

SUMMARY
Rapid release of neurotransmitters in synchrony with action potentials is considered a key hardwired property of synapses. Here, in glutamatergic synapses formed between induced human neurons, we show that action potential-dependent neurotransmitter release becomes progressively desynchronized as synapses mature and age. In this solely excitatory network, the emergence of NMDAR-mediated transmission elicits endoplasmic reticulum (ER) stress leading to downregulation of key presynaptic molecules, synaptotagmin-1 and cysteine string protein α, that synchronize neurotransmitter release. The emergence of asynchronous release with neuronal maturity and subsequent aging is maintained by the high-affinity Ca2+ sensor synaptotagmin-7 and suppressed by the introduction of GABAergic transmission into the network, inhibition of NMDARs, and ER stress. These results suggest that long-term disruption of excitation-inhibition balance affects the synchrony of excitatory neurotransmission in human synapses.
In brief
Uzay et al. show that long-term disruption of excitation-inhibition balance results in progressive desynchronization of excitatory neurotransmission in human synapses. In a purely excitatory human induced neuron network, NMDAR activation elicits endoplasmic reticulum stress leading to downregulation of Syt-1 and CSPα, key presynaptic proteins that synchronize neurotransmitter release.
Graphical Abstract
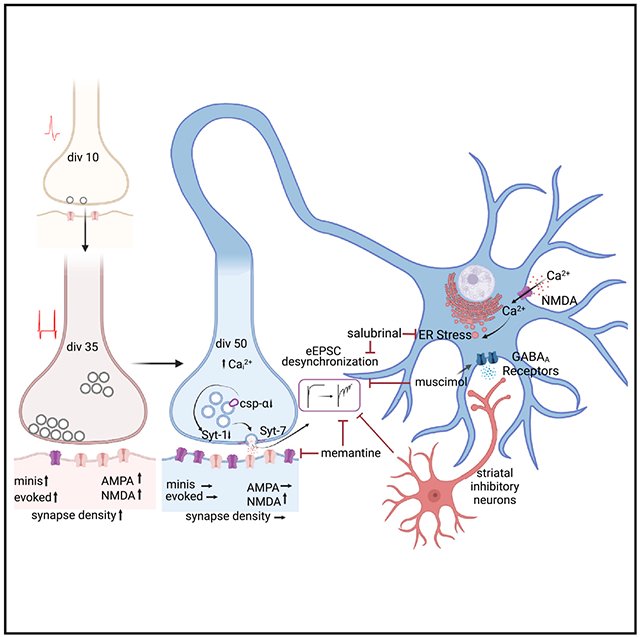
INTRODUCTION
The generation of mature human neurons by the differentiation of human embryonic stem cells (hESCs) or induced pluripotent stem cells (iPSCs) has revolutionized research of human cellular physiology and made it possible to overcome difficulties in investigating the pathophysiology of complex human diseases.1–3 Recent studies have demonstrated that overexpression of a single transcription factor, neurogenin-2 (Ngn2), results in the rapid induction of hESCs and iPSCs into synapse-forming cortical layer 2/3 excitatory human neurons (iN cells).4 In this study, we used Ngn2-induced excitatory human neurons to investigate how human synaptic maturation and in vitro aging affects neurotransmitter release by monitoring the progression of neurotransmitter release kinetics over time in this purely excitatory neuron preparation.
Numerous studies have detailed developmental changes in short-term synaptic plasticity, indicating synapse maturation as a critical factor that influences presynaptic function.5–11 Presynaptic properties such as emergence of spontaneous release, alterations in release probability, and short-term plasticity are determined by the functional availability of vesicles and corresponding active zone structures.8 These presynaptic properties, in turn, strongly impact synapse formation and assembly of synaptic circuits.12
In contrast to the substantial number of studies examining presynaptic changes associated with nascent synapses and synapse maturation, the presynaptic properties of aging synapses remain poorly understood. Extensive studies have been performed on the aging of postsynaptic properties in central synapses,13–16 where synapse aging has been shown to cause a significant decrease in postsynaptic density area,15 alterations in glutamate metabolism,17,18 and a preferential decrease in the thin spine density in axospinous synapses.19 Here, using Ngn2-induced human iN cells to investigate the progression of neurotransmitter release kinetics, we found that neurotransmitter release becomes progressively desynchronized as synapses mature and age in a purely excitatory network. This desynchronization was causally linked to the emergence of NMDAR-mediated transmission and associated endoplasmic reticulum (ER) stress, ultimately leading to the downregulation of synaptotagmin-1 (Syt-1) and cysteine string protein α (CSPα), which normally synchronize neurotransmitter release. Under these aging conditions, asynchronous release was maintained by the high-affinity Ca2+ sensor synaptotagmin-7 (Syt-7) and renormalized to earlier mature time points by introduction of GABAergic inputs into the solely excitatory network. Overall, our findings indicate that deficiencies in inhibitory transmission affect the synchrony of excitatory neurotransmission in human synapses.
RESULTS
Electrophysiological characterization of human iN cells
While the majority of studies investigating synaptic physiology is based on the data obtained from rodent neurons, the use of stem cell-derived systems in synapse research is relatively recent. Therefore, we first aimed to characterize the basic electrophysiological and synaptic release properties of human embryonic stem cell-derived neurons (iN cells) compared with rat hippocampal pyramidal neurons. We induced the differentiation of hESCs into iN cells by lentiviral delivery of Ngn2-expressing plasmid. Following transduction of hESCs with Ngn2 (days in vitro 0 [div0]), we selected and plated the transduced cells on a glial feeder layer on div3. To compare the basic electrophysiological properties of iN cells with rat hippocampal neurons, we prepared primary rat cultures from neonatal rats by hippocampal dissection and dissociation and assessed them at div14–18 when their synapses reach full maturity. To determine electrophysiological characteristics of human iN cells, we performed whole-cell patch clamp recordings to assess basic properties from div28 to div38 (Figure 1A). In comparison with primary rat hippocampal neurons, iN cells displayed significantly higher membrane resistance (Rm) and lower membrane capacitance (Cm) values (Figures 1B and 1C), consistent with previous studies.20 When we immune stained iN cells with antibodies against the presynaptic marker synapsin-1- and neuron-specific dendrite/soma marker MAP2, we observed that iN cells readily form synapses (Figures 1D and S2F–S2H). Quantification of soma size in MAP2-immunostained samples showed that rat hippocampal neurons are significantly larger than human iN cells, in line with their significantly higher Cm values (Figure 1E).
Figure 1. Electrophysiological characterization of human iN cells.
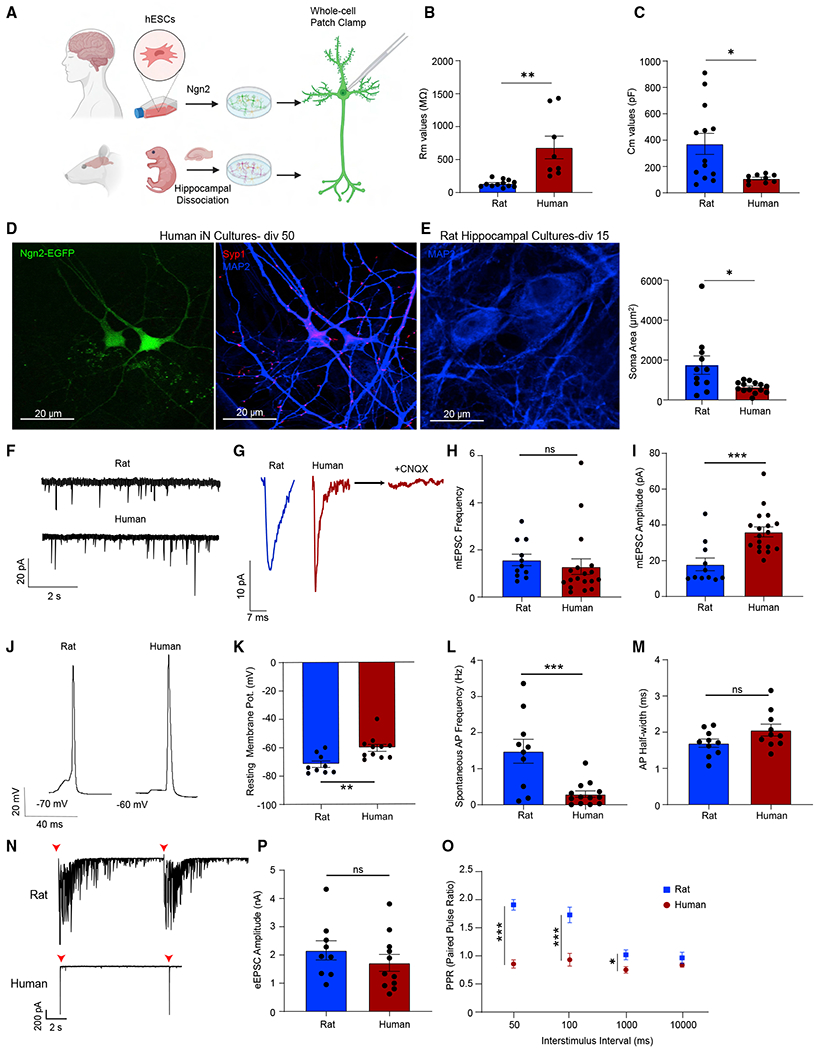
(A) Experimental design for the electrophysiological comparison of human iN and rat hippocampal cultures.
(B and C) Human iN cells have higher membrane resistance (Rm) values and lower capacitance values (Cm) compared with rat hippocampal neurons; dots show individual cells; bars show mean ± SEM; Rm, rat neurons, n = 12; human iN cells, n = 8; N = 2; p = 0.001; unpaired t test; Cm, rat neurons, n = 13; human iN cells, n = 9; N = 2; p = 0.0134; unpaired t test.
(D) Human iN cell immunostaining reveals robust synapse formation (green, Ngn2; blue, MAP2; red, synapsin1). Scale bar, 20 μm.
(E) MAP2 immunostaining reveals that human iN cells are significantly smaller than rat hippocampal neurons. Scale bar, 20 μm. Dots show individual cells; bars show mean ± SEM; rat, n = 11; human, n = 14; N = 2; p = 0.125; unpaired t test.
(F and G) Representative mEPSC traces revealing that iN cells demonstrate sharper miniature events.
(H and I) mEPSC frequency does not differ between human iN cells and rat hippocampal neurons while mEPSC amplitude is significantly higher in iN cells; dots show individual cells; bars show mean ± SEM; mEPSC frequency, rat neurons, n = 11; N = 3; human iN cells, n = 18; N = 3; p = 0.5499; unpaired t test; mEPSC amplitude, rat neurons, n = 11; N = 3; human iN cells, n = 18; N = 3; p = 0.0004; unpaired t test.
(J) Representative APs of human iN cells and rat hippocampal neurons.
(K) iN cell RMP are significantly more depolarized than rat hippocampal neurons; dots show individual cells; bars show mean ± SEM; rat neurons, n = 9; N = 3; human iN cells, n = 11; N = 3; p = 0.0036; unpaired t test.
(L and M) iN cells show lower spontaneous AP frequency compared with rat hippocampal neurons, whereas the AP half-width does not differ; dots show individual cells; bars show mean ± SEM; AP frequency, rat neurons, n = 10, human iN cells, n = 16; N = 3; p = 0.0009; unpaired t test; AP half-width rat neurons, n = 10; N = 3; human iN cells, n = 10; N = 3; p = 0.085; unpaired t test.
(N) Representative traces of eEPSCs of human iN cells and rat hippocampal cells revealing that rat cultures have a profound reverberatory activity upon stimulation.
(O and P) eEPSC amplitudes do not differ between human iN cells and rat hippocampal neurons while the PPRs of rat neurons are significantly higher than human iN cells upon 1, 10, and 20 Hz stimulation; dots show individual cells; bars show mean ± SEM; eEPSC amplitude, rat neurons, n = 9; N = 2; human iN cells, n = 11; N = 3; p = 0.3391; unpaired t test; PPR-50 ms rat n = 8; N = 3; human n = 7; N = 3; p < 0.000001; unpaired t test; PPR-100 ms rat n = 7; N = 3; human n = 9; N = 3; p = 0.000492; unpaired t test; PPR-1,000 ms rat n = 9; N = 3; p = 0.0206; unpaired t test; human n = 10; N = 3; PPR-10,000 ms rat n = 10; N = 3; human n = 11; N = 3; p = 0.234; unpaired t test. Significance are as follows: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; ns, non-significance. See also Figure S1 and Table S1.
Next, we focused on investigating the different modes of synaptic transmission. We first assessed spontaneous miniature excitatory postsynaptic currents (mEPSCs) in the presence of tetrodotoxin to suppress AP firing and detected a similar mEPSC frequency, larger mEPSC amplitudes, and sharper event kinetics, relative to rat hippocampal neurons (Figures 1F–1I and S1A). We then employed current clamp electrophysiology to assess the intrinsic properties and AP dynamics of iN cells and found that the shape, the half-width, and the amplitudes of APs were similar between iN cells and rat hippocampal neurons, although iN cells had a more depolarized resting membrane potential (RMP) and significantly less spontaneous AP firing (Figures 1J–1M and S1B). When we performed stepwise current injections to assess excitability, we found that human iN cells have significantly lower rheobase values indicating increased intrinsic excitability compared with rat hippocampal neurons (Figures S1C and S1D). Despite having a more depolarized RMP and increased excitability, the spontaneous AP frequency is significantly lower in human iN cells, possibly due to homeostatic adaptation to the lack of inhibitory neurotransmission in this purely excitatory network or alternatively due to potential differences in voltage-gated Na+ channel availability. We then assessed evoked excitatory postsynaptic currents (eEPSCs) and found that rat hippocampal neurons display reverberatory activity upon stimulation unlike human iN cells (Figure 1N). We observed that evoked responses are significantly sharper compared with rat hippocampal neurons and did not differ in amplitudes (Figures S1E, S1F, and 1P). Upon high-frequency stimulation, rat hippocampal neurons showed synaptic facilitation, whereas human iN cells show synaptic depression (Figures 1O, S1G, and S1H). This difference may stem from differences in release probability between human iN cells and rat hippocampal neurons.
Desynchronization of evoked neurotransmitter release
To systematically determine the time course of synapse maturation and how different modes of synaptic transmission develop over time, we performed whole-cell voltage clamp recordings at different time points throughout neuronal maturation and observed the first signs of functional synapses around div10, when both mEPSCs and eEPSCs were detectable. From div21 to div35, mEPSC amplitudes did not significantly change, whereas mEPSC frequency increased until div35, and remained unchanged thereafter (Figures 2A–2C). At earlier time points (div10–13), evoked synaptic responses demonstrated a high rate of synaptic failure upon stimulation and facilitated upon high-frequency stimulation compared with more mature time points (div35–38) where high-frequency stimulation resulted in synaptic depression (Figures S1A, S1B, and S1D). We further observed that eEPSC amplitudes increased until div35 and did not change from div35 to div50 (Figures 2D and 2E). The changes in mEPSC frequency and eEPSC amplitudes correlated with an increase in synapse density (Figures S1F–S1H) outlining the time course of iN cell maturation and synaptogenesis. Throughout maturation until approximately div35, we observed iN cells that were silent (66% at div10, 8% at div28) or with low-amplitude evoked responses (<300 pA) and failures, whereas after div35 we found that synaptic responses were more consistent in terms of eEPSC amplitudes and mEPSC frequencies (for coefficients of variation, see Table S1). Therefore, we determined div35 as the point of iN cell synaptic maturity and div50 as an advanced time point to determine the changes that occur in neurotransmitter release in a purely excitatory system upon “in vitro aging.”
Figure 2. Desynchronization of evoked neurotransmitter release.
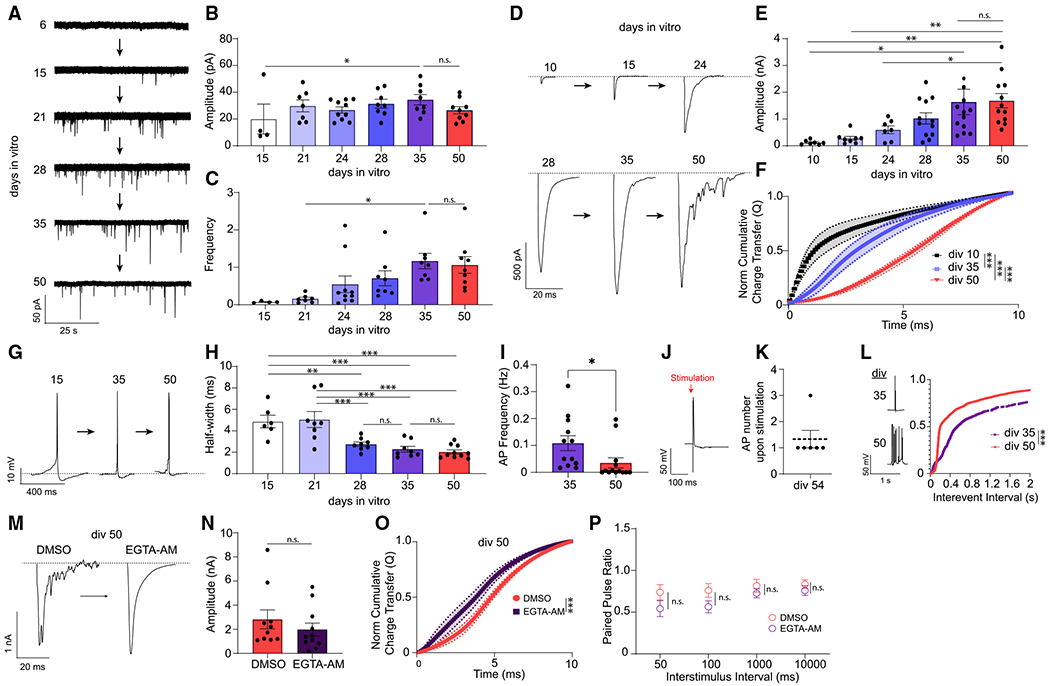
(A) mEPSCs representative traces from human iN cells from days in vitro 6 (div6) to div50.
(B) mEPSC amplitudes shows a significant difference between div15 and div35 but do not change from div21 to div50; dots show individual cells; bars show mean ± SEM; div15, n = 4; div21, n = 7; div24, n = 10; div28, n = 8; div35, n = 8; div50, n = 9; N = 2; p = 0.0423 div15 versus div35; p > 0.99, div35 versus div50; one-way ANOVA-Fisher’s LSD.
(C) mEPSC frequency increases from div21 to div35 and does not change from div35 to div50; dots show individual cells; bars show mean ± SEM; div15, n = 4; div21, n = 7; div24, n = 10; div28, n = 8; div35, n = 8; div50, n = 9;N = 2; p = 0.044 div21 versus div35; p = 0.819, div35 versus div50; one-way ANOVA-Fisher’s LSD.
(D) eEPSC representative traces from div10 to div50.
(E) eEPSC amplitudes increase from div10 to div35 but do not change from div35 to div50; dots show individual cells; bars show mean ± SEM; div10, n = 7; div15, n = 8; div24, n = 7; div28, n = 12; div35, n = 12; div50, n = 12; N = 2; p = 0.0219, div10 versus div35; p = 0.0011,div10 versus div50; p = 0.0021,div15 versus div50; p = 0.0207, div24 versus div50; p = 0.2203, div35 versus div50; one-way ANOVA-Fisher’s LSD.
(F) The NCCT(Q) of eEPSCs at div10, div28, and div50 demonstrating the progressive desynchronization of evoked neurotransmitter release upon in vitro aging; bars show mean ± SEM; div10, n = 6; div28, n = 8; div50, n = 10; N = 2; p < 0.0001; Kolmogorov-Smirnov test; p < 0.001; simple linear regression.
(G) AP representative traces at div10, div35, and div50.
(H) AP half-width decreases as iN cells mature from div15 to div35 but does not change from div35 to div50; dots show individual cells; bars show mean ± SEM; div15, n = 6; div21, n = 8; div28, n = 8; div35, n = 8; div50, n = 10; N = 2; p = 0.003, div15 versus div28; p < 0.001, div15 versus div35; p < 0.001, div15 versus div50; p < 0.001, div21 versus div28; p < 0.001, div21 versus div35; p < 0.001, div21 versus div50; p = 0.446, div28 versus div35; p = 0.661, div35 versus div50; one-way ANOVA-Fisher’s LSD.
(I) Spontaneous AP frequency decreases significantly from div35 to div50 in human iN cells; dots show individual cells; bars show mean ± SEM; div35, n = 12; div50, n = 13; N = 2; p = 0.037; unpaired t test.
(J and K) At div54, upon stimulation of human iN cells we observed single APs 1,000 ms following each stimulation; dots show individual cells; bars show mean ± SEM; n = 6; N = 1.
(L) The interevent intervals (ms) cumulative histogram of APs at div35 and div50 iN cells are different revealing a burst-like firing pattern at div50; bars show mean ± SEM; div35, n = 9; div50, n = 8; N = 2; p < 0.001; simple linear regression; p < 0.0001; Kolmogorov-Smirnov test.
(M) Representative traces of eEPSCs following acute (15 min) DMSO or EGTA-AM treatment at div50.
(N and P) EGTA-AM treatment does not significantly change eEPSC amplitude or PPRs; dots show individual cells; bars show mean ± SEM; eEPSC amplitude, DMSO, n = 10; EGTA-AM, n = 11; N = 2; p = 0.378; unpaired t test; PPR, 50 ms, DMSO, n = 8; EGTA-AM, n = 8; N = 2; p = 0.161; unpaired t test; 100 ms, DMSO, n = 8, EGTA-AM, n = 8; N = 2; p = 0.09; unpaired t test; 1,000 ms, DMSO, n = 9, EGTA-AM, n = 9;N = 2; p = 0.34; unpaired t test; 10,000 ms, DMSO, n = 9, EGTA-AM, n = 10; N = 2; p = 0.35; unpaired t test.
(O) NCCT of eEPSCs following acute (15 min) DMSO or EGTA-AM treatment at div50 reveal that EGTA-AM treatment makes eEPSCs significantly more synchronous; bars show mean ± SEM; DMSO, n = 10; EGTA-AM, n = 11; N = 2; p < 0.001; simple linear regression; p < 0.0001; Kolmogorov-Smirnov test. Significance levels are as follows: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; ns, non-significance. See also Figure S2 and Table S1.
With increasing days in vitro, we observed progressive desynchronization of evoked release, or an increase in asynchronous release, assessed by the cumulative time integral of eEPSC waveforms after a single stimulation (a measure also known as normalized cumulative charge transfer-NCCT(Q)). At div50, the NCCT showed a rightward shift relative to earlier days in vitro, indicating that neurotransmitter release becomes desynchronized upon the arrival of an AP at the presynaptic terminal. We observed this increase in asynchronous release most prominently at div50 (Figure 2F). We did not observe any accompanying changes in mEPSC kinetics, eEPSC amplitudes, total eEPSC charge transfer(Q), or in synapse density at div50 (Figures 2B, 2C, 2E, S2E, S1B, and S1G), demonstrating a specific robust effect of aging.
To determine if this desynchronization is a result distorted AP kinetics, we assessed the intrinsic firing properties of iN cells at different time points, as an increased AP width would result in a prolonged depolarization, potentially causing the desynchronization of release. However, we observed that the RMP of iN cells became more hyperpolarized with increasing days in vitro along with a narrowing of AP half-width, excluding prolongation of AP kinetics as a cause of desynchronization (Figures 2G, 2H, and S1E). In addition, we found that spontaneous AP frequency decreases from div35 to div50, indicating that the increase in asynchronous release is not related to an increase in network activity (Figure 2I). To determine if increased asynchronous activity is associated with an increase in reverberatory activity that could arise from the surrounding network upon stimulation, we quantified the presence of AP trains following stimulation. However, instead of AP trains following stimulation, a sign of reverberatory activity, we detected single APs up to 1 second following stimulation (Figures 2J and 2K). In addition, we did not observe a change in synapse density (Figure S2H) or mEPSC frequency between div35 and div50 (Figure 2C), providing further evidence that an increase in reverberatory network activity does not account for the changes we observe in release synchrony. Interestingly, we observed an increase in burst-like firing at div50, that could be a result of Ca2+ dysregulation in aged iN cells and may be associated with desynchronized neurotransmitter release (Figure 2L).
To determine if the desynchronization of evoked release is a result of dysregulated presynaptic Ca2+, we chelated intracellular calcium in div50 iN cells using the slow Ca2+ buffer EGTA-AM. Upon calcium chelation for 15 min, we observed a significantly more synchronous evoked release without a significant change in eEPSC amplitudes or paired-pulse ratios (PPRs), suggesting that desynchronization of evoked synaptic release is a Ca2+-sensitive process (Figures 2M–2P) as it is classically associated with asynchronous release.21,22 EGTA-AM treatment also causes a decreasing trend in PPR upon high-frequency (10 and 20 Hz) stimulations, suggesting that calcium chelation could reverse the presynaptic effects of in vitro aging and increase release probability (Figure 2P).
The role of NMDA receptor activation and excitatory/inhibitory balance on the desynchronization of evoked neurotransmitter release
The dysregulation of neuronal Ca2+ signaling via aberrant NMDA receptor activation has been associated with several forms of synaptic pathology.23,24 To determine if the desynchronization of evoked release is an NMDAR-dependent process, we investigated evoked NMDA currents at different time points by clamping the membrane potential at +40 mV to remove Mg2+ block of synaptic NMDA receptors. We did not observe an NMDAR-mediated current in iN cells until div35. From div35 to div65, we observed an increase in evoked NMDAR currents, which correlated with the desynchronization of evoked release (Figures 3A, 3B, S3A, and S3B). We therefore hypothesized that desynchronization is influenced by increased NMDAR activation and tested this hypothesis by incubating iN cells with memantine, an NMDAR antagonist, for 15 days after iN cells reached synaptic maturity (div35) (Figure 3C). Chronic memantine treatment (10 μM) resulted in significantly more synchronous evoked release at div50 when compared with untreated neurons, while leaving eEPSC amplitude and PPR unchanged (Figures 3D–3G).
Figure 3. Activation of NMDA receptors mediates the desynchronization of evoked neurotransmitter release.
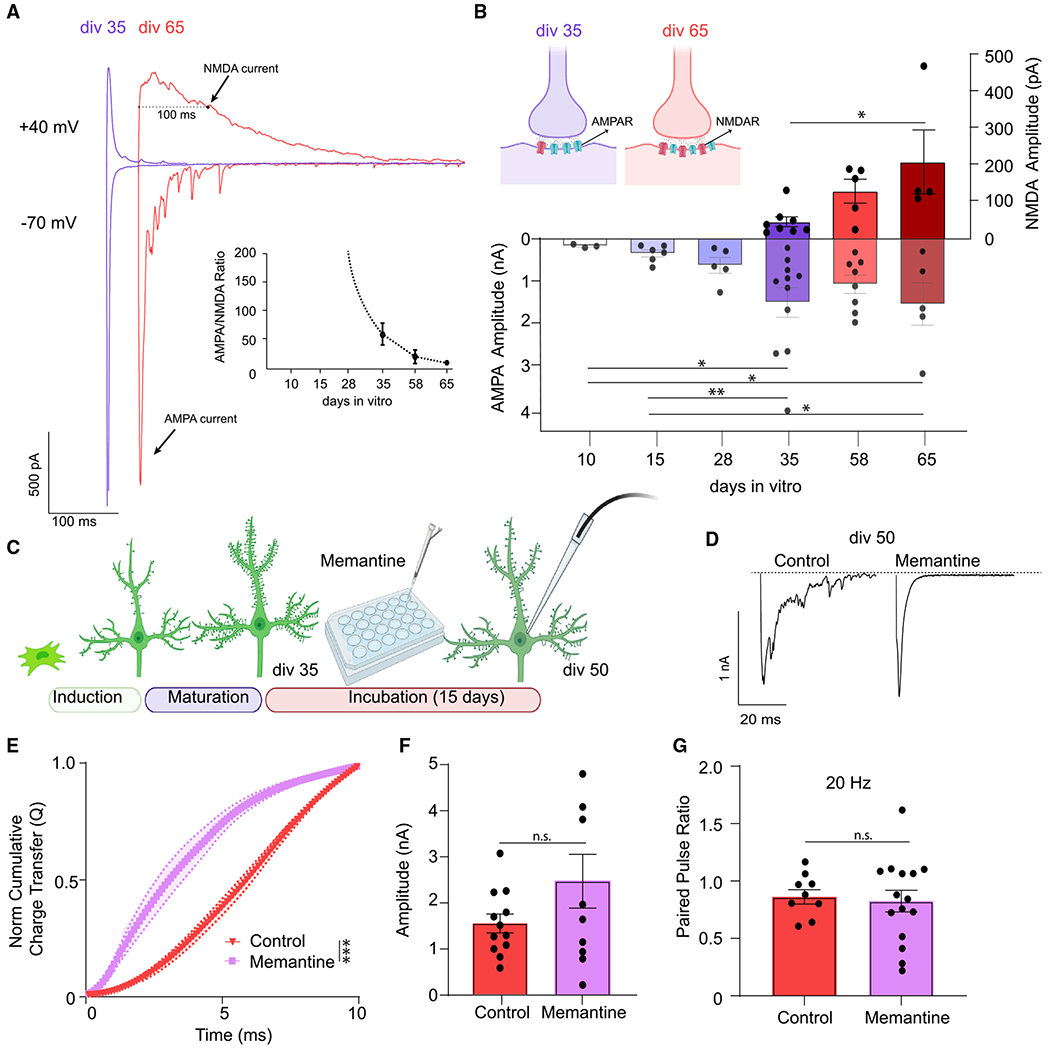
(A) Evoked AMPA currents (at −70 mV) and NMDA currents (at +40 mV) representative traces from iN cells at div35 and div65.
(B) iN cells develop NMDA currents after div35 that significantly increase to div65 correlating with the desynchronization of evoked neurotransmitter release; dots show individual cells; bars show mean ± SEM; div35(NMDA), n = 8; div58(NMDA), n = 5; div65(NMDA), n = 4; N = 3; p = 0.013 div35 versus div65; one-way ANOVA-Fisher’s LSD; div10(AMPA), n = 3; div15(AMPA), n = 6; div28(AMPA), n = 5; div35(AMPA), n = 11; div58(AMPA), n = 8; div65(AMPA), n = 5; N = 3; p = 0.019 div10 versus div35; p = 0.03 div10 versus div65; p = 0.01 div15 versus div35; p = 0.023 div15 versus div65; one-way ANOVA-Fisher’s LSD.
(C) Experimental design for chronic (15 days) 10 μM memantine treatment of div50 iN cells.
(D) eEPSC representative traces following chronic 10 μM memantine treatment versus control at div50.
(E) The NCCT(Q) of eEPSCs following memantine treatment versus controls at div50 reveals that memantine treatment makes evoked release significantly more synchronous; bars show mean ± SEM; control, n = 12; memantine, n = 10; N = 2; p < 0.001; simple linear regression; p < 0.0001; Kolmogorov-Smirnov test.
(F and G) The PPR or the eEPSC div50 amplitudes do not change following chronic 10 μM memantine treatment; dots show individual cells; bars show mean ± SEM; eEPSC amplitude, control, n = 12; memantine, n = 10; N = 2; p = 0.127; unpaired t test; PPR, control, n = 9; memantine, n = 15; N = 2; p = 0.783; unpaired t test. Significance levels are as follows: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; ns, non-significance. See also Figure S3 and Table S1.
Overactivation of NMDARs under conditions where excitatory and inhibitory inputs onto neurons are imbalanced (excitatory/inhibitory [E/I] imbalance) is a hallmark of various neurodegenerative processes and is the initiator of pathological signaling cascades.23,25 In our system, we have an extreme case, where excitatory glutamatergic transmission develops in the absence of concomitant inhibition. To evaluate the role of overexcitation and maladaptive NMDAR signaling in progressive desynchronization of release, we tested if increasing the inhibitory tone by chronic incubation with muscimol, a GABAA receptor agonist, would restore the E/I imbalance and correct the desynchronization of evoked release (Figure 4A). Here, induced human cortical neurons express GABAA receptors but lack the molecular machinery to produce GABA4(Figures S3C and S3D). Similar to memantine, chronic (15 day) incubation with muscimol (10 μM) synchronized evoked release and resulted in a decrease in the PPR at 50 ms interstimulus interval, without significantly affecting the eEPSC amplitudes (Figures 4B–4E).The decrease in PPR upon chronic muscimol treatment suggests an increase in release probability, which may reflect reversal of presynaptic effects of in vitro aging.
Figure 4. Muscimol treatment and introduction of inhibitory input renormalizes desynchronized evoked neurotransmitter release.
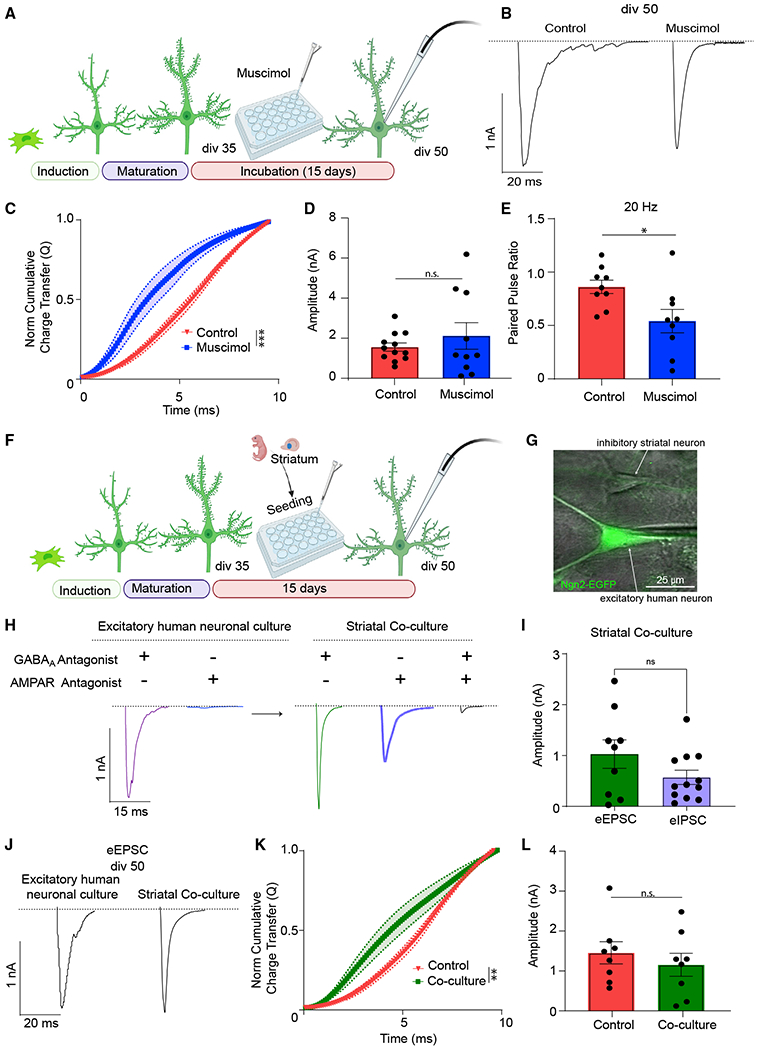
(A) Experimental design for chronic (15 days) 10 μM muscimol treatment of div50 iN cells.
(B) Representative traces of eEPSCs following chronic muscimol treatment versus control at div50.
(C) The NCCT(Q) of eEPSCs following muscimol treatment versus controls at div50 reveals that muscimol treatment makes evoked release significantly more synchronous; bars show mean ± SEM; control, n = 10; muscimol, n = 7; N = 2; p < 0.001; simple linear regression; p < 0.0001; Kolmogorov-Smirnov test.
(D and E) The amplitude of eEPSCs does not change following muscimol treatment, whereas the PPR at 20 Hz stimulation becomes significantly decreased in treated cultures; dots show individual cells; bars show mean ± SEM; eEPSC amplitude, control, n = 12; muscimol, n = 10; N = 2; p = 0.396; unpaired test; PPR, control, n = 9; muscimol, n = 9; N = 2; p = 0.02; unpaired t test.
(F) Experimental design for rat striatal-human iN cell co-cultures.
(G) Representative image of whole-cell patch clamping of EGFP(+) iN cells in proximity of an inhibitory striatal neuron. Scale bar, 25 μm.
(H) Representative traces of eEPSC currents of iN cells showing the absence of inhibitory currents in pure iN cell cultures and presence upon rat striatal coculturing. The representative postsynaptic responses were recorded from the same cell in each group, upon perfusion of blockers.
(I) iN cells that are co-cultured with rat striatal neurons show robust inhibitory transmission where eEPSC and eIPSC amplitudes do not differ (recordings were performed from different iN cells); dots show individual cells; bars show mean ± SEM; eEPSC n = 9; eIPSC n = 12; N = 2; p = 0.131; unpaired t test.
(J) div50 eEPSCs representative traces following rat striatal co-culturing versus pure iN cell cultures.
(K and L) The NCCT(Q) of eEPSCs following rat striatal co-culturing versus pure iN cultures at div50 reveals that inhibitory innervation makes evoked release significantly more synchronous with no change in eEPSC amplitudes; bars show mean ± SEM; NCCT, control, n = 6; striatal co-cultures, n = 9; N = 2; p = 0.002; simple linear regression; p < 0.0001; Kolmogorov-Smirnov test; eEPSC amplitudes, control, n = 8; striatal co-cultures, n = 8; N = 2; p = 0.466; unpaired t test. Significance levels are as follows: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; ns, non-significance.
As muscimol partially restored the E/I imbalance and rescued the desynchronization of evoked glutamate release, we asked if this imbalance could also be restored by introducing inhibitory neurons and reconstituting GABAergic neurotransmission in this excitatory network. For this purpose, we used primary rat striatal neurons, which are inhibitory medium spiny neurons and require excitatory innervation to maintain viability in the long term.26–28 After dissection and dissociation of striatum from neonate rat pups, we plated striatal cells on human iN cells at div35 (Figure 4F) and assessed the contribution of inhibitory innervation to iN cell synaptic function at div50. EGFP allowed us to isolate and record from iN cells under a fluorescence microscope in a co-culture of human iN cells and rat striatal neurons (Figure 4G). The presence of functional synapses between the two neuron types was validated by recording evoked inhibitory postsynaptic currents (eIPSCs), which were not detectable prior to rat striatal neuron seeding (Figures 4H, 4I, and S3E). In this co-culture system, eEPSC amplitudes did not change relative to purely excitatory cultures (Figures 4J–4L) but became more synchronous, providing further evidence that the desynchronization of evoked glutamate release is precipitated by E/I imbalance.
The molecular mechanisms underlying desynchronization of evoked neurotransmitter release
In a majority of central presynaptic terminals, Syt-1 acts as the main calcium sensor responsible for fast synchronous release, whereas Syt-7 regulates asynchronous release.29–31 Therefore, we hypothesized that Syt-7 may act as the predominant Ca2+ sensor in in-vitro-aged synapses sustaining asynchronous release. Knockdown of Syt-7 using lentivirus-mediated transduction of Syt-7-shRNA resynchronized eEPSCs without changing eEPSC amplitudes (Figures 5A–5F). To determine the changes in presynaptic release machinery that render Syt-7 predominant in mediating desynchronization, we measured protein levels of the soluble NSF attachment protein receptors (SNAREs) by immunoblotting. We did not observe a change in Syt-7 protein levels upon in vitro aging, nor did we observe a change in syntaxin-1, synaptosomal-associated protein 25 (SNAP25), or synaptobrevin-2 assessed by their respective band densities (Figures 5G–5L). However, we observed a decrease in Syt-1, the main presynaptic Ca2+ sensor regulating synchronous release and a decrease in CSPα expression, a presynaptic chaperone protein that is responsible for proper SNARE maintenance in the presynaptic terminal30,32 (Figures 5J and 5M). This decrease in both CSPα and Syt-1 was rescued when iN cells were treated with memantine or with muscimol, which resynchronize evoked release in aging iN cells (Figures 3E, 4C, 5J, and 5M). To determine if the lentivirus-mediated expression of Syt-1 or CSPα could directly rescue the desynchronization of evoked release, we transduced iN cells with Syt-1- or CSPα-expressing vectors at div35 and assessed evoked release at div50 (Figure 5N). Increasing Syt-1 levels by 31% made evoked release significantly more synchronous (Figure 5S). We did not observe a change in CSPα levels following Syt-1 overexpression, whereas CSPα overexpression significantly increased both Syt-1 and CSPα levels, suggesting that the decrease in CSPα acts upstream of the decrease in Syt-1, which eventually results in the observed increase in desynchronized glutamate release (Figures 5P–5S).
Figure 5. The molecular mechanisms underlying desynchronized neurotransmitter release.
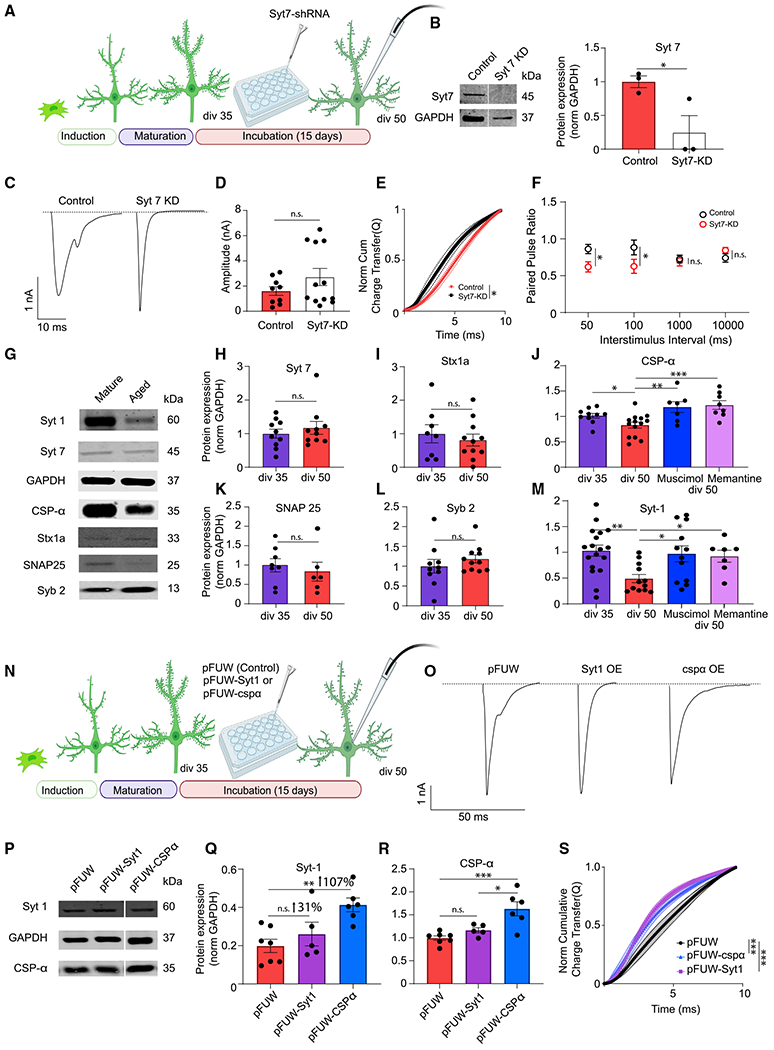
(A) Experimental design for iN Syt-7 knockdown (KD) experiment.
(B) Representative western blots and Syt-7 protein levels in iN cell lysates transduced with control or Syt-7-KD lentivirus; dots show individual coverslips; bars show mean ± SEM; control, n = 3; Syt-7-KD, n = 3; N = 2; p = 0.04; unpaired t test.
(C) Representative traces of control or Syt-7-KD eEPSCs at div50.
(D) eEPSC amplitude does not change significantly following Syt-7-KD; dots show individual cells; bars show mean ± SEM; control, n = 9; Syt-7-KD, n = 12; N = 2; p = 0.201; unpaired t test.
(E) The NCCT of eEPSC following Syt-7-KD shows synchronization of evoked release suggesting that Syt-7 acts as the calcium sensor in asynchronous release at div50; bars show mean ± SEM; control, n = 7; Syt-7-KD, n = 12; N = 2; p = 0.03; simple linear regression; p < 0.0001, Kolmogorov-Smirnov test.
(F) Syt-7-KD does not affect the PPR at low-frequency stimulation (0.1–1 Hz) but significantly decreases the PPR at high-frequency stimulation (10–20 Hz); bars show mean ± SEM; 50 ms-control, n = 9; Syt-7-KD, n = 5; N = 2; p = 0.04; unpaired t test; 100 ms-control, n = 10; Syt-7-KD, n = 5; N = 2; p = 0.02; unpaired t test; 1,000 ms-control, n = 11; Syt-7-KD, n = 5; N = 2; p = 0.846; unpaired t test; 10,000 ms-control, n = 12; Syt-7-KD, n = 6; N = 2; p = 0.32; unpaired t test.
(G, H, I, K, and L) Representative western blots and Syt-7, syntaxin-1a, SNAP25, and synaptobrevin-2 protein levels in iN cell lysates at div35 and div50; dots show coverslips; bars show mean ± SEM; Syt-7, div35, n = 10; div50, n = 6; N = 3; p = 0.447; unpaired t test; syntaxin-1a, div5, n = 8; div50, n = 11; N = 3; p = 0.5519; unpaired t test; SNAP25, div35, n = 8; div50, n = 6; N = 3; p = 0.557; unpaired t test; Syb2, div35, n = 10; div50, n = 11; N = 3; p = 0.356; unpaired t test.
(J–M) Western blot quantification reveals a decrease of Syt-1 and CSPα at div50 compared with div35. The decrease in both CSPα and Syt-1 were rescued by memantine or muscimol treatment; dots show coverslips; bars show mean ± SEM; Syt-1; div35, n = 15; div50, n = 12; div50 + muscimol, n = 12; div50 + memantine, n = 7; N = 3; p = 0.002, div35 versus div50; unpaired t test; p = 0.024, div50 versus div50 + muscimol; p = 0.041, div50 versus div50 + memantine; one-way ANOVA-Fisher’s LSD; CSPα, div35, n = 10; div50, n = 14; div50 + muscimol, n = 7; div50 + memantine, n = 8; N = 3; p = 0.03, div35 versus div50; unpaired t test; p = 0.001, div50 versus div50 + muscimol; p < 0.001, div50 versus div50 + memantine; one-way ANOVA-Fisher’s LSD.
(N) Experimental design for Syt-1, CSPα rescue experiments.
(O) Representative traces of eEPSC at div50 following infection with control (pFUW) virus, Syt-1-expressing virus, or CSPα-expressing virus.
(P–R) Representative western blots and Syt-1 and CSPα protein levels in iN cell lysates at div50 following infection with control virus, Syt-1-expressing virus, or CSPα-expressing virus; dots show coverslips; bars show mean ± SEM; Syt-1, pFUW, n = 7; pFUW-Syt-1, n = 5; pFUW-CSPα, n = 6; N = 3; p = 0.329, pFUW versus pFUW-Syt-1; p = 0.002, pFUW versus pFUW-CSPα; p = 0.03, pFUW-CSPα versus pFUW-Syt-1; one-way ANOVA-Fisher’s LSD; CSPα, pFUW, n = 7; pFUW-Syt-1, n = 5; pFUW-CSPα, n = 6; N = 3; p = 0.729, pFUW versus pFUW-Syt-1; p < 0.001, pFUW versus pFUW-CSPα; p = 0.018, pFUW-CSPα versus pFUW-Syt-1; one-way ANOVA-Fisher’s LSD.
(S) The NCCT of eEPSCs at div50 following infection with Syt-1 or CSPα rescue vectors show that increased expression of either Syt-1 or CSPα make evoked release more synchronous; bars show mean ± SEM; pFUW, n = 12; pFUW-Syt-1, n = 12; pFUW-CSPα, n = 11; N = 2; p < 0.01 for both pFUW versus pFUW-Syt-1 and pFUW versus pFUW-CSPα; simple linear regression; p < 0.0001 for both pFUW versus pFUW-Syt-1 and pFUW versus pFUW-CSPα, Kolmogorov-Smirnov test. When the representative bands in the figures belong to different membranes or different lanes on the same membrane, they are separated by a blank space. When they are in the neighboring lanes, they are presented without a blank space. Significance levels are as follows: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; ns, non-significance. See also Table S1.
The role of ER stress response on the desynchronization of evoked neurotransmitter release
Our data suggest that the desynchronization of neurotransmitter release in aging iN cells in vitro is precipitated by NMDAR overactivation due to the excessive E/I imbalance, which in turn decreases levels of Syt-1 and CSPα expression, rendering Syt-7 as the prominent Ca2+ sensor mediating evoked release. However, we do not know how NMDAR overactivation could lead to a decrease in Syt-1 and CSPα expression. NMDAR overactivation is known to disrupt intracellular Ca2+ homeostasis and cause Ca2+ overload in the cytoplasm that impairs ER function.33,34 This perturbation of ER function leads to the accumulation of unfolded or misfolded proteins in the ER lumen which activates an adaptive cellular program, the ER stress response.34–36 Intraluminal accumulation of misfolded proteins in the ER results in an increase in ER chaperones to maintain proper protein folding through activating Ire1α and PERK proteins.37 Activation of Ire1α and PERK proteins initiate signaling cascades, through activation of XBP1 and phosphorylation of eIF2α, that in turn modulate the transcription of various proteins (ER chaperones, transcription factors) to ensure cellular homeostasis37 (Figure 6A). We therefore hypothesized that components of the ER stress response may mediate the changes that we observe in the presynaptic terminal upon iN cell aging.
Figure 6. The role of ER stress response in the desynchronization of evoked neurotransmitter release.
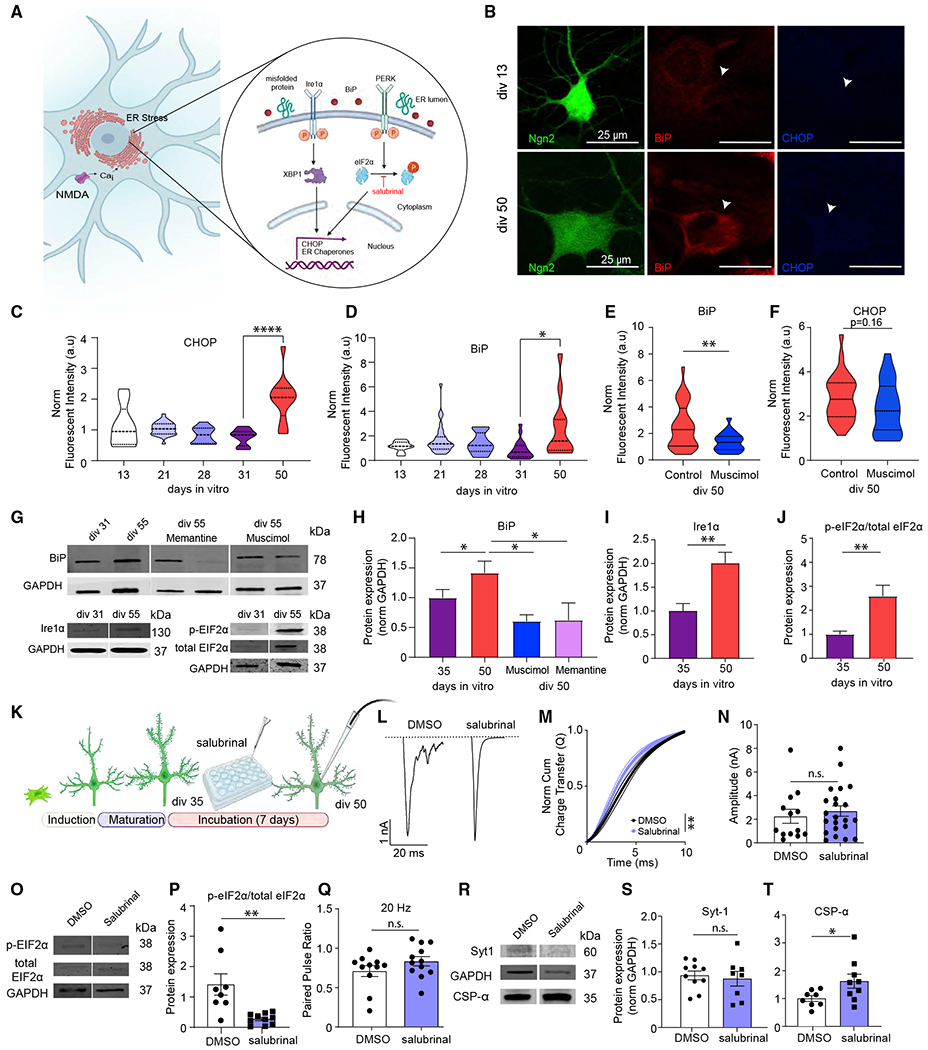
(A) Schematic image outlining the endoplasmic reticulum (ER) stress response.
(B) Representative images of immunofluorescent staining of iN cells at div13 and div50 against ER stress markers BiP and CHOP (green, Ngn2; red, BiP; blue, CHOP). Scale bar, 20 μm.
(C and D) Fluorescent intensity of BiP and CHOP immunofluorescent signal throughout maturation from div13 to div50 reveals increased ER stress markers at div50; bars show mean ± SEM; CHOP, div3, n = 9; div21, n = 28; div28, n = 13; div31, n = 19; div0, n = 12; N = 2; p < 0.0001, div31 versus div50; one-way ANOVA-Tukey’s multiple comparison test; BiP, div13, n = 9; div21,n = 28; div28, n = 13; div31, n = 19; div50, n = 12; N = 2; p = 0.03, div31 versus div50; one-way ANOVA-Tukey’s multiple comparison test.
(E and F) Fluorescence intensity of BiP and CHOP immunofluorescent signal at div50 upon chronic (15 days) muscimol treatment reveals significantly decreased BiP fluorescence intensity and a decreasing trend for CHOP fluorescence intensity; bars show mean ± SEM; BiP, div50 control, n = 29; div50 muscimol, n = 22; N = 2; p = 0.0056; Mann-Whitney U test; CHOP, div50 control, n = 29; div50 muscimol, n = 24; N = 2; p = 0.16; Mann-Whitney U test.
(G–J) Representative western blots, BiP and Ire1 α protein levels, and eIF2α phosphorylation at div31 and div55; bars show mean ± SEM; BiP, div35, n = 10; div50, n = 9; div50 + muscimol, n = 4; div50 + memantine, n = 3; N = 2; p = 0.04, div35 versus div50; p = 0.01, div50 versus div50 + muscimol; p = 0.02, div50 versus div50 + memantine; one-way ANOVA-Fisher’s LSD; Ire1α, div35, n = 10; div50, n = 13; N = 2; p = 0.003; unpaired t test; eIF2α phosphorylation, div35, n = 8; div50, n = 9; N = 2; p = 0.0048; unpaired t test.
(K) Experimental design for iN cell treatment with the ER stress inhibitor salubrinal.
(L) eEPSC representative traces following 7–10 day incubation with either DMSO, or salubrinal.
(M) The NCCT of eEPSCs following 7–10 day incubation with DMSO or salubrinal reveals that salubrinal makes evoked neurotransmitter release significantly more synchronous; bars show mean ± SEM; DMSO, n = 13; salubrinal, n = 12; N = 3; p = 0.003; simple linear regression; p < 0.0001, Kolmogorov-Smirnov test.
(N and Q) The amplitude and PPR (20 Hz) does not change upon incubation with salubrinal treatment; dots show individual cells; bars show mean ± SEM; eEPSC amplitude, DMSO, n = 13; salubrinal, n = 22; N = 2; p = 0.55; unpaired t test; PPR, DMSO, n = 11; salubrinal, n = 12; N = 2; p = 0.18; unpaired t test.
(O and P) Representative western blots and quantification reveal that eIF2α phosphorylation is decreased upon 7-to 10-day-long incubation with salubrinal; dots show individual coverslips; bars show mean ± SEM; eIF2α phosphorylation, DMSO, n = 8; salubrinal, n = 10; N = 2; p = 0.0014; Mann-Whitney U test.
(R–T) Representative western blots and Syt-1 and CSPα quantification reveal that salubrinal treatment does not change Syt-1, whereas it increases CSPα expression; dots show coverslips; bars show mean ± SEM; Syt-1, DMSO n = 10, salubrinal n = 8; N = 2; p = 0.6938; unpaired t test. CSPα, DMSO n = 8, salubrinal n = 9; N = 2; p = 0.045; unpaired t test. When the representative bands in the figures belong to different membranes or different lanes on the same membrane, they are separated by a blank space. When they are in the neighboring lanes, they are presented without a blank space. Significance levels are as follows: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; ns, non-significance. See also Table S1.
To test this premise, we immune stained iN cells at different time points for binding immunoglobulin protein (BiP), a chaperone protein that increases during ER stress response and C/EBP homologous protein (CHOP), the transcription factor that activates pro-apoptotic cascades upon prolonged ER stress response.37,38 We observed a significant increase in BiP and CHOP signals at div50 compared with div35 (Figures 6B–6D). When we immune stained human iN cells that were treated with muscimol, we observed a decrease in BiP and a trending decrease in CHOP (Figures 6E and 6F). We further confirmed the increase in BiP with in vitro aging by immunoblotting and observed that memantine and muscimol treatment cause a remarkable decrease in BiP levels (Figures 6G and 6H). To determine if different ER stress pathways become activated upon in vitro aging of human iN cells, we immunoblotted iN cell samples at div35 and div50 for Ire1α, phospho-eIF2α, and total eIF2α. We observed a significant increase in both Ire1α and eIF2α phosphorylation, suggesting the activation of both Ire1α- and PERK-mediated pathways upon in vitro aging (Figures 6I and 6J). To determine if the ER stress response plays a role in evoked release desynchronization, we treated iN cells with salubrinal (5 μM), an inhibitor of the PERK-mediated phosphorylation of eIF2α (Figure 6K). Under these conditions, we observed significant resynchronization of glutamate release in salubrinal-treated neurons compared with the controls at div50, providing further evidence that components of the ER stress response act as an intermediary that leads to eEPSC desynchronization (Figures 6L and 6M). We immunoblotted salubrinal- and DMSO-treated iN cell samples with phoshpho-eIF2α and total eIF2α antibodies and confirmed the pharmacological action of salubrinal in inhibiting e-IF2α phosphorylation (Figures 6O and 6P). Salubrinal treatment did not have a significant effect on eEPSC amplitude or PPR at the 50 ms interevent interval (Figures 6N–6Q). Finally, we immunoblotted salubrinal- and DMSO-treated iN cell samples with Syt1 or CSPα antibodies and found that salubrinal treatment increased CSPα levels without significantly affecting Syt1 levels (Figures 6R–6T).
Age-related changes in the presynaptic machinery translates to postmortem human cortex
So far, in a purely excitatory network, we found that in vitro aging results in desynchronization of evoked neurotransmitter release mediated by Syt-7, which is precipitated by a decrease in presynaptic Syt-1 and CSPα protein levels as a result of E/I imbalance and NMDAR overactivation. In addition, the ER stress response acts as a mechanism that alters the presynaptic neurotransmitter release machinery. The changes in presynaptic machinery and desynchronization of eEPSCs in synaptic aging could explain the prodromal signs of the synaptic changes in the aged human brain and ageing-related cognitive decline. Therefore, we hypothesized that the decrease in Syt-1 and CSPα protein levels upon in vitro aging of human iN cells also takes place in the aging human brain. To test this prediction, we immunoblotted postmortem human cortical samples of different ages for different SNARE proteins (Figure 7A). We dissected and immunoblotted three separate samples from each subject and normalized the band intensities to the synapse density by using synaptophysin as an internal control. When normalized to the synaptic vesicle protein synaptophysin, we observed a significant decrease in Syt-1, Syt-7, and CSPα protein levels with increased age, whereas age did not significantly correlate with levels of key SNARE proteins syntaxin-1, SNAP25, or synaptobrevin-2, suggesting that our molecular in vitro aging findings could be relevant to the aged human cortex, underlining the potential involvement of desynchronized neurotransmitter release in the aging human brain, possibly mediating ageing-associated cognitive decline (Figures 7B–7H and S4A–S4F).
Figure 7. Age-related changes in the presynaptic machinery translates to postmortem human cortex.
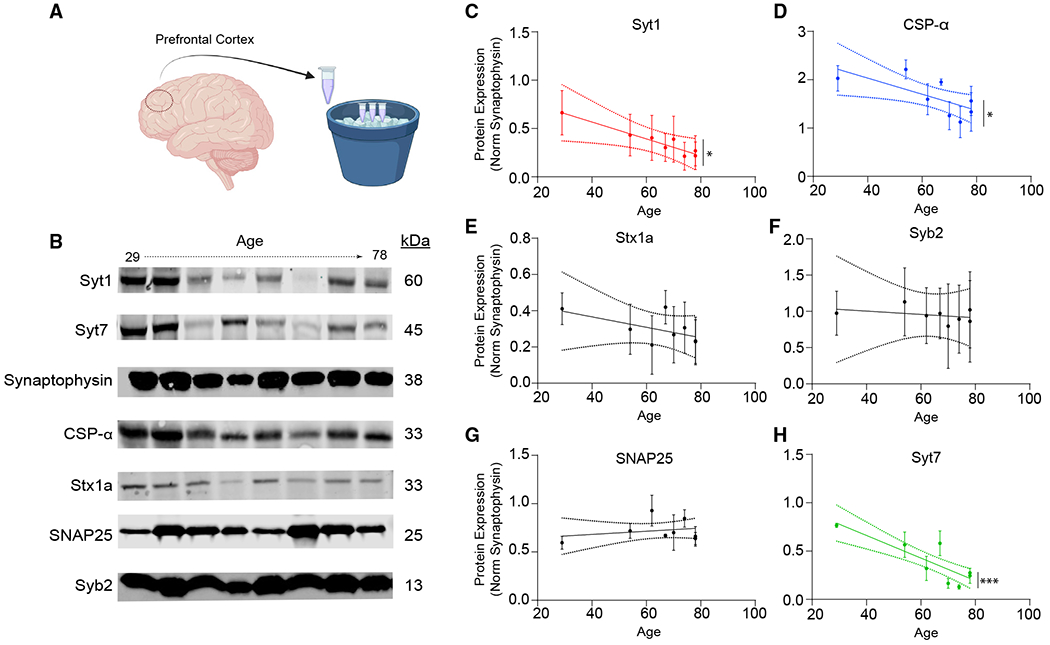
(A and B) The experimental design and representative western blots showing SNARE protein levels in postmortem human cortex as a function of age.
(C, D, and H) Syt-1, CSPα, and Syt-7 levels reveal that these proteins significantly decrease with age; bars show mean ± SEM; n = 8; experiments were replicated using three distinct cortical samples from the same subjects; N = 3; p = 0.027 (Syt-1); p = 0.022 (CSPα); p < 0.001 (Syt-7); simple linear regression.
(E–G) Stx1a, Syb2, and SNAP25 quantification reveals no change in expression with age; bars show mean ± SEM; n = 8; experiments were replicated using three distinct cortical samples from the same subjects; N = 3; p = 0.302 (Stx1a); p = 0.806 (Syb2); p = 0.497 (SNAP25); simple linear regression. When the representative bands in the figures belong to different membranes or different lanes on the same membrane, they are separated by a blank space. When they are in the neighboring lanes, they are presented without a blank space. Significance levels are as follows: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; ns, non-significance. See also Figure S4 and Table S1.
DISCUSSION
In this study, we monitored the progression of synaptic properties following initial formation and maturation of synaptic contacts between human iN cells. These experiments revealed that, as these synapses age in vitro, their evoked release kinetics transition to asynchronous release mediated by Syt-7 following a decrease in presynaptic Syt-1 and CSPα protein levels. This change in synaptic transmission is a result of unopposed NMDAR activation due to a severely disrupted E/I balance favoring excitation. Our data further suggest that ER stress acts as a key pathway that mediates these synaptic changes by linking NMDA receptor activation to a decrease in Syt-1 and CSPα and subsequent desynchronization.
Age-related desynchronization of neurotransmitter release has been described previously in mice at the endbulb of Held synapse.39,40 This desynchronization was associated with an age-related hearing loss phenotype and was proposed to be the result of ineffective calcium buffering.39 Here, we describe the desynchronization of evoked neurotransmitter release as a precursor to aging in human synapses. The data we obtained from our in vitro model causally link this desynchronization to NMDAR overactivation due to E/I imbalance. We believe these observations are significant because various studies have highlighted the importance of E/I balance in proper neural functioning.41–43 Aging is associated with an increase in excitatory signaling, resulting in an E/I imbalance, which has been linked to shorter lifespans in many species.44–46 In humans, suppression of genes that are involved in excitatory neurotransmission is correlated with extended longevity.47,48 In addition, impairments in inhibition has been associated with increased susceptibility to unprovoked seizures in Alzheimer disease (AD) patients.49–51 Our findings delineate the effect of NMDAR activation and E/I balance on synaptic neurotransmitter release and show that the desynchronization of evoked release is an NMDAR-mediated event, which can be rescued by chronic treatment with memantine, an NMDA antagonist that has wide clinical use in the treatment of mild cognitive impairment and AD.52,53
We found that synaptic aging is associated with a decrease in Syt-1 and CSPα levels, suggesting that these synaptic vesicle proteins act as key molecules underlying desynchronized neurotransmitter release. Synchronous release occurs when a presynaptic AP results in a rapid increase in presynaptic Ca2+ concentration that catalyzes vesicle fusion.54,55 The canonical SNARE complex is zippered through the vesicle SNARE synaptobrevin-2 and the plasma membrane SNAREs syntaxin-1 and SNAP25. Syt-1 acts as the major Ca2+ sensor for synchronous neurotransmitter release.30 Syt-1 knockout (KO) mice demonstrate a robust profile that shifts the release time into a delayed and desynchronized form in response to a stimulated increase in presynaptic Ca2+.56 We observed an approximately 50% decrease in Syt-1 levels upon in vitro aging, which was associated with an increase in asynchronous release. In a recent study, 50% decrease in Syt-1 levels in Syt-1(+/−) heterozygous mouse neuron preparations was not associated with a change in eEPSC release kinetics until Syt-1 was further reduced by approximately 75%.57 We believe that this discrepancy stems from the additional effects of E/I balance and ER stress on synaptic release machinery in our system. In particular, the decrease in CSPα levels likely exacerbate the desynchronization of evoked release as CSPα overexpression by itself is sufficient to re-establish release synchrony. The asynchronous release seen following Syt-1 loss is significantly suppressed when Syt-7 is knocked down,29,58 underlining the role of Syt-7 as the Ca2+ sensor predominately regulating asynchronous release. In line with previous reports, when we knocked down Syt-7 in aged iN cells we observed a more synchronized form of evoked release, indicating that Syt-7 mediates desynchronized release in aged synapses following a decrease in Syt-1. Contrary to the homogeneous loss of Syt-1 in the entire neuronal network in Syt-1 KO cultures, in vitro aging could result in a network that has a heterogeneous population of synapses that has decreased Syt-1. Although we found a decrease in Syt-1 expression in iN cell populations at div50, a number of these synapses may still contain Syt-1 and, likely, other synaptotagmins that may compete with Syt-7 for Ca2+-mediated release of neurotransmitters. This notion may also explain why eEPSC amplitudes are largely preserved and why the synchronous component of evoked release is still detectable.
Continuous function of the fusion machinery requires preservation of presynaptic proteostasis via proper folding and maintenance of synaptic vesicle fusion and endocytosis machinery components.32 CSPα is a presynaptic chaperone protein that serves to maintain synaptic proteostasis, and the loss of CSPα causes synapse degeneration.59 CSPα interacts with syntaxin,60 dynamin 1,61,62 SNAP25,63 and Syt-1.64 In CSPα KO mice, evoked neurotransmitter release at the calyx of Held becomes desynchronized.59 This desynchronization is consistent with the synaptic dysfunction and neurodegenerative profile of CSPα KO mice, underscoring the function of CSPα in maintaining the presynaptic machinery that mediates release synchrony.59 In our system, Syt-1 or CSPα overexpression in aging synapses re-synchronizes release, with Syt-1 overexpression producing a more robust effect. Moreover, CSPα overexpression resulted in an increase in Syt-1 levels, suggesting that CSPα may act upstream to regulate Syt-1 expression. Our in vitro molecular findings translate to the aged human brain as increased chronological age in postmortem human cortical samples negatively correlated with Syt-1 and CSPα protein levels irrespective of synapse density, as assessed by synaptophysin protein levels. A decrease in Syt-1 and CSPα was previously reported in postmortem brain samples of AD patients. However, the functional relevance of Syt-1 and CSPα expression to synaptic aging remained unclear.65,66 Interestingly, we also found a decrease in Syt-7 levels upon aging, which increases the possibility of altered release synchrony in the aged human cortex. Differential changes in the Syt-1/Syt-7 ratio may regulate the synchrony of neurotransmitter release in aged human brain and could be one of the contributing factors to aging-associated cognitive changes. Moreover, the decrease in Syt-7 may also be a compensatory mechanism to limit desynchronization of neurotransmitter release during aging.
We propose the ER stress response as a potential aging-associated pathway that mediates the link between NMDAR overactivation and decreased Syt-1 and CSPα levels. The ER functions to sequester Ca2+ to maintain calcium homeostasis in the cell. NMDAR overactivation is known to cause a potentially deleterious level of Ca2+ influx that disrupts ER homeostasis.36,67–69 Perturbations in ER homeostasis disrupt protein folding and lead to misfolded protein accumulation.37 To limit the accumulation of these proteins, cells initiate an adaptive response by activating a signaling pathway termed the ER stress response or the unfolded protein response.37 The ER stress response can lead to transcriptional changes in an effort to alleviate ER stress. However, prolonged ER stress could be maladaptive and trigger apoptosis.38 The relative amounts of unfolded proteins are increased with cellular aging70 and are associated with a breakdown of protein quality control systems that eventually results in the ER stress response.71 In line with these findings, we observed an increase in BiP, an ER chaperone, and CHOP, a transcription factor associated with activation of apoptotic pathways. Furthermore, inhibiting eIF2α phosphorylation, one of the main pathways mediating the ER stress response,71 resulted in a more synchronous form of release and increased CSPα levels, suggesting that ER stress may represent one of the pathways that transcriptionally affects the composition of the presynaptic machinery in synaptic aging. Syt-1 levels did not change upon salubrinal treatment, although our data suggest that CSPα acts upstream of Syt-1 in synchronizing release in aged iN cells. This discrepancy may stem from the possibility that resolution of Syt-1 immunoblotting may not be sufficient to detect the changes in synaptic Syt-1 levels upon salubrinal treatment.
The use of human iN cells differentiated from hESCs or iPSCs by Ngn-2 overexpression has become more common in the recent years.72 Our results demonstrate a robust ubiquitous pathway that may confound results in studies that use the iN cell systems. Investigators should consider pathophysiological changes that occur as a result of E/I imbalance at advanced days in vitro when using purely excitatory human iN cells as models of disease. The emergence of ER stress and the subsequent desynchronized neurotransmitter release are important factors that should be considered when exploring neurological disease processes.
Taken together, our results indicate that E/I balance and NMDA receptors are vital components of synaptic functional homeostasis and that E/I imbalance coupled with an increase in excitation can directly impact synaptic vesicle release machinery and result in desynchronization of neurotransmitter release. The desynchronization of release was reversible in response to direct manipulations that target NMDAR activity, E/I balance, Syt-1, CSPα, and Syt-7 levels. Moreover, targeting the ER stress response using small molecules also reversed the progressive desynchronization seen in aging synapses. Therefore, these observations not only point toward a potentially deleterious pathway that desynchronizes release kinetics prior to synapse degeneration during aging and related neurodegenerative disorders but also a possible plasticity pathway where alterations in E/I balance may impact release synchrony in the short term and alter information coding in synaptic networks. Future studies could aim to determine the changes in the presynaptic release machinery and release synchrony and in neurological and psychiatric disorders where NMDAR overactivation and ER stress play a role. In addition, further investigations may determine how ER stress causes these changes in the presynaptic terminal and whether ER stress inhibitors can be used as therapeutics to alleviate the dysregulation of neurotransmitter release.
Limitations of the study
Although we identify the desynchronization of neurotransmitter release and its underlying mechanisms in purely excitatory iN cell cultures, additional experiments would be needed to relate our findings to the aging human brain. The functional relevance of our findings to brain aging could be investigated through electrophysiological recordings of post-operational brain slices of young and aged subjects to test whether similar processes take place. Moreover, in the immunoblotting of the postmortem cortical tissue, the sex of the subjects is not optimally matched because of limited availability of younger healthy female brain tissue as opposed to younger male samples during the time of the experiments. Further investigations are to be performed to determine if sex affects synaptic release machinery protein levels. Finally, in this study we show that the decrease in Syt-1 and CSPα are precipitated by ER stress and results in the desynchronization of neurotransmitter release. However, Ca2+ dysregulation and other mechanisms that arise due to of E/I imbalance may also directly affect neurotransmitter release kinetics.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ege T. Kavalali (ege.kavalali@vanderbilt.edu).
Materials availability
There is one plasmid, pFUW-CSPα, which was generated in this manuscript. All plasmids used in the present manuscript are available for sharing via request to ETK or BU.
Data and code availability
All data supporting the findings of this study will be shared by the lead author Ege T. Kavalali upon request.
This study does not report any unique code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
For the rat hippocampal cultures and striatal-co-culture experiments, postnatal day 2–3 Sprague-Dawley rats of either sex were used. Pregnant Sprague-Dawley rats (Charles River) were housed individually until they give birth to a litter and were provided with treats and environmental enrichment. Postnatal day 2-3 littermates were used to prepare primary dissociated neuronal cultures. To provide a glial feeder layer for human neural progenitor cells, glial cultures were prepared from postnatal 3-4 CD1 mice. All animal procedures were performed in accordance with the guide for the care and use of laboratory animals and were approved by the Institutional Animal Care and Use Committee at Vanderbilt University. Health status of the live animals were periodically checked and confirmed by the veterinary staff of animal facilities of the Vanderbilt University.
Cell lines
H1 ESCs (WiCell Research Sources) were maintained in mTESR1 plus medium (Stem Cell Technologies) and were used to generate human cortical neurons by overexpressing Ngn-24,79. Human embryonic kidney-293T (HEK293T) cells (ATCC) were used to produce lentiviral particles to transduce hESCs or iN cells. All cell cultures were kept in humidified incubators at 37°C with 95% O2 and 5% CO2. Cells were passaged at approximately 80% confluency. The culture medium consisted of 10% FBS containing Dulbecco’s Modified Eagle Medium supplemented with penicillin and streptomycin.
Generation of human induced neurons from primary human embryonic stem cell (hESC) cultures
Human embryonic stem cells were transduced when they reached 80-90% confluency by a lentiviral vector with the Ngn2-rtTA construct accompanied by polybrene (8 μg/μL) and 2 μM thiazovivin (Day 0). The following day, cell medium was changed to N2/DMEM/F12/NEAA (induction medium) supplemented with human BDNF (10 μg/ml, Peprotech), human NT-3 (10 μg/ml, Peprotech), mouse laminin (0.2 mg/L, Invitrogen), and doxycycline (2 μg/mL). On day 2, puromycin selection was performed for 24 h using induction medium + 1 μg/ml puromycin. On day 3, the surviving cells were washed with PBS and treated with Accutase to be plated on mouse glia at a density of 1-2 x 105 cells per well of a 24-well plate. Differentiating iN cells were incubated in Neurobasal Plus medium supplemented with B27/Glutamax (Invitrogen), BDNF, NT-3 and AraC (1 g/L, Sigma) and 2.5% FBS. Half of the media in each well was changed every 2 days up to div13, followed by weekly media changes.
METHOD DETAILS
Primary mouse glia culture
Briefly, hippocampal and cortical tissue were digested in papain (Worthington) for 30 min followed by harsh dissociation and plated onto T75 flasks in DMEM supplemented with 10% FBS. Upon reaching confluence, cells were trypsinized with 0.025% trypsin and plated at a lower density. Glial cells were passaged at least twice before they were plated on 12 mm coverslips coated with a 1:50 MEM:Matrigel solution and poly-D-lysine at a density of 5 × 104 cell/well.
Primary dissociated neuronal cultures
Rat hippocampi or striata were dissected in ice-cold 20% fetal bovine serum (FBS) containing Hanks’ balanced salt solution. Tissues were then washed and treated with 10 mg/mL trypsin and 0.5 mg/mL DNase at 37°C for 10 min. The tissues were washed again, dissociated using a filtered P1000 tip and centrifuged at 1000 rpm for 10 min at 4°C. Pellet containing neurons was resuspended either in plating medium (for hippocampal cultures) containing MEM (no phenol red), 5 g/L D-glucose, 0.2 g/L NaHCO3, 0.1 g/L transferrin, 10% FBS, 2 mM L-glutamine and 20 mg/L insulin or in neurobasal medium (for striatal co-culture experiments) supplemented with GlutaMAX-I and B27 supplement (Invitrogen). Neurons were plated onto 12 mm coverslips coated with either 1:50 MEM:Matrigel solution (for hippocampal cultures) or on mature iN cell cultures (for striatal co-cultures). Cultures were kept in humidified incubators at 37°C with 95% O2 and 5% CO2. For the hippocampal cultures, on div1, the plating medium was changed with 4 μM cytosine arabinoside containing growth medium (MEM, 5 g/L D-glucose, 0.2 g/L NaHCO3, 0.1 g/L transferrin, 5% FBS, 0.5 mM L-glutamine and B27). On div4, the cytosine arabinoside concentration was dropped 2 μM by changing half of the media. All experiments were performed after div14, when synapses reached maturity. Sample size was not predetermined using statistical methods prior to experimentation.
Cloning and lentivirus preparation
Lentiviral particles were generated in HEK293T cells by cotransfection with three packaging plasmids (pRSV-rev, pMD2.G, pMDLg/pRRE)73,74 and lentiviral vectors (rtTA, Tet-Ngn2-puro, pFUW-Syt-1, pFUW-CSPα, L307-Syt-7)29,75–77(10 μg of lentiviral vector DNA and 5 μg of each packaging plasmid DNA per 75 cm2 culture area) via FuGENE transfection reagent (Promega). 24 h following transfection, the medium was changed to mTESR and harvested 36 h later, centrifuged briefly and filtered through 0.45 μm cellulose acetate filter. For iN cell induction, virus preparations with high infection efficacy (>60%), assessed by EGFP fluorescence in HEK293T cells, were directly added to transduce human embryonic stem cells (hESCs).
Electrophysiology and data analysis
Whole-cell patch clamp recordings were performed on iN cells using CV203BU headstage, Axopatch 200B amplifier, Digidata 1320 digitizer and Clampex 8.0 software (Molecular Devices). Recordings were filtered at 1 kHz and sampled at 100 μs for evoked recordings and filtered at 2 kHz and sampled at 20 μs for miniature post synaptic current recordings. Experiments were conducted at room temperature. For external bath solution, a modified Tyrode’s solution containing the followings was used: (in mM): 150 NaCl, 4 KCl, 1.25 MgCl2, 2 CaCl2, 10 D-glucose, 10 HEPES at pH 7.4. To isolate mEPSCs, 1 μM TTX, 50 μM PTX, and 50 μM D-AP5 were added. To isolate eEPSCs, 50 μM PTX and 50 μM D-AP5 were added. To evoke EPSCs, a parallel bipolar electrode (FHC) was immersed in the external bath solution, delivering 35 mA pulses via a stimulus isolation unit. For EGTA-AM experiments, coverslips were incubated either in 100 μM EGTA-AM in 0.1% DMSO containing external bath solution with 0 mM Ca2+ or in vehicle for 15 min at room temperature. For voltage clamp experiments, the membrane potential was held at −70 mV and 3-5 MΩ borosilicate glass patch pipettes were filled with internal solution contained the following (in mM): 115 Cs-MeSO3, 10 CsCl, 5 NaCl, 10 HEPES, 0.6 EGTA, 20 tetraethylammonium-Cl, 4 Mg-ATP, 0.3 Na3GTP, and 10 QX-314 [N-(2,6-dimethylphenylcarbamoylmethyl)-triethylammonium bromide] at pH 7.35 and 300 mOsm. For current clamp experiments, 4-6 MΩ borosilicate glass patch pipettes were filled with internal solution contained the following (in mM): 110 K-Gluconate, 20 KCl, 10 NaCl, 10 HEPES, 4 Mg-ATP, 0.3 Na3GTP, 0.6 EGTA at pH 7.3 and 284 mOsm. For all recordings included for the analysis, the membrane resistance was greater than 100 MΩ, the access resistance was less than 20 MΩ and time constant (τ) was less than 3 ms. Miniature events and spontaneous APs were recorded for 4-5 min mEPSC frequencies and amplitudes along with spike analysis were conducted. Membrane resistance values (Rm) are calculated through whole-cell current clamp experiments from the change in membrane voltage (Vm) upon current injection (Im) [ Vm = Im x Rm ]. The change in Vm was fitted on a first-degree exponential function on Clampfit (Molecular Devices) and the time constant of membrane depolarization, tau (m) was calculated. Membrane capacitance values (Cm) was a calculated from the formula [Tm = Rm x Cm], which is the solution of the first-order differential equation for current flow across the membrane.80
Normalized Cumulative Charge Transfer(NCCT) of evoked responses were calculated by taking the first 10 ms of the response, since human iN cells have considerably sharper responses (Figures S1E and S1F) that reach baseline in 10 miliseconds. To include additional asynchronous events that could take place after the first 10 ms, we performed an additional NCCT analysis with an extended period, by taking the first 40 ms of the response (Figures S5A–S5I). The cumulative charge transfer graphs were compared by comparing the slopes by calculating a p value (two-tailed) testing the null hypothesis that the slopes are all identical (the lines are not different). The p value indicates that when the slopes really were identical, the chance that randomly selected data points would have slopes as different81. We also validated these results by comparing the lines with Kolmogorov-Smirnov Test (Table S1).
Western blotting
To quantify protein levels, western blotting was performed. Briefly, protein samples were prepared from coverslips using Laemmli Buffer containing protease and phosphatase inhibitor cocktails (Roche) and beta-mercaptoethanol. Samples were sonicated and boiled for 5 min at 95°C to dissociate SNARE complexes and loaded on SDS-PAGE gels that were transferred to nitrocellulose membranes. Membranes were incubated with primary antibodies at 4°C overnight. After incubation with fluorescent secondary anti-rabbit and anti-mouse antibodies (IRDye Secondary Antibodies, Li-Cor), membranes were imaged using an Odyssey CLx imaging system (Li-Cor). Band intensities were analyzed using ImageJ and normalized to loading controls. When the representative bands in the figures belong to different membranes or different lanes on the same membrane, they are separated by a blank space. When they are in the neighboring lanes, they are presented without a blank space. All raw images of Western blot membranes are shown in Figure S6.
Immunofluorescence staining, confocal microscopy and image analysis
Human iN cell cultures were fixed with 1% para-formaldehyde (PFA) and 7.5% sucrose in phosphate buffered saline (PBS) and permeabilized using 0.0075% Triton X −100 in PBS. After blocking (1% BSA, 3% goat serum and 0.2% fish gelatin in PBS), primary antibodies were diluted in blocking buffer and incubated overnight at 4°C in a humid chamber. After incubation with fluorescent secondary antibodies, confocal images were acquired using an LSM 510 META confocal microscope (Carl Zeiss) with a 63X (NA1.4) objective at 1024× 1024-pixel resolution. The synapse densities were analyzed using Intellicount.78 The ER Stress markers (BiP and CHOP) fluorescent intensity measurements were made on ImageJ following subtraction of the background and measurement of the signal that overlaps with Ngn2-EGFP signal to specifically measure signals from iN cells.
Acquisition of human postmortem cortical samples
Human postmortem cortical tissue was provided by NIH Neurobiobank. The protocol to obtain postmortem cortical samples is as follows. Following the death of the subjects, cortical tissue was rapidly extracted where one hemisphere is dissected and frozen with vaporized nitrogen while the other hemisphere is fixed with formaldehyde. Postmortem interval (PMI) refers to the amount of time (in hours) that passed from the time of death to tissue collection. Following tissue collection, a pathologist investigates the fixed samples for a neuropathological assessment. In addition, this assessment is complemented with a thorough retrospective analysis of the clinical information of the subjects. In this study, we used samples that does not have any neurological or psychiatric diagnoses and that does not have an unexpected abnormality in the neuropathological assessment despite their lack of any diagnosis.
QUANTIFICATION AND STATISTICAL ANALYSIS
The data was presented as mean ± standard error of mean, unless stated otherwise in the figure legends. The sample size for each experiment was stated in the legend and was not predetermined using statistical methods prior to experimentation. For patch-clamp electrophysiology experiments, the sample size (n number) corresponds to number of cells patched, whereas for immunoblotting experiments, the sample size corresponds to the number of coverslips used. The results of power analysis of crucial experiments that assesses the NNCT is presented in Table S1. To ensure reproducibility of experimental findings, each set of experiments was performed at least two times and in different sets if iN cell cultures to validate the results. Prism 9 (Graphpad) was used to perform statistical analyses. When more than two independent groups were compared, ANOVA test or Kruskal-Wallis test was used decided upon the normal distribution parameters of the data. When normal distribution parameters were not met, Mann Whitney U test was used. When two independent groups were compared two-tailed non-paired t test or Mann Whitney U test was used decided upon the normal distribution parameters of the data. Outliers were identified with Robust regression and Outlier removal (ROUT) method. A P-value less than 0.05 was considered statistically significant. Significance levels were stated as follows: *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001. ns denotes non-significance.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-synapsin 1 (rabbit polyclonal) | Synaptic Systems | Catalog # 106 103; RRID:AB_11042000 |
| Anti-MAP2 (chicken polyclonal) | Synaptic Systems | Catalog # 188 006; RRID:AB_2619881 |
| Anti-synaptotagmin 7 (rabbit polyclonal) | Synaptic Systems | Cat# 105 173; RRID:AB_887838 |
| Anti-synaptotagmin 1 (rabbit polyclonal) | Synaptic Systems | Cat# 105 011; RRID:AB_887832 |
| Anti-GAPDH (rabbit monoclonal) | Cell signaling | Catalog # 14C10; RRID:AB_561053 |
| Anti-CSP-α (rabbit polyclonal) | Millipore | Catalog # AB1576; RRID:AB_90794 |
| Anti-syntaxin1 (mouse monoclonal) | Synaptic Systems | Catalog# 110 011; RRID:AB_887844 |
| Anti-SNAP25 (mouse monoclonal) | Synaptic Systems | Catalog #111 111; RRID:AB_887792 |
| Anti-synaptobrevin2 (mouse monoclonal) | Synaptic Systems | Catalog # 104 211; RRID:AB_887811 |
| Anti-BiP (rabbit polyclonal) | Abcam | Catalog # ab21685; RRID:AB_2119834 |
| Anti-CHOP (rabbit polyclonal) | Invitrogen | Catalog # PA5-86145; RRID:AB_2802940 |
| Anti-Ire1α (rabbit polyclonal) | Cell signaling | Catalog # 3294; RRID:AB_823545 |
| Anti-phospho-eIF2α (rabbit polyclonal) | Cell signaling | Catalog # 3597; RRID:AB_390740 |
| Anti-eIF2α (rabbit polyclonal) | Cell signaling | Catalog # 9722; RRID:AB_2230924 |
| Bacterial and virus strains | ||
| BL21-Gold(DE3) Competent E. coli | Agilent | Catalog #230132 |
| Chemicals, peptides, and recombinant proteins | ||
| mTESR Plus Medium | Stem Cell Technologies | Catalog# 100-0276 |
| DMEM-F12 Medium | Invitrogen | Catalog # 11330057 |
| N-2 Supplement | Invitrogen | Catalog # A1370701 |
| Insulin | Sigma | Catalog # I0516 |
| Puromycin | Stem Cell Technologies | Catalog # 73342 |
| DMEM Medium | Sigma | Catalog # D5796 |
| Fetal Bovine Serum | Fisher Scientific | Catalog # SH30070.03 |
| Penicillin-Streptomycin | Gibco | Catalog# 15140122 |
| 6-Cyano-7-nitroquinoxaline-2,3-dione disodium salt hydrate (CNQX) | Sigma-Aldrich | Catalog # C239 |
| D(−)-2-Amino-5-phosphonopentanoic acid (AP-5) | Sigma-Aldrich | Catalog # A8054 |
| Picrotoxin (PTX) | Sigma-Aldrich | Catalog # P1675 |
| Tetrodotoxin (TTX) | Enzo Life Sciences | Catalog # BML-NA120-0001 |
| Trypsin from bovine pancreas | Sigma-Aldrich | Catalog # T9935 |
| DNase I | Sigma-Aldrich | Catalog # D5025 |
| Matrigel | Corning | Catalog # 354234 |
| Neurobasal Plus Medium | GIBCO | Catalog # A3582901 |
| FuGENE 6 | Promega | Catalog # E2692 |
| QX-314 | EMD-Millipore | Catalog # 552233 |
| Transferrin | Calbiochem | Catalog # 616420 |
| Cytosine Arabinoside (Ara-C) | Sigma | Catalog # C6645 |
| B-27 supplement | GIBCO | Catalog # 17504-010 |
| Memantine | Sigma | Catalog # M9292 |
| Muscimol | Sigma | Catalog # M1523 |
| Kira6 | Selleck Chem | Catalog # S8658 |
| Salubrinal | Selleck Chem | Catalog # S2923 |
| Recombinant Human/Murine/Rat BDNF | PeproTech | Catalog # 45002 |
| Recombinant Human NT-3 | PeproTech | Catalog # 45003 |
| Mouse Laminin | Invitrogen | Catalog #23017015 |
| Growth-factor reduced matrigel | Corning | Catalog # 354230 |
| DMSO | Sigma | Catalog # D2650 |
| Experimental models: Cell lines | ||
| Human Embryonic Stem Cells (hESCs) | WiCell | Catalog #WA01 |
| Human embryonic kidney-293 (HEK293) cells | ATCC | Catalog # CRL-1573; RRID: CVCL_0045 |
| Experimental models: Organisms/strains | ||
| Sprague-Dawley rat pups (P2–P3) | Charles River | Strain code: 400 |
| CD1 mice pups (P2-P3) | Charles River | Strain code: 022 |
| Recombinant DNA | ||
| Plasmid: pRSV-REV (lentiviral packaging) | Dull etal., 199873 | Addgene# 12253 |
| Plasmid: pCMV-VSV-G (lentiviral packaging) | Stewart et al., 200374 | Addgene # 8454 |
| Plasmid: pMDLg/pRRE (lentiviral packaging) | Dull etal., 199873 | Addgene# 12251 |
| Plasmid: pFUW-TetO-hNgn2-EGFP-puroR | Ho et al, 201575 | Addgene #79823 |
| Plasmid:pFUW-rRTA | Hockemeyer et al, 200876 | Addgene #20342 |
| Plasmid:L307-Syt7-shRNA | Bacaj et al, 201329 | N/A |
| Plasmid:pFUW-Syt1 | Alten et al, 202277 | N/A |
| Plasmid:pFUW-CSPα | This paper | N/A |
| Software and algorithms | ||
| MiniAnalysis | Synaptosoft | http://www.synaptosoft.com/MiniAnalysis |
| Clampfit | Molecular Devices | https://www.moleculardevices.com |
| Axopatch | Molecular Devices | https://www.moleculardevices.com |
| Prism | GraphPad | https://www.graphpad.com:443/ |
| Intellicount | Fantuzzo et al., 201778 | N/A |
Highlights.
In human synapses, chronic E/I imbalance desynchronizes glutamate release
E/I imbalance results in ER stress, precipitated by NMDAR-mediated transmission
ER stress leads to downregulation of key presynaptic proteins, Syt-1 and CSPα
Desynchronized release is maintained by high-affinity Ca2+ sensor Syt-7
ACKNOWLEDGMENTS
We thank Natalie J. Guzikowski for her valuable opinions on the manuscript and Brent Trauterman for his expert help in molecular cloning. This research was supported by NIH grants MH66198 and AG055577 to E.T.K. Graphical depictions were prepared at biorender.com. Human postmortem tissue was obtained from the NIH NeuroBioBank (NBB). The postmortem donations to NIH NBB are obtained from individuals who register before death and/or next-of-kin who approve the postmortem donation. The consent is obtained and documented by trained individuals in the Brain and Tissue Repositories (BTRs) participating in the NBB program according to the policies and procedures approved by their respective institutional review boards (IRBs).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112042.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Hanna JH, Saha K, and Jaenisch R (2010). Pluripotency and cellular reprogramming: facts, hypotheses, unresolved issues. Cell 143, 508–525. 10.1016/j.cell.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Okita K, and Yamanaka S (2011). Induced pluripotent stem cells: opportunities and challenges. Philos. Trans. R. Soc. Lond. B Biol. Sci 366, 2198–2207. 10.1098/rstb.2011.0016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blanpain C, Daley GQ, Hochedlinger K, Passegué E, Rossant J, and Yamanaka S (2012). Stem cells assessed. Nat. Rev. Mol. Cell Biol 13, 471–476. 10.1038/nrm3371. [DOI] [PubMed] [Google Scholar]
- 4.Zhang Y, Pak C, Han Y, Ahlenius H, Zhang Z, Chanda S, Marro S, Patzke C, Acuna C, Covy J, et al. (2013). Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 78, 785–798. 10.1016/j.neuron.2013.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bolshakov VY, Golan H, Kandel ER, and Siegelbaum SA (1997). Recruitment of new sites of synaptic transmission during the cAMP-dependent late phase of LTP at CA3-CA1 synapses in the hippocampus. Neuron 19, 635–651. 10.1016/s0896-6273(00)80377-3. [DOI] [PubMed] [Google Scholar]
- 6.Hsia AY, Malenka RC, and Nicoll RA (1998). Development of excitatory circuitry in the hippocampus. J. Neurophysiol 79, 2013–2024. 10.1152/jn.1998.79.4.2013. [DOI] [PubMed] [Google Scholar]
- 7.Kay L, Humphreys L, Eickholt BJ, and Burrone J (2011). Neuronal activity drives matching of pre- and postsynaptic function during synapse maturation. Nat. Neurosci 14, 688–690. 10.1038/nn.2826. [DOI] [PubMed] [Google Scholar]
- 8.Mozhayeva MG, Sara Y, Liu X, and Kavalali ET (2002). Development of vesicle pools during maturation of hippocampal synapses. J. Neurosci 22, 654–665. 10.1523/JNEUR0SCI.22-03-00654.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patzke C, Brockmann MM, Dai J, Gan KJ, Grauel MK, Fenske P, Liu Y, Acuna C, Rosenmund C, and Südhof TC (2019). Neuromodulator signaling bidirectionally controls vesicle numbers in human synapses. Cell 179, 498–513.e22. 10.1016/j.cell.2019.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pouzat C, and Hestrin S (1997). Developmental regulation of basket/stellate cell/purkinje cell synapses in the cerebellum. J. Neurosci 17, 9104–9112. 10.1523/JNEUR0SCI.17-23-09104.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reyes A, and Sakmann B (1999). Developmental switch in the short-term modification of unitary EPSPs evoked in layer 2/3 and layer 5 pyramidal neurons of rat neocortex. J. Neurosci 19, 3827–3835. 10.1523/JNEUR0SCI.19-10-03827.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andreae LC, and Burrone J (2018). The role of spontaneous neurotransmission in synapse and circuit development. J. Neurosci. Res 96, 354–359. 10.1002/jnr.24154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Durand GM, Kovalchuk Y, and Konnerth A (1996). Long-term potentiation and functional synapse induction in developing hippocampus. Nature 381, 71–75. 10.1038/381071a0. [DOI] [PubMed] [Google Scholar]
- 14.Wu G, Malinow R, and Cline HT (1996). Maturation of a central glutamatergic synapse. Science 274, 972–976. 10.1126/science.274.5289.972. [DOI] [PubMed] [Google Scholar]
- 15.Nicholson DA, Yoshida R, Berry RW, Gallagher M, and Geinisman Y (2004). Reduction in size of perforated postsynaptic densities in hippocampal axospinous synapses and age-related spatial learning impairments. J. Neurosci 24, 7648–7653. 10.1523/JNEUR0SCI.1725-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Serra M, Ghiani CA, Foddi MC, Motzo C, and Biggio G (1994). NMDA receptor function is enhanced in the hippocampus of aged rats. Neurochem. Res 19, 483–487. 10.1007/BF00967328. [DOI] [PubMed] [Google Scholar]
- 17.Banay-Schwartz M, Lajtha A, and Palkovits M (1989). Changes with aging in the levels of amino acids in rat CNS structural elements. I. Glutamate and related amino acids. Neurochem. Res 14, 555–562. 10.1007/BF00964918. [DOI] [PubMed] [Google Scholar]
- 18.Saransaari P, and Oja SS (1995). Age-related changes in the uptake and release of glutamate and aspartate in the mouse brain. Mech. Ageing Dev 81, 61–71. 10.1016/0047-6374(95)01583-1. [DOI] [PubMed] [Google Scholar]
- 19.Dumitriu D, Hao J, Hara Y, Kaufmann J, Janssen WGM, Lou W, Rapp PR, and Morrison JH (2010). Selective changes in thin spine density and morphology in monkey prefrontal cortex correlate with aging-related cognitive impairment. J. Neurosci 30, 7507–7515. 10.1523/JNEUR0SCI.6410-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eyal G, Verhoog MB, Testa-Silva G, Deitcher Y, Lodder JC, Benavides-Piccione R, Morales J, DeFelipe J, de Kock CP, Mansvelder D, and Segev I (2016). Unique membrane properties and enhanced signal processing in human neocortical neurons. Elife 5, e16553. 10.7554/eLife.16553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hefft S, and Jonas P (2005). Asynchronous GABA release generates long-lasting inhibition at a hippocampal interneuron-principal neuron synapse. Nat. Neurosci 8, 1319–1328. 10.1038/nn1542. [DOI] [PubMed] [Google Scholar]
- 22.Otsu Y, Shahrezaei V, Li B, Raymond LA, Delaney KR, and Murphy TH (2004). Competition between phasic and asynchronous release for recovered synaptic vesicles at developing hippocampal autaptic synapses. J. Neurosci 24, 420–433. 10.1523/JNEUR0SCI.4452-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hardingham GE, and Bading H (2003). The Yin and Yang of NMDA receptor signalling. Trends Neurosci. 26, 81–89. 10.1016/S0166-2236(02)00040-1. [DOI] [PubMed] [Google Scholar]
- 24.Papadia S, and Hardingham GE (2007). The dichotomy of NMDA receptor signaling. Neuroscientist 13, 572–579. 10.1177/10738584070130060401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li V, and Wang YT (2016). Molecular mechanisms of NMDA receptor-mediated excitotoxicity: implications for neuroprotective therapeutics for stroke. Neural Regen. Res 11, 1752–1753. 10.4103/1673-5374.194713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brown LE, Fuchs C, Nicholson MW, Stephenson FA, Thomson AM, and Jovanovic JN (2014). Inhibitory synapse formation in a co-culture model incorporating GABAergic medium spiny neurons and HEK293 cells stably expressing GABAA receptors. J. Vis. Exp, e52115. 10.3791/52115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paraskevopoulou F, Herman MA, and Rosenmund C (2019). Glutamatergic innervation onto striatal neurons potentiates GABAergic synaptic output. J. Neurosci 39, 4448–4460. 10.1523/JNEURO-SCI.2630-18.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Segal M, Greenberger V, and Korkotian E (2003). Formation of dendritic spines in cultured striatal neurons depends on excitatory afferent activity. Eur. J. Neurosci 17, 2573–2585. 10.1046/j.1460-9568.2003.02696.x. [DOI] [PubMed] [Google Scholar]
- 29.Bacaj T, Wu D, Yang X, Morishita W, Zhou P, Xu W, Malenka RC, and Südhof TC (2013). Synaptotagmin-1 and synaptotagmin-7 trigger synchronous and asynchronous phases of neurotransmitter release. Neuron 80, 947–959. 10.1016/j.neuron.2013.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Geppert M, Goda Y, Hammer RE, Li C, Rosahl TW, Stevens CF, and Südhof TC (1994). Synaptotagmin I: a major Ca2+ sensor for transmitter release at a central synapse. Cell 79, 717–727. 10.1016/0092-8674(94)90556-8. [DOI] [PubMed] [Google Scholar]
- 31.Südhof TC (2002). Synaptotagmins: why so many? J. Biol. Chem 277, 7629–7632. 10.1074/jbc.R100052200. [DOI] [PubMed] [Google Scholar]
- 32.Donnelier J, and Braun JEA (2014). CSPα—chaperoning presynaptic proteins. Front. Cell. Neurosci 8, 116. 10.3389/fncel.2014.00116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Choe ES,Ahn SM, Yang JH, Go BS, and Wang JQ (2011). Linking cocaine to endoplasmic reticulum in striatal neurons: role of glutamate receptors. Basal Ganglia 1, 59–63. 10.1016/j.baga.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dong Y, Kalueff AV, and Song C (2017). N-methyl-d-aspartate receptor-mediated calcium overload and endoplasmic reticulum stress are involved in interleukin-1beta-induced neuronal apoptosis in rat hippocampus. J. Neuroimmunol 3O7,7–13. 10.1016/j.jneuroim.2017.03.005. [DOI] [PubMed] [Google Scholar]
- 35.Huang X-T, Liu W, Zhou Y, Sun M, Sun C-C, Zhang C-Y, and Tang S-Y (2019). Endoplasmic reticulum stress contributes to NMDA-induced pancreatic β-cell dysfunction in a CHOP-dependent manner. Life Sci. 232, 116612. 10.1016/jJfs.2019.116612. [DOI] [PubMed] [Google Scholar]
- 36.Nosyreva E, and Kavalali ET (2010). Activity-dependent augmentation of spontaneous neurotransmission during endoplasmic reticulum stress. J. Neurosci 30, 7358–7368. 10.1523/JNEUROSCI.5358-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harding HP, Calfon M, Urano F, Novoa I, and Ron D (2002). Transcriptional and translational control in the Mammalian unfolded protein response. Annu. Rev. Cell Dev. Biol 18, 575–599. 10.1146/annurev.cellbio.18.011402.160624. [DOI] [PubMed] [Google Scholar]
- 38.Szegezdi E, Logue SE, Gorman AM, and Samali A (2006). Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 7, 880–885. 10.1038/sj.embor.7400779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xie R, and Manis PB (2017). Synaptic transmission at the endbulb of Held deteriorates during age-related hearing loss. J. Physiol 595, 919–934. 10.1113/JP272683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang Y-M, and Wang L-Y (2017). Ageing brains attend a symphony with asynchronous transmitter release. J. Physiol 595, 613–614. 10.1113/JP273421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sengupta B, Laughlin SB, and Niven JE (2013). Balanced excitatory and inhibitory synaptic currents promote efficient coding and metabolic efficiency. PLoS Comput. Biol 9, e1003263. 10.1371/jour-nal.pcbi.1003263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wehr M, and Zador AM (2003). Balanced inhibition underlies tuning and sharpens spike timing in auditory cortex. Nature 426, 442–446. 10.1038/nature02116. [DOI] [PubMed] [Google Scholar]
- 43.Yizhar O, Fenno LE, Prigge M, Schneider F, Davidson TJ, O’Shea DJ, Sohal VS, Goshen I, Finkelstein J, Paz JT, et al. (2011). Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature 477, 171–178. 10.1038/nature10360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Caspary DM, Ling L, Turner JG, and Hughes LF (2008). Inhibitory neurotransmission, plasticity and aging in the mammalian central auditory system. J. Exp. Biol 211, 1781–1791. 10.1242/jeb.013581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Richardson BD, Ling LL, Uteshev VV, and Caspary DM (2013). Reduced GABAA receptor-mediated tonic inhibition in aged rat auditory thalamus. J. Neurosci 33, 1218–127a. 10.1523/JNEURO-SCI.3277-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rozycka A, and Liguz-Lecznar M (2017). The space where aging acts: focus on the GABAergic synapse. Aging Cell 16, 634–643. 10.1111/acel.12605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ghosh I, Liu CS, Swardfager W, Lanctôt KL, and Anderson ND (2021). The potential roles of excitatory-inhibitory imbalances and the repressor element-1 silencing transcription factor in aging and aging-associated diseases. Mol. Cell. Neurosci 117, 103683. 10.1016/j.mcn.2021.103683. [DOI] [PubMed] [Google Scholar]
- 48.Zullo JM, Drake D, Aron L, O’Hern P, Dhamne SC, Davidsohn N, Mao C-A, Klein WH, Rotenberg A, Bennett DA, et al. (2019). Regulation of lifespan by neural excitation and REST. Nature 574, 359–364. 10.1038/s41586-019-1647-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu G-Q, Kreitzer A, et al. (2007). Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron 55, 697–711. 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Palop JJ, and Mucke L (2009). Epilepsy and cognitive impairments in Alzheimer disease. Arch. Neurol 66, 435–440. 10.1001/archneurol.2009.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pandis D, and Scarmeas N (2012). Seizures in alzheimer disease: clinical and epidemiological data: seizures in alzheimer disease. Epilepsy Curr. 12, 184–187. 10.5698/1535-7511-12.5.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Robinson DM, and Keating GM (2006). Memantine: a review of its use in Alzheimer’s disease. Drugs 66, 1515–1534. 10.2165/00003495-200666110-00015. [DOI] [PubMed] [Google Scholar]
- 53.Winblad B, and Jelic V (2003). Treating the full spectrum of dementia with memantine. Int. J. Geriatr. Psychiatry 18, S41–S46. 10.1002/gps.937. [DOI] [PubMed] [Google Scholar]
- 54.Rizo J, and Südhof TC (2012). The membrane fusion enigma: SNAREs, sec1/munc18 proteins, and their accomplices–guilty as charged? Annu. Rev. Cell Dev. Biol 28, 279–308. 10.1146/annurev-cellbio-101011-155818. [DOI] [PubMed] [Google Scholar]
- 55.Südhof TC (2013). Neurotransmitter release: the last millisecond in the life of a synaptic vesicle. Neuron 80, 675–690. 10.1016/j.neuron.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Maximov A, and Südhof TC (2005). Autonomous function of synaptotagmin 1 in triggering synchronous release independent of asynchronous release. Neuron 48, 547–554. 10.1016/j.neuron.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 57.Bouazza-Arostegui B, Camacho M, Brockmann MM, Zobel S, and Rosenmund C (2022). Deconstructing synaptotagmin-1’s distinct roles in synaptic vesicle priming and neurotransmitter release. J. Neurosci 42, 2856–2871. 10.1523/JNEUROSCI.1945-21.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li YC, Chanaday NL, Xu W, and Kavalali ET (2017). Synaptotagmin-1-and synaptotagmin-7-dependent fusion mechanisms target synaptic vesicles to kinetically distinct endocytic pathways. Neuron 93, 616–631.e3. 10.1016/j.neuron.2016.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fernández-Chacón R, Wölfel M, Nishimune H, Tabares L, Schmitz F, Castellano-Murñoz M, Rosenmund C, Montesinos ML, Sanes JR, Schneggenburger R, et al. (2004). The Synaptic Vesicle Protein CSP Prevents Presynaptic Degeneration hanced CSP function could attenuate neurodegenerative diseases. Neuron 42, 237–251. 10.1016/S0896-6273(04)00190-4. [DOI] [PubMed] [Google Scholar]
- 60.Nie Z, Ranjan R, Wenniger JJ, Hong SN, Bronk P, and Zinsmaier KE (1999). Overexpression of cysteine-string proteins in Drosophila reveals interactions with syntaxin. J. Neurosci 19, 10270–10279. 10.1523/JNEUROSCI.19-23-10270.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rozas JL, Gómez-Sánchez L, Mircheski J, Linares-Clemente P, Nieto-González JL, Vázquez ME, Luján R, and Fernández-Chacón R (2012). Motorneurons require cysteine string protein-α to maintain the readily releasable vesicular pool and synaptic vesicle recycling. Neuron 74, 151–165. 10.1016/j.neuron.2012.02.019. [DOI] [PubMed] [Google Scholar]
- 62.Zhang Y-Q, Henderson MX, Colangelo CM, Ginsberg SD, Bruce C, Wu T, and Chandra SS (2012). Identification of CSPα clients reveals a role in dynamin 1 regulation. Neuron 74, 136–150. 10.1016/j.neuron.2012.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sharma M, Burré J, and Südhof TC (2011). CSPα promotes SNARE-complex assembly by chaperoning SNAP-25 during synaptic activity. Nat. Cell Biol 13, 30–39. 10.1038/ncb2131. [DOI] [PubMed] [Google Scholar]
- 64.Evans GJO, and Morgan A (2002). Phosphorylation-dependent interaction of the synaptic vesicle proteins cysteine string protein and synaptotagmin I. Biochem. J 364, 343–347. 10.1042/BJ20020123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sze CI, Bi H, Kleinschmidt-DeMasters BK, Filley CM, and Martin LJ (2000). Selective regional loss of exocytotic presynaptic vesicle proteins in Alzheimer’s disease brains. J. Neurol. Sci 175, 81–90. 10.1016/s0022-510x(00)00285-9. [DOI] [PubMed] [Google Scholar]
- 66.Tiwari SS, d’Orange M, Troakes C, Shurovi BN, Engmann O, Noble W, Hortobágyi T, and Giese KP (2015). Evidence that the presynaptic vesicle protein CSPalpha is a key player in synaptic degeneration and protection in Alzheimer’s disease. Mol. Brain 8, 6. 10.1186/s13041-015-0096-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chanaday NL, Nosyreva E, Shin O-H, Zhang H, Aklan I, Atasoy D, Bezprozvanny I, and Kavalali ET (2021). Presynaptic store-operated Ca2+ entry drives excitatory spontaneous neurotransmission and augments endoplasmic reticulum stress. Neuron 109, 1314–1332.e5. 10.1016/j.neuron.2021.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chanaday NL, and Kavalali ET. (2022). Role of the endoplasmic reticulum in synaptic transmission. Curr. Opin. Neurobiol 73, 102538. 10.1016/j.conb.2022.102538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Concannon CG, Ward MW, Bonner HP, Kuroki K, Tuffy LP, Bonner CT, Woods I, Engel T, Henshall DC, and Prehn JHM (2008). NMDA receptor-mediated excitotoxic neuronal apoptosis in vitro and in vivo occurs in an ER stress and PUMA independent manner. J. Neurochem 105, 891–903. 10.1111/j.1471-4159.2007.05187.x. [DOI] [PubMed] [Google Scholar]
- 70.Berlett BS, and Stadtman ER. (1997). Protein oxidation in aging, disease, and oxidative stress. J. Biol. Chem 272, 20313–20316. 10.1074/jbc.272.33.20313. [DOI] [PubMed] [Google Scholar]
- 71.Higuchi-Sanabria R, Frankino PA, Paul JW 3rd, Tronnes SU, and Dillin A (2018). A futile battle? Protein quality control and the stress of aging. Dev. Cell 44, 139–163. 10.1016/j.devcel.2017.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Elder N, Fattahi F, McDevitt TC, and Zholudeva LV (2022). Diseased, differentiated and difficult: strategies for improved engineering of in vitro neurological systems. Front. Cell. Neurosci 16, 962103. 10.3389/fncel.2022.962103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, and Naldini L (1998). A third-generation lentivirus vector with a conditional packaging system. J. Virol 72, 8463–8471. 10.1128/JVI.72.11.8463-8471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Stewart SA, Dykxhoorn DM, Palliser D, Mizuno H, Yu EY, An DS, Sabatini DM, Chen ISY, Hahn WC, Sharp PA, et al. (2003). Lentivirus-delivered stable gene silencing by RNAi in primary cells. Rna 9, 493–501. 10.1261/rna.2192803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ho S-M, Hartley BJ, Tcw J, Beaumont M, Stafford K, Slesinger PA, and Brennand KJ (2016). Rapid Ngn2-induction of excitatory neurons from hiPSC-derived neural progenitor cells. Methods 101, 113–124. 10.1016/j.ymeth.2015.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hockemeyer D, Soldner F, Cook EG, Gao Q, Mitalipova M, and Jaenisch R (2008). A drug-inducible system for direct reprogramming of human somatic cells to pluripotency. Cell Stem Cell 3, 346–353. 10.1016/j.stem.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Alten B, Guzikowski NJ, Zurawski Z, Hamm HE, and Kavalali ET (2022). Presynaptic mechanisms underlying GABAB-receptor-mediated inhibition of spontaneous neurotransmitter release. Cell Rep. 38, 110255. 10.1016/j.celrep.2021.110255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fantuzzo JA, Mirabella VR, Hamod AH, Hart RP, Zahn JD, and Pang ZP (2017). Intellicount: high-throughput quantification of fluorescent synaptic protein puncta by machine learning. eNeuro 4, 17.2017, ENEURO.0219. 10.1523/ENEURO.0219-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Xu Y, Zhu X, Hahm HS, Wei W, Hao E, Hayek A, and Ding S (2010). Revealing a core signaling regulatory mechanism for pluripotent stem cell survival and self-renewal by small molecules. Proc. Natl. Acad. Sci. USA 107, 8129–8134. 10.1073/pnas.1002024107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Johnston D, and Wu Miao-Sin S (1995). Foundations of Cellular Neurophysiology (MIT Press; ). [Google Scholar]
- 81.Zar J (1984). Biostatistical Analysis, 2nd edition (Prentice-Hall; ). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data supporting the findings of this study will be shared by the lead author Ege T. Kavalali upon request.
This study does not report any unique code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.