

Abstract
In vitro display technologies have been successfully utilized for the discovery and evolution of monoclonal antibodies (mAbs) for diagnostic and therapeutic applications, with phage display and yeast display being the most commonly used platforms due to their simplicity and high efficiency. As their prokaryotic or lower eukaryotic host organisms typically have no or different post-translational modifications, several mammalian cell–based display and screening technologies for isolation and optimization of mAbs have emerged and are being developed. We report here a novel and useful mammalian cell display platform based on the PiggyBac transposon system to display mAbs in a single-chain Fab (scFab) format on the surface of HEK293F cells. Immune rabbit antibody libraries encompassing ~7 × 107 independent clones were generated in an all-in-one transposon vector, stably delivered into HEK293F cells and displayed as an scFab with rabbit variable and human constant domains. After one round of magnetic activated cell sorting and two rounds of fluorescence activated cell sorting, mAbs with high affinity in the subnanomolar range and cross-reactivity to the corresponding human and mouse antigens were identified, demonstrating the power of this platform for antibody discovery. We developed a highly efficient mammalian cell display platform based on the PiggyBac transposon system for antibody discovery, which could be further utilized for humanization as well as affinity and specificity maturation.
Keywords: in vitro display technologies, mammalian cell display, rabbit monoclonal antibodies, scFab, transposon
Statement of Significance: An efficient mammalian cell display platform for antibody discovery and development in an scFab format without requiring prior enrichment by microbial display technologies was developed based on PiggyBac transposition.
INTRODUCTION
Due to their high affinity and superb specificity, antibodies are widely used in basic research, as well as in diagnostic and therapeutic applications. Impressively, antibody-based therapeutics are the most rapidly growing drug class over the last three decades and have demonstrated a striking impact on human health, particularly in cancer, infectious disease and autoimmune disease [1–3]. As of 30 June 2022, 115 therapeutic monoclonal antibodies (mAbs) have been approved worldwide and hundreds more are currently under evaluation in various phases of clinical development worldwide [4]. To date, a variety of techniques have been developed for the discovery, engineering and evolution of antibodies with desired biological properties from non-human, human and transgenic human antibody repertoires, including hybridoma technology, single B cell sorting coupled with antibody gene cloning, as well as library-based antibody display approaches [1, 5–7]. Taking advantage of the capacity of performing high throughput screening or selection in vitro and the potential to avoid issues associated with in vivo immunization, such as immune tolerance to conserved antigens, toxicity and immunodominant epitopes, a variety of different antibody display systems have been exploited [8, 9]. For example, ribosome display and mRNA display are cell-free methods useful for antibody affinity maturation due to the large size of libraries (1013–1014), yet the high background and instability of RNA are inevitable drawbacks in these systems [8]. Prokaryotic display, especially phage display, is the most commonly used display technology due to its simplicity, high efficiency and low cost, but problems with codon usage, protein folding and post-translational modification limit the successful discovery and development of therapeutic mAbs [10–13]. For these reasons, eukaryotic display, such as yeast display, has been developed for antibody library selection [14–17]. However, post-translational modification with significantly different N-glycosylated carbohydrate composition in yeast compared to mammalian cells may still impact the physicochemical properties of mAbs, which are largely manufactured in mammalian cells for therapeutic and diagnostic applications in humans and other mammals. To curtail these limitations, substantial efforts have been devoted to better align antibody discovery and antibody manufacturing by developing mammalian cell display systems [18–24].
In contrast to prokaryotic or lower eukaryotic cells, mammalian cells are more difficult to engineer to stably display antibodies on the cell surface [19]. Thus far, different approaches (transiently expressed plasmids, episomally replicating plasmids, Sindbis virus, vaccinia virus, retrovirus, stable expression using the Flp-In system, transposon and CRISPR-Cas9) have been tried to deliver antibody genes into certain host mammalian cells (CHO cells, HEK293T cells and immortalized B cells) to display different formats of antibody fragments or full-length IgG [18, 22, 23, 25–29]. These technologies have their own advantages and disadvantages and need further improvements to rival phage and yeast display systems. For example, Flp-In and CRISPR-Cas9 systems can control the genome integration site to ensure monoclonality, i.e. one antibody gene per host cell as in phage and yeast display systems. Yet, they are much less efficient than viral systems with respect to antibody gene delivery to the genome. Viral systems, on the other hand, require time- and cost-consuming production of viral particles and an advanced biosafety level infrastructure. An efficient non-viral system, PiggyBack transposition, was previously employed to display full-length IgG on the surface of B cells [23]. Here, we describe a highly robust antibody display platform that uses PiggyBac transposition to stably express a single-chain Fab (scFab) [30] on the surface of HEK293F cells.
HEK293F cells are easy to transfect with polyethyleneimine (PEI), which is of low cost and toxicity and can be cultured either in suspension to high density in bulk without serum or with serum to support adherent culture for single colony growth. This human cell line, together with an all-in-one PiggyBac transposon vector (PB2.0), which has a >20% stable integration rate into the HEK293F genome after transfection, allowed us to readily make a mammalian cell display library with a size close to 108 independent stable transfectants. An scFab format was chosen for mammalian cell display in this study because it is more stable than scFv and retains a structure and activity more similar to the antigen-binding site of natural full-length IgG. Using this system, we made an immune library against complement protein C3d. Complement proteins are important for both the innate and adaptive immune systems, providing critical protection against infectious pathogens, while also contributing to the pathogenesis of a number of autoimmune and inflammatory diseases, as well as to the rejection reaction against transplanted organs. Since C3d is the final proteolytic fragment generated from C3 protein upon activation of the complement cascade and is covalently deposited on nearby pathogen surfaces, pathogen-infected cell surfaces, transplanted organs and tumor cell surfaces through an ester or an amide bond [31], it has been investigated as an attractive target for therapeutic and diagnostic antibodies [32–34].
In this study, variable domains of antibody light and heavy chains were amplified from the bone marrow and spleen of two b9 allotype rabbits immunized with human C3d (hC3d), fused to human constant domains and then cloned into PB2.0 to afford chimeric rabbit/human scFab transposon libraries. After transfection into HEK293F cells, integration of the scFab genes into the genome was mediated by PB2.0-encoded PiggyBac transposase at a high rate and stable cell clones displaying scFabs with subnanomolar affinity and cross-reactivity to both hC3d and mouse C3d (mC3d) were selected by one round of magnetic activated cell sorting (MACS) and two rounds of fluorescence activated cell sorting (FACS).
RESULTS
Transposon-mediated antibody display on mammalian cells
In order to stably display antibodies on mammalian cells without virus handling, we used an all-in-one PiggyBac transposon vector PB2.0-DGFP (DNA2.0) in which a hyperactive PiggyBac transposase [35, 36] and a protein of interest, the latter flanked by two inverted terminal repeat (ITR) sequences, are encoded on the same plasmid [37–39] (Fig. 1A). A puromycin resistance gene enabled selection of cells harboring stably integrated transposons in their genome. After transfection and one week of selection with puromycin (10 days post-transfection), the fluorescent protein Dasher GFP (DGFP), serving as protein of interest encoded between the ITRs of PB2.0-DGFP, was stably expressed in HEK293F cells and no significant loss of fluorescence was observed when puromycin was removed for 3 weeks (Fig. 1B).
Figure 1.

Transposon vectors and maintenance of gene of interest integrated in genome upon transposition. (A) All all-in-one PiggyBac transposon vectors were constructed based on PB2.0-DGFP from DNA 2.0, which combines in one vector a hyperactive transposase driven by a CMV promoter and the expression cassettes of the gene of interest and the resistance marker positioned between the inverted terminal repeat (ITR) elements. Genes of interest, here as Dasher GFP (DGFP), single-chain Fab (scFab) and secreted scFab (sscFab) are under the control of an EF1α promoter. The expression cassette of the puromycin-resistant gene (PuroR) enables selection of stable cells with transposon integrated into their genomes (PGK, 3-phosphoglycerate kinase promoter; SAR, scaffold-attached regions; WPRE, woodchuck hepatitis virus post-transcriptional regulatory element; BGH, bovine growth hormone; pA, polyadenylation). Displayed scFab with a 60-aa linker (Linker60) between light (rbVL-huCL) and heavy chains (rbVH-huCH1) (rb, rabbit; hu, human) are fused to a (G4S)3 linker (GSL) and an HA tag, followed by the transmembrane domain of human PDGFRβ (TM). sscFab are tagged with an octahistidine (8 × His) tag for purification. (B) Stability of transposed DGFP in cells in the presence or absence of 1.5 μg/ml puromycin (gray, unstained cells; green, GFP-stained cells). (C) Stability of transposed scFab in cells in the presence or absence of 1.5 μg/ml puromycin. Cell surface scFab (clone 3 N07) expression was analyzed by flow cytometry using biotinylated rat anti-HA mAb 3F10 followed by PE-conjugated streptavidin for staining (gray, unstained cells; red, PE-stained (scFab-positive) cells).
Considering that Fab is more stable than scFv and more reliably converted to IgG without affinity and specificity loss, we chose a Fab format for antibody display on HEK293F cells. However, we found that in pilot experiments, which used small chimeric rabbit/human Fab libraries in PB2.0 in which the two polypeptide (light-chain and heavy- chain fragment)-encoding cassettes were separated by IRES or T2A sequences, the Fab format could not be displayed efficiently and its display level on stably transfected HEK293F cells decreased significantly over time even in the presence of puromycin (data not shown).
As scFabs of human antibodies have been shown to be well displayed on phage [40] and yeast surfaces [41], we next switched the expression format to scFabs with a 60 amino-acid linker connecting the light chain C-terminus to the N-terminus of the heavy chain fragment [40] (Fig. 1A). In this vector, PB2.0-scFab, only one signal peptide was required for light chain and heavy chain fragment. A (G4S)3 linker and a hemagglutinin (HA) tag followed by a transmembrane domain derived from human PDGFRβ were fused to the C-terminus of the heavy chain fragment. To make cloning convenient for library construction and compatible with our Fab-phage display vector pC3C [42], the signal peptide in PB2.0-scFab was modified to have the same upstream SfiI site as pC3C, enabling asymmetric SfiI cloning of VL-CL-VH cassettes with a downstream SfiI site that separates VH and CH1 (Fig. 1A). Flow cytometry data shown in Fig. 1C demonstrated that the scFab format was highly expressed in HEK293F cells and removal of puromycin after one week selection did not diminish the positive cell population, indicating that the cells were stable once the transposons had integrated into their genome. Furthermore, 8 out of 12 randomly picked clones from two small pilot chimeric rabbit/human scFab libraries with κ and λ light chains, respectively, stably displayed scFabs on HEK293F cells after transfection and selection, and the small pilot κ and λ libraries revealed 57.6% and 53.7% of stably scFab-displaying cells, respectively (Supplementary Fig. S1).
Generation of immune chimeric rabbit/human scFab library displayed on mammalian cells
The high rate of successful display of scFabs on HEK293F cells tested with the two small pilot libraries encouraged us to construct an scFab library in a large scale. Using a new set of oligonucleotides we used for generating a large naïve chimeric rabbit/human Fab library [43] and an additional rbVH reverse primer (rbIgG-CH1-R) annealing to the 5′ end of the rabbit IgG constant domain CH1, the rabbit light and heavy chain variable domains (rbVκ, rbVλ and rbVH) were PCR-amplified from reverse transcribed RNA extracted from the bone marrow and spleen of two b9 allotype rabbits [44], which had been immunized with human C3d (hC3d) and developed a strong polyclonal antibody (pAb) response (Supplementary Fig. S2). Note that rabbits have only one IgG isotype and the additional primer rbIgG-CH1-R was employed with the intention of biasing the library with the secondary antibody repertoire generated by class switch recombination, somatic gene conversion and somatic hypermutation in response to the immunogen [45]. The variable domains were then assembled to chimeric rabbit/human scFab-encoding sequences with different middle fragments (huCκ or huCλ), followed by asymmetric SfiI-cloning into the mammalian cell display vector PB2.0-scFab (Fig. 1A). Transformation of the ligation products into Escherichia coli strain ER2738 by electroporation yielded approximately 3.6 × 107 and 3.7 × 107 independent transformants for κ library and λ library, respectively.
In order to generate mammalian cell display libraries covering most of the clones in both κ and λ PB2.0-scFab libraries, pilot experiments were performed with PB2.0-DGFP and PB2.0-scFab-A. The latter encodes an scFab with a κ light chain to determine the transfection efficiency of HEK293F cells cultured in suspension using PEI, which has low toxicity to cells and the integration rate meditated by the hyperactive PiggyBac transposase in the vector. As shown in Fig 2A, 75%-80% transfection produced 34.4% cells stably expressing DGFP and 18.1% cells stably displaying scFabs, indicating an integration rate of ~45 and ~23% of HEK293F cells, respectively. To retain the complexity of the transposon libraries, we transfected 3 × 108 HEK293F cells for both κ and λ PB2.0-scFab libraries, with 18.1% of 3 × 108 cells (= 5.4 × 107 cells) exceeding 3.6 × 107 (κ) and 3.7 × 107 (λ) independent transformants. After selection with puromycin, 52.7% of κ library- and 63.6% λ library-transfected cells displayed scFabs (Fig. 2B), consistent with the small pilot libraries (Supplementary Fig. S1).
Figure 2.
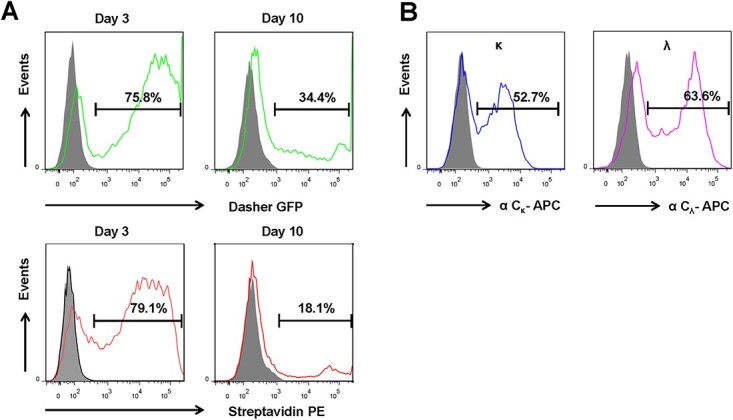
Transfection and integration efficiency of transposons into HEK293F cells. (A) Transfection and integration efficiency of DGFP (top, green) and scFab (bottom, red) were determined on day 3 and day 10 after transfection, respectively. Cells were maintained in culture without puromycin. (B) Cells displaying scFab after stable κ and λ library transposition were stained by a mouse anti-human Cκ mAb conjugated to BV421 (left, blue) or a mouse anti-human Cλ antibody conjugated to APC (right, purple) after 10-day culture in the presence of puromycin.
Enrichment of mammalian cells displaying high-affinity scFab
To enrich and select scFab-displaying HEK293F cells binding to hC3d, one round of MACS and two rounds of FACS were conducted (Fig. 3A). Before enrichment, neither κ nor λ libraries revealed detectable hC3d binders by flow cytometry (Fig. 3B). To prepare the capturing antigen for MACS, we deposited hC3d on the surface of magnetic streptavidin-coated beads through opsonization in the presence of normal human serum. After one round of selection by MACS, 3.0 × 107 and 2.7 × 107κ cells and λ cells, respectively, were recovered from 3 × 108 library-transfected HEK293F cells. As shown in Fig. 3C, 0.27% κ and 0.46% λ cells were positive after the first round of MACS. FACS was then used to further increase the abundance of positive clones, and 6 009 and 8 600 cells, respectively, were harvested and pooled from the top 0.1% of positive clones of κ and λ libraries. After expansion, we found that 9.3 and 6.2% cells of the two populations were positive for hC3d, and 48 single cells from the top 1.9% κ cells and top 0.7% λ cells were sorted separately by a second FACS in the presence of 10% pooled normal human complement serum (PNHCS), which was added to compete off cells displaying scFabs able to bind C3 in human serum (Fig. 3D).
Figure 3.
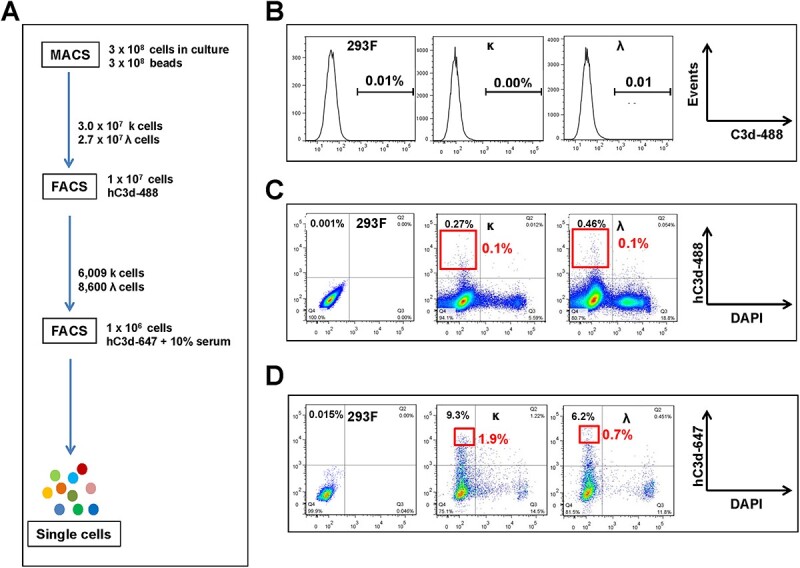
Enrichment of HEK293F cells displaying high-affinity antibodies against hC3d by MACS and FACS. (A) Scheme illustration of the enrichment process of scFab-displaying cells against hC3d by one round of MACS and two rounds of FACS. (B) Libraries before enrichment were stained with hC3d conjugated to Alexa Fluor 488. (C) Sorting of the top 0.1% positive cells from MACS-enriched libraries by staining with hC3d conjugated to Alexa Fluor 488. (D) Sorting of the top 1.9% κ and top 0.7% λ single cells, respectively, from FACS-enriched pools by staining with hC3d conjugated to Alexa Fluor 647 in presence of 10% PNHCS.
Following 2-week adherent culture without puromycin, 44 out of 48 single cells from the κ library (N1–N48) and 31 out of 48 single cells from the λ library (N49—N96) survived and all of them expressed scFabs that recognized hC3d. We then ranked the cell clones based on the ratio of the binding signal to the expression level and pursued eight clones for each library, including several top clones and randomly ranked clones (Fig. 4A). Using total RNA extracted from the cells, we amplified the scFab genes by RT-PCR with primers annealing to the signal peptide and HA tag sequences. After recloning into PiggyBac transposon vector PB2.0-sscFab, which has no transmembrane domain but an octahistidine tag at the C-terminus of scFabs for secretion expression and Immobilized Metal Ion Affinity Chromatography (IMAC) purification (Fig. 1A), we randomly picked three colonies after E. coli transformation and identified one to three (mode 2) copies of different scFab genes by DNA fingerprinting (Fig. 4A–C). Transient expression of individual PB2.0-sscFabs enabled us to easily uncover the right genes encoding and secreting responsible anti-hC3d scFabs for most single cell clones (Fig. 4B and C). However, for cell clones N4 and N68, none of the two different scFabs alone revealed hC3d binding, yet cotransfection of the two PB2.0-scFab plasmids produced a binding signal (Fig. 4B and C). Considering that the linker between the light chain and the heavy chain contains 60 amino acids, it is possible that the light chains and heavy chains of two different scFabs on the same cell cross-pair and reassemble into a functional Fab. To test this idea, we split the light chain and heavy chain of anti-hC3d mAb clone 3 N07, previously obtained through phage display, into two different vectors, PB2.0-scFab-M (3N07_HC + X_λC) and PB2.0-scFab-N (Y_HC + 3N07_κC). Co-transfection of the two plasmids, but not transfection of either plasmid alone, showed that the cells were indeed positively stained by hC3d, providing strong evidence for our cross-pairing hypothesis (Fig. 4D). Furthermore, sequence analysis revealed that the heavy chain of N4-1 and the light chain of N4-3 were similar to that of the functional clone N5-2, prompting us to put them together in one vector to form a new potential functional clone N4c. As shown in Fig. 4E, N4c indeed bound to hC3d deposited on zymosan. Similarly, cloning of the heavy chain of N68-2 and the light chain of N68-1 into one vector also produced a functional clone, N68c (Fig. 4E). Interestingly, for some of the positive cell clones, it was difficult to rescue the responsible scFab genes; e.g. for cell clone N93, 24 individual colonies that had been picked from the plates and confirmed to have the same DNA fingerprint showed no binding to hC3d when tested by ELISA (data not shown).
Figure 4.

Recovery of antibody genes from positive cell clones. (A) Following the three enrichment steps, 44 (out of 48 κ cell colonies, N1–N48) and 31 (out of 48 λ cell colonies, N49–N96) single cells that were enriched from the κ and λ libraries and survived expansion were pursued based on the ratio of binding signal to expression level as analyzed by flow cytometry using an HA antibody conjugated to Alexa Fluor 488 (row HA-488 giving median fluorescence intensity values) and hC3d labeled with Alexa Fluor 647 (row c3d-647 giving median fluorescence intensity values). (B) Antibody genes recovered from positive κ cell clones were cloned into PB2.0-sscFab and transfected alone or in combination into HEK293F cells to identify the responsible scFab binding to hC3d by ELISA using supernatants harvested 48 h after transfection. (C) Antibody genes recovered from positive λ cell clones were cloned into PB2.0-sscFab and transfected alone or in combination into HEK293F cells to identify the responsible scFab binding to hC3d by ELISA using supernatants harvested 48 h after transfection. (D) The heavy and light chains of anti-hC3d mAb 3 N07 (a phage display–derived clone) were split into plasmid PB2.0-scFab-M (3 N07/HC + X/LC) and plasmid PB2.0-scFab-N (Y/HC + 3 N07/LC), which were cotransfected into HEK93F cells to get stable cells for flow cytometry analysis of binding activity using biotinylated hC3d followed by PE-conjugated streptavidin for staining. (E) Responsible scFabs, including the two reassembled clones N4c and N68c, were expressed in HEK293F cells and tested for binding to hC3d and mC3d by ELISA using supernatants harvested 48 h after transfection.
Next, the kinetic and thermodynamic parameters of an assortment of clones binding to hC3d were determined by surface plasmon resonance (SPR). Since clone N66 was ranked first based on the ratio of the binding signal to the expression level, it was hypothesized to have high affinity to hC3d. As shown in Fig. 5, scFab N66 indeed revealed subnanomolar binding with a Kd of 0.311 nM. Furthermore, the affinity of the second ranked clone N5 was also high (Kd = 7.06 nM), validating the capability of our mammalian cell display platform to yield monovalent binders of high affinity. Note that N68c was ranked lower based on its binding/expression ratio but had higher affinity (Kd = 2.04 nM) compared to N5. Its lower ranking is probably due to a diluted expression signal of the responsible cross-paired scFab (N68c) that is assembled from two non-binding scFabs, N68-1 and N68-2. The amino acid sequence of scFab N68c is shown in Supplementary Fig. S3.
Figure 5.

Analysis of purified scFabs by SPR. (A) Biacore X100 sensorgrams obtained for the binding of each IMAC-purified scFab to hFc-hC3d or hFc-mC3d captured by the anti-human Fcγ antibody immobilized on a CM5 chip after instantaneous background depletion. scFabs were injected at six different concentrations. The highest concentrations used (e.g. 100 nM) are given in the top row of the table below, and the other five concentrations were prepared by two-fold serial dilutions from the highest concentration (e.g. 100, 50, 25, 12.5 and 6.25 nM). (B) Table with SPR-measured kinetic and thermodynamic parameters. The equilibrium dissociation constant (Kd) was calculated from koff/kon (kon, association rate constant; koff, dissociation rate constant; chi2 indicates the closeness of fit).
Enrichment of cells displaying cross-reactive antibody
In addition to identifying cells displaying scFabs with high affinity, we next demonstrated that mammalian cell display of scFabs could also be used to screen mAbs with cross-reactivity to both human and mouse C3d (hC3d and mC3d) by FACS in real time. To achieve this, recombinant fusion proteins hFc-hC3d and hFc-mC3d labeled with fluorescence Alexa Fluor 488 and Alexa Fluor 647, respectively, were used for dual staining of cells for sorting. From 1 × 106 of the expanded 6 009 κ cells and 8 600 λ cells that were previously retrieved by tandem MACS and FACS (Fig. 4), 18 κ single cells among the top 0.196% and 24 λ single cells among the top 0.658% were collected (Fig. 6A). After 3-week culture without puromycin, five κ colonies and six λ colonies were observed, two of each did not survive expansion. Two κ cell clones (κm2 and κm3) and three λ cell clones (λm1, λm2 and λm4) were confirmed to be reactive to both hC3d and mC3d, while one κ cell clone (κm1) only reacted with mC3d and one λ cell clone (λm3) was negative for both hC3d and mC3d (Fig. 6B). Antibody gene recovery and sequencing results revealed that κm2 and κm3 cell clones had identical inserts of only one functional scFab clone. Similarly, cell clone κm1 also had only one scFab clone. Among the λ cell clones, λm1, λm2 and λm4 were identical to the previously identified cell clone N68 and the responsible scFab was N68c. SPR showed that both N68c (Kd = 38.4 nM) and κm2 (Kd = 19.3 nM) had double-digit nanomolar affinity for mC3d. Interestingly, N5, which was identified as a high-affinity clone to hC3d (Kd = 7.06 nM), bound to mC3d with a Kd of 26.4 nM but did not emerge as a hit in the cross-activity selection, probably due to the small number of positive cell clones we pursued. Because of the bivalent binding of hFc-mC3d to cell surface scFabs, even cell clones with low affinity, such as cell clone κm1 (Kd = 302 nM), were sorted.
Figure 6.
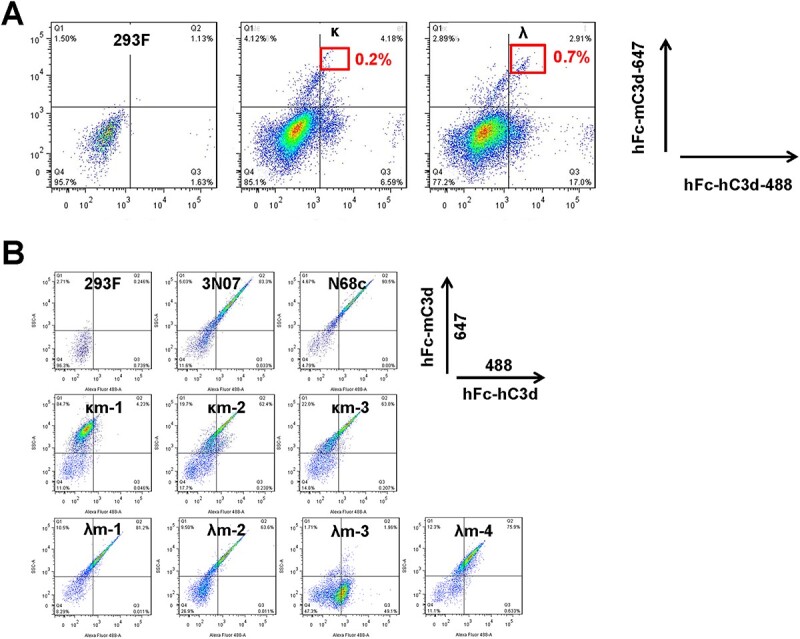
Enrichment and screen of cells displaying antibodies with cross-reactivity. (A) The top 0.2% and top 0.7% double-positive cells from both sublibraries were sorted to obtain single cells after staining by hFc-hC3d conjugated to Alexa Fluor 488 and hFc-mC3d conjugated to Alexa Fluor 647 in presence of 10% PNHCS. (B) Single-cell clones with binding activity to both hC3d and mC3d were verified by dual staining with hFc-hC3d conjugated to Alexa Fluor 488 and hFc-mC3d conjugated to Alexa Fluor 647 in presence of 10% PNHCS.
Collectively, six (N4c; N5-2 = N25-2 = N36-1 = N45-2; N41-2; N44-1; κm1 and κm2 = κm3) and three (N66-3; N68c = λm1 = λm2 = λm4 and N92) rabbit mAbs were rescued from the κ and λ libraries, respectively. However, only N66-3 had a λ light chain, while N66-3, N68c and all six clones derived from the κ library had κ light chains. Of these nine clones total, three (N5-2; κm2 and N68c) recognized both human and mouse C3d. Sequence alignments showed that six mAbs had identical heavy chain complementarity determining region 3 (HCDR3) sequences but harbored numerous mutations in their germline sequences (i.e. framework regions (FR) 1 through 3) likely due to in vivo affinity maturation during immunization (Supplementary Fig. S4).
DISCUSSION
Antibody-based therapeutics are one of the most successful and rapidly growing classes of pharmaceuticals across numerous indications. To meet the rising public health demand, a variety of technologies that allow the discovery, evolution and engineering of mAbs of therapeutic utility are being developed. Recent studies reported that mAbs discovered by phage display can have unfavorable biophysical features diminishing their chance to advance to large-scale manufacturing required for clinical trials when compared to mAbs derived from a mammalian cell source [10, 12, 46, 47]. Whereas mouse and rabbit hybridoma technologies, along with human antibody repertoires from transgenic animals, are highly valuable sources of mammalian cell-derived mAbs, they rely on immunization and are not suitable for conserved antigens with low immunogenicity or for toxic antigens. To address this shortcoming, in vitro library-based mammalian cell display technologies have been used to discover mAbs against numerous antigens of interest and shown to be versatile for incorporating the screening for certain desired properties in high-throughput methods, such as MACS and FACS [48]. In addition, compared to hybridoma cells, antibody genes are easier to recover from mammalian cell display libraries because of the tight coupling of the antibody phenotype and genotype and, as in our study, the confined nature of the introduced antibody genes with defined flanking sequences for facile RT-PCR recovery. As such, mammalian cell display–derived mAbs contribute to a great need of providing reliable recombinant antibodies with defined amino acid sequences for basic research, diagnostic and therapeutic applications [49, 50].
Here, we describe a new and highly efficient mammalian cell display platform that employs a PiggyBac transposon system for antibody discovery, evolution and engineering. Using this platform, we were able to generate mAbs of high affinity and desired cross-reactivity from an immune rabbit antibody repertoire.
In this study, we initially tried displaying conventional Fab on mammalian cells. However, with low efficiency of display of Fab with IRES or T2A to co-transcribe or co-translate, respectively, the light- and heavy-chain polypeptides in the constructs made us switch to scFabs [30, 40]. There were several reasons to choose this format. First, scFabs have been successfully displayed on phage. Second, to display scFabs on a mammalian cell surface, only one signal peptide, compared to two for conventional Fab, is needed to export the antibody. Third, light and heavy chains are expressed at the same level without interruption of translation. Fourth, combined in a single polypeptide, light and heavy chains can pair with each other more efficiently. Collectively, our results show that the change from Fab to scFab greatly improved the display efficiency from <10 to ~55%. The underlying mechanism for the remaining ~45% stable cells not displaying scFabs is unknown and needs to be further investigated. It is possible that the random combination of rbVL and rbVH we used to build the κ and λ libraries generates incompatible light- and heavy-chain pairs that are misfolded and prevented from reaching the cell surface through, e.g. the action of GRP78/BiP [51, 52].
The stable introduction of genes of interest into mammalian cells can be achieved through viral and non-viral approaches [24]. While virus-based approaches are costly, tedious and time-consuming, a variety of non-viral tools for gene integration, such as zinc fingers, TALENs, Flp-In and CRISPR-Cas9 systems, have been developed [22, 53]. However, due to the ability to mediate gene integration with site-specific precision, such non-viral systems usually have low efficiency for stable gene transfer into mammalian cells and are therefore not suitable for the generation of large libraries required for antibody discovery. In contrast, transposon systems are more efficient and have emerged as gene delivery and mutagenesis tools [38, 54, 55]. Among these, the PiggyBac transposon system showed higher efficiency of stable gene transfer compared to Sleeping Beauty, Tol2 and Mos1 systems [56]. Our study shows that using an all-in-one PiggyBac transposon vector in a single transfection step, integration rates can reach >20% of scFab-displaying HEK293F cells, which is considerably higher than the 6% reported for full-length IgG using a PiggyBac system for cotransfection of heavy chain, light chain and transposase split into three different vectors [23]. Combining the all-in-one PiggyBac transposon vector with the HEK293F cell culture system, which is easy to transfect and culture in suspension to high density, we readily got mammalian-cell display libraries encompassing close to 108 clones having cell surface scFabs. While still two to three orders of magnitude smaller than large naïve and synthetic libraries displayed on phage, we show that this library size is sufficient for successfully selecting an immune library and it should also be sufficient to evolve antibody affinity, specificity and manufacturability. Because of the hyperactive PiggyBac transposase in the vector [35, 36], we found that most cell clones harbored more than one scFab gene, which increased the complexity of the libraries on the one hand but somewhat complicated the recovery of the responsible scFab gene on the other hand. To overcome this issue, the all-in-one vector could be split into two vectors, one to express the gene-of-interest and the other to express the transposase, allowing for cotransfection at optimal ratios that promote single-copy scFab gene integration into the host cell genome. Interestingly, the 60-aa linker we used allowed the light chain (or heavy chain) from one scFab to cross-pair with the heavy chain (or light chain) of another scFab displayed on the same cell, probably due to preferential pairing of certain rbVL and rbVH. Reducing the linker length may decrease the chance of scFab cross-pairing on the HEK293F cell surface.
A significant advantage of mammalian cell display over phage display is the use of FACS to screen antibodies with desired properties in real time by defined gating and quantitative analysis. For instance, in our studies, we were able to identify clones with high affinity and cross-reactivity by dual staining. After normalization of the binding signal to the expression level, mAb clone N66 ranked first with a subnanomolar Kd (0.311 nM). In addition, the mammalian cell surface display level has been suggested to be indicative of the downstream developability of antibodies produced in mammalian cells [57]. Cross-reactivity of antibodies to ortholog antigens of different species is critical for preclinical studies in animal models for an early detection of on-target toxicities. C3d is highly conserved among different species. The amino acid sequence identity between human and cynomolgus C3d is 95.7%, while mouse and rabbit C3d are 84.1 and 81.8% identical to human C3d, respectively. Due to immunotolerance, it has been difficult to generate mouse mAbs to hC3d that cross-react with mC3d. Thurman et al. used C3 knock-out mice to generate mouse mAbs that recognize both hC3d and mC3d and demonstrated their therapeutic utility in mouse models of renal and ocular disease [33]. Considering the relative dissimilarity of rabbit C3d to mC3d (82.1% amino acid sequence identity) and hC3d (81.8%), we used rabbits for immunization with hC3d and generated an immune rabbit antibody library displaying chimeric rabbit/human scFabs on HEK293F cells. Using this mammalian cell display library coupled with dual staining in FACS, we successfully screened for mAbs with cross-reactivity for hC3d and mC3d. It is conceivable that this strategy can be applied to generating cross-reactive mAbs to other ortholog antigen pairs and panels as part of antibody discovery, evolution or engineering.
Finally, since the variable domains of the mAbs we retrieved by mammalian cell display are derived from rabbits, they may require humanization for further development as pharmaceuticals. Notably, the same mammalian cell display platform could be readily used to make small libraries displaying scFabs with rabbit CDR sequences grafted onto different human germline sequences with tailored FR diversification [58–60], followed by selection with MACS and FACS. Using similar specialized libraries, the platform can also be used for affinity maturation. However, it should be cautioned that a loss of affinity is possible when converting scFabs to IgG [61]. In summary, we developed an efficient and highly versatile platform based on an all-in-one transposon vector to display scFabs on HEK293F cells for antibody discovery and adaptable for humanization and affinity maturation. Although the platform does not require prior enrichment by microbial display technologies, it is compatible with our Fab-phage display platform based on phagemid pC3C, which also uses a VL-CL-VH cassette flanked by asymmetric SfiI sites [43]. As such, it facilitates high-throughput screening of pre-selected Fab or scFab pools from large naïve or synthetic antibody libraries.
MATERIALS AND METHODS
Cell lines
HEK293F cells (Thermo Fisher Scientific) were maintained in chemically defined, protein-free FreeStyle 293 Expression Medium supplemented with 1% (v/v) heat-inactivated FBS to support adherent culture or without FBS for suspension culture and 1 × penicillin–streptomycin (all from Thermo Fisher Scientific).
C3d proteins
Human C3d protein
Human complement protein C3d (hC3d) derived from C3 after a series of proteolytic cleavages was purchased from Complement Technology. Biotinylation of hC3d was performed using the Biotin-Tag Micro Biotinylation kit (Sigma-Aldrich). Labeling of hC3d with Alexa Fluor 488 and 647 was achieved following the instructions of the labeling kits (Thermo Fisher Scientific).
hFc-hC3d and hFc-mC3d proteins
DNA encoding hC3d (aa1002-1303 of hC3) with a mutation at position 1010 from cysteine to alanine to avoid thioester bond formation with glutamine at position 1013 was custom-synthesized as gBlock and cloned into pCEP4-hFc via HindIII/XhoI as described [62]. Analogously, mouse C3d (mC3d, aa1002-1303) with the same mutation was cloned into pCEP4-hFc. The resulting pCEP4-hFc-hC3d and pCEP4-hFc-mC3d plasmids were confirmed by DNA sequencing and transiently transfected into HEK293F cells using PEI (919012, Sigma-Aldrich). Transfected cells were cultured in FreeStyle 293 Expression medium, and hFc-hC3d and hFc-mC3d proteins were purified from supernatants by Protein A affinity chromatography as described [43]. The quality and quantity of purified proteins were analyzed by SDS-PAGE and A280 absorbance, respectively. Subsequently, hFc-hC3d and hFc-mC3d were labeled with Alexa Fluor 488 and 647 as described above.
Transposon vectors
All-in-one PiggyBac transposon vectors were constructed based on PB2.0-DGFP (DNA 2.0), which combines in one vector a hyperactive transposase driven by a CMV promoter and the expression cassettes of the gene of interest and the resistance marker positioned between the two inverted terminal repeat (ITR) elements. Genes of interest, i.e. Dasher GFP (DGFP), single-chain Fab (scFab) and secreted scFab (sscFab), are under the control of an EF1α promoter. The expression cassette of a puromycin-resistant gene enables selection of stable cells with transposon integrated into their genomes. The displayed scFab has a 60-aa linker [40] between light and heavy chain. As its C-terminus, the scFab is fused to a (G4S)3 linker followed by an HA tag (YPYDVPDYAS) and a human PDGFRβ segment (aa 513-561) that includes the transmembrane domain (aa 533-553). sscFabs are tagged with an octahistidine (HHHHHHHH) tag for purification. For cloning, two asymmetric BsaI sites flanking DGFP in PB2.0-DGFP were used to replace DGFP with scFabs, while two asymmetric SfiI sites were used to replace the rbVL-hCL-rbVH segment of different chimeric rabbit/human scFabs.
Library generation
All rabbit handling was carried out by veterinary personnel at R & R Research (Stanwood, WA). Two b9 allotype rabbits [44, 45] were immunized with 100 μg hC3d and Freund’s complete adjuvant, followed by three boosts with 50 μg hC3d and Freund’s incomplete adjuvant in 3-week intervals. The serum antibody response to the immunogen was monitored during the vaccination process by ELISA. Spleen and bone marrow from the two b9 allotype rabbits were collected 5 days after the last boost and separately processed for total RNA preparation and RT-PCR amplification of rbVκ, rbVλ and rbVH encoding cDNA using established protocols [43]. A degenerate reverse primer rbIgG-CH1-R (5’gaagactgaYggagccttaggttg3’, Y = t or c) that anneals to the 5′ end of the rabbit IgG constant domain CH1 was used to amplify IgG-derived rbVH encoding sequences enriched in the immune antibody repertoire. Subsequently, rbVκ/hCκ/rbVH and rbVλ/hCλ/rbVH segments were assembled by overlap extension PCR and cloned into PB2.0-scFab via SfiI. Transformation of E. coli strain ER2738 (Lucigen) by electroporation yielded approximately 3.6 × 107 and 3.7 × 107 independent transformants for library κ and library λ, respectively. To generate the stable mammalian cell display libraries, 300 μg maxi-prepped plasmids and 900 μl PEI were used to transfect 3 × 108 HEK293F cells in 100 ml culture and selected by 1.5 μg/ml puromycin 72 h after transfection for 1 week. Both libraries were kept in puromycin-containing medium until selection.
Library sorting by MACS and FACS
MACS
3 × 108 magnetic streptavidin-coated Dynabeads from the CELLection Biotin Binder Kit (Thermo Fisher Scientific) were washed and resuspended in 3 ml PBS, followed by incubation with 3 ml pooled normal human complement serum (PNHCS, Innovative Research) at 37°C for 1 h. After washing with 1% (w/v) BSA/DPBS, opsonized beads with freshly deposited natural hC3d were added to 300 ml suspension cultures of the mammalian cell display libraries. After 1 h, 6 × 50 ml cells were placed on a magnetic stand for 2 min. Magnetically separated cells bound by the beads were resuspended in fresh FreeStyle 293 Expression Medium and cultured without puromycin for 1 week before FACS.
FACS
1 × 107 MACS enriched sub-library cells were washed and resuspended in 10 ml ice-cold FACS buffer (DPBS containing 1% (w/v) BSA, 1 × penicillin–streptomycin and 1 mM EDTA), followed by staining with 10 μg Alexa Fluor 488 labeled C3d for 1 h in a cold room at 4°C and rotating at 10 rpm/min. After washing three times with ice-cold FACS buffer, cells were resuspended in DPBS containing 1% (w/v) BSA, 1 × penicillin–streptomycin and 2.5 mM Mg2+, 1 mM Ca2+, 20 μl DNaseI from the CELLection Biotin Binder Kit (Thermo Fisher Scientific) and 100 ng/ml 4′,6-diamidino-2-phenylindole (DAPI; Cell Signaling) and then filtered through 40-μm cell strainers. The cells were then sorted on a BD FACSAria™ Fusion instrument and pooled into a 6-well plate containing 2 ml FreeStyle 293 Expression Medium supplemented with 1% (v/v) heat-inactivated FBS to support adherent culture. After expansion, 1 × 106 pooled cells were stained with either 10 μg Alexa Fluor 647 labeled hC3d alone or with a mixture of 5 μg Alexa Fluor 488 labeled hFc-hC3d and 5 μg Alexa Fluor 647 labeled hFc-mC3d, in the presence of 10% PNHCS, for sorting of single cells with high affinity to hC3d or hC3d/mC3d cross-reactivity.
Antibody gene recovery
Single cells after sorting were cultured for 2–3 weeks for colony formation before expansion, and 1 × 106 cells were then used to extract RNA following the instructions of the RNeasy Mini Kit (Qiagen). After reverse transcription using the SuperScript III First-Strand Synthesis System (Thermo Fisher Scientific), scFab-encoding DNA was PCR-amplified with forward primer Sig-SfiI-F (5’tagctgctgcaactggggcccag3’) and reverse primer HA-R (5’agcgtaatctggaacatcgtatgggta3’) annealing to the signal peptide and HA tag encoding sequences, respectively, in the PB2.0-scFab vector. PCR products were purified, digested by SfiI and cloned into PB2.0-sscFab. To identify the right clones producing responsible scFabs, three transformants were picked for each cell colony and unique scFab-encoding sequences were Sanger-sequenced after DNA fingerprinting with AluI, for which DNA sequences encoding the rbVL-huCL-rbVH cassette were PCR-amplified, digested with AluI (frequent recognition site AG^CT) and analyzed by electrophoresis on a 4% (w/v) agarose gel. Subsequently, unique clones were transiently transfected into HEK293F adherent cells and the supernatants were harvested 48 h later. Clones revealing a positive signal in ELISA were then sequenced and pursued further.
scFab expression and purification
PB2.0-sscFab vectors were transiently transfected into suspension HEK293F cells using PEI and purified by Immobilized Metal Ion Affinity Chromatography (IMAC) using a 1-ml HisTrap column (Cytiva) as described [43]. The quality and quantity of purified scFabs were analyzed by SDS-PAGE and A280 absorbance, respectively.
ELISA
Rabbit antibody response to hC3d
Each well of a 96-well Costar 3690 plate (Corning) was coated with 100 ng hC3d protein in 30 μl coating buffer (0.1 M Na2CO3, 0.1 M NaHCO3, pH 9.6) for 1 h at 37°C. After blocking with 150 μl 5% (w/v) milk/PBS for 1 h at 37°C and washing three times with 150 μl PBS, 50 μl of 1:500 or 1:2 000 dilution of rabbit serum in 1% (w/v) milk/PBS was applied to each well. Following incubation for 2 h at 37°C and washing as before, 50 μl of a 1:1 000 dilution of donkey anti-rabbit IgG (H + L) pAb conjugated to horse radish peroxidase (HRP) (Jackson ImmunoResearch) in 1% (w/v) BSA/TBS was added and incubated for 1 h at 37°C. The wells were washed four times, and colorimetric detection was performed using 2,2′-azino-bis (3-ethylbenzothiazoline)-6-sulfonic acid (ABTS; Roche) as a substrate according to the manufacturer’s directions. The absorbance was measured at 405 nm using a SpectraMax M5 microplate reader (Molecular Devices) and SoftMax Pro software (Molecular Devices).
scFab binding assay to hC3d and mC3d
Each well of a 96-well Costar 3690 plate (Corning) was coated with 100 ng streptavidin (Sigma-Aldrich) in 30 μl coating buffer (see above) for 1 h at 37°C. After blocking with 150 μl 3% (w/v) BSA/PBS for 1 h at 37°C, 100 ng biotinylated hC3d or mC3d in 50 μl 1% (w/v) BSA/PBS was captured by incubation for 1 h at 37°C. The wells were washed three times with 150 μl PBS. Next, 50 μl supernatants harvested after 48 h transfection of HEK293F cells were applied to each well. Following incubation for 2 h at 37°C and washing as before, 50 μl of a 1:1 000 dilution of a mouse anti-His tag mAb conjugated to HRP (R&D Systems) in 1% (w/v) BSA/TBS was added and incubated for 1 h at 37°C. The wells were then washed four times, and detection with ABTS was carried out as described above.
Flow cytometry
Cells were stained using standard flow cytometry methodology. Briefly, 0.1–1 × 106 cells in 100 μl flow cytometry buffer (PBS containing 1% (w/v) BSA, 0.1% (w/v) sodium azide and 1 mM EDTA) were stained in a V-shaped 96-well plate (BrandTech) with 5 μl mouse anti human light chain κ or λ mAb conjugated to BV 421 or APC (BioLegend), or a 1:500 dilution of the biotinylated rat anti-HA mAb 3F10 (Roche) in conjunction with 2 μg/ml PE-conjugated streptavidin (BD Biosciences) to detect the light chains or the heavy chains displayed on cell surface. Binding activity was detected by 100 ng/100 μl C3d labeled with Alexa Fluor 488 or 647. All staining was performed on ice in dark, and DAPI (Cell Signaling) was added to a final concentration of 100 ng/ml to exclude dead cells. Cells were analyzed using a FACSCalibur instrument (BD Biosciences) and FlowJo analytical software (Tree Star).
SPR
SPR for the measurement of kinetic and thermodynamic parameters of the binding of purified scFabs to hC3d or mC3d proteins was performed on a Biacore X100 instrument using Biacore reagents and software (Cytiva). A mouse anti-human IgG CH2 mAb was immobilized on a CM5 sensor chip using reagents and instructions supplied with the Human Antibody Capture Kit (Cytiva). hFc-hC3d and hFc-mC3d fusion proteins were captured at a density not exceeding 400 RU. Each sensor chip included an empty flow cell for instantaneous background depletion. All binding assays used 1× HBS-EP+ running buffer (10 mM HEPES, 150 mM NaCl, 3 mM EDTA (pH 7.4) and 0.05% (v/v) Surfactant P20) and a flow rate of 30 μl/min. All scFabs were injected at five different concentrations. The sensor chips were regenerated with 3 M MgCl2 from the Human Antibody Capture Kit without any loss of binding capacity. Calculation of association (kon) and dissociation (koff) rate constants was based on a 1:1 Langmuir binding model. The equilibrium dissociation constant (Kd) was calculated from koff/kon.
FUNDING
We gratefully acknowledge support of this study by National Institutes of Health (NIH) grants R01 CA174844, R01 CA181258, R01 CA204484, R21 CA229961 and R21 CA263240 and by the Klorfine Foundation.
CONFLICT OF INTEREST STATEMENT
C.R. holds the position of Editorial Board Member for Antibody Therapeutics and is blinded from reviewing or making decisions for the manuscript. All authors declare no conflict of interest.
AUTHORS’ CONTRIBUTIONS
H.P. and C.R. conceived and designed the study. J.C. and H.P. conducted and analyzed all experiments. J.C, H.P. and C.R. wrote the manuscript.
CRediT AUTHOR STATEMENT
Jing Chang (Data curation-Lead, Investigation-Lead, Methodology-Lead, Resources-Lead, Validation-Lead, Visualization-Lead, Writing—original draft-Lead, Writing—review & editing-Lead), Christoph Rader (Conceptualization-Lead, Funding acquisition-Lead, Investigation-Lead, Project administration-Lead, Resources-Lead, Supervision-Lead, Writing—original draft-Lead, Writing—review & editing-Lead), Haiyong Peng (Conceptualization-Lead, Data curation-Lead, Formal analysis-Lead, Investigation-Lead, Methodology-Lead, Project administration-Lead, Resources-Lead, Supervision-Lead, Validation-Lead, Visualization-Lead, Writing—original draft-Lead, Writing—review & editing-Lead).
DATA AVAILABILITY
Four supplementary figures are provided. The data underlying this article will be shared on reasonable request to the corresponding authors.
ETHICS AND CONSENT STATEMENT
Consent was not required in this work.
ANIMAL RESEARCH STATEMENT
All rabbit handling was carried out by veterinary personnel at R & R Research (Stanwood, WA) in compliance with the NIH Guide for the Care and Use of Laboratory Animals.
Supplementary Material
Contributor Information
Jing Chang, Department of Immunology and Microbiology, The Herbert Wertheim UF Scripps Institute for Biomedical Innovation & Technology, University of Florida, Jupiter, FL 33458, USA.
Christoph Rader, Department of Immunology and Microbiology, The Herbert Wertheim UF Scripps Institute for Biomedical Innovation & Technology, University of Florida, Jupiter, FL 33458, USA.
Haiyong Peng, Department of Immunology and Microbiology, The Herbert Wertheim UF Scripps Institute for Biomedical Innovation & Technology, University of Florida, Jupiter, FL 33458, USA.
References
- 1. Mullard, A. FDA approves 100th monoclonal antibody product. Nat Rev Drug Discov 2021; 20: 491–5. [DOI] [PubMed] [Google Scholar]
- 2. Goydel, RS, Rader, C. Antibody-based cancer therapy. Oncogene 2021; 40: 3655–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kaplon, H, Chenoweth, A, Crescioli, Set al. Antibodies to watch in 2022. MAbs 2022; 14: 2014296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lyu, X, Zhao, Q, Hui, Jet al. The global landscape of approved antibody therapies. Antib Ther 2022; 5: 233–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Carter, PJ, Lazar, GA. Next generation antibody drugs: pursuit of the 'high-hanging fruit'. Nat Rev Drug Discov 2018; 17: 197–223. [DOI] [PubMed] [Google Scholar]
- 6. Chen, WC, Murawsky, CM. Strategies for generating diverse antibody repertoires using transgenic animals expressing human antibodies. Front Immunol 2018; 9: 460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Carter, PJ, Rajpal, A. Designing antibodies as therapeutics. Cell 2022; 185: 2789–805. [DOI] [PubMed] [Google Scholar]
- 8. Valldorf, B, Hinz, SC, Russo, Get al. Antibody display technologies: selecting the cream of the crop. Biol Chem 2022; 403: 455–77. [DOI] [PubMed] [Google Scholar]
- 9. Laustsen, AH, Greiff, V, Karatt-Vellatt, Aet al. Animal immunization, in vitro display technologies, and machine learning for antibody discovery. Trends Biotechnol 2021; 39: 1263–73. [DOI] [PubMed] [Google Scholar]
- 10. Kaleli, NE, Karadag, M, Kalyoncu, S. Phage display derived therapeutic antibodies have enriched aliphatic content: insights for developability issues. Proteins 2019; 87: 607–18. [DOI] [PubMed] [Google Scholar]
- 11. Alfaleh, MA, et al. Phage display derived monoclonal antibodies: from bench to bedside. Front Immunol 2020; 11: 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Almagro, JC, Pedraza-Escalona, M, Arrieta, HIet al. Phage display libraries for antibody therapeutic discovery and development. Antibodies (Basel) 2019; 8: 44–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ledsgaard, L, Ljungars, A, Rimbault, Cet al. Advances in antibody phage display technology. Drug Discov Today 2022; 27: 2151–69. [DOI] [PubMed] [Google Scholar]
- 14. Boder, ET, Wittrup, KD. Yeast surface display for screening combinatorial polypeptide libraries. Nat Biotechnol 1997; 15: 553–7. [DOI] [PubMed] [Google Scholar]
- 15. Sivelle, C, Sierocki, R, Ferreira-Pinto, Ket al. Fab is the most efficient format to express functional antibodies by yeast surface display. MAbs 2018; 10: 720–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Oh, EJ, Liu, R, Liang, Let al. Multiplex evolution of antibody fragments utilizing a yeast surface display platform. ACS Synth Biol 2020; 9: 2197–202. [DOI] [PubMed] [Google Scholar]
- 17. Krohl, PJ, Spangler, JB. A hybrid adherent/suspension cell-based selection strategy for discovery of antibodies targeting membrane proteins. Methods Mol Biol 2022; 2491: 195–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bowers, PM, Horlick, RA, Kehry, MRet al. Mammalian cell display for the discovery and optimization of antibody therapeutics. Methods 2014; 65: 44–56. [DOI] [PubMed] [Google Scholar]
- 19. Dangi, AK, Sinha, R, Dwivedi, Set al. Cell line techniques and gene editing tools for antibody production: a review. Front Pharmacol 2018; 9: 630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Robertson, N, Lopez-Anton, N, Gurjar, SAet al. Development of a novel mammalian display system for selection of antibodies against membrane proteins. J Biol Chem 2020; 295: 18436–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Luo, R, Zhao, Y, Fan, Yet al. High efficiency CHO cell display-based antibody maturation. Sci Rep 2020; 10: 8102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mason, DM, Weber, CR, Parola, Cet al. High-throughput antibody engineering in mammalian cells by CRISPR/Cas9-mediated homology-directed mutagenesis. Nucleic Acids Res 2018; 46: 7436–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Waldmeier, L, Hellmann, I, Gutknecht, CKet al. Transpo-mAb display: transposition-mediated B cell display and functional screening of full-length IgG antibody libraries. MAbs 2016; 8: 726–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Breous-Nystrom, E, Schultze, K, Meier, Met al. Retrocyte display® technology: generation and screening of a high diversity cellular antibody library. Methods 2014; 65: 57–67. [DOI] [PubMed] [Google Scholar]
- 25. Doerner, A, Rhiel, L, Zielonka, Set al. Therapeutic antibody engineering by high efficiency cell screening. FEBS Lett 2014; 588: 278–87. [DOI] [PubMed] [Google Scholar]
- 26. Smith, ES, Zauderer, M. Antibody library display on a mammalian virus vector: combining the advantages of both phage and yeast display into one technology. Curr Drug Discov Technol 2014; 11: 48–55. [DOI] [PubMed] [Google Scholar]
- 27. Bowers, PM, Horlick, RA, Neben, TYet al. Coupling mammalian cell surface display with somatic hypermutation for the discovery and maturation of human antibodies. Proc Natl Acad Sci U S A 2011; 108: 20455–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Taube, R, Zhu, Q, Xu, Cet al. Lentivirus display: stable expression of human antibodies on the surface of human cells and virus particles. PloS One 2008; 3: e3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Beerli, RR, Bauer, M, Buser, RBet al. Isolation of human monoclonal antibodies by mammalian cell display. Proc Natl Acad Sci U S A 2008; 105: 14336–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hust, M, Jostock, T, Menzel, Cet al. Single chain fab (scFab) fragment. BMC Biotechnol 2007; 7: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Toapanta, FR, Ross, TM. Complement-mediated activation of the adaptive immune responses: role of C3d in linking the innate and adaptive immunity. Immunol Res 2006; 36: 197–210. [DOI] [PubMed] [Google Scholar]
- 32. Rogers, LM, Veeramani, S, Weiner, GJ. Complement in monoclonal antibody therapy of cancer. Immunol Res 2014; 59: 203–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Thurman, JM, Kulik, L, Orth, Het al. Detection of complement activation using monoclonal antibodies against C3d. J Clin Invest 2013; 123: 2218–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Paek, JH, Kwon, J, Lim, Jet al. Clinical significance of C3d assay in kidney transplant recipients with donor-specific anti-human leukocyte antigen antibodies. Transplant Proc 2022; 54: 341–5. [DOI] [PubMed] [Google Scholar]
- 35. Yusa, K, Zhou, L, Li, MAet al. A hyperactive piggyBac transposase for mammalian applications. Proc Natl Acad Sci U S A 2011; 108: 1531–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Burnight, ER, Staber, JM, Korsakov, Pet al. A hyperactive transposase promotes persistent gene transfer of a piggyBac DNA transposon. Mol Ther Nucleic Acids 2012; 1: e50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chen, Q, Luo, W, Veach, RAet al. Structural basis of seamless excision and specific targeting by piggyBac transposase. Nat Commun 2020; 11: 3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sandoval-Villegas, N, Nurieva, W, Amberger, Met al. Contemporary transposon tools: a review and guide through mechanisms and applications of Sleeping Beauty, piggyBac and Tol2 for Genome Engineering. Int J Mol Sci 2021; 22: 5084–5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li, X, Burnight, ER, Cooney, ALet al. piggyBac transposase tools for genome engineering. Proc Natl Acad Sci U S A 2013; 110: E2279–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Koerber, JT, Hornsby, MJ, Wells, JA. An improved single-chain fab platform for efficient display and recombinant expression. J Mol Biol 2015; 427: 576–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Walker, LM, Bowley, DR, Burton, DR. Efficient recovery of high-affinity antibodies from a single-chain fab yeast display library. J Mol Biol 2009; 389: 365–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hofer, T, Tangkeangsirisin, W, Kennedy, MGet al. Chimeric rabbit/human fab and IgG specific for members of the Nogo-66 receptor family selected for species cross-reactivity with an improved phage display vector. J Immunol Methods 2007; 318: 75–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Peng, H, Nerreter, T, Chang, Jet al. Mining naive rabbit antibody repertoires by phage display for monoclonal antibodies of therapeutic utility. J Mol Biol 2017; 429: 2954–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Popkov, M, Mage, RG, Alexander, CBet al. Rabbit immune repertoires as sources for therapeutic monoclonal antibodies: the impact of kappa allotype-correlated variation in cysteine content on antibody libraries selected by phage display. J Mol Biol 2003; 325: 325–35. [DOI] [PubMed] [Google Scholar]
- 45. Weber, J, Peng, H, Rader, C. From rabbit antibody repertoires to rabbit monoclonal antibodies. Exp Mol Med 2017; 49: e305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jain, T, Sun, T, Durand, Set al. Biophysical properties of the clinical-stage antibody landscape. Proc Natl Acad Sci U S A 2017; 114: 944–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tiller, KE, Tessier, PM. Advances in antibody design. Annu Rev Biomed Eng 2015; 17: 191–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lerner, RA. Combinatorial antibody libraries: new advances, new immunological insights. Nat Rev Immunol 2016; 16: 498–508. [DOI] [PubMed] [Google Scholar]
- 49. Bradbury, A, Pluckthun, A. Reproducibility: standardize antibodies used in research. Nature 2015; 518: 27–9. [DOI] [PubMed] [Google Scholar]
- 50. Bradbury, AR, Pluckthun, A. Getting to reproducible antibodies: the rationale for sequenced recombinant characterized reagents. Protein Eng Des Sel 2015; 28: 303–5. [DOI] [PubMed] [Google Scholar]
- 51. Feige, MJ, Groscurth, S, Marcinowski, Met al. An unfolded CH1 domain controls the assembly and secretion of IgG antibodies. Mol Cell 2009; 34: 569–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Stoyle, CL, Stephens, PE, Humphreys, DPet al. IgG light chain-independent secretion of heavy chain dimers: consequence for therapeutic antibody production and design. Biochem J 2017; 474: 3179–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bak, RO, Gomez-Ospina, N, Porteus, MH. Gene editing on Center stage. Trends Genet 2018; 34: 600–11. [DOI] [PubMed] [Google Scholar]
- 54. Tipanee, J, VandenDriessche, T, Chuah, MK. Transposons: moving forward from preclinical studies to clinical trials. Hum Gene Ther 2017; 28: 1087–104. [DOI] [PubMed] [Google Scholar]
- 55. Wei, M, Mi, CL, Jing, CQet al. Progress of transposon vector system for production of recombinant therapeutic proteins in mammalian cells. Front Bioeng Biotechnol 2022; 10: 879222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wu, SC, Meir, YJJ, Coates, CJet al. piggyBac is a flexible and highly active transposon as compared to sleeping beauty, Tol2, and Mos1 in mammalian cells. Proc Natl Acad Sci U S A 2006; 103: 15008–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Jin, YJ, Yu, D, Tian, XLet al. A novel and effective approach to generate germline-like monoclonal antibodies by integration of phage and mammalian cell display platforms. Acta Pharmacol Sin 2022; 43: 954–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Goydel, RS, Weber, J, Peng, Het al. Affinity maturation, humanization, and co-crystallization of a rabbit anti-human ROR2 monoclonal antibody for therapeutic applications. J Biol Chem 2020; 295: 5995–6006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zhang, YF, Ho, M. Humanization of rabbit monoclonal antibodies via grafting combined Kabat/IMGT/Paratome complementarity-determining regions: rationale and examples. MAbs 2017; 9: 419–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Rader, C, Ritter, G, Nathan, Set al. The rabbit antibody repertoire as a novel source for the generation of therapeutic human antibodies. J Biol Chem 2000; 275: 13668–76. [DOI] [PubMed] [Google Scholar]
- 61. Steinwand, M, Droste, P, Frenzel, Aet al. The influence of antibody fragment format on phage display based affinity maturation of IgG. MAbs 2014; 6: 204–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yang, J, Baskar, S, Kwong, KYet al. Therapeutic potential and challenges of targeting receptor tyrosine kinase ROR1 with monoclonal antibodies in B-cell malignancies. PloS One 2011; 6: e21018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Four supplementary figures are provided. The data underlying this article will be shared on reasonable request to the corresponding authors.