

Abstract
Next-generation sequencing technology (NGS), including whole-exome or whole-genome sequencing and target gene sequencing, has allowed the molecular characterization of somatic mutation spectrums in hematologic diseases. Mutations in Additional sex combs-like 1 (ASXL1), a chromatin regulator, are identified in clonal hematopoiesis of indeterminate potential (CHIP), indicating ASXL1 mutations as early events in leukemogenesis. Not surprisingly, they occur at high frequency in myeloid malignancies and associated with poor prognosis. Therefore, understanding how mutant ASXL1 drives clonal expansion and leukemogenesis will serve as the basis for future development of preventative and/or therapeutic strategies for myeloid diseases with ASXL1 mutations. Here, we discuss the biology of ASXL1 and its role in controlling normal and malignant hematopoiesis. In addition, we review the clinical relevance of ASXL1 mutations in CHIP and myeloid diseases.
Introduction of ASXL1 gene and protein
Mammalian ASXL family genes (ASXL1, ASXL2 and ASXL3) are the mammalian homologs of Drosophila Additional sex combs Asx (1). Asx deletion leads to a homeotic phenotype characteristic of both Polycomb group (PcG, repressive complex associated with H3K27me3) and Trithorax group (TrxG, activating function associated with H3K4me3) gene deletions (1–3). Both Asxl1 and Asxl2 expression is virtually ubiquitous throughout embryogenesis and in adult tissues, whereas Asxl3 expression is more restricted and only detectable in lymph node, eye, lung, skin, brain, and pituitary gland (4).
The human ASXL1 gene is located on the chromosome 20q11.21 and encodes a 1541 amino acid protein (Figure 1) (5). ASXL1 contains an ASXN domain in the N-terminal region, an ASX homology (ASXH) domain in the N-terminal adjoining region, and a plant homeodomain (PHD) in the C-terminal region. ASXL family proteins share highly conserved ASXN, ASXH and PHD domains. The ASXN and PHD domains are putative DNA and histone binding domains, respectively. The ASXH domain (also referred to as DEUBAD, deubiquitinase adaptor) interacts with a partner protein BAP1 to confer deubiquitinase activity, leading to gene repression (6). At the endogenous level, truncated ASXL1 proteins resulting from ASXL1 mutations are rapidly degraded, and the ASXL1-BAP1 complex is undetectable (7). By contrast, overexpression of truncated ASXL1 increases the stability of BAP1 and enhances the deubiquitination activity of ASXL1-BAP1 complex. It is unclear whether overexpression of mutant ASXL1 recapitulates its function at the physiological level (8, 9).
Figure 1.

Schematic representation of structure of wildtype ASXL1 and mutant ASXL1.
In addition to BAP1, ASXL1 interacts with core polycomb repressive complex 2 (PRC2) components EZH2 and SUZ12, which are involved in the deposition of H3K27me3 histone repressive marks (7). The functions of the long stretch of amino acids between the ASXH and PHD domains have been poorly understood. A recent study revealed that the C-terminal intrinsic disordered region is important for the formation of nuclear paraspeckles. Deletion of this region disrupts these paraspeckles, leading to the attenuated repopulation capability of hematopoietic stem cells (HSCs) (10). Interestingly, ASXL1 mutations identified in myeloid diseases are predominantly located within this region, generating C-terminal truncated proteins (see below).
The functions of the other ASXL proteins are less known. ASXL2 has been shown to be essential for cardiac function and bone development (11–13). Recent studies revealed a high-frequency of ASXL2 mutations in acute myeloid leukemia (AML) patients bearing the RUNX1::RUNX1T1 (AML/ETO) fusion. Loss of Asxl2 in mouse leads to development of myelodysplastic syndrome (MDS)-like disease and promotes leukemogenesis driven by RUNX1::RUNX1T1 (14, 15). Unlike ASXL1 and ASXL2 mutations, ASXL3 mutations have not been detected in AML patients (16).
ASXL1 mutations in CHIP and myeloid diseases (clinal relevance)
Mutations in ASXL1 are identified in clonal hematopoiesis of indeterminate potential (CHIP) and significantly associated with smoking (17, 18). CHIP initially referred to the expansion of peripheral blood cells derived from hematopoietic stem cells (HSCs) with at least one somatic driver mutation in healthy elderly individuals (19–21). CHIP is strongly linked to aging and confers to an increased risk for blood cancers, non-hematological diseases (e.g. cardiovascular disease), and all-cause mortality (19–23). Although CHIP confers an approximately 10-fold increased risk to develop hematologic malignancies, such risk remains low (0.5-1% per year) (19). Therefore, it cannot explain the increased overall mortality associated with CHIP. A cause-specific mortality analysis revealed that non-leukemic mortality (e.g. cardiovascular diseases) in CHIP patients is higher than that due to blood cancers (21).
DNMT3A, TET2, and ASXL1 are among the most frequently mutated genes in CHIP. They are associated with initiation of acute myeloid leukemia (AML) and other myeloid diseases. Corroborating the human data, HSCs with Tet2 or Dnmt3a mutations robustly undergo expansion in transplant recipient mice (reviewed in (24)). Subsequently, CHIP was identified in patients previously treated for solid tumors and myeloid malignancy-associated CHIP mutations were also present in patients with lymphoid malignancies (24). The CHIP mutation spectrum in these patients is distinct from that in healthy individuals.
The selection and expansion of preleukemic-HSC clones precede the development of myeloid leukemia. Not surprisingly, ASXL1 mutations (and 20q deletion) are frequently identified in myeloid malignancies, in particular ~20% in MDS, ~45% in chronic myelomonocytic leukemia (CMML), ~10% in myeloproliferative neoplasms (MPNs), and ~20% in AML (25–28). Interestingly, ASXL1 mutations identified in CHIP are enriched around codons R404 (nonsense), Y591 (nonsense/frameshift), H630 (frameshift), and R693 (nonsense/frameshift). By contrast, ASXL1 mutations identified in myeloid diseases (including MDS and CMML) are predominantly frameshift mutations around codon G646 (G646: 18%; codon 630-660: 42%) (Figure. 2). Controversy has surrounded molecular testing of c.1934dupG p.Gly646fs ASXL1 variant. Its location within an 8 base-pair guanine mononucleotide repeat sequence made it suspicious for an artifact of PCR and/or sequencing rather than a true somatic mutation (29). However, the variant allele frequency of this mutation is >5% in many cases, arguing against PCR artefacts. Moreover, subsequent reports using NGS sequencing confirmed that the ASXL1 c.1934dupG is only detected in leukemia cells, but not in matched germline samples or healthy controls (30–33).
Figure 2. Distinct ASXL1 mutation spectrums in CHIP vs myeloid diseases.
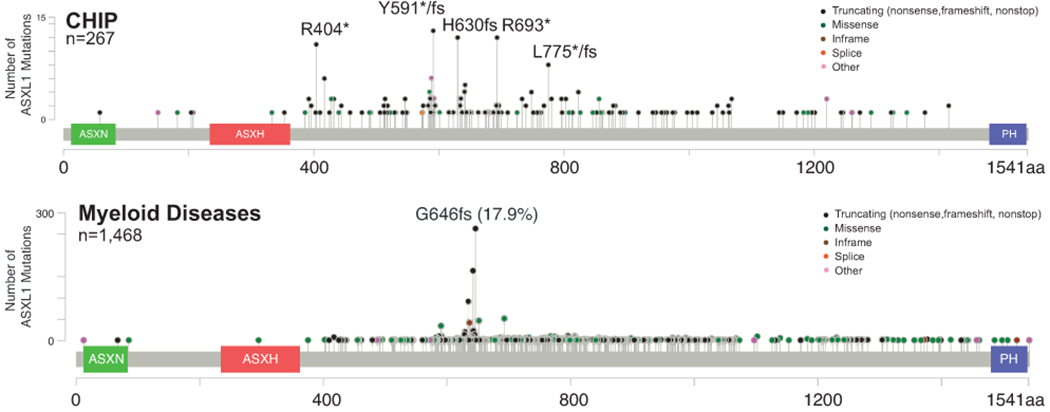
This figure summarizes published datasets of CHIP in healthy elderlies and cancer patients and of patients with myeloid diseases (including MPN, MDS, CMML, and AML).
Clearly, the ASXL1 mutations around codon G646 are prevalent in myeloid diseases but much less common in CHIP. Similarly, the hotspot DNMT3A R882H mutation in AML is rarely seen in CHIP (34). We and others hypothesize that unlike majority of CHIP mutations that are fairly stable and less pathogenic in elderly patients, the hotspot ASXL1 and DNMT3A mutations represent pathological CHIP mutations with high risk for accumulating additional driver mutations and developing myeloid diseases. In support of this idea, CMML cells with hotspot ASXL1 mutations around G646 display distinct transcriptomic changes from normal BM cells and these changes are absent in CMML cells with non-hotspot ASXL1 mutations (35). Asxl1−/− and Asxl1 G643Wfs (corresponding to human G646Wfs) knockin mice develop MDS and a fraction of them transform to myeloid leukemia (36, 37) (see below).
ASXL1 germline mutations and Bohring-Opitz syndrome
Bohring-Opitz syndrome (BOS) is a rare genetic disorder first reported by Bohring et al. in 1999, to describe four individuals with Opitz trigonocephaly (C)-like syndrome (38). BOS is a clinically recognizable syndrome characterized by facial dysmorphism, microcephaly, limb anomalies, postnatal failure to thrive, severe developmental delays and intellectual disability. To date ~100 cases have been described, almost half of which were molecularly confirmed to carry a heterozygous constitutive ASXL1 mutation, suggesting that constitutive mutations in ASXL1 are a major cause of BOS. Similar as CHIP and myeloid diseases-associated ASXL1 mutations, most of BOS-related ASXL1 mutations are de novo nonsense or frameshift mutation. Emma Bedoukiann and colleagues presented the first report of BOS caused by a pathogenic ASXL1 mutation inherited from a germline mosaic mother (39). Later, Karen Seiter and colleagues reported that a father and son were found to have the identical ASXL1 mutation (40), supporting the diagnosis of a germline mutation of ASXL1. Both of them developed AML without BOS symptoms. Therefore, how the same germline ASXL1 mutations cause different diseases remains unknown.
Biological function of Asxl1 (mouse work)
To evaluate the functions of Asxl1 in hematopoiesis and leukemogenesis, five different mouse models have been generated using different approaches (36, 37, 41–43). Conditional Asxl1 knockout mice were created to study loss of Asxl1 function in adult hematopoietic system (36). Asxl1−/− bone marrow (BM) cells display increased number of HSCs and decreased re-plating capability as compared to wildtype (WT) cells. Upon transplantation, Asxl1−/− BM cells show reduced reconstitution in young recipients. Deletion of Asxl1 leads to significant down-regulation of H3K27me3 due to loss of ASXL1-mediated recruitment of PRC2 key components, such as EZH2, to the chromatin (7, 36). In addition, a novel ASXL1-OGT(O-GlcNAc transferase) axis was identified to regulate H3K4 methylation in myeloid malignancies (44). ASXL1 interacts with HCFC1 and OGT and is stabilized via OGT-mediated O-GlcNAcylation. Disruption of this novel axis inhibits myeloid differentiation and H3K4 methylation(44). Consistent with the previous results, we reported that global H3K4me1, H3K4me3, and H3K27me3 levels were significantly decreased in Asxl1−/− BM cells (45). Although global H3K27Ac level in Asxl1−/− BM cells was comparable to that in control cells, H3K27Ac level was increased at specific gene loci.
Two transgenic overexpression models use different exogenous promoters (Rosa26 vs Vav1) to drive the transcription of different Asxl1 mutants (E635RfsX15 vs Y558X) (42, 43). Therefore, it is difficult to compare and interpret their results. Nonetheless, these transgenic overexpression models and in vitro overexpression studies (46) suggest that ASXL1 mutations may be dominant negative or gain-of-function. However, it is questionable whether these overexpression studies truefully reflects the physiology function of truncated ASXL1 proteins.
To overcome this problem, two groups independently generated Asxl1tm knock-in mouse models (37, 41). In both models, the same Asxl1 guanine duplication was introduced into the endogenous Asxl1 locus, closely resembling patient-derived ASXL1 G646WfsX12 mutation. This hotspot frameshift mutation creates a truncated protein of 655aa (658aa in human) in contrast to the full length ASXL1 protein of 1514aa (1541aa in human). Studies with these two knock-in mouse models yielded highly consistent results, some of which are distinct from Asxl1−/− data. For example, in comparison to WT cells, Asxl1tm/+ BM cells exhibit reduced number of HSCs, increased re-plating capability, and largely comparable reconstitution in young recipients, suggesting that in addition to losing part of WT ASXL1 functions, Asxl1tm instills some new functions. However, it remains unclear what epigenetic alterations this mutation causes and how this mutation could drive CH in humans.
Genetic interaction of ASXL1 with NRAS
ASXL1 mutations frequently coexist with other mutations, such as TET2 (47), RUNX1 (48), SETBP1 (49–51) and NRAS (25–28). Asxl1 loss in mice results in MDS that could transform to myeloid leukemia with age, suggesting that Asxl1 deficiency cooperates with additional mutations to induce myeloid leukemias.
ASXL1 mutations predict inferior outcomes in all myeloid diseases (26, 52, 53). They significantly co-occur with NRAS mutations in CMML (25–28). We showed that concurrent ASXL1 and NRAS mutations define a population of CMML patients with shorter leukemia-free survival compared to patients with ASXL1 mutation only (45). Corroborating these human data, we discovered that Asxl1−/− accelerates CMML progression and promotes CMML transformation to AML (secondary AML, sAML) in NrasG12D/+ mice. Although NrasG12D/+; Asxl1−/− (NA) model shares common genetic mutations with the published NrasG12D/+; Ezh2−/− (54) and Nf1+/−; Asxl1+/− (55) models, it displays distinct phenotypes and molecular mechanisms from the other two. NrasG12D/+; Asxl1−/− (NA) leukemia cells exhibited hyperactivation of MEK/ERK signaling and increased global level of H3K27Ac, a histone mark bound by bromodomain and extra-terminal domain (BET) proteins for gene transcriptional activation (45). NA-sAML cells were more immunosuppressive than NA-CMML cells and overexpressed all the major inhibitory immune checkpoint ligands, PD-L1/L2, CD155, and CD80/86 (45). Among them, overexpression of PD-L1 and CD86 correlated with upregulation of AP-1 transcription factors (TFs) in NA-sAML cells (45). An AP-1 inhibitor and shRNAs against AP-1 TF Jun decreased PD-L1 and CD86 expression in NA-AML cells. Once NA-sAML cells were transplanted into syngeneic recipients, NA-derived T cells were not detectable (45). Host-derived wildtype T cells overexpressed inhibitory immune checkpoint receptors, PD-1 and TIGIT, and displayed an exhausted T cell phenotype (45). Combined inhibition of MEK and pan-BET proteins led to downregulation of AP-1 TF expression, mitigation of the suppressive immune microenvironment, enhancement of CD8 T cell cytotoxicity, and prolonged survival in NA-sAML mice. Given the distinct phenotypes observed in Asxl1−/− and Asxl1tm/+ mice, it would be interesting to determine if NrasG12D/+; Asxl1tm/+ mice display similar phenotypes as NA mice and if the underlying mechanisms are distinct.
Treatment response of ASXL1 mutant leukemic cells
Recent studies revealed that patients with ASXL1 mutations are associated with distinct sensitivity to drug treatment. Hypomethylating agents (HMA) have been a standard treatment for CMML. A retrospective study of 177 CMML patients revealed that ASXL1 mutations predict a lower overall response rate to HMAs (azacitidine or decitabine) (ASXL1mt 42% versus ASXL1wt 60%, p = 0.02) (58). This clinical observation may be explained, at least partially, by the increased expression of anti-apoptotic gene BCL2 and elevated global cytosine methylation in ASXL1mt leukemia cells (59). Not surprisingly, combined veneteoclax, a selective BCL2 inhibitor, and azacitidine effectively inhibit ASXL1mt leukemia cell growth in vitro (59). This combination was approved by FDA to treat AML in 2018. It would be interesting to see if it treats ASXL1mt CMML patients better than HMA alone.
In summary, ASXL1 hotspot mutations around codon G646 are prevalent in myeloid diseases but rarely identified in CHIP, suggesting that they are highly pathogenic and confers higher risk to develop myeloid diseases. The nature of these mutations remains elusive. While mouse genetic studies suggest that they are loss-of-function and gain-of-new function at the physiological level, overexpression studies in transgenic mice and cell lines indicate that they are dominant negative and gain-of-function. Additional rigorous investigations are needed to provide a definitive answer to this question. ASXL1 mutations are associated with poor prognosis in all myeloid diseases, perhaps due to the reduced response to current treatment options (e.g. HMA in CMML). We recently discover that concurrent ASXL1 and NRAS mutations define dismal outcomes in CMML patients. Correspondingly, Asxl1 loss cooperates with oncogenic Nras in mice to reprogram immune microenvironment and drive leukemic transformation. Our study provides a strong rational to develop combined targeted therapy and immunotherapy for treating leukemia patients with concurrent ASXL1 and NRAS mutations.
References
- 1.Sinclair DA et al. , The Additional sex combs gene of Drosophila encodes a chromatin protein that binds to shared and unique Polycomb group sites on polytene chromosomes. Development 125, 1207–1216 (1998). [DOI] [PubMed] [Google Scholar]
- 2.Schuettengruber B, Bourbon HM, Di Croce L, Cavalli G, Genome Regulation by Polycomb and Trithorax: 70 Years and Counting. Cell 171, 34–57 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Fisher CL et al. , Additional sex combs-like 1 belongs to the enhancer of trithorax and polycomb group and genetically interacts with Cbx2 in mice. Dev Biol 337, 9–15 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fisher CL, Randazzo F, Humphries RK, Brock HW, Characterization of Asxl1, a murine homolog of Additional sex combs, and analysis of the Asx-like gene family. Gene 369, 109–118 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Fisher CL, Berger J, Randazzo F, Brock HW, A human homolog of Additional sex combs, ADDITIONAL SEX COMBS-LIKE 1, maps to chromosome 20q11. Gene 306, 115–126 (2003). [DOI] [PubMed] [Google Scholar]
- 6.Scheuermann JC et al. , Histone H2A deubiquitinase activity of the Polycomb repressive complex PR-DUB. Nature 465, 243–247 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abdel-Wahab O et al. , ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer Cell 22, 180–193 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Asada S et al. , Mutant ASXL1 cooperates with BAP1 to promote myeloid leukaemogenesis. Nat Commun 9, 2733 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Balasubramani A et al. , Cancer-associated ASXL1 mutations may act as gain-of-function mutations of the ASXL1-BAP1 complex. Nat Commun 6, 7307 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamamoto K et al. , A histone modifier, ASXL1, interacts with NONO and is involved in paraspeckle formation in hematopoietic cells. Cell Rep 36, 109576 (2021). [DOI] [PubMed] [Google Scholar]
- 11.Baskind HA et al. , Functional conservation of Asxl2, a murine homolog for the Drosophila enhancer of trithorax and polycomb group gene Asx. PLoS One 4, e4750 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Farber CR et al. , Mouse genome-wide association and systems genetics identify Asxl2 as a regulator of bone mineral density and osteoclastogenesis. PLoS Genet 7, e1002038 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Izawa T et al. , ASXL2 Regulates Glucose, Lipid, and Skeletal Homeostasis. Cell Rep 11, 1625–1637 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li J et al. , Loss of Asxl2 leads to myeloid malignancies in mice. Nat Commun 8, 15456 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Micol JB et al. , ASXL2 is essential for haematopoiesis and acts as a haploinsufficient tumour suppressor in leukemia. Nat Commun 8, 15429 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duployez N et al. , Unlike ASXL1 and ASXL2 mutations, ASXL3 mutations are rare events in acute myeloid leukemia with t(8;21). Leuk Lymphoma 57, 199–200 (2016). [DOI] [PubMed] [Google Scholar]
- 17.Dawoud AAZ, Tapper WJ, Cross NCP, Clonal myelopoiesis in the UK Biobank cohort: ASXL1 mutations are strongly associated with smoking. Leukemia 34, 2660–2672 (2020). [DOI] [PubMed] [Google Scholar]
- 18.van Zeventer IA et al. , Prevalence, predictors, and outcomes of clonal hematopoiesis in individuals aged >/=80 years. Blood Adv 5, 2115–2122 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Genovese G et al. , Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 371, 2477–2487 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xie M et al. , Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med 20, 1472–1478 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jaiswal S et al. , Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 371, 2488–2498 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jaiswal S, Clonal hematopoiesis and nonhematologic disorders. Blood 136, 1606–1614 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jaiswal S et al. , Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N Engl J Med 377, 111–121 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Asada S, Kitamura T, Clonal hematopoiesis and associated diseases: A review of recent findings. Cancer Sci 112, 3962–3971 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen TC et al. , Dynamics of ASXL1 mutation and other associated genetic alterations during disease progression in patients with primary myelodysplastic syndrome. Blood cancer journal 4, e177 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Elena C et al. , Integrating clinical features and genetic lesions in the risk assessment of patients with chronic myelomonocytic leukemia. Blood 128, 1408–1417 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Itzykson R et al. , Prognostic score including gene mutations in chronic myelomonocytic leukemia. J Clin Oncol 31, 2428–2436 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Patnaik MM et al. , ASXL1 and SETBP1 mutations and their prognostic contribution in chronic myelomonocytic leukemia: a two-center study of 466 patients. Leukemia 28, 2206–2212 (2014). [DOI] [PubMed] [Google Scholar]
- 29.Abdel-Wahab O, Kilpivaara O, Patel J, Busque L, Levine RL, The most commonly reported variant in ASXL1 (c.1934dupG;p.Gly646TrpfsX12) is not a somatic alteration. Leukemia 24, 1656–1657 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Montes-Moreno S et al. , Clinical molecular testing for ASXL1 c.1934dupG p.Gly646fs mutation in hematologic neoplasms in the NGS era. PLoS One 13, e0204218 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alberti MO et al. , Discriminating a common somatic ASXL1 mutation (c.1934dup; p.G646Wfs*12) from artifact in myeloid malignancies using NGS. Leukemia 32, 1874–1878 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Metzeler KH et al. , ASXL1 mutations identify a high-risk subgroup of older patients with primary cytogenetically normal AML within the ELN Favorable genetic category. Blood 118, 6920–6929 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thol F et al. , Prognostic significance of ASXL1 mutations in patients with myelodysplastic syndromes. J Clin Oncol 29, 2499–2506 (2011). [DOI] [PubMed] [Google Scholar]
- 34.Patnaik MM et al. , DNMT3A mutations are associated with inferior overall and leukemia-free survival in chronic myelomonocytic leukemia. Am J Hematol 92, 56–61 (2017). [DOI] [PubMed] [Google Scholar]
- 35.Binder M et al. , Oncogenic gene expression and epigenetic remodeling of cis-regulatory elements in ASXL1-mutant chronic myelomonocytic leukemia. Nat Commun 13, 1434 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abdel-Wahab O et al. , Deletion of Asxl1 results in myelodysplasia and severe developmental defects in vivo. J Exp Med 210, 2641–2659 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Uni M et al. , Modeling ASXL1 mutation revealed impaired hematopoiesis caused by derepression of p16Ink4a through aberrant PRC1-mediated histone modification. Leukemia 33, 191–204 (2019). [DOI] [PubMed] [Google Scholar]
- 38.Bohring A et al. , Severe end of Opitz trigonocephaly (C) syndrome or new syndrome? Am J Med Genet 85, 438–446 (1999). [DOI] [PubMed] [Google Scholar]
- 39.Bedoukian E, Copenheaver D, Bale S, Deardorff M, Bohring-Opitz syndrome caused by an ASXL1 mutation inherited from a germline mosaic mother. Am J Med Genet A 176, 1249–1252 (2018). [DOI] [PubMed] [Google Scholar]
- 40.Seiter K, Htun K, Baskind P, Liu Z, Acute myeloid leukemia in a father and son with a germline mutation of ASXL1. Biomark Res 6, 7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hsu YC et al. , The distinct biological implications of Asxl1 mutation and its roles in leukemogenesis revealed by a knock-in mouse model. J Hematol Oncol 10, 139 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nagase R et al. , Expression of mutant Asxl1 perturbs hematopoiesis and promotes susceptibility to leukemic transformation. J Exp Med 215, 1729–1747 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang H et al. , Gain of function of ASXL1 truncating protein in the pathogenesis of myeloid malignancies. Blood 131, 328–341 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Inoue D et al. , A novel ASXL1-OGT axis plays roles in H3K4 methylation and tumor suppression in myeloid malignancies. Leukemia 32, 1327–1337 (2018). [DOI] [PubMed] [Google Scholar]
- 45.You X et al. , Asxl1 loss cooperates with oncogenic Nras in mice to reprogram the immune microenvironment and drive leukemic transformation. Blood 139, 1066–1079 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Inoue D et al. , Truncation mutants of ASXL1 observed in myeloid malignancies are expressed at detectable protein levels. Exp Hematol 44, 172–176 e171 (2016). [DOI] [PubMed] [Google Scholar]
- 47.Bejar R et al. , Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med 364, 2496–2506 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schnittger S et al. , ASXL1 exon 12 mutations are frequent in AML with intermediate risk karyotype and are independently associated with an adverse outcome. Leukemia 27, 82–91 (2013). [DOI] [PubMed] [Google Scholar]
- 49.Inoue D et al. , SETBP1 mutations drive leukemic transformation in ASXL1-mutated MDS. Leukemia 29, 847–857 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Makishima H et al. , Somatic SETBP1 mutations in myeloid malignancies. Nat Genet 45, 942–946 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meggendorfer M et al. , SETBP1 mutations occur in 9% of MDS/MPN and in 4% of MPN cases and are strongly associated with atypical CML, monosomy 7, isochromosome i(17)(q10), ASXL1 and CBL mutations. Leukemia 27, 1852–1860 (2013). [DOI] [PubMed] [Google Scholar]
- 52.Gelsi-Boyer V et al. , Mutations in ASXL1 are associated with poor prognosis across the spectrum of malignant myeloid diseases. J Hematol Oncol 5, 12 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pratcorona M et al. , Acquired mutations in ASXL1 in acute myeloid leukemia: prevalence and prognostic value. Haematologica 97, 388–392 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gu Z et al. , Loss of EZH2 Reprograms BCAA Metabolism to Drive Leukemic Transformation. Cancer Discov 9, 1228–1247 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang P et al. , Chromatin regulator Asxl1 loss and Nf1 haploinsufficiency cooperate to accelerate myeloid malignancy. J Clin Invest 128, 5383–5398 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Guo Y et al. , Reduced BAP1 activity prevents ASXL1 truncation-driven myeloid malignancy in vivo. Leukemia 32, 1834–1837 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Saika M et al. , ASXL1 and SETBP1 mutations promote leukaemogenesis by repressing TGFbeta pathway genes through histone deacetylation. Sci Rep 8, 15873 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Duchmann M et al. , Prognostic Role of Gene Mutations in Chronic Myelomonocytic Leukemia Patients Treated With Hypomethylating Agents. EBioMedicine 31, 174–181 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rahmani NE et al. , ASXL1 mutations are associated with distinct epigenomic alterations that lead to sensitivity to venetoclax and azacytidine. Blood Cancer J 11, 157 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]