

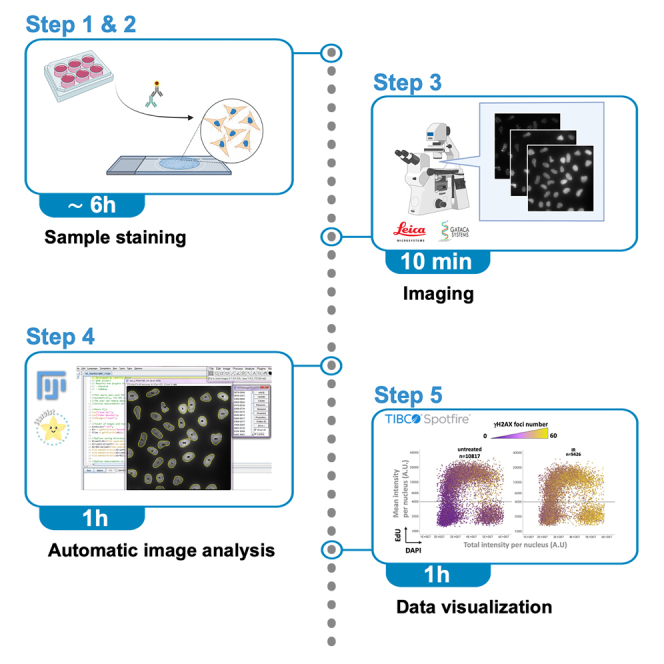
Summary
Here, we present a protocol for multivariate quantitative-image-based cytometry (QIBC) analysis by fluorescence microscopy of asynchronous adherent cells. We describe steps for the preparation, treatment, and fixation of cells, sample staining, and imaging for QIBC. We then detail image analysis with our open source Fiji script developed for QIBC and present multiparametric data visualization. Our QIBC Fiji script integrates modern artificial-intelligence-based tools, applying deep learning, for robust automated nuclei segmentation with minimal user adjustments, a major asset for efficient QIBC analysis.
For complete details on the use and execution of this protocol, please refer to Besse et al. (2023).1
Subject areas: Cell-based Assays, Microscopy, Signal Transduction
Graphical abstract
Highlights
-
•
Pipeline solution for typical quantitative-image-based cytometry (QIBC) analysis
-
•
Cost-effective widefield fluorescence microscope configuration
-
•
Open source Fiji script for QIBC integrating modern artificial-intelligence-based tools
-
•
Automated individual-cell multi-parameter extraction
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Here, we present a protocol for multivariate quantitative-image-based cytometry (QIBC) analysis by fluorescence microscopy of asynchronous adherent cells. We describe steps for the preparation, treatment, and fixation of cells, sample staining, and imaging for QIBC. We then detail image analysis with our open source Fiji script developed for QIBC and present multiparametric data visualization. Our QIBC Fiji script integrates modern artificial-intelligence-based tools, applying deep learning, for robust automated nuclei segmentation with minimal user adjustments, a major asset for efficient QIBC analysis.
Before you begin
The development of high-content fluorescence microscopy approaches for cell-cycle staging of adherent cells from large asynchronous population,2,3,4,5,6,7 referred to as Quantitative Image-Based Cytometry (QIBC),6,7 have allowed to extensively examine stress response at a single cell level.8,9,10,11,12,13,14 However, QIBC mainly relies on proprietary dedicated high-content microscopy and image analysis platforms. The challenges of QIBC are the implementation of automatic image acquisition and analysis that requires reliable single nucleus segmentation together with multi-parameters information extraction in an unbiased manner. For this, modern artificial intelligence-based tools applying deep-learning for robust automated nuclei segmentation are major assets for efficient QIBC analysis.
Here we describe an open-source QIBC pipeline solution for widefield fluorescence microscope configuration for automated multi-color acquisition and in house developed Fiji-scripts (macro) for parameters extraction. The first step of the analysis is based on StarDist, a modern deep-learning tool trained to recognize nuclei. Unlike the classical threshold method that uses only the fluorescence intensity parameter to segment nuclei, the StarDist15 segmentation calls for multiple nuclei features, which provides highly accurate and fully automated segmentation of crowded nuclei with low user intervention. The segmentation step is followed by automated individual cell multi-parameter extraction, including nuclei DNA content and EdU mean signal intensity, allowing for cell-cycle staging. In addition, extracted parameters for the proteins of interest include mean signal intensity and/or foci count per nucleus. Nuclear area (and nuclear volume) does not fully correlate with the nuclear DNA content during cell-cycle progression.16 Indeed, a significant increase in nuclear volume occurs in a variety of mammalian cells during the G1 phase before the entry into the S phase and DNA replication initiation.17,18,19,20 For this reason, the measurement of the DAPI total intensity of the nucleus parameter (annotated “RawIntDen” in Fiji), is used as a proxy for nuclei DNA content.
Using a data visualization software, the protein parameters can be cross-analyzed in the context of the cell-cycle at the individual cell level, thus providing in depth information from asynchronous cell populations. The detection by immunofluorescence technics of γH2AX and 53BP1 signals according to cell-cycle stage has provided important information about the molecular processes underlying the DNA damage response.21,22 For this the QIBC pipeline solution was applied to γH2AX and 53BP1 signal analysis.
Cell culture
Timing: 3–5 days (for steps 1 to 10)
Keep the cells at an optimal density for continued growth.
-
1.
Thaw U-2 OS cells in DMEM Complete Medium equilibrated at 37°C (DMEM Complete Medium cell culture media can be stored at 4°C for a maximum of three months).
-
2.
Remove the cryovial containing the cells from liquid nitrogen storage and immediately place it into a 37°C water bath.
-
3.
Quickly thaw the cells by gently swirling the vial in the water bath for 1 min.
-
4.
Transfer the vial into a biosafety cabinet.
-
5.
Transfer the vial’s content into a 15 mL centrifuge tube containing 10 mL of pre-warmed DMEM Complete Medium.
-
6.
Centrifuge the cell suspension at 200 × g for 5 min at 22°C.
-
7.
Discard the supernatant and resuspend the cells in 5 mL of DMEM Complete Medium.
-
8.
Transfer the contents into a T25 flask and incubate at a 37°C in a 5% CO2 incubator for 24 h.
-
9.
Aspirate out medium from the T25 flask and refresh with 5 mL pre-warmed DMEM Complete Medium the following day.
-
10.
Maintain the cultures and allow cells to grow until 90% confluency is reached.
Fiji installation and plugins implementation
The Fiji software is installed and set for the Stardist plugin.
-
11.
Download the Fiji software at https://imagej.net/software/fiji/downloads.
-
12.
Download Fiji updates, go to Menu>Help>Update.
-
13.
Click on Manage update sites.
-
14.
Scroll down through the list and tick the checkboxes for CSBDeep and StarDist. Note that if StarDist is missing from the list, click Update URLs to refresh the list.
-
15.
Click the Close button.
-
16.
ImageJ 1.53d or later versions must be used, if necessary, update ImageJ, go to Help>Update ImageJ>upgrade to select version 1.53 or later.
Download the in house developed Fiji-scripts for QIBC analysis
Images are analyzed by Fiji with in-house developed scripts (Macros) available for download on the Zenodo platform (https://doi.org/10.5281/zenodo.8014997). Multiple parameters are extracted from the automatically defined nuclei ROI on the selected channels. Then, for the protein of interest, the mean signal intensity per nucleus is extracted along with the protein foci count per nucleus, with the find maxima function in Fiji, when discrete foci are formed. The measured parameters are exported in an Excel file.
-
17.
Download the “Macros.zip” file containing the Fiji-script “00_Convert Files to TIF”.
Note: Fiji-scripts for QIBC analysis require “.tif” or “.TIF” image files extensions. Microscope company provide different type of image file saving formats. For this reason we propose the open source Fiji-script “00_Convert Files to TIF” adapted from a script developed by Olivier Burri, BioImaging and Optics Platform – EPFL, to convert your microscope file (.LIF; .CZI; .VSI; .ND2) into individual “.tif” image files to fit the requirement for QIBC analysis by two in house developed Fiji-scripts. Each image channel is saved as a separate image file which name must include the letter “C” or “c” followed by the channel number (C1, C2, C3, C4 or c1, c2, c3, c4). Image analysis for QIBC experiment is done on 2D images (no Z series are accepted).
-
18.
Download the “Macros.zip” file containing the Fiji-script “01_Shading-Dark correction_v2.ijm”.
Note: The in house developed Fiji-script (01_Shading-Dark correction_v2.ijm) applies dark and illumination corrections on all images.23,24 The Dark-current image is first subtracted from each sample image and from the shading image. In a second step, an illumination correction image is generated for which an illumination factor for each pixel is determined. For this, the intensity of each pixel of the shading image is divided by the mean pixel intensity of the shading image. The illumination correction image is used to calculate the corrected sample image obtained by dividing the sample image by the illumination correction image. The corrected image files automatically generated by the Fiji-script (01_Shading-Dark correction_v2.ijm) are copied in a new folder distinct from the folder containing the raw image files.
-
19.
Download the “Macros.zip” file containing the Fiji-script “02_StarDist-QIBC_v12.ijm”.
Note: The in house developed Fiji-script (02_StarDist-QIBC_v12.ijm) is applied on all corrected sample images. DAPI channel is used to automatically extract nuclei as region of interest (ROI) with the «Versatile fluorescent nuclei » model present in the Fiji plugin for StarDist15 and are added to the ROI manager window. Training automated nucleus segmentation tools using modern deep-learning approaches requires collecting annotated examples. The 2018 Data Science Bowl created a resource of diverse nuclei images contributed by numerous biological laboratories and manually annotated by a team of expert biologists at the Broad Institute. The StarDist versatile fluorescent nuclei model was trained on a subset of images and is well suited for robust U-2 OS nuclei segmentation.
-
20.
Download the Spotfire® canvas template.
-
21.
Download the Spotfire® tutorial video for help on how to execute QIBC multiparametric data visualization.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal antibody anti-H2AX-phospho-serine139 (anti-γH2AX) IgG1κ, clone JBW301, use at 1:500 dilution | Merck Sigma-Aldrich | Cat# 05-636-I; RRID:AB_2755003 |
| Mouse monoclonal antibody anti-p53 Binding Protein 1 (anti-53BP1) IgG1, clone BP13, use at 1:750 dilution | Merck Sigma-Aldrich | Cat# MAB3802; RRID:AB_2206767 |
| Mouse monoclonal anti-Tubulin, clone DMA1, ascites fluid, use at 1:1000 dilution | Merck Sigma-Aldrich | Cat# T-9026; RRID:AB_477593) |
| Donkey anti-Mouse IgG (H + L) Alexa Fluor 647 conjugated, use at 1:500 dilution | Thermo Fisher Scientific | Cat# A-31571; RRID: AB_162542 |
| Peroxidase-AffiniPure Goat Anti-Mouse IgG (H + L), use at 1:10 000 dilution | Jackson ImmunoResearch Labs | Cat# 115-035-003; RRID: AB_10015289 |
| Chemicals, peptides, and recombinant proteins | ||
| Phosphate-buffered saline (PBS) tablets | EUROMEDEX | Cat# EU1-2052-100 |
| (S)-(+)-Camptothecin (CPT) | Merck Sigma-Aldrich | Cat# C9911-100 mg; CAS: 7689-03-4 |
| Dimethyl sulfoxide (DMSO) | Merck Sigma-Aldrich | Cat# D8418-100 mL; CAS: 67-68-5 |
| 5′-Ethylene-2′-deoxyuridine (EdU) | Thermo Fisher Scientific | Cat# A10044 |
| PIPES | Merck Sigma-Aldrich | Cat# P1851; CAS: 5625-37-6 |
| Sucrose | Merck Sigma-Aldrich | Cat# S0389; CAS: 57-50-1 |
| TritonTM X-100 | Merck Sigma-Aldrich | Cat# 1.08603.1000; CAS: 9036-19-5 |
| MgCl2 1 M | Thermo Fisher Scientific | Cat# AM9530G |
| NaCl | Merck Sigma-Aldrich | Cat# 31434-1KG-M; CAS: 7647-14-5 |
| Tris-HCl 1 M, pH 7.5 | Thermo Fisher Scientific | Cat# 15567027 |
| Tris-HCl 0.5 M, pH 8.5 | Thermo Fisher Scientific | Cat# J62131.AP |
| cOmpleteTM, Mini, EDTA-free Protease Inhibitor Cocktail, Roche | Merck Sigma-Aldrich | Cat# 4693159001 |
| Paraformaldehyde (PFA), 16% (v/v) solution | Electron Microscopy Sciences | Cat# EM-15710 |
| Bovine serum albumin (BSA), standard grade | Interchim | Cat# UPQ84170; CAS: 9048-46-8 |
| (+) Sodium L-ascorbate | Merck Sigma-Aldrich | Cat# A4034; CAS: 134-03-2 |
| Copper (II) sulfate, anhydrous, 98%, Alfa Aesar | Thermo Fisher Scientific | Cat# A13986.30; CAS: 7758-98-7 |
| Alexa Fluor 488 Azide (Alexa Fluor 488 5-Carboxamido-(6-Azidohexanyl), Bis(Triethylammonium Salt)), 5-isomer) | Thermo Fisher Scientific | Cat# A10266 |
| 4′,6-Diamidine-2′-phenylindole dihydrochloride (DAPI) | Merck Sigma-Aldrich | Cat# D9542-5MG; CAS: 28718-90-3 |
| 1,4-Diazobicyclo-[2.2.2]-octane (DABCO) | Merck Sigma-Aldrich | Cat# D27802-25G; CAS: 280-57-9 |
| Glycerol, anhydrous, ≥99.5% (GC) | Honeywell | Cat# 49770; CAS: 56-81-5 |
| Mowiol® 4-88 | Merck Sigma-Aldrich | Cat# 813381-50G |
| UltraPure™ DNase/RNase-Free Distilled water | Thermo Fisher Scientific | Cat# 10977-035 |
| Accutase | Biowest | Cat# L0950 |
| LipofectamineTM RNAiMAX | Thermo Fisher Scientific | Cat# 13778075 |
| Deposited data | ||
| Raw and analyzed images | This paper | Zenodo DOI (https://doi.org/10.5281/zenodo.8014997) |
| Fiji-script to convert files to TIF images (00_Convert Files to TIF) | This paper, adapted from the script (Export_Images_Folder.ijm) from Olivier Burri, EPFL | https://gist.github.com/lacan/16e12482b52f539795e49cb2122060cc |
| Fiji-script (01_Shading-Dark correction_v2.ijm) | This paper | Zenodo DOI (https://doi.org/10.5281/zenodo.8014997 |
| Fiji-script (02_StarDist-QIBC_v12.ijm) | This paper | Zenodo DOI (https://doi.org/10.5281/zenodo.8014997 |
| Canava (SpotFire) for QIBC multivariate representations | This paper | Zenodo DOI (https://doi.org/10.5281/zenodo.8014997 |
| Step-by-step Fiji-script tutorials and video | This paper | Zenodo DOI (https://doi.org/10.5281/zenodo.8014997 |
| Experimental models: Cell lines | ||
| U-2 OS | RRID: CVCL_0042 | |
| Oligonucleotides | ||
| Non-targeting Pool siRNA (siCTL) | Dharmacon | Cat# D-001810-10-05 |
| ON-TARGET plus Smart pool Human TP53BP1 siRNA (si53BP1) | Dharmacon | Cat# L-003548-00-0010 |
| Software and algorithms | ||
| MetaMorph Microscopy Automation and Image Analysis Software (Version 7.10.4.407) | Molecular Devices | RRID:SCR_002368 |
| Scan slide interface for MataMorph | Gataca Systems® | nD-SCAN |
| Fiji (ImageJ version > 1.53d)ji | RRID:SCR_002285 https://imagej.net/software/fiji/downloads | |
| Fiji plugin for StarDist - Object Detection with Star-convex Shapes15 |
https://github.com/StarDist/StarDist https://imagej.net/StarDist |
|
| Fiji plugin for CSBDeep (Require to use StarDist) | https://github.com/CSBDeep/CSBDeep_fiji | |
| SpotFire (Desktop Version 11.4.2) | Tipco | RRID:SCR_008858 |
| Prism (Version v9.4.0) | GraphPad | RRID:SCR_005375 |
| Other | ||
| CO2 incubator, temperature 37°C, 5% CO2, 95% humidity, HeraCell 150 | Thermo Fisher Scientific | Cat# 51026282 |
| DMEM with 4,5 g/L of glucose | Biowest | Cat# L0101-500 |
| Fetal bovine serum (FBS) | Thermo Fisher Scientific | Cat# 10270 |
| 2 mM L-glutamine | EUROBIO | Cat# CSTGLU00-0U |
| 10 UI/mL Penicillin, 100 μg/mL Streptomycin | EUROBIO | Cat# CABPES01-0U |
| Accutase | Biowest | Cat# L0950 |
| Automated cell counter TC20 | Bio-Rad | Cat# 1450102 |
| Cell counting dual-chamber slide | Bio-Rad | Cat# 1450003 |
| 6-well tissue culture plates | TPP | Cat# 9206 |
| Conical tubes, 15 mL and 50 mL | TPP | Cat# 191015 and 19050 |
| Pipets P1000, P200, P20, P10 | Gilson | |
| Vacuum pump | Rocker | Cat# 169300-22 |
| Round coverslip, 22 mm diameter, #1½ (0.16–0.19 mm) | Electron Microscopy Sciences | Cat# EM-72224-01 |
| GSR D1® gamma irradiator | Gamma-Service Medical GmbH | |
| Humidity chamber | StainTray M920 | Cat# 631-1923 |
| Microscopy slides Epredia™ SuperFrost Plus™ Adhesion slides | Thermo Fisher Scientific | Cat# 10149870 |
| Autofluorescent plastics slides | Chroma | Cat# 92001 |
| Transparent nail polish | Mavala | colorless N°43 |
| NuPAGE Novex 4%–12% Bis-Tris pre-cast gels | Thermo Fisher Scientific | Cat# NW04120BOX |
| NuPAGE Transfer Buffer | Thermo Fisher Scientific | Cat# NP0006 |
| Nitrocellulose membrane | Amersham Protran | Cat# 10600001 |
| Widefield inverted microscope DMI6000B with Adaptive Focus Control system | Leica Microsystems | Cat# 11888945 |
| HC PL APO 40×/0,95 CORR objective | Leica Microsystems | Cat# 506414 |
| Lumen 200S Fluorescence Illumination System | Prior Scientific | Cat# L200S |
| Motorized scanning stage for DMI6000B | Märzhäuser | Cat# SCAN IM 127x83 |
| Two fast filter wheel Lambda 10-3, one for excitation and one for emission | Sutter Instrument® | Cat# LB10-3 |
| Quad Sedat D/F/Cy3/Cy5 band dichroic mirror | Semrock®/AHF analysentechnik AG | Cat# F66-988 |
| Band-pass filter for DAPI excitation AT350/50× | Chroma Technology/AHF analysentechnik AG | Cat# F34-000SG |
| Band-pass filter for FITC excitation ET490/20× | Chroma Technology/AHF analysentechnik AG | Cat# F49-490SG |
| Band-pass filter for CY3 excitation ET555/25× | Chroma Technology/AHF analysentechnik AG | Cat# F49-555SG |
| Band-pass filter for CY5 excitation ET645/30× | Chroma Technology/AHF analysentechnik AG | Cat# F49-645SG |
| Band-pass filter for DAPI emission ET455/50m | Chroma Technology/AHF analysentechnik AG | Cat# F47-456 |
| Band-pass filter for FITC emission ET525/36m | Chroma Technology/AHF analysentechnik AG | Cat# F47-526 |
| Band-pass filter for CY3 emission ET605/52m | Chroma Technology/AHF analysentechnik AG | Cat# F47-606 |
| Band-pass filter for CY5 emission ET705/72m | Chroma Technology/AHF analysentechnik AG | Cat# F47-705 |
| ORCA-Fusion Digital sCMOS camera | Hamamatsu® | Cat# C14440-20UP |
Materials and equipment
Image acquisition system
-
•
A widefield inverted microscope allowing for the acquisition of large number of nuclei per image. It is equipped with an adaptive focus control system that detects the surface of the coverslip to correct focal drift throughout multidimensional acquisition.
-
•
A dry 40× objective, plan Apo correction, numerical aperture 0.95 was selected. The use of a dry objective facilitates XY stage displacement. The high numerical aperture for the dry objective allows for smaller structures to be visualized, protein foci in this particular protocol. Coverslips with 0.16–0.19 mm (#1½) thickness are used to match the objective coverslip correction.
-
•
The Lumen 200S Fluorescence Illumination System is the high-power metal halide fluorescent lamp used as the light source.
-
•To reduce pixel shift between wavelengths and optimize multi-wavelength acquisition, our system is equipped with two fast filter wheels and a quad band dichroic mirror mounted on an empty bloc filter adapted for the widefield inverted microscope. Excitation and emission filters bandpass are:
-
•DAPI filters (AHF-Chroma, excitation 350/50, emission 455/50).
-
•FITC filters (AHF-Chroma, excitation 490/20, emission 525/36).
-
•Cy3 filters (AHF-Chroma, excitation 555/25, emission 605/52).
-
•Cy5 filters (AHF-Chroma, excitation 645/30, emission 705/72).
-
•
-
•
ORCA-Fusion Digital sCMOS camera with pixel size of 6.5 μm2 was combined with the selected objective for maximum image resolution. The effective pixel size on the image is 0.1625 μm.
-
•
Motorized stage enabling automatic capture of multiple fields of view (XY dimension, no Z series are captured). The nD-SCAN module is the software implemented in the imaging software MetaMorph® (Version 7.10.4.407) to set up the 2D scanning area during multidimensional acquisition.
Alternatives: Acquisition systems including widefield inverted microscope implemented with an autofocus system, a motorized scanning stage, fast filter wheel and a high-resolution digital camera is compatible with multidimensional acquisition for QIBC.
DMEM Complete Medium for cell culture
| Reagent | Final concentration | Amount |
|---|---|---|
| DMEM with 4,5 g/L of glucose | N/A | 440 mL |
| Fetal Bovine Serum (FBS) | 10% (v/v) | 50 mL |
| 2 mM L-glutamine | 2 mM | 5 mL |
| mix of Penicillin and Streptomycin | 10 UI/mL and 100 μg/mL respectively | 5 mL |
| Total | N/A | 500 mL |
-
•
10 mM EdU stock solution: dissolve 50 mg of EdU in 20 mL of DMSO.
-
•
10 mM camptothecin (CPT) stock solution: dissolve 100 mg of CPT in 28.7 mL of DMSO.
-
•
4% and 3% Paraformaldehyde (PFA) Fixation solution: dilute 1 volume of PFA 16% (v/v) in 3 volumes of Phosphate Buffered Saline (PBS) or in 4.33 volumes of PBS respectively. The PAF-PBS fixation solution requires extemporaneous preparation. Unused 16% PFA can be stored at -20°C for several weeks and should not be refrozen after thawing.
CRITICAL: paraformaldehyde is toxic. Please read the MSDS before working with this chemical. Gloves and safety glasses should be worn and solutions must be handled inside a fume hood.
CSK buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| PIPES pH 6.8 (100 mM) | 10 mM | 5 mL |
| NaCl (2.5 M) | 100 mM | 2 mL |
| Sucrose (2 M) | 300 mM | 7.5 mL |
| MgCl2 (1 M) | 3 mM | 150 μL |
| cOmpleteTM, Mini, EDTA-free Protease Inhibitor Cocktail | N/A | 1 tablet |
| H2O | N/A | 35.35 mL |
| Total | N/A | 50 mL |
-
•
CSK-Triton buffer: CSK buffer supplemented with 0.5% Triton X-100.
-
•
1 mM Alexa-Fluor-488-Azide stock solution: dissolve 5 mg of Alexa-Fluor-488-Azide in 0.58 mL of DMSO. Store in aliquots at −20°C for up to one year, protect from light.
-
•
1 M Sodium ascorbate solution: dissolve 198.11 mg of sodium ascorbate in 1 mL PBS, requires extemporaneous preparation. Limit air exposure and place on ice until use.
Click-it reaction mix
| Reagent | Final concentration | Amount |
|---|---|---|
| PBS | N/A | 897.5 μL |
| Sodium ascorbate (1 M) | 100 mM | 100 μL |
| CuSO4 (1 M) | 2 mM | 2 μL |
| Alexa-Fluor-488-Azide (1 mM) | 0.5 μM | 0.5 μL |
| Total | N/A | 1000 μL |
-
•
2% BSA saturation buffer: dissolve 2 g of BSA in 100 mL PBS, requires extemporaneous preparation.
Mowiol-mounting media
| Reagent | Final concentration | Amount |
|---|---|---|
| Glycerol | 25% (w/v) | 6 g |
| Mowiol 4-88 | 100 g/L | 2.4 g |
| Tris-HCl 0.2 M, pH 8.5 | 0.1 M | 12 mL |
| DABCO | 2.5% (w/v) | 0.6 g |
| UltraPure™ DNase/RNase-free distilled water | N/A | 6 mL |
| Total | N/A | 24mL |
Note: put 6 g of glycerol in a 50 mL plastic centrifuge tube and a small stir bar, add 2.4 g Mowiol 4–88 and stir to mix. While stirring, add 6 mL of UltraPure™ DNase/RNase-free distilled water and leave for 2 h at 18°C–22°C. Add 12 mL of 0.2 M Tris-HCl buffer, pH 8.5. The final volume will be 24 mL. Incubate the tube in a 50°C water bath for 10 min, repeat several times until the majority of the Mowiol powder goes into solution (this can take several hours). Add 2.5% (w/v) DABCO and vortex until dissolved. Centrifuge at 5000 g for 15 min to remove any undissolved solids. Carefully remove the supernatant and aliquot into 1.5 mL tubes. Store in aliquots at −20°C. The refractive index (RI) of Mowiol-mounting media should be close to 1.4. For quality control, RI of frozen Mowiol-mounting media should be compared with a refractometer to that of the freshly prepared batch with a refractometer. Mowiol-mounting media RI varies between batches and increases during hardening.
-
•
DAPI stock solution: dissolve 5 mg of DAPI in 1 mL of UltraPure™ DNase/RNase-free distilled water. Store in aliquots at −20°C for up to one year.
-
•
1 μg/mL DAPI working solution: dilute the DAPI stock solution by a factor of 5000 in PBS, requires extemporaneous preparation.
Step-by-step method details
Part 1: Cells preparation
Plate cells on coverslips for the negative controls and the experimental samples.
Note: Activation of the DNA-damage response (DDR) consists in a cascade of events initiated by the loss of DNA integrity. At the center of the DDR is the chromatin component histone 2A variant (H2AX) that is phosphorylated on the serine residue 139 forming the DNA damage mark γH2AX. Gamma irradiation-induced DNA damages activates PI3K-like kinases, a family including ataxia telangiectasia mutated (ATM), ataxia telangiectasia and Rad3-related (ATR) and DNA-dependent protein kinase (DNA-PK),21,25 which in turn leads to γH2AX foci formation (Figure 1). The histone mark γH2AX is also induced by perturbation of the DNA replication process, known as replication stress. Treatment with agents that deplete the nucleotide pool (hydroxyurea), inhibit DNA polymerases (aphidicolin), induce DNA crosslinks and bulky lesions (cisplatin) or target topoisomerase I and II (camptothecin and etoposide, respectively), impair the progression of DNA replication forks. The pattern of γH2AX signal can vary from the formation of discrete foci to global nuclear staining referred to as pan-nuclear staining (Figure 2). Spreading of the γH2AX mark over large chromosomic regions surrounding an unrepaired double-strand break (DSB) promotes chromatin binding of other DDR factors including the TP53-binding protein 1 (53BP1)26,27 (Figures 3 and 4). The recruitment of 53BP1 at DSB involves several reader domains interacting with specific histone marks.22,28 53BP1 recruitment at damaged DNA involves direct interaction of the two 53BP1 BRCA-carboxyterminal (BRCT) repeats with locally induced γH2AX29,30 and the recognition by the ubiquitin-dependent motif (UDR) of the DNA damage-induced ubiquitylated H2AK15 marks.31 The TUDOR domain of 53BP1 can interact with the di-methylated histone H4 (H4K20me2)32,33 that are unmasked in response to DNA damage34,35 and the dilution of H4K20me2 with H4K20me0 during DNA replication reduces the recruitment 53BP1 to DBSs in the S/G2 phase.36,37 These DNA damage specific histone marks along with the 53BP1 oligomerization38,39 domain and liquid–liquid phase separation (LLPS) condensates40,41,42 stimulate the assembly of 53BP1 compartments around the damaged chromatin, and these biomolecular condensates are proposed to provide an environment protecting from DNA end-resection that favors DSB repair by non-homologous end-joining (NHEJ) during the G1 phase of the cell-cycle.
-
1.
Start with a logarithmically growing U-2 OS cells culture.
-
2.
Remove the medium and rinse once with PBS.
-
3.
Cover the cells with Accutase to dissociate the cells.
-
4.
Count the cells with a BioRad cell counter.
-
5.
Place one 22 mm diameter coverslip per well of a 6 wells plate.
-
6.
Rinse the coverslip with 2 mL of PBS, aspirate the PBS so that the coverslips hold by capillary action to the bottom of the well. Seed 105 cells per well in 2 mL of cell culture medium.
-
7.
Incubate the cells in a CO2 incubator for 48 h.
Note: The number of plated cells need to be optimized for each cell line to ensure a log-phase growth on the day of the EdU pulse.
Figure 1.

IR-induced γH2AX foci number is dependent on the cell’s DNA content
Asynchronously growing U-2 OS cells were left untreated or treated with 2 Gy of IR and allowed to recover for 30 min. Cells were subsequently treated with extraction buffer (CSK-Triton 0.5%) before fixation with paraformaldehyde, staining for DNA content, EdU and γH2AX signal for quantitative image-based cytometry analysis. (A) Representative images for DAPI, EdU and γH2AX (invert LUT) detection are presented. Right panels: nuclei segmented with the in house developed Fiji-script (02_StarDist-QIBC_v12.ijm) are outlined in yellow and the nuclei cell-cycle phase is indicated. The full sizes of acquired images is 2034 × 2034 pixels, an 800 × 800 pixels sector is presented for each wavelength. Scale bar: 20 μM. (B) Multivariate scatter plots displaying QIBC analysis from (A) representing the relationship among nuclei total DAPI intensity (X-axis), mean EdU intensity (Y-axis) and γH2AX foci number (color gradient). The number of analyzed nuclei (n) is indicated for each sample. Samples for which the EdU pulse was omitted are used to position the horizontal line delineating the EdU negative nuclei (G1 and G2) from the EdU positive (S-phase). (C) Number of γH2AX foci per nuclei as a function of the cells-cycle phase from untreated cells or cells treated with 2 Gy of IR. The bounds of the box are 25–75th percentile, center shows the median, whiskers indicate the 5–95 percentiles and the data points outside of this range are drawn as individual dots. Statistical analysis was performed with Prism (GraphPad) software. Data were analyzed with the two-tailed non-parametric Kruskal-Wallis test followed by a two-stage linear set-up procedure of Bonferroni, Kreiger and Yekutieliwas.
Figure 2.

γH2AX activation in response to camptothecin is restricted to S-phase cells
Asynchronously growing U-2 OS cells were left untreated or treated with 1 μM of camptothecin (CPT) for 1 h. Cells were subsequently treated with extraction buffer (CSK-Triton 0.5%) before fixation with paraformaldehyde and staining for DNA content, EdU and γH2AX signal for quantitative image-based cytometry analysis. (A) Representative images for DAPI, EdU and γH2AX (invert LUT) staining are presented. Right panels: nuclei segmented with the in house developed Fiji-script (02_StarDist-QIBC_v12.ijm) are outlined in yellow and nuclei cell-cycle phase is indicated. The acquired images full size of acquired images is 2034 × 2034 pixels, an 800 × 800 pixels sector is presented for each wavelength. Scale bar: 20 μM. (B) Multivariate scatter plots displaying QIBC analysis from (A) representing the relationship among nuclei total DAPI intensity (X axis), mean EdU intensity (Y axis) and γH2AX mean intensity (color gradient). Number of analyzed nuclei (n) is indicated for each sample. (C) γH2AX signal intensity as a function of the cell-cycle phase in nuclei of untreated cells or cells treated with CPT. The bounds of the box are 25–75th percentile, center shows the median, whiskers indicate the 5–95 percentiles and the data points outside of this range are drawn as individual dots. Statistical analysis was performed with Prism (GraphPad) software. Data were analyzed with the two-tailed non-parametric Kruskal-Wallis test followed by a two-stage linear set-up procedure of Bonferroni, Kreiger and Yekutieliwas.
Figure 3.

53BP1 foci formation does not affect the 53BP1 signal intensity
Asynchronously growing U-2 OS cells were left untreated or treated with 2 Gy of IR and allowed to recover for 30 min. Cells were subsequently fixed with paraformaldehyde and then permeabilization with PBS-Triton 0.2% and stained for DNA content, EdU and 53BP1 signal for quantitative image-based cytometry analysis. (A) Representative images for DAPI, EdU and 53BP1 (invert LUT) staining are presented. The full size of acquired images is 2034 × 2034 pixels, an 800 × 800 pixels sector is presented for each wavelength. Scale bar: 20 μM. (B and C) Multivariate scatter plots displaying QIBC analysis representing the relationship among nuclei total DAPI intensity (X axis), mean 53BP1 intensity (Y axis) and 53BP1 foci number (color gradient) for cells left untreated or treated with 2 Gy of IR as indicated. The negative controls (in B) are the samples incubated only with the anti-Mouse Alexa-Fluor-647 secondary antibody, whereas the 53BP1 antibody staining (in C) corresponds to the experimental samples first incubated with the 53BP1 primary antibody followed by an incubation with the anti-Mouse Alexa-Fluor-647 secondary antibody. The number of analyzed nuclei (n) is indicated for each sample. (E) For the same cells shown in (D), the 53BP1 foci counts per nucleus were normalized at the single cell level to DNA content to account for increasing damage load with increasing DNA amount. (F) Box-plot representation of the number of 53BP1 foci counts per nucleus normalized to the nucleus DNA content in the indicated condition and cell-cycle phase. The bounds of the box are 25–75th percentile, center shows the median, whiskers indicate the 5–95 percentiles and the data points outside of this range are drawn as individual dots. Statistical analysis was performed with Prism (GraphPad) software. Data were analyzed with the two-tailed non-parametric Kruskal-Wallis test followed by a two-stage linear set-up procedure of Bonferroni, Kreiger and Yekutieliwas.
Figure 4.

decreased 53BP1 foci formation by 53BP1 siRNA
Asynchronously growing U-2 OS cells were left untransfected or transfected with control siRNA (siCTL) or 53BP1 siRNA (si53BP1) Individual siRNA transfections were performed for 72 h with siRNA from Dharmacon using Lipofectamine RNAiMAX. The siRNAs used were the Non-targeting Pool (siCTL); ON-TARGET plus Smart pool Human TP53BP1 siRNA (si53BP1). Cells were subsequently fixed with paraformaldehyde and then permeabilization with PBS-Triton 0.2% and stained for DNA content, EdU and 53BP1 signal for quantitative image-based cytometry analysis. (A) Representative images for DAPI, EdU and 53BP1 (invert LUT) staining are presented. The full size of acquired images is 2034 × 2034 pixels, an 800 × 800 pixels sector is presented for each wavelength. Scale bar: 20 μM. (B) 53BP1 level assessed by immunoblotting. Individual siRNA transfections were performed for 72 h with the indicated siRNA. Cells were lysed in 8 M urea, 50 mM Tris HCl, pH 7.5 and 150 mM β-mercaptoethanol, sonicated and heated at 90°C for 10 min. Samples were subjected to electrophoresis in NuPAGE Novex 4–12% Bis-Tris pre-cast gels (Thermo Fisher Scientific, Catalog # NW04120BOX), proteins were subjected to western transfer onto the nitrocellulose membrane with the NuPAGE Transfer Buffer. Primary antibodies for immunoblotting were used the following concentrations: mouse anti-53BP1 antibody (1:1000), mouse anti-α-Tubulin antibody (1:1000) and secondary horseradish peroxidase conjugated goat anti-mouse IgG antibody (1:10000). (C) Multivariate scatter plots of QIBC analysis presenting the relationship among nuclei total DAPI intensity (X-axis) and 53BP1 foci counts per nucleus normalized at the single cell level to DNA content (Y-axis). The untransfected sample was incubated only with the anti-Mouse Alexa-Fluor-647 secondary antibody, whereas the siCTL and the si53BP1 experimental samples were first incubated with the 53BP1 primary antibody followed by an incubation with the anti-Mouse Alexa-Fluor-647 secondary antibody. Number of analyzed nuclei (n) is indicated for each sample. (D) Box-plot representation of the number of 53BP1 foci counts per nucleus normalized to nucleus DNA content for the indicated condition and cell cycle phase. The bounds of the box are 25–75th percentile, center shows the median, whiskers indicate the 5–95 percentiles and the data points outside of this range are drawn as individual dots. Statistical analysis was performed with Prism (GraphPad) software. Data were analyzed with the two tailed non-parametric Kruskal-Wallis test followed by a two-stage linear set-up procedure of Bonferroni, Kreiger and Yekutieliwas.
Part 2: Cells treatments and fixation
The cells are treated and fixed to prepare the negative controls and experimental samples.
-
8.EdU pulseCRITICAL: To position the delimitation between the EdU negative and EdU positive nuclei, an EdU-pulse free negative control should be included (Figure 1).
-
a.Replace the medium with 2 mL of 37°C pre-warm medium containing 25 μM EdU. Store the 10 mM EdU stock in aliquots at −20°C for up to one year, protect from light.
-
b.Incubate the cells for 8 min in the CO2 incubator.
-
c.Remove the EdU containing medium.
-
d.Rince the cells once with 37°C pre-warmed fresh medium and proceed with cells treatment with camptothecin (step 9a.) or cells irradiation (step 9b.).
-
a.
-
9.Cells treatment.
-
a.Cells treatment with camptothecin (CPT).CRITICAL: Camptothecin is a carcinogen and toxic if swallowed.
-
i.Add 2 mL of pre-warmed fresh medium supplemented with 1 μM CPT for the treated cells or 2 mL of pre-warmed fresh medium without CPT for untreated (control) sample. Store the 10 mM CPT stock in aliquots at −20°C for up to three months, protect from light.
-
ii.Incubate the cells for 1 h in the CO2 incubator at 37°C.
-
iii.Remove the medium.
-
iv.Place the culture plate on an ice bed and proceed with step 10.
-
i.
-
b.Cells irradiation at 2 Gy.
-
i.Add 2 mL of pre-warmed fresh medium and proceed with the 2 Gy irradiation, the irradiation step is omitted for the unirradiated (control) sample.
-
ii.Incubate the cells for 30 min in the CO2 incubator.
-
iii.Remove the medium.
-
iv.Place the culture plate on an ice bed and proceed with step 10.
-
i.
-
a.
-
10.Cell fixation for γH2AX detection or 53BP1 detection.Note: culture plate containing the coverslip should remain on ice bed for step 10.CRITICAL: For the detection of strongly chromatin-bound proteins or chromatin components including γH2AX, nuclear soluble-proteins could be pre-extracted by treating the cells with Triton-containing buffer prior to paraformaldehyde fixation. For this procedure, users are invited to continue with step 10.a. For the detection of proteins that are forming nuclear condensates and/or, are associated with chromatin (including 53BP1), or for the detection of nuclear soluble proteins, cells should be fixed with paraformaldehyde prior to permeabilization to avoid protein wash out (also see the troubleshooting section). For this, users are invited to continue with step 10.b.
-
a.Pre-extraction and fixation for γH2AX detection.
-
i.Rince the cells twice with 2 mL of ice-cold PBS.
-
ii.Rince the cells with 2 mL of ice-cold CSK buffer. Store the CSK buffer in aliquots at −20°C for several weeks.
-
iii.Cells are permeabilized for 5 min with 2 mL of ice-cold CSK-T buffer with agitation on orbital shaker at 45 rpm. Store the CSK-T buffer at −20°C for several weeks.
-
iv.Wash the cells with 2 mL of ice-cold CSK buffer for 1 min with agitation (45 rpm).
-
v.Rince the cells once with 2 mL of PBS at 18°C–22°C.
-
vi.Incubate the cells in 1.5 mL of freshly prepared PFA 4% (v/v) fixation solution for 20 min at 18°C–22°C without shaking.
-
vii.Wash the cells three times for 5 min with 2 mL of PBS at 18°C–22°C .
-
viii.Proceed with cells staining (Part 2).
 Pause point: Alternatively, fixed cells can be preserved in PBS at 4°C for up to a week.
Pause point: Alternatively, fixed cells can be preserved in PBS at 4°C for up to a week.
-
i.
-
b.Fixation and permeabilization for 53BP1 detection.
-
i.Rince the cells twice with 2 mL of ice-cold PBS.
-
ii.Incubate the cells in 1.5 mL of freshly prepared PFA 3% (v/v) fixation solution for 15 min at 18°C–22°C without shaking.
-
iii.Wash the cells three times for 5 min with 2 mL of PBS at 18°C–22°C .
-
iv.Cells are permeabilized for 5 min with 2 mL of PBS supplemented with 0.2% Triton X-100 at 18°C–22°C with agitation (45 rpm).
-
v.Wash the cells with 2 mL of PBS with agitation (45 rpm) at 18°C–22°C .
-
vi.Proceed with cells staining (Part 2).Pause point: Fixed cells can be preserved in PBS at 4°C for up to a week.
-
i.
-
a.
Part 3: Sample staining
EdU staining and immunolabeling of the negative controls and the experimental samples.
-
11.Click-it reaction for EdU detection.Note: Samples are protected from light during incubation time.
-
a.Transfer coverslips in the humidity chamber with the side containing the cells facing up.
-
b.Cover the sample with 250 μL of freshly prepared Click-it solution, incubate for 1 h at 18°C–22°C.
-
c.Wash the coverslip once with PBS (0.5 mL/cm2) for 5 min with agitation (45 rpm).Note: For convenience washes are performed in 6-well tissue culture plates in 2 mL of PBS for 22 mm diameter coverslip.
-
a.
-
12.
Samples are saturated with 250 μL of freshly prepared 2% BSA saturation buffer for 16 h at 4°C.
Note: Alternatively, the samples can be incubated 1 h at 18°C–22°C in 2% BSA saturation buffer.
-
13.
Wash 2 times with 2 mL PBS for 5 min with agitation (45 rpm).
-
14.
When γH2AX is detected, cover the sample with 250 μL of anti-γH2AX antibody at 1:500 dilution in freshly prepared 2% BSA saturation buffer. When 53BP1 is detected, cover the sample with 250 μL of anti-53BP1 antibody at 1:750 dilution in freshly prepared 2% BSA saturation buffer.
-
15.
Sample covered with the primary antibody dilution are incubated in the humidity chamber for 1 h at 18°C–22°C .
Note: Alternatively, when samples are incubated in 2% BSA saturation buffer for 1 h (step 12), then incubate the samples 16 h at 4°C with the primary antibody dilution.
-
16.
Wash 2 times with 2 mL PBS for 5 min with agitation (45 rpm).
-
17.
Incubate the sample with 250 μL of freshly prepared anti-Mouse Alexa Fluor 647 secondary antibody diluted at 1:500 in 2% BSA saturation buffer, for 1 h at 18°C–22°C, in the humidity chamber.
Note: The present protocol describes single protein immunofluorescence staining. However, the same protocol can be applied for dual protein staining if the cell preparation procedure for immunostaining is compatible with two proteins detection (step 10). For immunostaining, the sample is incubated with a mix of the two primary antibodies specifically directed against each protein and raised in different host species (step 14). The sample is then incubated with a mix of the two secondary antibodies (step 17) directed against the corresponding host and coupled to distinct fluorophore.
-
18.
Wash with 2 mL of PBS 2 times for 5 min with agitation (45 rpm).
-
19.
Label the nuclei with 1.5 mL of freshly prepared 1 μg/mL DAPI working solution for 20 min at 18°C–22°C in the dark. Store the 5 mg/mL DAPI stock solution in aliquots at −20°C for up to one year.
-
20.
Wash with 2 mL PBS 2 times for 5 min with agitation (45 rpm).
-
21.
Dispense 20 μL of Mowiol mounting medium for fluorescence on the top of a microscope slide and mount the coverslips with cells in contact with the mounting medium. Mowiol mounting media is stored in aliquots at −20°C for up to 1 year.
-
22.
Seal the edges of the slides with nail polish to prevent drying.
-
23.
Proceed with sample imaging step 24.
Part 4: Sample imaging for quantitative image-based cytometry
-
24.Start the acquisition system.
-
a.Turn on the fluorescence lamp, the microscope and the camera at least 20 min before starting the session to allow for the system to stabilize.
-
b.Let the sample slides equilibrate at 18°C–22°C for 20 min before the acquisition to reduce drift due to temperature variation between the microscope room and the slides.
-
c.Select the 40× dry objective and adjust the focus on the DAPI signal.
-
d.Run the stage calibration.
-
a.
Note: Performing the stage calibration prevents contiguous images overlapping during X and Y displacement of the stage when performing software-controlled automatic acquisition of multiple fields of view.
-
25.
Select the appropriate filters for sequential fluorescent signal acquisition starting with the longest and ending with the shortest wavelength.
Note: To reduce pixel shift between wavelengths and optimize multiwavelength acquisitions, two filter wheels equipped with single-band filters are used in combination with a quad dichroic mirror.
-
26.
Adjust the exposition time for each wavelength such that the maximum dynamic range is filled to 80%.
Note: For a camera in 16-bits, the grayscale pixel values should range from 0 to 52 000.
-
27.
Move the objective to the center of the coverslip.
-
28.
Randomly define 10 × 10 images acquisition zone around this position.
-
29.
Activate the hardware adaptive focus control.
-
30.
Start the acquisition.
-
31.
Sample shading images are generated for each experimental wavelength at the end of the acquisition session.
Note: Heterogeneous field illumination due to widefield acquisition needs to be corrected for accurate quantitative QIBC analysis. Field illumination correction requires shading images for the execution of the Fiji-script “01_Shading-Dark correction_v2.ijm” (step 35). The shading images are generated for each experimental wavelength by averaging three frames and are obtained by imaging an autofluorescent plastic slide (Chroma®) at an exposure time and lamp power adjusted such that grayscale pixel values do not exceed 52 000 for a camera in 16 bits.
-
32.
Dark-current images are generated for each experimental wavelength at the end of the acquisition session.
Note: Correction to remove the noise intensity value coming from the electronic of the camera requires dark-current images for the execution of the Fiji-script “01_Shading-Dark correction_v2.ijm” (step 35). Dark-current images are generated for each experimental wavelength by averaging three frames and are acquired for each wavelength with the fluorescent lamp switch OFF with the exposure time used during samples acquisition.
Part 5: Image analysis and data extraction
Note: Proceed directly to step 34 if the images files extensions are “.tif” or “.TIF” and that each channel is saved as a separate image file which name includes the letter “C” or “c” followed by the channel number (C1, C2, C3, C4 or c1, c2, c3, c4).
-
33.Execute the Fiji-script “00_Convert Files to TIF”.
-
a.drag and drop the script file in Fiji and click on the RUN button.
-
b.Select the folder containing the microscope file.
-
c.Select from the list the type of microscope image files.
-
d.A folder is automatically created where each channel image file is saved individually with the extension (c1, c2, c3, c4). From this step, you can start the analysis with the two Fiji-scripts for QIBC analysis.
-
a.
Note: If foci count per nucleus is to be extracted with the “Number of Foci/Spots” Fiji-script function, then proceed with step 34 to determine the prominence value that is the value used to define local maxima where a maxima is considered as a foci/spot. Otherwise go directly to step 35.
-
34.Determine the prominence value for foci/spot count.
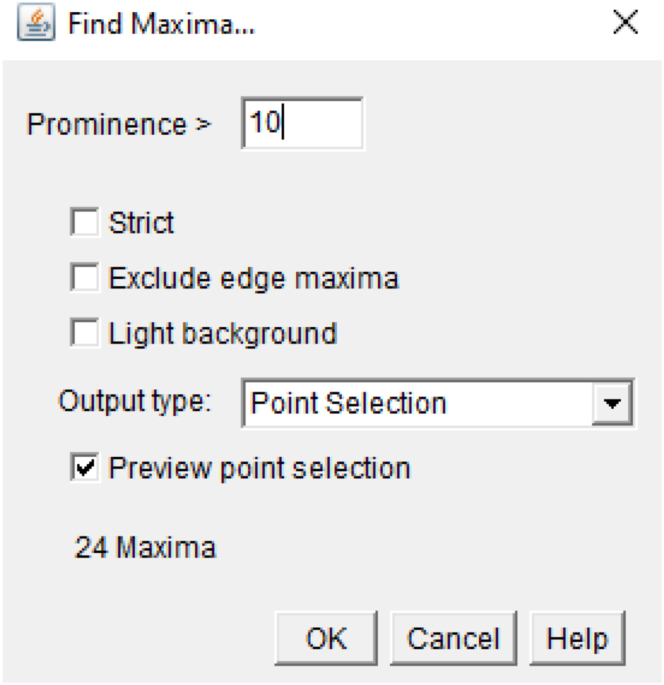
-
a.Open a channel image from which the foci/spot count is to be extracted.
-
b.Apply background subtraction: go to Process>Subtract background (rolling ball: 20; uncheck light background).
-
c.Draw a region of interest (ROI) containing one nucleus, zoom in to facilitate foci/spot visualization.
-
d.Open the maxima function: go to Process>Find Maxima, uncheck light background, for the output type select Point Selection, check Preview point selection to directly highlight the maxima from the selected nucleus.
-
e.Adjust the prominence value such that all visualized foci/spots are adequately detected by the Find Maxima function.
-
f.Repeat this process on 4 to 5 channel images of an experimental condition to confirm that the selected prominence value allows for the detection of foci/spot.
-
g.Verify that the selected prominence value is not leading to fauls positive foci/spot by running the Find Maxima function on the experimental negative control images (samples where the EdU pulse was omitted or sample where the primary antibody was omitted).
-
h.Repeat steps 34 for all channel images from which the foci/spot count is to be extracted.
-
a.
-
35.Execute the Fiji-script “01_Shading-Dark correction_v2.ijm”.
-
a.drag and drop the script file in Fiji and click on the RUN button.
-
b.Define the analysis settings in the opening dialogue box:
-
i.Select the image folder using the "Browse" menu.
-
ii.Check the image channel corresponding to the experiment.
-
iii.Specify the channels name. Keep the text box empty for unused channels.
-
iv.If the images are not calibrated, tick the calibration option and specify the pixel size in microns. Otherwise, skip this step.
-
v.Click OK.
-
vi.Users are prompted to "Select a Shading image" and click OK. This step is repeated for each channel.
-
vii.Users are then prompted to "Select a Dark-current image" and click OK. This step is repeated for each channel.
-
viii.Images correction is completed when the message "Done" appears.
-
i.
-
a.


Note: The corrected image files are copied in the sub-folder “Corrected” automatically created by the Fiji-script in the folder containing the raw image files.
-
36.Execute the Fiji-script “02_StarDist-QIBC_v12.ijm”.
-
a.Drag and drop the script file in Fiji and click on the RUN button.
-
b.Define the analysis settings in the opening dialogue box:
-
i.In the “Image directory” select the “Corrected” image folder generated step 35 using the "Browse" menu.
-
ii.Specify the name of the analyzed condition (e.g., Untreated, Irradiated).Note: The selected name is attributed to the results containing excel spread sheet generated by the analysis.
-
iii.From the list of numbers, select the number corresponding to the DAPI signal channel.
-
iv.Specify the minimum nucleus diameter, only objects with a calculated surface area above the indicated specification are included in the analysis.Note: Defaults minimum nucleus diameter is set at 8 μm and objects area smaller than 50.2 μm2 are ignored from the analysis.
-
v.When "Add manual step for selection of nucleus ROI" is ticked, users are prompted to manually validate, for each image of the folder the nucleus ROI detected by StarDist.Note: This step allows for manual elimination from the nuclei ROI list of any miss-segmented objects. Untick this box for fully automated analysis. Information about the objects included in the nuclei ROI list are automatically exported in the results table.
-
vi.Select the channels number (1 corresponding to the image C1) to be analyzed, the DAPI channel must not be included.
-
vii.Click OK.Note: in addition to object size filtration using the minimum nucleus diameter parameters (iv), objects that are in contact with the edge of the image are automatically excluded.
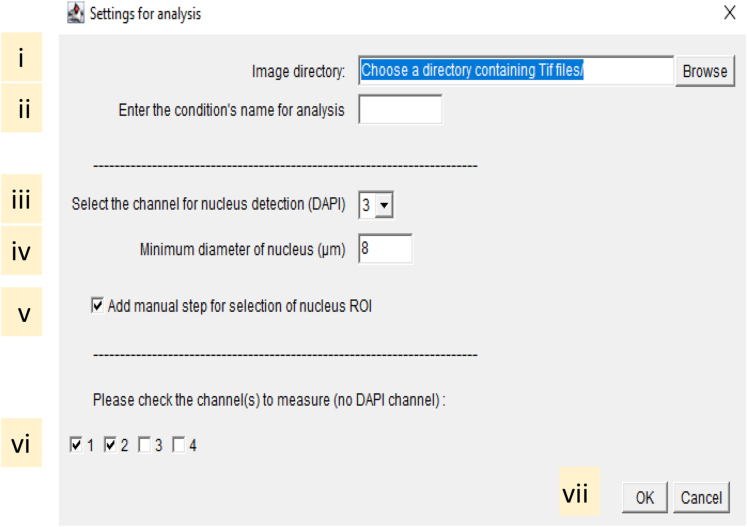
-
i.
-
a.
-
37.
Define the settings for StarDist nuclei segmentation in the opening dialogue box.


Note: From the scrolling menu, select the StarDist model to be applied for nuclei segmentation. Robust nuclei segmentation from U-2 OS cells is obtained by applying the “versatile (fluorescent nuclei)” built-in model, a model adapted for the automated segmentation ovoid-round nuclei, with the default parameters (Percentile low 0, Percentile high 100, Probability score 0.5, 0verlap Threshold 0.4, Number of tiles 1 and Boundary Exclusion 2). Users are referred to the Fiji plugin for StarDist15 web page (https://imagej.net/StarDist) parameters guidelines. Alternatively, users can apply the QIBC analysis on cell lines requiring StarDist integrated models “DSB 2018 (from StarDist 2D paper)”15 or any other open source available model by selecting the “Model (.zip) from File”.
-
38.In the next “Settings for C1 analysis” dialogue window users are prompted to define additional settings related to the analysis of the corresponding channel image.
-
a.In the Channel 1 name (C1), users are prompted to specify a name for the signal analyzed in channel 1 (e.g., EdU, γH2AX).Note: The indicated name is included in the column header’s name in the results excel spread sheet.
-
b.Tick the boxes to validate the parameters to be extracted.
-
c.Tick “Number of Foci/Spots” if necessary.
-
d.When the box “Number of Foci/Spots” is ticked, users must indicate the previously determined prominence value.CRITICAL: Follow the prerequisite section (step 34) to determine the prominence value.
-
e.Click OK.
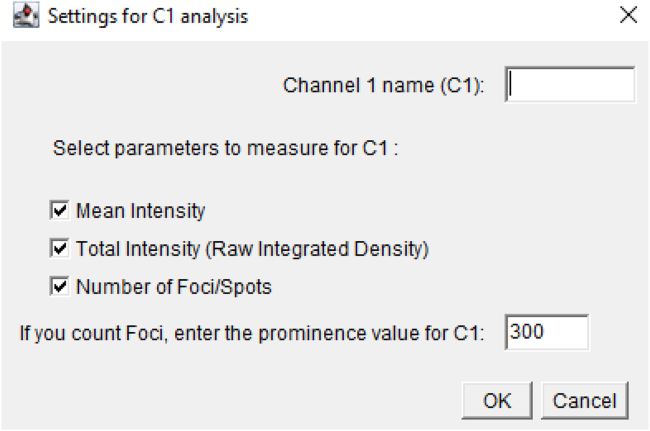
-
a.
-
39.
Repeat step 38 for all other image channels selected (step 36.b.vi.).
-
40.
The Fiji-script automatically executes the QIBC analysis for all corrected images of the selected folder.

Note: When the "Add manual step for selection of nucleus ROI" is ticked (step 36.b.v.), users are invited to validate the segmented objects. Objects from the ROI list can be removed. For this, the object ROI is selected on the image and the “delete” button of the ROI manager window is clicked. The manual editing process is repeated for all images.
-
41.
The fully automated analysis is complete when the message "All images in the folder have been analyzed with StarDist plugin" is displayed.
Note: Automatically extracted parameters are saved in a new Excel spread sheet called “Results.xls” generated during the Fiji-script execution. The analyzed images QIBC Fiji-script extracted parameters are detailed in the Table 1. The “Results.xls” file is saved in the folder “image folder” under the "QIBC_analysis" folder.
-
42.
The Fiji-script execution generates a new “Label-Image” folder.
Note: The “Label-Image” folder generated by the execution of the Fiji-script contains for each analyzed image an image on which StarDist segmented object are colored. The “ROI” folder contains for each image the ROI list in a “.Zip“ extension file. To open the ROI, drag and drop the “.Zip” file into Fiji. The ROI will automatically be loaded to the ROI manager window.
Table 1.
Extracted image parameters reported in the “Results.xls” table
| Column name | Definition | |
|---|---|---|
| General information | Image_Name | The image name |
| Nucleus_Name | ROI number of each segmented nucleus | |
| Measurements in the DAPI image | DAPI_Nucleus_Mean Intensity | Average gray value within the nucleus ROI obtained by taking the sum of the gray values of all pixels in the nucleus ROI divided by the number of pixels |
| DAPI_Nucleus_Total Intensity | Values are the sum of all pixel intensity over the ROI area, this value corresponds the raw integrated density (“RawIntDen”) parameter in Fiji | |
| Nucleus_Area (μm2) | Area of the nucleus | |
| Nucleus_Circularity | Circularity of the nucleus | |
| Nucleus_Aspect Ratio | Aspect ratio of the nucleus | |
| Nucleus_Round | Round of the nucleus | |
| Nucleus_Fit Ellipse Major axis | Major axis length after fitting the nucleus with an ellipse (μm) | |
| Nucleus_Fit Ellipse Minor axis | Minor axis length after fitting the nucleus with an ellipse (μm) | |
| measurements in the other channels according to the selected parameters in the Fiji-script | Channel_Nucleus_Mean Intensity | Average gray value within the nuclei ROI obtained by taking the sum of the gray values of all the pixels in the nuclei ROI divided by the number of pixels |
| Channel_Nucleus_Total Intensity | The sum of all pixel intensity over the ROI area, this value corresponds the raw integrated density (“RawIntDen”) parameter in Fiji | |
| Channel_Nucleus_Number of Spots: Prominance value: | The extracted value is obtained with the maxima function according to the chosen prominence value (step 34) |
Part 6: Multiparametric data visualization with Spotfire® software (TIBCO)
The use of data visualization tool, such as Spotfire, allows users to cross-analyze the protein parameters in the context of the cell cycle at the individual cell level, thus providing in depth information from asynchronous cell populations.
Note: The dot plots of asynchronous cell populations presented in Figures 1, 2, 3, and 4 were generated with Spotfire® software (TIBCO). Cell cycle staging based on total DAPI intensities and Mean EdU intensities was performed to discriminate cells into 3 sub-populations (G1, S and G2). A second multidimensional analysis was then applied on the number of 53BP1 or γH2AX foci or on the mean γH2AX intensities in the context of the cellcycle stage of the cell. For visualizing discrete data in scatterplots, mild jittering (random displacement of data points along the discrete data axes) was applied in order to demerge overlapping data points. A template (canvas) of Spotfire® dedicated to QIBC experiment is available for download on the Zenodo platform. Follow the instructions provided in the video tutorial to execute the QIBC multiparametric data visualization for QIBC experiment (https://doi.org/10.5281/zenodo.8014997).
Expected outcomes
The QIBC pipeline protocol was applied to γH2AX and 53BP1 in the context of the ionizing radiation (IR) induced DNA Damage Response, for which we quantified about twice as many γH2AX foci in G2 irradiated cells as compared with G1 irradiated cells (Figure 1), as previously reported.12 This is consistent with direct relationship between the cell’s DNA content which doubles from G1 to G2 and the numbers of γH2AX foci formation after DNA damage induced by IR. In addition, camptothecin-induced replication stress specifically activated γH2AX in S-phase cells from an asynchronous treated population (Figure 2). We also observed that the 53BP1 protein signal level remains stable after IR and yet the protein is redistributed within the nucleus to form foci (Figure 3). This clearly illustrates the usefulness of the multiparametric comparison offered by the QIBC approach. Finally, our observations recapitulate previous studies12,37 showing that the number of 53BP1 foci normalized to nucleus DNA content is higher in G1 and gradually decline as cells progress through the cell cycle, in both untreated and IR-treaded cells (Figures 3 and 4). Overall, our QIBC pipeline protocol including publicly made available QIBC-scripts for Fiji, is providing a rapid and automated analysis with minimal end-user intervention for unbiased nuclei segmentation and multi-parameter extraction.
Limitations
Unbiased multi-parameters extraction for QIBC analysis relies on robust single nucleus segmentation a function provided by the Fiji-plugin for StarDist that is implemented in the open source QIBC-script for Fiji presented here. StarDist includes multiple deep-learning based models for automated objects segmentation. Applying the “versatile (fluorescent nuclei)” built-in model with default parameters is well adapted for the automated nuclei segmentation of cell lines with ovoid-round nuclei such as the U-2 OS or RPE1-hTERT cells. However, the authors have not yet applied the script to other cell lines. Users who wish to apply this QIBC pipeline to cell lines with non-ovoid-round nuclei are invited to refer to the StarDist plugin web page (https://imagej.net/StarDist) for StarDist parameters and/or for StarDist models selection.
The cross-analysis of DAPI total intensity per nucleus and mean EdU intensity per nucleus parameters allows for cell-cycle staging of individual cells from asynchronously growing adherent cells. Additional information about proteins of interest such as the mean signal intensity per nucleus or protein foci count per nucleus can be extracted and cross analyzed with cell-cycle parameters. Foci count per nucleus can be extracted using the Fiji Find Maxima function with a user defined prominence value. However, the identification of discrete foci in samples showing high intensity, high foci density or heterogeneous foci intensity and density is challenging. This is illustrated in the camptothecin treated sample resulting in global nuclear staining for γH2AX in S-phase cells (see Figure 2). Therefore, users are advised to apply the “Number of Foci/Spots” Fiji-script function with careful consideration.
In this protocol, the Spotfire® software (TIBCO) was used to generate the Multivariate data visualizations presented in the figures. However other analytic platforms for business (including GoodData, Oracle Analytics Server, Zoho Analytics) or the free software environment for statistical computing and graphics R or Google Charts could also be used.
Troubleshooting
Problem 1
The detected DAPI intensity is outside the optimum dynamic range (related to step 26).
Potential solution
-
•
For samples showing high DAPI intensity, the concentration of the DAPI working solution is reduced to meet users working environment and cell line requirements (step 19).
-
•
For samples showing low DAPI intensity, users are invited to increase the exposition time and if necessary, increase the lamp intensity output %.
-
•
The exposition time for all the wavelengths must be adapted such that the maximum dynamic range is filled to 80% (for a camera in 16 bits, the grayscale pixel values should be ranging from 0 to 52 000).
Problem 2
Proteins pre-extraction before fixation step can introduce a bias in the analysis (related to step 10).
Potential solution
-
•
The detection of strongly chromatin-bound proteins or chromatin components including γH2AX, nuclear soluble protein can be pre-extracted by treating the cells with Triton-containing buffer prior to paraformaldehyde fixation (step 10a).
-
•
The detection of proteins that are forming nuclear condensates and/or are associated with chromatin (including 53BP1), or for the detection of nuclear soluble proteins, cells are fixed with paraformaldehyde prior to permeabilization to avoid protein wash out (step 10b).
-
•
The authors have compared the 53BP1 mean intensity per nucleus from samples pre-extracted before fixation (step 10a) and from samples fixed before permeabilization (step 10b). It was observed that the 53BP1 mean intensity per nucleus was nearly 50% lower in untreated samples pre-extracted before the fixation as compared to that of samples fixed before permeabilization (53BP1 mean signal intensity per nuclei is 2172 A.U. and 4007 A.U., respectively).
Problem 3
Unexpected distribution of the cells in the G1, S and G2 phases of the cell cycle. Be aware that when performing extended EdU pulse, the progression of the G1 cells into S phase and of the S phase cells into G2 will result in miss cell cycle staging (related to step 8).
Potential solution
-
•
The incubation time after the EdU pulse (step 8), prior to cell fixation should not exceed 1 h (step 10). The cell cycle staging precision is improved by reducing the timing between the EdU pulse and the cells fixation.
-
•
Verify that, more than one log difference in the EdU signal between the EdU negative and the EdU positive nuclei is obtained with 1 h incubation in fresh media after the EdU pulse (step 8).
Problem 4
Focus loss during multipositioning acquisition (related to step 30).
Potential solution
-
•
Air bubbles in the mounting media will interfere with the automatic focusing. If bubbles are present in the Mowiol solution, wait until the bubbles rise to the surface before using for coverslip mounting at step 21.
-
•
Avoid using excess mounting media. Typically, 20 μL of Mowiol is dispensed on a glass slide for a 22 mm2 coverslip mounting at step 21.
-
•
Mowiol is self-hardening once it is applied to the sample. Allow to harden for 6–16 h at 18°C–22°C before samples acquisition or placing the samples at 4°C after step 21.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by Vincent Pennaneach (Vincent.Pennaneach@curie.fr).
Materials availability
No unique material were generated during this study, any information on the equipment reported in this study should be directed to Vincent Pennaneach (Vincent.Pennaneach@curie.fr).
Data and code availability
The open-source Fiji-script reported in this study (00_Convert Files to TIF) and the in house developed Fiji-scripts (01_Shading-Dark correction_v2.ijm) and (02_StarDist-QIBC_v12.ijm) are available from the open repository Zenodo. Additional files available from the open repository Zenodo, include corrected image files from each of the experimental condition described in this protocol article and generated from the raw images using the Fiji-script (01_Shading-Dark correction_v2.ijm). Corrected images are ready for data extraction using the Fiji-script (02_StarDist-QIBC_v12.ijm), the corresponding excel file generated by this Fiji-script (Corrected Images and QIBC analysis.zip) are also available. A solution with ready to use template (Canevas Spotfire) accompanied by instructions (Spotfire_Application to QIBC) applicable for data visualization and cross parameter analysis is proposed. All the forementioned files are downloadable from the Zenodo platform (https://doi.org/10.5281/zenodo.8014997).
Acknowledgments
This study was supported by grants from the Institut Curie, the CNRS, and the Fondation LIGUE Nationale Contre Le Cancer “Equipe Labellisée 2020 (EL2020LNCC/Sal) to S.A.E.L. The equipment used in this work was supported by the Fondation pour la Recherche Médicale (FRM N° DGE20111123020), by the "Dispositif d’aide urgente aux laboratoires pour financement immédiat, 2021", Faculty of Sciences, Université Paris-Saclay and by the Unit UMR 3348. The authors are very grateful to Frédéric Coquelle, head of the Multimodal Imaging Center Imaging Facility, for support and useful advice on the microscope and image processing. We thank Pierre-Marie Girard and Frédérique Megnin-Chanet for their help with sample irradiation.
Author contributions
T.R. performed the experiments, analyzed the data, prepared the multiparameter data visualizations, and wrote the protocol; L.B and M-N.S. set the microscopy equipment; L.B. generated the QIBC-macro for Fiji, analyzed the data, and wrote the tutorials; C.M. helped on QIBC-macro generation; C.M. and M-N.S. read the manuscript; A.R-A. set up the protocol and analyzed the data; V.P. analyzed the data, prepared the multiparameter data visualizations, wrote the manuscript, and wrote the tutorials; S.A.E.L designed the experiments and edited the manuscript.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Sarah A.E. Lambert, Email: sarah.lambert@curie.fr.
Vincent Pennaneach, Email: vincent.pennaneach@curie.fr.
References
- 1.Besse L., Rumiac T., Reynaud-Angelin A., Messaoudi C., Soller M.-N., Lambert S.A.E., Pennaneach V. Fiji-Script for QIBC: Automated Pipeline for Multivariate Quantitative Image-Based Cytometry (QIBC) Analysis by Fluorescence Microscopy of Asynchronous Adherent Cells. Star Protoc. 2023 doi: 10.1016/j.xpro.2023.102446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bruhn C., Kroll T., Wang Z.-Q. Systematic Characterization of Cell Cycle Phase-dependent Protein Dynamics and Pathway Activities by High-content Microscopy-assisted Cell Cycle Phenotyping. Dev. Reprod. Biol. 2014;12:255–265. doi: 10.1016/j.gpb.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lyman S.K., Crawley S.C., Gong R., Adamkewicz J.I., McGrath G., Chew J.Y., Choi J., Holst C.R., Goon L.H., Detmer S.A., et al. High-Content, High-Throughput Analysis of Cell Cycle Perturbations Induced by the HSP90 Inhibitor XL888. PLoS One. 2011;6 doi: 10.1371/journal.pone.0017692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ozaki Y.i., Uda S., Saito T.H., Chung J., Kubota H., Kuroda S. A Quantitative Image Cytometry Technique for Time Series or Population Analyses of Signaling Networks. PLoS One. 2010;5 doi: 10.1371/journal.pone.0009955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roukos V., Pegoraro G., Voss T.C., Misteli T. Cell cycle staging of individual cells by fluorescence microscopy. Nat. Protoc. 2015;10:334–348. doi: 10.1038/nprot.2015.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Toledo L.I., Altmeyer M., Rask M.-B., Lukas C., Larsen D.H., Povlsen L.K., Bekker-Jensen S., Mailand N., Bartek J., Lukas J. ATR Prohibits Replication Catastrophe by Preventing Global Exhaustion of RPA. Cell. 2013;155:1088–1103. doi: 10.1016/j.cell.2013.10.043. [DOI] [PubMed] [Google Scholar]
- 7.Michelena J., Altmeyer M. In: Poly(ADP-Ribose) Polymerase: Methods and Protocols Methods in Molecular Biology. Tulin A.V., editor. Springer; 2017. Cell Cycle Resolved Measurements of Poly(ADP-Ribose) Formation and DNA Damage Signaling by Quantitative Image-Based Cytometry; pp. 57–68. [DOI] [PubMed] [Google Scholar]
- 8.Bhowmick R., Lerdrup M., Gadi S.A., Rossetti G.G., Singh M.I., Liu Y., Halazonetis T.D., Hickson I.D. RAD51 protects human cells from transcription-replication conflicts. Mol. Cell. 2022;82:3366–3381.e9. doi: 10.1016/j.molcel.2022.07.010. [DOI] [PubMed] [Google Scholar]
- 9.Ochs F., Somyajit K., Altmeyer M., Rask M.-B., Lukas J., Lukas C. 53BP1 fosters fidelity of homology-directed DNA repair. Nat. Struct. Mol. Biol. 2016;23:714–721. doi: 10.1038/nsmb.3251. [DOI] [PubMed] [Google Scholar]
- 10.Lezaja A., Panagopoulos A., Wen Y., Carvalho E., Imhof R., Altmeyer M. RPA shields inherited DNA lesions for post-mitotic DNA synthesis. Nat. Commun. 2021;12:3827. doi: 10.1038/s41467-021-23806-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saldivar J.C., Hamperl S., Bocek M.J., Chung M., Bass T.E., Cisneros-Soberanis F., Samejima K., Xie L., Paulson J.R., Earnshaw W.C., et al. An intrinsic S/G2 checkpoint enforced by ATR. Science. 2018;361:806–810. doi: 10.1126/science.aap9346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Michelena J., Pellegrino S., Spegg V., Altmeyer M. Replicated chromatin curtails 53BP1 recruitment in BRCA1-proficient and BRCA1-deficient cells. Life Sci. Alliance. 2021;4 doi: 10.26508/lsa.202101023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zonderland G., Vanzo R., Gadi S.A., Martín-Doncel E., Coscia F., Mund A., Lerdrup M., Benada J., Boos D., Toledo L. The TRESLIN-MTBP complex couples completion of DNA replication with S/G2 transition. Mol. Cell. 2022;82:3350–3365.e7. doi: 10.1016/j.molcel.2022.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Somyajit K., Spies J., Coscia F., Kirik U., Rask M.-B., Lee J.-H., Neelsen K.J., Mund A., Jensen L.J., Paull T.T., et al. Homology-directed repair protects the replicating genome from metabolic assaults. Dev. Cell. 2021;56:461–477.e7. doi: 10.1016/j.devcel.2021.01.011. [DOI] [PubMed] [Google Scholar]
- 15.Schmidt U., Weigert M., Broaddus C., Myers G. In: Medical Image Computing and Computer Assisted Intervention – MICCAI 2018 Lecture Notes in Computer Science. Frangi A.F., Schnabel J.A., Davatzikos C., Alberola-López C., Fichtinger G., editors. Springer International Publishing); 2018. Cell Detection with Star-Convex Polygons; pp. 265–273. [DOI] [Google Scholar]
- 16.Maeshima K., Iino H., Hihara S., Funakoshi T., Watanabe A., Nishimura M., Nakatomi R., Yahata K., Imamoto F., Hashikawa T., et al. Nuclear pore formation but not nuclear growth is governed by cyclin-dependent kinases (Cdks) during interphase. Nat. Struct. Mol. Biol. 2010;17:1065–1071. doi: 10.1038/nsmb.1878. [DOI] [PubMed] [Google Scholar]
- 17.Fidorra J., Mielke T., Booz J., Feinendegen L.E. Cellular and Nuclear Volume of Human Cells during the Cell Cycle. Radiat. Environ. Biophys. 1981;19:205–214. doi: 10.1007/BF01324188. [DOI] [PubMed] [Google Scholar]
- 18.Maul G.G., Maul H.M., Scogna J.E., Lieberman M.W., Stein G.S., Hsu B.Y., Borun T.W. Time sequence of nuclear pore formation in phytohemagglutinin-stimulated lymphocytes and in HeLa cells during the cell cycle. J. Cell Biol. 1972;55:433–447. doi: 10.1083/jcb.55.2.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang L., Guan T., Gerace L. Integral Membrane Proteins of the Nuclear Envelope Are Dispersed throughout the Endoplasmic Reticulum during Mitosis. J. Cell Biol. 1997;137:1199–1210. doi: 10.1083/jcb.137.6.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yen A., Pardee A.B. Role of nuclear size in cell growth initiation. Science. 1979;204:1315–1317. doi: 10.1126/science.451539. [DOI] [PubMed] [Google Scholar]
- 21.Bonner W.M., Redon C.E., Dickey J.S., Nakamura A.J., Sedelnikova O.A., Solier S., Pommier Y. GammaH2AX and cancer. Nat. Rev. Cancer. 2008;8:957–967. doi: 10.1038/nrc2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Panier S., Boulton S.J. Double-strand break repair: 53BP1 comes into focus. Nat. Rev. Mol. Cell Biol. 2014;15:7–18. doi: 10.1038/nrm3719. [DOI] [PubMed] [Google Scholar]
- 23.Model M. Intensity Calibration and Flat-Field Correction for Fluorescence Microscopes. Curr. Protoc. Cytom. 2014;68:10.14.1–10.14.10. doi: 10.1002/0471142956.cy1014s68. [DOI] [PubMed] [Google Scholar]
- 24.Model M.A., Burkhardt J.K. A standard for calibration and shading correction of a fluorescence microscope. Cytometry. 2001;44:309–316. doi: 10.1002/1097-0320(20010801)44:4<309::AID-CYTO1122>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 25.Rothkamm K., Barnard S., Moquet J., Ellender M., Rana Z., Burdak-Rothkamm S. DNA damage foci: Meaning and significance. Environ. Mol. Mutagen. 2015;56:491–504. doi: 10.1002/em.21944. [DOI] [PubMed] [Google Scholar]
- 26.Lee C.-S., Lee K., Legube G., Haber J.E. Dynamics of yeast histone H2A and H2B phosphorylation in response to a double-strand break. Nat. Struct. Mol. Biol. 2014;21:103–109. doi: 10.1038/nsmb.2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clouaire T., Rocher V., Lashgari A., Arnould C., Aguirrebengoa M., Biernacka A., Skrzypczak M., Aymard F., Fongang B., Dojer N., et al. Comprehensive Mapping of Histone Modifications at DNA Double-Strand Breaks Deciphers Repair Pathway Chromatin Signatures. Mol. Cell. 2018;72:250–262.e6. doi: 10.1016/j.molcel.2018.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rass E., Willaume S., Bertrand P. 53BP1: Keeping It under Control, Even at a Distance from DNA Damage. Genes. 2022;13:2390. doi: 10.3390/genes13122390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baldock R.A., Day M., Wilkinson O.J., Cloney R., Jeggo P.A., Oliver A.W., Watts F.Z., Pearl L.H. ATM Localization and Heterochromatin Repair Depend on Direct Interaction of the 53BP1-BRCT2 Domain with γH2AX. Cell Rep. 2015;13:2081–2089. doi: 10.1016/j.celrep.2015.10.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kleiner R.E., Verma P., Molloy K.R., Chait B.T., Kapoor T.M. Chemical proteomics reveals a γH2AX-53BP1 interaction in the DNA damage response. Nat. Chem. Biol. 2015;11:807–814. doi: 10.1038/nchembio.1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fradet-Turcotte A., Canny M.D., Escribano-Díaz C., Orthwein A., Leung C.C.Y., Huang H., Landry M.-C., Kitevski-LeBlanc J., Noordermeer S.M., Sicheri F., Durocher D. 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature. 2013;499:50–54. doi: 10.1038/nature12318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Charier G., Couprie J., Alpha-Bazin B., Meyer V., Quéméneur E., Guérois R., Callebaut I., Gilquin B., Zinn-Justin S. The Tudor tandem of 53BP1: a new structural motif involved in DNA and RG-rich peptide binding. Structure. 2004;12:1551–1562. doi: 10.1016/j.str.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 33.Botuyan M.V., Lee J., Ward I.M., Kim J.-E., Thompson J.R., Chen J., Mer G. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell. 2006;127:1361–1373. doi: 10.1016/j.cell.2006.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Acs K., Luijsterburg M.S., Ackermann L., Salomons F.A., Hoppe T., Dantuma N.P. The AAA-ATPase VCP/p97 promotes 53BP1 recruitment by removing L3MBTL1 from DNA double-strand breaks. Nat. Struct. Mol. Biol. 2011;18:1345–1350. doi: 10.1038/nsmb.2188. [DOI] [PubMed] [Google Scholar]
- 35.Mallette F.A., Mattiroli F., Cui G., Young L.C., Hendzel M.J., Mer G., Sixma T.K., Richard S. RNF8- and RNF168-dependent degradation of KDM4A/JMJD2A triggers 53BP1 recruitment to DNA damage sites. EMBO J. 2012;31:1865–1878. doi: 10.1038/emboj.2012.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Simonetta M., de Krijger I., Serrat J., Moatti N., Fortunato D., Hoekman L., Bleijerveld O.B., Altelaar A.F.M., Jacobs J.J.L. H4K20me2 distinguishes pre-replicative from post-replicative chromatin to appropriately direct DNA repair pathway choice by 53BP1-RIF1-MAD2L2. Cell Cycle. 2018;17:124–136. doi: 10.1080/15384101.2017.1404210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pellegrino S., Michelena J., Teloni F., Imhof R., Altmeyer M. Replication-Coupled Dilution of H4K20me2 Guides 53BP1 to Pre-replicative Chromatin. Cell Rep. 2017;19:1819–1831. doi: 10.1016/j.celrep.2017.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lottersberger F., Bothmer A., Robbiani D.F., Nussenzweig M.C., de Lange T. Role of 53BP1 oligomerization in regulating double-strand break repair. Proc. Natl. Acad. Sci. USA. 2013;110:2146–2151. doi: 10.1073/pnas.1222617110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zgheib O., Pataky K., Brugger J., Halazonetis T.D. An oligomerized 53BP1 tudor domain suffices for recognition of DNA double-strand breaks. Mol. Cell Biol. 2009;29:1050–1058. doi: 10.1128/MCB.01011-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kilic S., Lezaja A., Gatti M., Bianco E., Michelena J., Imhof R., Altmeyer M. Phase separation of 53BP1 determines liquid-like behavior of DNA repair compartments. EMBO J. 2019;38 doi: 10.15252/embj.2018101379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ghodke I., Remisova M., Furst A., Kilic S., Reina-San-Martin B., Poetsch A.R., Altmeyer M., Soutoglou E. AHNAK controls 53BP1-mediated p53 response by restraining 53BP1 oligomerization and phase separation. Mol. Cell. 2021;81:2596–2610.e7. doi: 10.1016/j.molcel.2021.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Spegg V., Altmeyer M. Biomolecular condensates at sites of DNA damage: More than just a phase. DNA Repair. 2021;106 doi: 10.1016/j.dnarep.2021.103179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roukos V., Pegoraro G., Voss T.C., Misteli T. Cell cycle staging of individual cells by fluorescence microscopy. Nat. Protoc. 2015;10:334–348. doi: 10.1038/nprot.2015.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The open-source Fiji-script reported in this study (00_Convert Files to TIF) and the in house developed Fiji-scripts (01_Shading-Dark correction_v2.ijm) and (02_StarDist-QIBC_v12.ijm) are available from the open repository Zenodo. Additional files available from the open repository Zenodo, include corrected image files from each of the experimental condition described in this protocol article and generated from the raw images using the Fiji-script (01_Shading-Dark correction_v2.ijm). Corrected images are ready for data extraction using the Fiji-script (02_StarDist-QIBC_v12.ijm), the corresponding excel file generated by this Fiji-script (Corrected Images and QIBC analysis.zip) are also available. A solution with ready to use template (Canevas Spotfire) accompanied by instructions (Spotfire_Application to QIBC) applicable for data visualization and cross parameter analysis is proposed. All the forementioned files are downloadable from the Zenodo platform (https://doi.org/10.5281/zenodo.8014997).