

Abstract
An increased understanding of how the acceptor site in Gcn5‐related N‐acetyltransferase (GNAT) enzymes recognizes various substrates provides important clues for GNAT functional annotation and their use as chemical tools. In this study, we explored how the PA3944 enzyme from Pseudomonas aeruginosa recognizes three different acceptor substrates, including aspartame, NANMO, and polymyxin B, and identified acceptor residues that are critical for substrate specificity. To achieve this, we performed a series of molecular docking simulations and tested methods to identify acceptor substrate binding modes that are catalytically relevant. We found that traditional selection of best docking poses by lowest S scores did not reveal acceptor substrate binding modes that were generally close enough to the donor for productive acetylation. Instead, sorting poses based on distance between the acceptor amine nitrogen atom and donor carbonyl carbon atom placed these acceptor substrates near residues that contribute to substrate specificity and catalysis. To assess whether these residues are indeed contributors to substrate specificity, we mutated seven amino acid residues to alanine and determined their kinetic parameters. We identified several residues that improved the apparent affinity and catalytic efficiency of PA3944, especially for NANMO and/or polymyxin B. Additionally, one mutant (R106A) exhibited substrate inhibition toward NANMO, and we propose scenarios for the cause of this inhibition based on additional substrate docking studies with R106A. Ultimately, we propose that this residue is a key gatekeeper between the acceptor and donor sites by restricting and orienting the acceptor substrate within the acceptor site.
Keywords: acetylation, aspartame, docking visualization, enzyme kinetics, Gcn5‐related N‐acetyltransferase, GNAT, molecular docking, polymyxin B, substrate docking
1. INTRODUCTION
One important family of enzymes that utilizes a distinctive protein scaffold to catalyze diverse acylation reactions is the Gcn5‐related N‐acetyltransferase (GNAT) superfamily. These enzymes transfer an acyl group from a donor molecule to an acceptor molecule, which bind within specific sites of the protein. A wide range of acceptor substrates are acylated by GNATs, including proteins, antibiotics, RNA, polyamines, and other metabolites (Burckhardt & Escalante‐Semerena, 2020), yet many protein members within this family remain uncharacterized. Major hurdles to studying GNAT functions include inconsistent conservation of these enzymes across diverse genomes and limited genomic annotations of functions. GNATs can also exhibit low sequence identities even between proteins with similar functions, which adds to the complexity of computational annotation of genomes. Additionally, small molecule substrates can sometimes be identified in vitro, but they are not necessarily reflective of enzyme physiological function in vivo. Structurally, GNATs adopt a common fold where the donor site has remained relatively conserved; however, the acceptor site can vary dramatically, which enables these enzymes to acylate quite diverse substrates. An additional level of complexity of GNATs is that many of them are also capable of modifying more than one substrate, and the promiscuous nature of some GNATs makes it more challenging to determine their native substrates in vitro.
A GNAT that exhibits substrate promiscuity and has become a model enzyme for exploring GNAT substrate specificity and mechanisms in our laboratory is the PA3944 enzyme from Pseudomonas aeruginosa. We previously showed that this enzyme acetylates multiple substrates, including polymyxins B and E (colistin) on the 3‐Dab residue (Czub et al., 2018), a synthetically designed and prepared polymyxin substrate analog (N‐(2‐aminoethyl)‐N‐methyloctanamide, NANMO) (Baumgartner et al., 2021), and the dipeptide sweetener aspartame (Czub et al., 2018; Kuhn et al., 2013). Although some GNATs provide a link between antibiotic acetylation and antibiotic resistance, PA3944's ability to acetylate polymyxin B does not enable P. aeruginosa to survive in its presence, nor does polymyxin B appear to be a physiological substrate for the enzyme (Czub et al., 2018). Regardless, we were able to use these substrates to elucidate a novel chemical and kinetic mechanism for this enzyme (Baumgartner et al., 2021), wherein we showed that S148 was critical for catalysis and acts as an acyl intermediate during the hybrid ping–pong kinetic reaction. Additionally, we revealed that a specific residue in the acceptor site (E102) is important for substrate specificity toward polymyxin B and NANMO, despite the fact that E102 does not act as a general base in the reaction (Baumgartner et al., 2021).
Herein, we expanded our study of the role of several PA3944 acceptor site residues and assessed their significance for substrate specificity toward polymyxin B, NANMO, and aspartame. Our experiments involved in vitro enzyme kinetic assays, which were used to evaluate the importance of seven acceptor site residues for discriminating between three different known substrates. Additionally, we used in silico molecular docking studies to observe the binding modes of these substrates in the acceptor site in the presence or absence of AcCoA/CoA or an acyl enzyme intermediate (AcS148). We also used an approach to visualize docking results of complex molecules in a simplified manner, and we designed a method for selecting poses of molecules that were near specific active site residues. These tools were also useful for easily assessing whether the docked molecules were located in the donor or acceptor sites of the protein. Our results provide a much clearer picture of the acceptor site of the PA3944 enzyme and expand our knowledge of the diverse architectures of GNAT acceptor sites, which is critical in enabling directed evolution of GNAT enzymes for tailored industrial or chemical purposes.
2. RESULTS
2.1. Kinetic characterization of the WT PA3944 enzyme toward aspartame
Our previous screening assays showed that the PA3944 enzyme acetylates polymyxin B/colistin, aspartame, and a synthetic substrate analog of polymyxin B called NANMO (N‐(2‐aminoethyl)‐N‐methyloctanamide hydrochloride) (Czub et al., 2018). Given our unexpected results that the PA3944 enzyme uses a hybrid ping–pong mechanism for catalysis (Baumgartner et al., 2021), we wanted to fully characterize the WT enzyme toward aspartame as well. Therefore, we performed enzyme kinetics to obtain substrate saturation curves of the WT PA3944 enzyme toward aspartame and compared its activity to our previous results with polymyxin B and NANMO (Baumgartner et al., 2021). Overall, the WT enzyme exhibited a similar catalytic efficiency toward all three substrates (Table 1); its activity toward aspartame was slightly higher than polymyxin B and NANMO, but it had a ~1.5‐fold higher apparent affinity toward NANMO (Figure 1a,b, Table 1). Similar to our previous results with polymyxin B and NANMO substrates, the S148A mutant was inactive toward aspartame (data not shown), indicating S148 is critical for PA3944 WT activity toward all three substrates. Since the dipeptide aspartame substrate is structurally distinct from NANMO and polymyxin B, we investigated whether the enzyme would utilize a similar kinetic mechanism when aspartame was the substrate. In theory, a kinetic mechanism should not be altered for different substrates, but we did observe some small discrepancies in our previous study (Baumgartner et al., 2021). For example, the best kinetic model that fits the data we previously collected when polymyxin B was the acceptor substrate was hybrid ping–pong, whereas we could not discriminate between ping–pong and hybrid ping–pong models due to their similar AICc and relative likelihood values when NANMO was the acceptor substrate. To determine the kinetic model that best fit the kinetic data when aspartame is the acceptor substrate, we generated a series of kinetic curves at varying concentrations of AcCoA and aspartame and fitted the data to a set of kinetic models as described previously (Baumgartner et al., 2021). Similar to our earlier data with polymyxin B, the kinetic model that best fit the data when aspartame was the acceptor substrate was a hybrid ping–pong kinetic mechanism (Table 2, Figure S1). Therefore, the hybrid ping–pong model explains how the PA3944 enzyme can acetylate all three structurally distinct substrates using a combination of direct transfer and ping–pong mechanistic capabilities.
TABLE 1.
PA3944 wild‐type and mutant enzyme kinetic parameters toward polymyxin B, NANMO, and aspartame.
| Enzyme | Polymyxin B | NANMO | Aspartame | ||||||
|---|---|---|---|---|---|---|---|---|---|
| K m (mM) | k cat (s−1) | k cat/K m (M−1 s−1) | K m (mM) | k cat (s−1) | k cat/K m (M−1 s−1) | K m (mM) | k cat (s−1) | k cat/K m (M−1 s−1) | |
| WT a | 1.68 ± 0.07 | 0.51 | 3.0 × 102 | 1.07 ± 0.03 | 0.41 | 3.8 × 102 | 1.68 ± 0.07 | 0.61 | 3.6 × 102 |
| R59A | 0.708 ± 0.058 | 0.44 | 6.2 × 102 | 1.34 ± 0.09 | 0.39 | 2.9 × 102 | 2.00 ± 0.15 | 0.32 | 1.5 × 102 |
| E102A a | 1.05 ± 0.05 | 0.06 | 5.7 × 101 | 0.101 ± 0.013 | 0.15 | 1.5 × 103 | 0.346 ± 0.050 | 0.09 | 2.6 × 102 |
| R106A b | 0.107 ± 0.007 | 0.11 | 1.0 × 103 | 0.083 ± 0.007 | 0.16 | 1.9 × 103 | 7.74 ± 0.53 | 0.06 | 7.8 × 100 |
| T141A | 2.12 ± 0.18 | 0.53 | 2.5 × 102 | 1.40 ± 0.09 | 0.32 | 2.3 × 102 | 4.17 ± 0.20 | 0.19 | 4.3 × 101 |
| H167A | 0.122 ± 0.010 | 0.21 | 1.7 × 103 | 0.323 ± 0.027 | 0.14 | 4.3 × 102 | 3.38 ± 0.23 | 0.48 | 1.4 × 102 |
| L169A | 0.332 ± 0.039 | 0.42 | 1.3 × 103 | 0.871 ± 0.047 | 0.37 | 4.3 × 102 | 1.83 ± 0.12 | 0.65 | 3.5 × 102 |
| H179A | 0.250 ± 0.048 | 0.20 | 8.0 × 102 | 0.329 ± 0.035 | 0.27 | 8.2 × 102 | 1.64 ± 0.10 | 0.34 | 1.9 × 102 |
Data for WT and E102A toward polymyxin B and NANMO were previously reported (Baumgartner et al., 2021).
Significant substrate inhibition of the R106A mutant toward NANMO was observed and data were fitted to the biphasic Hill (Bi‐Hill) equation where y is velocity, x is concentration of substrate, P m is the maximal velocity, K a is the concentration of substrate at half the maximal activity for activation (reported as K m in the table), K i is the concentration of substrate at half the maximal activity for inhibition, H a is the Hill coefficient for activation, and H i is the Hill coefficient for inhibition. K i was calculated as 0.332 ± 0.066 mM, H a as 2.02 ± 0.33, and H i as 0.47 ± 0.06 from the fitting.
FIGURE 1.

Substrate saturation curves for PA3944 WT and mutant proteins toward aspartame, NANMO, and polymyxin B. The average activity for each protein as a function of substrate concentration (polymyxin B or N‐(2‐aminoethyl)‐N‐methyloctanamide (NANMO) or aspartame) are shown in panels a, c, e, g, i, k, m, o, and q; the standard deviation reflects at least two biological replicates in duplicate. Normalized activity is shown in panels b, d, f, h, j, l, n, p, and r. Panels a–f show all aspartame data with triangles, NANMO with circles, and polymyxin B with squares. WT data are indicated with filled shapes and mutant data are with unfilled shapes. Panels g–l have WT data in black squares, R59A in red circles, and L169A in blue triangles. Panels m–r have WT data in black squares, H167A in green triangles, H179A in orange inverted triangles, and R106A in purple circles. Substrate inhibition of the R106A mutant toward NANMO is shown and data were fitted to the Bi‐Hill equation using non‐linear regression in Origin 2017. All other curves were fitted to the Michaelis–Menten equation in the same program. See Section 4 for more details about the enzyme assays. The WT and E102A data toward polymyxin B and NANMO were previously reported (Baumgartner et al., 2021).
TABLE 2.
Results of fitting selected kinetic models to a series of PA3944 WT enzyme kinetic curves toward aspartame.
| Acceptor substrate | Model | AICc | ΔAICc | Relative likelihood |
|---|---|---|---|---|
| Aspartame | Random | −346 | 103 | 4.30 × 10−23 |
| Ordered AB | −278 | 171 | 7.38 × 10−38 | |
| Ordered BA | −305 | 144 | 5.38 × 10−32 | |
| Ping–pong | −433 | 16 | 3.35 × 10−4 | |
| Hybrid | −449 | 0 | 1 |
Note: Enzyme activity data were collected by reacting WT enzyme with varying concentrations of aspartame from 0 to 7.5 mM and different concentrations of AcCoA (0.1, 0.25, 0.5, and 1 mM). Models used for fitting were described previously (Baumgartner et al., 2021). The hybrid ping–pong model is bolded because it is the one with the lowest corrected Akaike Information Criterion (AICc) and ΔAICc values and the highest relative likelihood, which was calculated using the equation .
2.2. Molecular docking studies with the PA3944 WT enzyme and three acceptor substrates
To learn more about how the PA3944 enzyme recognizes these three diverse substrates and map key residues in the acceptor site that are important for substrate recognition, we performed a series of molecular docking studies described below. We used the crystal structure of the PA3944 enzyme in complex with CoA in the donor site (PDB ID: 6EDV) and docked acceptor substrate molecules into the acceptor site; the donor and acceptor sites of the protein are located in distinct pockets (Figure 2a). Since we know the enzyme acetylates substrates by direct transfer or via an acyl‐enzyme intermediate, we docked the three fully protonated substrates aspartame, NANMO, and polymyxin B into this structure under three different conditions: (1) in the presence of AcCoA, (2) in the presence of CoA and AcS148 (AcS148 + CoA), and (3) in the absence of CoA and in the presence of AcS148 (AcS148 − CoA). These conditions represent different possible occupancies for the donor/product AcCoA/CoA during various steps of the kinetic mechanism and how AcCoA/CoA may influence acceptor substrate binding.
FIGURE 2.

Location of donor and acceptor sites of the PA3944 WT enzyme and modeled tetrahedral AcCoA‐polymyxin B intermediate. (a) WT PA3944 crystal structure with CoA occupying the donor site (PDB ID: 6EDV). Donor and acceptor sites and CoA are labeled. (b) Simplified diagram showing the general location of selected amino acids in acceptor site of PA3944. Amino acids are represented as circles using the color scheme of L169 in blue, H167 in green, H179 in orange, T141 in pink, E102 in yellow, S148 in black, R106 in purple, and R59 in red. (c) PA3944 crystal structure with the modeled tetrahedral AcCoA‐polymyxin B intermediate (cyan sticks) spanning both donor and acceptor sites. (d) Key acceptor site residues surrounding the modeled AcCoA‐polymyxin B tetrahedral intermediate. All residues are colored as described in panel b and the tetrahedral intermediate is colored in cyan. (e) Interactions between residues of the acceptor site and the modeled AcCoA‐polymyxin B tetrahedral intermediate. Atoms of the tetrahedral intermediate that form H‐bonds with residues lining the acceptor site are indicated with four‐digit numbers in purple. Residues of the cyclic peptide polymyxin B are labeled in red. H‐bonding interactions between the tetrahedral intermediate and acceptor site residues are indicated with black arrows. Amino acids selected for mutation and further study are shown in rainbow colors as described in panel b and also denoted with stars.
In this study, we were most interested in identifying docking poses of substrates that would reveal information about how they may bind during catalysis and which residues could be important for substrate recognition and specificity. While docked molecules were generally located in the acceptor site of the protein, we found that many of them were located in regions that were not amenable to a productive acetylation reaction, for example, on the surface, away from the active site, or were located within the donor site. Therefore, we utilized the following approach to analyze the docking data. First, we sorted all of the docked substrate molecules in the PA3944 WT protein by lowest (best/most stable) S scores (Table S1 ) and selected the top 10 poses by lowest S score for further analysis. Next, we measured the distance between the acceptor substrate amine nitrogen atom and the acetyl donor (either AcCoA or AcS148) carbonyl carbon atom (see Section 4 for details) for these 10 poses and compared their distances and S scores across all docking conditions (Figure 3). We found the distances between these atoms for each condition were widely distributed and placed the acceptor amine further from the acetyl donor regardless of docking condition (Figure 3). On average, we found NANMO docked closer to the acetyl donor than aspartame or polymyxin but most poses were still far from the donor (Figure 3). These results showed that focusing on poses with lowest S scores did not reveal binding modes of acceptor substrates that would help us identify residues important for substrate specificity and instead were more reflective of possible inhibitor binding modes.
FIGURE 3.

Box plots of substrate docking and selection of top 10 poses by either closest distance or lowest S score in the PA3944 WT and R106A enzymes. Three substrates (aspartame, NANMO, and polymyxin B) were docked into the WT crystal structure (PDB ID: 6EDV) and R106A mutant generated in MOE based on the WT structure. The top 10 poses by closest distance between the acceptor amine and the donor carbonyl carbon and by lowest S score were selected for further analysis. The data for polymyxin B were sorted based on the distance between the 3‐Dab amine and carbonyl carbon of the acetyl donor. The distances between the acceptor amines and donor carbonyl carbon and their corresponding S scores were plotted as a function of the presence or absence of AcCoA/CoA and non‐acetylated/acetylated S148 (AcS148) during docking experiments. The box plots are colored by ligand with solid fill for the WT enzyme and no fill for the R106A enzyme distances. S scores are shown with a diagonal pattern in the box. Data for aspartame are in pink, NANMO are in teal, and polymyxin B are in purple. Distance and S score data were automatically binned in Origin. Data points located on a horizontal line belong to the same bin. Boxes show the span of quartile 1 (lower bound of box) to quartile 3 (upper bound of box). Medians are shown as a solid line across a box and whiskers are drawn from the top of the box to the highest point and the bottom of the box to the lowest point.
To identify docking poses that were closer to the donor, we changed strategies and sorted all of the docked substrates by distance between the acceptor substrate amine nitrogen atom and the acetyl donor carbonyl carbon atom (Table S1). We selected the top 10 poses by distance between these two atoms for further analysis and found that on average the three substrates were located closer to the acetyl donor molecule than when we selected poses by lowest S score (Figures 3 and S2). Our results showed on average aspartame docked closer to the acetyl donor when AcCoA was present compared to the two AcS148 conditions but exhibited less favorable S scores. This indicates aspartame can bind in close enough proximity to AcCoA for a sequential transfer to occur. Additionally, the presence or absence of CoA in the AcS148 docking conditions influenced how close aspartame docked to the donor; when CoA was absent, 40% of the aspartame poses docked closer than when CoA was present (Figure 3). This result follows what is generally expected for a ping–pong kinetic mechanism where CoA leaves prior to acceptor substrate binding. Thus, the collective docking results with aspartame mirror our kinetic data that show the PA3944 enzyme utilizes a hybrid ping–pong kinetic mechanism with sequential and ping–pong paths for transfer. Furthermore, sorting docking poses by distance between acceptor amine and donor yields more relevant binding modes of aspartame for acetyl transfer and thus can be used to identify residues that may be important for substrate specificity.
Next, we applied this same strategy of sorting docking poses by distance rather than S score to analyze the docking results for NANMO and polymyxin B. Compared to aspartame, a different trend in the proximity of NANMO to the donors was observed. We found that all three docking conditions placed the acceptor amine atom of NANMO within ~3–4 Å from the carbonyl carbon atom of all donors (Figure 3). This indicates the identity of the donor and/or presence of CoA product does not significantly influence the ability of NANMO to dock close to the donors and matches our previous kinetic data that showed we could not discriminate between a ping–pong or hybrid ping–pong mechanism for this substrate. When we analyzed the polymyxin B docking results, we found the distance between the polymyxin B 3‐Dab amine nitrogen atom and the acetyl donor carbonyl carbon atom across all conditions was furthest compared to aspartame and NANMO (Figure 3). While some polymyxin B docking poses in the AcCoA docking condition were closer to the donor, none of the poses across all conditions were close enough for acetyl transfer. These poses were, however, closer than when we sorted the data by lowest S score. Based on the collective results across all three substrates and docking conditions, we concluded that sorting docking data by distance rather than S score provided poses that placed the substrates in a more advantageous location for acetylation to occur. Furthermore, this strategy reveals more relevant amino acids that should be queried for their possible contributions to PA3944 substrate specificity.
2.3. Comparative analysis of locations of docked molecules sorted by distance using x, y, z coordinates of specific substrate atoms
Due to the difficulty of analyzing numerous docking poses of complex molecules like polymyxin B within a protein structure, we designed an approach to more easily and quickly visualize trends in docking poses using x, y, z, coordinates of specific atoms within the pdb files (Figure S3). For substrates like polymyxin B that have multiple primary amines, we used a different colored‐coded point in the plot for each Dab nitrogen atom. We also used different colored points to track the terminal carbon of the octanoyl tail of NANMO compared to its primary amine nitrogen to assess the position of the hydrocarbon tail in 3D space. Furthermore, we added a translucent plane to the plots to symbolize the boundary between the donor and acceptor sites of the PA3944 protein and assess where docked substrates bound. While other methods like interaction fingerprints have been used in the past (Deng et al., 2004; Da & Kireev, 2014), our approach allowed us to more easily monitor trends in locations of specific substituents on substrates as well as whether acceptor substrates docked fully into the acceptor site or traversed into the donor site. Our docking results for NANMO showed that the locations of the terminal carbon atom of the octanoyl hydrocarbon chain of NANMO remained in the acceptor site except when CoA was removed (AcS148 − CoA docking condition); the octanoyl tail of some NANMO molecules docks within the donor site in this condition. In contrast, for polymyxin B we observed that the locations of different Dab residues of polymyxin B were scattered throughout the acceptor site with some docking conditions producing poses with more ordering of specific Dab residues than others (Figure S3). None of the docking conditions had polymyxin B in a consistent conformation, and none of the poses were located in the donor site. It appears polymyxin B binding is variable in the PA3944 acceptor site regardless of acetyl donor and can orient itself in multiple conformations within the acceptor site. In total, these results show that tracking specific substituents of complex molecules in a simplified way enables trends across numerous docking poses to be more easily identified.
2.4. Enzyme kinetics with PA3944 acceptor site mutant proteins
Since the polymyxin B substrate is much larger and more complex than NANMO and aspartame, it is possible that the protein requires a significant conformational change to bind polymyxin B in a proper position for acetyl transfer. Therefore, we built a tetrahedral intermediate of AcCoA and polymyxin B, docked it into the PA3944 WT protein, and performed molecular dynamics to help identify an appropriate binding mode for this substrate (Figure 2c). Next, we analyzed this model to determine which acceptor site residues interact with the polymyxin B portion of the tetrahedral intermediate (Figure 2c–e). Based on this analysis, we generated the following mutants that were grouped based on three main areas of the acceptor site: near the active site (E102A, T141A), toward the entrance of the acceptor site (R59A, L169A), and other residues lining the internal region of the acceptor pocket (H167A, H179A, and R106A) (Figure 2b,d). We screened all mutants for activity toward all three substrates (polymyxin B, NANMO, and aspartame) and generated substrate saturation curves to compare kinetic parameters.
2.4.1. E102A and T141A
The E102 and T141 residues are located near the active site of the protein where the donor and acceptor sites meet. First, we examined the E102A mutant activity toward aspartame since our previous studies indicated that the E102A mutant was important for substrate binding and specificity rather than playing a catalytic role (Baumgartner et al., 2021). In this study, we found that the E102A mutant exhibited an improved apparent affinity for aspartame compared to WT by ~5‐fold, and the catalytic efficiency toward aspartame was greater than polymyxin B but less than NANMO for the E102A mutant (Figure 1c,d, Table 1). Thus, NANMO was the preferred substrate for the E102A enzyme, and the apparent affinity of the enzyme toward all substrates was improved by mutating the E102 residue to a smaller and more hydrophobic residue. Next, we tested activity of the T141A mutant toward all three substrates and found that it displayed an ~8‐fold decrease in catalytic efficiency compared to WT when aspartame was the substrate (Figure 1e,f, Table 1). The catalytic efficiencies of the T141A enzyme toward both polymyxin B and NANMO were comparable (~1‐ and ~2‐fold decrease compared to WT, respectively). We concluded that residues near the active site of the protein that do not participate directly in the chemical mechanism can alter the substrate specificity and apparent affinity of the enzyme when mutated (E102A), but not all residues at this location (e.g., T141A) exhibit dramatic effects on activity or substrate specificity.
2.4.2. R59A and L169A
The R59 and L169 residues are located on opposite sides of the entrance to the acceptor site and are approximately 7 Å away from each other in the PDB ID 6EDV crystal structure. R59 is on an alpha helix (α2) and L169 is on a mobile loop between β8 and β9 strands (Figure S4). When we assayed these mutant enzymes for activity, we saw several changes in kinetic parameters depending on the acceptor substrate. For example, the most dramatic alterations to catalytic efficiency or apparent affinity toward the three substrates was observed with polymyxin B. Both R59A and L169A mutants exhibited an increased catalytic efficiency (~2‐ and ~4‐fold, respectively) and an improved apparent affinity (~2‐fold and ~ 5‐fold, respectively) compared to WT when polymyxin B was the substrate (Figure 1g,h, Table 1). Relatively minor changes in catalytic efficiency and apparent affinity were observed when NANMO or aspartame was the acceptor substrate (Figure 1i–l, Table 1). Therefore, replacing a charged residue with a smaller and/or hydrophobic residue (R59A) or reducing the size of hydrophobic residue in the entrance of the acceptor site (L169A) leads to an improved apparent affinity for a large substrate like polymyxin B, but not for the smaller substrates aspartame or NANMO.
2.4.3. H167A and H179A
The H167 and H179 residues line the internal surface of the acceptor site and are located between the T141 and L169 residues on one side of the acceptor pocket (Figure 2b,d). H167 is the first residue on the mobile loop between β8 and β9 while H179 is the second residue on β9 (Figure S4). The H179A mutation improved the catalytic efficiency of the enzyme toward both polymyxin B and NANMO by ~3‐fold and ~ 2‐fold, respectively compared to the WT protein, but decreased catalytic efficiency of the enzyme toward aspartame by ~2‐fold (Figure 1m–r, Table 1). This effect on catalytic efficiency was mainly driven by the improved apparent affinity of the H179A protein for polymyxin B and NANMO compared to WT. The greatest improvement was observed toward polymyxin B (~7‐fold increase). When the adjacent H167 residue was mutated to alanine, this increase in apparent affinity for polymyxin B nearly doubled compared to the H179A mutant (~14‐fold increase compared to WT). The catalytic efficiency was also nearly doubled compared to the H179A (~6‐fold higher than WT) for the H167A mutant. In contrast, when NANMO was the substrate, the apparent affinities of the H167A and H179A mutants were nearly identical and the catalytic efficiencies for the WT and H167A proteins were similar (Figure 1o,p, Table 1). On the other hand, when aspartame is the substrate, a different effect was observed. The catalytic efficiencies of the H179A and H167A mutants decreased ~2‐ and ~3‐fold compared to WT, respectively, and the apparent affinity of H179A for aspartame was nearly identical to WT, whereas H167A showed a 2‐fold decrease compared to WT (Figure 1q,r, Table 1). Therefore, mutating either H167 or H179 to the small hydrophobic alanine residue significantly improves the apparent affinity of the enzymes for both polymyxin B and NANMO, with the greatest improvement toward polymyxin B.
2.4.4. R106A
The R106 residue is located on the opposite side of the acceptor pocket from the H167 and H179 residues and is further inside the pocket than R59 (Figure 2b,d). Surprisingly, the R106A mutant exhibited a ~3‐fold and ~5‐fold increase in catalytic efficiency and an ~16‐fold and ~13‐fold increase in apparent affinity for polymyxin B and NANMO, respectively, compared to WT. However, it had a ~46‐fold decrease in catalytic efficiency and ~5‐fold decrease in apparent affinity toward aspartame (Figures 1m–r and 4, Table 1). In fact, this R106A mutation exhibited the most significant decrease in catalytic efficiency and apparent affinity of all the mutants tested toward aspartame. Additionally, the R106A mutation displayed the highest increased apparent affinity toward both polymyxin B and NANMO than any other mutant. Therefore, reducing the size and/or removing the positive charge in this region of the acceptor site enhances catalytic efficiency and the apparent affinity of the enzyme toward more hydrophobic and complex molecules than the small peptide aspartame; this positive charge is critical for activity toward aspartame. Additionally, the R106A mutant appears to exhibit substrate inhibition at higher concentrations of NANMO (Figure 4). The K i for NANMO was calculated as 0.332 ± 0.066 mM (Table 1).
FIGURE 4.

Normalized activity of PA3944 WT and R106A toward aspartame, NANMO, and polymyxin B. (a) WT activity toward polymyxin B is shown with filled black squares and R106A in open black squares. (b) WT activity toward NANMO is shown with filled purple circles and R106A in open purple circles. (c) WT activity toward aspartame is shown with filled magenta triangles and R106A in open magenta triangles.
2.5. NANMO substrate inhibition and double docking in R106A
Since our kinetic studies showed the R106A enzyme exhibited an enhanced apparent affinity for NANMO but substrate inhibition at higher concentrations, we performed docking studies with the R106A enzyme to generate hypotheses as to how substrate inhibition could occur. We found that when R106 is mutated to alanine, the interior of the acceptor pocket increases in volume and hydrophobicity (Figure 5a). Additionally, the acceptor substrates did not dock identically into the AcSer148 − CoA condition in the R106A model compared to WT (Figures S2, S3, S5). Often, the acceptor substrate was found within the donor site when the top 10 poses by distance were examined. We hypothesized that the observed substrate inhibition was due to NANMO binding in the donor site. Therefore, we focused on a deeper examination of the AcS148 − CoA docking results with NANMO. For docking poses sorted by distance, NANMO docked in a more restricted conformation in the WT structure compared to the R106A model where the conformation of NANMO varies and it can explore alternative conformations in this pocket (Figure 5b,c). However, NANMO is also pulled deeper into the donor site in the R106A model than for WT (Figure 5b). A comparison of docked NANMO molecules sorted by S score showed that NANMO docks into two different locations in the acceptor pocket for the WT protein, whereas all of the NANMO molecules dock almost exclusively in the donor site for the R106A model (Figure 5b,c). Based on these results, we propose three different scenarios for the cause of the substrate inhibition for the R106A enzyme. The first scenario is that the decreased activity may be due to competition between NANMO and AcCoA for binding to the donor site when R106 is mutated. The second scenario is that NANMO may bind close to the alanine residue once R106 is removed and be more difficult to release due to the change in hydrophobicity. The third scenario is that at higher concentrations of NANMO, more than one substrate molecule may bind to the acceptor site.
FIGURE 5.
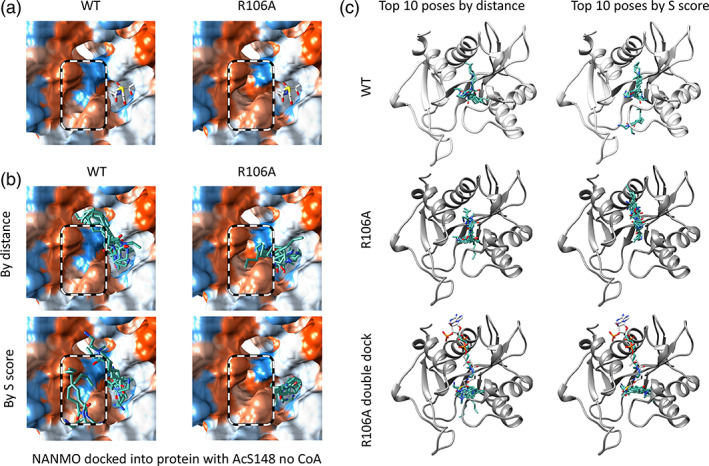
Comparison of WT and R106A acceptor site hydrophobicity and location of docked NANMO molecules. (a) Surface representations of the hydrophobicity of the acceptor site with hydrophilic regions colored in blue and hydrophobic regions colored in orange. AcCoA is shown in white sticks to indicate the location of the donor site of the PA3944 protein. (b) Comparison of top 10 NANMO dockings in AcS148 − CoA condition when sorted by distance between its primary amine and the carbonyl carbon of AcS148 and S score. NANMO is shown in teal sticks. A black and white dashed circle indicates the location of the R106 position for the WT and R106A proteins. (c) Comparison of docked NANMO locations for top 10 docking poses by distance between NANMO primary amine and carbonyl carbon of AcS148 − CoA and S scores for PA3944 WT, R106A, and doubly docked R106A proteins (ribbon representations). A second round of NANMO docking experiments were performed using the R106A enzyme containing a single molecule of NANMO that was selected based on the lowest S score and closest distance to AcS148 in the AcS148 − CoA condition. AcCoA is shown as white sticks to indicate the location of the donor site.
To determine whether a second NANMO could bind to the acceptor site of the R106A protein once another molecule of NANMO was already bound, we performed a second round of docking we termed a “double dock” (Table S2). In this experiment, we selected the pose for NANMO that had the acceptor amine closest to the AcS148 residue from the top 10 poses sorted by S score in the R106A AcS148 − CoA docking condition. This pose happened to be present in the AcCoA donor site. We then performed a second round of NANMO docking into this model and found additional molecules of NANMO could be docked into the acceptor site. When the “double‐docked” poses of NANMO are sorted by distance and S score we saw the conformations of the poses when sorted by S score were elongated across one side of the acceptor pocket (Figure 5c). This more elongated conformation forms significant van der Waals interactions with the alanine residue of R106A and lies in the newly formed hydrophobic pocket. Ultimately, R106 may act as a gatekeeper of the conformation of NANMO in the absence of CoA and prevent possible substrate inhibition through binding multiple substrate molecules due to increased access to van der Waals interactions when R106 is mutated to alanine.
2.6. Interactions of docked substrates with PA3944 protein acceptor site residues as a mechanism for understanding mutant enzyme kinetics results
We next explored the interactions of docked substrates into the WT protein with acceptor site residues that we mutated and screened for kinetic activity (Figure 6, Table S3). The goal was to shed light on how the enzyme uses these residues for substrate recognition and specificity and whether the residue interactions change based on presence of acetyl donor and/or CoA product.
FIGURE 6.

Comparison of catalytic efficiency and K m for WT and mutant PA3944 enzymes toward aspartame, NANMO, and polymyxin B. (a) Fold increase or decrease in catalytic efficiency (CE) compared to WT for each substrate rounded to the nearest whole number. (b) Fold increase or decrease in K m compared to WT for each substrate rounded to the nearest whole number. Red numbers indicate fold decreases and black numbers are fold increases. The shapes for each substrate (triangle, circle, square) are consistent with the substrate saturation curves in Figure 1. (c) Cartoon representation of the total number of interactions (e.g., van der Waals, H‐bonding, and ionic interactions) identified between PA3944 residues and docked aspartame or NANMO using Chimera. The thickness of lines between residues and substrate were based on the total number of combined interactions identified from analyzing the top 10 poses sorted by distance. Line thicknesses range between 1.5 and 16.5 pt where the thickness increases 1 pt for every 10 interactions identified. No line is shown if interactions were not present between the docked molecule and specific residue. Interactions with S148 are shown protruding toward the donor pocket.
2.6.1. Aspartame
We found that aspartame formed considerable interactions with a variety of acceptor site residues when it was docked into the AcS148 − CoA condition compared to the other two conditions (Figure 6). In the AcCoA condition, we observed mostly ionic interactions between R59 and the aspartate sidechain of the dipeptide aspartame. These interactions with R59 became more prominent when the acetyl donor was AcS148. Most of the interactions observed with aspartame and L169 were van der Waals interactions with the peptide backbone of aspartame and were consistent across all three conditions. Mutating R59 to alanine does not eliminate activity toward aspartame, but rather reduces turnover by approximately half (Table 1, Figure 1k). The interactions with L169 may stabilize the aspartame molecule sufficiently for acetylation to occur when R59 is mutated, but L169 is not required for effective turnover or apparent affinity since the catalytic efficiency of L169A is nearly identical to that of WT when aspartame is the substrate. The interactions with R59 and L169 in the presence of AcCoA or CoA may help to orient the acceptor amine of aspartame toward the active site of the protein, but when the donor pocket is unoccupied, these interactions are not sufficient to maintain a proper orientation for acetyl transfer.
2.6.2. NANMO
In contrast to aspartame, NANMO formed significant interactions with a variety of acceptor site residues in all docking conditions. Additionally, with a vacant donor site, NANMO exhibited more frequent interactions with a select set of residues on one side of the acceptor site (Figure 6). In the AcCoA docking results, the orientation of the octanoyl tail of NANMO varies and forms van der Waals interactions with a variety of residues we mutated and screened for kinetic activity throughout the acceptor site. These included R59, E102, R106, T141, and H179. Very few interactions occurred with H167, and no interactions with L169 were observed. All of these residues formed van der Waals interactions with NANMO in the WT AcS148 + CoA docking condition as the molecule adopted a more elongated structure within the acceptor pocket. However, when CoA is removed, NANMO is pushed toward one side of the acceptor pocket and forms interactions with R59, E102, R106, T141. We did not observe any significant change in kinetic activity toward NANMO when R59 or L169 residues were mutated to alanine. However, when H167 and H179 residues were mutated to alanine, we saw a small increase in catalytic efficiency, which was primarily due to an improved K m for NANMO. This improvement may be due to increasing the hydrophobicity of this region of the acceptor pocket to bind NANMO.
3. DISCUSSION
3.1. GNAT acceptor site tunability as a chemical tool
One of the most limiting aspects in predicting protein function from sequence and structure for GNAT enzymes is the lack of understanding of how acceptor substrates are recognized by and bind to the active site. Moreover, key residues required for substrate specificity are not well understood, especially in enzymes with unknown or promiscuous functions and due to varying sizes and amino acid residue compositions of acceptor sites. In this study, we utilized several approaches to learn about roles of specific residues within the acceptor site of the PA3944 enzyme for recognizing and acetylating three diverse substrates aspartame, NANMO, and polymyxin B. Our combinatorial approach reveals remarkable promiscuity of the PA3944 enzyme and residues that enable often significant improvements in apparent affinities and catalytic efficiencies toward these substrates, especially toward polymyxin B and NANMO. These results provide support for the tunability of GNATs for specific functions and highlight the promise of these enzymes as acylating reagents in the chemists’ toolkit with broad, basal activity that can be manipulated through readily prepared mutants.
3.2. Molecular docking and coordinate map approaches to identify catalytically relevant substrate poses
Molecular docking is a powerful tool for predictive ligand binding to proteins. While it is typically utilized to identify energy minima that mimic enzyme‐inhibitor complexes, its use as a tool to explore catalytically relevant substrate binding is not as widespread. The standard approach is to identify the “best pose” in docking studies which relies on a calculated S score that is not necessarily reflective of reactive enzyme‐substrate complexes. Therefore, we compared and contrasted docking S scores representing low energy poses with calculations of distance between reacting groups, specifically between a nucleophilic amine of acceptor substrates and the electrophilic carbonyl carbon in an acyl donor in the PA3944 enzyme. Collectively, our analysis of the two sorting methods across all docking conditions showed that poses sorted based on distance between the acceptor amine nitrogen atom and donor carbonyl carbon atom were closer to the active site but generally had less favorable S scores compared to poses sorted by S score. Additionally, when the poses were sorted by S score instead of distance, the location of the acceptor amine nitrogen atoms in the various poses were more variable for all three substrates in all three docking conditions. These results show that when the substrates dock closer to the active site of the protein where catalysis occurs, the S score can often be less favorable. Thus, selecting docking poses based on distance provides potentially catalytically relevant substrate poses that are less reliant on stable occupancy typical of inhibitor binding. Our results add additional insight to prior studies that have called into question the selection criteria that are used for identifying the “best pose” in molecular docking experiments (Udatha et al., 2012; Ramírez & Caballero, 2018), predictive substrate identification for proteins of unknown function (Hermann et al., 2006) and a greater understanding substrate specificity (Juhl et al., 2009) with docking approaches. However, additional targeted docking studies to more deeply address these issues may be required. Furthermore, our coordinate mapping approach to help track locations of key moieties of complex molecules simplifies data analysis of docking data and provides a more global perspective of possible conformations of substrates in the acceptor sites of GNATs.
3.3. Relationship of docking data to substrate specificity and architecture of acceptor site
Our enzyme kinetics and molecular docking data provide new insight into key acceptor site residues that are critical for substrate specificity and binding and validate our previous kinetic mechanistic data. For example, we found that the specific docking condition greatly impacts the binding interactions between the enzyme and substrate for both aspartame and NANMO (Figure 6). Moreover, performing docking studies under three possible conditions (AcCoA, AcS148 + CoA, and AcS148 − CoA) show that for some substrates the presence of the donor influences the binding location of the acceptor substrate and highlights possible substrate binding modes for the sequential or ping–pong paths of the hybrid kinetic mechanism. In some cases, removal of CoA enabled the acceptor amine nitrogen to dock closer to the acetyl donor than when CoA was present, and for some poses portions of the acceptor molecule protrude into the donor site when CoA is absent. It is possible that the ping–pong path of acetyl transfer where CoA leaves prior to acceptor substrate binding may itself provide a strategy to enable greater substrate promiscuity through acceptor substrate binding that extends into the donor site. On the other hand, residues in a similar location as R106 within GNAT acceptor sites likely contribute to the orientation of and restrict acceptor substrate binding within this site to prevent substrate inhibition and enhance turnover.
4. MATERIALS AND METHODS
4.1. Materials
Acetyl coenzyme A (AcCoA) trilithium salt (Cat#A2181), polymyxin B sulfate salt (Cat#P4932), and aspartame (N‐(L‐α‐aspartyl)‐L‐phenylalanine methyl ester; Cat#A5139) were purchased from Millipore Sigma. N‐(2‐Aminoethyl)‐N‐methyloctanamide hydrochloride (NANMO) was synthesized as described previously (Baumgartner et al., 2021). Substrates were dissolved in nanopure water to create 100 mM stocks for polymyxin B and NANMO and a 25 mM stock for aspartame for kinetic experiments.
4.2. Clones and site directed mutagenesis
The plasmid containing the Pseudomonas aeruginosa PAO1 pa3944 gene (UniProt ID: Q9HX72) previously sub‐cloned into the ampicillin resistant p11 vector (Czub et al., 2018) was used as a template for constructing point mutations of the PA3944 enzyme. The T141A mutant was a generous gift from Karolina Majorek and Wladek Minor at the University of Virginia Charlottesville, and the S148A and E102A mutants were the same as those previously reported (Baumgartner et al., 2021). The remaining mutants (R59A, R106A, N145A, H167A, L169A, and H179A) were constructed using the Phusion Site‐Directed Mutagenesis Kit from Thermo Fisher Scientific and phosphorylated primers at the 5′ position from Integrated DNA Technologies (IDT) following the manufacturer's instructions.
4.3. Protein expression and purification
All WT and mutant proteins were expressed and purified as described previously (Czub et al., 2018). All proteins were purified to near homogeneity as assessed by SDS‐PAGE, and protein concentrations were determined using Beer's Law based on the absorbance at A 280nm and an extinction coefficient of 37,470 M−1 cm−1. The N‐terminal polyhistidine tag was retained on all proteins.
4.4. Steady‐state enzyme kinetics
All enzyme kinetics were performed using a colorimetric assay as previously described (Czub et al., 2018; Kuhn et al., 2013). Reactions (50 μL total) for substrate saturation curves contained varying concentrations of acceptor substrates ranging from 0 to 6 mM polymyxin B, 0 to 4 mM NANMO, and 0 to 10 mM aspartame at a constant concentration of AcCoA (0.5 mM) in 100 mM Tris–HCl pH 8.0 and 100 mM NaCl. The enzymatic reactions were initiated with 10 μL of enzyme, allowed to proceed for 10 min at 37°C, and then were terminated and reacted with DTNB as described (Czub et al., 2018; Kuhn et al., 2013). Enzyme concentrations in each reaction were 1.3 μM of WT toward all three substrates; 0.86, 1.1, and 2.6 μM for R59A; 4.4, 1.8, and 4.4 μM for E102A; 1.5, 1.9, and 9.2 μM for R106A; 0.90, 1.8, and 4.5 μM for T141A; 0.64, 1.3, and 0.4 μM for H167A; 1.3, 1.4 and 1.2 μM for L169A; and 1.3, 1.4 and 1.9 μM for H179A toward polymyxin B, NANMO, and aspartame, respectively. All data were fitted to the Michaelis–Menten equation using non‐linear regression in Origin 2017 with the exception of data for R106A toward NANMO, which was fitted to the Bi‐Hill equation. Details are reported in Table 1. Values for k cat were calculated using the protein molecular weight of 21.9 kDa.
4.5. Kinetic mechanism
A series of enzyme kinetic assays with the WT enzyme were performed as described above as well as previously (Baumgartner et al., 2021) with the following modifications. The concentration of aspartame was varied from 0 to 10 mM at four different concentrations of AcCoA (0.1, 0.25, 0.5, and 1 mM) and 1.3 μM WT enzyme. Data were fitted to several models (random bisubstrate, ordered AB bisubstrate, ordered BA bisubstrate, ping–pong bisubstrate, and hybrid ping–pong bisubstrate steady state) as described before (Baumgartner et al., 2021).
4.6. Protein and small molecule preparation for molecular docking studies
The Molecular Operating Environment (MOE) (Anon, 2013) and the X‐ray crystal structure of the WT PA3944 protein (PDB ID: 6EDV) were used for docking studies. The structure was saturated with requisite hydrogens and protonated at pH 7.4 and 310 K using a previously described protocol (Heath et al., 2018). The enzyme was then energy minimized in the gas phase using an Amber14:EHT force field with crystallographic water molecules set as non‐rigid. A similar approach was used for the R106A mutant protein, in which the MOE Sketch feature with ChemDraw was used to manually mutate the R106 residue to alanine and then energy minimized. To create the AcCoA ligand in the energy minimized WT and R106A protein models, an acetyl group was added to the sulfur atom of CoA using the MOE Builder Utility, followed by energy minimization. A similar procedure was used to create the AcS148 version of the enzymes in the presence and absence of the CoA ligand. Compounds were protonated at pH 7.4 and 310 K followed by energy minimization in the gas phase with the MMFF94x forcefield (Halgren, 1996). The different energy minimized, liganded WT and R106A structures (non‐AcS148 with AcCoA, AcS148 with CoA, AcS148 without CoA) were used to dock the individual, fully protonated substrates into the acceptor sites using automatic and manual pocket detection strategies and employing the AMBER14:EHT forcefield. If CoA or AcCoA was present in the donor site, automatic detection of the acceptor site was used by MOE. Alternatively, if these ligands were absent, we manually defined the acceptor site as the Pocket site by populating it with inactive dummy atoms with the MOE utility Site Finder (Figure S5); the wall constraint option in MOE was not selected so docked molecules were not rigidly restricted. To dock a second molecule of NANMO into the R106A protein model acceptor site in the absence of CoA, we used the structure corresponding to the lowest S score pose from the single NANMO substrate docking result. Ligand placement for the docking protocol utilized the Alpha Triangle method (Udatha et al., 2012) with Affinity dG scoring to generate 1000 data points per unique ligand. These were further refined using the Induced Fit method with GBVI/WSA (Labute, 2008) dG scoring to obtain docked poses, and were further analyzed. An S score is computed by MOE to indicate the GBVI/WSA free energy of the pose.
4.7. Generation of an AcCoA‐polymyxin B tetrahedral intermediate
MOE was used to generate a hypothetical tetrahedral intermediate between the 3‐Dab of polymyxin B and AcCoA. Starting with the tetrahedral carbon atom formed from polymyxin B and AcCoA, the remainder of the polymyxin B molecule was manually built into the acceptor site using the MOE Builder utility. All atoms in the system were optimized with a short, localized molecular minimization process in the gas phase with atoms further than 8 Å from the tetrahedral intermediate held in a fixed position. System refinement continued until an RMS Gradient of 0.1 kcal/mol/Å was obtained. The tetrahedral intermediate PA3944 model was resolvated in a simple water box at pH 7.4 and treated with NaCl counterions to balance the charge. Periodic boundary conditions were enabled, and the hydrogen bonding network of the model was optimized by automatically sampling different tautomer/protomer states using Protonate3D (Shoichet, 2004). The model was then subjected to a local minimization in the liquid phase followed by a 1 ns MD equilibration run using an NPA algorithm with an Amber12:EHT force field. MD experiments utilized an initial heating phase from 0 to 300 K over 100 ps followed by equilibration for 100 ps at 300 K, 700 ps production run, and finally a 100 ps cooling from 300 to 0 K. Simulation results were minimized once more before final binding poses for the bisubstrate tetrahedral intermediate model were obtained for analysis.
4.8. Selection criteria for selecting and sorting docking poses for further analysis
We used the following strategy to select docked poses that were located adjacent to the acyl donor. To begin, we selected the top 10 docking poses of NANMO and aspartame substrates for further analysis based on the lowest S scores. These poses were then visually inspected to determine which poses had the free amine oriented toward the acyl donor. Since polymyxin B has multiple primary amines, we did not exclude any poses from the analysis. Next, we adapted the selection strategy to decrease bias based on appearance by measuring distances between the substrate primary amine nitrogen and the carbonyl carbon of the acyl donor for all poses. The location of these atoms was determined from the x, y, z coordinates in pdb files generated from the docking output database. We used the diaminobutyric acid primary amine nitrogens (1, 3, 5, 8, 9‐Dab) of polymyxin B, the primary amine nitrogen of NANMO, and the primary amine nitrogen of aspartame. For each docking pose, the distance between the primary amine nitrogen of the acceptor substrate and the carbonyl carbon of the acetyl donor was determined using the equation: where are the coordinates of the primary amine nitrogen atom and are the coordinates of the carbonyl carbon atom of the donor in Å. These poses were then ranked by distance and the top 10 poses were plotted as box plots in Origin with the following configurations: Box 25th to 75th percentile, data points centered, whiskers 5th percentile lower boundary, 95th percentile upper boundary.
AUTHOR CONTRIBUTIONS
Cillian Variot: Investigation; Visualization; Writing ‐ review & editing; Writing ‐ original draft; Formal analysis; Methodology; Data curation. Daniel Capule: Investigation; Writing ‐ original draft; Writing ‐ review & editing; Visualization; Methodology; Formal analysis; Data curation. Xhulio Arolli: Investigation; Writing ‐ review & editing; Formal analysis; Data curation. Jackson Baumgartner: Investigation; Writing ‐ review & editing; Data curation; Formal analysis. Cory Reidl: Investigation; Writing ‐ review & editing. Charles Houseman: Investigation; Writing ‐ review & editing. Miguel A. Ballicora: Investigation; Formal analysis; Writing ‐ review & editing. Daniel P. Becker: Investigation; Funding acquisition; Writing ‐ review & editing; Methodology; Formal analysis; Project administration; Supervision; Data curation; Resources. Misty L. Kuhn: Conceptualization; Investigation; Funding acquisition; Writing ‐ original draft; Methodology; Visualization; Writing ‐ review & editing; Formal analysis; Project administration; Data curation; Supervision; Resources; Validation.
Supporting information
Appendix S1.
TABLE S1. Molecular docking results sorted by pose number for each substrate into PA3944 WT and R106A proteins. Docking S scores in kcal/mol and distances (Å) between the acceptor substrate amine nitrogen atom and carbonyl carbon of the acetyl donor are shown for all poses and each substrate. Tabs within the Excel sheet are named using the following convention: enzyme (WT or R106A), docking condition (AcCoA, AcS148 + CoA, AcS148 − CoA), and substrate.
TABLE S2. Molecular docking results for docking a second molecule of NANMO substrate into the PA3944 R106A model in the AcS148 − CoA condition. Docking S scores in kcal/mol and distances (Å) between the acceptor substrate amine nitrogen atom of a second NANMO molecule and carbonyl carbon of AcS148 in the R106A model are shown.
TABLE S3. PA3944 WT and R106A enzyme‐substrate interactions. The Protein‐Ligand Interaction Profiler (PLIP) utility in MOE was used to identify interactions between docked substrates and modeled enzymes for each docking set. Columns with formulas were used to parse enzyme, substrate, condition, and hydrogen bonds from the PLIP data. A pivot table was generated from the PLIP data with the following layers (collapsible/expandable), in order, for rows: enzyme, substrate, condition, interaction, and docking set. The table columns are residue or water IDs. Each table cell displays the count of residue or water ID. When each row of the pivot table is fully expanded, cells with numbers denote the number of interactions between ligand and substrate for each pose under the various combinations.
ACKNOWLEDGMENTS
Federal funds from the National Science Foundation (CHE‐1708863 to Misty L. Kuhn and CHE‐1708927 to Daniel P. Becker) were used to perform scientific studies presented in this manuscript. We thank Karolina Majorek and Wladek Minor at the University of Virginia Charlottesville for the PA3944 T141A mutant plasmid, David Tran, Melissa Law, and Layton Joe at San Francisco State University and Sarah Witzke at the Chemical Computing Group for their technical assistance.
Variot C, Capule D, Arolli X, Baumgartner J, Reidl C, Houseman C, et al. Mapping roles of active site residues in the acceptor site of the PA3944 Gcn5‐related N‐acetyltransferase enzyme. Protein Science. 2023;32(8):e4725. 10.1002/pro.4725
Cillian Variot and Daniel Capule contributed equally to this work.
Review Editor: Nir Ben‐Tal
Contributor Information
Daniel P. Becker, Email: dbecke3@luc.edu.
Misty L. Kuhn, Email: mkuhn@sfsu.edu.
REFERENCES
- Anon . Molecular Operating Environment (MOE). Montreal: Chemical Computing Group Inc.; 2013. [Google Scholar]
- Baumgartner JT, Habeeb Mohammad TS, Czub MP, Majorek K, Arolli X, Variot C, et al. Gcn5‐related N‐acetyltransferases (GNATs) with a catalytic serine residue can play ping–pong too. Front Mol Biosci. 2021;8:646046. 10.3389/fmolb.2021.646046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burckhardt RM, Escalante‐Semerena JC. Small‐molecule acetylation by GCN5‐related N‐acetyltransferases in bacteria. Microbiol Mol Biol Rev. 2020;84:e00090‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czub MP, Zhang B, Chiarelli MP, Majorek KA, Joe L, Porebski PJ, et al. A Gcn5‐related N‐acetyltransferase (GNAT) capable of acetylating polymyxin B and colistin antibiotics in vitro. Biochemistry. 2018;57:7011–7020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da C, Kireev D. Structural protein–ligand interaction fingerprints (SPLIF) for structure‐based virtual screening: method and benchmark study. J Chem Inf Model. 2014;54:2555–2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Z, Chuaqui C, Singh J. Structural interaction fingerprint (SIFt): a novel method for analyzing three‐dimensional protein‐ligand binding interactions. J Med Chem. 2004;47:337–344. [DOI] [PubMed] [Google Scholar]
- Halgren TA. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J Comput Chem. 1996;17:490–519. [Google Scholar]
- Heath TK, Lutz MR, Reidl CT, Guzman ER, Herbert CA, Nocek BP, et al. Practical spectrophotometric assay for the dapE‐encoded N‐succinyl‐L,L‐diaminopimelic acid desuccinylase, a potential antibiotic target. PLoS One. 2018;13:e0196010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann JC, Ghanem E, Li Y, Raushel FM, Irwin JJ, Shoichet BK. Predicting substrates by docking high‐energy intermediates to enzyme structures. J Am Chem Soc. 2006;128(49):15882–15891. 10.1021/ja065860f [DOI] [PubMed] [Google Scholar]
- Juhl PB, Trodler P, Tyagi S, Pleiss J. Modelling substrate specificity and enantioselectivity for lipases and esterases by substrate‐imprinted docking. BMC Struct Biol. 2009;9:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn ML, Majorek KA, Minor W, Anderson WF. Broad‐substrate screen as a tool to identify substrates for bacterial Gcn5‐related N‐acetyltransferases with unknown substrate specificity. Protein Sci Publ Protein Soc. 2013;22:222–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labute P. The generalized born/volume integral implicit solvent model: estimation of the free energy of hydration using London dispersion instead of atomic surface area. J Comput Chem. 2008;29:1693–1698. [DOI] [PubMed] [Google Scholar]
- Ramírez D, Caballero J. Is it reliable to take the molecular docking top scoring position as the best solution without considering available structural data? Mol J Synth Chem Nat Prod Chem. 2018;23:1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoichet BK. Virtual screening of chemical libraries. Nature. 2004;432:862–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udatha DBRKG, Sugaya N, Olsson L, Panagiotou G. How well do the substrates KISS the enzyme? Molecular docking program selection for feruloyl esterases. Sci Rep. 2012;2:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1.
TABLE S1. Molecular docking results sorted by pose number for each substrate into PA3944 WT and R106A proteins. Docking S scores in kcal/mol and distances (Å) between the acceptor substrate amine nitrogen atom and carbonyl carbon of the acetyl donor are shown for all poses and each substrate. Tabs within the Excel sheet are named using the following convention: enzyme (WT or R106A), docking condition (AcCoA, AcS148 + CoA, AcS148 − CoA), and substrate.
TABLE S2. Molecular docking results for docking a second molecule of NANMO substrate into the PA3944 R106A model in the AcS148 − CoA condition. Docking S scores in kcal/mol and distances (Å) between the acceptor substrate amine nitrogen atom of a second NANMO molecule and carbonyl carbon of AcS148 in the R106A model are shown.
TABLE S3. PA3944 WT and R106A enzyme‐substrate interactions. The Protein‐Ligand Interaction Profiler (PLIP) utility in MOE was used to identify interactions between docked substrates and modeled enzymes for each docking set. Columns with formulas were used to parse enzyme, substrate, condition, and hydrogen bonds from the PLIP data. A pivot table was generated from the PLIP data with the following layers (collapsible/expandable), in order, for rows: enzyme, substrate, condition, interaction, and docking set. The table columns are residue or water IDs. Each table cell displays the count of residue or water ID. When each row of the pivot table is fully expanded, cells with numbers denote the number of interactions between ligand and substrate for each pose under the various combinations.