

Abstract
Background
Proteorhodopsin (pR) is a light-activated proton pump homologous to bacteriorhodopsin and recently discovered in oceanic γ-proteobacteria. One perplexing difference between these two proteins is the absence in pR of homologues of bR residues Glu-194 and Glu-204. These two residues, along with Arg-82, have been implicated in light-activated fast H+ release to the extracellular medium in bR. It is therefore uncertain that pR carries out its physiological activity using a mechanism that is completely homologous to that of bR.
Results
A pR purification procedure is described that utilizes Phenylsepharose™ and hydroxylapatite columns and yields 85% (w/w) purity. Through SDS-PAGE of the pure protein, the molecular weight of E.-coli-produced pR was determined to be 36,000, approximately 9,000 more than the 27,000 predicted by the DNA sequence. Post-translational modification of one or more of the cysteine residues accounts for 5 kDa of the weight difference as measured on a cys-less pR mutant. At pH 9.5 and in the presence of octylglucoside and diheptanoylphosphotidylcholine, flash photolysis results in fast H+ release and a 400-nm absorbing (M-like) photoproduct. Both of these occur with a similar rise time (4–10 μs) as reported for monomeric bR in detergent.
Conclusions
The presence of fast H+ release in pR indicates that either different groups are responsible for fast H+ release in pR and bR (i.e. that the H+ release group is not highly conserved); or, that the H+ release group is conserved and is therefore likely Arg-94 itself in pR (and Arg-82 in bR, correspondingly).
Background
Proteorhodopsin is a 249-amino acid membrane protein native to several uncultured species of γ-proteobacteria, which are a component of marine plankton [1]. Addition of retinal to E. coli expressing pR was shown to cause a reddish coloration of the bacteria with an absorption maximum near 520 nm. The pR contained in the bacterial membranes was shown to act as a light-activated proton pump, but only when retinal is present. Time-resolved UV/vis studies at pH 8 also revealed that the protein undergoes a photocycle, similar to that of wild type bacteriorhodopsin, but with a predominance of the O intermediate instead of M.
The bR photocycle has been characterized by spectroscopic methods as having six principal photointermediates: bR, K, L, M, N and O. Each intermediate has a distinct absorbance maximum; the most studied are bR (570 nm), M (412 nm), and O (640 nm) since these are the ones that can be produced in the highest concentration at physiological pH values. Monitoring of the absorbance at individual wavelengths after photoexcitation is used to determine the relative concentrations and decay times of each of these photointermediates. The L → M transition in bR is characterized by the deprotonation of the Schiff base to Asp-85, producing the distinctive 412 nm absorbance maximum of M, and by so-called fast proton release, the ejection of a proton from a different (unknown) residue into the external medium on the ~10–100 μs time scale, depending on pH. Reprotonation of the Schiff base from Asp-96 occurs during the M → N transition with an absorbance maximum of 560 nm [2]. The N → O transition involves the reprotonation of the Asp-96 from the cytoplasmic space.
Like bR, pR consists of seven transmembrane α-helices that include in the membrane interior all of the residues conserved among archaeal rhodopsin proton pumps. In particular, analogues of Asp-85, Asp96, Arg-82, and Lys-216 of bR are present in pR. Conspicuously absent are analogues for Glu-194 and Glu-204 of bR. The latter, as well as Arg-82, have been implicated in fast proton release. In particular, mutagenesis of Glu-194 or Glu-204 in bR results in loss of fast proton release [3,4]. The absence of homologs for these residues in pR leaves open the question of whether it carries out fast H+ release.
Experiments described here demonstrate that pR does indeed undergo fast H+ release, at least under elevated pH conditions that resemble somewhat those of the γ-proteobacteria's native open ocean environment. We also demonstrate that there is a post-translational modification of at least one of the three native cysteines when pR is expressed in E. coli. Both of these discoveries were made possible through purification methods for pR described herein.
Results
Purification
PR was obtained in 85% purity, assuming that values of ε280 and ε546 for pR are the same as for bR solubilized in DMPC/cholate/SDS mixtures at pH 8 (ε280= 7.85 × 104 cm-1 M-1 and ε551 = 4.8 × 104 cm-1 M-1) [5]. This assumption is actually expected to underestimate the purity of pR produced, by up to ~20%, since the pR we expressed has 10 tryptophan and 14 tyrosine residues, as compared to 8 tryptophans and 11 tyrosines in bR from H. salinarum. The absorbance of contaminant proteins was assumed to be 1.1 for a 1 mg/mL solution. By using these assumptions, the relative concentrations of pR and other proteins can be determined from the absorbance spectra of the various fractions (fig. 1). The resulting purity values correlate well with those Coomasie-stained SDS-PAGE gels (see below). The OG extract of cholate-washed membrane pellets starts out at a pR content of 7% total protein (w/w). The Phenylsepharose column increases the purity level to 24%, with approximately 5% loss. The final purification step by hydroxylapatite column chromatography produces pR with ~85% purity and a further loss of ~60%, i.e. the overall yield of the two column procedure was ~30%.
Figure 1.
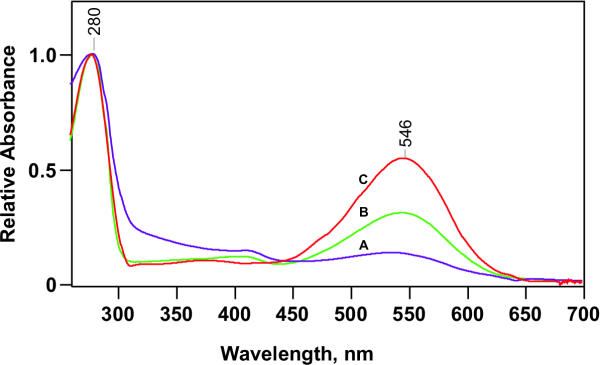
UV/visible absorption spectra of pR in octylglucoside solution (1–3%) at three stages of purification. All three spectra were measured in the presence of octylglucoside at pH 8, and are normalized to the 280-nm protein peak. Spectrum A, the OG extract of cholate-washed E. coli membranes; spectrum B, pooled 546-nm absorbing fractions from Phenylsepharose column; spectrum C, same material after hydroxylapatite column.
Polyacrylamide gel electrophoresis
Relative to protein standards, the apparent molecular weight of bR is 25,000 while the apparent molecular weights of pR-wt and pR-TCM are 36,000 and 31,000, respectively (fig. 2, lanes E and C, respectively). SDS-PAGE (fig. 2) also confirms the estimates of purity level based on the assumed ε280/ε546 ratio identical with that of detergent solubilized bR. Interestingly, the pR appears to be a doublet band whose relative concentrations remain almost unchanged during purification. This doublet is also present in the less-purified sample of pR-TCM, with both bands shifted down by approximately the same amount (fig. 2, lane C).
Figure 2.
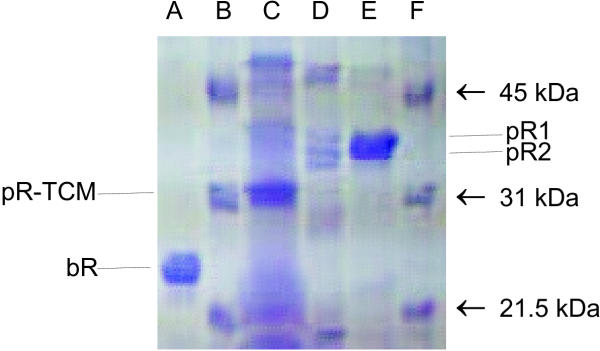
SDS-PAGE of pR (wild type and pR-triple cysteine mutant). Lane A contains bacteriorhodopsin (bR). Lanes B and F contain BioRad protein molecular weight markers including labeled bands at 21.5 (trypsin inhibitor), 31 (bovine carbonic anhydrase), and 45 (ovalbumin) kDa. Lane C is of the pR triple cysteine mutant (TCM). Lane D contains the Phenylsepharose™-purified pR wild type protein, corresponding to spectrum B of fig. 1. Lane E contains the hydroxylapatite-purified pR wild type protein, corresponding to spectrum C in fig. 1.
Subsequent SDS-PAGE analysis of pR samples that had been stored for periods of time up to several months indicate that after sitting for several weeks in octylglucoside solution at 4°C, the largest post-translational modification on wild-type pR is eliminated – presumably hydrolysed off of the cysteine(s) – leaving only a 31,000-MW band indistinguishable from that seen for pR-TCM (data not shown). Furthermore, after boiling for several min in gel loading solution, this cleaved wild-type protein, as well as the TCM, both give an extra artifactual band near 36,000 dalton. The latter band, a singlet, is coincidentally at almost the same apparent MW as the doublet from the uncleaved post-translationally-modified wild type pR (fig. 2, lane E). These potential artifacts should be taken into consideration in any attempt to reproduce the results in Fig. 2.
Photocycle kinetics and flash-induced proton concentration changes
Photocycle kinetics were measured at 400, 500. and 580 nm in the presence of the short-chain lipid DHPC. This lipid does not support the formation of closed bilayer vesicles, but rather forms micelles like a detergent. The time-resolved measurements showed no positive 400-nm absorbance signals at pH 8.0 or lower (Fig. 3). This is somewhat in disagreement with Béjà et al [1], who detected small 400-nm transient absorbance increases upon photolysis at pH 8.0. However, we observed a transient 400-nm absorbance increase at an elevated pH of 9.5 (fig. 3).
Figure 3.
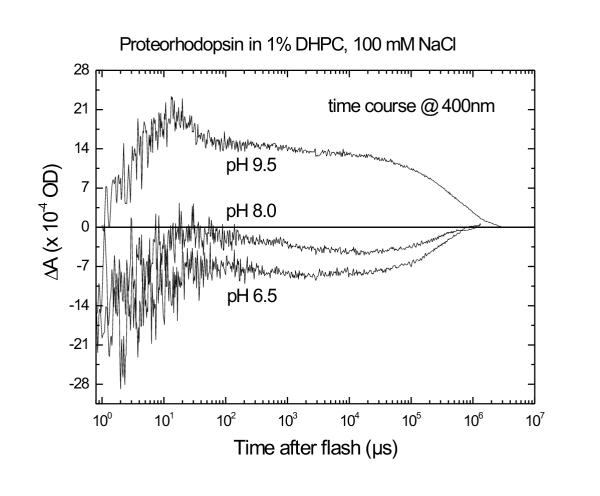
Dependence on pH of the M-like intermediate of pR. Time courses of flash-induced absorbance changes measured at 400 nm and 22°C for pR in 1% DHPC/100 mM NaCl solution at pH 6.5, 8.0 and 9.5. A positive differential absorbance at 400 nm is indicative of the presence of the M intermediate. The logarithmic time scale ranges from 100–107 μs after photolysis by a 10-ns laser pulse at 500 nm, with an energy of 3–6 mJ.
At pH 9.5 in the presence of DHPC, and observing transient changes at 500 nm (fig. 4), pR undergoes a 2-phase decay after the initial unresolved absorbance decrease. Multiexponential fits show that the first decay phase has a time constant of 4 μs, in good agreement with the 4-μs rise time of the 400 nm signal (Fig. 4). The amplitude of this decay represents about 80% of the initial absorbance depletion. The second phase of the 500 nm absorbance decay occurs with a substantially slower time constant of 0.5 s, returning the remaining 20% of initial absorbance change. The slowest decay components of the positive 400-nm signal and the negative 500-nm signal follow similar kinetics, although the amplitudes of these components differ by a factor of 3. At pH 9.5, the 580 nm trace has no significant positive values indicative of an O-like intermediate, although, in agreement with earlier measurements [1], at lower pH values a red-shifted transient is the predominant positive absorbance signal (data not shown).
Figure 4.
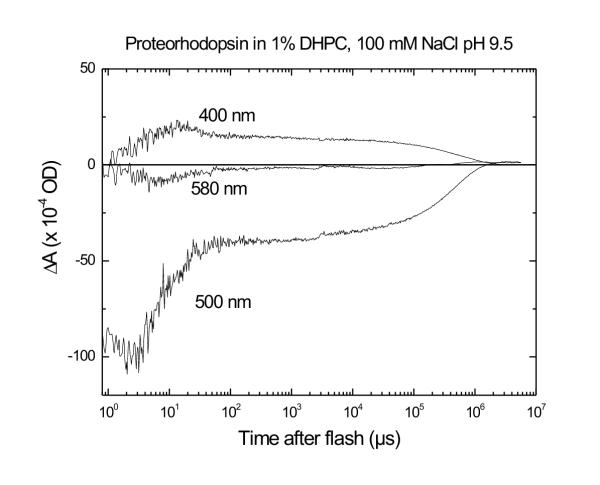
Photocycle kinetics of pR at selected wavelengths at pH 9.5. Time traces were measured at 400, 500, and 580 nm. The 400-nm trace shows the kinetics of the M intermediate, i.e. the deprotonated Schiff base, as in Fig. 3. The 500-nm trace shows the depletion signal of pR at the earliest times, and then the time course of the N intermediate as well as return of the pR resting state. The 580-nm trace is indicative of an O-like intermediate. The conditions are 1% DHPC, 100 mM NaCl, pH9.5 at 22°C. The laser excitation is as in fig. 3.
Figure 5 shows a different type of time-resolved measurement, probing not the pR chromophore, but rather pH changes in the protein environment. Proton concentration changes in the aqueous bulk phase were measured with the pH sensitive dye cresol red, which has a pKa of 8.2–8.5. The bottom trace in Figure 5 shows the absorbance change of the indicator during the pR photocycle. The negative signal is indicative of a pH decrease, corresponding to transient H+ release from the protein into the solution. The best-fit time constant for the release phase is 6 μs. The positive 400 nm trace in fig. 5 (reproduced from fig. 3) shows that the proton release and uptake follow kinetics very similar to the apparent formation and decay of M, as is typically seen in bR near neutral pH [6,7,21]. However, no proton release signal could be observed for pR at pH 6 or 8 (data not shown).
Figure 5.
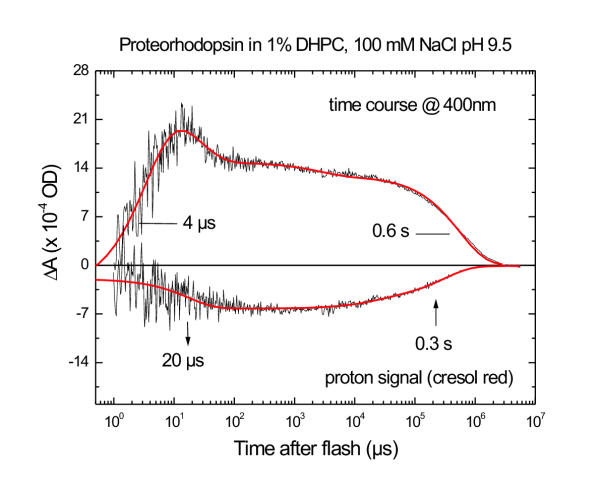
Comparison of the kinetics of M formation and decay with kinetics of ET release and uptake. The time trace of the M-like intermediate was measured at 400 nm (upper panel). Time-resolved H+ concentration changes (lower panel) were measured with the pH indicator dye Cresol Red. A negative Cresol Red absorbance change at 580 nm is indicative of a transient decrease in the pH of the solution, i.e. of H+ release by pR. Solid lines represent multiexponential fits, with the main rise and decay times indicated for the M intermediate. The H+ release and uptake time constants obtained from the fit are marked with arrows pointing down for release and pointing up for uptake. Sample and excitation conditions are as in fig. 4.
Discussion
Purification of pR
Meaningful comparison of intrinsic physiological properties of pR and bR depends on the purification of pR. The E. coli expression system can easily be used to prepare pR at 85% purity with similar or less effort and time than required for bR (purple membrane) production from S9 H. salinarum.
The initial purity level of the OG-solubilized cholate-extracted membrane is about 7% pR by weight. Phenylsepharose™ column chromatography separates proteins on the basis of hydrophobicity, and has been used previously in hR purification [8-10]. As an initial purification step, the Phenylsepharose™ column achieved a substantial increase in protein purity to 25%, along with removal of most of the lipid. The final purification step, utilizing a hydroxylapatite column, has been previously used with rhodopsin [11]. This column proved to be more efficient as a final step in the purification than as a preliminary one, because large amounts of contaminant protein tended to slow the flow rate drastically. This step of the purification yielded an increase in protein purity to >85%. The overall yield of pR from membrane through the column purifications is ~30%. Most of this loss, ~65%, occurs during the hydroxylapatite column.
Molecular weight differences
Béjà et al. reported a molecular weight for wild type pR of 27 kDa based on the predicted amino acid sequence of the protein [1]. Its 249 amino acids barely exceed the 248 of mature bR, which has a molecular weight of 26,000. However, we observed a significantly higher apparent molecular weight (~36,000) for wild type pR on SDS-PAGE gels (Fig. 2, lanes D and E). Post-translational modification of pR must almost certainly account for some of the observed molecular weight difference between pR and bR. Lipids or sugars covalently bound to the protein surface would not be removed during the purification procedure and could cause a higher apparent molecular weight. Cysteines are frequently a site of lipid association with membrane proteins, (e.g. mammalian rhodopsin, which has two palmitoyl molecules attached to cysteine residues). Therefore we compared the SDS-PAGE gel mobility of wild-type pR to that of a cys-less mutant (pR-TCM). The elimination of the three possible sulfhydryl attachment sites lowers the apparent molecular weight of the pR-TCM by 5,000 (Fig. 2, lane C). From this, we conclude that at least one of the three cysteines in pR is probably modified post-translationally.
However, this by itself does not account fully for the anomalous mobility of pR on SDS-PAGE gels, because pR-TCM is still approximately 5,000 higher in molecular weight than bR according to SDS-PAGE (fig. 2, lanes A & C). Only ~2,600 of this can be accounted for by the V5 eiptope and poly-histidine tail that are appended to the C-terminus of pR by the pBAD-TOPO expression vector that we used [1]. There is undoubtedly a further post-translational modification of unknown nature.
Spectral comparisons
Béjà et al. reported an absorbance maximum for pR of 520 nm in E. coli membranes, using a difference bleaching technique to remove interfering absorbance bands from other membrane components in the impure pR sample [1]. We confirmed this result using crude E. coli membranes without detergent present (data not shown). However, we observed an absorbance maximum of 546 ± 5 nm for pR in OG at pH 7 at all stages of purification (Fig. 1). Small blue shifts were observed for pR samples in OG when measured at pH 8 and 9.5 (8 and 16 nm, respectively; data not shown).
For the pR samples reconstituted in DHPC, which were used for flash photolysis experiments, the absorbance maxima were similar to those measured in OG (spectra not shown). A chromophore absorption maximum near 540 nm was also obtained by using difference spectroscopy of pR in crude E. coli membranes solubilized in OG (spectrum not shown). However, for pR measured directly in crude E. coli membranes, i.e. not solubilized in OG, we obtained the same value (520 nm) as reported previously [1].
Solubilization in detergent presumably leads to structural distortions of the native protein conformation, and therefore a change in the absorbance properties of the chromophore. However, the direction that λmax for pR would have to shift upon solubilization in OG is inconsistent with the pattern for bR, whose λmax decreases when it is solubilized in OG [12]. Furthermore, pR in OG showed resonance Raman spectra (D. Dunmire, R. A. Krebs and M. S. Braiman, unpublished data) indicative of a chromophore structure very close to native light-adapted (i.e. all-trans) bR in purple membrane. However, there is one major difference: pR in OG exhibits an upshifted, doublet C=N Schiff base band consisting of two components of nearly-equal intensity. These appear to correspond to the presence of two distinct subpopulations of pR, at least when expressed in E. coli and solubilized in OG micelles. The different values of λmax for pR in membrane state [1] and OG solution might be related to the presence of these multiple subpopulations, but this connection remains unclear.
Principal photointermediates of pR
Of the six principal photointermediates present in bR, four can be discerned from the time-dependent visible absorbance traces from pR in fig. 4, along with previously published time traces at 600 nm [1]: the resting state (pR), M, N and O. The resting state, with an absorbance maximum of 546 nm (see above), provides the baseline spectrum for the difference time courses reported. The M intermediate of bR has an unprotonated Schiff base group, giving rise to a blue-shifted λmax (400 nm). Likewise in pR, an increase in the 400 nm absorbance should indicate formation of a deprotonated Schiff base, and therefore the presence of an M-like intermediate.
Interpretation of the 500 nm time course trace is more complicated. As in bR, it likely involves decay of M to N, as well as from N back to bR. The absorbance maximum of the N intermediate in bR is 560 nm, corresponding to a protonated Schiff base. This is not very different from λmax for the resting state of pR (546 nm). The likely spectral overlap between pR and its own postulated N photoproduct complicates the determination of amounts of each that are present. The slow (~500 ms) decay observed in both the 400 nm and 500 nm time courses indicates an equilibrium between the M and N intermediates that remains until pR returns to its initial resting state.
Béjà et al. reported a strong positive 580-nm transient absorbance increase in suspensions of membranes prepared from E. coli expressing pR [1]. This positive absorbance difference is indicative of O intermediate formation. We looked for its presence in partially-purified samples reconstituted in DHPC in the pH range 6–11. (Data are shown only at pH 9.5; see Fig. 4). Our observations at pH 9.5 do not show any evidence of O intermediate formation (Fig. 4). Only at lower pH values was a clear positive 580-nm absorption change observed (data not shown). This is in agreement with results on bR, for which O formation is also enhanced at lower pH values, and becomes small or nearly unobservable in the alkaline range.
Dependence of the M intermediate on pH
Deprotonation of the Schiff base linkage of the retinal and Lys-216 is dependent on its pKa which changes between photointermediate states. The Schiff base readily undergoes deprotonation in the M intermediate. However, no M intermediate formation occurs below a pH of ~9 (figure 3); instead the O intermediate predominates [1]. Predominance of O at lower pH values is also observed in bR. However, in bR the M intermediate is detectable at low and high pHs, but has a longer lifetime at higher pH due to a long-lived equilibrium between M and another intermediate, N [13]. It seems likely that an N intermediate of pR is similarly in equilibrium with its M intermediate, based on the fact that the transient positive (400 nm) absorbance increase is smaller than the negative 500 nm bleach, and the time course at 500 nm shows a partial return to baseline on a timescale of ~50 μs.
The difference in pH dependence between the pR and bR photocycles can likely be attributed to differences in the microenvironment of the Schiff base, and is perhaps related to absence of Glu194/204 in pR. These adaptive differences presumably optimize the proteins to operate at maximum efficiency in the niches that their respective organisms occupy. In the case of the λ-proteobacteria, which in the open ocean (pH 7.8–8.0) occupy a signficantly more alkaline environment than halobacteria, perhaps the proton-release group is simply not under any evolutionary pressure to be capable of deprotonating in the M state at neutral pH. In this view, the principal role of E194 and E204 in bR may to modulate the pKa of the H+-release group in the M state to a value lower than the pH of the organism's external environment.
Fast proton release in pR
Under the same conditions where M is observed (pH 9.5 and in 1% DHPC), pR undergoes fast proton release during its photocycle (Fig. 5). The pH indicator dye Cresol Red was used to detect pH changes in the bulk aqueous phase. These turn out to be similar to those observed for bR in the pH range 5.5–10. After photoexcitation, pR (like bR, presumably) ejects a H+ from a residue near its extracellular surface decreasing the pH of the solution. When the N → O transition takes place in bR, H+ is taken up from the medium, raising the pH once again. The H+ signals from pR measured with Cresol Red occur on a time scale similar to that assignable to M and N decay, returning to baseline about 1 s after photolysis.
There is a clear kinetic correlation between M (and/or N) intermediate formation and fast H+ release in pR. The linkage between these two phenomena is further supported by the observation that neither a transient 400-nm absorbance increase, nor fast H+ release, is shown to occur at pH 8.0 and below. Nor is either observed in the absence of a reconstituting lipid (DHPC in these experiments).
In bR, the ejected proton is thought to originate from a triad of amino acids, R82-E194-E204. However, in pR a homolog of only one of these three residues (the arginine) is present. This raises doubts about previous conclusions regarding the specific roles of these 3 residues in fast H+ proton release, in both pR and bR. In particular, the apparently obligatory roles of E204 and E194 in fast H+ release in bR are not matched in pR. Therefore, even in bR it is less likely that these groups themselves change protonation state between bR and M to provide the H+ released to the bulk medium. Instead, it now seems more likely that E204 and E194 merely help to lower the pKa of the H+ release group from above 8, the apparent value for pR, into the vicinity of 6 for bR. It also seems very unlikely that the specific structural configuration of 2 carboxylic acid groups and arginine in bR could be conserved in pR, even if, as suggested previously [1], other surface carboxylic acids in pR could substitute in some ways for the roles of E194 and E204 in bR.
The only way that the H+ release mechanism can be strongly conserved between bR and pR is if arginine itself serves as the principal donor group for fast H+ release in both, with nearby residues (such as E194 and E204) merely modulating the pKa of the arginine in the M intermediate. However, it remains unclear how the pKa of Arg-82 in bR could be made sufficiently low in its M intermediate to serve as the H+ release group at pH values down to 6.0.
Alternatively, it is possible that the fast H+ release we observe from pR at pH 9.5 may differ from that in bR. One possibility is that in pR, the released proton could come directly from the chromophore counterion, Asp-97. This would be consistent with a proposed mechanism for fast H+ release that has been observed above pH 10 in the bR mutant E194Q [14]. In this mutant, Asp-85 was detected by low-temperature infrared difference spectroscopy to be deprotonated only in the N intermediate, and not in M [15]. It is not clear yet whether Asp-85 deprotonation in an N-like state could account for the proton-release kinetics of pR (Fig. 5), or whether the M intermediate itself might have a partially-deprotonated Asp-85.
The reason for the requirement of DHPC in M intermediate formation and fast proton release is unclear. Delipidated bR in octylglucoside is fully capable of M formation and presumably proton release, although with altered kinetics [12,16]. The requirement for pR to be in lipid to show fast H+ release and M formation stems either from a protein/lipid interaction needed to establish a stable, active tertiary structure, or from the need for the phosphate group in DHPC to act as a proton release group. The latter seems unlikely due to the DHPC molecule being zwitterionic at pH 9.5, with no proton on the trimethyl-modified nitrogen of the choline. Hence, the DHPC most likely interacts with the protein to effect minor structural changes needed to place the active site residues in their functional configuration.
Conclusions
A comparison of the primary sequences of pR and bR at first glance seems as to preclude fast H+ release as part of the proton-pumping mechanism of pR due to the absence of residues analogous to Glu194 and Glu 204 of bR. However, fast H+ release is indeed observed in pR under conditions where an M intermediate is formed. Glu194 and Glu 204 in bR play a role in fast H+ release that is apparently not required for the mechanism of the bR family of proton transporters. It is therefore necessary to conclude that either the H+-release groups in pR and bR are non-homologous surface carboxylic acid residues (as suggested previously [1]), or else that a conserved non-carboxylic acid residue, i.e. Arg82/94 or Asp85/97, is the H+-release group in pR. The higher pH requirement for the M intermediate of pR presumably corresponds with adaptation to the more alkaline oceanic environment in which the γ-proteobacteria are found.
The necessity of reconstituting pR with some lipid before it is capable of photocycling shows that the presence of lipids facilitates pR in assuming its fully active structure. E.-coli-expressed pR has post-translational modifications, including ~4000 daltons of substituents at one or more of its three cysteines. Such post-translational modifications might also play a role in explaining the different physiological properties of pR and bR.
Materials & methods
Protein expression and detergent extraction
Proteorhodopsin was expressed by E. coli strain UT5600 containing an additional plasmid encoding for the first-reported pR gene (accession #AF279106, obtained from the uncultured proteobacterium EBAC31A08 clone BAC [1]) with an Ara promoter and ampicillin resistance (kindly provided by O. Béjà). Single colonies were selected and grown overnight in LB/amp media (200 ml, 37°C, 300 rpm). This culture was then diluted 10× into several 500-ml cultures. After a further 2 h incubation in the shaker bath, a stock solution of 20% L-arabinose was added, to give a final concentration of 0.2% L-arabinose. This culture was then incubated for 4 h (37°C, 300 rpm). The cells (~20 ml wet volume) were then collected by centrifugation (6000 rpm × 30 min) and washed 3× with 100 mM HEPES, pH 7.1 (buffer A). The cells were then resuspended in buffer A and incubated at 4°C with 50 μg all-trans-retinal (added as a concentrated ethanol solution) for 3 h. The cells were collected by centrifugation (6000 rpm × 30 min), then resuspended in 60 mL buffer A containing 0.3 mg/mL lysozyme, and stirred for 4 h at room temperature. The cells were again collected by centrifugation (6000 rpm × 30 min), then lysed with 20 ml of 20% sodium cholate, pH 7.1 (30 min., 4°C). The cells were centrifuged again (6000 rpm × 30 min), and the supernatants collected. After extracting 3× more with the same cholate solution, the pooled supernatants were diluted 10× with buffer A and centrifuged at 180,000 g for 45 min to collect the membrane pellet. This cholate-washed membrane pellet was then further extracted 3× with 3.0% β-octyl-D-glucoside (OG) in buffer A (30 min, with stirring, 4°C). The pooled supernatants, containing OG-solubilized pR (~15 mg), were then diluted 6× with buffer A.
Column purification
The diluted OG-solubilized membrane extract (10 mg in 300 mL total volume of 0.5% OG) was loaded on a 25 × 1 cm column containing Phenylsepharose™ (6 fast flow high sub; Amersham Pharmacia Biotech). The column was eluted with a 0.5%-2.0% OG gradient in buffer A (300 mL total volume, 0.5 mL flow rate). The pR eluted at an OG concentration of 1.5–2.0%. Fractions having an A280/A546 ratio of 4.0 or lower were pooled (9.5 mg pR recovered in all) and concentrated using Vivaspin™ 20 concentrators having a 5000 MW cutoff (Vivascience, Westford, MA). A portion of the Phenylsepharose™-purified pR (1.5 mg) was diluted to an OG concentration of 0.5% with 0.5 M KCl, 100 mM acetate. It was then loaded on a 10 cm × 1 cm hydroxylapatite (BioGel HTP, BioRad) column and eluted under pressure with a 0–600 mM phosphate gradient (200 mL total volume, flow rate 0.5 ml/min). Fractions with an A280/A546 ratio of 2.5 or lower were pooled and concentrated for subsequent experiments (0.5 mg).
Mutagenesis
Methodology for the site-directed mutagenesis of pR is discussed in detail elsewhere (R. Parthasarathy, T. Caterino, R.A. Krebs, M.S. Braiman, manuscript in preparation). The triple cysteine mutant (pR-TCM) has all three of its native cysteines (Cys-107, Cys-156, and Cys-175) replaced with serines, and was prepared using the same E. coli expression system and purification methods as the wild type.
Polyacrylamide gel electrophoresis
A 12% discontinuous SDS/polyacrylamide gel was used for molecular weight and purity analysis [17].
Flash photolysis
Time-resolved UV/vis spectroscopy methods were as described previously [18]. A Phenylsepharose™-purified pR sample was reconstituted into mixed micelles containing 1,2-diheptanoyl-SN-glycero-3-phosphocholine (DHPC), by adding a 1% solution of the short-chain lipid and then removing most of the detergent on a Sephadex G-25 column equilibrated with 1% DHPC in 100 mM NaCl. Proton release and uptake in the aqueous bulk medium were detected from the pR-containing micelles suspended in 1% DHPC, 100 mM NaCl, with 45 μM Cresol Red pH indicator dye. Flash-induced absorbance changes at 580 nm of samples with and without the Cresol Red were subtracted to determine the transient signals due to proton concentration changes. Photoexcitations were performed with 10-ns laser pulses of 3–6 mJ at 500 nm. The time courses in Figs. 3,4 are an average of 40 cycles with the exception of the Cresol Red experiments averaging 100 cycles (Fig. 5, bottom trace) [19-21].
Abbreviations
pR, proteorhodopsin; bR, bacteriorhodopsin; OG, β-octyl-D-glucoside; SDS, sodium dodecylsulfate; PAGE, polyacrylamide gel electrophoresis; pR-TCM, pR triple cysteine mutant [C(107,156,175)S]; DHPC, 1,2-diheptanoyl-SN-glycero-3-phosphocholine; DMPC, dimyristoylphosphatidylcholine; HEPES, N-2-hydroxyethylpiperazine-N'-2-ethanesulfonic acid
Acknowledgments
Acknowledgements
This work was supported by Syracuse University, and by a grant of the Deutsche Forschungsgemeinschaft (Sfb 449-TPA5 to U. Alexiev and M. P. Heyn). A.-M. DeVita was supported by an NSF-REU award to Syracuse University Chemistry Department.
Contributor Information
Richard A Krebs, Email: rakrebs@syr.edu.
Ulrike Alexiev, Email: alexiev@physik.fu-berlin.de.
Ranga Partha, Email: rdpartha@syr.edu.
Anne Marie DeVita, Email: devi3777@fredonia.edu.
Mark S Braiman, Email: mbraiman@syr.edu.
References
- Béjà O, Aravind L, Koonin EV, Suzuki MT, Hadd A, Nguyen LP, Jovanovich SB, Gates CM, Feldman RA, Spudich JL, Spudich EN, DeLong EF. Bacterial rhodopsin: Evidence for a new type of phototrophy in the sea. Science. 2000;289:1902–1906. doi: 10.1126/science.289.5486.1902. [DOI] [PubMed] [Google Scholar]
- Kouyama T, Nasuda-Kouyama A, Ikegami A, Mathew MK, Stoeckenius W. Bacteriorhodopsin photoreaction: identification of a long-lived intermediate N (P,R350) at high pH and its M-like photoproduct. Biochemistry. 1988;27:5855–5863. doi: 10.1021/bi00416a006. [DOI] [PubMed] [Google Scholar]
- Balashov SP, Imasheva E, Ebrey TG, Chen N, Menick DR, Crouch RK. Glutamate-194 to cysteine mutation inhibits fast light-induced-proton release in bacteriorhodopsin. Biochemistry. 1997;36:8671–8676. doi: 10.1021/bi970744y. [DOI] [PubMed] [Google Scholar]
- Brown LS, Sasaki J, Kandori H, Maeda A, Needleman R, Lanyi JK. Glutamic acid 204 is the terminal proton release group at the extracellular surface of bacteriorhodopsin. J Biol Chem. 1995;270:27122–27126. doi: 10.1074/jbc.270.45.27122. [DOI] [PubMed] [Google Scholar]
- London E, Khorana HG. Denaturation and renaturation of bacteriorhodopsin in detergents and lipid-detergent mixtures. J Biol Chem. 1982;257:7003–7011. [PubMed] [Google Scholar]
- Sherman WV, Slifkin MA, Caplan SR. Kinetic studies of phototransients in bacteriorhodopsin. Biochim Biophys Acta. 1976;423:238–248. doi: 10.1016/0005-2728(76)90182-1. [DOI] [PubMed] [Google Scholar]
- Grzesiek S, Dencher NA. Time-course and stoichiometry of light-induced proton release and uptake during the photocycle of bacteriorhodopsin. FEBS Lett. 1986;208:337–341. [Google Scholar]
- Steiner M, Oesterhelt D. Isolation and properties of the native chromoprotein halorhodopsin. EMBO J. 1983;2:1379–1385. doi: 10.1002/j.1460-2075.1983.tb01595.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duschl A, McCloskey MA, Lanyi JK. Functional reconstitution of halorhodopsin. Properties of halorhodopsin-containing proteoliposomes. J Biol Chem. 1988;263:17016–17022. [PubMed] [Google Scholar]
- Walter TJ, Braiman MS. Anion-protein interactions during halorhodopsin pumping: halide binding at the protonated Schiff base. Biochemistry. 1994;33:1724–1733. doi: 10.1021/bi00173a015. [DOI] [PubMed] [Google Scholar]
- Hong K, Knudsen PJ, Hubbell WL. Purification of rhodopsin on hydroxyapatite columns, detergent exchange, and recombination with phospholipids. Methods Enzymol. 1982;81:144–150. doi: 10.1016/s0076-6879(82)81024-0. [DOI] [PubMed] [Google Scholar]
- Dencher NA, Heyn MP. Formation and properties of bacteriorhodopsin monomers in the non-ionic detergents octyl-β-D-glucoside and triton X-100. FEBS Lett. 1978;96:322–325. doi: 10.1016/0014-5793(78)80427-x. [DOI] [PubMed] [Google Scholar]
- Váró G, Lanyi JK. Protonation and deprotonation of the M, N, and O intermediates during the bacteriorhodopsin photocycle. Biochemistry. 1990;29:6858–6865. doi: 10.1021/bi00481a015. [DOI] [PubMed] [Google Scholar]
- Koyama K, Miyasaka T, Needleman R, Lanyi JK. Photoelectrical verification of proton-releasing groups in bacteriorhodopsin. Photochem Photobiol. 1998;68:400–406. [Google Scholar]
- Lazarova T, Sanz C, Querol E, Padrós E. Fourier Transform Infrared Evidence for Early Deprotonation of Asp85 at Alkaline pH in the Photocycle of Bacteriorhodopsin Mutants Containing E194Q. Biophys J. 2000;78:2022–2030. doi: 10.1016/S0006-3495(00)76749-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dencher NA, Heyn MP. Bacteriorhodopsin monomers pump protons. FEBS lett. 1979;108:307–310. doi: 10.1016/0014-5793(79)80552-9. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Otto H, Marti T, Holz T, Mogi T, Lindau M, Khorana HG, Heyn MP. Aspartic acid-96 is the internal proton donor in the reprotonation of the Schiff base of bacteriorhodopsin. Proc Natl Acad Sci USA. 1989;86:9228–9232. doi: 10.1073/pnas.86.23.9228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexiev U, Mollaaghababa R, Scherrer P, Khorana HG, Heyn MP. Rapid long-range proton diffusion along the surface of the purple membrane and delayed proton transfer into the bulk. Proc Natl Acad Sci USA. 1995;92:372–376. doi: 10.1073/pnas.92.2.372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexiev U, Mollaaghababa R, Khorana HG, Heyn MP. Evidence for long range allosteric interactions between the extracellular and cytoplasmic parts of bacteriorhodopsin from the mutant R82A and its second site revertant R82A/G231C. J Biol Chem. 2000;275:13431–13440. doi: 10.1074/jbc.275.18.13431. [DOI] [PubMed] [Google Scholar]
- Moltke S, Alexiev U, Heyn MP. Kinetics of light-induced intramolecular charge transfer and proton release in bacteriorhodopsin. Isr J Chem. 1995;35:401–414. [Google Scholar]