy, followed by calcitriol supplementation. 10
Our findings might be explained by exposure to severe hypercalcemia before parathyroidectomy, since hypercalcemia has been reported to induce cerebral vasoconstriction and subsequently, ischemia. 11 At the present follow‐up, the brain MRI findings were consistent with hypoxic–ischemic damage which may have been caused by the vasoconstriction induced by hypercalcemia during the neonatal period. Additionally, it has been hypothesized that CaSR plays a role in regulating the growth and development of the brain and inactivating mutations of CaSR may result in neurodevelopmental abnormalities. 12
Another explanation for hypoxic–ischemic damage in this patient is sepsis‐associated encephalopathy. The pathophysiology of sepsis‐associated encephalopathy is complex and multifactorial, including a number of intertwined mechanisms such as vascular damage, endothelial activation, breakdown of the blood brain barrier, altered brain signaling, brain inflammation, and apoptosis. 13
Based on these findings, we suggest that the neurodevelopmental impairment caused by NSHPT may be multifactorial in nature. Therefore, we recommend a strict follow‐up program in order to minimize the comorbidities and complications associated with the disease. Moreover, a pediatric neurologist should be involved in the management of these children in order to facilitate early detection and reduce the neurologic complications that may arise. The major limitation of our study is the absence of genetic testing for the CaSR gene due to the high cost and difficulty of accessing this test in our country. To our knowledge, this is the first case report and the longest follow‐up of NSHPT in Iran.
AUTHOR CONTRIBUTIONS
Nahid Khosroshahi: Project administration; supervision; writing – review and editing. Zahra Haghshenas: Data curation; writing – review and editing. Arya Afrooghe: Writing – original draft. Elham Ahmadi: Writing – original draft. Mahdieh Mousavi Torshizi: Writing – review and editing.
FUNDING INFORMATION
No funding was received for the study.
CONFLICT OF INTEREST STATEMENT
There is no conflict of interest to declare.
ETHICS STATEMENT
None.
CONSENT STATEMENT
Written informed consent was obtained from the patient's parents to publish this report in accordance with the journal's patient consent policy.
ACKNOWLEDGMENTS
None.
Khosroshahi N, Haghshenas Z, Afrooghe A, Ahmadi E, Torshizi MM. Ten‐year follow‐up report and neurologic sequelae in a case of neonatal severe primary hyperparathyroidism. Clin Case Rep. 2023;11:e7626. doi: 10.1002/ccr3.7626
DATA AVAILABILITY STATEMENT
Data available on request from the authors.
REFERENCES
- 1. Marx SJ, Sinaii N. Neonatal severe hyperparathyroidism: novel insights from calcium, PTH, and the CASR gene. J Clin Endocrinol Metab. 2020;105(4):1061‐1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Abdullayev T, Korkmaz M, Kul M, Koray N. A rare cause of neonatal hypercalcemia: neonatal severe primary hyperparathyroidism: a case report and review of the literature. Int J Surg Case Rep. 2020;66:365‐369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Özgüç Çömlek F, Demir S, Gürkan H, et al. The efficiency of cinacalcet treatment in delaying parathyroidectomy in a case with neonatal severe hyperparathyroidism caused by homozygous mutation in the CASR gene. Pediatr Endocrinol Diabetes Metab. 2022;28(2):168‐174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fisher MM, Cabrera SM, Imel EA. Successful treatment of neonatal severe hyperparathyroidism with cinacalcet in two patients. Endocrinol Diabetes Metab Case Rep. 2015;2015:150040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Capozza M, Chinellato I, Guarnieri V, et al. Case report: acute clinical presentation and neonatal management of primary hyperparathyroidism due to a novel CaSR mutation. BMC Pediatr. 2018;18(1):340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gupta P, Tak SA, Viswanath SA, et al. A case of neonatal severe hyperparathyroidism: challenges in management. Indian J Pediatr. 2022;89:1025‐1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Naseri M, Kaveh M, Kamrani K, Habibi M, Haghshenas Z. Severe hypercalcemia due to primary neonatal hyperparathyroidism in an 8‐day‐old neonate: a case report. Int J Clin Pediatr. 2014;3(3):94‐96. [Google Scholar]
- 8. Schnabel D, Letz S, Lankes E, Mayr B, Schofl C. Severe but not neonatally lethal. A homozygous inactivating CaSR mutation in a 3 year old child. Exp Clin Endocrinol Diabetes. 2014;122(3):P041. [Google Scholar]
- 9. Corrado KR, Andrade SC, Bellizzi J, D'Souza‐Li L, Arnold A. Polyclonality of parathyroid tumors in neonatal severe hyperparathyroidism. J Bone Miner Res. 2015;30:1797‐1802. [DOI] [PubMed] [Google Scholar]
- 10. Savas‐Erdeve S, Sagsak E, Keskin M, et al. Treatment experience and long‐term follow‐up data in two severe neonatal hyperparathyroidism cases. J Pediatr Endocrinol Metab. 2016;29(9):1103‐1110. [DOI] [PubMed] [Google Scholar]
- 11. Nardone R, Brigo F, Trinka E. Acute symptomatic seizures caused by electrolyte disturbances. J Clin Neurol. 2016;12(1):21‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sfar S, Bzeouich AA, Kerkeni E, Bouaziz S, Najjar MF, et al. A novel CASR mutation in a Tunisian FHH/NSHPT family associated with a mental retardation. Mol Biol Rep. 2012;39:2395‐2400. [DOI] [PubMed] [Google Scholar]
- 13. Chaudhry N, Duggal AK. Sepsis associated encephalopathy. Adv Med. 2014;2014:762320. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data available on request from the authors.