

Abstract
Identifying function-enhancing enzyme variants is a ‘holy grail’ challenge in protein science because it will allow researchers to expand the biocatalytic toolbox for late-stage functionalization of drug-like molecules, environmental degradation of plastics and other pollutants, and medical treatment of food allergies. Data-driven strategies, including statistical modeling, machine learning, and deep learning, have largely advanced the understanding of the sequence–structure–function relationships for enzymes. They have also enhanced the capability of predicting and designing new enzymes and enzyme variants for catalyzing the transformation of new-to-nature reactions. Here, we reviewed the recent progresses of data-driven models that were applied in identifying efficiency-enhancing mutants for catalytic reactions. We also discussed existing challenges and obstacles faced by the community. Although the review is by no means comprehensive, we hope that the discussion can inform the readers about the state-of-the-art in data-driven enzyme engineering, inspiring more joint experimental-computational efforts to develop and apply data-driven modeling to innovate biocatalysts for synthetic and pharmaceutical applications.
Keywords: automation, beneficial mutation, machine learning, new-to-nature reactions
Introduction
Enzyme engineering is the process of optimizing enzyme sequences for enhanced physical (e.g. thermal stability, cold adaption, solubility, and complex stoichiometry), chemical (e.g. activity, substrate specificity, promiscuity, and selectivity), and biological (e.g. expressibility) functions. Typical strategies used in enzyme engineering include directed evolution, truncation, ancestral sequence reconstruction, site-directed mutagenesis, and terminal fusion (Bruggink et al., 2003; Yi et al., 2021; Ali et al., 2020). Enzyme engineering has enabled the development of new enzymes and enzyme variants for synthetic, medicinal, and energy uses (Bruggink et al., 2003; Yi et al., 2021; Ali et al., 2020; Knott et al., 2020; Rorrer et al., 2019). Biocatalysts have been developed to accelerate stereoselective and regioselective chemical transformations. Well-known examples include cytochrome P450 for chemical oxidations (Li et al., 2020), CALB for hydrolytic kinetic resolution (Xia et al., 2017), and halide methyltransferase for the synthesis of non-native S-adenosyl methionine analogs (Tang et al., 2021). Enzymes have also been engineered for therapeutics—for example, microbial transglutaminases have been used as ‘protein glue’ in tissue engineering, α-gliadin peptidases in gluten degradation (Gordon et al., 2012; Wolf et al., 2015), lysosomal enzymes in the treatment of Hunter syndrome (Hendrikse et al., 2021) and metachromatic leukodystrophy (Simonis et al., 2019), and Cas9 enzymes for handling the off-target effects in gene editing (Schindele and Puchta, 2020; Yin et al., 2018). Additionally, enzymes have been used to catalyze difficult reactions for biofuel production and polymer upcycling. These enzymes include xylanase (Min et al., 2021) and endo-beta-1,4-glucanase (Cecchini et al., 2018) for biomass conversion, lipases for depolymerization of polyesters with nano-dispersion (DelRe et al., 2021), and PET depolymerase for plastic recycling and upcycling (Tournier et al., 2020).
Despite the success of enzyme engineering, a priori computational identification of function-enhancing enzymes and enzyme variants remains a ‘holy grail’ challenge due to the unknown sequence–structure–function relationship for enzymes. The natural occurrence of beneficial mutations has been reported to be below 1%, which presents an urgent need for the development of computational tools to optimize enzyme sequence in silico (Romero and Arnold, 2009; Melnikov et al., 2014; Fowler and Fields, 2014; Araya and Fowler, 2011; Kries et al., 2013; Hilvert, 2013; Bunzel et al., 2018; Zeymer and Hilvert, 2018). Different types of statistical, machine learning (ML), and deep learning (DL) models have been developed to guide directed evolution (Wittmann et al., 2021a; Singh et al., 2021; Siedhoff et al., 2020). Unlike molecular simulations that help understand the electronic and dynamic nature of biocatalytic reactions, data-driven modeling indicates predictive descriptors and design principles that assist the discovery of enzymes with enhanced functions or even new functions. Specifically, supervised learning models enable in silico screening of enzyme sequences for desired functions. Unsupervised learning models help avoid non-functional variants a priori. Semi-supervised learning models leverage unsupervised pretraining (i.e. extracted from large number of protein sequences) to build supervised learning models with low number of sequence-function data. Generative models produce artificial protein sequences that bear similar functions to those used for model training.
Among numerous applications, data-driven methods have been used to predict enzyme EC number, catalytic site, optimal operating conditions, solubility, substrate promiscuity or specificity, reaction selectivity, turnover number, and reaction pathway (Feehan et al., 2021; Mazurenko et al., 2020). They have also been used to design new orthologous enzymes and gain-of-function enzymes from originally non-functional structural scaffolds. Considering the extensive integration of data-driven modeling with enzyme engineering, this review does not intend to provide a comprehensive discussion on all active fronts of data-driven approaches for enzyme engineering. Rather, we emphasize the models published in the past five years that facilitate the discovery of enzymes with enhanced catalytic functions. In the following sections, we first introduce the common numerical features and models used in data-driven enzyme engineering. We then talk about the applications of data-driven modeling in accelerating free energy simulations for enzymes, predicting enzyme catalytic properties, and designing new enzyme sequences. Finally, we discuss the challenges faced by the community to further develop multi-objective, generalizable, and trust-worthy data-driven models. We hope the discussion can inspire more researchers to develop and apply data-driven modeling to innovate biocatalysts for synthetic and medicinal uses.
Features and models used in data-driven enzyme engineering
Building a predictive model starts from choosing numerical features to characterize enzymes and choosing models to map the relationship between enzyme features and observational data (Fig. 1). In this section, we will briefly introduce some common features and models used in data-driven enzyme engineering.
Fig. 1.
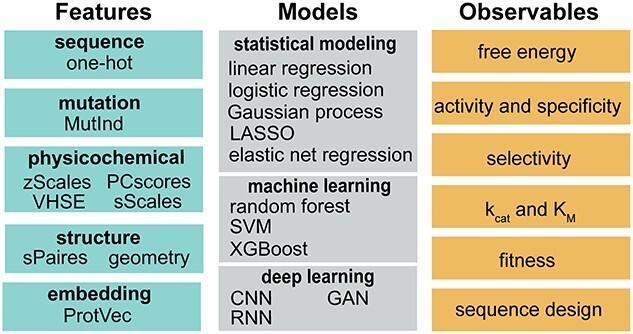
Features, models, and observables used in data-driven enzyme engineering. MutInd: mutation indicator; VHSE: principal components score vectors of hydrophobic, steric, and electronic properties; ProtVec: protein-vectors; LASSO: least absolute shrinkage and selection operator; SVM: support vector machines; XGBoost: extreme gradient boosting; CNN: convolutional neural network; GAN: generative antagonistic networks; RNN: recurrent neural network
Numerical features can be derived from enzyme’s amino acid sequence or three-dimensional structure. For sequence-based features, one-hot encoding is arguably the simplest form of descriptor that encodes amino acid-level information—it uses a binary vector (0 or 1) to indicate a certain residue as one of the twenty natural amino acids. Similarly, to represent mutation, the binary vector can be used to indicate the presence of specific mutations in a sequence (i.e. MutInd; Xu et al., 2020). Despite the simplicity, one-hot encoding does not carry much information of amino acids that could be physically or chemically relevant to enzyme functions. As such, physicochemical feature vectors are used. In the amino acid index (AA-index) databases, hundreds of amino acid descriptors can be found that involve amino acids’ geometric, hydrophobic, steric, and electronic properties. To represent a residue, a physicochemical feature vector can take the sum of a subset of carefully selected AA-indices (sScales; Xu et al., 2020). The feature vector can also consist of multiple AA-indices chosen based on domain-knowledge (zScales; Sandberg et al. 1998) or multiple extracted features derived from the dimension reduction of large number of AA-indices using principal component analysis (e.g. VHSE, Mei et al., 2005; PCscores, Xu et al., 2020). Besides the physicochemical feature vectors, language embedding models are increasingly used for representing enzyme sequences (e.g. ProtVec, Asgari and Mofrad, 2015; UniRep, Alley et al., 2019). In contrast, physicochemical feature vectors can encode local amino acid information in a physically intuitive fashion, whereas the embedding obtained from millions of sequences is more likely to embed global and evolutionary information.
For structure-based features, geometric descriptors (e.g. distance, angle, and dihedral) are widely used to describe the spatial relationship among functionally important residues (e.g. active site residues) (Lodola et al., 2010). These features can be incorporated in the model as distance map or AA-index amino acid pairwise contact potential (e.g. sPairs (Xu et al., 2020)). Notably, it has been shown that protein structures can be predicted using sequence information alone (Jumper et al., 2021; Morcos et al., 2011). This implies that replacing sequence-based features by structure-based features might not necessarily result in improved prediction performance. However, structure-based features have the advantage of easy incorporation of protein dynamics and substrate-enzyme interaction information—these can be crucial for engineering enzymes to catalyze new-to-nature reactions.
Data-driven models can be roughly classified as statistical model, machine learning, and deep learning models. Statistical models are designed for inferring the association or causal relationships between enzyme features and observables. Some common forms of statistical modeling are linear regression, which uses a linear combination of features to fit the data according to mathematical constraints; logistic regression, which describes the probability of one event by having the logarithm of the odds for the event be a linear combination of features; LASSO, which performs feature selection and regularization to enhance the prediction accuracy and interpretability of the statistical model (e.g. linear regression); and Gaussian process regression, which uses an ensemble of feature-dependent curves to fit the observational data where the parameters associated with features are randomized with normal distribution (i.e. Bayesian approach). These models facilitate researchers to investigate the quantitative relationship between enzyme sequence or structure and their functions, elucidating the physical or chemical principles that can be used to identify function-enhancing enzyme variants (detailed in Section 3).
Different from statistical modeling that is used to infer the feature-observable relationship, ML and DL models are designed to make accurate prediction about enzyme observables using sequence or structure features as input. ML models are usually built by leveraging physically, chemically, or statistically meaningful descriptors as features; while in contrast, DL models are built by employing multiple artificial neural network layers to progressively derive high-dimensional tensors as features. Some broadly used machine learning models include random forests, which employs an ensemble of decision trees to conduct classification based on majority-voting or regression based on the average prediction of individual trees; support vector machine, which finds a maximum-margin hyperplane in the feature space that separates the data points into different categories and predicts which category the new data points should fall into; and XGBoost, which leverages an ensemble of weak learning models to conduct prediction and is known to outperform random forests when the weak learning model adopts the form of a decision tree. DL model is technically part of a broader family of ML models—they are featured by using artificial neural networks to encode enzyme features and then conduct classification or regression task. In enzyme engineering, models based on convolutional neural network (i.e. CNN), recurrent neural network (i.e. RNN), and graph neural network (i.e. GNN) have been developed. CNN is designed to learn spatial hierarchies of enzyme sequence or structure features using multiple building blocks including convolution layers, pooling layers, and fully connected layers. Most CNN models are not invariant to translation. An SE(3)-invariant transformer has been recently developed to encode enzyme structure information with preserved molecular symmetry and chirality (Adams et al., 2021). RNN models use a series of feedforward networks to learn sequence or time-series information. The model has been widely applied in natural language processing and speech recognition. The architecture of RNN naturally fits to the task of learning enzyme’s sequence-function relationship and predict function-enhancing mutation for enzyme engineering. GNN is designed to perform enzyme prediction tasks because enzyme structures can be represented as graph (e.g. representing each residue as a node). GNN adopts pairwise message-passing architecture, where graph nodes iteratively update their representations by exchanging information with neighboring nodes. The permutation-equivariant layers used in message passing also help preserve the geometric symmetry of enzymes.
Due to the page limit, the discussion in this section does not involve the mathematical foundations and technical implementation of different models. Interested readers should refer to recent books and literatures for more technical details (Bishop, 2006; Goodfellow et al., 2016; Jurtz et al., 2017). For the prediction performance and time consumption of different feature-model combination, we would recommend a recent benchmark study by Xu et al. (2020) that comprehensively investigated 44 combinations of enzyme features and models in different enzyme engineering tasks.
Applications of machine-learning and deep-learning models in enzyme catalysis
In this section, we will talk about examples that employed these data-driven models to gain physical insight into enzyme catalysis or to develop predictive models that identify function-enhancing biocatalysts. Specifically, we will discuss models for efficient enzyme design and engineering applications (Table I). We will discuss research works to (i) accelerate QM/MM-based free energy simulations (section 3.1; Pan et al., 2021; von der Esch et al., 2019; Bonk et al., 2019), (ii) predict enzyme activity and substrate specificity (section 3.2; Masso and Vaisman, 2011; Masso and Vaisman, 2014; Saito et al., 2021; Shroff et al., 2020; Xu et al., 2022; Voutilainen et al., 2020; Mou et al., 2021; Robinson et al., 2020), (iii) predict enzyme enantioselectivity (section 3.3; Xu et al., 2020; Cadet et al., 2018), (iv) predict enzyme kinetic and thermodynamic parameters (section 3.4; Goldman et al., 2022; Kroll et al., 2021; Heckmann et al., 2018; Carlin et al., 2016; Mellor et al., 2016; Li et al., 2022), (v) predict functional fitness in enzyme evolution (section 3.5; Wittmann et al., 2021b; Figliuzzi et al., 2016; Teze et al., 2021; Hon et al., 2020; Luo et al., 2021; Favor and Jayapurna, 2020; Biswas et al., 2021; Hsu et al., 2022), and (vi) design new functional enzyme sequences (section 3.6; Russ et al., 2020; Repecka et al., 2021; Madani et al., 2021).
Table I.
Summary of statistical, machine learning, and deep learning models for biocatalyst engineering reported in the past 5 yearsa
| Section | Input | Predictive Model | Output | Performance | Paper |
|---|---|---|---|---|---|
| 3.1 | Structural feature from trajectories of QM/MM transition interface sampling | Logistic regression | Reactive trajectory classifier |
|
2019-Bonk (Bonk et al., 2019) |
| 3.1 | QM coordinate | Elastic net regression | Activation free energy |
|
2019-Esch (von der Esch et al. (2019)) |
| 3.1 |
|
ANN | Energy | RMSD = 0.69 kcal/mol | 2021-Pan (Pan et al., 2021) |
| 3.2 | Substrate structure | Support vector machine | Activity score | Accuracy~80% | 2017-Pertusi (Pertusi et al., 2017) |
| 3.2 | Sequence | Supervised machine learning decision tree | Substrate specificity | Accuracy = 0.94 | 2017-Chevrette (Chevrette et al., 2017) |
| 3.2 |
|
|
Multipoint mutation |
|
2018-Khersonsky (Khersonsky et al., 2018) |
| 3.2 |
|
Decision tree | Activity classifier | Accuracy~90% | 2018-Yang (Yang et al., 2018) |
| 3.2 | Graph kernel derived from protein coordinates | Gaussian process | Activity | Pearson r = 0.81 | 2020-Voutilainen (Voutilainen et al., 2020) |
| 3.2 | AA descriptor | CNN | AA type probability | ~70% in predicting the natural AA type | 2020-Shroff (Shroff et al., 2020) |
| 3.2 | Physicochemical feature of enzyme-substrate pairs |
|
|
|
2020-Robinson (Robinson et al., 2020) |
| 3.2 |
|
|
|
|
2021-Mou (Mou et al., 2021) |
| 3.2 |
|
|
|
|
2022-Xu (Xu et al., 2022) |
| 3.3 | Sequence | Partial least squares regression | Activation free energy | R2 = 0.96 | 2018-Cadet (Cadet et al., 2018) |
| 3.3 | Sequence | Gradient boosting | Enantioselectivity | Pearson r = 0.65 | 2019-Wu (Wu et al., 2019) |
| 3.3 |
|
CNN | Activity | AUROC = 0.88 | 2020-Xu (Xu et al., 2020) |
| 3.4 |
|
|
|
|
2016-Mellor (Mellor et al., 2016) |
| 3.4 |
|
Elastic net regularization | kcat/KM |
|
2016-Carlin (Carlin et al., 2016) |
| 3.4 |
|
|
kapp,max | R2 = 0.76 | 2018-Heckmann (Heckmann et al., 2018) |
(continue)
Table I.
Continued.
| Section | Input | Predictive Model | Output | Performance | Paper |
|---|---|---|---|---|---|
| 3.4 |
|
Gradient boost model regression | KM |
|
2021-Kroll (Kroll et al., 2021) |
| 3.4 |
|
CNN | kcat | Pearson r = 0.94 (log10-scale) | 2022-Li (Li et al., 2022) |
| 3.4 |
|
Feed forward network | KD classifier | AUROC = 0.89 | 2022-Goldman (Goldman et al., 2022) |
| 3.5 | Sequence | Ridge regression | Fitness | MSE = 0.74 | 2020-Favor (Favor and Jayapurna, 2020) |
| 3.5 | Sequence |
|
Fitness | Spearman ρ = 0.61 | 2021-Wittmann (Wittmann et al., 2021b) |
| 3.5 | Sequence | Iterative MSA and conservation analysis | Conserved AA | N/A | 2021-Teze (Teze et al., 2021) |
| 3.5 | Sequence | RNN |
|
|
2021-Luo (Luo et al., 2021) |
| 3.5 | Sequence | Regularized linear regression | Fitness |
|
2021-Biswas (Biswas et al., 2021) |
| 3.5 | Sequence | Ridge regression | Fitness | Spearman ρ ~ 0.66 | 2022-Hsu (Hsu et al., 2022) |
| 3.6 | Sequence | Generative adversarial network | Artificial enzyme sequence | 24% with catalytic activity | 2021-Repecka (Repecka et al., 2021) |
| 3.6 | Sequence | Protein language model | Artificial enzyme sequence | AUC = 0.85 | 2021-Madani (Madani et al., 2021) |
| 3.6 | Sequence | Direct coupling statistical analysis of sequence MSA | Artificial enzyme sequence | Hit rate = 30% | 2020-Russ (Russ et al., 2020) |
| 3.6 | Sequence | Variational autoencoder model of Blast sequence | Artificial enzyme sequence | Pearson R2 = 0.99 | 2022-Giessel (Giessel et al., 2022) |
aFor each research work, only the best-performing models are shown. Abbreviations: artificial neural network (ANN), convolutional neural network (CNN), deep neural network (DNN), recurrent neural network (RNN).
bQ(von der Esch et al., 2019): Leave-one-out cross-fold validation score.
Acceleration of free energy simulation for enzyme catalysis
Free energy simulations, augmented with multiscale quantum mechanics/molecular mechanics (QM/MM) methods, have been widely applied to evaluate the activation barriers and reaction energies of enzymatic reactions. From classical molecular dynamics (MD)-sampled conformers, QM/MM calculations can be conducted to evaluate the activation-free energies. However, it has been an unsolved puzzle in the community regarding what geometric features of an enzyme conformer determine the activation barrier height of the catalyzed chemical reaction. To answer this question, Lodola et al. (2010) conducted statistical analyses (i.e. multivariate linear regression and principal component analysis) to elucidate the correlation between conformational fluctuation and QM/MM-determined activation barrier height using the fatty acid amide hydrolase as the model system. Among 36 conformers, the authors identified that the nucleophile attacking distance, substrate binding, and stabilization of the general base Lys142 are most associated with the energetic fluctuation of the activation barrier. As far as we know, this study represents the first systematic application of data-driven modeling to understand the determining geometric factors behind the fluctuation of QM/MM-calculated enzyme potential energy barrier heights among enzyme conformers.
Bonk et al. (2019) reported the use of machine learning algorithms to elucidate the geometric features that enable an enzyme conformer capable of activating the substrate to undergo a reactive event in ketol-acid reductoisomerase. From reactive trajectories calculated using QM/MM transition interface sampling, the authors collected 68 different geometric features in the active site that represent elements of the local conformation (e.g. distances, planar angles, and dihedral angles). Employing the LASSO method, they identified important descriptors of the starting conformation that leads to reactive trajectories, which are substrate conformation, substrate bond polarization, and metal coordination geometry. Based on the selected features, they trained a logistic regression model to infer the probability of a specific trajectory in the reactive portion of the conformational space. The model exhibits an accuracy of 81.6% and an area under the curve of the receiver operating characteristic (AUROC) of 89.0%. This model can be potentially applied to enhance the sampling efficiency for QM/MM transition interface sampling of ketol-acid reductoisomerase.
As a follow-up to the works of Lodola et al. (2010) and Bonk et al. (2019), von der Esch et al. (2019) reported an elastic net regression model to predict the activation energy for enzymatic reactions. Besides understanding the molecular details behind conformational dependence of enzyme catalysis, the model can also be leveraged to determine suitable starting conformation for reaction path calculations. The model was trained using 150 activation free energy values derived from QM/MM calculations of 150 MD-sampled enzyme conformational snapshots of Class III histone deacetylase sirtuin 5. For each conformation, the input feature involves 15 structural features describing the distances between nonhydrogen atoms and nearby water molecules within the QM region. Although the model exhibits moderate accuracy (i.e. RMSD = 4.46 kcal/mol; cross-validated R2 = 0.28) in the prediction task, the author proposed strategies to further enhance the model by using quality data of transition barrier that are derived from free energy simulations and higher-level quantum mechanical calculations.
Besides inferring or predicting enzyme structure-barrier height relationships using statistical modeling or machine learning, deep learning has been applied to accelerate free energy calculations for enzymatic reactions based on QM/MM methods. Pan et al. (2021) reported the first neural network model to accelerate simulations of free energy paths using QM/MM methods in enzymatic reactions. They built a machine learning potential model (i.e. MLP) to predict the energy and force of QM/MM calculations using the cartesian coordinates and MM point charges as input. In addition, they developed a delta learning model (called ∆MLP) to predict the correction factors that improve the accuracy of semiempirical QM/MM free energies to the ab initio level. Compared to the existing framework, these two models include an MM environment, incorporate long-range electrostatics in the model training, and use a single set of descriptors. Using chorismate mutase as a test case, both MLP and PM3* + ΔMLP methods show similar accuracy to the B3LYP/6-31G*/MM method in generating free energy profiles (i.e. less than 1 kcal/mol difference). The MLP and PM3* + ΔMLP methods are 32- and 46-fold faster than the B3LYP/6-31G*/MM calculations, respectively. The methods thus provide new approaches to compute free energy profiles of enzyme-catalyzed reactions with balanced accuracy and efficiency. Looking forward, deep learning algorithms, such as neural network potential and deep generative models, are expected to further innovate the strategies for enhancing free energy simulations in enzyme-catalytic processes.
Prediction of enzyme activity and substrate specificity
Data-driven modeling has been extensively used to predict enzyme activity and substrate specificity. Different from biophysical properties (i.e. stability), enzyme activity or substrate specificity depends on not only the sequence context that determines protein structural fold, but also specific functional amino acids that determine or tune the chemical reactions catalyzed in the enzyme active site. To predict enzyme activity, earlier works primarily use sequence alone as input; the models developed in recent decade tend to incorporate more catalytically relevant information, such as substrate and enzyme structure, to enhance the predictive accuracy.
Using multiple sequence alignment as input, Casari et al. (1995) used principal component analysis to identify enzyme residues that modulate substrate specificity of biological function; Hannenhalli and Russell (2000) employed positional entropy analysis to identify sequence regions that confer specificity of known enzyme sub-types and to predict sub-type for unclassified sequences. Pertusi et al. (2017) developed the first support vector machine-coupled active learning approach, SimAL, to predict high-probability promiscuous enzymatic reactions for metabolic engineering. SimAL identified active substrates for four different enzymes with ~80% accuracy. With the capability of producing active compounds for experimental testing, SimAL provides a computational tool for the design of new metabolic pathways.
In addition to models using sequence-based features, structure has also been augmented with sequence information to enhance prediction performance. For example, Röttig et al. (2010) developed a support vector machine model, named active site classification, to achieve functional annotation of enzymes within an enzyme family. The model is not only capable of predicting enzyme activity (i.e. the type of reactions catalyzed by the enzyme) but also substrate specificity due to the incorporation of structural information. Masso and Vaisman (2014) developed a random forests classifier that predicts whether a single amino acid substitution affects or unaffacts enzyme activity (Masso and Vaisman, 2011, 2014). The input data consist of 1417 high-resolution PDB structures with a diverse range of sequence and structure. The structure of the protein was represented by the tessellation of all Cα coordinates, which describes local environment of each residue in protein structures. In the 10-fold cross-validation test, the successful hit rate is 84%. The model has been embedded in a software package AUTO-MUTE (Masso and Vaisman, 2011, 2014). Khersonsky et al. (2018) developed FuncLib as a web application to automate design of efficiency-enhancing enzyme variants with multipoint mutation. Although the prediction scoring is based on Rosetta calculations (Fleishman et al., 2011), phylogenetic analysis (a statistical modeling approach) was employed to identify residues that are more likely to modulate substrate specificity. Using phosphotriesterase and an acetyl-CoA synthetase as model enzymes, FuncLib identifies several dozen designs with three to six active-site mutations. These variants were tested to involve 10- to 4000-fold higher efficiencies with a range of alternative substrates, including hydrolysis of the nerve agents soman and cyclosarin, and synthesis of butyryl-CoA. Unlike most models that predict experimentally-characterized enzyme activities, Shroff et al. (2020) developed a CNN model to predict the structurally ‘optimal’ residue type in a protein fold environment; they applied the model to identify mutations that enhance enzyme activity. The model was trained using 19 136 protein PDB structures. Two types of features were used for each amino acid, including the local structural motif (i.e. 20 Å cube for each centered residue) and physicochemical properties (e.g. atomic charge, surface area, and so on). The model accuracy is 87% in predicting the type of natural amino acid. The model enabled the identification of beneficial mutations in TEM-1 β-lactamase and Candida albicans phosphomannose isomerase (CaPMI).
Different from the above-mentioned generalist models that predict enzyme activity and substrate specificity across different types of enzymes, specialist models have also been advanced to engineer, design, and discover new enzyme variants with desired substrate specificity. Chevrette et al. (2017) developed a computational method, named SANDPUMA, for ensemble prediction of substrate specificity of adenylation domains in nonribosomal peptide synthases. They compiled experimentally validated substrate specificity data from the MIBGC database (i.e. minimum information about a biosynthetic gene cluster) and scientific literature. Using the dataset, they benchmarked the accuracy of multiple existing algorithms (including the support vector machine method by Röttig et al. 2010). Through analyzing 83589 adenylation domains in the genomes across Actinobacteria, the study revealed 458 distinct nonribosomal peptide synthases superfamilies. Yang et al. (2018) developed a decision tree-based classifier, GT-Predict, to predict glycosyltransferase activity. The model incorporates local sequence information with the physicochemical properties of substrate donor and acceptor molecules. Superior to sequence-alone models, GT-Predict exhibits an accuracy of ~90% in the task of functional prediction over the 107 sequences from the glycosyltransferase superfamily 1 of the plant Arabidopsis thaliana. The model is expected to guide the streamlined design and engineering of new glycosyltransferases. Besides nonribosomal peptide synthases and glycosyltransferases, specificity prediction models have been developed for engineering synthetic biology enzymes, including 2-deoxy-d-ribose 5-phosphate aldolase variants for catalyzing smaller non-phosphorylated acceptor substrates in the aldol addition reaction with acetaldehyde (Voutilainen et al., 2020), OleA thiolase variants for the production of desired metabolites (Robinson et al., 2020), and bacterial nitrilases for hydrolysis of nitrile compounds to the corresponding carboxylic acids and ammonia (Mou et al., 2021).
As a summary, in Table II, we listed the activity- or specificity-enhancing enzymes and enzyme variants that were engineered with the help of data-driven methods and reported in the past five years. These examples demonstrate the enormous potential of data-driven modeling to be incorporated as a routine strategy for the bioengineering of synthetically useful enzymes.
Table II.
Summary of enzymes used in the development of date-driven enzyme engineering models for enhanced activity or specificitya
| Enzyme | Substrate | Mutation | Performance | Paper |
|---|---|---|---|---|
| 2-succinyl-5-enolpyruvyl-6-hydroxy-3-cyclohexene-1-carboxylic acid synthase | aldehyde | NA | new substrate specificity | 2017-Pertusi (Pertusi et al., 2017) |
| carboxylic acid reductase | carboxylic acid | NA | new substrate specificity | 2017-Pertusi (Pertusi et al., 2017) |
| amino acid ester hydrolase | amide, ester | NA | new substrate specificity | 2017-Pertusi (Pertusi et al., 2017) |
| 4-hydroxyacetophenone monooxygenase | keton | NA | new substrate specificity | 2017-Pertusi (Pertusi et al., 2017) |
| phosphotriesterase | phosphotriester, ester, lactone | I106L/F132L/H254R/H257W/L303T | 741664-fold specificity enhancement (paraoxon) | 2018- Khersonsky (Khersonsky et al., 2018) |
| acetyl-CoA synthetase | CoA, aliphatic acid | V310I/T311V/S314T/Y355F/V386L/F421A | 7-fold activity enhancement (butyrate) | 2018- Khersonsky (Khersonsky et al., 2018) |
| glycosyltransferase | sugar donor, glycosyl acceptor | NA | new sequence for new substrate | 2018-Yang (Yang et al., 2018) |
| phosphomannose isomerase | phosphomannose | D229W/N272K/L335A/N388S/S425T | 5-fold activity enhancement | 2020-Shroff (Shroff et al., 2020) |
| TEM-1 β-lactamase | carbenicillin | N52, F60, Q88, Q99, T114, M182, E197 | gain of new function | 2020-Shroff (Shroff et al., 2020) |
| 2-deoxy-D-ribose 5-phosphate aldolase | acetaldehyde | C47V/G204A/S239D | ~3-fold activity enhancement (acetaldehyde), abolishment of natural activity (deoxyribose-5-phosphate, deoxyribose) | 2020-Voutilainen (Voutilainen et al., 2020) |
| thiolase | p-nitrophenyl ester | NA | Specificity-determining residue, structural/chemical feature influencing activity | 2020-Robinson (Robinson et al., 2020) |
| nitrilase | nitriles | NA | substrate scope expansion | 2021-Mou (Mou et al., 2021) |
aIn the column of Mutation and Performance, only the best-performing enzyme mutants are shown. Mutations separated by slash ‘/’ indicate multiple mutations in one variant. Mutations separated by comma ‘,’ indicate different variants with single amino acid substitution.
Prediction of enzyme stereoselectivity
Enzyme stereoselectivity is the property of an enzyme to favor the formation of one over other possible stereoisomeric products. Enhancing enzyme stereoselectivity is critical for the biocatalytic production of compounds with high stereochemical purity—this is a prerequisite for the pharmaceutical and fine chemical industry. Due to its intrinsic chiral binding cavity, enzyme naturally fits to the task of stereoselective catalysis. However, due to the diversity of substrate structures, wild-type enzymes usually need to be engineered for effectively catalyzing a specific stereoselective reaction.
Statistical and machine learning models have been developed to identify efficiency-enhancing variants for transforming a certain stereoselective reaction or to identify variants that can alter the stereoselectivity based on actual synthetic needs. As an early attempt, Fox et al. (2003) from Codexis Inc. reported a statistical approach, named protein sequence activity relationships (ProSAR), to help identify beneficial mutants in directed evolution. Later, Fox et al. (2007), using ProSAR, identified a highly efficient bacterial halohydrin dehalogenase variant (i.e. > 4000 fold rate acceleration) for the production of (R)-4-cyano-3-hydroxybutyrate with high enantioselective purity.
As a new approach to describing enzyme sequence-function relationship, Cadet et al. (2018) reported an innovative sequence-activity relationship (innov’SAR) methodology that first employs digital signal processing (fast Fourier transform) to encode sequence as protein energy spectrum and then use the energy spectrum to perform regression with wet-lab functional data (e.g. enantiomeric excess value). Trained with experimentally characterized ∆∆G values obtained from the wild-type Aspergillus niger epoxide hydrolases and nine single-point mutants, innov’SAR exhibits an R2 of 0.96 in the leave-one-out cross-validation. Using the model, the authors identified L202W mutant with higher enantioselectivity than the wild-type hydrolase for kinetic resolution. Notably, the application of digital signal processing to encode sequence information is distinct from most existing enzyme feature engineering strategies that are based on one-hot encoding, physicochemical descriptors, or contextual embedding (Fig. 1). The learning efficiency of this encoding algorism against other methods remains to be compared in the future studies.
Wu et al. (2019) reported a machine learning-guided directed evolution approach to assist the construction of ‘smart’ library for protein engineering. The model was demonstrated in the task of identifying enzyme variants for enantiomeric catalysis of a new-to-nature reaction. Machine-learning models were trained with sequencing and enantiomeric information. A wide range of machine learning models were tested to determine the optimal one that best fits the specific fitness landscape of selected mutation positions. The model was applied to evolve P450 enzymes for producing each of the two possible enantiomeric products for a carbene Si–H insertion reaction. Combined with experimental screening, the approach identifies seven mutations in two rounds of iterative mutagenesis and finally pinpoints variants for enantiomeric formation of products with 93 and 79% ee. Notably, the Si–H insertion reaction is a new-to-nature activity evolved in the laboratory. Besides demonstrating the model accuracy, this study also highlights the use of machine learning for predicting non-natural functions for enzymes.
Xu et al. (2020) conducted a benchmark over 44 combinations of sequence features and machine learning methods to investigate what combination provides the best prediction performance in protein engineering. As one of the prediction tasks, stereoselectivity prediction was conducted using the dataset from the Reetz group (Gumulya et al., 2012) (epoxide hydrolases) and the Arnold group (Wu et al., 2019) (P450 enzymes). The benchmark shows that the CNN model combined with the mutation one-hot-encoding binary vector gives the best prediction accuracy (i.e. median scaled-RMSE = 1) in identifying enantioselectivity-enhancing epoxide hydrolases for glycidyl phenyl ether; XGBoost model combined with the PCscores gives the best prediction accuracy (i.e. median scaled-RMSE = 1) in identifying P450 enzyme variants that mediate stereoselective carbene Si–H insertion. In general, the authors recommended CNN models built with amino acid property descriptors as the most widely applicable choice for prediction tasks. This study serves as an important future guide to select the features and models for data-driven stereoselective enzyme engineering. However, we should note that the generality of the conclusion might be limited by the low diversity of enzyme types and relatively small number of data points in the testing set (i.e. 16 epoxide hydrolase variants and 318 P450 enzyme variants). The limitation on the dataset will be further discussed in a later section (i.e., Challenges).
As a summary, in Table III, we listed the stereoselectivity-enhancing enzymes and enzyme variants that were resulted from data-driven enzyme engineering in the past five years. These strategies will guide the engineering of enzymes for stereoselective biocatalysis.
Table III.
Summary of enzymes with the enhanced stereoselectivity through the date-driven enzyme engineeringa
| Enzyme | Substrate | Mutation | Performance | Paper |
|---|---|---|---|---|
| Epoxide hydrolase | Glycidyl phenyl ether | L215F/A217N/L249Y/T317W/T318V/M329P/C350V | E-value = 253 | 2018-Cadet (Cadet et al., 2018) |
| Nitric oxide dioxygenase | Phenyldimethyl silane, ethyl 2-diazopropanoate | R51V | % ee = 93 | 2019-Wu (Wu et al., 2019) |
aIn the column of Mutation and Performance, only the best-performing enzyme mutants are shown.
Prediction of thermodynamic and kinetic properties for enzymatic reactions
Enzyme kinetic and thermodynamic parameters inform enzyme’s capability of binding (e.g. Michaelis constant KM and dissociation constant KD) and converting (e.g. turnover number kcat) substrates in reactions. Unlike enzyme activities and substrate specificity, these parameters are rigorously defined and are normalized over enzymes’ concentration and expression level. Predicting these parameters are central to establish metabolic models (Heckmann et al., 2018; Li et al., 2022) and to pinpoint rate- or efficiency-enhancing enzyme mutants (Goldman et al., 2022; Kroll et al., 2021; Carlin et al., 2016).
Heckmann et al. (2018) pioneered in the development of elastic net regression, random forests, and deep neural network models to conduct genome-scale kcat prediction in Escherichia coli. The training and testing data consist of 172 in vitro kcat values of and 106 in vivo kapp,max values (i.e. the maximal effective turnover rate), which were curated from BRENDA, Metacyc, and SA-BIORK. The input features involve genome-scale metabolic parameters, enzyme structures, biochemistry properties, and kinetic assay conditions. In different models, the cross-validated R2 is around 0.31 for the kcat and 0.76 for the kapp,max. Enabled by the predicted kcat values, the authors improved the accuracy of metabolic models in the prediction of quantitative proteome data than previous approaches. Li et al. (2022) applied DL models to predict genome-scale kcat values for over 300 yeast species. The DL model encodes substrate connectivity using 2 layers of graphic neural network (GNN) and protein sequences using 3 layers of CNN; and then predict kcat using a CNN model. The model was trained on 7822 unique enzyme sequences and 2672 unique substrates with an overall 16 838 kcat data points. The model shows a Pearson r of 0.94 in predicting the kcat of well-studied enzyme-substrate pairs from literatures and original datasets. Compared to the ML models used by Heckmann et al. (2018), the DL model reported by Li et al. (2022) substantially improved the prediction accuracy for not only the native enzymes but also the enzyme mutants. Besides, the DL model only took protein sequence and substrate connectivity as input, which is in contrast to the use of a diverse range of metabolic, biochemical, and structural features in Heckmann et al.’s ML models. Notably, it remains an open question regarding whether Li et al. (2022) DL model can be used to identify function-enhancing enzymes or enzyme variants for engineering uses. Despite a wide variety of substrates involved in the kcat dataset, the amount of substrate data for each single enzyme is rather small. In addition, the magnitude to what the DL model learns the catalytically essential substrate-enzyme interaction also remains unknown. Nonetheless, both works demonstrate the capability of data-driven modeling in large-scale prediction of turnover values. Importantly, they shared well-curated datasets to the community for the development of future machine learning models.
Besides models to predict turnover numbers, Mellor et al. (2016) built a semi-supervised Gaussian process model to predict KM. The KM data were collected from BRENDA and other public datasets with 7318 reaction labels. The EC numbers range from 1.1.1 to 6.2.1 (Schomburg et al., 2002). The input features involve k-mer vector representation of enzyme sequence and binary reaction feature vectors that describe the change of atomic connectivity in bonding rearrangement. The Q2 scores of the leave-one-out cross-validation for the three datasets were between 0.5 and 0.8. The model was applied to identify enzymes used in the synthesis of N-Acetyl-L-Leucine and flavonoids, respectively. Kroll et al. (2021) developed a deep learning framework to predict KM. The authors curated a dataset of 5158 KM values from BRENDA. They employed a GNN to encode substrate and enzyme sequence, leading to a 120-dimensional feature vector for substrates and 769-dimensional binary vector for enzymes. The feature vectors were then used to predict KM in a gradient boost regression model. The cross-validated mean-squared error (MSE) and R2 between the experimental and predicted log10-scale KM values are 0.72 and 0.42, respectively.
To guide enzyme-substrate specificity screenings, Goldman et al. (2022) constructed a self-supervised learning framework to predict enzyme’s binding affinity to a substrate (i.e. KD values). The model was trained on 36 000 enzyme-substrate pairs from six different types of enzymes including halogenase, glycosyltransferase, thiolase, beta-keto acid cleavage enzyme, esterase, and phosphatase. The input features are represented using a substrate autoencoder and protein encoder. Specifically, the authors compared between autoencoder (JT-VAE), Morgan circular fingerprints, and compound-protein interaction model in encoding substrate information; they applied a pretrained model, ESM-1b, to represent proteins. The studies showed the incapability of existing compound-protein interaction models to learn interactions between compounds and proteins across various families of enzymes. The authors introduced an active-site pooling strategy for enhanced representation of enzyme-substrate interactions, which can be potentially used to guide developments of future machine learning models for predicting substrate-protein interactions. We should note that this work provides a working solution to a fundamental problem in deep learning-facilitated enzyme engineering, which involves the representation of physical interactions between enzyme and ligand in a physically meaningful fashion. For enzyme engineering, however, the problem is likely to be more complex, because both enzyme and substrate can adopt reactive conformation, instead of their ground-state conformation, to accomplish the barrier-crossing events.
Besides generalist models for large-scale kcat, KM, or KD prediction, Carlin et al. (2016) developed a multivariate regression model to predict kcat and KM of glycoside hydrolases. Instead of curating kinetic parameters from publicly available databases, the authors experimentally characterized kcat and KM values for 100 mutants of a glycoside hydrolase enzyme, BglB. They extracted the structural features of these enzyme mutants based on their computationally optimized molecular models. They employed the elastic net regularization method to identify statistically significant structural features; using these features, they built a multivariate linear regression model to predict kcat and KM values. The model exhibits a cross-validated Pearson r of 0.76 and Spearman ρ of 0.55. The model also identified the hydrogen bonding energy of the substrate as the most informative feature to predict kcat/KM. Besides the predictive models, the quality kcat and KM data of mutant glycoside hydrolases reported in this work are valuable for future work of data-driven modeling. As the advancement of microfluidic strategies for high-throughput kinetic parameter measurement (Markin et al., 2021), we expect to see more quality specialist models emphasizing accurate prediction of enzyme kinetics.
Prediction of fitness or mutational landscape in enzyme evolution
Predicting the impact of mutation on enzyme functional fitness is a central challenge in protein engineering. Unlike physical or chemical observables, functional fitness is an evolutionary property that is relevant to the survival rate of the biological host in a certain environment. Higher functional fitness is usually correlated to better enzyme expressibility, activity, thermostability, or solubility. In the past years, statistical (Figliuzzi et al., 2016; Teze et al., 2021; Hon et al., 2020), ML (Wittmann et al., 2021b), and DL models (Luo et al., 2021; Favor and Jayapurna, 2020; Biswas et al., 2021; Hsu et al., 2022) have been significantly advanced to encode enzyme sequence information to predict functional fitness in enzyme evolution. For example, using multiple sequence alignment as an input, Hon et al. (2020) established a bioinformatics web server, EnzymeMiner, to select enzymes with enhanced solubility while preserving enzyme activity. Teze et al. (2021) conducted clustering analysis of protein family sequences to identify conserved mutations as candidate beneficial mutations for trans-glycosylation by glycoside hydrolases (GH). Both works essentially take advantage of the evolutionary information to infer the mutational landscape for beneficial mutation selection.
Wittmann et al. (2021b) reported a machine learning-assisted directed evolution (i.e. MLDE) method that encodes enzyme variants using Rosetta scoring (i.e. REF2015) to describe protein mutation effect on stability and physicochemical features to represent protein sequences (e.g. Georgiev protein sequences embeddings). Tested with 384 variants derived from GB1 datasets, the MLDE experiment exhibits a maximum normalized discounted cumulative gain (i.e. NDCG) value of 0.91. Notably, the use of Rosetta scoring embeds both physical and statistical information in an empirical fashion. Different from typical physicochemical features derived from individual amino acids in solution or vacuum, the Rosetta scoring can account for electrostatic and van der Waals interactions between residues in the protein, thus encoding many-body effects into the model.
To extract the coevolutionary and other context information from protein sequence, language-based DL models have been developed, including UniRep (Alley et al., 2019), MSA transformer (Rao et al., 2021), EVMutation (Hopf et al., 2017), DeepSequence (Riesselman et al., 2018), Autoregressive (Shin et al., 2021), and so on. These models extract biophysical and evolutionary ‘embeddings’ from large amount of protein sequences to guide enzyme engineering. Favor and Jayapurna (2020) developed an end-to-end pipeline, eUniRep, to guide enzyme engineering. The eUniRep model involves a semi-supervised learning architecture consisting of unsupervised pretrained sequence embedding and supervised regression models. The pretrained embedding was trained on Uniref50 with more than 20 million raw protein sequences—they served as an evolutionary description of the protein sequences. The model was tested on three enzymes, TEM-1 β-lactamase, IsPETase and MS2 bacteriophage’s capsid protein. The best predictive accuracy for protein fitness score involves a mean square error (MSE) of 0.74 on double mutations of MS2 bacteriophage’s capsid protein. eUniRep outperforms other neural network models in predicting the enzyme fitness landscape.
Enabled by eUniRep, Biswas et al. (2021) developed a fitness predictor using low-number protein sequences as training data (e.g. tens of protein sequences). A major contribution of this work is to demonstrate the power of pretrained embeddings for significantly enhancing the data efficiency in the process of model training. As a proof of concept, the model was trained on 96 TEM-1 β-lactamase single mutant sequences and applied to identify mutants with 5- to 10-fold higher hit rate than the full-sequence one-hot encoding in the plate-based antibiotic selection experiment. Different from DL sequence encoders trained from a large amount of sequences, Luo et al. (2021) developed ECNet to predict the functional fitness of enzymes using local coevolution features generated from direct coupling analysis. The incorporation of local coevolution features enables ECNet to prioritize high-order, high-performing variants for fitness prediction. As a proof of concept, ECNet was applied to identify 37 TEM-1 β-lactamase variants covering 22 residue positions with improved resistance to ampicillin. A 4-point mutation, E26K/N98S/L100V/A182V, has the highest resistance. Despite the gigantic number of sequences used in the model training, the lack of substrate information likely limits the model’s generalizability to predict function-enhancing enzymes for transforming non-native substrates and reactions. In addition, most data-driven models apply β-lactamase as their testing system—new types of enzymes with greater chemical and functional diversity should be experimentally assessed.
Functional enzyme sequence design
In recent years, statistical and generative models have been advanced to computationally design artificial enzyme sequences that involve similar or superior functions to natural enzymes (Russ et al., 2020; Repecka et al., 2021; Madani et al., 2021). For instance, Russ et al. (2020) developed statistical models to design new chorismate mutases. The model adopts direct coupling analysis to identify conserved amino acids and evolutionarily correlated pairs of amino acids for a family of enzyme sequences. By incorporating these conserved residues and residue pairs, artificial sequences can be designed to mimic natural enzyme function. As a proof of concept, the authors computationally designed and experimentally characterized a new artificial chorismate mutase. The overall hit rate of the prediction is 30%.
Repecka et al. (2021) built a generative adversarial networks (GAN) model, ProteinGAN, to generate artificial protein sequences with natural-like functional properties. The self-attention-based GAN consists of a customized temporal convolutional network with an additional self-attention layer to encode information of enzyme catalytic residues. The model was trained on malate dehydrogenase (MDH) with a total of 16,706 mutant sequences. The catalytic performance of the resulting sequences was experimentally validated. The results indicate that 24% of artificial enzymes show catalytic activity comparable to natural MDH. Notably, the average sequence identity between the generated sequences and the native MDH sequence is 64.6%; this result highlights the diversity of the generated sequences. Similar to ProteinGAN, Madani et al. (2021) adapted a general protein language model, ProGen, to generate novel lysozyme sequences. ProGen is a self-supervised protein language model that was trained on millions of raw protein sequences across protein families and functions. The model generates lysozyme sequences by using transformer architecture with functional annotation. In the following wet lab evaluation, the author used ProGen to generate 100 artifact protein sequences and received an activity AUROC of 0.85 compared with experimental data. Giessel et al. (2022) reported a variational autoencoder model (VAE) to generate human ornithine transcarbamylase (hOTC) sequences with enhanced catalytic activity. The VAE model was applied to generate 87 variants. Experimental tests show that these enzyme variants involve an average enhancement of specificity by 1.4-fold relative to the wild-type hOTC. Despite the capability of generating artificial sequences with natural functions, most of the generated sequences only exhibit comparable catalytic competency to the wild-type enzyme. As such, it remains an open question regarding how to generate sequences with substantially improved catalytic activity by exploiting the latent space. In addition, it remains unknown how to develop a model for generating artificial sequences for enzymes with human-desired new-to-nature functions. This capability requires the model to master the chemical and physical principles behind enzyme catalysis and to understand the role of reactive intermediate in chemical transformation. This likely calls for a feature-embedding form that incorporates the contextual, structural, and evolutionary information for sequence and substrate.
Challenges
In this section, we will discuss three challenges that are involved in developing accurate and generalizable data-driven models for enzyme engineering—curation of enzyme structure and function data, generation of predictive features from first-principle simulations, and incorporation of substrate information.
The roadblock for data curation manifests in data collection, cleaning, and joining. First, data collection is hard because enzyme structure and functional data are stored in different databases, such as PDB for enzyme structure (Berman et al., 2000), BRENDA and SA-BIORK for enzyme kinetics (Jeske et al., 2019; Wittig et al., 2012), M-CSA for enzyme catalytic mechanisms (Ribeiro et al., 2018), UniProt for sequences (UniProt, 2019; Apweiler et al., 2004), ProThermDB for thermostability (Nikam et al., 2021), eSOL for solubility (Niwa et al., 2009), ProtaBank for designed and engineered enzymes (Wang et al., 2018), and so on (Carlin et al., 2016). These databases involve a variety of hierarchical structures for data storage and algorithms for data query. To train holistic and multi-objective data-driven models, significant efforts are needed to search and collect data from these sources. Second, data cleaning is difficult because enzyme databases adopt various data standard, format, and validation mechanism. In many enzyme entries, essential parameters are missing, such as mutational spot labeling and experimental conditions for kinetic assays. In addition, inconsistency exists between the stored data and original literature, which might be caused by manual input and other numerical rounding errors. To clean the data, significant manual curation and validation are needed. Third, data joining is difficult because no unified primary or foreign keys exists across various enzymology databases to allow one-to-one mapping of sequence, enzyme-substrate complex structure, and catalytic function data. For example, kinetic databases lack the information of enzyme structure identifiers (e.g. PDB ID), but structure database typically do not have sufficient quantitative functional annotation. Efforts are needed to map different types of enzymology data. As a first step to address this challenge, we are building an integrated enzyme structure–function database, IntEnzyDB, which stores clean and tabulated structure and kinetics data by adopting a relational architecture with the flattened data structure (Yan et al., 2021; Yan et al., 2022) In addition, we expect that the use of AlphaFold-2 in enzyme structure prediction will expand the pool of quality structural data (Jumper et al., 2021). Based on the well-curated dataset, a gold standard benchmark set can be established that comprehensively incorporate biocatalytic performance of enzymes under different mutations, substrates, and experimental conditions. This will make it possible to provide an objective assessment on the ML and DL models constructed from different research groups.
Generating features from first-principles molecular simulations is essential but challenging. Structural and physicochemical descriptors have been widely used to represent enzyme residues, but they are incapable of describing unique aspects of enzyme catalysis, including chemical bonding in enzyme catalytic cycle (Petchey and Grogan, 2019), enzyme interior long-range electrostatics (Sagui and Darden, 1999; Baker, 2010; Yang et al., 2021), charge transfer between reactive species and active-site residues (Yang et al., 2019a; Yang et al., 2019b; Kulik, 2018; Acosta-Silva et al., 2020), and protein dynamics for substrate positioning and thermal activation (Mehmood et al., 2021; Khersonsky et al., 2012; Gao et al., 2020). Generating features from molecular simulations, such as classical molecular dynamics and quantum chemistry, will provide descriptors of electronic structure and protein dynamics, which are critical to physical description of enzyme catalysis. However, automatic workflows are needed to enable generation of large number of simulation-derived features for enzymes and their mutants. To address this, we are developing a software suite, EnzyHTP, for automating enzyme model construction, mutant generation, and molecular simulations (Shao et al., 2021). We expect that this tool will be further advanced to generate molecular features for building predictive data-driven models.
Incorporating substrate information represents a distinct challenge in building predictive models for enzyme catalysis. Sequence-alone models have been developed to predict enzyme activity (Xu et al., 2022). However, due to the lack of generalizable substrate representation, these models are likely to fail in predicting enzymes for new-to-nature reactions with non-native substrates. Although different representations have been used to embed substrate in the model, including similes string, Morgan fingerprint, or 3D fingerprint, these representations typically fail to manifest enzyme-substrate interaction in enzymatic reactions. Goldman et al. (2022) showed that the compound–protein interaction model, which augments the substrate Morgan fingerprint with protein sequence features, does not show superior performance to sequence-alone model in the task of predicting enzyme promiscuity. Rather, labeling enzyme active-site residues can improve the predictive accuracy. These results highlight the urgent need of developing strategies to incorporate substrate-enzyme interactions. In addition, chirality is frequently missed in substrate representation. The models without substrate chirality annotation will not predict enzyme’s preference toward a certain substrate stereoisomer. The advancement of chirality-resolved 3D representation of substrate structure is expected to address the challenge.
Conclusion
In this mini-review, we discussed recent development of statistical, ML, and DL models to identify function-enhancing enzymes or enzyme mutants. Models have been developed to accelerate QM/MM-based free energy and reactive trajectory simulations, predict substrate specificity in various enzymes (e.g. nonribosomal peptide synthases, bacterial nitrilases, OleA thiolases, and SrtA), predict stereoselectivity of epoxide hydrolases and P450 enzymes, evaluate thermodynamic and kinetic parameters such as kcat, KM, and KD, predict the functional fitness score to facilitate directed evolution, and even design new enzyme sequences with similar or superior functions to natural enzymes. In addition, we discussed challenges in developing accurate and generalizable data-driven models for enzyme engineering, including curation of enzyme structure and function data, generation of predictive features from first-principles simulations, and incorporation of substrate information. We hope that this review can inform the readers of the recent progresses of data-driven modeling in enzyme engineering. As more and more quality enzymology data are available to the public, we believe that the upcoming years will witness a rapidly growing impact of data-driven strategies on enzyme engineering.
Acknowledgments
We thank Chris Jurich for proofreading the whole article.
Edited by: Timothy Whitehead
Contributor Information
Yaoyukun Jiang, Department of Chemistry, Vanderbilt University, Nashville, TN 37235, USA.
Xinchun Ran, Department of Chemistry, Vanderbilt University, Nashville, TN 37235, USA.
Zhongyue J Yang, Department of Chemistry, Vanderbilt University, Nashville, TN 37235, USA; Center for Structural Biology, Vanderbilt University, Nashville, TN 37235, USA; Vanderbilt Institute of Chemical Biology, Vanderbilt University, Nashville, TN 37235, USA; Data Science Institute, Vanderbilt University, Nashville, TN 37235, USA; Department of Chemical and Biomolecular Engineering, Vanderbilt University, Nashville, TN 37235, USA.
Funding
National Institute of General Medical Sciences of the National Institutes of Health (award number R35GM146982 to Z.J.Y., Y.J., and X.R.).
Conflict of Interest
The authors declare no conflict of interest.
References
- Acosta-Silva, C., Bertran, J., Branchadell, V. and Oliva, A. (2020) ChemPhysChem, 21, 295–306. [DOI] [PubMed] [Google Scholar]
- Adams, K., Pattanaik, L. and Coley, C.W. (2021) CoRR, abs/2110.04383. [Google Scholar]
- Ali, M., Ishqi, H.M. and Husain, Q. (2020) Biotechnol. Bioeng., 117, 1877–1894. [DOI] [PubMed] [Google Scholar]
- Alley, E.C., Khimulya, G., Biswas, S., AlQuraishi, M. and Church, G.M. (2019) Nat. Methods, 16, 1315–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apweiler, R., Bairoch, A., Wu, C.H. et al. (2004) Nucleic Acids Res., 32, 115D–1119D.14704348 [Google Scholar]
- Araya, C.L. and Fowler, D.M. (2011) Trends Biotechnol., 29, 435–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asgari, E. and Mofrad, M.R. (2015) PLoS One, 10, e0141287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker, D. (2010) Protein Sci., 19, 1817–1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman, H.M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T.N., Weissig, H., Shindyalov, I.N. and Bourne, P.E. (2000) Nucleic Acids Res., 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop, C. (2006) Pattern Recognition and Machine Learning. New York City, New York, Springer. [Google Scholar]
- Biswas, S., Khimulya, G., Alley, E.C., Esvelt, K.M. and Church, G.M. (2021) Nat. Methods, 18, 389–396. [DOI] [PubMed] [Google Scholar]
- Bonk, B.M., Weis, J.W. and Tidor, B. (2019) J. Am. Chem. Soc., 141, 4108–4118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruggink, A., Schoevaart, R. and Kieboom, T. (2003) Org. Process Res. Dev., 7, 622–640. [Google Scholar]
- Bunzel, H.A., Garrabou, X., Pott, M. and Hilvert, D. (2018) Curr. Opin. Struct. Biol., 48, 149–156. [DOI] [PubMed] [Google Scholar]
- Cadet, F., Fontaine, N., Li, G., Sanchis, J., Ng Fuk Chong, M., Pandjaitan, R., Vetrivel, I., Offmann, B. and Reetz, M.T. (2018) Sci. Rep., 8, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlin, D.A., Caster, R.W., Wang, X. et al. (2016) PLoS One, 11, e0147596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casari, G., Sander, C. and Valencia, A. (1995) Nat. Struct. Biol., 2, 171–178. [DOI] [PubMed] [Google Scholar]
- Cecchini, D.A., Pepe, O., Pennacchio, A., Fagnano, M. and Faraco, V. (2018) AMB Express, 8, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevrette, M.G., Aicheler, F., Kohlbacher, O., Currie, C.R. and Medema, M.H. (2017) Bioinformatics, 33, 3202–3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DelRe, C., Jiang, Y., Kang, P. et al. (2021) Nature, 592, 558–563. [DOI] [PubMed] [Google Scholar]
- Favor, A. and Jayapurna, I. (2020) Authorea Preprints. [Google Scholar]
- Feehan, R., Montezano, D. and Slusky, J.S.G. (2021) Protein Eng. Des. Sel., 34, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figliuzzi, M., Jacquier, H., Schug, A., Tenaillon, O. and Weigt, M. (2016) Mol. Biol. Evol., 33, 268–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleishman, S.J., Leaver-Fay, A., Corn, J.E. et al. (2011) PLoS One, 6, e20161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler, D.M. and Fields, S. (2014) Nat. Methods, 11, 801–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox, R., Roy, A., Govindarajan, S., Minshull, J., Gustafsson, C., Jones, J.T. and Emig, R. (2003) Protein Eng. Des. Sel., 16, 589–597. [DOI] [PubMed] [Google Scholar]
- Fox, R.J., Davis, S.C., Mundorff, E.C. et al. (2007) Nat. Biotechnol., 25, 338–344. [DOI] [PubMed] [Google Scholar]
- Gao, S., Thompson, E.J., Barrow, S.L., Zhang, W., Iavarone, A.T. and Klinman, J.P. (2020) J. Am. Chem. Soc., 142, 19936–19949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giessel, A., Dousis, A., Ravichandran, K., Smith, K., Sur, S., McFadyen, I., Zheng, W. and Licht, S. (2022) Sci. Rep., 12, 1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman, S., Das, R., Yang, K.K. and Coley, C.W. (2022) PLoS Comput. Biol., 18, e1009853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodfellow, I., Bengio, Y. and Courville, A. (2016) Deep Learning. Cambridge, Massachusetts, MIT Press. [Google Scholar]
- Gordon, S.R., Stanley, E.J., Wolf, S., Toland, A., Wu, S.J., Hadidi, D., Mills, J.H., Baker, D., Pultz, I.S. and Siegel, J.B. (2012) J. Am. Chem. Soc., 134, 20513–20520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumulya, Y., Sanchis, J. and Reetz, M.T. (2012) Chem. Bio. Chem., 13, 1060–1066. [DOI] [PubMed] [Google Scholar]
- Hannenhalli, S.S. and Russell, R.B. (2000) J. Mol. Biol., 303, 61–76. [DOI] [PubMed] [Google Scholar]
- Heckmann, D., Lloyd, C.J., Mih, N., Ha, Y., Zielinski, D.C., Haiman, Z.B., Desouki, A.A., Lercher, M.J. and Palsson, B.O. (2018) Nat. Commun., 9, 5252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrikse, N.M., Sandegren, A., Andersson, T. et al. (2021) iScience, 24, 102154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilvert, D. (2013) Annu. Rev. Biochem., 82, 447–470. [DOI] [PubMed] [Google Scholar]
- Hon, J., Borko, S., Stourac, J., Prokop, Z., Zendulka, J., Bednar, D., Martinek, T. and Damborsky, J. (2020) Nucleic Acids Res., 48, W104–W109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopf, T.A., Ingraham, J.B., Poelwijk, F.J., Scharfe, C.P., Springer, M., Sander, C. and Marks, D.S. (2017) Nat. Biotechnol., 35, 128–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu, C., Nisonoff, H., Fannjiang, C. and Listgarten, J. (2022) Nat. Biotechnol., 40, 1114–1122. [DOI] [PubMed] [Google Scholar]
- Jeske, L., Placzek, S., Schomburg, I., Chang, A. and Schomburg, D. (2019) Nucleic Acids Res., 47, D542–D549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumper, J., Evans, R., Pritzel, A. et al. (2021) Nature, 596, 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurtz, V.I., Johansen, A.R., Nielsen, M., Almagro Armenteros, J.J., Nielsen, H., Sonderby, C.K., Winther, O. and Sonderby, S.K. (2017) Bioinformatics, 33, 3685–3690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khersonsky, O., Kiss, G., Rothlisberger, D., Dym, O., Albeck, S., Houk, K.N., Baker, D. and Tawfik, D.S. (2012) Proc. Natl. Acad. Sci. USA., 109, 10358–10363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khersonsky, O., Lipsh, R., Avizemer, Z. et al. (2018) Mol. Cell., 72, 178–186.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott, B.C., Erickson, E., Allen, M.D. et al. (2020) Proc. Natl. Acad. Sci., 117, 25476–25485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kries, H.; Blomberg, R.; Hilvert, D., De Curr. Opin. Chem. Biol. 2013, 17 , 221–228. [DOI] [PubMed] [Google Scholar]
- Kroll, A., Engqvist, M.K.M., Heckmann, D. and Lercher, M.J. (2021) PLoS Biol., 19, e3001402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulik, H.J. (2018) Phys. Chem. Chem. Phys., 20, 20650–20660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, F., Yuan, L., Lu, H., Li, G., Chen, Y., Engqvist, M.K.M., Kerkhoven, E.J. and Nielsen, J. (2022) Nat. Catal., 5, 662–672. [Google Scholar]
- Li, Z., Jiang, Y., Guengerich, F.P., Ma, L., Li, S. and Zhang, W. (2020) J. Biol. Chem., 295, 833–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodola, A., Sirirak, J., Fey, N., Rivara, S., Mor, M. and Mulholland, A.J. (2010) J. Chem. Theory Comput., 6, 2948–2960. [DOI] [PubMed] [Google Scholar]
- Luo, Y., Jiang, G., Yu, T., Liu, Y., Vo, L., Ding, H., Su, Y., Qian, W.W., Zhao, H. and Peng, J. (2021) Nat. Commun., 12, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madani, A., Krause, B., Greene, E.R. et al. (2021) bioRxiv., 2021.2007.2018.452833.
- Markin, C.J., Mokhtari, D.A., Sunden, F., Appel, M.J., Akiva, E., Longwell, S.A., Sabatti, C., Herschlag, D. and Fordyce, P.M. (2021) Science, 373, eabf8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masso, M. and Vaisman, I.I. (2011) Annu. Int. Conf. IEEE Eng. Med. Biol. Soc., 2011, 3221–3224. [DOI] [PubMed] [Google Scholar]
- Masso, M. and Vaisman, I.I. (2014) Adv. Bioinform., 2014, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazurenko, S., Prokop, Z. and Damborsky, J. (2020) ACS Catal., 10, 1210–1223. [Google Scholar]
- Mehmood, R., Vennelakanti, V. and Kulik, H.J. (2021) ACS Catal., 11, 12394–12408. [Google Scholar]
- Mei, H., Liao, Z.H., Zhou, Y. and Li, S.Z. (2005) Biopolymers, 80, 775–786. [DOI] [PubMed] [Google Scholar]
- Mellor, J., Grigoras, I., Carbonell, P. and Faulon, J.L. (2016) ACS Synth. Biol., 5, 518–528. [DOI] [PubMed] [Google Scholar]
- Melnikov, A., Rogov, P., Wang, L., Gnirke, A. and Mikkelsen, T.S. (2014) Nucleic. Acids. Res., 42, e112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min, K., Kim, H., Park, H.J., Lee, S., Jung, Y.J., Yoon, J.H., Lee, J.S., Park, K., Yoo, Y.J. and Joo, J.C. (2021) Bioresour. Technol., 340, 125737. [DOI] [PubMed] [Google Scholar]
- Morcos, F., Pagnani, A., Lunt, B., Bertolino, A., Marks, D.S., Sander, C., Zecchina, R., Onuchic, J.N., Hwa, T. and Weigt, M. (2011) Proc. Natl. Acad. Sci., 108, E1293–E1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mou, Z., Eakes, J., Cooper, C.J., Foster, C.M., Standaert, R.F., Podar, M., Doktycz, M.J. and Parks, J.M. (2021) Proteins, 89, 336–347. [DOI] [PubMed] [Google Scholar]
- Nikam, R., Kulandaisamy, A., Harini, K., Sharma, D. and Gromiha, M.M. (2021) Nucleic Acids Res., 49, D420–D424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa, T., Ying, B.W., Saito, K., Jin, W., Takada, S., Ueda, T. and Taguchi, H. (2009) Proc. Natl. Acad. Sci. USA., 106, 4201–4206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan, X., Yang, J., Van, R., Epifanovsky, E., Ho, J., Huang, J., Pu, J., Mei, Y., Nam, K. and Shao, Y. (2021) J. Chem. Theory. Comput., 17, 5745–5758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertusi, D.A., Moura, M.E., Jeffryes, J.G., Prabhu, S., Walters Biggs, B. and Tyo, K.E.J. (2017) Metab. Eng., 44, 171–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petchey, M.R. and Grogan, G. (2019) Adv. Synth. Catal., 361, 3895–3914. [Google Scholar]
- Rao, R., Liu, J., Verkuil, R., Meier, J., Canny, J.F., Abbeel, P., Sercu, T. and Rives, A. (2021) bioRxiv.
- Repecka, D., Jauniskis, V., Karpus, L., Rembeza, E., Rokaitis, I., Zrimec, J., Poviloniene, S., Laurynenas, A., Viknander, S. and Abuajwa, W. (2021) Nat. Mach. Intell., 3, 324–333. [Google Scholar]
- Ribeiro, A.J.M., Holliday, G.L., Furnham, N., Tyzack, J.D., Ferris, K. and Thornton, J.M. (2018) Nucleic Acids Res., 46, D618–D623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riesselman, A.J., Ingraham, J.B. and Marks, D.S. (2018) Nat. Methods, 15, 816–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, S.L., Smith, M.D., Richman, J.E., Aukema, K.G. and Wackett, L.P. (2020) Synth. Biol., 5, 1–12. [Google Scholar]
- Romero, P.A. and Arnold, F.H. (2009) Nat. Rev. Mol. Cell Biol., 10, 866–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rorrer, N.A., Nicholson, S., Carpenter, A., Biddy, M.J., Grundl, N.J. and Beckham, G.T. (2019) Joule, 3, 1006–1027. [Google Scholar]
- Röttig, M., Rausch, C. and Kohlbacher, O. (2010) PLoS Comput. Biol., 6, e1000636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russ, W.P., Figliuzzi, M., Stocker, C. et al. (2020) Science, 369, 440–445. [DOI] [PubMed] [Google Scholar]
- Sagui, C. and Darden, T.A. (1999) Annu. Rev. Biophys. Biomol. Struct., 28, 155–179. [DOI] [PubMed] [Google Scholar]
- Saito, Y., Oikawa, M., Sato, T., Nakazawa, H., Ito, T., Kameda, T., Tsuda, K. and Umetsu, M. (2021) ACS Catal., 11, 14615–14624. [Google Scholar]
- Sandberg, M., Eriksson, L., Jonsson, J., Sjostrom, M. and Wold, S. (1998) J. Med. Chem., 41, 2481–2491. [DOI] [PubMed] [Google Scholar]
- Schindele, P. and Puchta, H. (2020) Plant Biotechnol. J., 18, 1118–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schomburg, I., Chang, A. and Schomburg, D. (2002) Nucleic Acids Res., 30, 47–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao, Q., Jiang, Y. and Yang, Z.J. (2021) J. Chem. Inf. Model., 62, 647–655. [DOI] [PubMed] [Google Scholar]
- Shin, J.-E., Riesselman, A.J., Kollasch, A.W., McMahon, C., Simon, E., Sander, C., Manglik, A., Kruse, A.C. and Marks, D.S. (2021) Nat. Commun., 12, 2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shroff, R., Cole, A.W., Diaz, D.J., Morrow, B.R., Donnell, I., Annapareddy, A., Gollihar, J., Ellington, A.D. and Thyer, R. (2020) ACS Synth. Biol., 9, 2927–2935. [DOI] [PubMed] [Google Scholar]
- Siedhoff, N. E.; Schwaneberg, U.; Davari, M. D. (2020) Methods in Enzymology, Vol.643, Cambridge, Massachusetts, Academic Press, pp. 281–315. [DOI] [PubMed] [Google Scholar]
- Simonis, H., Yaghootfam, C., Sylvester, M., Gieselmann, V. and Matzner, U. (2019) Hum. Mol. Genet., 28, 1810–1821. [DOI] [PubMed] [Google Scholar]
- Singh, N., Malik, S., Gupta, A. and Srivastava, K.R. (2021) Emerging Top. Life Sci., 5, 113–125. [DOI] [PubMed] [Google Scholar]
- Tang, Q., Grathwol, C.W., Aslan-Uzel, A.S., Wu, S., Link, A., Pavlidis, I.V., Badenhorst, C.P.S. and Bornscheuer, U.T. (2021) Angew. Chem. Int. Ed. Engl., 60, 1524–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teze, D., Zhao, J., Wiemann, M. et al. (2021) Chemistry–A. Eur. J. Dermatol., 27, 10323–10334. [DOI] [PubMed] [Google Scholar]
- Tournier, V., Topham, C.M., Gilles, A. et al. (2020) Nature, 580, 216–219. [DOI] [PubMed] [Google Scholar]
- UniProt, C. (2019) Nucleic Acids Res., 47, D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von der Esch, B., Dietschreit, J.C.B., Peters, L.D.M. and Ochsenfeld, C. (2019) J. Chem. Theory. Comput., 15, 6660–6667. [DOI] [PubMed] [Google Scholar]
- Voutilainen, S., Heinonen, M., Andberg, M. et al. (2020) Appl. Microbiol. Biotechnol., 104, 10515–10529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, C.Y., Chang, P.M., Ary, M.L., Allen, B.D., Chica, R.A., Mayo, S.L. and Olafson, B.D. (2018) Protein Sci., 27, 1113–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittig, U., Kania, R., Golebiewski, M. et al. (2012) Nucleic Acids Res., 40, D790–D796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittmann, B.J., Johnston, K.E., Wu, Z. and Arnold, F.H. (2021a) Curr. Opin. Struct. Biol., 69, 11–18. [DOI] [PubMed] [Google Scholar]
- Wittmann, B.J., Yue, Y. and Arnold, F.H. (2021b) Cell Syst., 12, 1026–1045.e7. [DOI] [PubMed] [Google Scholar]
- Wolf, C., Siegel, J.B., Tinberg, C., Camarca, A., Gianfrani, C., Paski, S., Guan, R., Montelione, G., Baker, D. and Pultz, I.S. (2015) J. Am. Chem. Soc., 137, 13106–13113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, Z., Kan, S.B.J., Lewis, R.D., Wittmann, B.J. and Arnold, F.H. (2019) Proc. Natl. Acad. Sci., 116, 8852–8858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia, B., Xu, J., Xiang, Z., Cen, Y., Hu, Y., Lin, X. and Wu, Q. (2017) ACS Catal., 7, 4542–4549. [Google Scholar]
- Xu, Y., Verma, D., Sheridan, R.P., Liaw, A., Ma, J., Marshall, N.M., McIntosh, J., Sherer, E.C., Svetnik, V. and Johnston, J.M. (2020) J. Chem. Inf. Model., 60, 2773–2790. [DOI] [PubMed] [Google Scholar]
- Xu, Z., Wu, J., Song, Y.S. and Mahadevan, R. (2022) Machine Learning in Computational Biology. PMLR, pp. 78–87. [Google Scholar]
- Yan, B., Ran, X., Jiang, Y., Torrence, S.K., Yuan, L., Shao, Q. and Yang, Z.J. (2021) J. Phys. Chem. B, 125, 10682–10691. [DOI] [PubMed] [Google Scholar]
- Yan, B., Ran, X., Gollu, A., Cheng, Z., Zhou, X., Chen, Y. and Yang, Z.J. (2022) J. Chem. Inf. Model., In Press. 10.1021/acs.jcim.2c01139. [DOI] [PMC free article] [PubMed]
- Yang, M., Fehl, C., Lees, K.V., Lim, E.-K., Offen, W.A., Davies, G.J., Bowles, D.J., Davidson, M.G., Roberts, S.J. and Davis, B.G. (2018) Nat. Chem. Biol., 14, 1109–1117. [DOI] [PubMed] [Google Scholar]
- Yang, Z., Hajlasz, N., Steeves, A. and Kulik, H. (2021) ChemRxiv., 1, 362–373. [Google Scholar]
- Yang, Z., Liu, F., Steeves, A.H. and Kulik, H.J. (2019a) J. Phys. Chem. Lett., 10, 3779–3787. [DOI] [PubMed] [Google Scholar]
- Yang, Z., Mehmood, R., Wang, M., Qi, H.W., Steeves, A.H. and Kulik, H.J. (2019b) React. Chem. Eng., 4, 298–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi, D., Bayer, T., Badenhorst, C.P.S., Wu, S., Doerr, M., Hohne, M. and Bornscheuer, U.T. (2021) Chem. Soc. Rev., 50, 8003–8049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin, Y., Wang, Q., Xiao, L. et al. (2018) J. Biomed. Nanotechnol., 14, 456–476. [DOI] [PubMed] [Google Scholar]
- Zeymer, C. and Hilvert, D. (2018) Annu. Rev. Biochem., 87, 131–157. [DOI] [PubMed] [Google Scholar]