

Abstract
Examination of a series of naturally-occurring trypsin inhibitor proteins, led to identification of a set of three residues (which we call the “interface triplet”) to be determinant of trypsin binding affinity, hence excellent templates for small molecule mimicry. Consequently, we attempted to use the Exploring Key Orientation (EKO) strategy developed in our lab to evaluate small molecules that mimic the interface triplet regions of natural trypsin inhibitors, and hence potentially might bind and inhibit the catalytic activity of trypsin. A bis-triazole scaffold (“TT-mer”) was the most promising of the molecules evaluated in silico. Twelve such compounds were synthesized and assayed against trypsin, among which the best showed a Kd of 2.1 μM. X-ray crystallography revealed a high degree of matching between an illustrative TT-mer’s actual binding mode and that of the mimics that overlaid the interface triplet in the crystal structure. Deviation of the third side chain from the PPI structure seems to be due to alleviation of an unfavorable dipole–dipole interaction in the small molecule’s actual bound conformation.
Introduction
High affinity inhibitors to cationic trypsin-1 are desirable because overactivation of trypsin, and inactivation of endogenous trypsin inhibitors, causes inflammation, fibrosis, and pancreatic dysfunction.1 Furthermore, trypsin inhibitors are emblematic of drugs used to treat maladies involving other members of the trypsin family. For example, Momordica cochinchinensis trypsin inhibitor II (MCoTI-II), a natural trypsin-inhibitory miniprotein, has been adapted to inhibit pathogenic proteases including β-tryptase (implicated in asthma),2 neutrophil elastase (chronic obstructive pulmonary disorder),3 coagulation factor XIIa (thrombosis),4 kallikrein-related peptidase 4 (KLK4, prostate cancer),5 and matriptase (cancers).6 Novartis adapted a small molecule trypsin ligand to inhibit complement factor D, a structurally similar protease and a major component in immune activation;7 dysfunction of factor D leads to disorders including age-related macular degeneration. Other trypsin-family proteases of therapeutic interest include urokinase plasminogen activator (uPA) and plasmin (important in cancer metastasis), thrombin, factor VII, and factor X (regulate blood coagulation). In view of this therapeutic impact, we set out to develop peptidomimetic inhibitors to the flagship, trypsin. Trypsin was chosen because its active site structure is well understood, and there are numerous high affinity natural ligands to serve as design starting points. However, our prime motivation was as a test case for our Exploring Key Orientations (EKO) strategy.8 EKO compares PPI interface regions with favored small molecule conformations that present three amino acid side chains, based on degree of fit of side chain Cα and Cβ coordinates. Validation for EKO has been reported for the HIV-1 protease dimer,8 antithrombin dimer,9 PCSK9·LDLR,10 and uPA·uPAR.11,12 However, none of those cases are supported by crystallographic data to confirm the actual binding mode of the small molecules. Trypsin is relatively easy to crystallize, so we anticipated it could be co-crystalized to obtain the first structure of a small molecule evaluated by EKO bound to a protein receptor.
Results and discussion
Structural commonalities between naturally-occurring trypsin inhibitors.
We examined interface regions of natural trypsin inhibitors (Fig. 1) and observed a conformational similarity in a trypsin-interface binding segment common to all. This is somewhat surprising in view of the diverse global structures and evolutionary origins of the parent protein. This common segment, which we refer to as the “interface triplet”, comprises residues occupying the S1, S1′, and S2′ pockets in trypsin’s active site, which appear to dominate the interaction energy in the protein–protein interaction (PPI).
Fig. 1.
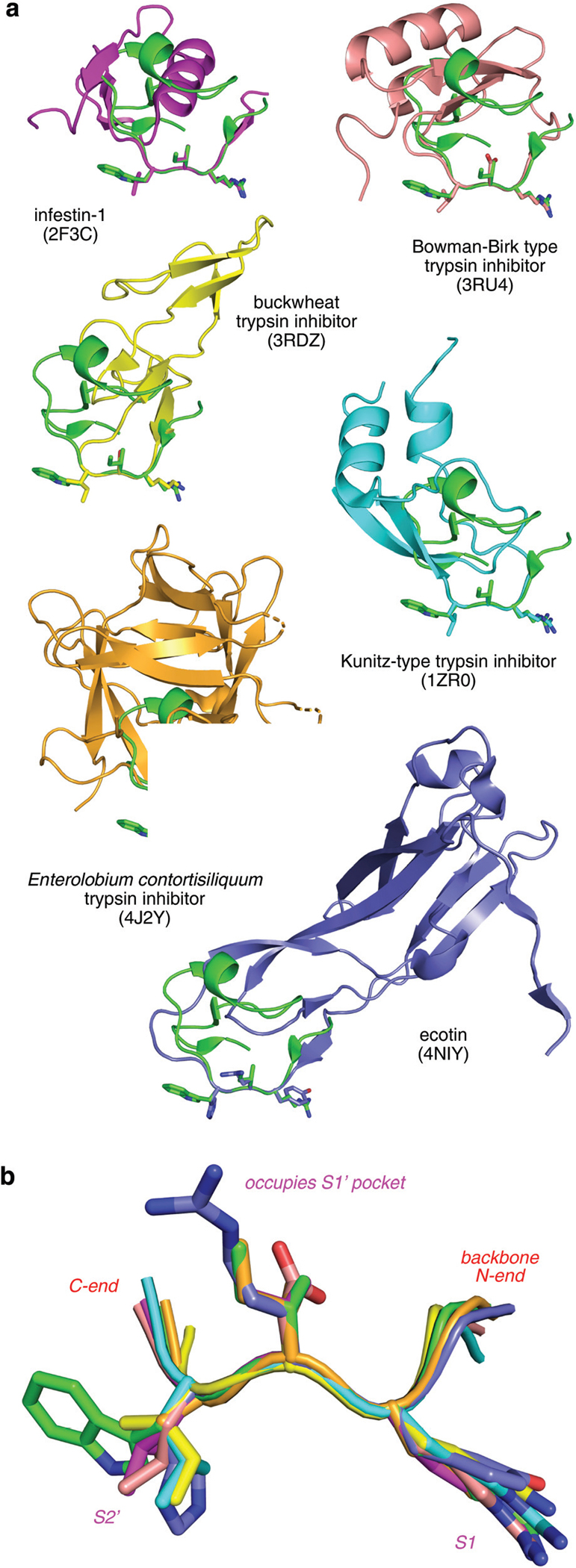
Overlay of the interface regions of natural trypsin inhibitors showing similarities in the trypsin-binding segment, ie the highly conserved PPI interface. (a) Momordica charantia trypsin inhibitor A (MCTI-A, green, PDB ID 1F2S) is overlaid on six other trypsin inhibitors. (b) Enlarged view of the overlay with side chains of the P1, P1’, P2’ residues are highlighted as sticks.
Peptidomimetic design.
Amides between amino acids in dipeptides can often be effectively substituted by five-membered ring motifs that replace four consecutive peptide backbone atoms, either C–Cα–N–C or N–C–Cα–N, and “stitching” them together by an additional fifth atom (Fig. 2a). These five-membered rings may be saturated (illustrative13–24) or unsaturated (e.g. ref. 25–27) where the optimal for is determined by the degree of curvature in the bound state of the parent peptide. Tripeptides can be similarly mimicked by joining two such five-membered rings.
Fig. 2.

Design and EKO evaluation of target compounds. (a) The peptide backbone is replaced with two 5-membered aromatic rings. (b) The structure and retrosynthesis of TT-mer. (c) A simulated conformer of TT-mer overlays on the interface triplet found on bovine pancreatic trypsin inhibitor (BPTI) with an RMSD of 0.37 Å.
The strategy explored in this work featured two chemically synthesizable five-membered rings, such as triazole, hydantoin, oxazole, or oxazolidinone to mimic the interface triplet referred to above. EKO was deployed to evaluate how well these scaffolds can adopt conformations corresponding to inhibitor· trypsin PPI interface triplet region. Thus, each tripeptide mimic was installed with three methyl side chains (as the R groups in Fig. 2a) before a molecular dynamics routine (quenched molecular dynamics, QMD)28,29 was employed to sample its conformers. These conformers were clustered based on methyl side chains orientations, and the lowest energy conformer of each cluster was selected as a representative. The potential energy of each representative conformer was calculated, and those more than 3 kcal mol−1 higher than the lowest-energy one were discarded because they are unlikely to be populated. The remaining stable conformers were then overlaid on the peptide template using the Kabsch algorithm. Overlay “goodnesses of fit” were quantified in terms of the root mean square deviation (RMSD) of the Cα–Cβ vectors of the three methyl side chains to that of the peptide-segment template. In our experience, an RMSD less than 0.5 Å may be considered a good overlay.
Among all the tripeptide mimics evaluated (summary in ESI†), one that consists of two triazoles, which we colloquially refer to as the “TT-mer”, emerged as a fine target for experimental evaluation since it appeared easy to synthesize (Fig. 2b) and overlaid on the interface triplet with a RMSD of 0.37 Å (Fig. 2c).
Binding affinities of TT-mers to trypsin.
Twelve TT-mers with various side chain combinations were synthesized and their binding affinities to bovine cationic trypsin measured by a kinetic assay (Zimmerman et al.,30 and ESI†). Bovine cationic trypsin is typically used as a substitute for the human variant in early stage inhibitor discovery (example1) as they have high sequence and structural similarity, and segments around the active site (residues 189–215) are almost completely the same. The compounds that gave greatest inhibition against bovine trypsin (1k and 1l) were assayed against human trypsin using a spectrophotometric probe, H-Glu-Gly-Arg-pNA (Table 1, and ESI†); data from the two assays (using bovine and human trypsin) correlate within reasonable ranges. These data were referenced to benzamidine positive control, known to bind trypsin with a Kd 18.4 μM, which compares well to the value of 22.2 μM in our assay H-Glu-Gly-Arg-pNA.31
Table 1.
Binding affinities of TT-mers to bovine cationic trypsin

|
Trends in the binding constants of the test molecules are largely uninteresting since better trypsin inhibitors exist. More importantly, the data pointed to superior binders might therefore be good candidates for co-crystallization studies. After some experimentation, a co-crystal of 1l with trypsin obtained and the crystal structure was solved.
Binding mode and of 1l to trypsin.
EKO evaluations will only reveal “hits” corresponding to compounds that can attain low energy conformations that overlay on the appropriate protein ligand segment. For example, in the case featured here, a conformation of 1l overlaid R1, R2, R3 side chains on the P2′, P1′, and P1 residues of the interface triplet, hence that compound was selected for experimental evaluation.
Compound 1l·trypsin bound in the targeted trypsin pocket, but in a slightly different conformation (Fig. 3). Side chains R2 and R3 occupied the S1′ and S1 pockets that correspond to the interface triplet in the parent PPI used for the EKO analysis. However, the benzamidine group on R3 formed a salt bridge with Asp189, as well as hydrogen bonds with Ser190 and the backbone carbonyl of Gly219, similar to arginine in naturally-occurring trypsin inhibitors and other benzamidine derivatives (examples32,33). EKO would not have evaluated this pose since it only considers small molecule conformers that overlay on interface segments of the protein ligand.
Fig. 3.

(a) X-ray structure of 1l bound to bovine trypsin (PDB ID 7jwx). (b) Actual solid-state conformation (green) compared with interface triplet one as illustrated by the overlay of 1l on this generated by EKO (cyan), (structure of the trypsin inhibitor removed for clarity).
Conformational preference of 1l.
In retrospect, the experimentally observed orientation of the R1 side chain in the solid state structure can be attributed to a positive factor in an alternative binding mode, and a negative one which would have existed if the mimic had bound in the conformation that overlaid the triplet interface region well. Binding of the lysine ammonium cation in the R1 residue is positively stabilized by hydrogen bonding with the Gln192 carbonyl (Fig. 3a). Conversely, the two triazoles of 1l may adopt “cis” and “trans” orientations (Fig. 4). In the cis-conformation that overlays with the interface triplet, the triazole dipoles are unfavorably aligned, while in the trans they are favorably opposed. In other words, lone pairs on the closest nitrogen atoms in each ring repel each other in the cis-state, destabilizing that conformation, while this effect is alleviated in the trans-form.
Fig. 4.
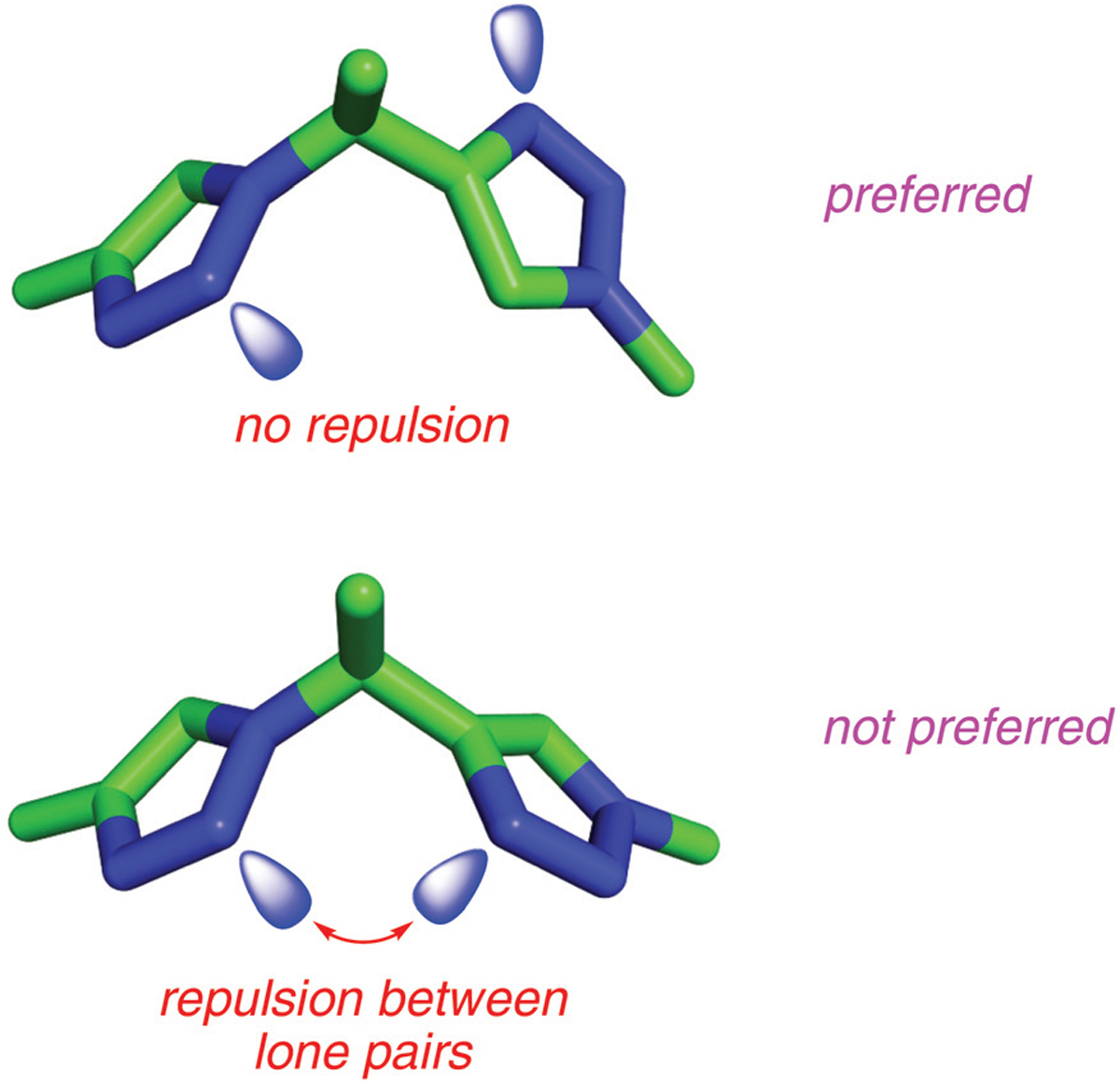
Trans (top) and cis (bottom) conformers of TT-mers. The repulsion between the lone pairs of triazole nitrogens makes cis-conformer less favorable compared to trans-conformers.
EKO samples populated solution state conformations of the small molecule core backbone (represented by the case where R1–R3 are all methyl) to see if they will overlay with interface protein ligand orientations in the PPI solid state structure. The key hypothesis underlying EKO is that conformations that do overlay well will be reinforced in the protein receptor when R1–R3 correspond to appropriate regions of the protein ligand interface. Justification for the assertions they are reinforced is that the protein receptor interacts with those particular side chains in the bound conformation. Consequently, calculations of relative energies of alternative conformations of the small molecule core backbone in the absence of the protein receptor are not directly relevant to EKO. Nevertheless, those types of calculations were performed here to assess stability differences for the cis- and trans-orientations of the TT-mers in the gas phase.
Calculations using the Merck molecular force field (MMFF94) estimated the trans-conformer is ~5.86 kcal mol−1 more stable than the cis; QM calculations (DFT at B3LYP/3–21 g, Gaussian 16) of the same compound were also performed and these experiments gave a similar difference to the molecular mechanics experiments: 4.845 kcal mol−1 (gas phase, throughout). The force field used for molecular mechanics in EKO (AMBER10) also indicatedtrans was more stable than cis but indicated an energy gap between the two forms that is below the cutoff threshold that was applied in the featured EKO analysis (3 kcal mol−1); consequently, both forms were classified as “populated”. The EKO analyses are performed using a continuous dielectric medium of 80 wherein the negative effects of aligned dipoles would be dampened, so it is unsurprising that the predicted energy difference between the cis- and trans-TTmer states was less for the AMBER10 calculations than in the gas phase ones.
Conclusions
In summary, we identified a highly conserved tripeptide conformation in a number of naturally-occurring trypsin inhibitors bound to the trypsin active site. A bis-triazole scaffold (“TT-mer”) was designed to mimic this triplet and was verified by EKO. A library of only 18 TT-mers were synthesized and their binding affinities against trypsin were measured. Co-crystallization of 1l and trypsin revealed that in the solid state the bound trans-TT-mer conformation had two of the three side chains mimicking those of the interface tripeptide. In that trans-TT-mer conformation, R3 could not overlay with the interface triplet. This alternative conformation was favored because it leads to a favorable hydrogen-bonding interaction, and alleviates unfavorable dipole alignments in the cis-form.
This work successfully highlights how EKO can be used in design of small molecule drug candidates that have three side chains corresponding to protein ligand interface segments. The structural commonality between interface segments in the natural trypsin-family inhibitor proteins (Fig. 1) was leveraged using EKO to validate small molecule mimicry featuring one conceptual type of core structure but different side chains to impart selectivity.
Supplementary Material
Acknowledgements
Financial support for this project was provided by DoD BCRP Breakthrough Award (BC141561), CPRIT (RP170144), Robert A. Welch Foundation (A-1121), National Science Foundation (NSF; CHE1608009), NIH R01EY029645, Texas A&M University T3-Grants Program (246292–00000), and NIH DP2GM123486.
We thank Dr Lisa M. Perez and Dr Yohannes Rezenom for useful discussions, and Dr Zhe Gavin Gao for help with the supporting material. NMR instrumentation at Texas A&M University was supported by a grant from the National Science Foundation (DBI-9970232) and the Texas A&M University System.
Footnotes
Conflicts of interest
The authors declare no competing financial interests. The study was designed by RL and KB, syntheses of the compounds was performed by SJ and RL, assays and modeling was performed by RL, and CP with AL formed the co-crystals and solved the structure.
Electronic supplementary information (ESI) available: Protocols for EKO modeling, synthesis, assays, and X-ray crystallography; characterization data of compounds synthesized.
References
- 1.Brandl T, Simic O, Skaanderup PR, Namoto K, Berst F, Ehrhardt C, Schiering N, Mueller I and Woelcke J, Bioorg. Med. Chem. Lett, 2016, 26, 4340–4344. [DOI] [PubMed] [Google Scholar]
- 2.Sommerhoff CP, Avrutina O, Schmoldt H-U, Gabrijelcic-Geiger D, Diederichsen U and Kolmar H, J. Mol. Biol, 2010, 395, 167–175. [DOI] [PubMed] [Google Scholar]
- 3.Thongyoo P, Bonomelli C, Leatherbarrow RJ and Tate EW, J. Med. Chem, 2009, 52, 6197–6200. [DOI] [PubMed] [Google Scholar]
- 4.Swedberg JE, Mahatmanto T, Abdul Ghani H, de Veer SJ, Schroeder CI, Harris JM and Craik DJ, J. Med. Chem, 2016, 7287–7292. [DOI] [PubMed] [Google Scholar]
- 5.Swedberg JE, Ghani HA, Harris JM, de Veer SJ and Craik DJ, ACS Med. Chem. Lett, 2018, 9, 1258–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Quimbar P, Malik U, Sommerhoff CP, Kaas Q, Chan LY, Huang Y-H, Grundhuber M, Dunse K, Craik DJ, Anderson MA and Daly NL, J. Biol. Chem, 2013, 288, 13885–13896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maibaum J, Liao S-M, Vulpetti A, Ostermann N, Randl S, Rudisser S, Lorthiois E, Erbel P, Kinzel B, Kolb FA, Barbieri S, Wagner J, Durand C, Fettis K, Dussauge S, Hughes N, Delgado O, Hommel U, Gould T, Mac Sweeney A, Gerhartz B, Cumin F, Flohr S, Schubart A, Jaffee B, Harrison R, Risitano AM, Eder J and Anderson K, Nat. Chem. Biol, 2016, 12, 1105–1110. [DOI] [PubMed] [Google Scholar]
- 8.Ko E, Raghuraman A, Perez LM, Ioerger TR and Burgess K, J. Am. Chem. Soc, 2013, 135, 167–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xin D, Holzenburg A and Burgess K, Chem. Sci, 2014, 5, 4914–4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taechalertpaisarn J, Zhao B, Liang X and Burgess K, J. Am. Chem. Soc, 2018, 140, 3242–3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arancillo M, Taechalertpaisarn J, Liang X and Burgess K, Angew. Chem., Int. Ed, 2021, 60, 6653–6659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arancillo M, Lin CM, Churion K and Burgess K, J. Med. Chem, 2021, submitted. [Google Scholar]
- 13.Borg S, Estenne-Bouhtou G, Luthman K, Csoregh I, Hesselink W and Hacksell U, J. Org. Chem, 1995, 60, 3112–3120. [Google Scholar]
- 14.Luthman K, Borg S and Hacksell U, Methods Mol. Med, 1999, 23, 1–23. [DOI] [PubMed] [Google Scholar]
- 15.Boeglin D, Cantel S, Heitz A, Martinez J and Fehrentz J-A, Org. Lett, 2003, 5, 4465–4468. [DOI] [PubMed] [Google Scholar]
- 16.Brik A, Alexandratos J, Lin Y-C, Elder JH, Olson AJ, Wlodawer A, Goodsell DS and Wong C-H, ChemBioChem, 2005, 6, 1167–1169. [DOI] [PubMed] [Google Scholar]
- 17.Zhang Z and Fan E, Tetrahedron Lett, 2006, 47, 665–669. [Google Scholar]
- 18.Angelo NG and Arora PS, J. Org. Chem, 2007, 72, 7963–7967. [DOI] [PubMed] [Google Scholar]
- 19.Bock VD, Speijer D, Hiemstra H and Van Maarseveen JH, Org. Biomol. Chem, 2007, 5, 971–975. [DOI] [PubMed] [Google Scholar]
- 20.Jochim AL, Miller SE, Angelo NG and Arora PS, Bioorg. Med. Chem. Lett, 2009, 19, 6023–6026. [DOI] [PubMed] [Google Scholar]
- 21.Narendra N, Vishwanatha TM and Sureshbabu VV, Int. J. Pept. Res. Ther., 2010, 16, 283–290. [Google Scholar]
- 22.Petit S, Fruit C and Bischoff L, Org. Lett, 2010, 12(21), 4928–4931. [DOI] [PubMed] [Google Scholar]
- 23.Doak BC, Scanlon MJ and Simpson JS, Org. Lett, 2011, 13, 537–539. [DOI] [PubMed] [Google Scholar]
- 24.Ke Z, Chow H-F, Chan M-C, Liu Z and Sze K-H, Org. Lett, 2012, 14, 394–397. [DOI] [PubMed] [Google Scholar]
- 25.Hoekstra WJ, Hulshizer BL, McComsey DF, Andrade-Gordon P, Kauffman JA, Addo MF, Oksenberg D, Scarborough RM and Maryanoff BE, Bioorg. Med. Chem. Lett, 1998, 8, 1649–1654. [DOI] [PubMed] [Google Scholar]
- 26.Garcia-Reynaga P and VanNieuwenhze MS, Org. Lett, 2008, 10, 4621–4623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Biron E, Chatterjee J and Kessler H, Org. Lett, 2006, 8, 2417–2420. [DOI] [PubMed] [Google Scholar]
- 28.Pettitt BM, Matsunaga T, Al-Obeidi F, Gehrig C, Hruby VJ and Karplus M, Biophys. J, 1991, 60, 1540–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.O’Connor SD, Smith PE, Al-Obeidi F and Pettitt BM, J. Med. Chem, 1992, 35, 2870–2881. [DOI] [PubMed] [Google Scholar]
- 30.Zimmerman M, Yurewicz E and Patel G, Anal. Biochem, 1976, 70, 258–262. [DOI] [PubMed] [Google Scholar]
- 31.Mares-Guia M and Shaw E, J. Biol. Chem, 1965, 240, 1579–1585. [PubMed] [Google Scholar]
- 32.Muley L, Baum B, Smolinski M, Freindorf M, Heine A, Klebe G and Hangauer DG, J. Med. Chem, 2010, 53, 2126–2135. [DOI] [PubMed] [Google Scholar]
- 33.Said AM, Parker MW and Vander Kooi CW, Bioorg. Chem, 2020, 100, 103856. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.