

Abstract
Antiviral therapeutics to treat SARS‐CoV‐2 are needed to diminish the morbidity of the ongoing COVID‐19 pandemic. A well‐precedented drug target is the main viral protease (MPro), which is targeted by an approved drug and by several investigational drugs. Emerging viral resistance has made new inhibitor chemotypes more pressing. Adopting a structure‐based approach, we docked 1.2 billion non‐covalent lead‐like molecules and a new library of 6.5 million electrophiles against the enzyme structure. From these, 29 non‐covalent and 11 covalent inhibitors were identified in 37 series, the most potent having an IC50 of 29 and 20 μM, respectively. Several series were optimized, resulting in low micromolar inhibitors. Subsequent crystallography confirmed the docking predicted binding modes and may template further optimization. While the new chemotypes may aid further optimization of MPro inhibitors for SARS‐CoV‐2, the modest success rate also reveals weaknesses in our approach for challenging targets like MPro versus other targets where it has been more successful, and versus other structure‐based techniques against MPro itself.
Keywords: major protease, SARS‐COV‐2, structure‐based inhibitor, discoverydockinganti‐viral
1. INTRODUCTION
SARS‐CoV‐2 encodes two cysteine proteases that have essential roles in hydrolyzing viral polyproteins into nonstructural proteins, enabling virus replication. The main protease (MPro, also known as 3CL protease) cleaves 11 different sites in the viral polyprotein (Jin et al., 2020; Pillaiyar et al., 2016). While MPro is highly conserved across other coronaviruses such as SARS‐CoV‐1 and MERS, it has no close human homolog (Anand et al., 2003; Rut et al., 2021; Zhang, Lin, Kusov, et al., 2020). This makes it attractive for potential pan‐coronavirus targeting, and for selective action.
MPro is the fifth nonstructural protein (Nsp5) encoded by SARS‐CoV‐2 and is a homodimeric cysteine protease with a catalytic diad comprised of Cys145 and His41. MPro has a P1 primary specificity determinant of glutamine and a preference for aliphatic residues in the P4 and P2 positions, while alanine and serine are preferred in the P1′ position (Schechter & Berger, 1967) (Figure 1D). The catalytic cycle is typical of many cysteine proteases, with the catalytic Cys145 primed by proton transfer to His41 and formation of an acyl enzyme intermediate via nucleophilic attack of Cys145 at the scissile peptide carbonyl function. The thioester intermediate is then hydrolyzed by an attacking water to free the catalytic cysteine and initiate another catalytic cycle (Wang et al., 2020).
FIGURE 1.
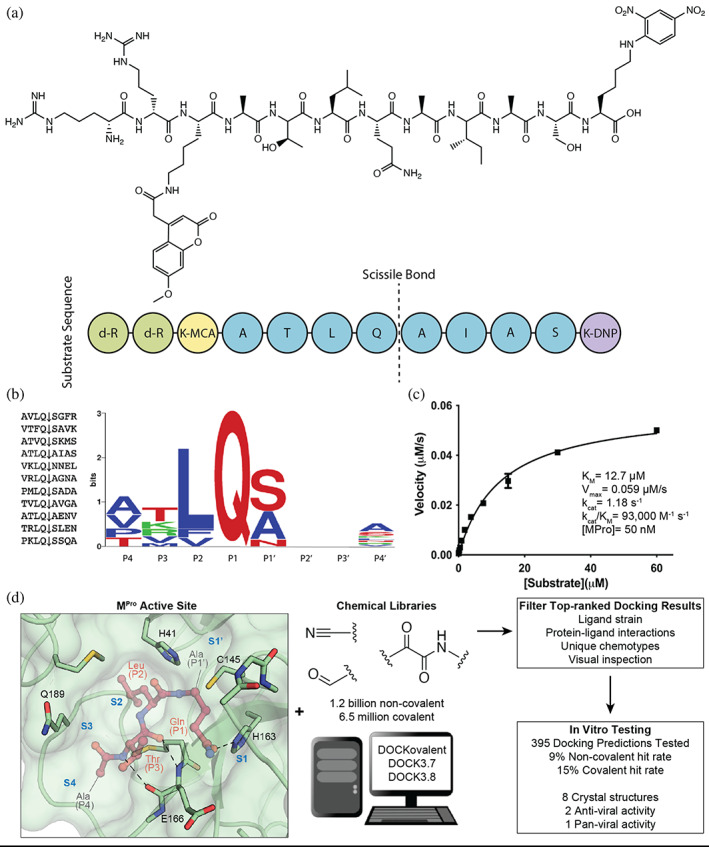
Substrate design and assay development allows structure‐based inhibitor discovery. (a) The chemical structure of the optimized NSP7 substrate shown as a schematic (top) of the substrate sequence highlights the role of each residue (bottom). The substrate contains the P4‐P4′ NSP7 extended substrate sequence (blue), the fluorophore (yellow), the fluorescent quencher (purple), and the residues for increasing solubility (green). (b) A list of the viral polypeptide NSP sequences (P4‐P4′) that are cleaved by MPro (left). The sequence logo highlighting the substrate specificity of MPro, yielding a P4‐P4′ consensus sequence: ATLQ(S/A)XXA (right). (c) The Michaelis‐Menten kinetics for the NSP7 substrate with MPro yield parameters indicative of an optimized, efficient substrate. (d) SARS‐CoV‐2 MPro active site (PDB 6Y2G) (Zhang, Lin, Sun, et al., 2020) (green; sub‐pockets S1′, S1, S2, S3, S4), shown here with substrate preferences (pink; P1′, P1, P2, P3, P4) (modeled after PDB 3SNE) (Zhu et al., 2011), was used to dock 1.2 billion non‐covalent molecules and 6.5 million electrophile molecules. Top‐ranked molecules were filtered and 395 were synthesized for in vitro testing. Some docking hits were prioritized for compound optimization, crystallography, pan‐viral enzymatic activity, and cell‐based antiviral activity. For C, experiments were performed in triplicate.
The therapeutic potential of MPro inhibitors was substantiated by the approval of Paxlovid in December 2021. The treatment combines nirmatrelvir, which covalently inhibits MPro, with ritonavir, which slows nirmatrelvir's metabolism (Owen et al., 2021). Nirmatrelvir was optimized from PF‐00835231, an inhibitor of the SARS‐CoV‐1 MPro developed in response to the 2002 SARS outbreak (Hoffman et al., 2020). Meanwhile, other potent MPro inhibitors are advancing through the drug development pipeline. Among them is the orally active MPro inhibitor S‐217622 (Unoh et al., 2022), which has entered clinical trials. Other inhibitors show much promise (Dai et al., 2020; Ge et al., 2022; Lu et al., 2022; Ma et al., 2020; Moon et al., 2022; Qiao et al., 2021; Vuong et al., 2021; Zhang, Lin, Kusov, et al., 2020; Zhang, Spasov, et al., 2021; Zhang, Stone, et al., 2021), including a non‐covalent MPro inhibitor from the international Covid‐19 Moonshot consortium, an advanced pre‐clinical candidate (Chodera et al., 2020; Douangamath et al., 2020; The COVID Moonshot Consortium et al., 2020), and more experimental molecules that show promise (Luttens et al., 2022).
Notwithstanding these successes, both the resistance that may be expected to emerge (Flynn et al., 2022; Shaqra et al., 2022) and the inevitable liabilities of the early drugs support the discovery of new scaffolds. Accordingly, we targeted the structure of MPro for large library docking, seeking new starting points for lead discovery. Docking a library of over 1.2 billion “tangible” (make‐on‐demand) lead‐like molecules and 6.5 million tangible lead‐like electrophiles from Enamine REAL space (https://enamine.net/compound-collections/real-compounds) led to MPro inhibitors from 37 scaffolds, with affinities ranging from the low μM to 200 μM. Crystal structures for eight of the new inhibitors bound to MPro largely confirmed the docking predictions, while cell‐based antiviral activity for two of the new inhibitors supports their further optimization (Figure 1).
Crucial to inhibitor testing was the design and synthesis of an optimal substrate, as was done previously for SARS CoV MPro (Figure 1) (Goetz et al., 2007). The endogenous Nsp substrates of MPro were compiled and a consensus sequence was observed that closely matched the individual sequence of the Nsp7 cleavage site (ATLQAIAS) (Figure 1B). This sequence was flanked with an N‐terminal Lysine‐MCA fluorophore and a C‐terminal DNP‐quencher. Noting the preference for nonpolar residues at multiple sites, we were concerned that this substrate would have low solubility. Accordingly, two D‐Arginines were coupled to the N‐terminal Lysine‐MCA to increase solubility (Figure 1A). This Nsp7‐like substrate yielded a favorable Km of 12 μM and a kcat/Km of 93,000 M−1 s−1 , 3.5‐fold better than that of the commonly used commercial substrate (Nsp4: AVLQSGFR; kcat/Km = 26,500 M−1 s−1) (Jin et al., 2020); this substrate was used in all enzyme inhibition assays (Figure 1C). This more efficient Nsp7‐like peptide is readily synthesized and provides the field with an optimized MPro substrate.
While the multiple chemotypes explored here, and their crystallographic structures, may template further optimization of MPro inhibitors, a feature of these studies was docking hit rates between 7% and 15%, with hits often in the mid‐μM range. These rates and affinities are substantially worse than observed in many GPCRs, integral membrane proteins, transporters, and enzymes like β‐lactamase (Alon et al., 2021; Carlsson et al., 2011; Fink et al., 2022; Kaplan et al., 2022; Levit Kaplan et al., 2022; Lyu et al., 2019; Manglik et al., 2016; Singh et al., 2022; Stein et al., 2020; Wang et al., 2018). Meanwhile, the optimized affinities reached here were meaningfully weaker than those achieved by approaches such as fragment‐based discovery, both against MPro itself and against other SARS‐2 targets, like macrodomain (Gahbauer et al., 2022; Schuller et al., 2021). What we might learn from these differences for future studies will be discussed.
2. RESULTS
2.1. Assay development and substrate design
In early proof‐of‐concept testing, we observed an intolerance of MPro activity to high concentrations of DMSO, introduced when evaluating inhibitors and substrate itself from DMSO stocks, perhaps reflecting oxidation of the catalytic cysteine. The increased solubility of the D‐Arginine‐modified substrate mitigated the DMSO effect by reducing the volume of DMSO needed in substrate aliquots. In addition, we found that ethanol and acetonitrile were better tolerated by the enzyme, though these solvents have issues with volatility (Figure S1A). These observations highlight the importance of controlling and minimizing the addition of organic solvents in MPro activity assays and provide alternatives when DMSO is unsuitable. We also found that small amounts of non‐ionic detergent were crucial for retaining MPro activity in our in vitro assays. Removing the 0.05% Tween‐20 we used in our assays resulted in no observed substrate cleavage. Activity could be recovered by increasing addition of bovine serum albumin (BSA); these effects may reflect MPro sequestration by the polymer of the reaction‐well and highlight the need of detergent or enzyme stabilizing additives (Figure S1B). This is similar to literature (Moghadasi, Esler, et al., 2022; Rut et al., 2021; Zaidman et al., 2021).We tested three previously reported compounds under our assay conditions. The covalent inhibitor nirmatrelvir had a similar IC50 as reported (Owen et al., 2021), and while two non‐covalent inhibitors (PET‐UNK‐29afea89‐2 and VLA‐UCB‐1dbca3b4‐15) had IC50 values 2‐ to 5‐fold higher versus reported values (The COVID Moonshot Consortium et al., 2020), this likely reflects simply different substrate concentrations and Km values in the different assays (Table S1). These rates provide a reference for comparing the different inhibitors.
2.2. Non‐covalent docking screen and compound optimization for MPro inhibitors
Seeking new inhibitors, we began with a SARS‐CoV‐2 MPro crystal structure in complex with an α‐ketoamide covalent inhibitor (PDB 6Y2G) (Zhang, Lin, Sun, et al., 2020). To define hot‐spots for ligand docking in the active site, we modeled a complex of SARS‐CoV‐2 MPro bound to a non‐covalent SARS‐CoV MPro inhibitor (PubChem SID87915542) (Jacobs et al., 2013) (non‐covalent inhibitor complex crystal structures of the enzyme from SARS‐CoV‐2 were at that time unavailable). The crystal structure of the non‐covalently ligated SARS‐CoV MPro (PDB 3V3M) (Jacobs et al., 2013) was structurally aligned onto the SARS‐CoV‐2 structure, the atomic coordinates of the α‐ketoamide inhibitor were replaced with those of the non‐covalent SARS‐CoV MPro inhibitor SID87915542 (IC 50 = 4.8 μM) (Jacobs et al., 2013), and the complex was energy‐minimized (Methods). After calibration of the docking parameters (Bender et al., 2021) (Methods), approximately 225 million neutral molecules, mainly from the lead‐like subset of the ZINC15 library (Sterling & Irwin, 2015) (molecular weight (MWT) ranging from 250 to 350 amu and clogP ≤4.5) were docked against MPro. Another 110 million molecules with 350 < MWT ≤ 500 were docked in a separate screen. Docked molecules were filtered for intramolecular strain (Gu et al., 2021) and selected for their ability to hydrogen bond with Gly143, His163, or Glu166, and to make favorable non‐polar contacts with Met49 and Asp187. Ultimately, 220 molecules were prioritized, of which 194 (88%) were successfully synthesized by Enamine. Enzymatically, compounds were first tested at a concentration of 100 μM using the fluorescence‐based substrate cleavage assay, and 19 showed >30% inhibition of enzyme activity and were prioritized for full concentration‐response curves. Concentration‐response experiments revealed 12 molecules with IC50 values ≤300 μM, a hit rate of 6% (12 hits/194 molecules tested); IC50s ranged from 97 to 291 μM (Tables 1 and S1; Figures S2.1 and S2.2).
TABLE 1.
Hits from the first non‐covalent docking screen.
| Chemical structure | Compound ID | IC50 (μM) (solvent) | Chemical Structure | Compound ID | IC50 (μM) (solvent) |
|---|---|---|---|---|---|
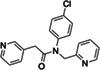
|
ZINC346371112 |
214 (DMSO) 98 (ACN) |
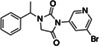
|
ZINC813360541 |
275 (DMSO) 94 (ACN) |
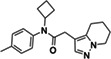
|
ZINC894230117 |
225 (DMSO) 164 (ACN) |
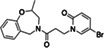
|
ZINC553840273 |
200 (DMSO) 88 (ACN) |
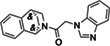
|
ZINC1339780091 |
224 (DMSO) 121 (ACN) |
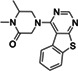
|
ZINC336912805 |
250 (DMSO) 177 (ACN) |
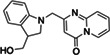
|
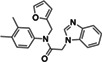
ZINC433294115 | 97 (DMSO) |
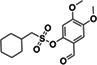
|
ZINC271072260 |
115 (DMSO) 143 (ACN) |
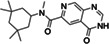
|
ZINC618071006 |
290 (DMSO) 200 (EtOH) |
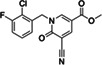
|
ZINC338540162 |
281 (DMSO) <30 (ACN) |
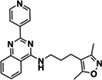
|
ZINC301553312 |
122 (DMSO) 63 (EtOH) |
|
ZINC915668084 |
291 (DMSO) 184 (ACN) |
As DMSO had been observed to lower enzyme activity, the actives, initially tested from 10 mM DMSO stocks, were re‐tested against MPro from 30 mM acetonitrile (ACN) or ethanol (EtOH) stocks. Eleven compounds showed clear dose‐response with IC50 values ranging from 30 to 200 μM. Although covalent docking was not employed in this campaign, we noted three initial docking hits (ZINC338540162: IC50[ACN] = 30 μM, ZINC271072260: IC50[ACN] = 143 μM and ZINC795258204: IC50[DMSO] = 177 μM) could, in principle, inhibit MPro covalently as they contain warheads (nitrile, aldehyde) known to react with catalytic cysteines. Several initial docking hits were tested for colloidal aggregation using dynamic light scattering (DLS) and off‐target counter screens against malate dehydrogenase (MDH) and AmpC β‐lactamase (McGovern et al., 2003; O'Donnell et al., 2021) (Figure S3). In DLS experiments, some scattering higher than 106 was observed indicating potential aggregation. While a few compounds, for example, ‘3312 showed unspecific inhibition of MDH, off‐target activities were reversed by addition of 0.01% Triton X‐100. As the inhibition of MPro was observed in the presence of 0.05% Tween‐20, an even stronger disruptor of colloidal aggregation than 0.01% Triton‐X 100, we deemed the weak aggregation of these compounds irrelevant to their activity on MPro.
We focused on four initial hits (ZINC346371112: IC50[ACN] = 98 μM, ZINC301553312: IC50[EtOH] = 63 μM, ZINC813360541: IC50[ACN] = 90 μM and ZINC553840273: IC50[ACN] = 88 μM) for structure‐based optimization. We used the SmallWorld search engine (NextMove Software, Cambridge UK) (Irwin et al., 2020) to identify purchasable analogs of these inhibitors within a 12 billion compound version of the REAL library (https://enamine.net/compound‐collections/real‐compounds/real‐space‐navigator), docking each analog into the MPro structure to assess complementarity. Between 10 and 20 analogs of each of the four inhibitors were selected for testing in the initial round of optimization (Figure 2, Table S1). For two initial hits, ‘0541 and ‘0273, more potent analogs were identified in two to three rounds of this analog‐by‐catalog approach (Table S1). The ‘0273 analogs Z4924562413 and Z4946671001 had IC50 values of 13 and 5 μM, respectively (Figure 2A). Analogs of the initial docking hit ‘0541, such as Z4929615577 and Z4929616137, reached similar potencies of 10 and 8 μM, respectively (Figure 2G).
FIGURE 2.
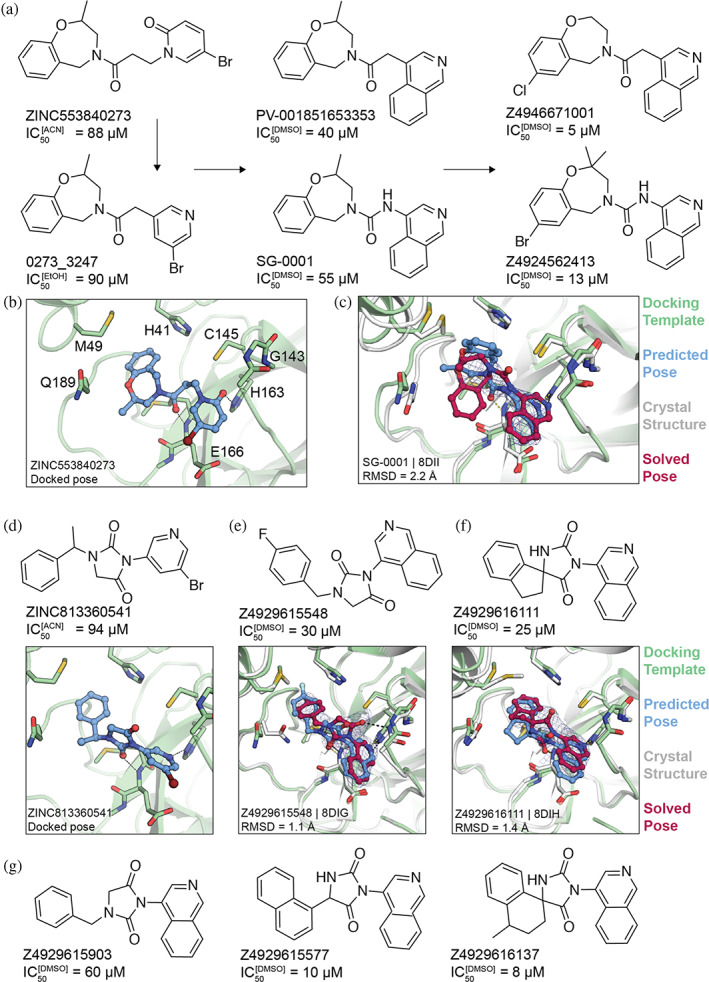
Non‐covalent compound optimization to low‐μM potencies. (a) Progression of the ‘0273 scaffold. (b) Predicted binding pose of ‘0273. (c) Comparison of crystal structure (grey protein, red compound) and docked complex (green protein, blue compound) of SG‐0001 (PDB 8DII). (d) Predicted binding pose of ‘0541. (e), (f) Comparison of crystal structures and docked complexes of ‘5548 (PDB 8DIG) and ‘6111 (PDB 8DIH), respectively. (g) Additional ‘0541 analogs with improved affinities. The 2fo‐fc ligand density maps (blue contour) are shown at 1 σ. Hungarian root mean square deviations (RMSD) were calculated with DOCK6.
2.3. Crystal structures of the non‐covalent inhibitors
To investigate how the docked poses of the new inhibitors corresponded to their true binding modes, and to inform further optimization, crystal structures of three of the optimized non‐covalent inhibitors were determined with resolutions ranging from 2.12 to 2.59 Å. For the ‘0273 analog, SG‐0001 (IC50 = 55 μM, Figure 2A–C), the crystal structure revealed only moderate density for the ligand. Still, the predicted binding pose compared well with the experimentally determined pose, with a Hungarian (symmetry corrected) root mean square deviation (RMSD) of 2.2 Å. The isoquinoline group of SG‐0001 is inserted in the S1 subpocket, hydrogen‐bonding with His163; this was also predicted for the pyridone carbonyl in the parent molecule ‘0273 (Figures 2B,C and S4). However, the tetrahydrobenzoxazepine ring, predicted to bind in the S2 subpocket in ‘0273, appeared much less buried in the SG‐0001 experimental structure. The crystal structure of MPro in complex with the ‘0541 analog ‘5548 superimposed with high fidelity to the docking‐predicted pose, with an RMSD of 1.1 Å (Figures 2E and S4). Here, the compound's hydantoin core hydrogen bonds with the backbone amine of Glu166 and Gly143. In addition, the crystal structure of MPro in complex with ‘6111 confirms the predicted biding pose (RMSD = 1.4 Å) with the isoquinoline placed in the S1 subpocket and the hydrophobic spirocyclic indane group occupying the S2 pocket (Figures 2F and S4).
2.4. A second docking screen for non‐covalent inhibitors of MPro
As our studies progressed, other groups identified potent inhibitors with scaffolds resembling our own (Luttens et al., 2022). We therefore performed a second docking campaign, seeking to incorporate insights emerging from our own results and those from other studies (Methods) emphasizing the discovery of novel chemotypes.
The new docking screen targeted the SARS‐CoV‐2 MPro crystal structure in complex with MAT‐POS‐b3e365b9‐1 (MPro‐x11612.pdb) (The COVID Moonshot Consortium et al., 2020), a non‐covalent ligand reported by the COVID‐19 Moonshot consortium. Compared to the previous docking template (PDB 6Y2G), the MAT‐POS‐b3e365b9‐1‐bound site is slightly smaller, with the 2‐turn alpha helix between Thr45 and Leu50, and the loop between Arg188 and Ala191, shifted inwards by roughly 2 Å, constricting the P2 sub‐pocket. After calibration of docking parameters, ensuring the model prioritizes 15 previously reported MPro inhibitors against different decoy sets (Bender et al., 2021; Stein et al., 2021), we used the ZINC library (https://cartblanche22.docking.org/) to dock 862 million neutral compounds with 18‐29 non‐hydrogen atoms from the Enamine REAL database (Methods).
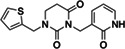
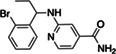
High‐ranking docked molecules were filtered for novelty by removing those with ECFP4‐based Tanimoto coefficients (Tc) greater than 0.35–1,716 SARS‐CoV‐2 MPro inhibitors (Methods). Roughly 9,500 of these were graphically evaluated for favorable contacts, and 146 compounds were de novo synthesized by Enamine Ltd. Of these, 17 inhibited MPro with IC50 values ≤200 μM (Table 2; Figures S2.3 and S2.4) for a hit rate of 12% (17 hits/146 tested). To our knowledge, none of the new actives fell into scaffolds that have been previously reported for MPro. Compared to the first docking screen, several initial hits from the second screen showed slightly higher activity, such as Z3535317212, with an IC50 value of 29 μM. For ‘7212, the docked pose suggests hydrogen bonds between the compound's dihydrouracil core and Glu166 as well as Gly143, in addition to hydrogen bonds between the compound's pyridinol group (Figure S2.3). Five docking hits (Z5420225795: IC50 = 40 μM, Z1669286714: IC50 = 110 μM, Z1355254448: IC50 = 110 μM, ZINC5420738300: IC50 = 160 μM, Z2195811405: IC50 ~200 μM) share a common ketoamide functional group predicted to form one hydrogen bond to Glu166; however, we note that ketoamide might also inhibit MPro through covalent linkage to Cys145. As in the first docking campaign, hits were tested for colloidal aggregation. A few compounds (‘7900, ‘8488, ‘1405, ‘8300) had higher DLS scattering or caused >50% inhibition of MDH in the absence of detergent, which was reversed by 0.01% Triton X‐100 (Figure S3). Here again, we believe that the measured activities of those compounds at MPro, in presence of 0.05% Tween‐20, originate from specific on‐target actions, but care should be taken when using related scaffolds in detergent‐free experiments.
TABLE 2.
Hits from the second non‐covalent docking screen.
| Chemical structure | Compound ID | IC50 (μM) | Chemical Structure | Compound ID | IC50 (μM) |
|---|---|---|---|---|---|
|
Z3535317212 | 29 |
|
Z1425997900 | 110 |
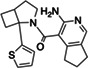
|
Z4124468376 | 33 |
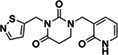
|
Z3541227016 | 130 |
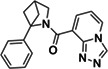
|
Z3555684465 | 33 |
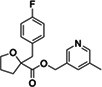
|
Z3382155230 | 140 |
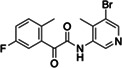
|
Z5420225795 | 40 |
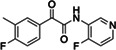
|
Z5420738300 | 160 |
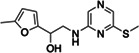
|
Z1716270280 | 60 |
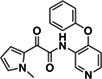
|
Z2195811405 | 200 |
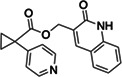
|
Z5420228488 | 60 |
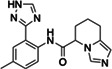
|
Z4289708272 | 200 |
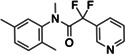
|
Z3079159560 | 90 |
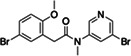
|
Z5385490967 | 200 |
|
Z1669286714 | 110 |
|
Z4335534517 | 200 |
|
Z1355254448 | 110 |
Taken together, the active compounds from this campaign explored ten different scaffold classes with IC50 values better than 150 μM. These scaffolds represent new points of departure for MPro inhibitor discovery.
2.5. A covalent docking screen targeting MPro Cys145
In addition to non‐covalent inhibitors, we also sought electrophiles that could covalently modify the catalytic Cys145. We searched the 1.4 billion molecules in the ZINC15/ZINC20 (Irwin et al., 2020; Sterling & Irwin, 2015) databases for three Cys‐reactive covalent warheads: aldehydes, nitriles, and α‐ketoamides. Precedence for covalent inhibitor design is seen with a range of targets, including KRAS G12C anticancer therapeutics (sotorasib, GDC‐6036) (Canon et al., 2019; Purkey, 2022), FGFR4 inhibitor (roblitinib) with an reversible aldehyde warhead in clinical trials (Fairhurst et al., 2020), antivirals for HCV with ketoamides (boceprevir, telaprevir) (Lin et al., 2006; Venkatraman, 2012), a sickle cell hemoglobin stabilizer aldehyde inhibitor (GBT440) (Metcalf et al., 2017), and the SARS‐CoV‐2 nitrile MPro covalent inhibitor nirmatrelvir (Owen et al., 2021). While aldehydes, especially, are reactive—if reversible—electrophiles, previous work has demonstrated the viability of electrophile swapping to less reactive groups, or to irreversible groups for development goals (Adams et al., 1998; Boike et al., 2022; Vankadara et al., 2022). Dockable 3D molecules were built for covalent docking with DOCKovalent (London et al., 2014; Wan et al., 2020) (Methods). The molecules and their DOCKovalent files for the final 6.5 million molecules are openly available at http://covalent2022.docking.org.
We then docked 3.6 million nitriles, 1.5 million aldehydes, and 1.4 million α‐ketoamides against MPro (PDB 6Y2G) (Zhang, Lin, Sun, et al., 2020). The top‐ranked molecules were filtered for torsional strain (Gu et al., 2021), for favorable enzyme interactions, and clustered for chemical diversity using an ECFP4‐based best first clustering algorithm (Methods). Remaining molecules were visually prioritized for favorable interactions with His41, Cys145, Gly143, Thr26, or Glu166. Ultimately, 35 aldehydes, 41 nitriles, and 21 α‐ketoamides were selected for synthesis, of which 27, 31, 16, respectively, were successfully made and tested for activity against MPro (Table S1). Those compounds with single‐point percent inhibition >50% at 100 μM—a more stringent criterion than we had used earlier—were prioritized for full concentration‐dose‐response assays.
Defining active compounds as molecules with IC50 ≤ 150 μM, the hit rate for covalent docking was 15% (11 actives/74 compounds tested); the most potent had an IC50 of 20 μM (Figures 3 and S5). Eight others had IC50 values 25–100 μM. Initial nitriles and aldehyde docking hits had activities as low as 20 μM in compound ‘5103, and 55 μM in compound ‘3620, respectively. None of the α‐ketoamides were potent enough to be considered active (Table S1). Initial docking hits were evaluated for potential MPro inhibition through colloidal aggregation as described above (Figure S3). Some DLS scattering or non‐specific inhibition is observed in the AmpC and MDH enzymatic assays. However, adding 0.01% Triton X‐100 in the MDH inhibition assay largely recovered enzymatic activity and eliminated any non‐specific inhibition, suggesting that the measured activities in the detergent‐containing MPro enzymatic assays are not caused by aggregation (also confirmed by subsequent crystallography, see below). We note that while the covalent nature of the aldehydes was subsequently confirmed by crystallography (below), it remains possible that the nitriles, though modeled to be reversible covalent, were in fact non‐covalent.
FIGURE 3.
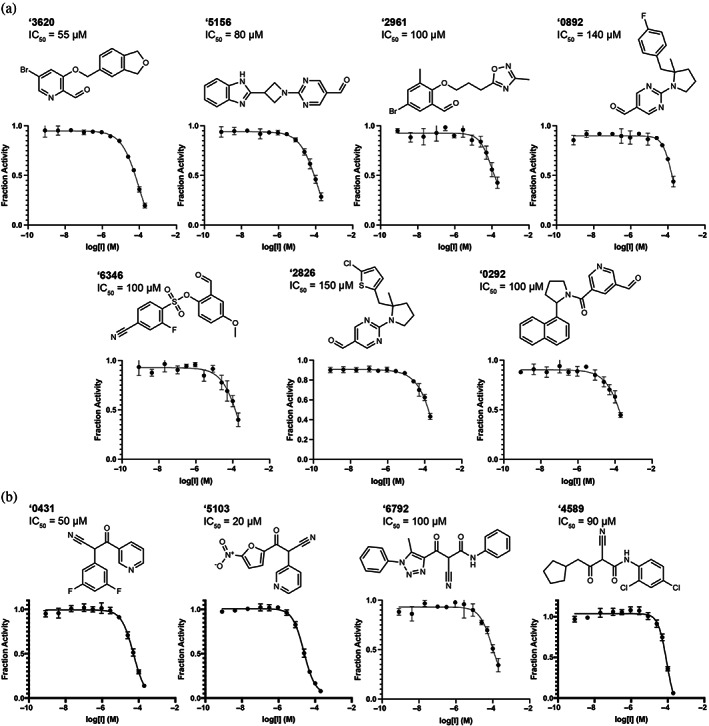
Covalent hits from 6.5 million virtual screen. Dose response curves for (a) aldehyde and (b) nitrile docking hits. IC50 values shown. All measurements were done in triplicate.
The covalent inhibitors had diverse chemotype and their docked poses explored different enzyme sub‐pockets (Figures 1, 3 and S5). In the S1′ pocket, hydrophobic interactions were made by compounds ‘3620, ‘6345, ‘6792 in their docked poses. Hydrogen bonding with His163 in the S1 pocket was made by ‘5103, ‘0431, ‘2961 in their docked poses. Several compounds, such as ‘0892 and ‘0292, docked to occupy the S2 and S3 pockets, making non‐polar interactions with Met49 and Phe181. Other compounds appeared to span the binding site between the S1 and S2/S3 pockets, for example, ‘5156 hydrogen‐bonding with Glu166. Many compounds, such as ‘3620 and ‘6792, formed hydrogen‐bonds with the peptide backbone atoms of Cys145, Ser144 and Gly143.
We sought to optimize several of the new covalent inhibitors, focusing on the aldehyde ‘3620 with an IC50 of 55 μM (Table S1). These analogs were identified through multiple strategies, including simply seeking readily available “make‐on‐demand” congeners that fit in the enzyme site, using SmallWorld and Arthor (NextMove Software, Cambridge, UK) (Irwin et al., 2020), or testing perturbations to what seemed to be key interactions. From these studies emerged 39 analogs with IC50 values better than ‘3620. The most potent analog ‘7021 had an IC50 of 1 μM acted as a reversible inhibitor (Figure S6). Other analogs ranging from 2 to 48 μM had changes to different benzene substituents or bicyclic systems of ‘3620 (Figures 4 and S7; Table S2).
FIGURE 4.
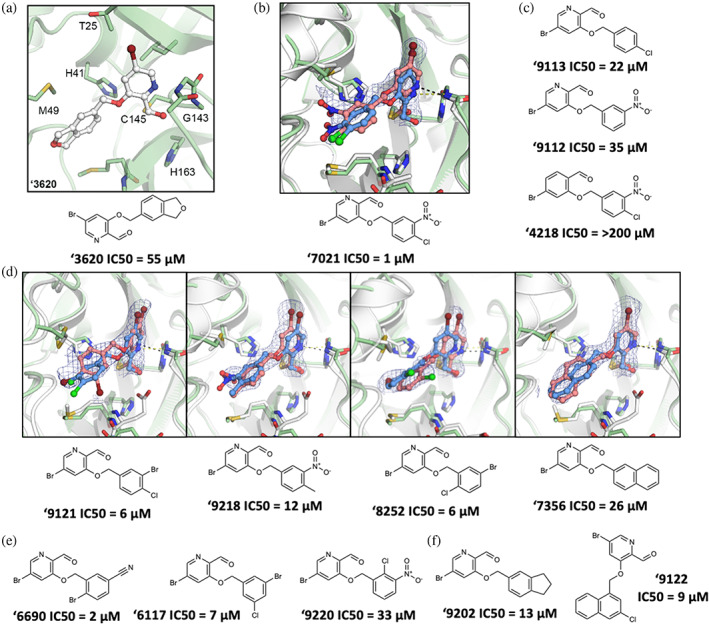
Compound optimization of aldehyde ‘3620. (a) Docked pose of docking hit ‘3620. (b) Crystal structure (pink carbons) and docked pose (blue carbons) comparison for analog ‘7021 (RMSD 1.29 Å; PDB 8DIB). (c) Hypothesis testing analogs of ‘7021 included removing the nitro in ‘9113 and the chlorine in ‘9112, both with weaker inhibition. Analog ‘4218 replaced the pyridine with a benzene eliminating inhibition. (d) Crystal structures of additional ‘3620 analogs comparing experimental (pink carbons) and docked (blue carbons) poses (RMSDs of 1.75, 0.78, 1.18, and 0.84 Å, respectively; PDB 8DIC, 8DIE, 8DID, 8DIF, respectively). (e) Analogs with different benzene substituent orientations (‘6690, ‘6117) inhibit MPro at similar potencies. Substituents oriented like ‘9220 were weaker inhibitors. (f) Examples of the most potent larger hydrophobic analogs of ‘3620. For A‐F, MPro protein structure is PDB 6Y2G (green carbons) used in docking or from the solved structures (white carbons). Hydrogen bonds shown with dashed lines. The 2fo‐fc ligand density maps (blue contour) are shown at 1 σ. IC50 values are shown with concentration response curves in Figure S7. All measurements were done in triplicate.
In its docked pose, the pyridine nitrogen of ‘7021 hydrogen bonds to Gly143 (Figure 4B). To test the importance of this interaction, the phenyl analog of the pyridine, compound ‘4218, was synthesized and tested. This molecule lost all measurable activity (IC50 > 200 μM), consistent with the importance of the pyridine hydrogen bonds (Figure 4C). The more electro‐deficient pyridine ring also may make the aldehyde more reactive. Meanwhile, removing non‐polar groups from the distal phenyl ring of ‘7021, as in analogs ‘9313 and ‘9112, increased IC50 values to 22 μM and 35 μM, respectively, indicating more hydrophobic bulk was preferred in the shallow subsite in which this substituted phenyl was docked. Additional analogs were synthesized changing the aldehyde to other electrophiles, however none inhibited MPro (Figure S8).
2.6. Crystal structures of the covalent inhibitors
To investigate how the docked poses of the covalent inhibitors corresponded to true binding modes, and to aid further optimization, crystal structures of five aldehyde inhibitors complexed with MPro were determined: ‘7021 (IC50 = 1 μM), ‘9121 (IC50 = 6 μM), ‘8252 (IC50 = 6 μM), ‘9218 (IC50 = 12 μM), and ‘7356 (IC50 = 26 μM), with resolutions ranging from 1.90 to 2.17 Å (Figures 4B,D and S9). The structures of these compounds recapitulated the docking predictions with high fidelity, with all‐atom Hungarian RMSD values ranging from 0.78 to 1.75 Å (Figure 4B). Consistent with the docking and with the results of the analogs, the pyridine nitrogen in each inhibitor hydrogen bonds with Gly143 and the thioacetal adduct hydrogen bonds with the backbone of Cys145 in the oxyanion hole of the enzyme. The hydrophobic groups on the distal aryl ring interact with residues in the S2/S3 pockets, including Met49 and Phe181 (Figures 4B,D and S9).
2.7. Lead compounds inhibit SARS‐CoV‐2 MPro variants and additional coronaviral MPro
With the progression of covalent and non‐covalent inhibitor optimization, we tested ‘7021 for its ability to inhibit MPro of other coronaviruses, SARS‐CoV‐1 and MERS (Figure S10). ‘7021 inhibited the SARS‐CoV‐1 MPro with an IC50 of 8 μΜ, similar to its SARS‐CoV‐2 MPro IC50 of 1 μM, however it was a weaker inhibitor for the MERS MPro with an IC50 of 50 μM (Figure 5A, Table S3).
FIGURE 5.
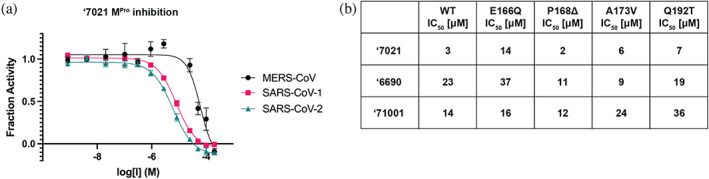
Docking‐derived compounds inhibit MPro of other coronaviruses and SARS‐CoV‐2 MPro mutants. (a) ‘7021 inhibits SARS‐CoV‐1 and MERS‐CoV MPro. (b) IC50 values for ‘7021, ‘6690, and ‘71001 against a panel of MPro mutants. WT, wildtype. All measurements were done in triplicate.
Several MPro variants have arisen over the course of the pandemic and have been associated with Paxlovid (nirmatrelvir and ritonavir) resistance. MPro A173V (Moghadasi, Heilmann, et al., 2022) has been previously associated with over 10‐fold resistance to nirmatrelvir. Valine at position 173 is naturally occurring in other coronaviruses (including human coronavirus 229E and NL63) and has arisen independently in some clinical isolates of SARS‐CoV‐2 (n = 231 occurrences of 14,195,429 genomes, GISAID database accessed December 12, 2022) (Moghadasi, Heilmann, et al., 2022). P168Δ (Moghadasi, Heilmann, et al., 2022) has been previously associated with a roughly 5‐fold resistance to nirmatrelvir and is the among the most prevalent changes at that amino acid position (n = 167 occurrences), though other substitutions have not been found to confer resistance (i.e., P168S, n = 524 occurrences). Several reports found substitutions at E166 (Hu et al., 2022; Iketani et al., 2022; Jochmans et al., 2022; Moghadasi, Heilmann, et al., 2022) and neighboring residues confer differing degrees of resistance, though only E166Q has arisen with meaningful frequency in multiple SARS‐CoV‐2 sublineages (n = 4,792 occurrences in distinct Delta and Omicron clades). Finally, substitutions at Q192 have been found to confer resistance to nirmatrelvir, including Q192T (Hu et al., 2022; Sasi et al., 2022) (n = 227 occurrences) and Q192R (Heilmann et al., 2023) (n = 40 occurrences), though Q192Δ is the most prevalent (n = 1,304 occurrences, nearly all Omicron).
Focusing on four naturally occurring variants that have been previously shown to confer nirmatrelvir resistance (E166Q, ΔP168, A173V, and Q192T), we tested their resistance to compounds ‘7021, ‘6690, and ‘71001 (Figure 5B and S11). The ΔP168 mutation had minimal to no impact on compound potencies compared to wild type MPro, reflecting this residues relatively distant position from the active site. A173V and Q192T had minimal effect on the potency of the covalent compounds (‘7021, ‘6690), but conferred resistance to ‘71001. On the contrary, E166Q conferred some resistance to ‘7021 and ‘6690, but not ‘71001. The differential effects on nirmatrelvir likely reflect the greater distances of the substituted residues from the new inhibitors, compared to the drug (comparing the nirmatrelvir complex in PDB 8B2T to the crystal structures determined here). For instance, taking the Mpro/‘7021 complex as representative (PDB 8DIB), the carboxylate of Glu166 forms a polar interactions with the lactam moiety of nirmatrelvir, at a distance of 3.4 Å, but this residue is no closer than 4.5 Å from ‘7021, and makes no polar interactions with the new inhibitor. Similarly, Pro168 is within 3.9 Å of the trifluoromethyl group of nirmatrelvir, and its deletion would be expected to disrupt drug recognition. Conversely, this residue is no closer than 7.7 Å to ‘7021, too distant to have a direct interaction. While Ala173 is 6.5 Å from the nirmatrelvir trifluoromethyl, its mutation to Val will likely disrupt packing with nearby residues such as Leu167 and Phe181. These perturbations are less likely to be felt by ‘7021, whose closest atom is 7.9 Å away from this residue. Finally, the Cβ atom of Gln192 also forms van der Waals contacts with the terminal trifluoromethyl of nirmatrelvir, and the substitution of this residue with a β‐branched threonine is likely to disrupt this; meanwhile, this site of perturbation is 7.4 Å from the closest atom in ‘7021. Overall, the smaller size of the new inhibitors versus nirmatrelvir seem to make them more resistant to these drug‐induced mutations, most of which, intriguingly, affect contacts on at the site of the drug furthest from the catalytic center. Naturally, it would be premature to draw many conclusions from these observations, not least because the mutations have occurred in an environment exposed to nirmatrelvir, and not these newer inhibitors. Still, it may be that molecules that are smaller than nirmatrelvir, and more compact around the catalytic center, may be less sensitive to mutations than the larger Mpro inhibitors.
3. DISCUSSION
From this study emerged 132 MPro inhibitors with IC50 values less than 150 μM, covering 37 different scaffold classes (Figure 3, Tables 1 and 2). Of these, 15 inhibitors in 3 scaffolds inhibited the enzyme with IC50 values less than 10 μM. The best covalent inhibitor, '7021, was reversible (Figure S6) reflecting the fast‐on/fast‐off kinetics characteristic of aldehyde covalent inhibitors. To dock the electrophile library, we first had to create it, drawing on aldehydes, nitriles, and α‐ketoamides in the expanding library of tangible molecules. This resulted in a library of over 6.5 million new electrophiles, which is openly available to the community at https://covalent2022.docking.org. Crystal structures of eight of the new inhibitors closely corresponded to the docking predictions (Figures 3 and 4). Inhibitors could also inhibit other viral MPro enzymes with similar potencies, as well as MPro mutants appearing in patients (Figures 5 and S11).
While the strengths of this study were the identification of multiple new MPro inhibitor scaffolds, with subsequent crystal structures supporting the docking predictions, the work also revealed liabilities of our docking strategy. Docking large libraries of lead‐like molecules against G protein‐coupled receptors (Levit Kaplan et al., 2022; Lyu et al., 2019; Stein et al., 2020; Wang et al., 2017) and other integral membrane proteins (Alon et al., 2021; Singh et al., 2022), with well‐formed orthosteric sites and well‐defined ligand‐recognition hot‐spots, can have hit rates in the 25%–60% range, whereas the hit rates against MPro were in the 6%–15% range. A similar trend is true for the potency of the docking hits against MPro, which were in the 20–150 μM range, not the low‐ to mid‐nM range found against the integral membrane proteins. The low lead‐like hit‐rates against MPro resemble those against other soluble proteins such as β‐lactamase (Babaoglu et al., 2008; Lyu et al., 2019) and the macrodomain of SARS‐CoV‐2 (Gahbauer et al., 2022; Schuller et al., 2021), or even against allosteric sites or peptide binding sites in GPCRs (Huang et al., 2015; Korczynska et al., 2018; Mysinger et al., 2012). Like MPro, these targets are characterized by more open, solvent‐exposed binding sites, lacking the high‐complementarity typical of the small molecule GPCRs and other membrane receptors. This deficit can be partly overcome in sites with well‐defined ligand‐recognition hot spots, which can guide docking campaigns. Thus, in lead‐like non‐covalent and covalent (London et al., 2014) docking versus β‐lactamase it has been possible to find relatively potent hits directly out of docking, notwithstanding sometimes low hit rates, and to optimize these to mid‐nM potency. The same may be true for typically more challenging peptide (Manglik et al., 2016) and lipid receptors (Sadybekov et al., 2022), where hot‐spot recognition can help lead‐like docking and optimization. Confronted with a target like MPro, with its solvent‐exposed, relatively flat interface and, when this study began, lack of ligands to define receptor hot spots, a more fragment‐based approach may have advantages to the larger and more complicated lead‐like molecules prioritized here. As was observed in studies by the Covid Moonshot (The COVID Moonshot Consortium et al., 2020), and in our own experience versus the orphan SARS‐CoV‐2 enzyme Mac1 (Schuller et al., 2021), fragment screens and fragment docking had much higher hit rates and could be used directly for optimization, but they also could be used to define and identify hot‐spots enabling subsequent docking of lead‐like libraries with better hit‐rates and affinities (Gahbauer et al., 2022). Such fragment based approaches may find broad use against structurally‐enabled viral enzymes that are under‐characterized for ligand recognition. We do note that, notwithstanding the liabilities of Mpro as a target, others have seen better success with large library docking screens, even using methods similar to those used here (Luttens et al., 2022).
These caveats should not distract from the key observations of this study. Large library docking of both lead‐like molecules and covalent electrophiles has revealed 11 scaffold families of MPro inhibitors (Figure 3, Tables 1 and 2), the best of which act in the low μM range (Figures 2 and 4). Whereas neither hit rates nor affinities rose to levels seen against targets with well‐defined binding sites, eight crystal structure of characteristic lead molecules confirmed the docking poses (Figures 2 and 4), suggesting that, notwithstanding the lower hit rates, when the docking was right it was right for the right reasons. These structures may template the further optimization of these new MPro inhibitors.
4. METHODS
4.1. Expression and purification of MPro
All reducing agents were added to buffers immediately before use. We received nsp5 cloned into pGEX6p‐1 with a N‐terminal GST tag and MPro cleavage‐site SAVLQ↓SGFRK and an HRV‐3C‐cleavable C‐terminal 6xHis tag from Rolf Hilgenfeld. This bacterial expression plasmids were transformed into One Shot™ BL21(DE3)pLysS Chemically Competent E. coli (Thermo). The expression for MPro in E. coli was modified from the expression previously described (Zhang, Lin, Sun, et al., 2020). In brief, a transformed clone of BL21(DE3)pLysS E. coli was added to a 50 mL culture of 2xYT media supplemented with 2% glucose and 100 μg/mL ampicillin grown overnight at 37°C. A total of 30 mL of overnight culture were used to inoculate 1 L of 2xYT media supplemented with 100 μg/mL ampicillin The inoculated culture was shaken at 225 rpm at 37°C and then induced when culture OD600 reached 0.8 (after ~3 h) by adding 1 mL of 1 M IPTG. After 5 h of expression at 37°C, the culture was pelleted through centrifugation at 9,000 rpm for 15 min. Supernatant was discarded and cell pellet stored at −80°C. The frozen cell pellet was thawed on ice in 30 mL of 20 mM Tris 150 mM NaCl pH 7.4 buffer. The resuspended sample was sonicated until lysis was complete. Sonicated cell lysate was clarified by centrifugation at 15,000 rpm for 30 min. 3 mL of Super Ni‐NTA Affinity HP Resin beads (Protein Ark) equilibrated with wash buffer (20 mM TRIS 150 mM NaCl 20 mM imidazole) were incubated with 57 mL of the clarified supernatant for 1 h at 4°C. Beads were centrifuged at 200 rpm for 2 min and the supernatant was decanted. The Ni‐NTA beads were washed with ~3 column volumes of wash buffer. Hexahistidine tagged protein was eluted with 1 mL fractions of elution buffer (20 mM Tris 150 mM NaCl 350 mM Imidazole). Fractions containing protein were pooled and then buffer exchanged into 20% Glycerol 20 mM Tris 150 mM NaCl pH 7.4 using Amicon concentrators. 3C protease was added in a 5:1 ratio of MPro to 3C protease and incubated overnight at 4°C. A 2 L of culture yielded 2.28 mg of MPro following 3C cleavage. 3C protease and 6xHis‐tag were removed by incubation with Ni‐NTA beads. The active dimer was isolated with a MonoQTM 5/50 GL column (GE Healthcare). MonoQ column equilibrated with buffer A (20 mM Tris 1 mM DTT pH 8) and the protein sample was then loaded onto the column and eluted with a linear gradient of buffer B (1 M NaCl 20 mM Tris 1 mM DTT pH 8) 0–500 mM NaCl over 20 column volumes. MPro variants were cloned using the Q5® mutagenesis kit (NEB) according the manufacturer's protocol. Primers used to generate these variants were as follows:
E166Q_F (5′GCACCACATGCAGTTGCCGACTG3′),
E166Q_R (5′ATATAGCAGAAGCTAACGC3′),
Q192T_F (5′CCAAACAGCCACCGCCGCAGGAAC3′),
Q192T_R (5′CGATCGACAAACGGACCA3′),
ΔP168_R (5′CAATTCCATGTGGTGCATATAGCAG3′),
ΔP168_F (5′FORACTGGTGTACATGCCGGG3′),
A173V_R (5′ATGTACACCAGTCGGCAATTC3′),
A173V_F (5′GTCGGGACGGACTTAGAAGG3′).
The variant proteins were purified using the same protocol as wild‐type.
4.2. MPro inhibition assay
A fluorescence‐quenched substrate with the sequence H2N(d‐Arg)(d‐Arg)‐K(MCA)‐ATLQAIAS‐K(DNP)‐COOH was synthesized via the Fmoc solid‐phase peptide synthesis as previously described (Zhao et al., 2021). Kinetic measurements were carried out in Corning black 384‐well flat‐bottom plates and read on a BioTek H4 multimode plate reader. The quenched fluorogenic peptide had a final concentration of KM = 12.7 μM, and MPro had a final concentration of 50 nM. The reaction buffer was 20 mM Tris, 150 mM NaCl, 1 mM EDTA, 0.05% Tween‐20 (v/v), and 1 mM DTT, pH 7.4. Compounds were incubated with protease prior to substrate addition at 37°C for 1 h. After incubation, the substrate was added, and kinetic activity was monitored for 1 h at 37°C. Initial velocities were calculated at 1–30 min in RFU/s. Velocities were corrected by subtracting the relative fluorescence of a substrate‐only control, and fraction activity was calculated using a substrate‐corrected no‐inhibitor control where DMSO was added instead of a drug. Kinetics measurements were carried out in triplicate. SARS‐CoV‐1 and MERS MPro were both purchased from Bio‐Techne (catalogue #: E‐718‐050 and E‐719‐050, respectively). KM was derived with the NSP7 substrate for each protease (Figure S10), which was the substrate concentration used for each protease for comparative dose‐response curves. Enzyme concentration was 50 nM for SARS‐CoV‐1 and 100 nM for MERS. The same assay buffer described above was used for all kinetic assays with each protease. Since the MPro variants were not as active as WT on the NSP7 substrate, inhibition assays were performed similarly with the following alterations: 150 nM protease, and 5 μM substrate. The variants were incubated with the indicated compound for 30 min at 37°C, and then the reaction was initiated with the addition of substrate. The resulting traces were analyzed in the same manner as the wildtype.
4.3. Non‐covalent molecular docking
The protein template was modeled based on the crystal structure of the MPro dimer in complex with a covalent alpha‐ketoamide inhibitor (PDB 6Y2G) (Zhang, Lin, Sun, et al., 2020). All water molecules except for HOH 585 and HOH 602, which are located at the dimeric interface, were deleted. The binding pocket of the crystal structure's chain A was selected for docking. The alpha‐ketoamide inhibitor was replaced by the non‐covalent SARS‐CoV inhibitor SID87915542 (Jacobs et al., 2013). Here, the SID87915542‐bound MPro crystal structure (PDB 3V3M) was aligned onto the SARS‐CoV‐2 MPro crystal structure in order to project SID87915542 into the SARS‐CoV‐2 MPro binding site. Next, the modeled protein‐ligand complex and selected water molecules were prepared for docking with the protein prepwizard protocol of Maestro (Schrödinger, Inc. v. 2019‐3) (Madhavi Sastry et al., 2013). Protons were added with Epik and protonation states were optimized with PropKa at pH 7. The C‐terminus (Ser301) of each protein monomer structure was capped with N‐methyl groups while the N‐termini (Ser1) were positively charged. Subsequently, the modeled complex was energetically minimized using the OPLS3e force field. To better accommodate the modeled non‐covalent ligand SID87915542, the CE atom of Met49 was displaced by 1.7 Å from its initial position in the covalently ligated crystal structure (PDB 6Y2G).
Computational docking was performed using DOCK3.7 (Coleman et al., 2013). Precomputed scoring grids for efficient quantification of van der Waals interaction between MPro and docked molecules were generated with CHEMGRID (Meng et al., 1992). Using the AMBER united‐atom partial charges (Weiner et al., 1984), electrostatic potentials within the binding pocket were computed following the numerical solution of the Poisson‐Boltzmann equation with QNIFFT (Gallagher & Sharp, 1998). The partial charges of the hydrogen at the epsilon nitrogen of His163, as well as the hydrogen atoms of the backbone amines of Gly143 and Glu166 were increased by 0.4 elementary charge units (e). In turn, the partial charges of oxygen atoms of the corresponding backbone carbonyl groups were decreased by 0.4e to maintain the initial net charge of each residue (Bender et al., 2021). The low dielectric protein environment was extended by 1.2 Å from the protein surface, as previously described (Mysinger et al., 2012). Similarly, the low dielectric boundary was extended by 0.7 Å from the protein surface for the calculation of ligand desolvation scoring grids with SOLVMAP (Mysinger & Shoichet, 2010). The atomic coordinates of SID87915542 (PDB 3V3M) (Jacobs et al., 2013), the alpha‐ketamide inhibitor of the initial crystal structure (PDB 6Y2G) (Zhang, Lin, Sun, et al., 2020), BDBM512845 (PDB 4MDS) (Turlington et al., 2013), as well as fragment hits MAT‐POS‐7dfc56d9‐1 (MPro‐x0161) (The COVID Moonshot Consortium et al., 2020) and AAR‐POS‐d2a4d1df‐5 (MPro‐x0305) (The COVID Moonshot Consortium et al., 2020) obtained from the Covid‐19 Moonshot screening efforts, were used to generate 80 matching spheres (Coleman et al., 2013) for ligand placement in the docking calculations.
The obtained docking parameters were evaluated based on their ability to prioritize 34 previously reported ligands of SARS‐CoV MPro obtained from the Chembl database (Gaulton et al., 2012), against a background of 1,805 property matched decoys generated with the DUDE‐Z approach (Stein et al., 2021). In addition, an “Extrema” set (Bender et al., 2021; Stein et al., 2020) of 194,921 molecules, including compounds with net‐charges ranging from −2 to +2, was screened against the docking model in order to assess the parameters’ ability to prioritize neutral molecules.
Using the ZINC15 database (Sterling & Irwin, 2015), 225,327,212 neutral molecules mainly from the lead‐like chemical space, that is, molecular weight (MWT) between 250 and 350 amu and calculated (c)logP ≤4.5, from the make‐on‐demand compound libraries from Enamine Ltd. and WuXi Appetec. (Shanghai, China), were screened. Thereby, 219,305,079 molecules were successfully scored with each molecule sampling on average 3,588 orientations and 425 conformations which resulted in the evaluation of approximately 148 trillion complexes in roughly 70 h on a 1,000‐core computer cluster. In addition, 110,898,461 molecules with 350 < MWT ≤ 500 and clogP ≤ 4.5 from ZINC15 were screened in a separate docking campaign. 107,486,710 compounds were successfully scored, each exploring on average 4,175 orientations and 540 conformations within the binding pocket. Nearly 90 trillion complexes were scored in roughly 45 h using a 1,000‐core cluster.
From each docking screen, the predicted binding poses of the 500,000 top‐ranked molecules were analyzed for internal molecular strain (Gu et al., 2021). Molecules that passed the strain criteria (total strain <6.5 tortion energy units (TEU); maximum single torsion <1.8 TEU), were judged by their ability to form hydrogen bonds with Gly143, His163 (S1 subpocket) or Glu166 and proximity to residues forming the S2 subpocket such as Met49 or Asp187. Finally, 120 compounds, selected from the lead‐like docking screen, were ordered from Enamine Ltd., of which 105 were successfully synthesized (87.5%) in addition to 100 molecules of larger MWT that were ordered from the second docking screen, 89 of which were successfully synthesized by Enamine Ltd.
A second docking campaign for non‐covalent inhibitors was performed against the crystal structure of MPro in complex with MAT‐POS‐b3e365b9‐1 (MPro‐x11612) (The COVID Moonshot Consortium et al., 2020) from the Covid‐19 Moonshot consortium. All water molecules except HOH6 and HOH300 were removed and the protein‐ligand complex structure was prepared for docking following the protein prepwizard protocol of Maestro (Schrödinger v. 2019‐3) as described above.
As described above in the previous docking campaign, the partial charges of the hydrogen atoms at the epsilon nitrogen of His163 and the backbone amine of Glu166 were increased by 0.4e, whereas the partial charges of corresponding backbone carbonyl oxygen atoms were decreased by 0.4e to maintain the net charge of each residue. For calculating electrostatic scoring grids, the low‐dielectric volume of the protein was extended by 1.9 Å from the protein surface (based on surface mapping spheres generated by Sphgen). In addition, the low dielectric boundary was extended by 1.0 Å from the protein surface for calculating ligand desolvation scoring grids with SOLVMAP. The atomic coordinates of MAT‐POS‐b3e365b9‐1 were used to generate 45 matching spheres for ligand placement with DOCK3.8. The performance of the obtained docking grids was evaluated by their ability to enrich 15 previously reported SARS‐CoV‐2 MPro inhibitors over 650 property‐matched decoys or an Extrema set containing 153,256 molecules with net charges ranging from −2 to +2, molecular weight between 300 and 500 amu. Finally, 862,382,088 neutral compounds with 18–29 heavy atoms from the Enamine REAL chemical library were screened using the ZINC22 database (http://files.docking.org/zinc22/). Molecules with strained conformations (total strain >8 TEU, maximum single strain >3 TEU), were excluded by the docking program. 778,517,250 molecules were successfully scored, each sampled in approximately 836 conformations and 3,439 orientations, leading to the evaluation of roughly 905.8 trillion complexes within 481h on a 1000‐core computer cluster.
21,284,498 compounds scored lower than −35 kcal/mol and the poses of top scoring 5,004,192 compounds were extracted. 214,580 compounds formed favorable interactions with key residues such as His163, Glu166 and the P2 subpocket, 181,866 of which obtained ECFP4‐based TC coefficients of less than 0.35 to the 1,716 known SARS‐CoV and SARS‐CoV‐2 MPro inhibitors reported in the literature (Anand et al., 2003; Blanchard et al., 2004; Breidenbach et al., 2021; Chen, Gui, et al., 2005; Chen, Wang, et al., 2005; Clyde et al., 2022; Dai et al., 2020; Ghosh et al., 2008; Han et al., 2022; Hoffman et al., 2020; Jacobs et al., 2013; Jin et al., 2020; Kim et al., 2012; Luttens et al., 2022; Ma et al., 2020; Mukherjee et al., 2008; Qiao et al., 2021; Rathnayake et al., 2020; The COVID Moonshot Consortium et al., 2020; Westberg et al., 2020; Wu et al., 2006; Xue et al., 2008; Yang et al., 2006; Yang et al., 2021; Zhang et al., 2008; Zhang, Lin, Kusov, et al., 2020; Zhang, Lin, Sun, et al., 2020; Zhang, Spasov, et al., 2021; Zhang, Stone, et al., 2021). Finally, roughly 9,000 top‐ranking compounds were visually inspected, and 167 molecules were ordered from Enamine Ltd., 146 of which (87.4%) were successfully synthesized.
4.4. Covalent molecular docking
Cysteine‐reactive warheads of aldehydes, nitriles, and alpha‐ketoamides were searched in the ZINC20/Enamine REAL databases of 1.4 billion molecules using their respective SMARTS patterns (ketoamides O=[CR0]([#6])[CR0](=O)N[#6]; aldehydes [CX3H1](=O)[#6]; nitriles [CX4]‐C#N). This returned 25.7 million nitriles, 2.5 million aldehydes, and 1.5 million ketoamides. Molecules were filtered to have at least one ring, and to be fragment to lead‐like molecular weights (<350). Three‐dimensional “dockable” conformations were generated with molecules in their transition‐state form and a dummy atom in place for the covalent docking algorithm to indicate which atom should be modeled covalently bound to the Cysteine sulfur (London et al., 2014; Wan et al., 2020). Overall, 6.5 million molecules were docked—3.6 million nitriles, 1.4 million ketoamides, and 1.5 million aldehydes.
The protein was prepared in DOCK3.7 (Coleman et al., 2013). Pose reproduction of the truncated covalent molecule of PDB 6Y2G (Zhang, Lin, Sun, et al., 2020) (smiles of dockable ligand: O=C1NCC[C@H]1CC[C@]([SiH3])(C(NCC2=CC=CC=C2)=O)O) was checked for the docking setup. Default generated grids were used for electrostatic (radius size 1.9 Å) and VDW scoring, and no matching spheres were used in docking calculations as they are not used by the covalent docking DOCKovalent (London et al., 2014; Wan et al., 2020) algorithm. For covalent docking, the Cys145 SH group was indicated as the anchor for molecules screened. The distance was slightly relaxed from the C‐C bond distance to 1.85 Å. For His41 protonation, aldehydes, nitriles, and neutral ketoamides used HID, while negative ketoamides used HIP. Each warhead was docked separately with a total 6.5 million molecules screened. Accordingly, each warhead was also processed separately.
For the aldehydes, the top 300,000 ranked molecules were evaluated for torsional strain (Gu et al., 2021), and those with a total torsional strain greater than 9.8 (around 3.7 incurred due to strain on atom types on the warhead and this was disregarded, therefore total energy was 6) and single torsional strain greater than 2.5 were excluded (155,386 left). Molecules making more than 1 hydrogen bond to the protein, having no hydrogen bond clashes, no unpaired hydrogen bond donors (56,969 left) were prioritized. Remaining molecules were clustered for chemical similarity based on ECFP4‐based Tanimoto coefficient (Tc) of 0.5. Viable poses filling the S1’, S1 or S2 sites were selected during visual inspection. A total of 35 aldehydes were selected for make‐on‐demand synthesis of which 27 were successfully synthesized. For the nitriles, the top 100,000 ranked molecules were evaluated for torsional strain (17,424 left), then filtered for favorable interactions (6,201 left). Lastly, we visually inspected remaining molecules for favorable hydrogen bonds formed with His41, Gly143, Thr26, Glu166, or Cys145. Finally, 41 compounds were ordered for synthesis (31 were successfully obtained). For the ketoamides the top 393,000 ranked molecules with scores less than 0.0 were evaluated for torsional strain (121,234 left), and favorable interactions with the enzyme (37,267 remained). Visual inspection focused on those making hydrogen bonds with His41, Cys145, Gly143, Thr26. In total 21 molecules were prioritized and 16 were successfully synthesized.
4.5. Compound synthesis
Make‐on‐demand non‐covalent and covalent compounds purchased from docking screens, as well as analogs, were synthesized by Enamine Ltd. (Table S1). Purities of molecules were at least 90% and most active compounds were at least 95% (based on LC/MS data) (Figure S12).
4.6. Synthesis of RLA‐5573
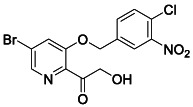
To a solution of NaOH (66 mg, 1.60 mmol, 4.5 equiv.) in 5 mL MeOH was added RLA‐5575 (140 mg, 0.36 mmol, 1 equiv.). Then PhI(OAc)2 (175 mg, 0.55 mmol, 1.5 equiv.) was added portion‐wise at 0°C. After stirring for 5 h at room temperature, the reaction was quenched by the addition of water. Remove the MeOH under the vacuum. The product was extracted with EtOAc (50 mL × 3), and the combined organic extracts were washed with brine and dried over Na2SO4. After removal of the solvents under reduced pressure, crude acetal intermediate was obtained as a white solid without further purification. The resulting solid was dissolved in 5 mL MeOH and TFA (0.56 mL, 7.26 mmol, 20 equiv.) was added the mixture. After 20 h, the reaction was concentrated and purified by reverse phase column chromatography (0–100% water/MeCN) to yield the desired compound (58 mg, 40%). 1 H NMR (400 MHz, DMSO) δ 8.41 (d, J = 1.4 Hz, 1H), 8.29 – 8.14 (m, 1H), 8.10 (d, J = 1.4 Hz, 1H), 7.89‐7.80 (m, 2H), 5.39 (s, 2H), 4.73 (s, 2H). 13 C NMR (100 MHz, DMSO) δ 198.90, 153.46, 147.54, 141.59, 140.34, 137.32, 132.37, 131.88, 125.27, 124.38, 124.16, 123.98, 68.48, 66.29. LRMS (ESI) calcd for C14H11BrClN2O5 [M + H]+ m/z 400.95, found 401.08.
4.7. Synthesis of RLA‐5574
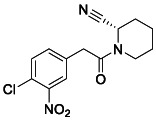
To a mixture of 2‐(4‐chloro‐3‐nitrophenyl) acetic acid (150 mg, 0.7 mmol, 1.0 equiv.), (2S)‐piperidine‐2‐carbonitrile; oxalic acid (139 mg, 0.7 mmol, 1.0 equiv.), HATU (529 mg, 1.4 mmol, 2.0 equiv.), and DMAP (26 mg, 0.2 mmol, 0.3 equiv.) in DCM, was added N‐ethyl‐N‐isopropylpropan‐2‐amine (0.35 mL, 2.1 mmol, 3.0 equiv.). The reaction was stirred at room temperature for 4 h. The mixture was quenched with NH4Cl aq. solution and extracted with ethyl acetate. The organic layers were dried over MgSO4 and concentrated. The resulting oil was purified by flash column chromatography (0–15% MeOH/DCM) to yield the desired compound (120 mg, 56%). 1 H NMR (400 MHz, CDCl3) δ 7.74 (d, J = 2.0 Hz, 1H), 7.50 (d, J = 8.3 Hz, 1H), 7.38 (dd, J = 8.3, 2.1 Hz, 1H), 5.86 – 5.65 (m, 1H), 3.86 (d, J = 13.2 Hz, 1H), 3.75 (s, 2H), 3.44 – 3.17 (m, 1H), 1.99 (d, J = 13.4 Hz, 1H), 1.88 – 1.63 (m, 4H), 1.41 (dtt, J = 17.4, 8.6, 3.8 Hz, 1H). 13 C NMR (100 MHz, CDCl3) δ 168.41, 147.74, 134.65, 134.36, 132.05, 126.56, 125.92, 117.32, 43.66, 41.64, 38.93, 28.20, 25.14, 20.36. LRMS (ESI) calcd for C14H15ClN3O3 [M + H]+ m/z 308.07, found 308.09.
4.8. Synthesis of RLA‐5575
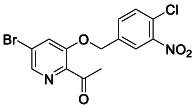
To a solution of 1‐(5‐bromo‐3‐hydroxypyridin‐2‐yl) ethan‐1‐one (300 mg, 1.4 mmol, 1.0 equiv.) in 10 mL THF was added sodium methoxide 0.5 M in MeOH (2.7 mL, 1.4 mmol, 1.0 equiv.). The mixture was stirred at room temperature for 15 min before the addition of 4‐(bromomethyl)‐1‐chloro‐2‐nitrobenzene (348 mg, 1.4 mmol, 1.0 equiv.). After 16 h, the mixture was quenched quenched with NH4Cl aq. solution and extracted with ethyl acetate. The organic layers were dried over MgSO4 and concentrated. The resulting oil was purified by flash column chromatography (0–10% MeOH/DCM) to yield the desired compound (78 mg, 15%). 1 H NMR (400 MHz, CDCl3) δ 8.38 (d, J = 1.7 Hz, 1H), 8.05 (d, J = 1.9 Hz, 1H), 7.77 (dd, J = 8.3, 2.0 Hz, 1H), 7.62 (d, J = 8.3 Hz, 1H), 7.55 (d, J = 1.7 Hz, 1H), 5.18 (s, 2H), 2.67 (s, 3H). 13 C NMR (100 MHz, CDCl3) δ 198.65, 153.68, 148.16, 142.60, 142.49, 135.96, 132.57, 131.54, 127.00, 124.87, 124.44, 123.95, 69.26, 28.64. LRMS (ESI) calcd for C14H11BrClN2O4 [M + H]+ m/z 384.95, found 385.07.
4.9. Synthesis of RLA‐5576
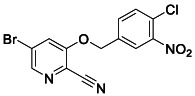
To a mixture of 5‐bromo‐2‐cyano‐3‐nitro pyridine (200 mg, 0.88 mmol, 1.0 equiv.), (4‐chloro‐3‐nitrophenyl) methanol (165 mg, 0.88 mmol, 1.0 equiv) in 10 mL THF, was added sodium hydride 60% dispersion in mineral oil (39 mg, 0.97 mmol, 1.1 equiv.). The reaction was stirred at room temperature for 16 h. The mixture was quenched with NH4Cl aq. solution and extracted with ethyl acetate. The organic layers were dried over MgSO4 and concentrated. The resulting oil was purified by flash column chromatography (10%–100% ethyl acetate/Hexanes) to yield the desired compound (158 mg, 49%). 1 H NMR (400 MHz, DMSO) δ 8.51 (d, J = 1.7 Hz, 1H), 8.29 (d, J = 1.7 Hz, 1H), 8.20 (d, J = 1.9 Hz, 1H), 7.88 (d, J = 8.3 Hz, 1H), 7.81 (dd, J = 8.3, 2.0 Hz, 1H), 5.47 (s, 2H). 13 C NMR (100 MHz, DMSO) δ 157.39, 147.53, 144.27, 136.28, 133.08, 132.13, 125.51, 125.23, 125.03, 124.79, 120.92, 114.93, 69.22. LRMS (ESI) calcd for C13H8BrClN3O3 [M + H]+ m/z 367.94, found 368.06.
4.10. Compound optimization
Optimization of docking hits ZINC346371112, ZINC301553312, ZINC813360541, ZINC553840273, ‘3620, ‘0431, ‘4589, ‘5103, ‘5156, ‘6246, ‘6792, ‘0292, ‘2826/’0892 were attempted (Table S1). Analogs were designed for desired chemical perturbations or searched in SmallWorld and Arthor catalogs and synthesized by Enamine Ltd. For ‘3620, compounds were also designed from the ‘7356 and ‘7021 crystal structures and were modeled with covalent docking or with Maestro (v. 2021‐2, Schrödinger, LLC) ligand alignment.
4.11. Protein crystallization
Both covalent and non‐covalent compounds including 7021, ‘9121, 8252, ‘9218, 7356, 5548, 6111 and SG‐0001 were co‐crystallized with SARS‐CoV2 MPro. Before setting up crystals, 10 mg/ml of protein was incubated with either 0.3 mM of covalent compounds or 1.5 mM of non‐covalent compounds on ice for 1 h. Crystals were set using vapor diffusion hanging drop method at 20°C in conditions including 0.1 M Tris pH 7.4 and 20% PEG 8000; and 0.1 M MES pH 6.5, 20% PEG 6000. Crystals took 3–4 days to grow for all compounds. Before data collection, crystals were cryo‐cooled in a solution containing reservoir solution and 25% glycerol.
4.12. Structure determination and refinement
The MPro‐inhibitor compound datasets were either collected at the Advanced Light Source beamline 8.3.1 (Lawrence Berkeley laboratory) or SSRL beamline 12‐2 beamline (Stanford, United States) at a temperature of 100K. The diffraction datasets were processed using XDS (Kabsch, 2010) and CCP4 software's suite(Collaborative Computational Project, Number 4, 1994). AIMLESS (Evans, 2006) was used for scaling and merging. Molecular replacement was performed either using PHASER (McCoy et al., 2007) using the protein model from PDB entry 7NG3 (Costanzi et al., 2021) as the search model. The bound ligand in the PDB 7NG3 was removed from the search model during molecular replacement, giving unbiased electron density for ligands in the initial electron density maps. The initial model fitting and addition of waters was done in COOT (Emsley et al., 2010) followed by refinement in REFMAC (Murshudov et al., 2011). Geometry restraints for the ligands were created in eLBOW‐PHENIX (Afonine et al., 2012) and following rounds of refinement were carried out in PHENIX. Geometry for each structure was assessed using Molprobidity and PHENIX polygon. Datasets have been deposited to the PDB with PDB IDs 8DIB, 8DIC, 8DID, 8DIE, 8DIF, 8DIG, 8DIH and 8DII. Statistics for data collection and refinement are in Table S4. The ligand symmetry accounted RMSDs between the docked pose and experimental pose were calculated by the Hungarian algorithm in DOCK6 (Allen & Rizzo, 2014).
4.13. Dynamic light scattering (DLS)
Samples were prepared in filtered 50 mM KPi buffer pH 7.0 with final DMSO concentration at 1% (v/v). Colloidal particle formation was detected using DynaPro Plate Reader II (Wyatt Technologies). All compounds were screened in triplicate at roughly 2‐fold higher concentration than reported IC50 (concentrations can be found in Table S1). Analysis was performed with GraphPad Prism software version 9.1.1 (San Diego, CA).
4.14. Enzyme inhibition assays for aggregation
Enzyme inhibition assays were performed at room temperature using using CLARIOstar Plate Reader (BMG Labtech). Samples were prepared in 50 mM KPi buffer pH 7.0 with final DMSO concentration at 1% (v/v). Compounds were incubated with 4 nM AmpC β‐lactamase (AmpC) or Malate dehydrogenase (MDH) for 5 min. AmpC reactions were initiated by the addition of 50 μM CENTA chromogenic substrate. The change in absorbance was monitored at 405 nm for 1 min 45 s. MDH reactions were initiated by the addition of 200 μM nicotinamide adenine dinucleotide (NADH) (54839, Sigma Aldrich) and 200 μM oxaloacetic acid (324427, Sigma Aldrich). The change in absorbance was monitored at 340 nm for 1 min 45 s. Initial rates were divided by the DMSO control rate to determine % enzyme activity. Each compound was screened at roughly 2‐fold higher concentration than reported the IC50 value in triplicate (concentrations can be found in Table S1). Data were analyzed using GraphPad Prism software version 9.1.1 (San Diego, CA).
For detergent reversibility experiments, inhibition was screened near IC75 with or without 0.01% (v/v) Triton X‐100 in triplicate. Enzymatic progress was performed/monitored as previously described (Lak et al., 2021; O'Donnell et al., 2021).
4.15. Statistical analyses
All statistical analyses were performed on the GraphPad Prism version 8.0 or 9.1.1 software. Changes only at the 95% confidence level (p < 0.05) were considered as statistically significant.
AUTHOR CONTRIBUTIONS
Stefan Gahbauer conducted non‐covalent docking screens and compound optimization, with input from Brian K. Shoichet, Jiankun Lyu, Seth Vigneron, and the 2021 CUBS Cohort. Elissa A. Fink, Shuo Gu, and Xiaobo Wan performed covalent database building with input from Jack Taunton and Brian K. Shoichet, covalent docking with input from Brian K. Shoichet, and compound optimization with input from Brian K. Shoichet, Adam R. Renslo, Stefan Gahbauer, and Isha Singh. Jun Chen designed and synthesized ‘7021 analogs chaning electrophiles, supervised by Adam R. Renslo. Enzymatic testing was conducted by Conner Bardine and Tyler C. Detomasi, assisted by Nicholas J. Young, and supervised by Charles S. Craik. Antiviral and cytotoxicity assays were performed by Kris White, with supervision by Adolfo García‐Sastre. Protein purification was done by Isha Singh, Beatrice Ary, Pavla Fajtová, with supervision by Brian K. Shoichet, Charles S. Craik, and Anthony J. O'Donoghue. Crystallography was done by Isha Singh assisted by Joseph O'Connell and with input from Brian K. Shoichet. Aggregation testing was performed by Isabella Glenn and Henry O'Donnell with input from Brian K. Shoichet. ZINC15 and ZINC22 databases were built by John J. Irwin. Yurii S. Moroz supervised compound synthesis of Enamine compounds, assisted by Ivan S. Kondratov. Charles S. Craik and Brian K. Shoichet supervised the project. Arghavan Alisoltani and Lacy M. Simons performed evolutionary analysis of mutations, supervised by Egon A. Ozer, Ramon Lorenzo‐Redondo and Judd F. Hultquist. Elissa A. Fink, Stefan Gahbauer, and Conner Bardine wrote the paper with input from all other authors, and primary editing from Charles S. Craik and Brian K. Shoichet. Charles S. Craik and Brian K. Shoichet conceived the project.
CONFLICT OF INTEREST STATEMENT
Brian K. Shoichet is the founder of Epiodyne Therapeutics, and with John J. Irwin co‐founder of Deep Apple Therapeutics and BlueDolphin Lead Discovery, a docking‐based CRO. The Adolfo García‐Sastre laboratory has received research support from Pfizer, Senhwa Biosciences, Kenall Manufacturing, Avimex, Johnson & Johnson, Dynavax, 7Hills Pharma, Pharmamar, ImmunityBio, Accurius, Nanocomposix, Hexamer, N‐fold LLC, Model Medicines, Atea Pharma, Applied Biological Laboratories and Merck, outside of the reported work. Adolfo García‐Sastre has consulting agreements for the following companies involving cash and/or stock: Vivaldi Biosciences, Contrafect, 7Hills Pharma, Avimex, Vaxalto, Pagoda, Accurius, Esperovax, Farmak, Applied Biological Laboratories, Pharmamar, Paratus, CureLab Oncology, CureLab Veterinary, Synairgen and Pfizer, outside of the reported work. Adolfo García‐Sastre has been an invited speaker in meeting events organized by Seqirus, Janssen, Abbott and Astrazeneca. Adolfo García‐Sastre is inventor on patents and patent applications on the use of antivirals and vaccines for the treatment and prevention of virus infections and cancer, owned by the Icahn School of Medicine at Mount Sinai, New York, outside of the reported work. Yurii S. Moroz is the scientific advisor at Enamine Ltd. Ivan S. Kondratov is the Director of Medicinal Chemistry at Enamine Ltd.
Supporting information
Data S1. Supporting Information.
ACKNOWLEDGMENTS
This work was supported by DARPA grant HR0011‐19‐2‐0020 (Brian K. Shoichet, John J. Irwin, Adolfo García‐Sastre), by NIH grant R35GM122481 (Brian K. Shoichet, John J. Irwin), and by NIH grant R21AI163912 (Judd F. Hultquist). This was also funded by NIAID grant U19AI171110 (Charles S. Craik, Brian K. Shoichet, Adolfo García‐Sastre, Adam R. Renslo) run by Principal Investigator Nevan Krogan. This work was also partly funded by CRIPT (Center for Research on Influenza Pathogenesis and Transmission), an NIAID funded Center of Excellence for Influenza Research and Response (CEIRR, contract #75N93021C00014) and by supplements to DoD grant W81XWH‐20‐1‐0270 and to NIAID grant U19AI135972 to Adolfo García‐Sastre. Beatrice Ary received the Covid Catalyst Award from the Center for Emerging and Neglected Diseases (CEND). We gratefully acknowledge OpenEye Software for Omega and related tools, and Schrodinger LLC for the Maestro package. We also thank Dr. Randy Albrecht for support with the BSL3 facility and procedures at the ISMMS as well as Richard Cadagan and Daniel Flores for excellent technical assistance. In addition, we thank Dr. Rolf Hilgenfeld for providing the SARS‐CoV‐2 MPro plasmid. We acknowledge the contributions of the UCSF Chemical Underpinnings of Biological Systems (CUBS) 2021 cohort which included Siyi Wang, Isabel Lee, Vineet Mathur, Sham Rampersaud, Luis Santiago, Sara Warrington, and Rose Yang.
Fink EA, Bardine C, Gahbauer S, Singh I, Detomasi TC, White K, et al. Large library docking for novel SARS‐CoV‐2 main protease non‐covalent and covalent inhibitors. Protein Science. 2023;32(8):e4712. 10.1002/pro.4712
Elissa A. Fink, Conner Bardine, Stefan Gahbauer and Isha Singh contributed equally to this study.
Review Editor: Nir Ben‐Tal
Contributor Information
Brian K. Shoichet, Email: bshoichet@gmail.com.
Charles S. Craik, Email: charles.craik@ucsf.edu.
DATA AVAILABILITY STATEMENT
All crystallographic structures have been deposited in the PDB as 8DIB (‘7021), 8DIC (‘9121), 8DID (8252), 8DIE (‘9218), 8DIF (‘7356), 8DIG (‘5548), 8DIH (‘6111), 8DII (SG‐0001). The identities of compounds docked in non‐covalent screens can be found at ZINC15/ZINC20 (http://zinc15.docking.org and http://zinc20.docking.org) and ZINC22 (http://files.docking.org/zinc22/). The covalent compounds have been deposited in http://covalent2022.docking.org along with their DOCKovalent files. Active compounds may be purchased from Enamine Ltd. All other data are available from the corresponding authors on request. DOCK3.7 and DOCK3.8 are freely available for non‐commercial research from the authors; commercial licenses are available via the UC Regents. An open‐source web‐based version of the program is available without restriction to all (https://blaster.docking.org), as are the Arthor and Small World analoging tools used in this study.
REFERENCES
- Adams J, Behnke M, Chen S, Cruickshank AA, Dick LR, Grenier L, et al. Potent and selective inhibitors of the proteasome: dipeptidyl boronic acids. Bioorg. Med. Chem. Lett. 1998;8(4):333–8. 10.1016/S0960-894X(98)00029-8 [DOI] [PubMed] [Google Scholar]
- Afonine PV, Grosse‐Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 2012;68(Pt 4):352–67. 10.1107/S0907444912001308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen WJ, Rizzo RC. Implementation of the hungarian algorithm to account for ligand symmetry and similarity in structure‐based design. J. Chem. Inf. Model. 2014;54(2):518–29. 10.1021/ci400534h [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alon A, Lyu J, Braz JM, Tummino TA, Craik V, O'Meara MJ, et al. Structures of the σ 2 receptor enable docking for bioactive ligand discovery. Nature. 2021;600(7890):759–64. 10.1101/2021.04.29.441652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand K, Ziebuhr J, Wadhwani P, Mesters JR, Hilgenfeld R. Coronavirus main proteinase (3CL pro ) structure: basis for design of anti‐sars drugs. Science. 2003;300(5626):1763–1767. 10.1126/science.1085658 [DOI] [PubMed] [Google Scholar]
- Babaoglu K, Simeonov A, Irwin JJ, Nelson ME, Feng B, Thomas CJ, et al. Comprehensive mechanistic analysis of hits from high‐throughput and docking screens against β‐lactamase. J. Med. Chem. 2008;51(8):2502–11. 10.1021/jm701500e [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender BJ, Gahbauer S, Luttens A, Lyu J, Webb CM, Stein RM, et al. A practical guide to large‐scale docking. Nat Protoc. 2021;16(10):4799–832. 10.1038/s41596-021-00597-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchard JE, Elowe NH, Huitema C, Fortin PD, Cechetto JD, Eltis LD, et al. High‐throughput screening identifies inhibitors of the SARS coronavirus main proteinase. Chem. Biol. 2004;11(10):1445–53. 10.1016/j.chembiol.2004.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boike L, Henning NJ, Nomura DK. Advances in covalent drug discovery. Nat. Rev. Drug Discov. 2022;21(12):881–898. 10.1038/s41573-022-00542-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breidenbach J, Lemke C, Pillaiyar T, Schäkel L, Al Hamwi G, Diett M, et al. Targeting the main protease of SARS‐CoV‐2: from the establishment of high throughput screening to the design of tailored inhibitors. Angew. Chem. Int. Ed Engl. 2021;60(18):10423–9. 10.1002/anie.202016961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti‐tumour immunity. Nature. 2019;575(7781):217–23. 10.1038/s41586-019-1694-1 [DOI] [PubMed] [Google Scholar]
- Carlsson J, Coleman RG, Setola V, Irwin JJ, Fan H, Schlessinger A, et al. Ligand discovery from a dopamine D3 receptor homology model and crystal structure. Nat. Chem. Biol. 2011;7(11):769–78. 10.1038/nchembio.662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Gui C, Luo X, Yang Q, Günther S, Scandella E, et al. Cinanserin Is an inhibitor of the 3C‐like proteinase of severe acute respiratory syndrome coronavirus and strongly reduces virus replication in vitro. J. Virol. 2005;79(11):7095–103. 10.1128/JVI.79.11.7095-7103.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L‐R, Wang Y‐C, Lin YW, Chou S‐Y, Chen S‐F, Liu LT, et al. Synthesis and evaluation of isatin derivatives as effective SARS coronavirus 3CL protease inhibitors. Bioorg. Med. Chem. Lett. 2005;15(12):3058–62. 10.1016/j.bmcl.2005.04.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chodera J, Lee AA, London N, von Delft F. Crowdsourcing drug discovery for pandemics. Nat. Chem. 2020;12(7):581–1. 10.1038/s41557-020-0496-2 [DOI] [PubMed] [Google Scholar]
- Clyde A, Galanie S, Kneller DW, Ma H, Babuji Y, Blaiszik B, et al. High‐throughput virtual screening and validation of a SARS‐CoV‐2 main protease noncovalent inhibitor. J. Chem. Inf. Model. 2022;62(1):116–28. 10.1021/acs.jcim.1c00851 [DOI] [PubMed] [Google Scholar]
- Coleman RG, Carchia M, Sterling T, Irwin JJ, Shoichet BK. Ligand pose and orientational sampling in molecular docking. PLoS One. 2013;8(10):e75992. 10.1371/journal.pone.0075992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4 . The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 1994;50(Pt 5):760–3. 10.1107/S0907444994003112 [DOI] [PubMed] [Google Scholar]
- Costanzi E, Kuzikov M, Esposito F, Albani S, Demitri N, Giabbai B, et al. Structural and biochemical analysis of the dual inhibition of MG‐132 against SARS‐CoV‐2 main protease (Mpro/3CLpro) and human Cathepsin‐L. Int. J. Mol. Sci. 2021;22(21):11779. 10.3390/ijms222111779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai W, Zhang B, Jiang X‐M, Su H, Li J, Zhao Y, et al. Structure‐based design of antiviral drug candidates targeting the SARS‐CoV‐2 main protease. Science. 2020;368(6497):1331–5. 10.1126/science.abb4489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douangamath A, Fearon D, Gehrtz P, Krojer T, Lukacik P, Owen CD, et al. Crystallographic and electrophilic fragment screening of the SARS‐CoV‐2 main protease. Nat. Commun. 2020;11(1):5047. 10.1038/s41467-020-18709-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66(Pt 4):486–501. 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 2006;62(Pt 1):72–82. 10.1107/S0907444905036693 [DOI] [PubMed] [Google Scholar]
- Fairhurst RA, Knoepfel T, Buschmann N, Leblanc C, Mah R, Todorov M, et al. Discovery of roblitinib (FGF401) as a reversible‐covalent inhibitor of the kinase activity of fibroblast growth factor receptor 4. J. Med. Chem. 2020;63(21):12542–73. 10.1021/acs.jmedchem.0c01019 [DOI] [PubMed] [Google Scholar]
- Fink EA, Xu J, Hübner H, Braz JM, Seemann P, Avet C, et al. Structure‐based discovery of nonopioid analgesics acting through the Α2A‐adrenergic receptor. Science. 2022;377(6614):eabn7065. 10.1126/science.abn7065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn JM, Samant N, Schneider‐Nachum G, Bakan DT, Yilmaz NK, Schiffer CA, et al. Comprehensive fitness landscape of SARS‐CoV‐2 Mpro reveals insights into viral resistance mechanisms. eLife. 2022;11:e77433. 10.7554/eLife.77433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gahbauer S, Correy GJ, Schuller M, Ferla MP, Doruk YU, Rachman M, et al. Iterative computational design and crystallographic screening identifies potent inhibitors targeting the Nsp3 macrodomain of SARS‐CoV‐2. Proc. Natl. Acad. Sci. 2023;120(2):e2212931120. 10.1101/2022.06.27.497816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher K, Sharp K. Electrostatic contributions to heat capacity changes of DNA‐ligand binding. Biophys. J. 1998;75(2):769–76. 10.1016/S0006-3495(98)77566-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaulton A, Bellis LJ, Bento AP, Chambers J, Davies M, Hersey A, et al. ChEMBL: a large‐scale bioactivity database for drug discovery. Nucleic Acids Res. 2012;40(D1):D1100–7. 10.1093/nar/gkr777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge R, Shen Z, Yin J, Chen W, Zhang Q, An Y, et al. Discovery of SARS‐CoV‐2 main protease covalent inhibitors from a DNA‐encoded library selection. SLAS Discov. 2022;27(2):79–85. 10.1016/j.slasd.2022.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh AK, Gong G, Grum‐Tokars V, Mulhearn DC, Baker SC, Coughlin M, et al. Design, synthesis and antiviral efficacy of a series of potent chloropyridyl ester‐derived SARS‐CoV 3CLpro inhibitors. Bioorg. Med. Chem. Lett. 2008;18(20):5684–8. 10.1016/j.bmcl.2008.08.082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz DH, Choe Y, Hansell E, Chen YT, McDowell M, Jonsson CB, et al. Substrate specificity profiling and identification of a new class of inhibitor for the major protease of the SARS coronavirus. Biochemistry. 2007;46(30):8744–52. 10.1021/bi0621415 [DOI] [PubMed] [Google Scholar]
- Gu S, Smith MS, Yang Y, Irwin JJ, Shoichet BK. Ligand strain energy in large library docking. J. Chem. Inf. Model. 2021;61(9):4331–4341. 10.1101/2021.04.06.438722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han SH, Goins CM, Arya T, Shin W‐J, Maw J, Hooper A, et al. Structure‐based optimization of ML300‐derived, noncovalent inhibitors targeting the severe acute respiratory syndrome coronavirus 3CL protease (SARS‐CoV‐2 3CLpro). J. Med. Chem. 2022;65(4):2880–904. 10.1021/acs.jmedchem.1c00598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilmann E, Costacurta F, Moghadasi SA, Ye C, Pavan M, Bassani D, et al. SARS‐CoV‐2 3CLpro mutations selected in a VSV‐based system confer resistance to Nirmatrelvir, Ensitrelvir, and GC376. Sci. Transl. Med. 2023;15(678):eabq7360. 10.1126/scitranslmed.abq7360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman RL, Kania RS, Brothers MA, Davies JF, Ferre RA, Gajiwala KS, et al. Discovery of ketone‐based covalent inhibitors of coronavirus 3CL proteases for the potential therapeutic treatment of COVID‐19. J. Med. Chem. 2020;63(21):12725–47. 10.1021/acs.jmedchem.0c01063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Lewandowski EM, Tan H, Zhang X, Morgan RT, Zhang X, et al. Naturally occurring mutations of SARS‐CoV‐2 main protease confer drug resistance to nirmatrelvir. BioRxiv Prepr. Serv. Biol. 2022;497978. 10.1101/2022.06.28.497978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X‐P, Karpiak J, Kroeze WK, Zhu H, Chen X, Moy SS, et al. Allosteric ligands for the pharmacologically dark receptors GPR68 and GPR65. Nature. 2015;527(7579):477–83. 10.1038/nature15699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iketani S, Hong SJ, Sheng J, Bahari F, Culbertson B, Atanaki FF, et al. The functional landscape of SARS‐CoV‐2 3CL protease. BioRxiv Prepr. Serv. Biol. 2022;497404. 10.1101/2022.06.23.497404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin JJ, Tang KG, Young J, Dandarchuluun C, Wong BR, Khurelbaatar M, et al. ZINC20—a free ultralarge‐scale chemical database for ligand discovery. J. Chem. Inf. Model. 2020;60(12):6065–73. 10.1021/acs.jcim.0c00675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs J, Grum‐Tokars V, Zhou Y, Turlington M, Saldanha SA, Chase P, et al. Discovery, synthesis, and structure‐based optimization of a series of N‐(Tert‐Butyl)‐2‐(N‐Arylamido)‐2‐(Pyridin‐3‐Yl) acetamides (ML188) as potent noncovalent small molecule inhibitors of the severe acute respiratory syndrome coronavirus (SARS‐CoV) 3CL protease. J. Med. Chem. 2013;56(2):534–46. 10.1021/jm301580n [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z, Du X, Xu Y, Deng Y, Liu M, Zhao Y, et al. Structure of Mpro from SARS‐CoV‐2 and discovery of its inhibitors. Nature. 2020;582(7811):289–93. 10.1038/s41586-020-2223-y [DOI] [PubMed] [Google Scholar]
- Jochmans D, Liu C, Donckers K, Stoycheva A, Boland S, Stevens SK, et al. The substitutions L50F, E166A and L167F in SARS‐CoV‐2 3CLpro are selected by a protease inhibitor in vitro and confer resistance to nirmatrelvir. MBio. 2023;14(1):e02815‐22. 10.1101/2022.06.07.495116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010;66(Pt 2):125–32. 10.1107/S0907444909047337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan AL, Confair DN, Kim K, Barros‐Álvarez X, Rodriguiz RM, Yang Y, et al. Bespoke library docking for 5‐HT2A receptor agonists with antidepressant activity. Nature. 2022;610(7932):582–591. 10.1038/s41586-022-05258-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Lovell S, Tiew K‐C, Mandadapu SR, Alliston KR, Battaile KP, et al. Broad‐spectrum antivirals against 3C or 3C‐like proteases of picornaviruses, noroviruses, and coronaviruses. J. Virol. 2012;86(21):11754–62. 10.1128/JVI.01348-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korczynska M, Clark MJ, Valant C, Xu J, Moo EV, Albold S, et al. Structure‐based discovery of selective positive allosteric modulators of antagonists for the M2 muscarinic acetylcholine receptor. Proc. Natl. Acad. Sci. 2018;115(10):E2419–E2428. 10.1073/pnas.1718037115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lak P, O'Donnell H, Du X, Jacobson MP, Shoichet BK. A crowding barrier to protein inhibition in colloidal aggregates. J. Med. Chem. 2021;64(7):4109–16. 10.1021/acs.jmedchem.0c02253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levit Kaplan A, Strachan RT, Braz JM, Craik V, Slocum S, Mangano T, et al. Structure‐based design of a chemical probe set for the 5‐HT 5A serotonin receptor. J. Med. Chem. 2022;65(5):4201–17. 10.1021/acs.jmedchem.1c02031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C, Kwong AD, Perni RB. Discovery and development of VX‐950, a novel, covalent, and reversible inhibitor of hepatitis C virus NS3.4A serine protease. Infect. Disord. Drug Targets. 2006;6(1):3–16. 10.2174/187152606776056706 [DOI] [PubMed] [Google Scholar]
- London N, Miller RM, Krishnan S, Uchida K, Irwin JJ, Eidam O, et al. Covalent docking of large libraries for the discovery of chemical probes. Nat. Chem. Biol. 2014;10(12):1066–72. 10.1038/nchembio.1666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Chen SA, Khan MB, Brassard R, Arutyunova E, Lamer T, et al. Crystallization of feline coronavirus Mpro with GC376 reveals mechanism of inhibition. Front. Chem. 2022;10:852210. 10.3389/fchem.2022.852210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luttens A, Gullberg H, Abdurakhmanov E, Vo DD, Akaberi D, Talibov VO, et al. Ultralarge virtual screening identifies SARS‐CoV‐2 main protease inhibitors with broad‐spectrum activity against coronaviruses. J. Am. Chem. Soc. 2022;144(7):2905–20. 10.1021/jacs.1c08402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyu J, Wang S, Balius TE, Singh I, Levit A, Moroz YS, et al. Ultra‐large library docking for discovering new chemotypes. Nature. 2019;566(7743):224–9. 10.1038/s41586-019-0917-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C, Sacco MD, Hurst B, Townsend JA, Hu Y, Szeto T, et al. Boceprevir, GC‐376, and calpain Inhibitors II, XII inhibit SARS‐CoV‐2 viral replication by targeting the viral main protease. Cell Res. 2020;30(8):678–92. 10.1038/s41422-020-0356-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhavi Sastry G, Adzhigirey M, Day T, Annabhimoju R, Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013;27(3):221–34. 10.1007/s10822-013-9644-8 [DOI] [PubMed] [Google Scholar]
- Manglik A, Lin H, Aryal DK, McCorvy JD, Dengler D, Corder G, et al. Structure‐based discovery of opioid analgesics with reduced side effects. Nature. 2016;537(7619):185–90. 10.1038/nature19112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40(Pt 4):658–74. 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGovern SL, Helfand BT, Feng B, Shoichet BK. A specific mechanism of nonspecific inhibition. J. Med. Chem. 2003;46(20):4265–72. 10.1021/jm030266r [DOI] [PubMed] [Google Scholar]
- Meng EC, Shoichet BK, Kuntz ID. Automated docking with grid‐based energy evaluation. J. Comput. Chem. 1992;13(4):505–24. 10.1002/jcc.540130412 [DOI] [Google Scholar]
- Metcalf B, Chuang C, Dufu K, Patel MP, Silva‐Garcia A, Johnson C, et al. Discovery of GBT440, an orally bioavailable R‐state stabilizer of sickle cell hemoglobin. ACS Med. Chem. Lett. 2017;8(3):321–6. 10.1021/acsmedchemlett.6b00491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moghadasi SA, Esler MA, Otsuka Y, Becker JT, Moraes SN, Anderson CB, et al. Gain‐of‐signal assays for probing inhibition of SARS‐CoV‐2 M pro /3CL pro in living cells. mBio. 2022;13(3):e00784–22. 10.1128/mbio.00784-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moghadasi SA, Heilmann E, Khalil A, Nnabuife C, Kearns F, Ye C, et al. Transmissible SARS‐CoV‐2 variants with resistance to clinical protease inhibitors. Sci. Adv. 2023;9:eade8778. 10.1126/sciadv.ade8778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon P, Boike L, Dovala D, Henning NJ, Knapp M, Spradlin JN, et al. Discovery of potent pyrazoline‐based covalent SARS‐CoV‐2 main protease inhibitors. ChemBioChem. 2023;24:e202300116. 10.1101/2022.03.05.483025 [DOI] [PubMed] [Google Scholar]
- Mukherjee P, Desai P, Ross L, White EL, Avery MA. Structure‐based virtual screening against SARS‐3CL(pro) to identify novel non‐peptidic hits. Bioorg. Med. Chem. 2008;16(7):4138–49. 10.1016/j.bmc.2008.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Skubák P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 2011;67(Pt 4):355–67. 10.1107/S0907444911001314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mysinger MM, Shoichet BK. Rapid context‐dependent ligand desolvation in molecular docking. J. Chem. Inf. Model. 2010;50(9):1561–73. 10.1021/ci100214a [DOI] [PubMed] [Google Scholar]
- Mysinger MM, Weiss DR, Ziarek JJ, Gravel S, Doak AK, Karpiak J, et al. Structure‐based ligand discovery for the protein‐protein interface of chemokine receptor CXCR4. Proc. Natl. Acad. Sci. 2012;109(14):5517–22. 10.1073/pnas.1120431109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donnell HR, Tummino TA, Bardine C, Craik CS, Shoichet BK. Colloidal aggregators in biochemical SARS‐CoV‐2 repurposing screens. J. Med. Chem. 2021;64(23):17530–9. 10.1021/acs.jmedchem.1c01547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen DR, Allerton CMN, Anderson AS, Aschenbrenner L, Avery M, Berritt S, et al. An oral SARS‐CoV‐2 Mpro inhibitor clinical candidate for the treatment of COVID‐19. Science. 2021;374(6575):1586–93. 10.1126/science.abl4784 [DOI] [PubMed] [Google Scholar]
- Pillaiyar T, Manickam M, Namasivayam V, Hayashi Y, Jung S‐H. An overview of severe acute respiratory syndrome–coronavirus (SARS‐CoV) 3CL protease inhibitors: peptidomimetics and small molecule chemotherapy. J. Med. Chem. 2016;59(14):6595–628. 10.1021/acs.jmedchem.5b01461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purkey H. Abstract ND11: discovery of GDC‐6036, a clinical stage treatment for KRAS G12C‐positive cancers. Cancer Res. 2022;82(Suppl 12):ND11–1. 10.1158/1538-7445.AM2022-ND11 [DOI] [Google Scholar]
- Qiao J, Li Y‐S, Zeng R, Liu F‐L, Luo R‐H, Huang C, et al. SARS‐CoV‐2 Mpro inhibitors with antiviral activity in a transgenic mouse model. Science. 2021;371(6536):1374–8. 10.1126/science.abf1611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathnayake AD, Zheng J, Kim Y, Perera KD, Mackin S, Meyerholz DK, et al. 3C‐like protease inhibitors block coronavirus replication in vitro and improve survival in MERS‐CoV‐infected mice. Sci. Transl. Med. 2020;12(557):eabc5332. 10.1126/scitranslmed.abc5332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rut W, Groborz K, Zhang L, Sun X, Zmudzinski M, Pawlik B, et al. SARS‐CoV‐2 Mpro inhibitors and activity‐based probes for patient‐sample imaging. Nat. Chem. Biol. 2021;17(2):222–8. 10.1038/s41589-020-00689-z [DOI] [PubMed] [Google Scholar]
- Sadybekov AA, Sadybekov AV, Liu Y, Iliopoulos‐Tsoutsouvas C, Huang X‐P, Pickett J, et al. Synthon‐based ligand discovery in virtual libraries of over 11 billion compounds. Nature. 2022;601(7893):452–9. 10.1038/s41586-021-04220-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasi VM, Ullrich S, Ton J, Fry SE, Johansen‐Leete J, Payne RJ, et al. Predicting antiviral resistance mutations in SARS‐CoV‐2 main protease with computational and experimental screening. Biochemistry. 2022;61(22):2495–505. 10.1021/acs.biochem.2c00489 [DOI] [PubMed] [Google Scholar]
- Schechter I, Berger A. On the size of the active site in proteases. I. papain. Biochem. Biophys. Res. Commun. 1967;27(2):157–62. 10.1016/S0006-291X(67)80055-X [DOI] [PubMed] [Google Scholar]
- Schuller M, Correy GJ, Gahbauer S, Fearon D, Wu T, Díaz RE, et al. Fragment binding to the Nsp3 macrodomain of SARS‐CoV‐2 identified through crystallographic screening and computational docking. Sci. Adv. 2021;7(16):eabf8711. 10.1126/sciadv.abf8711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaqra AM, Zvornicanin SN, Huang QYJ, Lockbaum GJ, Knapp M, Tandeske L, et al. Defining the substrate envelope of SARS‐CoV‐2 main protease to predict and avoid drug resistance. Nat. Commun. 2022;13(1):3556. 10.1038/s41467-022-31210-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh I, Seth A, Billesbølle CB, Braz J, Rodriguiz RM, Roy K, et al. Structure‐based discovery of conformationally selective inhibitors of the serotonin transporter. Cell. 2023;186(10):2160–2175. 10.1101/2022.06.13.495991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein RM, Kang HJ, McCorvy JD, Glatfelter GC, Jones AJ, Che T, et al. Virtual discovery of melatonin receptor ligands to modulate circadian rhythms. Nature. 2020;579(7800):609–14. 10.1038/s41586-020-2027-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein RM, Yang Y, Balius TE, O'Meara MJ, Lyu J, Young J, et al. Property‐unmatched decoys in docking benchmarks. J. Chem. Inf. Model. 2021;61(2):699–714. 10.1021/acs.jcim.0c00598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterling T, Irwin JJ. ZINC 15—Ligand discovery for everyone. J. Chem. Inf. Model. 2015;55(11):2324–37. 10.1021/acs.jcim.5b00559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The COVID Moonshot Consortium , Achdout H, Aimon A, Bar‐David E, Barr H, Ben‐Shmuel A, et al. Open science discovery of oral non‐covalent SARS‐CoV‐2 main protease inhibitor therapeutics; preprint. Biochemistry; 2020. 10.1101/2020.10.29.339317 [DOI] [Google Scholar]
- Turlington M, Chun A, Tomar S, Eggler A, Grum‐Tokars V, Jacobs J, et al. Discovery of N‐(Benzo[1,2,3]Triazol‐1‐Yl)‐N‐(Benzyl)AcetamidoPhenyl carboxamides as severe acute respiratory syndrome coronavirus (SARS‐CoV) 3CLpro inhibitors: identification of ML300 and noncovalent nanomolar inhibitors with an induced‐fit binding. Bioorg. Med. Chem. Lett. 2013;23(22):6172–7. 10.1016/j.bmcl.2013.08.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unoh Y, Uehara S, Nakahara K, Nobori H, Yamatsu Y, Yamamoto S, et al. Discovery of S‐217622, a noncovalent oral SARS‐CoV‐2 3CL protease inhibitor clinical candidate for treating COVID‐19. J. Med. Chem. 2022;65(9):6499–512. 10.1021/acs.jmedchem.2c00117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vankadara S, Dawson MD, Fong JY, Oh QY, Ang QA, Liu B, et al. A warhead substitution study on the coronavirus main protease inhibitor nirmatrelvir. ACS Med. Chem. Lett. 2022;13(8):1345–50. 10.1021/acsmedchemlett.2c00260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatraman S. Discovery of Boceprevir, a direct‐acting NS3/4A protease inhibitor for treatment of chronic hepatitis C infections. Trends Pharmacol. Sci. 2012;33(5):289–94. 10.1016/j.tips.2012.03.012 [DOI] [PubMed] [Google Scholar]
- Vuong W, Fischer C, Khan MB, van Belkum MJ, Lamer T, Willoughby KD, et al. Improved SARS‐CoV‐2 Mpro inhibitors based on feline antiviral drug GC376: structural enhancements, increased solubility, and micellar studies. Eur. J. Med. Chem. 2021;222:113584. 10.1016/j.ejmech.2021.113584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan X, Yang T, Cuesta A, Pang X, Balius TE, Irwin JJ, et al. Discovery of lysine‐targeted EIF4E inhibitors through covalent docking. J. Am. Chem. Soc. 2020;142(11):4960–4. 10.1021/jacs.9b10377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, He S, Deng W, Zhang Y, Li G, Sun J, et al. Comprehensive Insights into the catalytic mechanism of middle east respiratory syndrome 3C‐like protease and severe acute respiratory syndrome 3C‐like protease. ACS Catal. 2020;10(10):5871–90. 10.1021/acscatal.0c00110 [DOI] [PubMed] [Google Scholar]
- Wang S, Che T, Levit A, Shoichet BK, Wacker D, Roth BL. Structure of the D2 dopamine receptor bound to the atypical antipsychotic drug risperidone. Nature. 2018;555(7695):269–73. 10.1038/nature25758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Wacker D, Levit A, Che T, Betz RM, McCorvy JD, et al. D4 dopamine receptor high‐resolution structures enable the discovery of selective agonists. Science. 2017;358(6361):381–6. 10.1126/science.aan5468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner SJ, Kollman PA, Case DA, Singh UC, Ghio C, Alagona G, et al. A new force field for molecular mechanical simulation of nucleic acids and proteins. J. Am. Chem. Soc. 1984;106(3):765–84. 10.1021/ja00315a051 [DOI] [Google Scholar]
- Westberg M, Su Y, Zou X, Ning L, Hurst B, Tarbet B, et al. Rational design of a new class of protease inhibitors for the potential treatment of coronavirus diseases. bioRxiv. 2020. 10.1101/2020.09.15.275891 [DOI] [Google Scholar]
- Wu C‐Y, King K‐Y, Kuo C‐J, Fang J‐M, Wu Y‐T, Ho M‐Y, et al. Stable benzotriazole esters as mechanism‐based inactivators of the severe acute respiratory syndrome 3CL protease. Chem. Biol. 2006;13(3):261–8. 10.1016/j.chembiol.2005.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue X, Yu H, Yang H, Xue F, Wu Z, Shen W, et al. Structures of two coronavirus main proteases: implications for substrate binding and antiviral drug design. J. Virol. 2008;82(5):2515–27. 10.1128/JVI.02114-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang KS, Ma XR, Ma Y, Alugubelli YR, Scott DA, Vatansever EC, et al. A quick route to multiple highly potent SARS‐CoV‐2 main protease inhibitors*. ChemMedChem. 2021;16(6):942–8. 10.1002/cmdc.202000924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Chen S‐J, Hsu M‐F, Wu J‐D, Tseng C‐TK, Liu Y‐F, et al. Synthesis, crystal structure, structure−activity relationships, and antiviral activity of a potent SARS coronavirus 3CL protease inhibitor. J. Med. Chem. 2006;49(16):4971–80. 10.1021/jm0603926 [DOI] [PubMed] [Google Scholar]
- Zaidman D, Gehrtz P, Filep M, Fearon D, Gabizon R, Douangamath A, et al. An automatic pipeline for the design of irreversible derivatives identifies a potent SARS‐CoV‐2 Mpro inhibitor. Cell Chem. Biol. 2021;28(12):1795–1806.e5. 10.1016/j.chembiol.2021.05.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C‐H, Spasov KA, Reilly RA, Hollander K, Stone EA, Ippolito JA, et al. Optimization of triarylpyridinone inhibitors of the main protease of SARS‐CoV‐2 to low‐nanomolar antiviral potency. ACS Med. Chem. Lett. 2021;12(8):1325–32. 10.1021/acsmedchemlett.1c00326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C‐H, Stone EA, Deshmukh M, Ippolito JA, Ghahremanpour MM, Tirado‐Rives J, et al. Potent noncovalent inhibitors of the main protease of SARS‐CoV‐2 from molecular sculpting of the drug perampanel guided by free energy perturbation calculations. ACS Cent. Sci. 2021;7(3):467–75. 10.1021/acscentsci.1c00039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Huitema C, Niu C, Yin J, James MNG, Eltis LD, et al. Aryl methylene ketones and fluorinated methylene ketones as reversible inhibitors for severe acute respiratory syndrome (SARS) 3C‐like proteinase. Bioorg Chem. 2008;36(5):229–40. 10.1016/j.bioorg.2008.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Lin D, Kusov Y, Nian Y, Ma Q, Wang J, et al. α‐Ketoamides as broad‐spectrum inhibitors of coronavirus and enterovirus replication: structure‐based design, synthesis, and activity assessment. J. Med. Chem. 2020;63(9):4562–78. 10.1021/acs.jmedchem.9b01828 [DOI] [PubMed] [Google Scholar]
- Zhang L, Lin D, Sun X, Curth U, Drosten C, Sauerhering L, et al. Crystal structure of SARS‐CoV‐2 main protease provides a basis for design of improved α‐ketoamide inhibitors. Science. 2020;368(6489):409–12. 10.1126/science.abb3405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao N, Bardine C, Lourenço AL, Wang Y‐H, Huang Y, Cleary SJ, et al. In vivo measurement of granzyme proteolysis from activated immune cells with PET. ACS Cent. Sci. 2021;7(10):1638–49. 10.1021/acscentsci.1c00529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L, George S, Schmidt MF, Al‐Gharabli SI, Rademann J, Hilgenfeld R. Peptide aldehyde inhibitors challenge the substrate specificity of the SARS‐coronavirus main protease. Antiviral Res. 2011;92(2):204–12. 10.1016/j.antiviral.2011.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supporting Information.
Data Availability Statement
All crystallographic structures have been deposited in the PDB as 8DIB (‘7021), 8DIC (‘9121), 8DID (8252), 8DIE (‘9218), 8DIF (‘7356), 8DIG (‘5548), 8DIH (‘6111), 8DII (SG‐0001). The identities of compounds docked in non‐covalent screens can be found at ZINC15/ZINC20 (http://zinc15.docking.org and http://zinc20.docking.org) and ZINC22 (http://files.docking.org/zinc22/). The covalent compounds have been deposited in http://covalent2022.docking.org along with their DOCKovalent files. Active compounds may be purchased from Enamine Ltd. All other data are available from the corresponding authors on request. DOCK3.7 and DOCK3.8 are freely available for non‐commercial research from the authors; commercial licenses are available via the UC Regents. An open‐source web‐based version of the program is available without restriction to all (https://blaster.docking.org), as are the Arthor and Small World analoging tools used in this study.