

Abstract
SHOC2 acts as a strong synthetic lethal interactor with MEK inhibitors in multiple KRAS cancer cell lines. SHOC2 forms a heterotrimeric complex with MRAS and PP1C that is essential for regulating RAF and MAPK-pathway activation by dephosphorylating a specific phosphoserine on RAF kinases. Here we present the high-resolution crystal structure of the SHOC2–MRAS–PP1C (SMP) complex and apo-SHOC2. Our structures reveal that SHOC2, MRAS, and PP1C form a stable ternary complex in which all three proteins synergistically interact with each other. Our results show that dephosphorylation of RAF substrates by PP1C is enhanced upon interacting with SHOC2 and MRAS. The SMP complex forms only when MRAS is in an active state and is dependent on SHOC2 functioning as a scaffolding protein in the complex by bringing PP1C and MRAS together. Our results provide structural insights into the role of the SMP complex in RAF activation and how mutations found in Noonan syndrome enhance complex formation, and reveal new avenues for therapeutic interventions.
The mitogen-activated protein kinase (MAPK) signaling pathway comprises the RAF, MEK, and ERK protein kinases, constituting a critical effector cascade used by the RAS proteins to regulate cell growth, survival, proliferation, and differentiation1. Aberrant activation of the MAPK signaling pathway is one of the most common drivers of human cancer and is responsible for multiple developmental disorders, known as RASopathies2,3. Within this signaling pathway, the regulation of RAF kinases is a complex process that involves protein and lipid interactions, subcellular localization, and multiple phosphorylation and dephosphorylation events4. RAF kinases are held in an autoinhibited state by the 14–3-3 family of phosphoserineand phosphothreonine-binding proteins, which bind to RAF using two phosphorylation-dependent 14–3-3 binding sites5,6. In the RAF kinase, these two phosphorylation sites are found at the conserved regions (CRs) on either side of the kinase domain: CR2-pS (ARAF-S214, BRAF-S365, CRAF/RAF1-S259) and CR3-pS (ARAF-S582, BRAF-S729, CRAF-S621). RAF kinase activation requires both active RAS binding to the RAS-binding domain (RBD) and membrane-anchoring cysteine-rich domain (CRD) of RAF and dephosphorylation of CR2-pS to prevent 14–3-3 binding at this site7,8. Dephosphorylation allows the released kinase domain to form an active dimeric RAF complex stabilized by a 14–3-3 dimer bound to the CR3-pS sites of each RAF kinase. CRAF/RAF1 mutations (S257L and P261S) around the CR2-pS259 14–3-3 binding site are frequently detected in the RASopathy Noonan syndrome (NS)9. These mutations have been suggested to enhance CRAF activation by disrupting 14–3-3 binding to the S259 site, underscoring the critical role of this step in RAF and MAPK-pathway regulation.
The dephosphorylation of CR2-pS is mediated by a heterotrimeric complex comprising SHOC2, MRAS, and PP1C (the SMP complex), and each of the three proteins has indispensable roles in the proper function of the complex10,11. SHOC2 is a ubiquitously expressed protein composed primarily of predicted leucine-rich repeats (LRRs). Amino-terminal to the LRR domains, SHOC2 contains a ~90-residue-long sequence that is predicted to be intrinsically disordered and has been suggested to be necessary for complex formation with MRAS and PP1C11,12. Germline mutations in SHOC2 (S2G, M173I, and Q269H/H270Y) have been detected in NS11,13–15. SHOC2 plays a vital role in transformation, metastasis, epithelial-to-mesenchymal transition, and MAPK-pathway-inhibitor resistance16–19. Multiple genome-scale, single-gene CRISPR–Cas9 fitness screens in human cancer cells have suggested selective dependency of RAS mutant cells on SHOC2 (refs.18,20–22). SHOC2 has also been identified as the strongest synthetic lethal target in the presence of MEK inhibitors in KRAS-mutant lung and pancreatic cancer cell lines17. Thus, SHOC2 may provide a unique therapeutic opportunity within the RTK–RAS–MAPK pathway in oncogenic RAS cells.
SMP-complex formation is initiated following MRAS activation, as SHOC2 and PP1C bind only to GTP-bound MRAS23. The canonical RAS family members HRAS, KRAS, and NRAS (H/K/NRAS) also bind SHOC2, although with considerably lower affinity than does MRAS24. The nature of the selectivity for MRAS is not known. MRAS shares ~50% sequence identity with H/K/NRAS and contains an extra ten amino acids at the N terminus. Activating mutations in MRAS are rare in cancer; however, gain-of-function mutations (p.G23V, p.T68I, p.Q71R) in MRAS have been identified in people with NS25,26. In the SMP complex, PP1C provides the enzymatic activity for dephosphorylation. PP1C is a serine/threonine phosphatase with three highly conserved isoforms (PP1CA, PP1CB, and PP1CC, which have >90% sequence identity) that are ubiquitously expressed and catalyze the dephosphorylation of a substantial fraction of phosphoserine and phosphothreonine in eukaryotic cells27–29. Mutations in the PP1CB isoform have been found in NS, and these residues are conserved in other PP1C isoforms30–33.
To understand how SHOC2, MRAS, and PP1C proteins assemble to form a ternary complex that regulates dephosphorylation of the RAF CR2-pS and how RASopathy mutations impact complex formation, we solved the structure of the SMP complex. Structural and mutational analysis provide a rationale for MRAS selectivity versus H/K/NRAS and the impact of NS mutations on SMP complex assembly. Dephosphorylation activity of PP1C towards RAF substrates is enhanced upon complex formation with SHOC2 and MRAS. Analysis of the protein-protein interfaces in the SMP complex and mutagenesis studies provide insights into complex assembly and potential sites that could be exploited using structure-based drug-discovery approaches.
Results
Assembly of the SHOC2–MRAS–PP1CA complex
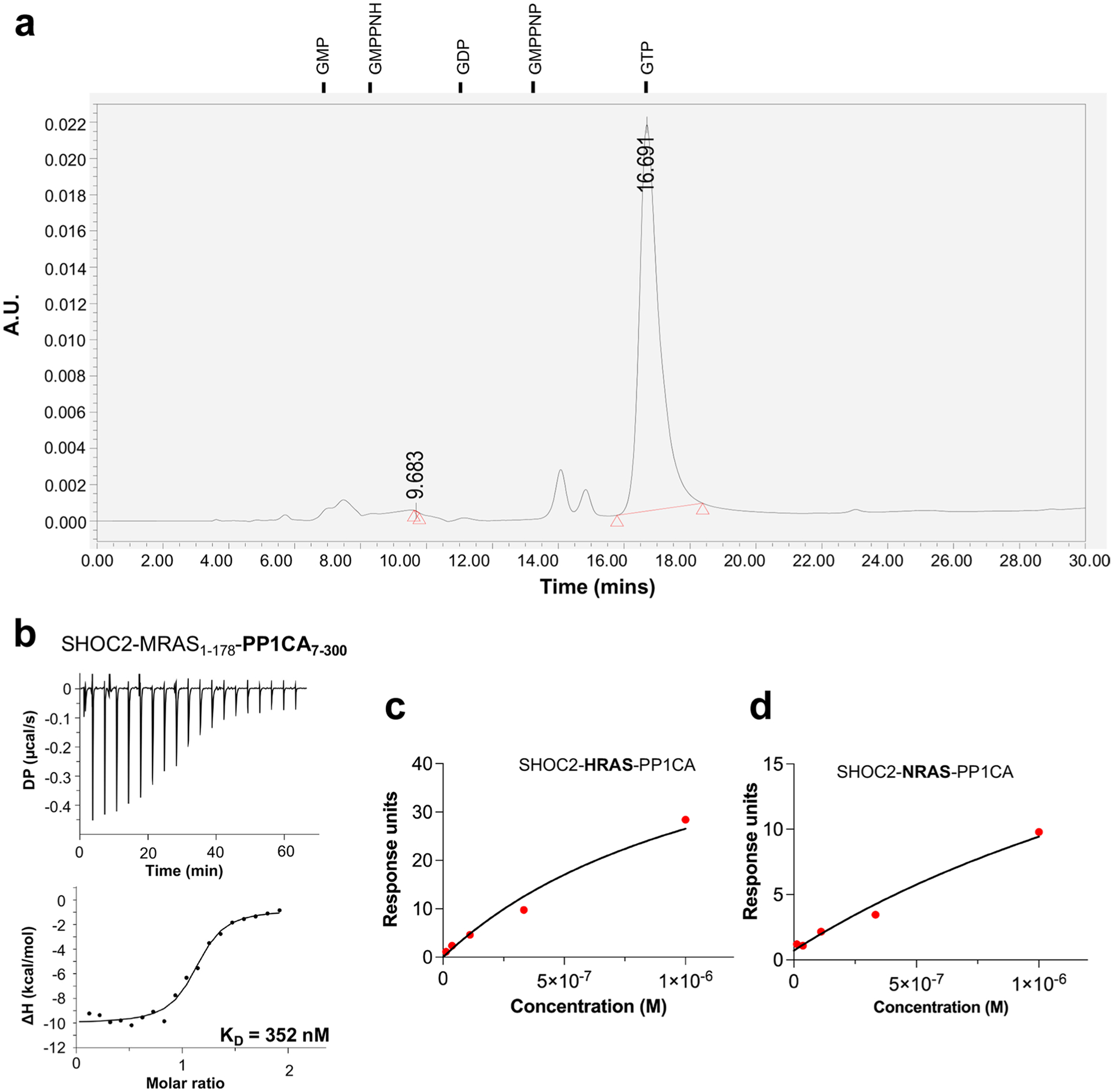
We purified the pre-assembled human SMP complex by co-expressing SHOC2, MRAS (MRAS-Q71L or wild type (WT)), and PP1CA from a single plasmid (Fig. 1a), along with the chaperone SUGT1, in baculovirus-infected insect cells, as described previously34. Nucleotide analysis of the purified SMP complex shows that MRAS is bound to GTP (Extended Data Fig. 1a). Using surface plasmon resonance (SPR), we measured the affinity of SMP complex formation with SHOC2 and PP1CA, and with MRAS in the GDP- or GMPPNP-bound state. Weak, transient association of PP1CA was observed with SHOC2 but could not be quantified (Fig. 1b,c). We observed stable SMP-complex formation only between SHOC2, PP1CA, and MRAS-GMPPNP, with a dissociation constant (KD) of ~120 nM (Fig. 1c); we observed no SMP-complex formation with MRAS-GDP (Fig. 1b). A similar binding affinity of ~350 nM for SMP-complex formation was observed by isothermal titration calorimetry (ITC) using MRAS-GMPPNP, despite the higher salt concentration that is required for the ITC experiments (Extended Data Fig. 1b). We used the MRAS-Q71L mutant for our structural work, as SPR measurements showed an approximately fivefold-higher affinity (KD = 26 nM) for SMP-complex formation with this mutant than for that with WT MRAS (Fig. 1d). SPR measurement using KRAS-GMPPNP, HRAS-GMPPNP, or NRAS-GMPPNP in place of MRAS-GMPPNP showed ternary complex formation (SKP, SHP, or SNP), with an apparent KD of 0.7 μM, 2 μM, and 4 μM, respectively, owing to a substantial increase in both the on-rate and off-rate (Fig. 1e–g and Extended Data Fig. 1c,d). Thus, a 7- to 40-fold-higher affinity of MRAS over H/K/NRAS for complex formation confirmed that MRAS is the preferred partner of SHOC2 and PP1C.
Fig. 1 |. Assembly, activity, and selectivity of the SMP complex.
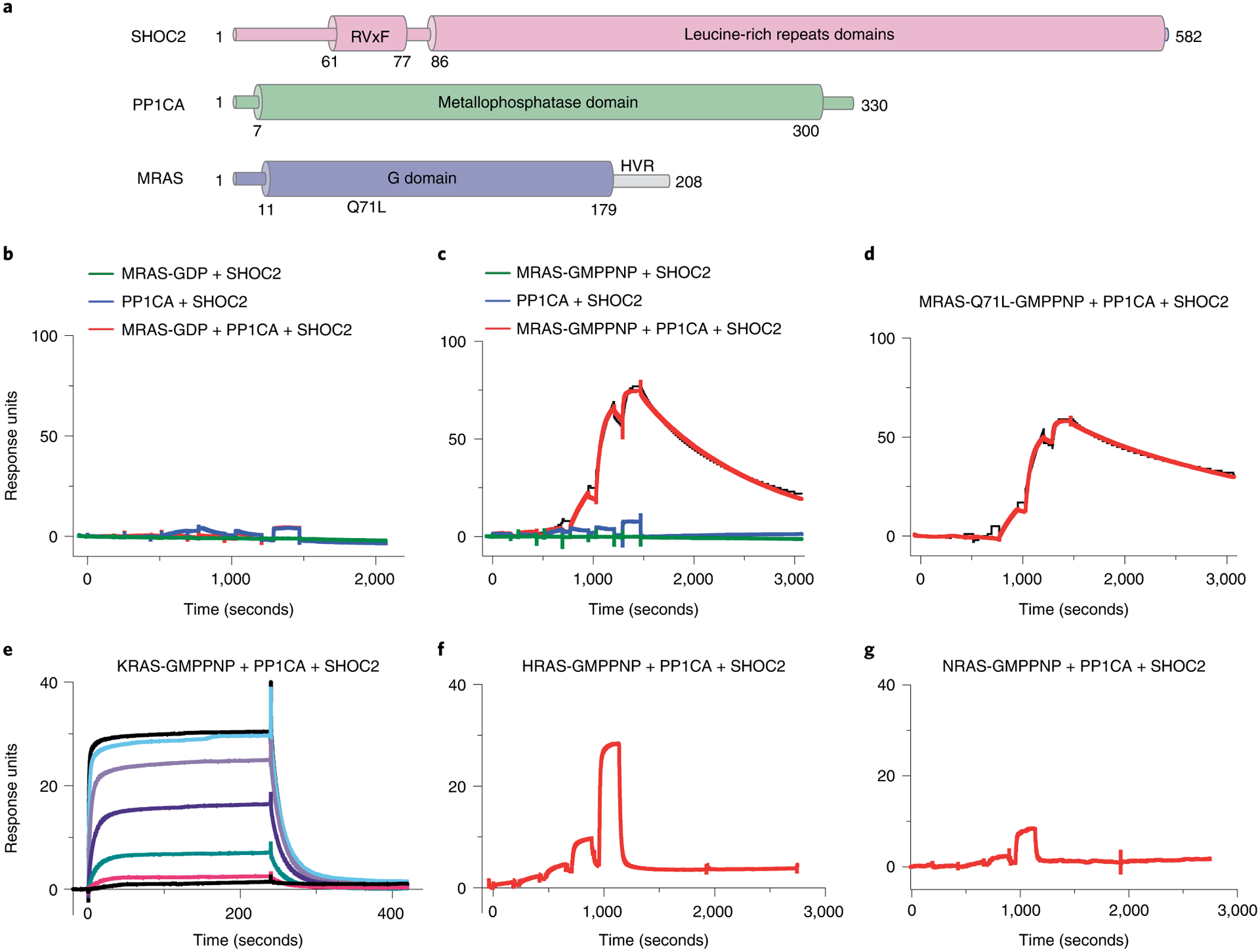
a, Domain architecture of SHOC2, MRAS, and PP1CA. Full-length SHOC2 and PP1CA and the G domain of MRAS (1–179) were used for structure determination. b,c, Single-cycle kinetic titration SPR binding experiments were performed on immobilized avi-tagged SHOC2 with threefold dilutions of 1 μM MRAS (green), PP1CA (blue), and MRAS with PP1CA (red). All experiments were either conducted with MRASGDP (b) or MRAS-GMPPNP (c). The data were fit to a 1:1 kinetic model (black). SMP-complex assembly occurred only with MRAS-GMPPNP and in the presence of PP1CA. d, Single-cycle kinetic analysis was performed on immobilized avi-tagged SHOC2 with threefold serial dilutions of 1 μM MRAS-Q71L-GMPPNP and 1 μM PP1CA (red). The data were fit to a 1:1 kinetic model (black). e, Assembly of the SKP (SHOC2–KRAS–PP1CA) complex was measured by SPR kinetic analysis. Twofold dilutions of 5 μM KRAS-GMPPNP and 5 μM PP1CA were injected over immobilized avi-tagged SHOC2. f,g, Assembly of the SHP (SHOC2–HRAS–PP1CA) or SNP (SHOC2–NRAS–PP1CA) complexes were measured by SPR single-cycle kinetic analysis. Twofold dilutions of 5 μM HRAS-GMPPNP (f) and 5 μM PP1CA or 5 μM NRAS-GMPPNP and 5 μM PP1CA (g) were injected over immobilized avi-tagged SHOC2.
Structural description of the SMP complex
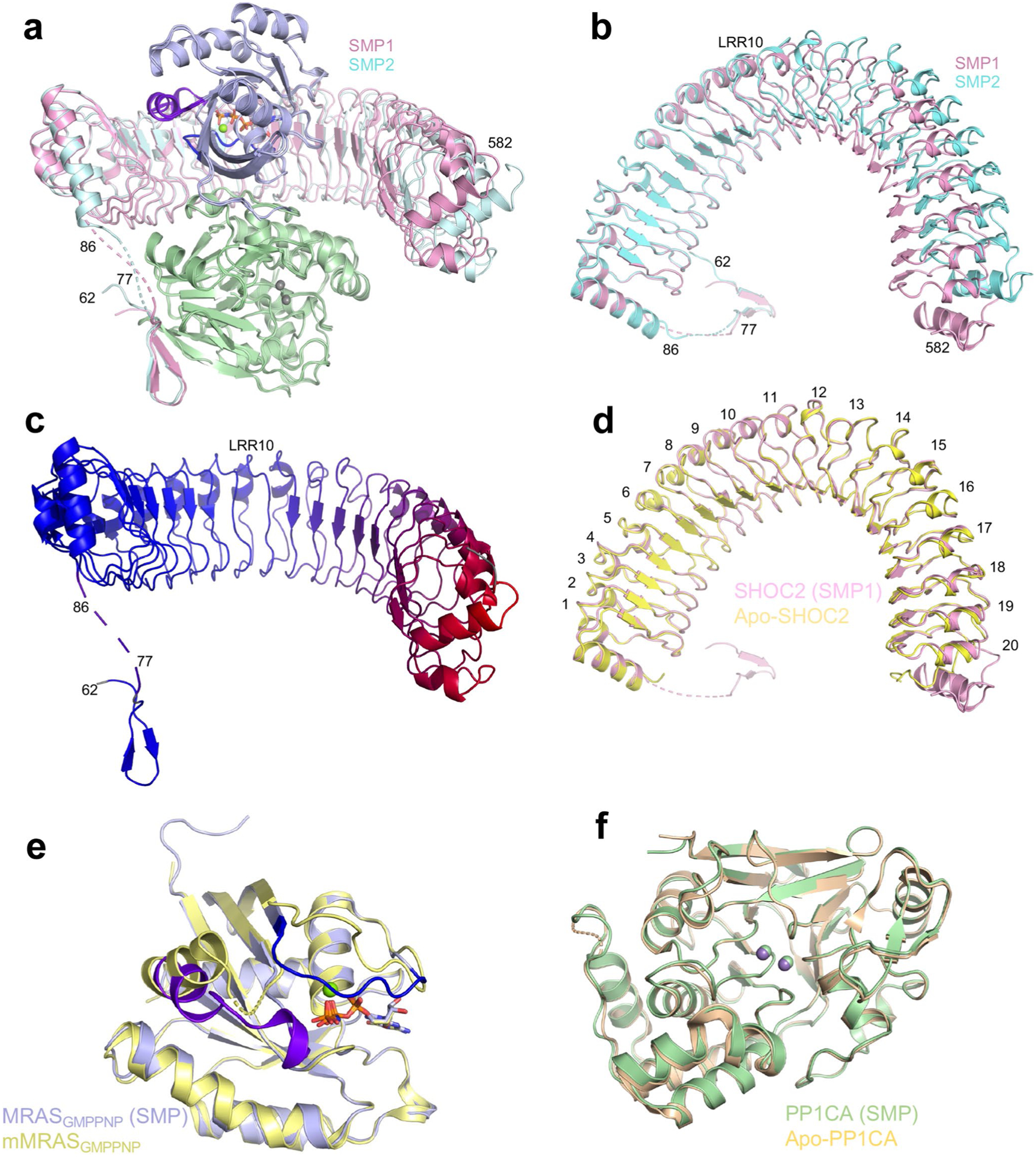
To understand how SHOC2, MRAS, and PP1CA interact with each other, the structure of the SMP complex was determined at 2.17 Å (Fig. 2a,b and Table 1). In the crystal, two copies of the SMP complex (SMP1 and SMP2) are present in the asymmetric unit. The superposition of these two complexes shows an almost identical arrangement of three proteins (Fig. 2c and Extended Data Fig. 2a). MRAS and PP1CA are nearly identical in the two complexes (Extended Data Fig. 2a); however, the carboxy termini of SHOC2 molecules differ between the two SMP complexes in the asymmetric unit, with SHOC2 of SMP1 forming additional contacts with MRAS and PP1CA (Fig. 2c and Extended Data Fig. 2a,b). This distortion in SHOC2 is propagated and amplified, causing the C-terminal helix at the end of the LRRs to move 10 Å towards MRAS (Fig. 2c and Extended Data Fig. 2a). Several SHOC2 structure predictions suggested a flexible hinge around LRRs 13–15 (refs.12,13,16,35). Our structure is consistent with this prediction, but the flexible hinge is within LRR 10 (Extended Data Fig. 2b,c).
Fig. 2 |. The 2.17-Å structure of the SMP complex.
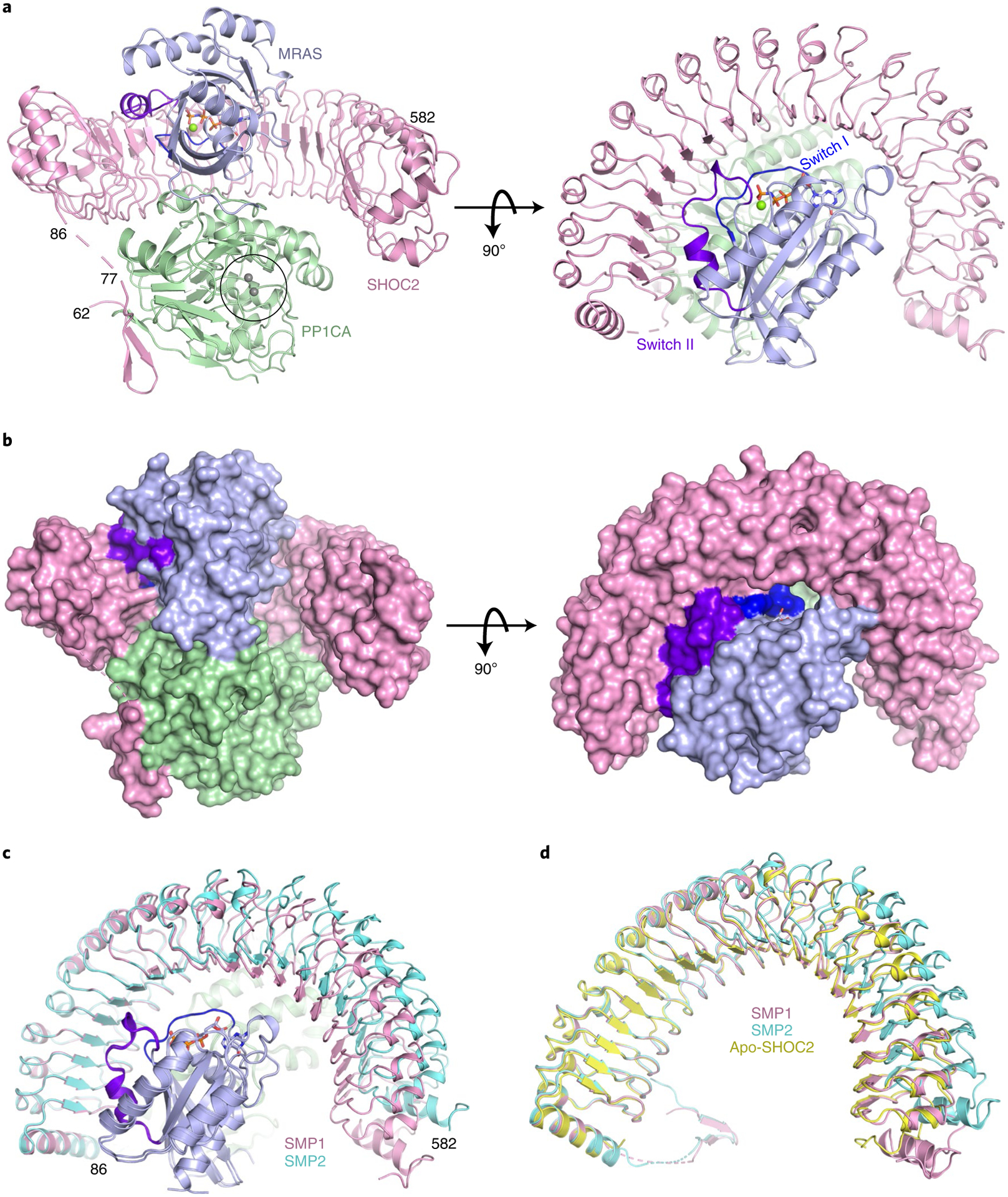
a,b, The overall structure of the SMP complex is shown as a cartoon (a) and in surface representation (b) in two different views. SHOC2 and PP1CA are colored pink and green, respectively. MRAS is colored blue, with the switch I and switch II regions highlighted in dark blue and purple, respectively. GMPPNP is shown as sticks, and Mg2+ (green) and Mn2+ (gray) ions as spheres. The active site containing Mn2+ ions is within the black circle. c, Superposition of the two SMP complexes present in the asymmetric subunit in cartoon form. Both chains of MRAS and PP1CA are in the same color, and the two SHOC2 chains are colored pink and cyan. d, Superposition of apo-SHOC2 (yellow) onto the two SHOC2 chains from the SMP complex.
Table 1 |.
Data collection and refinement statistics (molecular replacement)
| SMP complex | SHOC2 | |
|---|---|---|
| Data collection | ||
| Space group | P41 21 2 | P32 |
| Cell dimensions | ||
| a, b, c (Å) | 129.94, 129.94, 326.80 | 77.57, 77.57, 83.01 |
| a, β, y(°) | 90.0, 90.0, 90.0 | 90.0, 90.0, 120.0 |
| Resolution (Å) | 163.40–2.17 (2.20–2.17)* | 41.51–2.40 (2.49–2.40)* |
| R merge | 0.137 (2.007) | 0.036 (1.090) |
| R pim | 0.077 (1.228) | 0.024 (0.721) |
| I/σ/ | 8.5 (0.9) | 19.8 (1.7) |
| Completeness (%) | 99.9 (98.4) | 99.9 (100.0) |
| Redundancy | 7.9 (6.8) | 6.2 (6.4) |
| CC1/2 | 0.997 (0.340) | 1.000 (0.581) |
| Refinement | ||
| Resolution (Å) | 101.7–2.17 | 35.31–2.40 |
| No. reflections | 148,297 | 21,811 |
| Rwork / Rfree | 19.6/22.6 | 21.7/26.7 |
| No. atoms | ||
| Protein | 15,683 | 3,742 |
| Ligand/ion | 180 | 31 |
| Water | 1,046 | 36 |
| B factors | ||
| Protein | 51.3 | 86.6 |
| Ligand/ion | 70.0 | 122.2 |
| Water | 52.8 | 86.0 |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.002 | 0.004 |
| Bond angles (°) | 0.51 | 0.69 |
Values in parentheses are for highest-resolution shell.
To determine whether SHOC2 undergoes conformational changes upon assembly of the SMP complex, we solved the structure of uncomplexed apo-SHOC258–564 at 2.4 Å (Table 1). In the SHOC258–564 structure, all LRRs and the helix that caps the N-terminal end of LRR are well defined. We did not observe any residues (58–86) prior to the N-terminal capping helix, suggesting that these residues have no interactions with LRRs. Apo-SHOC258–564 superimposes with a lower root mean square deviation (0.57 Å) onto the SMP1 complex (Fig. 2d). SHOC2 in SMP1 forms extra contacts with MRAS and PP1CA (Fig. 2d and Extended Data Fig. 2a,d), which are important for binding and are described below. Our subsequent structural analysis is based on this SMP complex.
As expected, MRAS in the SMP complex adopts the conserved G-domain fold in an active state. We used the structure of mouse MRAS-GMPPNP (no human structure exists; PDB: 1X1S, 97% identity) to understand structural similarities and differences with human MRAS present in the SMP complex36. Superposition of mouse MRAS-GMPPNP with the MRAS of the SMP complex revealed differences in the two switch regions (Extended Data Fig. 2e). In the apo-MRAS structure, the switch I region (residues 40–48) is in the open solvent-exposed state I conformation, whereas in the SMP complex it is in the closed state II conformation, as has been observed in structures of RAS–effector complexes1,6,7. Multiple residues in the switch II region (residues 69–73), which are typically disordered in the apo-MRAS structure, are ordered in the SMP complex. MRAS-Q71L, which increases SMP-complex formation by approximately fivefold, likely aids the formation of a helical loop that contributes additional interactions from switch II to the SMP complex. Structural comparison of PP1CA in the SMP complex with human apo-PP1CA (PDB: 4MOV) showed no major structural changes in PP1CA upon formation of the SMP complex (Extended Data Fig. 2f)37. Among the PP1C isoforms, the C-terminal tail shows high sequence diversity (residues 300–330) and has been proposed to function as an inhibitor when phosphorylated at T320 (PP1CA) and in complex with other PP1C regulators38. The C-terminal tail of PP1CA is disordered in our structure, except for residues 318–322 in one molecule of PP1CA, which interact with a symmetry-related molecule of SHOC2. To test whether PP1C isoform specificity exists, we used ITC to measure the affinity of SMP-complex formation with PP1CA and PP1CB. We observed similar affinities for these two isoforms, signifying no PP1C isoform specificity (Extended Data Fig. 3a,b). We observed only residues 60–76 of the intrinsically disordered N-terminal domain of SHOC2, which folded into a β-hairpin and interacted with PP1CA. The C-terminal domain of SHOC2 contains 20 LRR domains, which are capped at the termini by helices, resulting in a horseshoe-shaped protein (Fig. 2a). In the SMP complex, each protein interacts with the other two proteins, resulting in a total of ~6,200 Å2 of buried surface area upon complex formation.
The SHOC2–PP1CA interface
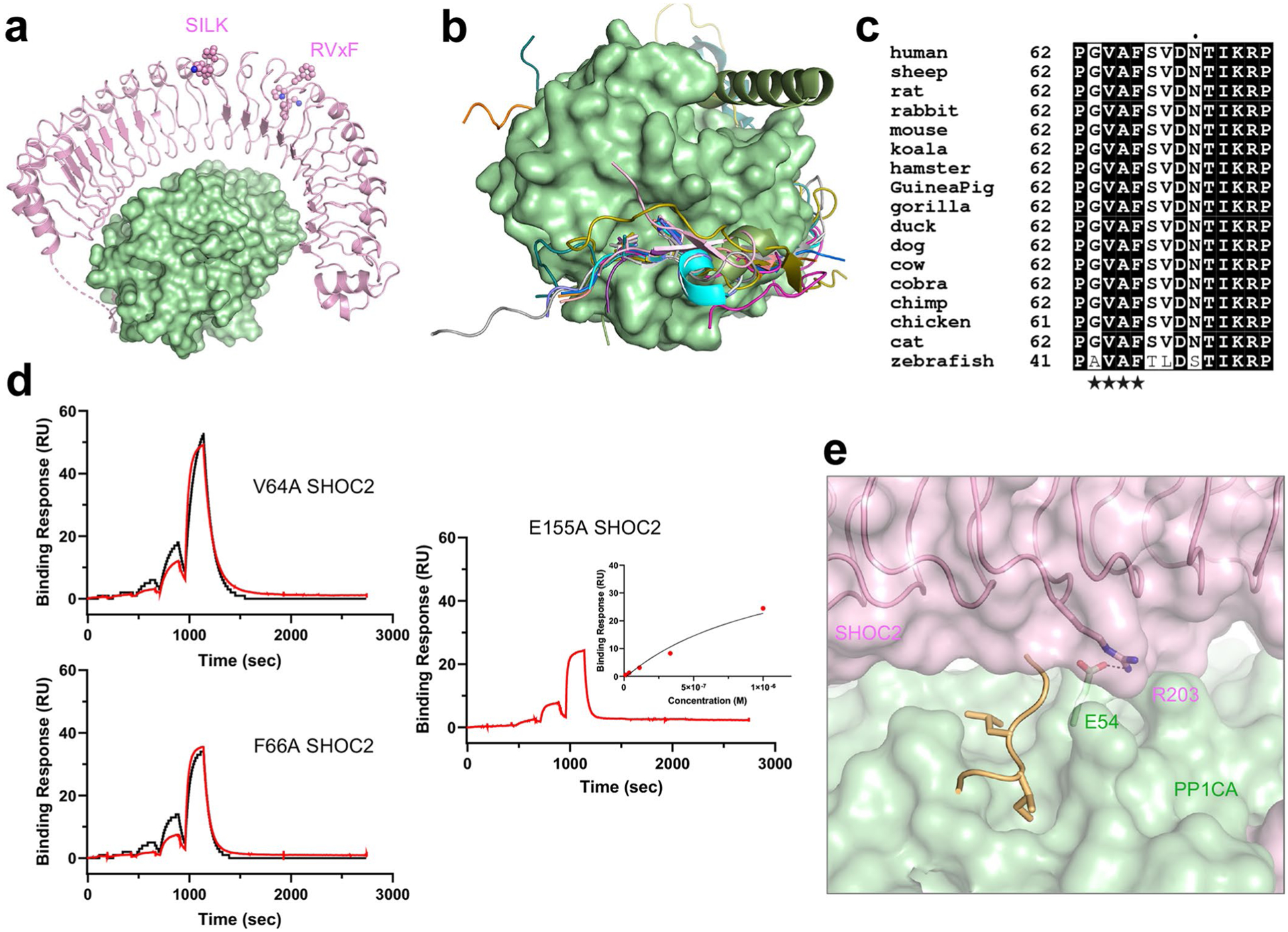
The SHOC2–PP1CA interface contributes the largest buried surface area, of 2,800 Å2. SHOC2 is predicted to interact with PP1CA through SILK and RVxF motifs identified in the folded region of SHOC2 between LRR 10–11 and within LRR 12 (Extended Data Fig. 4a)16. PP1CA does not contact these residues (Extended Data Fig. 4a); however, we did identify a genuine RVxF motif within the β-hairpin of the N-terminal intrinsically disordered domain of SHOC2 that contacts PP1CA (Figs. 2a and 3a,b). This RVxF motif (62PGVAF66) would not be recognized by RVxF prediction algorithms39,40. The RVxF motif buries V64 and F66 into the hydrophobic pockets on the surface of PP1CA (Fig. 3a,b), with the β-hairpin forming five hydrogen bonds and burying ~1,150 Å2 of surface area (Fig. 3c). The binding of the SHOC2 RVxF motif mimics all PP1CA–proteinRVxF complexes whose structures have been solved and does not alter the structure of PP1CA (Extended Data Fig. 4b). The RVxF motif is believed to function as an anchoring motif29. RVxF motif binding can be regulated through phosphorylation of the variable residue if a serine or threonine is present. The presence of alanine in that position in SHOC2 prevents its regulation directly. However, SHOC2-T71, which lies on the second strand of the β-hairpin, has been shown to be phosphorylated and may play a role in regulating the SMP complex41. The SHOC2 RVxF motif is conserved across higher eukaryotes (Extended Data Fig. 4c). Mutation of valine or phenylalanine drastically weakens the formation of the SMP complex, with nearly a 600-fold reduction in the apparent KD (Fig. 3d and Extended Data Fig. 4d). These SHOC2 mutants retain weakened SMP-complex formation owing to the extensive contacts through the LRRs to PP1CA that involves residues (shown in bold) preceding and/or succeeding the conserved asparagine of the highly conserved segment (HCS) motif (LXXLXLXXN(X)1–2L) in LRRs 2–5, 8–11, and 13–18 (Fig. 3e). As such, when mapped onto the surface of SHOC2, PP1CA contacts the underside of the LRR domain (Fig. 3e). This binding interface buries ~1,650 Å2 of surface area and the formation of nine hydrogen bonds and eight salt bridges (Fig. 3f–h). SHOC2-E155 forms a hydrogen bond with PP1CA-R188. The SHOC2-E155A mutant results in an approximately tenfold reduction in the apparent affinity (Fig. 3d and Extended Data Fig. 4d). SHOC2 does not contain a SILK motif; however, it does interact with the periphery of the SILK-binding pocket on PP1CA through van der Waals interactions and a hydrogen bond between SHOC2-R203 and PP1CA-E54 (Fig. 3f,h and Extended Data Fig. 4e). SHOC2 therefore restricts access to the SILK-binding pocket. The double mutation of the SILK-binding pocket in PP1CB-E53A L54A (or mutation of E54 and L55 in PP1CA; Extended Data Fig. 5a) has previously been shown to weaken complex formation11. This suggests that the hydrogen bond between SHOC2-R203 and PP1CA-E54 is important (PP1CA-L55 does not contact SHOC2). The LRR domain of SHOC2 acts as a tiara that interacts with the crown of PP1CA (Figs. 2a and 3e), with all SHOC2 residues >20 Å away from the PP1CA active site (Fig. 2a).
Fig. 3 |. Structural and mutational analysis of the SHOC2–PP1CA interface.
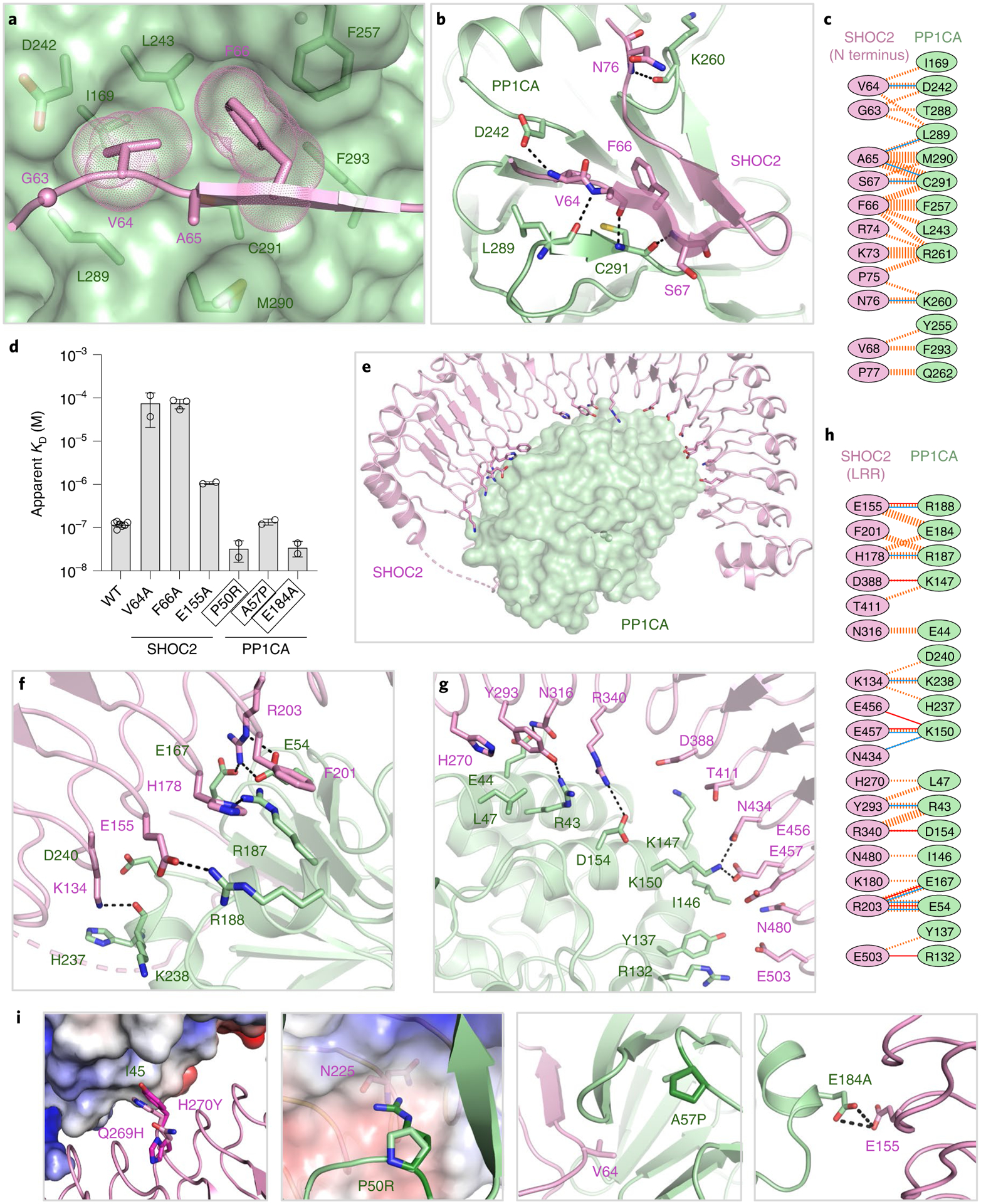
a, The RVxF motif of SHOC2 (GVAF, pink) bound to the surface of the RVxF-binding pocket of PP1CA (green). b, The interaction of the RVxF motif of SHOC2 (pink cartoon) with PP1CA (green cartoon) is shown. Hydrogen bonds are shown as black dashes. c, Schematic representation of the SHOC2 RVxF–PP1CA interaction interface, as analyzed by PDBSum. The interactions are colored using the following notations: hydrogen bonds as solid blue lines and non-bonded contacts as dashed orange lines (the width of the lines is proportional to the number of atomic contacts). d, Plots of the mean apparent KD measurements of SM-complex assembly for NS mutants and point mutants present at the RVxF motif. The s.d. is shown as error bars, with the number of independent replicates shown as circles (summarized in Supplementary Table 1). NS mutations are enclosed in black boxes. e, Overall view of the SHOC2 LRR interactions (pink cartoon) with PP1CA (green surface). f,g, Enlarged view of the N-terminal (f) and C-terminal (g) LRRs of SHOC2 with PP1CA, as depicted in e.h, Schematic representation of the SHOC2 LRRs–PP1CA interaction interface, as analyzed by PDBSum. Interactions are colored as described in c, with the addition of salt bridges as solid orange lines. i, NS mutations modeled onto the SMP structure. The SHOC2-Q269H H270Y double mutation increases contacts between SHOC2-Y270 and PP1CA-I45. The PP1CA-P50R mutation (P49R in PP1CB) would result in a de novo interaction with SHOC2-N225 (shown as an electrostatic surface). PP1CA-A57P (A56P in PP1CB) surrounds the residues that form the hydrophobic pocket that the RVxF motif interacts with. The PP1CAE184A mutation (E183A in PP1CB) relieves the charge-charge repulsion with SHOC2-E155.
Several NS mutations are present near the SHOC2–PP1CA interface, and structural analysis explains why they are gain-of-function mutations. Normally, SHOC2-H270 interacts with PP1CA-I45 through van der Waals interactions. The SHOC2-Q269H H270Y NS-associated double mutation15 potentially forms larger contacts between PP1CA-I45 and SHOC2-Y270 (Fig. 3i), which may increase the affinity of the complex. Three NS mutations have been identified in PP1CB42,43. These residues are conserved across all PP1C isoforms. One of these, P50R (P49R in PP1CB), appears to form a de novo hydrogen bond with SHOC2-N225 (Fig. 3i). This mutation increases the apparent affinity of SMP complex formation by about fourfold (Fig. 3d and Extended Data Fig. 5b). The NS mutation A57P (A56P in PP1CB) is found within a loop adjacent to the RVxF motif binding site of PP1CA, which may affect SHOC2 binding, although no measurable difference in SMP-complex formation was observed (Fig. 3d and Extended Data Fig. 5b). The NS mutation E184A (E183A in PP1CB) appears to relieve charge-charge repulsion to SHOC2-E155 (Fig. 3i). This NS mutation results in an approximately fourfold increase in apparent affinity, to 35 nM (Fig. 3d and Extended Data Fig. 5b).
The SHOC2–MRAS interface
The SHOC2–MRAS interface buries ~2,000 Å2 in the SMP complex. MRAS binds to the concave surface of SHOC2, adjacent to the PP1CA–SHOC2 (LRR) interface (Fig. 4a). Specifically, MRAS contacts LRRs 1–10, 12, and 14–16 (Extended Data Fig. 6a,b). Residues in the switch II region of MRAS contact LRRs 1–4 and 6–7. Switch II engages predominately through van der Waals interactions (Fig. 4b). MRAS switch I residues interact with SHOC2 LRRs 4–6 and 8–10, forming five hydrogen bonds and three salt bridges (Fig. 4b). In addition, several residues within the C-terminal region of MRAS interact with the C-terminal residues of SHOC2; specifically, MRAS-H132 forms a hydrogen bond with SHOC2-E428, and MRAS-K158 forms a salt bridge with SHOC2-E406 (Fig. 4c). In total, seven hydrogen bonds and six salt bridges are formed at the SHOC2–MRAS interface (Fig. 4d). A schematic of a single LRR is shown in Fig. 4e. The HCS motif (LXXLXLXXN(X)1–2L) forms the concave surface of SHOC2. All interactions of the switch I and II residues of MRAS with LRRs 1–10 are with the variable residues (shown in bold) preceding the conserved asparagine of the HCS motif (LXXLXLXXN(X)1–2L, Fig. 4e). The residues that MRAS contacts in the C-terminal half of the LRRs of SHOC2 are the earlier variable residues (shown in bold) in the HCS motif (LXXLXLXN(X)1–2L, Fig. 4e) and as such are found higher up on the molecular surface of SHOC2 (Fig. 4f). The three NS mutations, MRAS-G23V, MRAS-T68I, and MRAS-Q71R, are constitutively active variants (Extended Data Fig. 6c), but none of these residues directly contact SHOC2 or PP1CA25,44,45.
Fig. 4 |. Structural and mutational analysis of the SHOC2–MRAS interface.

a, Overall view of SHOC2 (pink cartoon) interacting with MRAS (blue surface). Switch I, switch II, nucleotides, and Mg2+ are shown in dark blue and purple and as sticks and the green sphere, respectively. b,c, Enlarged view of the LRRs of SHOC2 that interact with switch I and switch II of MRAS (b) and the C terminus of MRAS (c). d, Schematic representation of the SHOC2–MRAS interaction interface, as analyzed by PDBSum. The interactions are colored using the following notations: hydrogen bonds as solid blue lines, salt bridges as solid orange lines, and non-bonded contacts as dashed orange lines (the width of the lines is proportional to the number of atomic contacts). e, Schematic representation of a single LRR, with the LRR sequence motif mapped onto it. Interactions of the LRRs with MRAS and PP1C occur through the top and midriff residues of SHOC2, while PP1C interacts through the bottom residues of SHOC2. f, Surface of SHOC2 (pink), with the residues contacted by switch I (dark blue), switch II (purple), and the C terminus (blue) of MRAS and PP1CA highlighted. g, The SHOC2-M173I NS mutation potentially forms a new contact with MRAS-M77. h, A critical interaction of MRAS switch I (dark blue) and switch II (purple) with SHOC2 (pink) and PP1CA (green), either directly or indirectly through bridging water molecules (red spheres). i, Plots of the mean apparent KD measurements of SMP complex assembly for NS mutants and point mutants present at the SHOC2–MRAS interface. The s.d. is shown as error bars, with the number of independent replicates shown as circles (summarized in Supplementary Table 1). NS mutations are enclosed in black boxes. No binding is indicated as NB.
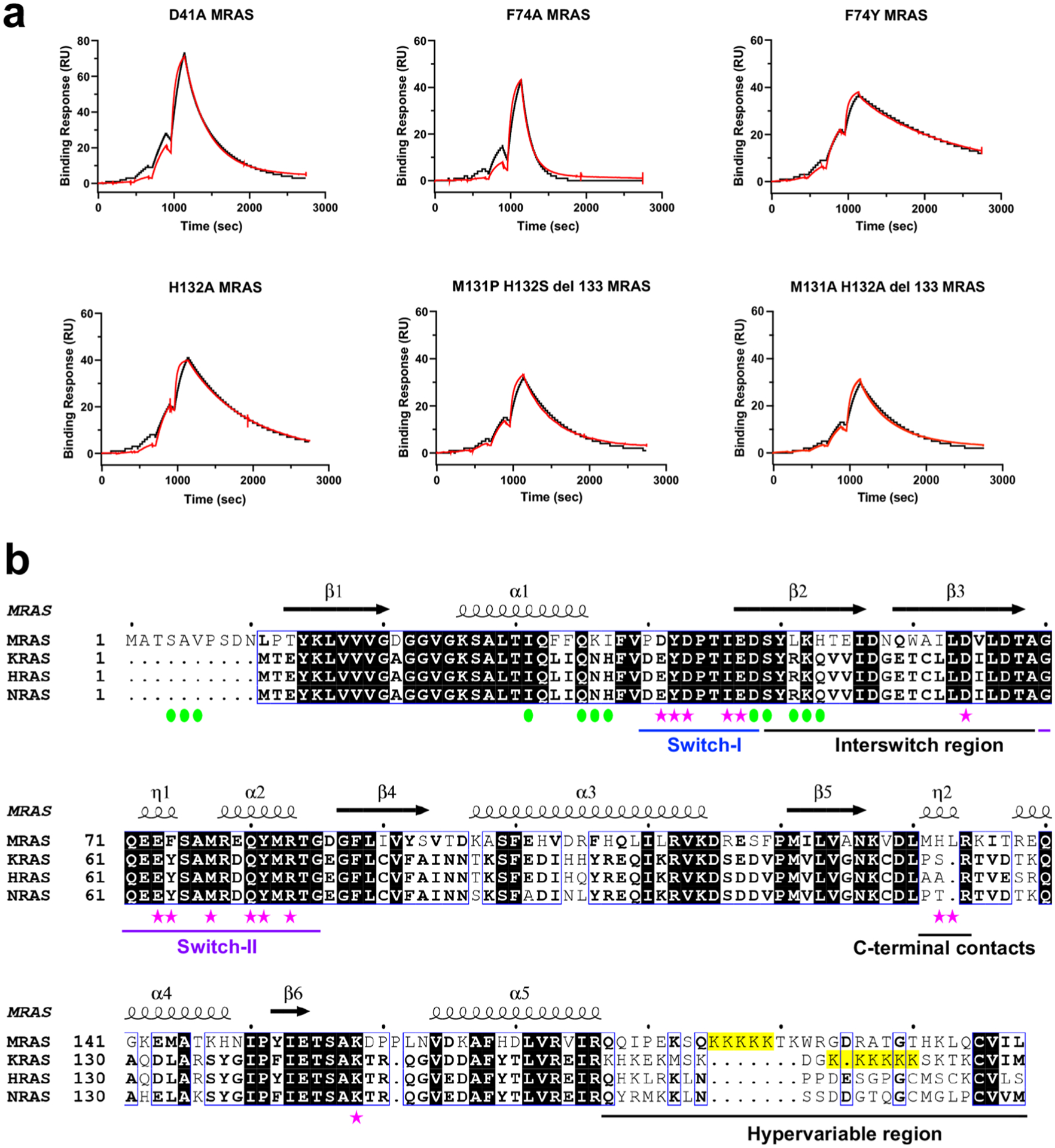
SPR measurements of the NS SHOC2-M173I mutation show a fivefold increase in the apparent affinity of SMP-complex formation (Extended Data Fig. 6d), demonstrating that this gain-of-function mutation stabilizes the complex. SHOC2-M173 does not contact MRAS. However, the substitution of methionine with isoleucine results in increased hydrophobicity and potentially forms a de novo contact with MRAS-M77 (Fig. 4g). A genetic screen of the SHOC2 homolog in Caenorhabditis elegans identified D175N as a loss-of-function mutation35. We believe the loss of function arises not from contact with MRAS-M77, but from the removal of hydrogen bonds to the guanidino head group of SHOC2-R177, which pre-orientates it to interact with MRAS-E47 and MRAS-Y81 of switch I and II, respectively (Fig. 4h). SHOC2-D175N or SHOC2-R177A mutations result in no complex formation (Fig. 4h,i and Extended Data Fig. 6d).
Additional mutations were made in SHOC2 and MRAS to identify key interactions at the SHOC2–MRAS interface. SHOC2-R223 interacts with MRAS-D43 (switch I), whereas SHOC2-Y129 and SHOC2-Y131 contact MRAS-Q80 and MRAS-Y81 (switch II). SHOC2-R223A results in complex formation that is ~300-fold weaker, whereas no binding is observed for SHOC2-Y129A Y131A (Fig. 4i and Extended Data Fig. 6d). MRAS-D41, present in the switch I region, interacts with SHOC2-R292. MRAS-D41A results in weakening of SMP-complex formation by tenfold (Fig. 4i and Extended Data Fig. 7a). MRAS-F74 of switch II protrudes towards switch I and interacts with SHOC2-T242. The MRAS-F74A mutation causes a ~300-fold weakening of complex formation (Fig. 4i and Extended Data Fig. 7a). MRAS-H132 is found within a helical loop and forms a hydrogen bond to the SHOC2-E428 only in SMP1 (Fig. 2d and Extended Data Fig. 2a). In H/K/NRAS, this helical loop is one residue shorter, and the histidine is replaced by shorter aliphatic residues, potentially resulting in the loss of this interaction (Extended Data Fig. 7b). The MRAS-H132A mutation results in a weakening of complex formation by about threefold (Fig. 4i and Extended Data Fig. 7a). Furthermore, mutation of this loop to its KRAS (131MHL133→131PS132) or HRAS (131MHL133→131AA132) equivalent also results in a similar threefold weakening of complex formation, suggesting that this region of MRAS contributes to its higher-affinity complex formation over that of H/K/NRAS, and that SHOC2 does interact with this region, as observed in the SMP1 complex (Figs. 2d and 4i and Extended Data Fig. 7a).
The MRAS–PP1CA interface
The MRAS–PP1CA interaction buries ~1,400 Å2 of surface area. MRAS binds to PP1CA adjacent to the PP1C–SHOC2 (LRR) interface such that all MRAS residues are >20 Å away from the PP1CA active site (Fig. 5a). PP1CA contacts three regions on MRAS (Fig. 5b). (1) N-terminal contacts: the unique N-terminal residues of MRAS interact with PP1CA, forming a hydrogen bond between MRAS-S4 and PP1CA-E218 and van der Waals interactions (Fig. 5b and Extended Data Fig. 7b). These residues occupy the myosin phosphatase N-terminal element (MyPhoNE) cleft on PP1CA. The myosin phosphatas-targeting subunit 1 (MYPT1) protein uses RVxF and MyPhoNE motifs to bind to PP1CA. Although we observe MRAS occupying the MyPhoNE cleft, it does so differently than does MYPT1 (Fig. 5c and Extended Data Fig. 8a)46. Deletion of these N-terminal residues in MRAS results in a sixfold weakening of SMP-complex formation, as observed by ITC, confirming their importance for binding and as a key region of specificity between MRAS and H/K/NRAS proteins (Extended Data Fig. 8b). (2) Pre-switch I contacts: residues 31–37 of MRAS interact with residues 189–198 of PP1CA, forming two hydrogen bonds and van der Waals interactions (Fig. 5b). (3) Interswitch contacts: residues 48–53 of MRAS interact with residues between 178–190 of PP1CA. MRAS-H53 interacts with PP1CA-D179. MRAS-H53A weakens SMP-complex formation by about tenfold (Fig. 5d and Extended Data Fig. 8c). PP1CA-R188 is the only residue in the entirety of the SMP complex that engages with the other two proteins by forming hydrogen bonds with SHOC2-E155, MRAS-D48, and MRAS-S49 (Figs. 4h and 5b). This potentially makes PP1CA-R188 the linchpin of the SMP complex. No SMP complex formation is observed with the PP1CA-R188A mutant (Fig. 5d and Extended Data Fig. 8d). Despite being the smallest protein-protein interface, PP1CA and MRAS form eight hydrogen bonds and two salt bridges (Fig. 5e).
Fig. 5 |. Structural and mutational analysis of the MRAS–PP1CA interface.
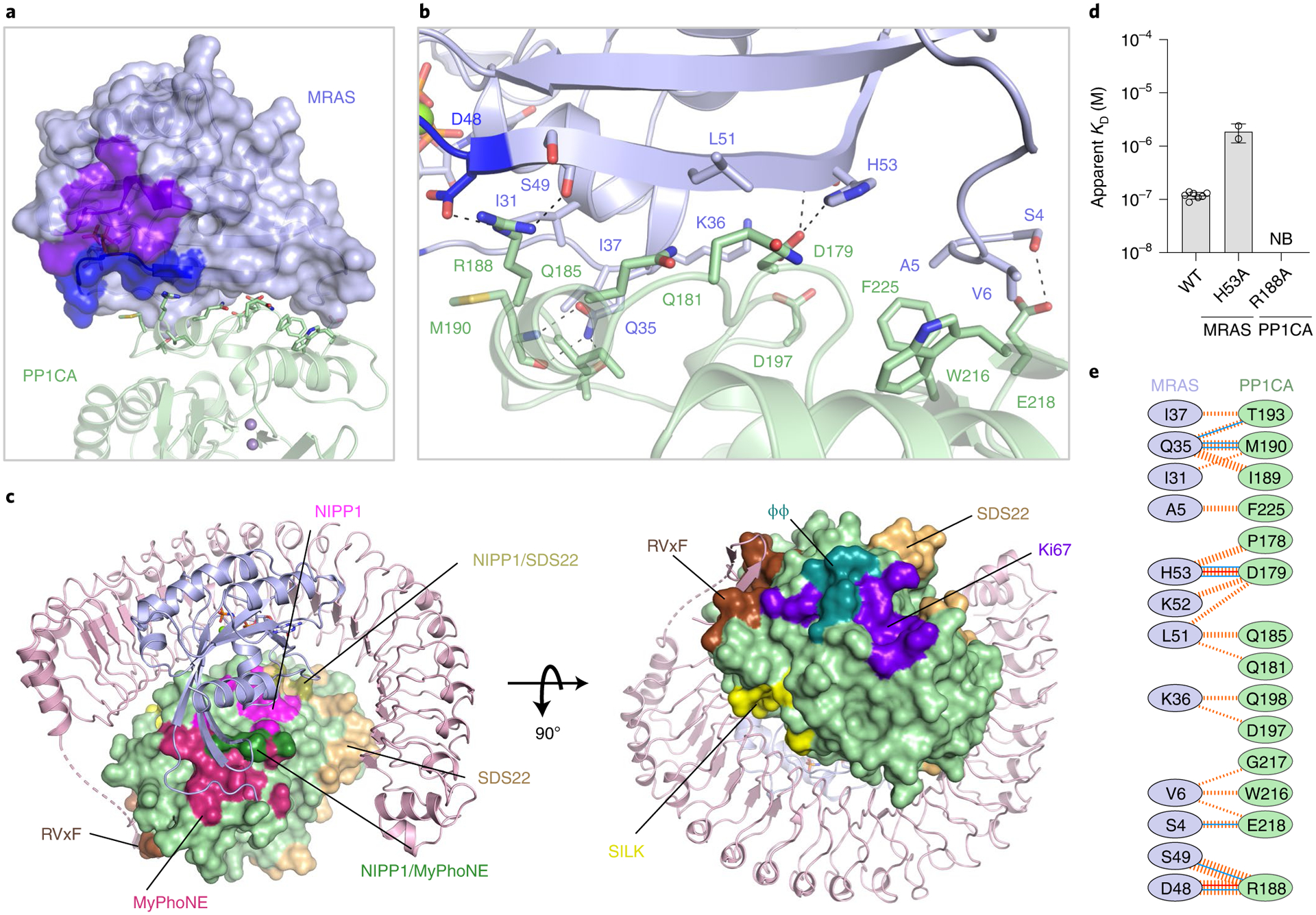
a, Overall view of PP1CA (green) interacting with the surface of MRAS (blue surface), with switch I, switch II, nucleotides, and Mg2+ shown in dark blue and purple and as sticks and the green sphere, respectively. The active site Mn2+ ions are shown as gray spheres. b, Zoomed-in view of the PP1CA–MRAS interaction interface, with side chains shown as sticks and hydrogen bonds as black dashed lines. c, PP1CA is shown as a surface in the context of the SMP complex. All known PP1C interaction sites are colored on the surface of PP1CA (green) in the SMP complex; RVxF (brown), SILK (yellow), SDS22-binding site (wheat), ϕϕ (teal), ki67-binding site (purple), MyPhoNE (dark red), NIPP1 helix (magenta), overlap of MyPhoNE and NIPP1 helix (dark green) and overlap of the NIPP1- and SDS22-binding sites (olive). d, Plots of the mean apparent KD measurements of SMP-complex assembly for point mutants present at the MRAS–PP1CA interface. The s.d. is shown as error bars, with the number of independent replicates shown as circles (summarized in Supplementary Table 1). No binding is indicated as NB. e, Schematic representation of the MRAS–PP1CA interaction interface, as analyzed by PDBSum. The interactions are colored using the following notations: hydrogen bonds as solid blue lines, salt bridges as solid orange lines, and non-bonded contacts as dashed orange lines (the width of the lines are proportional to the number of atomic contacts).
Recognition of RAF substrates by the SMP complex
PP1CA has three active-site channels/grooves, denoted the acidic, hydrophobic, and C-terminal channels (Fig. 6a)47. To understand how the SMP recognizes RAF substrates, we docked a 15-mer BRAF CR2-pS polypeptide using the CABS-Dock server. Most of the top clusters containing 202 docked peptides were placed with the CR2-pS S365 in the active site, with the N and C termini of the peptides occupying the acidic and hydrophobic channels, respectively (Fig. 6b)48. We observed similar docking poses with a 15-mer CRAF CR2-pS polypeptide (Extended Data Fig. 9a). Two NS mutations (D252Y and E274K in PP1CB) have been identified in the acidic and hydrophobic channels (Extended Data Fig. 9b). None of the top-scoring peptide models contact the equivalent residues in PP1CA, D251 and E273, suggesting that these residues may selectively prevent other substrates from competing with RAF in the SMP complex or fine tune the affinity for RAF.
Fig. 6 |. Model of recognition of RAF substrates by the SMP complex.
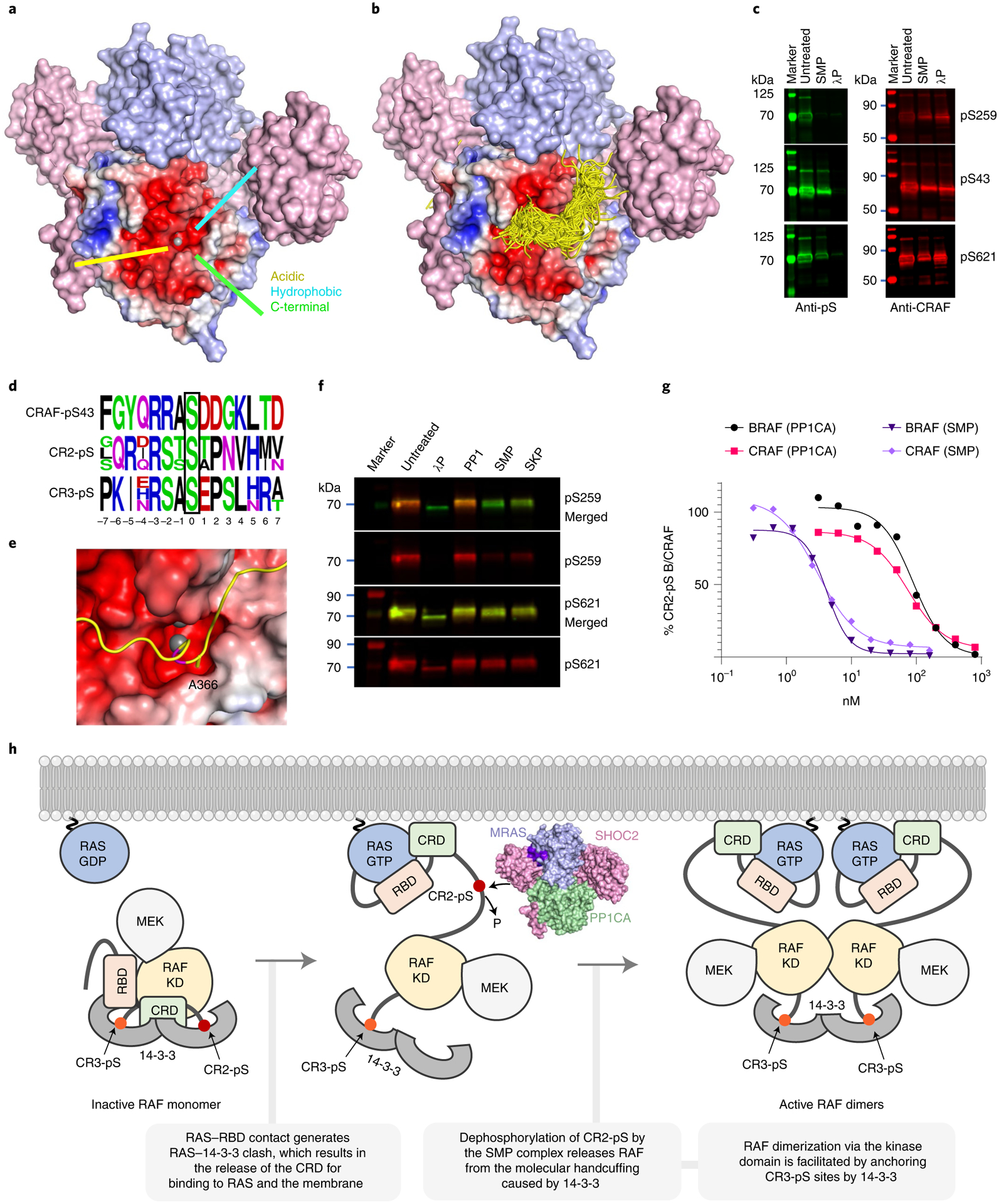
a, Structure of the SMP complex is shown as a surface. SHOC2 and MRAS are colored pink and blue, respectively. The surface of PP1CA is shown as an electrostatic surface. The three active-site channels—acidic, hydrophobic, and C-terminal—are shown. The Mn2+ ion is shown as a gray sphere. b, The CABS-dock server was used to generate a 15-mer polypeptide of the CR2-pS region of BRAF, which was docked into the PP1CA structure of the SMP complex. All 202 peptides from the top cluster of solutions are presented as ribbons. The vast majority were placed in the active site, and all peptides placed with their N and C termini in the acidic and hydrophobic active-site channels. c, Fluorescent western blot (representative of three independent experiments) of CRAF that was either untreated or treated with SMP or lambda phosphatase (λP). Right-hand panels show the total CRAF present (red), whereas left-hand panels reveal CRAF by specific phosphoserine antibodies (green) targeting pS259 (top), pS43 (middle), and pS621 (bottom). d, Sequence alignments of CRAF pS43, CR2-pS of ARAF, BRAF, and CRAF, and CR3-pS of ARAF, BRAF, and CRAF. The phosphoserine in each case is boxed in black at position 0. e, The top docked CR2-pS peptide of BRAF is displayed as a ribbon in the active site, with the PP1CA surface shown in electrostatic surface representation. S365 of BRAF present in the active site is colored magenta. The docked model suggests that BRAF-A366 would be placed inside the narrow negatively charged active-site channel. f, Fluorescent western blot (representative of three independent experiments) of CRAF that was either untreated or treated with λP, PP1CA, SMP, or SKP. Phosphoserine-specific antibodies for pS259 and pS621 are shown in red. Total CRAF is shown in green. SMP and SKP complexes specifically dephosphorylate CRAF-pS259. g, Comparison of dephosphorylation activity (half-maximal effective concentration, EC50) of the PP1CA and SMP complex on BRAF and RAF substrates derived from Li-COR quantification of bands from Extended Data Fig. 6c. h, Model showing the role of the SMP complex in the RAF activation process.
To validate the specificity of the SMP complex for various phosphorylation sites on BRAF and CRAF, we performed dephosphorylation assays. Treatment of BRAF or CRAF with lambda phosphatase non-specifically removes all phosphates (pS43, pS249, and pS621). However, treatment of BRAF or CRAF with the SMP complex shows that it selectively dephosphorylates the CR2-pS site (Fig. 6c and Extended Data Fig. 9c,d). Sequence composition around different pS sites provides a rationale for CR2-pS site specificity. The CR2-pS site contains either threonine or alanine residues at the +1 position in RAF substrates (Fig. 6d), while the CR3-pS site and the pS43 site in CRAF contain acidic residues at this position (Fig. 6d). Our docking results show that residues in the +1 position would be placed inside the restrictive, negatively charged active-site channel, suggesting a preference for small and non-acidic residues at this position (Fig. 6e), consistent with previous experiments49. Dephosphorylation of a phosphorylated BRAF 15-mer CR2-pS polypeptide by the SMP complex in which the +1 position is mutated to glutamic acid was slower than the WT sequence measured by matrix-assisted laser desorption/ionization coupled to time of flight (MALDI–TOF), supporting our docking results (Extended Data Fig. 10).
Comparison of dephosphorylation using the SKP and SMP complexes showed that the SKP complex has slightly weaker dephosphorylation activity for CRAF CR2-pS than does the SMP complex (Fig. 6f). To determine whether SHOC2 and MRAS have any effect on the dephosphorylation activity of PP1CA, we assessed dephosphorylation activity using apo-PP1CA and the SMP complex with BRAF and CRAF as substrates. Interestingly, the dephosphorylation activity of apo-PP1CA was 10- to 30-fold lower than that of the SMP complex, yet still displayed specificity to CR2-pS (Fig. 6g and Extended Data Fig. 9c,d), suggesting that MRAS and SHOC2 play a role in enhancing the dephosphorylation activity of PP1CA towards CR2-pS in RAF substrates.
Discussion
Our high-resolution structure of the heterotrimeric SMP complex provides insights into how SHOC2, MRAS, and PP1CA interact to form this ternary complex and to dephosphorylate RAF. Analyses of NS mutations in SHOC2, MRAS, and PP1C in the SMP complex structure suggest how these substitutions would result in additional interactions that lead to tighter complex formation, sustained dephosphorylation of RAF, and activation of MAPK/ERK signaling. Interestingly, none of the three proteins form stable, high-affinity, binary complexes with each other, despite multiple contacts with each protein, highlighting the strikingly synergistic nature of SMP-complex formation. We did observe a weak binary SHOC2–PP1CA interaction by SPR, suggesting this forms first. All PP1C regulators that rely on the RVxF motif to bind PP1C form high-affinity binary complexes. As SHOC2 contains an RVxF motif, it is unusual and unique to observe only a weak interaction with PP1C. This is distinct from SDS22 (an LRR protein without an RVxF motif) and RVxF-containing proteins, both of which form high-affinity binary complexes with PP1C. Ternary complex assembly is achieved only with active MRAS, indicating that MRAS plays a vital role in initiating and regulating the SMP complex assembly. As MRAS is anchored in the plasma membrane through its hypervariable region (HVR), it targets PP1C to the plasma membrane only in the presence of SHOC2, suggesting that SHOC2 functions as an adapter protein in this complex.
Our findings and previous results suggest that H/K/NRAS can substitute for MRAS in the SMP complex24. However, in vivo, MRAS is most likely to form part of the SHOC2–RAS–PP1C complex, for several reasons. Our SPR data show a 7- to 40-fold higher affinity of complex formation with MRAS than H/K/NRAS and that the SKP complex displays relatively weaker dephosphorylation activity than does SMP. This increase in affinity observed with MRAS comes from the additional interactions from the N and C termini (residues 4–6 and H132) and compositional differences in interacting residues present in the pre-switch and interswitch regions of MRAS. Previous studies have shown that substituting MRAS residues with corresponding residues in KRAS in the pre-switch-I and interswitch regions decreases MRAS affinity for SHOC2 and PP1C11. MRAS-L51R (equivalent to KRAS-R41) increased MRAS affinity to BRAF and CRAF, whereas it decreased its affinity for SHOC2 and PP1C, suggesting that MRAS and H/K/NRAS evolved to play different roles during the RAF activation process. This is supported by the observation that MRAS cannot activate RAF kinases to the same extent as H/K/NRAS, and it is likely due to differences in the interswitch region that affect MRAS interaction with the CRD of RAF proteins7,11. However, only SHOC2 has been repeatedly identified in synthetic lethality CRISPR–Cas9 screens17–20. Furthermore, MRAS knockout does not phenocopy SHOC2 knockout in mice50,51. It is therefore possible that in the absence of MRAS, the lower-affinity interaction of H/K/NRAS for SHOC2 and PP1C complex formation may be sufficient for CR2-pS RAF dephosphorylation in vivo. Similarly, it has recently been shown using mouse embryonic fibroblasts lacking H/K/NRAS that MRAS can substitute for H/K/NRAS to activate ERK by RAF inhibitors25,52. Thus, the lower affinity complexes of MRAS-RAF and SHOC2-H/K/NRAS-PP1C may be sufficient to provide redundancy in some contexts.
The SMP complex is responsible for dephosphorylation of CR2-pS sites and activation of RAF. Our results show that the interaction with MRAS and SHOC2 selectively enhances the dephosphorylation activity of PP1CA by about twentyfold against CR2-pS but not any other RAF phosphorylation sites, suggesting that MRAS and SHOC2 play a role in targeting and enhancing dephosphorylation of CR2-pS by PP1CA. SHOC2 and/or MRAS may aid in the direct recruitment of RAF through different mechanisms observed in other PP1C and PP1C-interacting protein (PIP) complexes. Several PIPs contain extra domains which interact with substrates either directly or indirectly. The muscle-glycogen-targeting (GM) regulatory subunit–PP1C complex can directly recruit the muscle-specific glycogen synthase substrate to the holoenzyme through a carbohydrate-binding domain found within GM53. This domain can also indirectly recruit phosphorylase-a, by binding to glycogen where phosphorylase-a co-localizes. The direct recruitment of RAF by MRAS, like H/K/NRAS binding to RAF, cannot occur as this interaction is weaker, and the pre-switch, switch I, and interswitch residues that bind RAF are buried by SHOC2 and PP1CA7. This would suggest that another region of MRAS aids in recruiting RAF or SHOC2. In addition to the direct substrate mechanism, an indirect substrate-recruitment mechanism could also occur at the plasma membrane. Both MRAS and KRAS share a similar HVR and lipidation profile, and co-localize within the disordered lipid regions of the plasma membrane54. Active KRAS would therefore bind and recruit RAF substrates both temporally and spatially with the active SMP complex at the disordered lipid regions of the plasma membrane.
PP1C forms complexes with >200 PIPs that bind through short linear motifs (SLIMs), including the RVxF, SILK, MyPhoNE, φφ, and SpiDoC motifs (Fig. 5c). PIPs use combinations of these SLIMs to form multivalent interactions with PP1C that enhance regulator binding avidity and create PP1C holoenzymes with unique properties and substrate specificity, although the exact molecular mechanisms of how they alter substrate specificity are unclear. This is true for the SMP complex, as both SHOC2 and MRAS bind >20 Å from the active site. We did not observe any alteration of residues or electrostatics of the active-site channels of PP1CA upon complex formation or extension of these active-site channels, as seen in the Phatcr1–PP1CA complex55. However, the entrance to the acidic channel may be partially blocked because of the disordered residues between the RVxF motif and LRRs of SHOC2, as seen in the NIPP1–PP1CA complex56. The formation of the membrane-bound SMP complex is likely to prevent the formation of other PP1C holoenzymes owing to SHOC2 and MRAS occluding several PIP-binding sites on PP1C, including the RVxF, SILK, SDS22, MyPhoNE, and NIPP1 helix-binding pockets (Fig. 5c)27,29,57.
The RAF activation cycle starts when active RAS interacts with RBD in the autoinhibited RAF complex (Fig. 6h). The RAS–RAF RBD interaction causes a steric clash between RAS and 14–3-3, resulting in conformational changes that dislodge the RBD and CRD from the autoinhibited RAF complex58. This action allows the CRD to interact with the plasma membrane and RAS to further stabilize the RAS–RAF interaction. The release of the CRD exposes and allows dephosphorylation of the CR2-pS site by the SMP complex (Fig. 6h). The exposed kinase domain dimerizes and is stabilized by binding a 14–3-3 dimer to the CR3-pS sites. Our in vitro assays and previous studies show that the SMP can dephosphorylate RAF without RAS, although this would not happen inside the cell. The membrane-bound SMP complex would not dephosphorylate RAF unless it is recruited to the plasma membrane by active RAS.
The SMP complex is a high-value target for regulating RAF and MAPK-pathway activation. Considering that SHOC2, MRAS, and PP1C do not form a high-affinity binary complex, and that several interface mutants described in this study disrupt complex formation, targeting any of the three interaction interfaces would likely disrupt the SMP complex formation. Targeting MRAS appears to be difficult, as it could be substituted by H/K/NRAS. Targeting PP1C would also be challenging owing to the large number of proteins that bind to PP1C (Fig. 5c). However, our results show that PP1C-R188 is important for SMP assembly, and it is not part of any known SLIM. A small molecule targeting this site could prevent SMP-complex formation specifically. Although the biology of SHOC2 is the least understood of the three proteins, SHOC2 does make an interesting target owing to its identification in several synthetic lethality CRISPR–Cas9 screens, though it is unknown whether the loss of the SMP complex or the loss of a different SHOC2 interaction causes the lethality. On the basis of other SHOC2 studies, the MRAS-binding region on SHOC2 is a unique interaction site and thus a promising druggable site59,60. Our data and previous studies support that altering the surface of SHOC2 in LRR2 and LRR4 prevents the formation of the SMP complex through disruption of the SHOC2–MRAS interface14,35,61, suggesting that these two regions of the SHOC2 surface could be exploited as druggable target sites. The question as to how the SMP complex interacts and dephosphorylates the RAF–RAS complex remains unanswered. A deeper understanding of how these two complexes interact with each other at the plasma membrane could lead to new approaches to target RAS–RAF-driven cancers and NS.
Methods
DNA
DNA constructs of human SHOC22–584, SHOC258–564, the SMP complex, PP1CA7–300, PP1CA2–330, PP1CAmutant 7–300, PP1CB2–327, MRAS1–179, MRASmutant 1–179, H/K/NRAS1–169, and H/K/NRASmutant 1–169 were synthesized as gene optimized fragments for insect cells (SHOC2 and SMP complex) or Escherichia coli (PP1C and H/K/M/NRAS) downstream of a tobacco etch virus (TEV) protease site (ENLYFQ/G). Entry clones were transferred to either baculovirus or E. coli expression clones containing amino-terminal His6-MBP (maltose-binding protein) fusions by Gateway LR recombination into pDest-636 (baculovirus, Addgene no. 159574) or pDest-566 (E. coli, Addgene no. 11517). Final baculovirus expression clones were used to generate bacmid DNA in strain DE95 using the Bac-to-Bac system (Thermo Fisher Scientific).
Protein expression and purification
SHOC2, SMP complex, and mutants were expressed in insect cells with the chaperone SUGT1, as described before34. Briefly, 1.5 × 106 cells/ml of Sf9 cells adapted for serum-freee suspension and grown in SF900 III medium in 100 ml were transfected with DNA–cellfectin II lipid complex (70 μl of bacmid DNA:250 μl cellfectin II:500 μl SF900 III medium). The culture was incubated for 120 hours at 27 °C before isolation of the cell culture supernatant. The baculovirus titer was measured by qPCR (TaqMan Gene Expression assay for the baculovirus GP64 protein). Then, 1–2 L of 7 × 105 Tni-FNL cells62 in SF900 III medium was grown for 24 hours at 21 °C to allowing doubling before baculovirus infection with a multiplicity of infection of 3. Infected cell cultures were grown for 72 hours at 21 °C before cells were collected. Cells were lysed in 20 mM HEPES, pH 7.4, 300 mM NaCl, and 1 mM TCEP (100 ml/L of cells), using a Microfluidizer. Lysates were clarified by ultracentrifugation (100,000g, 30 minutes at 4 °C) and filtration (0.45-μm high flow PES filter). Proteins were captured by immobilized metal affinity chromatography (IMAC) using a 5-ml Ni Sepharose High Performance column (GE Healthcare) equilibrated in 20 mM HEPES, pH 7.4, 300 mM NaCl, 1 mM TCEP, and 35 mM imidazole on an NGC chromatography system (Bio-Rad). The column was washed for 5 column volumes (CVs) with equilibration buffer before elution using a gradient of 7–100% buffer A (20 mM HEPES, pH 7.4, 300 mM NaCl, 1 mM TCEP, 500 mM imidazole) over 20 CVs. The protein peak was pooled, dialyzed into buffer A with no imidazole, and digested with His-tagged TEV at a ratio of 1:20 (vol/vol) protease:pooled protein overnight at 4 °C. His-tagged TEV and cleaved His-MBP were captured by a second IMAC purification step. Cleaved proteins eluted in the flow-through and by running a shallow gradient of 0–10% over 10 CVs using buffer A. Cleaved protein fractions were pooled, concentrated, and further purified by size-exclusion chromatography using a 16/600 Superdex 200 column (Cytiva) equilibrated with 20 mM HEPES, pH 7.4, 150 mM NaCl, 1 mM TCEP.
RAS proteins and mutants were expressed and purified as described for the Dynamite expression protocol63. Briefly, a culture of E. coli harboring the expression plasmid of interest was grown in non-inducing MDAG-135 medium, overnight at 37 °C. This was used to inoculate (1:50 dilution) 1–2 L of Dynamite media. Cells were grown at 37 °C until and OD600 nm of 6–8 was reached, before induction with 0.5 mM IPTG and incubation at 16 °C for 18–20 hours. Cells were collected by centrifugation. Proteins were purified as outlined for KRAS64, or as outlined for SHOC2 and the SMP complex described above, except the size-exclusion chromatography buffer contained 5 mM MgCl2.
PP1CA, PP1CA mutants, and PP1CB proteins were expressed and purified in a similar manner as the RAS proteins, but with some modifications. Specifically, the expression strain also included the GroEL-expressing plasmid pG-tf2 (Takara Bio USA), and expression was induced at 10 °C. The lysis buffer was 20 mM Tris-HCl, pH 8.0, 700 mM NaCl, 10% glycerol (wt/vol), 1 mM MnSO4 (or MnCl2), 1 mM TCEP, and 0.5% Triton X-100 (wt/vol). The same buffer without Triton X-100 was used in subsequent steps until the SEC/final buffer, which was 20 mM Tris-HCl, pH 8.0, 500 mM NaCl, 1.0 mM MnSO4, and 1 mM TCEP. Clarification of the lysate required extended conditions to overcome the presence of glycerol in the buffer (2 hours at 13,000g), and a 5 ml MBPTrap HP column (Cytiva) was placed in front of the preparative SEC column to capture undigested fusion protein. All mutant protein SEC elution profiles and measured thermal denaturation temperatures were similar to the wild-type proteins.
Nucleotide exchange
MRAS-GDP (the protein is normally in the GDP-bound state when purified from E. coli) was mixed with a 5-molar excess of non-hydrolysable GMPPNP (tetralithium salt, Jena Biosciences NU-401–50) in a reaction mixture of 200 mM ammonium sulfate and 100 μM ZnCl2. The final MgCl2 concentration in the reaction was less than 1 mM through dilution of the stock protein with the reaction mixture components. The typical protein concentration range in the reaction was 0.1–0.3 mM. Alkaline phosphatase-agarose beads (Sigma P0762–250UN) were added at a ratio of 1 U per mg of protein and the reaction was mixed at room temperature for 3 hours. The beads were then removed by centrifugation at 1,500g for 2 minutes. The sample was adjusted with an additional tenfold molar concentration of GMPPNP and incubated at 4 °C for 2 hours or overnight. Excess nucleotide was removed by passing over a PD-10 desalting column packed with Sephadex G-25 resin (cat. no. 17085101, Cytiva) in 20 mM HEPES, pH 7.4, 150 mM NaCl, 1 mM MgCl2, and 1 mM TCEP. Protein concentration was determined on a Nanodrop 2000C spectrophotometer (Thermo Fisher Scientific), reading at A280.
Crystallization and data collection
Purified SMP complex was concentrated to 15 mg/ml, and crystallization screening was carried out at 20 °C using the sitting-drop vapor diffusion method by mixing purified SMP complex with an equal volume of reservoir solution (200 nL:200 nL). Crystals of the SMP complex appeared within 24 hours in the crystallization condition containing 25% wt/vol PEG 1500, 0.1 M MIB pH 4.0. These crystals, cryoprotected with 20% vol/vol of glycerol, diffracted anisotropically to a resolution of ~3.7 Å. To improve the diffraction quality of these crystals and the stability of the SMP complex during the crystallization, GTP present in the MRAS of the SMP complex was exchanged with GMPPNP. Further optimization of the crystallization condition was carried out by increasing the pH (0.1 M MIB, pH 4.2) and reducing the concentration of PEG 1500 (15% wt/vol). However, these optimized crystals only diffracted to 3.2 Å and remained anisotropic. Matrix micro-seeding was performed to further improve the quality of diffraction65. Briefly, two drops worth of SMP crystals were transferred to a seed bead tube (Hampton Research) containing 100 μL of 15% PEG 1500, 0.1 M MIB, pH 4.2, vortexed for 30 seconds, before dilution to 1 mL with 15% PEG 1500, 0.1 M MIB, pH 4.2. Another round of extensive crystallization screening was carried out in which a ratio of 200 nL protein:133 nL reservoir:67 nl of seeds was used (7.5 mg/ml of the SMP complex). Approximately 30 new conditions were identified, though only one yielded isotropic diffracting crystals to 2.8 Å (20% wt/vol PEG 3350, 0.2 M sodium sulfate). Crystals were further optimized around this crystallization condition through a grid screen and seeding. The grid varied the concentration of PEG 3350 and sodium sulfate from 15–25% wt/vol (in steps of 1.43%) and 0–250 mM (in steps of 23 mM), respectively. A ratio of 200 nL protein:133 nL reservoir:67 nl of seeds was used (7.5 mg/ml SMP complex). Seeds were prepared fresh, as described above, using crystals from the original condition of 15% PEG 1500, 0.1 M MIB, pH 4.2 (frozen seeds failed to work). A 2.17-Å dataset was collected on beamline 24-ID-C at the Advanced Photon Source (Argonne) with a crystal grown from 17.9% wt/vol PEG 3350, 136 mM sodium sulfate, and 1:10 dilution of seeds in a ratio of 200 nL protein:133 nL reservoir:67 nl of seeds. The crystal was cryoprotected with 25% (vol/vol) glycerol.
To solve the structure of SHOC2, we carried out crystallization screening of two SHOC2 constructs (SHOC22–584, SHOC258–564) using commercial screens at 15 mg/ml protein concentration. SHOC258–564 produced crystals in multiple ammonium sulfate conditions at low pH. Optimization of SHOC258–564 crystals produced diffracting crystals in 1.5 M ammonium sulfate, 0.1 M sodium citrate pH 5.0. Crystals were cryoprotected with 30% glycerol, and a 2.4-Å dataset was collected on beamline 24-ID-C at the Advanced Photon Source (Argonne).
Structure determination and analysis
Crystallographic datasets were indexed and integrated using XDS66. The integrated data were then scaled, truncated, and converted to structure factors using the program Aimless present in the CCP4 suite (v7.1.014)67,68. Matthew’s coefficient suggested two copies of the SMP complex inside the asymmetric unit. The structure was determined using the molecular replacement program Phaser (Phenix v1.19) using mouse GMPPNP-bound MRAS (PDB: 1X1S) and human PP1CA (PDB: 6DNO)69. This helped in locating two copies of MRAS and PP1C inside the asymmetric unit. Since the structure of SHOC2 was not available at this time, we used a Rosetta-generated model of SHOC2 as a search model in our molecular replacement runs69. Although this approach did not work, the initial maps calculated after placing two copies of MRAS and PP1C allowed the manual placement of the Rosetta-generated model of SHOC2. This was followed by a rigid body refinement. The initial model of the SMP complex was iteratively rebuilt in COOT and refined with Refmac5, followed by Phenix.Refine (Phenix v1.19)68–71. During the final stages of model building and refinement, water molecules were identified by the automatic water-picking algorithm in COOT and Refmac5/Phenix.refine. The positions of these automatically picked waters were checked manually during model building. The structure of SHOC2 was determined using SHOC2 present in the SMP complex as a search model in the molecular replacement Phaser69. This search identified one copy of SHOC2 in the asymmetric unit. Model building and refinement of SHOC2 were carried out using the same protocol as described above for the SMP complex. Secondary structural elements were assigned using DSSP (https://swift.cmbi.umcn.nl/gv/dssp/). Figures were generated with PyMOL (v2.5.1), and surface electrostatics were calculated with APBS72,73. Crystallographic and structural analysis software support was provided by the SBGrid Consortium74. Data collection and refinement statistics are shown in Table 1.
Surface plasmon resonance measurements
CM5 chips (Cytiva Life Sciences) were preconditioned by injecting 0.5% SDS, 100 mM HCl, 0.85% H3PO4, and 50 mM NaOH in that order at 30 μL/minute for 60 seconds in PBS pH 7.4 running buffer. Then, 200 μg/mL neutravidin (Thermo Scientific) in 10 mM sodium acetate, pH 4.5, was amine coupled to the surface in PBS running buffer using standard EDC/NHS chemistry to a density of ~7,000 RU per flow cell. All buffers were vacuum filtered through 0.2-μm cellulose acetate membranes. Avi-tagged SHOC2 proteins were biotinylated in vitro using a procedure described previously75 and then captured by manual injection to an appropriate density in 10 mM HEPES, 150 mM NaCl, 2 mM MgCl2, 0.05% Tween 20, 1 mM TCEP, pH 7.4. Protein analytes MRAS and PP1CA were diluted equimolar to the highest concentration, typically 1 μM, in the buffer above then serially diluted threefold four times in the same buffer for a total of five concentrations. Single-cycle kinetic responses consisted of injections at 30 μL/minute with a contact time of 180 seconds and a dissociation time of 1,600 seconds for each concentration of analytes. Sensorgrams were double referenced by subtracting the signal from a reference channel of neutravidin alone and a buffer blank. The data were fit to a 1:1 kinetic model to calculate an apparent KD using the S200 evaluation software package (v1.1.27) or the Insight software package (v3.0.12.15655). All experiments were conducted at 25 °C on an S200 or 8K instrument (Cytiva Life Sciences). All binding data are tabulated in Supplementary Table 1, with the number of replicates indicated. The s.d. was calculated from multiple independent experiments and plotted in Prism9. Certain mutants could not be tested by SPR owing to non-specific binding to the reference channel. In these cases, affinity measurements were performed by ITC.
Isothermal titration calorimetry measurements
Proteins were extensively dialyzed against 30 mM HEPES, 500 mM NaCl, 1 mM MgCl2, 0.5 mM TCEP, 0.1 mM MnCl2, pH 7.5, 5% glycerol. Duplicate ITC measurements were performed on a MicroCal PEAQ-ITC instrument (Malvern Panalytical). An ITC experiment consisted of 15 μM of PP1C and MRAS in the cell with 175 μM of SHOC2 in the syringe. All measurements were carried out at 25 °C, with a stirring speed of 750 rpm and 19 injections of 2 μl injected at 210 s intervals. Data analysis was performed using a “one set of sites” model using the MicroCal PEAQ-ITC analysis software (v1.41, Malvern Panalytical). All binding data are tabulated in Supplementary Table 2 with the number of replicates indicated.
RAF kinase dephosphorylation assay by western blotting
PP1CA and SMP complex phosphatase activity was tested on purified His6-CRAF protein, GST-CRAF, or GST-BRAF. For His6-CRAF protein, substrate was diluted in 20 mM HEPES pH 7.4, 150 mM NaCl, 1 mM TCEP, 2 mM MgCl2, and 2 mM MnCl2 to a final concentration of 686 nM. Twenty microliters of diluted CRAF/BRAF sample was mixed with 20 μl of 204 nM SMP or PP1CA and incubated at 30 °C for 30 minutes. After 30 minutes, 40 μl of 2× NuPAGE LDS sample buffer (Thermo Fisher Scientific) was added to the tube, and samples were boiled for 5 minutes to stop the reaction. Western blots were prepared by electrophoresing 10 μl of each sample on an SDS–PAGE gel, transferring samples to a PVDF membrane via iBlot (Thermo Fisher Scientific) using standard manufacturer’s conditions, and probing for CRAF pS43 (Abcam no. ab150365, 1:1,000 dilution), pS259 (Abcam no. ab173539, 1:1,000 dilution), pS621 (Abcam no. ab4767, 1:1,000 dilution), and anti-His6 for total CRAF (Abcam no. ab18184, 1:500 dilution). For GST-CRAF and GST-BRAF proteins, 50 nM of the substrate was incubated with twofold dilutions of either 800 nM of PP1CA or 160 nM of SMP complex for 1 hour at 37 °C. Reactions were stopped by mixing with 4× LDS sample buffer. Western blots were prepared as described above and probed using antibodies against pS365 BRAF (in-house antibody, 1:1,000 dilution), pS259 CRAF (CST no. 9421 1:2,000), BRAF F7 (SC no. 5284, 1:6,000 dilution), CRAF (BD no. 610152, 1:10,000 dilution), SHOC2 (in-house antibody, 1:10,000 dilution), and PP1CA E-9 (SC no. 7482, 1:1,000 dilution). Final images were taken using an Odyssey CLx (LI-COR Biosciences).
RAF substrate docking
The CABS-Dock web server (http://biocomp.chem.uw.edu.pl/CABSdock) was used to dock BRAF and CRAF CR2-pS 15-mer peptides48. Briefly, ten RAF-substrate peptides were generated from a generic library and placed randomly approximately 20 Å from the surface of PP1CA. Each peptide underwent 50 annealing cycles of a Replica Exchange Monte Carlo Scheme. Snapshots (1,000) were taken of the trajectory of each starting peptide, resulting in 10,000 initial models. Non-binding peptide models were removed and then sorted by calculating their protein-peptide interaction energy. The lowest 10% (1,000 models, CA atoms only) were then clustered in a k-medoids procedure (k = 10). The root mean square deviation (r.m.s.d.) of peptides in each cluster was then calculated, and r.m.s.d. and cluster size were used as ranking parameters. The top model of each cluster is reconstructed to an all-atom complex using MODELLER.
BRAF CR2-pS dephosphorylation assay by matrix-assisted laser desorption/ionization–time of flight
SMP complex phosphatase activity was tested on two synthesized 15-mer phosphopeptides (Genscript) of BRAF (the N terminus was acetylated). One peptide was of the WT sequence (GQRDRSSpSAPNVHIN), and the second was mutated at the +1 position to glutamic acid (GQRDRSSpSEPNVHIN). Stock solutions of each peptide were made in water (~10 mM). A 50-μl reaction of 600 μM peptide with 100 nM of the SMP complex diluted in 20 mM HEPES, 150 mM NaCl, and 1 mM TCEP was carried out. Two microliters were taken at time points t = 0 hours, 2 hours, and 16 hours and mixed with 10 μl saturated sinapinic acid solution (10% acetonitrile, 0.1% TFA) and spotted onto 384-well sample MALDI-MS plate and allowed to air dry. Mass spectrometry covering the range 1,500–2,500 Da was carried out using a Bruker rapidfleX MALDI Tissuetyper in reflector mode with 2,000 laser shots per spectrum.
Extended Data
Extended Data Fig. 1 |. Assembly and selectivity of the SMP complex.
a Nucleotide analysis of the SMP complex by HPLC. Nucleotide standards and their retention time are shown above. b ITC experiment to measure the dissociation constant between SHOC2, MRAS1–178 and PP1CA7–300. c A steady-state plot of measured RU values from the formation of the SHOC2-HRAS-PP1CA complex against concentrations of HRASGMPPNP. d A steady-state plot of measured RU values from the formation of the SHOC2-NRAS-PP1CA complex against concentrations of NRASGMPPNP.
Extended Data Fig. 2 |. Comparison of the individual components of the SMP complex with their apo-forms.
a Superposition of the two SMP complexes found in the asymmetric subunit in cartoon form. Both chains of MRAS and PP1CA are in the same color, while the two SHOC2 chains are colored pink and cyan. The overall, SHOC2, MRAS and PP1CA RMSDs are 0.62 Å, 1.74 Å, 0.19 Å and 0.15 Å, respectively. b Top view as shown in panel a without MRAS and PP1CA present. LRR10 is marked, highlighting the hinge. c A cartoon of SHOC2 with a color gradient from blue to red showing the RMSD between the two SHOC2 molecules in the SMP complex, with blue and red representing low and high RMSD, respectively. LRR10 is marked highlighting the hinge. d Superposition of apo-SHOC2 (yellow) with SHOC2 from the SMP complex (pink) which was used for all subsequent analysis. All LRRs are labeled. e Superposition of mouse MRAS bound to GMPPNP (yellow; PDB ID 1X1S36) with human MRAS from the SMP complex (blue). Switch I, switch II, nucleotide and Mg2+ ions are shown in dark blue, purple, sticks and green spheres, respectively. The overall RMSD is 0.32 Å. f Superposition of apo-PP1CA (olive, PDB ID 4MOV37) with PP1CA from the SMP complex (green). Mn2+ ions from SMP and apo-PP1CA are shown in green and gray, respectively. The overall RMSD is 0.28 Å.
Extended Data Fig. 3 |. PP1C isoform specificity of the SMP complex.
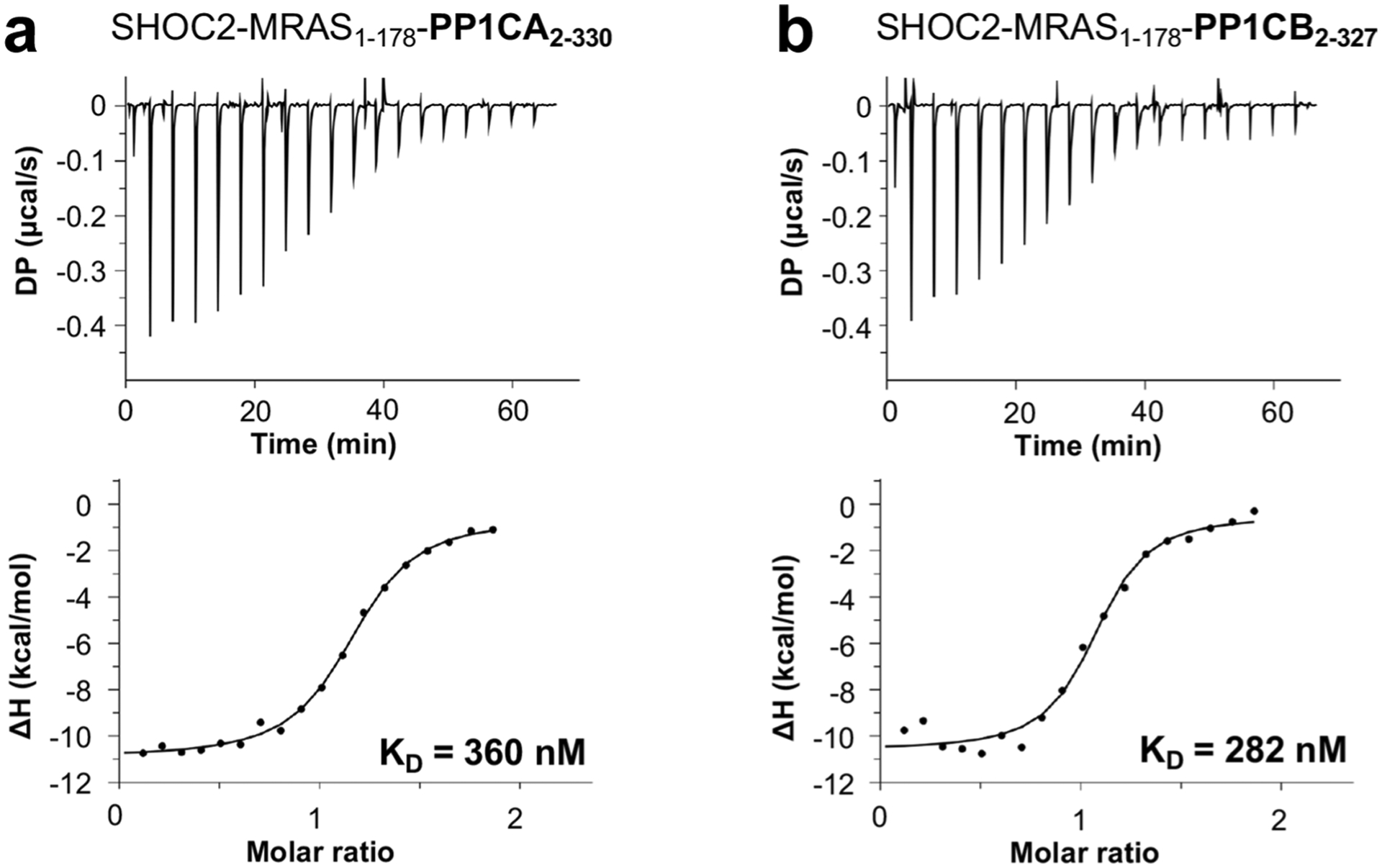
ITC experiments to measure the dissociation constant between a SHOC2, MRAS1–178 and PP1CA2–330 and b SHOC2, MRAS1–178 and PP1CB2–327.
Extended Data Fig. 4 |. Analysis of the SHOC2-PP1CA interface.
a The proposed SILK and RVxF binding motifs mapped onto SHOC2 (pink spheres) of the SMP complex. b Superposition of all RVxF-PP1C complexes present in the PDB onto the SMP complex. Surface of PP1CA (green) with the RVxF motif of SHOC2 (current work, pink), muscle glycogen-targeting subunit (PDB ID 6DNO, cyan53), RepoMan (PDB ID 5IOH, magenta76), cell-permeable peptide (PDB ID 4G9J, salmon77), PP1 regulatory subunit 3 A (PDB ID 5ZQV, light gray78), PP1 regulatory subunit 3B (PDB ID 5ZT0, violet78), Phactr1 (PDB ID 6ZEF, teal55), NIPP1 (PDB ID 3V4Y, orange56), Retinoblastoma-associated protein (PDB ID 3N5U, purple79), PP1 regulatory subunit 10 (PDB ID 4MOY, gray37), GADD34 (PDB ID 4XPN, dark blue80), Spinophilin (PDB ID 3EGG, gold81) and mouse-inhibitor 2 (PDB ID 2O8G, dark olive82). c Sequence alignment of the RVxF motif of SHOC2 across different species. Totally conserved residues are bold and highlighted in black, while similar residues are bold and highlighted in white. The RVxF motif is denoted with black stars. d Single-cycle kinetic analysis was performed on immobilized avi-tagged SHOC2 mutants as denoted in the figure with five injections of MRASGMPPNP and PP1CA (blue). The data were fit to a 1:1 kinetic model (black). e The SILK binding pocket on PP1C (green surface), as shown by the SILK of mouse inhibitor-2 (yellow cartoon) is occluded by SHOC2 (pink). A hydrogen bond forms between E54 of PP1CA at the periphery of the SILK binding pocket to R203 of SHOC2.
Extended Data Fig. 5 |. Isoforms and Noonan Syndrome mutations of PP1C.

a Sequence alignment of the three human isoforms of PP1C. Totally conserved residues are bold and highlighted in black, while similar residues are bold and highlighted in white. Non-conserved residues are only highlighted in white. The secondary structure of the PP1CA structure is shown above the alignment. α-helices and β-strands are labeled according to the nomenclature of Peti et al.29. Residues of PP1CA which interact with SHOC2 and MRAS are denoted with pink ovals and blue stars, respectively. R188 is the only residue of PP1CA which interacts with both SHOC2 and MRAS. b Single-cycle kinetic analysis was performed on immobilized avi-tagged SHOC2 with five injections of MRASGMPPNP and PP1CA mutants as denoted in the figure (blue). The data were fit to a 1:1 kinetic model (black).
Extended Data Fig. 6 |. Analysis of SHOC2 binding to the surface of MRAS.

a The N-terminal LRRs of SHOC2 (pink) are shown interacting with the switch I (dark blue) and switch II (purple) of MRAS (blue). b The C-terminal LRRs of SHOC2 (pink) are shown interacting with the C-terminus of MRAS (blue surface). c Residues of MRAS found mutated in NS highlighted as spheres on the structure of MRAS. d Single-cycle kinetic analysis was performed on immobilized avi-tagged SHOC2 mutants as denoted in the figure with five injections of MRASGMPPNP and PP1CA (blue). The data were fit to a 1:1 kinetic model (black).
Extended Data Fig. 7 |. Analysis of MRAS binding to the surface of SHOC2.
a Single-cycle kinetic analysis was performed on immobilized avi-tagged SHOC2 with five injections of MRASmutant-GMPPNP as denoted in the figure and PP1CA (blue). The data were fit to a 1:1 kinetic model (black). b Sequence alignment of human MRAS, KRAS, HRAS and NRAS sequences. Totally conserved residues are bold and highlighted in black, while highly conserved residues are bold and highlighted in white. Non-conserved residues are only highlighted in white. The secondary structure of the MRAS present in the SMP complex is shown above the alignment. Residues of MRAS which interact with SHOC2 and PP1CA are denoted with pink stars and green ovals, respectively.
Extended Data Fig. 8 |. Analysis of the MRAS-PP1CA interface.

a The N-terminus of MRAS (blue) occupies the MyPhoNE cleft (dark red) on PP1CA (green). The helical MyPhoNE motif of MYPT1 (PDB ID 1s70 (ref.46)) is shown in cyan. b ITC experiment to measure the dissociation constant between SHOC2, MRAS11–178 and PP1CA7–300. c Single-cycle kinetic analysis was performed on immobilized avi-tagged SHOC2 with five injections of MRASH53A-GMPPNP and PP1CA (blue). d Single-cycle kinetic analysis was performed on immobilized avi-tagged SHOC2 with five injections of MRASGMPPNP and PP1CAR188A (blue). In each case, the data were fit to a 1:1 kinetic model (black).
Extended Data Fig. 9 |. Docking of CRAF substrates and Noonan syndrome mutations found in the active site of PP1C.

a The CABS-dock server docked 15-mer peptides of the CR2-pS region of CRAF into the PP1CA structure of the SMP complex. All 167 peptides were placed in the active site, with all peptides placed with the N- and C-termini in the acidic and hydrophobic active site channels (magenta ribbons). PP1CA is shown as an electrostatic surface. b Two NS mutations are found to line the acidic and C-terminal channels of PP1CB. These were mapped onto the PP1CA surface with D253Y and E275K shown in blue (D252Y and E274K in PP1CB). c Fluorescent Western blot (representative of three independent experiments) of different concentrations of PP1CA or SMP incubated with either BRAF or CRAF substrates monitoring loss of CR2-pS signal (top gel, green bands). Total RAF loaded are shown as red bands (2nd from top gel). d Western blot (representative of three independent experiments) of different concentrations of PP1CA or SMP with either BRAF or CRAF substrates. Blots were probed at different phosphorylation sites in the substrates. SMP complex only dephosphorylates CR2-pS.
Extended Data Fig. 10 |. Dephosphorylation of BRAF CR2-pS phosphopeptides by the SMP complex.

Dephosphorylation of BRAF CR2-pS 15mer wild type peptide (top) and +1-position mutation to glutamic acid (bottom) as monitored by MALDI-TOF over 16 hours by the SMP complex. Sodium adducts of both dephosphorylated and phosphorylated peptides are denoted with *.
Supplementary Material
Acknowledgements
We thank B. Gillette, H. Ambrose, J. Cregger, P. Frank, B. Higgins, M. Hong, J. Mehalko, A. Mitchell, S. Perkins, N. Ramakrishnan, M. Sherekar, M. Smith, T. Taylor, V. Wall, and S. Widmeyer of the Protein Expression Laboratory (Frederick National Laboratory for Cancer Research) for their help in preparing recombinant proteins. We are grateful to S. Dharmaiah and D. Czyzyk for their help with crystallization, and to T. Waybright for nucleotide analysis (Frederick National Laboratory for Cancer Research). We thank M. Murphy (Cytiva Life Sciences) for his advice on fitting the SPR kinetic data. We thank L. Young for critical feedback on the manuscript (University of California San Francisco). We acknowledge W. Peti (University of Connecticut Health Center) for advice on the expression and purification of PP1CA. X-ray diffraction data were collected at the Northeastern Collaborative Access Team beamlines (24-ID-C/E), funded by the US National Institutes of Health (NIGMS P30 GM124165). The Pilatus 6M detector on the 24-ID-C beamline is funded by an NIH-ORIP HEI grant (S10 RR029205). This research used resources of the Advanced Photon Source, a US Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract DE-AC02-06CH11357. This project was funded in part with federal funds from the National Cancer Institute, National Institutes of Health Contract HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, and the mention of trade names, commercial products, or organizations does not imply endorsement by the US government.
Competing interests
F.M. is a consultant for Amgen, Daiichi, Frontiers Med, Exuma Biotech, Ideaya Biosciences, Kura Oncology, Leidos Biomedical Research, PellePharm, Pfizer, P.M.V. Pharma, and Quanta Therapeutics. F.M. is a consultant and co-founder for (with ownership interest including stock options) BridgeBio, Olema Pharmaceuticals, and Quartz. F.M. has received research grants from Daiichi Sankyo and Gilead Sciences and has a current grant from Boehringer-Ingelheim. The remaining authors declare no competing interests.
Footnotes
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Extended data is available for this paper at https://doi.org/10.1038/s41594-022-00841-4.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41594-022-00841-4.
Peer review information Nature Structural and Molecular Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available. Primary Handling Editors: Beth Moorefield and Carolina Perdigoto, in collaboration with the Nature Structural & Molecular Biology team.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41594-022-00841-4.
Data availability
The atomic coordinates and structure factors of the SMP complex and SHOC2 have been deposited in the Protein Data Bank and are available under accession numbers 7TVF and 7TVG, respectively. Structures under PDB accession codes 6DNO and 1X1S were used as initial models for molecular replacement. Structures used for superpositions are available in the PDB, including 4MOV, 5IOH, 4G9J, 5ZQV, 5ZT0, 6ZEF, 3V4Y, 3N5U,4MOY, 4XPN, 3EGG, 2O8G, and 1S70. The individual apparent KD values measured by SPR, mass spectrometry data, and full gel scans are accessible as source data published along this manuscript. Source data are provided with this paper.
References
- 1.Simanshu DK, Nissley DV & McCormick F RAS proteins and their regulators in human disease. Cell 170, 17–33 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yaeger R & Corcoran RB Targeting alterations in the RAF–MEK pathway. Cancer Discov. 9, 329–341 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Longo JF & Carroll SL The RASopathies: biology, genetics and therapeutic options. Adv. Cancer Res 153, 305–341 (2022). [DOI] [PubMed] [Google Scholar]
- 4.Lavoie H & Therrien M Regulation of RAF protein kinases in ERK signalling. Nat. Rev. Mol. Cell Biol 16, 281–298 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Park E et al. Architecture of autoinhibited and active BRAF–MEK1–14–3-3 complexes. Nature 575, 545–550 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Simanshu DK & Morrison DK A structure is worth a thousand words: new insights for RAS and RAF regulation. Cancer Discov. 12, 899–912 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tran TH et al. KRAS interaction with RAF1 RAS-binding domain and cysteine-rich domain provides insights into RAS-mediated RAF activation. Nat. Commun 12, 1176 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cookis T & Mattos C Crystal structure reveals the full Ras–Raf interface and advances mechanistic understanding of Raf activation. Biomolecules 11, 996 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Molzan M et al. Impaired binding of 14–3-3 to C-RAF in Noonan syndrome suggests new approaches in diseases with increased Ras signaling. Mol. Cell. Biol 30, 4698–4711 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rodriguez-Viciana P, Oses-Prieto J, Burlingame A, Fried M & McCormick F A phosphatase holoenzyme comprised of Shoc2/Sur8 and the catalytic subunit of PP1 functions as an M-Ras effector to modulate Raf activity. Mol. Cell 22, 217–230 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Young LC et al. SHOC2–MRAS–PP1 complex positively regulates RAF activity and contributes to Noonan syndrome pathogenesis. Proc. Natl Acad. Sci. USA 115, E10576–E10585 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jeoung M, Abdelmoti L, Jang ER, Vander Kooi CW & Galperin E Functional integration of the conserved domains of Shoc2 scaffold. PLoS ONE 8, e66067 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cordeddu V et al. Mutation of SHOC2 promotes aberrant protein N-myristoylation and causes Noonan-like syndrome with loose anagen hair. Nat. Genet 41, 1022–1026 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hannig V, Jeoung M, Jang ER, Phillips JA 3rd & Galperin E A novel SHOC2 variant in rasopathy. Hum. Mutat 35, 1290–1294 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Motta M et al. Clinical and functional characterization of a novel RASopathy-causing SHOC2 mutation associated with prenatal-onset hypertrophic cardiomyopathy. Hum. Mutat 40, 1046–1056 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Young LC et al. An MRAS, SHOC2, and SCRIB complex coordinates ERK pathway activation with polarity and tumorigenic growth. Mol. Cell 52, 679–692 (2013). [DOI] [PubMed] [Google Scholar]
- 17.Sulahian R et al. Synthetic lethal interaction of SHOC2 sepletion with MEK inhibition in RAS-driven cancers. Cell Rep. 29, 118–134 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jones GG et al. SHOC2 phosphatase-dependent RAF dimerization mediates resistance to MEK inhibition in RAS-mutant cancers. Nat. Commun 10, 2532 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaplan FM et al. SHOC2 and CRAF mediate ERK1/2 reactivation in mutant NRAS-mediated resistance to RAF inhibitor. J. Biol. Chem 287, 41797–41807 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Terai H et al. SHOC2 is a critical modulator of sensitivity to EGFR-TKIs in non-small cell lung cancer cells. Mol. Cancer Res 19, 317–328 (2021). [DOI] [PubMed] [Google Scholar]
- 21.Boned Del Rio I et al. SHOC2 complex-driven RAF dimerization selectively contributes to ERK pathway dynamics. Proc. Natl Acad. Sci. USA 116, 13330–13339 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Han K et al. CRISPR screens in cancer spheroids identify 3D growth-specific vulnerabilities. Nature 580, 136–141 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Young LC & Rodriguez-Viciana P MRAS: a close but understudied member of the RAS family. Cold Spring Harb. Perspect. Med 8, a033621 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kota P et al. M-Ras/Shoc2 signaling modulates E-cadherin turnover and cell-cell adhesion during collective cell migration. Proc. Natl Acad. Sci. USA 116, 3536–3545 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Higgins EM et al. Elucidation of MRAS-mediated Noonan syndrome with cardiac hypertrophy. JCI Insight 2, e91225 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Suzuki H et al. Severe Noonan syndrome phenotype associated with a germline Q71R MRAS variant: a recurrent substitution in RAS homologs in various cancers. Am. J. Med. Genet. A 179, 1628–1630 (2019). [DOI] [PubMed] [Google Scholar]
- 27.Verbinnen I, Ferreira M & Bollen M Biogenesis and activity regulation of protein phosphatase 1. Biochem. Soc. Trans 45, 89–99 (2017). [DOI] [PubMed] [Google Scholar]
- 28.Korrodi-Gregorio L, Esteves SL & Fardilha M Protein phosphatase 1 catalytic isoforms: specificity toward interacting proteins. Transl. Res 164, 366–391 (2014). [DOI] [PubMed] [Google Scholar]
- 29.Peti W, Nairn AC & Page R Structural basis for protein phosphatase 1 regulation and specificity. FEBS J. 280, 596–611 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bertola D et al. The recurrent PPP1CB mutation p.Pro49Arg in an additional Noonan-like syndrome individual: broadening the clinical phenotype. Am. J. Med. Genet. A 173, 824–828 (2017). [DOI] [PubMed] [Google Scholar]
- 31.Gripp KW et al. A novel rasopathy caused by recurrent de novo missense mutations in PPP1CB closely resembles Noonan syndrome with loose anagen hair. Am. J. Med. Genet. A 170, 2237–2247 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huckstadt V, Chinton J, Gomez A, Obregon MG & Gravina LP Noonan syndrome with loose anagen hair with variants in the PPP1CB gene: First familial case reported. Am. J. Med. Genet A 185, 1256–1260 (2021). [DOI] [PubMed] [Google Scholar]
- 33.Zambrano RM et al. Further evidence that variants in PPP1CB cause a rasopathy similar to Noonan syndrome with loose anagen hair. Am. J. Med. Genet A 173, 565–567 (2017). [DOI] [PubMed] [Google Scholar]
- 34.Snead K, Wall V, Ambrose H, Esposito D & Drew M Polycistronic baculovirus expression of SUGT1 enables high-yield production of recombinant leucine-rich repeat proteins and protein complexes. Protein Expr. Purif 193, 106061 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Selfors LM, Schutzman JL, Borland CZ & Stern MJ soc-2 encodes a leucine-rich repeat protein implicated in fibroblast growth factor receptor signaling. Proc. Natl Acad. Sci. USA 95, 6903–6908 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ye M et al. Crystal structure of M-Ras reveals a GTP-bound “off” state conformation of Ras family small GTPases. J. Biol. Chem 280, 31267–31275 (2005). [DOI] [PubMed] [Google Scholar]
- 37.Choy MS et al. Understanding the antagonism of retinoblastoma protein dephosphorylation by PNUTS provides insights into the PP1 regulatory code. Proc. Natl Acad. Sci. USA 111, 4097–4102 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Salvi F et al. Towards dissecting the mechanism of protein phosphatase-1 inhibition by its C-terminal phosphorylation. ChemBioChem 22, 834–838 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wakula P, Beullens M, Ceulemans H, Stalmans W & Bollen M Degeneracy and function of the ubiquitous RVXF motif that mediates binding to protein phosphatase-1. J. Biol. Chem 278, 18817–18823 (2003). [DOI] [PubMed] [Google Scholar]
- 40.Hendrickx A et al. Docking motif-guided mapping of the interactome of protein phosphatase-1. Chem. Biol 16, 365–371 (2009). [DOI] [PubMed] [Google Scholar]
- 41.Lee KH et al. Stabilization of Sur8 via PKCα/Δ degradation promotes transformation and migration of colorectal cancer cells. Oncotarget 8, 115596–115608 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meehan TF et al. Disease model discovery from 3,328 gene knockouts by The International Mouse Phenotyping Consortium. Nat. Genet 49, 1231–1238 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ferreira M, Beullens M, Bollen M & Van Eynde A Functions and therapeutic potential of protein phosphatase 1: insights from mouse genetics. Biochim. Biophys. Acta Mol. Cell. Res 1866, 16–30 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Burd CE et al. Mutation-specific RAS oncogenicity explains NRAS codon 61 selection in melanoma. Cancer Discov. 4, 1418–1429 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Endo T M-Ras is muscle-Ras, moderate-Ras, mineral-Ras, migration-Ras, and many more-Ras. Exp. Cell. Res 397, 112342 (2020). [DOI] [PubMed] [Google Scholar]
- 46.Terrak M, Kerff F, Langsetmo K, Tao T & Dominguez R Structural basis of protein phosphatase 1 regulation. Nature 429, 780–784 (2004). [DOI] [PubMed] [Google Scholar]
- 47.Goldberg J et al. Three-dimensional structure of the catalytic subunit of protein serine/threonine phosphatase-1. Nature 376, 745–753 (1995). [DOI] [PubMed] [Google Scholar]
- 48.Kurcinski M, Badaczewska-Dawid A, Kolinski M, Kolinski A & Kmiecik S Flexible docking of peptides to proteins using CABS-dock. Protein Sci. 29, 211–222 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hoermann B et al. Dissecting the sequence determinants for dephosphorylation by the catalytic subunits of phosphatases PP1 and PP2A. Nat. Commun 11, 3583 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yi J et al. Endothelial SUR-8 acts in an ERK-independent pathway during atrioventricular cushion development. Dev. Dyn 239, 2005–2013 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nunez Rodriguez N et al. Characterization of R-ras3/m-ras null mice reveals a potential role in trophic factor signaling. Mol. Cell. Biol 26, 7145–7154 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lai LP et al. Classical RAS proteins are not essential for paradoxical ERK activation induced by RAF inhibitors. Proc. Natl Acad. Sci. USA 119, e2113491119 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kumar GS et al. Identification of the substrate recruitment mechanism of the muscle glycogen protein phosphatase 1 holoenzyme. Sci. Adv 4, eaau6044 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ehrhardt A, Ehrhardt GR, Guo X & Schrader JW Ras and relatives–job sharing and networking keep an old family together. Exp. Hematol 30, 1089–1106 (2002). [DOI] [PubMed] [Google Scholar]
- 55.Fedoryshchak RO et al. Molecular basis for substrate specificity of the Phactr1/PP1 phosphatase holoenzyme. eLife 9, e61509 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.O’Connell N et al. The molecular basis for substrate specificity of the nuclear NIPP1:PP1 holoenzyme. Structure 20, 1746–1756 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Choy MS et al. SDS22 selectively recognizes and traps metal-deficient inactive PP1. Proc. Natl Acad. Sci. USA 116, 20472–20481 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Martinez Fiesco JA, Durrant DE, Morrison DK & Zhang P Structural insights into the BRAF monomer-to-dimer transition mediated by RAS binding. Nat. Commun 13, 486 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kwon JJ & Hahn WC A leucine-rich repeat protein provides a SHOC2 the RAS circuit: a structure–function perspective. Mol. Cell Biol 41, e00627–20 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jang H, Stevens P, Gao T & Galperin E The leucine-rich repeat signaling scaffolds Shoc2 and Erbin: cellular mechanism and role in disease. FEBS J. 288, 721–739 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sieburth DS, Sun Q & Han M SUR-8, a conserved Ras-binding protein with leucine-rich repeats, positively regulates Ras-mediated signaling in C. elegans. Cell 94, 119–130 (1998). [DOI] [PubMed] [Google Scholar]
- 62.Talsania K et al. Genome assembly and annotation of the Trichoplusia ni Tni-FNL insect cell line enabled by long-read technologies. Genes (Basel) 10, 79 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Taylor T, Denson JP & Esposito D Optimizing expression and solubility of proteins in E. coli using modified media and induction parameters. Methods Mol. Biol 1586, 65–82 (2017). [DOI] [PubMed] [Google Scholar]
- 64.Kopra K et al. Homogeneous dual-parametric-coupled assay for simultaneous nucleotide exchange and KRAS/RAF-RBd interaction monitoring. Anal. Chem 92, 4971–4979 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.D’Arcy A, Bergfors T, Cowan-Jacob SW & Marsh M Microseed matrix screening for optimization in protein crystallization: what have we learned? Acta Crystallogr. F. Struct. Biol. Commun 70, 1117–1126 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kabsch W XDS. Acta Crystallogr. D. Biol. Crystallogr 66, 125–132 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Evans PR & Murshudov GN How good are my data and what is the resolution? Acta Crystallogr. D. Biol. Crystallogr 69, 1204–1214 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Winn MD et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D. Biol. Crystallogr 67, 235–242 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liebschner D et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D. Struct. Biol 75, 861–877 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Emsley P, Lohkamp B, Scott WG & Cowtan K Features and development of Coot. Acta Crystallogr. D. Biol. Crystallogr 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Murshudov GN et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D. Biol. Crystallogr 67, 355–367 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schrodinger LLC The PyMOL Molecular Graphics System version 1.8 (2015). [Google Scholar]
- 73.Jurrus E et al. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 27, 112–128 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Morin A et al. Collaboration gets the most out of software. eLife 2, e01456 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Siddiqui FA et al. PDE6D inhibitors with a new design principle selectively block K-Ras activity. ACS Omega 5, 832–842 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kumar GS et al. The Ki-67 and RepoMan mitotic phosphatases assemble via an identical, yet novel mechanism. eLife 5, e16539 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chatterjee J et al. Development of a peptide that selectively activates protein phosphatase-1 in living cells. Angew. Chem. Int. Ed. Engl 51, 10054–10059 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yu J, Deng T & Xiang S Structural basis for protein phosphatase 1 recruitment by glycogen-targeting subunits. FEBS J. 285, 4646–4659 (2018). [DOI] [PubMed] [Google Scholar]
- 79.Hirschi A et al. An overlapping kinase and phosphatase docking site regulates activity of the retinoblastoma protein. Nat. Struct. Mol. Biol 17, 1051–1057 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Choy MS et al. Structural and functional analysis of the GADD34:PP1 eIF2α phosphatase. Cell Rep. 11, 1885–1891 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ragusa MJ et al. Spinophilin directs protein phosphatase 1 specificity by blocking substrate binding sites. Nat. Struct. Mol. Biol 17, 459–464 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hurley TD et al. Structural basis for regulation of protein phosphatase 1 by inhibitor-2. J. Biol. Chem 282, 28874–28883 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The atomic coordinates and structure factors of the SMP complex and SHOC2 have been deposited in the Protein Data Bank and are available under accession numbers 7TVF and 7TVG, respectively. Structures under PDB accession codes 6DNO and 1X1S were used as initial models for molecular replacement. Structures used for superpositions are available in the PDB, including 4MOV, 5IOH, 4G9J, 5ZQV, 5ZT0, 6ZEF, 3V4Y, 3N5U,4MOY, 4XPN, 3EGG, 2O8G, and 1S70. The individual apparent KD values measured by SPR, mass spectrometry data, and full gel scans are accessible as source data published along this manuscript. Source data are provided with this paper.