

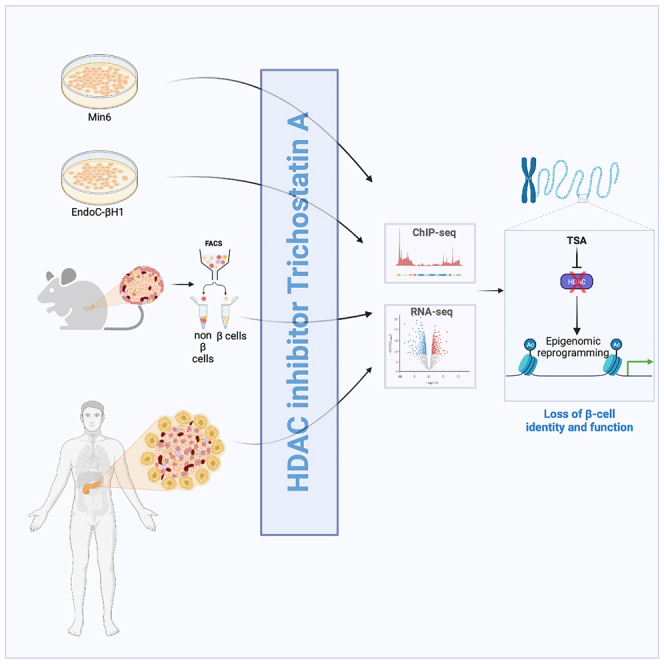
Summary
Histone deacetylases enzymes (HDACs) are chromatin modifiers that regulate gene expression through deacetylation of lysine residues within specific histone and non-histone proteins. A cell-specific gene expression pattern defines the identity of insulin-producing pancreatic β cells, yet molecular networks driving this transcriptional specificity are not fully understood. Here, we investigated the HDAC-dependent molecular mechanisms controlling pancreatic β-cell identity and function using the pan-HDAC inhibitor trichostatin A through chromatin immunoprecipitation assays and RNA sequencing experiments. We observed that TSA alters insulin secretion associated with β-cell specific transcriptome programming in both mouse and human β-cell lines, as well as on human pancreatic islets. We also demonstrated that this alternative β-cell transcriptional program in response to HDAC inhibition is related to an epigenome-wide remodeling at both promoters and enhancers. Our data indicate that HDAC activity could be required to protect against loss of β-cell identity with unsuitable expression of genes associated with alternative cell fates.
Subject areas: Natural sciences, Biological sciences, Molecular biology, Molecular mechanism of gene regulation, Epigenetics, Endocrinology
Graphical abstract
Highlights
-
•
The HDAC inhibitor trichostatin A (TSA) impairs insulin secretion
-
•
TSA alters the transcriptome of mouse and human β-cell lines and pancreatic islets
-
•
Pharmacological HDAC inhibition leads to an epigenome-wide remodeling
Natural sciences; Biological sciences; Molecular biology; Molecular mechanism of gene regulation; Epigenetics; Endocrinology.
Introduction
The endocrine pancreas is composed of distinct cell subtypes including α (producing and secreting glucagon), β (producing and secreting insulin), δ (producing and secreting somatostatin), ε (producing and secreting ghrelin), and PP (producing and secreting pancreatic polypeptide) cells that play a crucial role in the regulation of glucose homeostasis.1 A specific gene expression pattern defines each endocrine cell identity, but the underlying molecular network that controls this transcriptional specificity remains elusive. The roles of some tissue-specific transcription factors, such as PDX1 or MAFA, in the maintenance of expression of genes controlling β-cell phenotype is well known,2 but the contribution of chromatin modifiers in the maintenance of β-cell identity is less documented. Among these putative epigenomic regulators, there are histone acetyl transferases (HATs) and histone deacetylases (HDACs) that directly regulate gene expression through acetylation/deacetylation of lysine residues within specific histone and non-histone proteins.3
HDACs are zinc metalloenzymes divided into three main classes on the basis of their protein sequence homologies with yeast deacetylase enzymes.4 Briefly, class I HDACs, composed of HDAC1, HDAC2, HDAC3 and HDAC8, are closely related to yeast Rpd3 (reduced potassium dependency 3) transcriptional regulator. Class II HDACs, including HDAC4, HDAC5, HDAC7, HDAC9 (class IIa) and HDAC6, HDAC10 (class IIb), share domains with similarity to yeast HdaI (histone deacetylase I), whereas HDAC11 belongs to the class IV. All these HDACs have a conserved catalytic domain and are therefore considered as ancestral enzymes that play a crucial role in the regulation of gene expression. Although these enzymes have been considered as transcriptional inhibitors because of the resulting compaction of chromatin structure on histone deacetylation,5 studies have also demonstrated that HDAC enzymes can actively contribute to cell-specific gene expression,6 suggesting that HDACs could play a dual active and inhibiting role in the regulation of gene expression to maintain cell identity.
The pharmacological inhibition of HDAC has gained a strong interest following the demonstration that HDAC inhibitors (HDACi) harbor anticancer properties.7 HDACi suberoylanilide hydroxamic acid (SAHA, vorinostat) has been approved by the Food and Drug Administration for cancer therapy. HDACi may have potential as treatments for type 2 diabetes (T2D). Indeed, HDAC inhibition prevents cytokine-induced toxicity in β cells,8,9,10 and improves β-cell proliferation.11 Hdac3 β-cell specific knock down and pharmacological HDAC inhibition improve glucose tolerance12,13 and insulin sensitivity14 in mice.
However, the treatment of the rodent β-cell line β-TC3 with HDACi also induces a loss of cell identity through a decrease of β-cell markers, correlated with an increase of α-cell marker expression within β cells,15 but the impact of HDACi on chromatin remodeling and the subsequent modulation of gene expression within β cells has not yet been interrogated.
Here, we explored the molecular and functional effects of treating β-cell models with the HDACi trichostatin A (TSA) at the epigenomic and transcriptomic level. We show that TSA treatment leads to an epigenome-wide redistribution of histone marks that are enriched in active promoters and enhancers.
Results
TSA-mediated HDAC inhibition increases global histone acetylation associated to impaired insulin secretion and decreased expression of β-cell identity genes
We first characterized the role of HDAC inhibition in Min6 cells through a pharmacological approach using the pan-HDAC inhibitor TSA. A time-course TSA treatment (0.5 μM) of Min6 cells was performed from 15 min to 24 h and acetylation of lysine 27 (H3K27ac), lysine 9 (H3K9ac) and global acetylation (PanH3ac) levels of histone H3 were monitored by dot blot to validate global HDAC inhibition (Figure 1A). TSA induced a time-dependent increase of H3K27ac (Figures 1A and 1B), H3K9ac (Figures 1A and 1C) and PanH3ac (Figures 1A and 1D), indicating that Min6 cells were sensitive to HDAC inhibitor treatment. These results were further confirmed through western blot analysis (Figure S1A). To ensure that TSA did not induced cytotoxicity, the viability of Min6 cells on 16 h of TSA treatment was assessed by FACS through annexin V and propidium iodide labeling to identify both apoptosis events and cell viability, respectively. Neither annexin V (Figure S1B) nor propidium iodide (Figure S1C) positive cells were significantly overrepresented on TSA treatment compared to vehicle (DMSO 0.1%) treatment, indicating that TSA did not affect Min6 cell viability. Because we observed no toxicity and a significant increase of histone H3K27 and H3K9 acetylation signal 16 h after TSA treatment (Figures 1A, 1B, and 1C), subsequent experiments were performed at this time point. We also monitored the trimethylation level of lysine 27 (H3K27me3) by dot blot in Min6 cells after 16 h of TSA treatment. Compared to vehicle-treated cells, trimethylation level of lysine 27 of H3 (H3K27me3) was significantly decreased on TSA treatment (Figure 1E). Then, to evaluate the impact of TSA on functional properties of Min6 cells, insulin secretion was measured through glucose-stimulated insulin secretion (GSIS) assays. TSA treatment significantly increased insulin secretion in low glucose conditions (2.8 mM) and significantly decreased insulin secretion in high glucose conditions (20 mM, Figures 1F and S1D) without affecting insulin content (Figure S1E). These functional effects were associated with a decreased expression of genes involved in β-cell functions, such as Pdx1 or Mafa (Figure 1G). These results suggest that HDACs maintain functional properties of β cells through mechanisms that imply the modulation of histone acetylation and gene expression.
Figure 1.

The increase of histone H3 acetylation level on TSA treatment is correlated with alteration of functional properties of Min6 cells
(A) Dot blot kinetic analysis of H3K9ac, H3K27ac and pan-acetylated histone H3 (Pan H3ac) levels on TSA treatment (0.5 μM) in Min6 cells at the indicated time (n = 3). Vehicle (DMSO 0.1%) was used as negative control.
(B–D) Signals were normalized to total H3 signals and densitometry analysis of A was performed by comparing the mean of signal at each time point to mean signal in vehicle (n = 3).
(E) Dot blot analysis of H3K27me3 levels on TSA treatment (0.5 μM, 16 h) in Min6 cells. Vehicle (DMSO 0.1%) was used as negative control. Signals were normalized to total H3 signals and densitometry analysis is expressed as fold of H3K27me3 signals in TSA-treated cells compared to signals in vehicle-treated cells (n = 3).
(F) Glucose-stimulated insulin secretion of TSA-treated cells (0.5 μM, 16 h, n = 7). Vehicle (DMSO 0.1%) was used as a control. Results are presented as fold insulin secretion in response to 20 mM glucose compared to 2.8 mM glucose +/−SEM.
(G) Quantitative RT-PCR analysis of key β-cell identity genes in vehicle and TSA-treated Min6 cells (n = 3).
Results in B, C, D, E, F, G, are displayed as means +/−SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
See also Figure S1.
Epigenome-wide histone mark and transcriptome profiling identifies distinct functional genomic regions in Min6 cells
With the aim to restrict our analysis to the effects of HDAC inhibition on functional genomic regions, we first performed profiling of H3K4me3, H3K4me1, H3K27ac and H3K27me3 through next-generation sequencing of chromatin immunoprecipitation (ChIP-seq) experiments in untreated, control Min6 cells to delineate these specific chromatin features in basal conditions. As expected, H3K4me3 and H3K27ac were mostly enriched within gene promoter (74% and 58%, respectively, Figures S2A and S2B) whereas H3K4me1 and H3K27me3 were mostly enriched both within gene body (46% and 40%, respectively) and intergenic regions (25% and 44%, respectively, Figures S2C and S2D). Of intererst, this systematic histone mark profiling at the epigenome-wide level allowed us to precisely circumscribe 6 distinct functional genomic regions based on combinatorial histone mark enrichment within both promoter and distal intergenic regions. Indeed, at the promoter level, we defined active (i.e., both H3K4me3 and H3K27ac enriched; Figure 2A), bivalent (i.e., both H3K4me3 and H3K27me3 enriched; Figure 2B) and inactive (i.e., no histone marks enrichment; Figure 2C) promoters. Within distal intergenic regions, we were able to define active enhancers (i.e., both H3K27ac and H3K4me1 enriched; Figure 2D), poised enhancers (i.e., both H3K4me1 and H3K27me3 enriched; Figure 2E) and heterochromatin (i.e., only H3K27me3 enriched; Figure 2F).
Figure 2.

Histone marks profiling by ChIP-seq defines distinct functional genomic regions in Min6 cells
(A) Characterization of active promoters in Min6 cells. Heatmap displayed signal centered on transcription start site (TSS) +/− 2.5 kb for H3K4me3, H3K27ac and H3K27me3 within each active promoter (n = 8303 regions, 2549 genes). Mean signal centered on TSS +/− 2.5 kb for H3K4me3, H3K27ac and H3K27me3 within active promoters is displayed.
(B) Characterization of bivalent promoters in Min6 cells. Heatmap displayed signal centered on TSS +/− 2.5 kb for H3K4me3 and H3K27me3 within each bivalent promoter (n = 3685 regions, 1323 genes). Mean signal centered on TSS +/− 2.5 kb for H3K4me3 and H3K27me3 within bivalent promoters is displayed.
(C) Characterization of inactive promoters in Min6 cells. Heatmap displayed signal centered on TSS +/− 2.5 kb for H3K4me3, H3K27ac and H3K27me3 within each inactive promoter (n = 7632 genes). Mean signal centered on TSS +/− 2.5 kb for H3K4me3, H3K27ac and H3K27me3 within inactive promoters is displayed.
(D) Characterization of active enhancers in Min6 cells. Heatmap displayed signal centered on the center of the regions +/− 2.5 kb for H3K4me1 and H3K27ac within each active enhancer (n = 1597 regions). Mean signal centered on the center of the regions +/−2.5 kb for H3K4me1 and H3K27ac within active enhancers is displayed.
(E) Characterization of poised enhancers in Min6 cells. Heatmap displayed signal centered on the center of the regions +/−2.5 kb for H3K4me1, H3K27ac and H3K27me3 within each poised enhancer (n = 683 regions). Mean signal centered on centered on the center of the regions +/−2.5 kb for H3K4me1, H3K27ac and H3K27me3 within poised enhancers is displayed.
(F) Characterization of heterochromatin in Min6 cells. Heatmap displayed signal centered on the center of the regions +/−2.5 kb for H3K4me1, H3K27ac and H3K27me3 within heterochromatin (n = 21292 regions). Mean signal centered on centered on the center of the regions +/−2.5 kb for H3K4me1, H3K27ac and H3K27me3 within heterochromatin is displayed.
See also Figure S2.
To go further in the characterization of these distal functional intergenic regions, their associated genes were defined using Genomic Regions Enrichment of Annotations Tool (GREAT) software.16
Second, with the aim to link these chromatin features to the associated-gene expression level, transcriptomic analysis through RNA sequencing (RNA-seq) was performed in untreated Min6 cells. The genes associated with active regions such as active promoter as well as active enhancers were significantly found to be the most expressed compared to genes associated with other chromatin features (i.e., bivalent and inactive promoter, poised enhancers and heterochromatin, Figure 3A). The expression level of genes associated with poised enhancers was not significantly different from the expression level of genes associated with heterochromatin (Figure 3A). Then, we analyzed the gene ontology of genes associated with these functional genomic regions. This analysis showed that biological processes related to genes associated with functionally active genomic regions were enriched either in pathways involved in general biological functions such as mitotic cell cycle process (-Log10(q) = 22, active promoter, Figure 3B) or in β-cell specific functions such as the regulation of secretion ((-Log10(q) = 34, active enhancers, Figure 3C). Conversely, genes associated with inactive bivalent, poised genomic regions were rather enriched in pathways unrelated to β-cell functions such as embryonic morphogenesis (-Log10(q) = 35, bivalent promoters, Figure 3D; -Log10(q) = 325, heterochromatin, Figure 3F), dorsal spinal cord development (-Log10(q) = 19, poised enhancers, Figure 3E) or cell fate commitment (-Log10(q) = 29, bivalent promoters, Figure 3D; -Log10(q) = 325, heterochromatin, Figure 3F). Owing to the large number of genes associated with inactive promoters in Min6 cells, no specific pathway was identified for these associated genes (data not shown). Altogether, these results indicate that, based on the combinatorial enrichment of a selection of histone marks involved in transcriptional regulation in Min6 cells, these chromatin features were functional and directly related to gene expression level and the regulation of biological process involved in β-cell function.
Figure 3.

Functional genomic regions in Min6 cells are directly associated with gene expression level and distinct biological processes
(A) RNA-seq based expression level of genes associated with functional genomic regions in Min6 cells. Results are displayed as mean of Log2(TPM+1) for each functional genomic region associated–genes. The violin plot displays median and quartile values for each group of genes.
(B) Gene ontology (GO) analysis of active promoter associated-genes was performed using Metascape by filtering output only on GO Biological process. The 5 more enriched GO Biological processes are displayed as -Log10(q) (i.e., -Log10(Padj)).
(C) GO was performed using Genomic Regions Enrichment of Annotations Tool (GREAT) following the two nearest genes association rule (cut-off<1000 kb). The 5 more enriched GO biological processes are displayed as -Log10(q) (i.e., -Log10(Padj)).
(D) GO analysis was performed using Metascape by filtering output only on GO Biological process. The 5 more enriched GO Biological processes are displayed as -Log10(q) (i.e., -Log10(Padj)).
(E) GO analysis was performed using GREAT following the two nearest genes association rule (cut-off<1000 kb). The 5 more enriched GO biological processes are displayed as -Log10(q) (i.e., -Log10(Padj)).
(F) GO analysis was performed using GREAT following the two nearest genes association rule (cut-off<1000 kb). The 5 more enriched GO biological processes are displayed as -Log10(q) (i.e., -Log10(Padj)).
The functional genomic regions identified in Min6 cells overlap with functional chromatin segments found in mouse islets
To go further in the characterization of these functional genomic regions in Min6 cells, we next assessed whether they overlapped with functional genomic regions recently identified in mouse islets.17 Therefore, we first sought to determine the overlapping level between the genomic regions (peaks) independently enriched in a selection of histone marks in Min6 cells and the functional genomic segments resulting from functional chromatin segmentation (categorized from A to X) defined in mouse islets of Langerhans.17 Using these publicly available datasets,17 this analysis showed that most of the regions enriched in H3K4me3 in Min6 cells were mostly found in active (segments A to D) or bivalent (segment K to M) chromatin segments, suggesting that the active promoter regions of mouse β cells are conserved in Min6 cells (Figure 4A, H3K4me3). This result was corroborated by data obtained with the genomic regions enriched in H3K27ac in Min6 cells showing that these also mostly overlapped with the segments of active chromatin (segments A to D, Figure 4A, H3K27ac). Furthermore, the regions enriched with H3K4me1 in Min6 cells were also conserved because they predominantly overlapped with the active chromatin segments (segments from C to E) and to a lesser extent with the bivalent regions (segment K, Figure 4A, H3K4me1). Regarding the regions enriched in H3K27me3 in Min6 cells, these were mainly found in the segments of silent chromatin (segments N to P, Figure 4A, H3K27me3). Taken together, this analysis demonstrated that the regions enriched in histone marks involved in transcriptional regulation are, at least partially, shared between Min6 cells and mouse pancreatic islets.
Figure 4.

Functional genomic regions in Min6 cells are partly conserved with functional genomic segments in mouse islets of Langerhans
(A) Percentage overlap of H3K4me3, H3K27ac, H3K4me1 and H3K27me3 peaks in Min6 cells with the distinct genomic segments (A to J, active segments; K to U, silent segments; V to X, others segments) from mouse pancreatic islets chromatin segmentation.17 Peaks for each histone mark were intersected with each mouse islets chromatin segment to define the overlap associated-percentage. Results are displayed as heatmap scaled on this percentage of overlap.
(B) Percentage overlap of functional genomic regions in Min6 cells with the distinct genomic segments from mouse islets of Langerhans chromatin segmentation.17 Each specific functional genomic region was intersected with each mouse pancreatic islets chromatin segment to define the overlap associated-percentage. Results are displayed as heatmap scaled on this percentage of overlap.
(C–D) Examples of conserved functional genomic regions. Figures are adapted from the IGB genome browser screenshots and genomic coordinates are indicated.
(E) RNA-seq based expression level of genes associated with conserved functional genomic regions in Min6 cells (active promoters: 2370 genes, bivalent promoters: 1191 genes, inactive promoters: 3055 genes, active enhancers: 837 genes, poised enhancers: 648 genes, heterochromatin: 3308 genes). Results are displayed as mean of Log2(TPM+1) for each conserved functional genomic regions associated-genes. The violin plot displays median and quartile values for each group of genes.
This comparative analysis led us to consider the conservation level of the functional genomic regions characterized in Min6 cells (Figure 2) with the functional chromatin segments defined in mouse islets.17 The functional genomic regions of Min6 cells were also conserved compared to functional chromatin segments defined in mouse islets (Figure 4B). Indeed, we showed that the most active promoters in Min6 cells were related to active promoter segments (segments A and B, Figure 4B, active promoter) whereas bivalent promoters were related to bivalent segments (segments L and M, Figure 4B, bivalent promoter) and inactive promoters were mostly related to silent segments (segments N to Q segments, Figure 4B, inactive promoter). Regarding the distal intergenic functional regions, the active enhancers mainly overlapped with the distal active segments (segments C to E, Figure 4B, active enhancers) whereas the poised enhancers were mainly related to the bivalent segments (K and M segments, Figure 4B, poised enhancers). Regarding the heterochromatin regions, these were mostly related to the inactive segments (mainly segment N, Figure 4B, heterochromatin). Each conserved functional genomic region was exemplified through a series of selected chromatin features (gene promoter or intergenic region, Figures 4C and 4D), such as the β-cell marker Pdx1, the α-cell marker somatostatin receptor 1 (Sstr1) or the ameloblast specific gene, Amelotin (Amtn). Finally, we selected the functional chromatin regions that were conserved between Min6 cells and mouse islets. To reach this aim, we selected only the functional chromatin regions of Min6 cells with more than 30% overlap with each related functional chromatin segment within the mouse islets. Subsequently, the expression level of the genes associated with these conserved genomic functional regions was analyzed (Figure 4E). The results notably showed that the genes associated with the conserved active chromatin regions (active promoter and active enhancers) are most expressed compared to the genes associated with the inactive regions in line with our data generated without applying the conservation filter (Figure 4E). Altogether, these results showed that the functional chromatin regions are mostly shared and conserved between Min6 cells and mouse islets, indicating that Min6 cells represent a pertinent β-cell model to explore the effect of HDAC inhibition at the genome-wide level.
HDAC inhibition differentially alters acetylation level of conserved functional genomic regions in Min6 cells
The correlation between the alteration of β-cell function and the increase of histone acetylation in response to HDAC inhibition (Figure 1) prompted us to interrogate the effects of TSA treatment on histone acetylation within previously characterized conserved functional genomic regions in Min6 cells. To reach this aim, Min6 cells were treated either with vehicle (DMSO 0.1%) or TSA (0.5 μM) during 16 h and H3K9ac, H3K27ac and H3K27me3 were profiled through ChIP-seq experiments (Figure 5A). Compared to the vehicle condition, TSA treatment led to the detection of a higher number of peaks for both H3K9ac (27438 for TSA versus 13771 for DMSO) and H3K27ac (66347 for TSA versus 13451 for DMSO) (Figure 5B). Concomitantly, TSA treatment drastically reduced the number of peaks for H3K27me3 (17367 for DMSO versus 1253 for TSA) (Figure 5B). Consistent with the results from dot blot experiments (Figures 1A–1D), these data however indicated that the increase of H3K9ac and H3K27ac signal intensity in response to TSA treatment may not be only related to an increase of signal intensity within constitutively acetylated regions but also to an increase in the number of de novo acetylated genomic sites. This was corroborated by the analysis of the genomic distribution of H3K9ac and H3K27ac in response to TSA treatment showing a significant redistribution of H3K9ac and H3K27ac enriched regions especially toward gene bodies and distal genomic regions for H3K9ac (Figures S3A and S3B, H3K9ac) and gene bodies for H3K27ac (Figures S3C and S3D, H3K27ac). The genomic distribution of H3K27me3 was less modified suggesting that it was rather the loss of signal than the genomic redistribution affecting H3K27me3 in response to TSA treatment (Figures S3E and S3F, H3K27me3).
Figure 5.

TSA treatment leads to a genomic redistribution of H3K9ac, H3K27ac and H3K27me3
(A) Scheme representing the strategy used in Min6 cells.
(B) Number of H3K9ac, H3K27ac and H3K27me3 peaks in vehicle- and TSA-treated Min6 cells.
(C–F) H3K9ac, H3K27ac and H3K27me3 signal in conserved active promoters (C), bivalent promoters (D), poised enhancers (E) and active enhancers (F) in vehicle- and TSA-treated Min6 cells. Heatmaps and mean signals centered on TSS +/−2.5 kb are represented.
See also Figures S3 and S4.
Considering that a genomic redistribution of H3K9ac and H3K27ac occurred in response to TSA treatment, we then hypothesized that the levels of H3K9ac and H3K27ac acetylation could be altered within the conserved functional genomic regions described above (Figures 2 and S2). In addition, the H3K27me3 signal was also monitored in these regions. First, a promoter-focused analysis centered on the transcription start site (TSS) +/− 2.5 kb was performed. Surprisingly, this analysis revealed that H3K9ac signal was completely blunted after treating Min6 cells with TSA within shared active promoters whereas the H3K27ac signal was weakly affected or even slightly increased in the close vicinity of the promoters whereas H3K27me3 signal was not modulated (Figure 5C). Conversely, an increase of H3K9ac and, to a lesser extent, H3K27ac signal, associated with a decrease of H3K27me3, was detected within shared bivalent promoters on TSA condition (Figure 5D). Considering shared inactive promoters, neither H3K9ac, H3K27ac nor H3K27me3 signal was modulated by TSA treatment (Figure S4A). This promoter-based analysis at genome-wide level showed that TSA treatment led to distinct effects on histone acetylation depending on the basal activation level of the promoters. Acetylation level in response to TSA treatment was next interrogated within conserved functional distal intergenic regions at genome-wide level focusing on the center of the region +/− 2.5 kb. Within shared poised enhancers, both H3K9ac and H3K27ac signal was increased whereas H3K27me3 signal was decreased (Figure 5E) whereas shared active enhancers were weakly enriched with H3K9ac and H3K27ac without modulation of H3K27me3 signal on TSA treatment (Figure 5F). Finally, shared heterochromatin-focused analysis showed that H3K27me3 was decreased within these regions whereas H3K9ac and H3K27ac signals were weakly affected (Figure S4B). Taken together, these genome-wide analyses suggest that TSA treatment in Min6 cells differentially impacts acetylation profile depending on the type of functional genomic regions. This implied that gene expression level could be directly affected by these changes after genomic reprogramming of acetylation.
Epigenomic remodeling of histone acetylation on HDAC inhibition directly reprograms the transcriptome of key genes involved in insulin release in Min6 cells
We interrogated the functional outcomes of the modulation of histone acetylation profile within conserved functional genomic regions through transcriptome-wide analysis in Min6 cells. To reach this aim, RNA-seq was then performed on 16 h vehicle- and TSA-treated Min6 cells. By applying an adjusted p value cut-off threshold at 0.05 (Padj<0.05), 5636 genes were significantly downregulated and 6571 genes were significantly upregulated (Table S1). Given this high number of deregulated genes, we applied a second cut-off threshold based on Log2 fold change (Log2FC) to select only the most deregulated genes. Using these two cut-off thresholds, 2,626 genes were downregulated (Padj<0.05, Log2FC < −1) and 3877 genes were upregulated (Padj<0.05, Log2FC > 1) on TSA treatment (Figure 6A, Table S1). Ingenuity pathway analysis (IPA) revealed that downregulated genes were associated with canonical pathways involved in kinetochore metaphase signaling (-Log10(P) = 16), mitotic roles of Polo-like kinase (-Log10(P) = 9), but most interestingly, with insulin secretion (-Log10(P) = 9), Maturity Onset of Diabetes of Young (MODY, -Log10(P) = 7) and GPCR-mediated nutrient sensing (-Log10(P) = 5, Figure 6B). In line with this, several key β-cell genes were negatively affected by TSA treatment, such as Slc2a2, Nkx2-2, G6pc2, Kcnj11, Mafa, Pdx1 or P2ry1 (Figure S5A and Table S1). Conversely, upregulated genes were associated with pathways unrelated to β-cell functions, such as axonal guidance, pulmonary fibrosis or cardiac hypertrophy (Figure 6C). Based on these results, the deregulated genes were classified according to the conserved functional genomic region with which they were associated and the proportion of each group was plotted in a pie chart. Among the downregulated genes, this analysis showed that more than 25% were genes with an active promoter (Figures 6D) and 10% of the downregulated genes were associated with heterochromatin regions. Genes associated with other functional genomic regions were marginally represented (less than 15%). In addition, this analysis showed that 48% were not associated with any previously characterized functional genomic region, likely because of a lower analytical power related to the restricted number of histone marks profiled in this study. These results were directly correlated with the functional genomic data showing that the active promoters harbored a drastic decrease in the H3K9ac signal potentially responsible to gene expression downregulation (Figure 5C).
Figure 6.

Redistribution of histone acetylation at genome-wide level on TSA treatment in Min6 cells is associated with differential gene expression at transcriptome-wide level
(A) Volcano plot displaying downregulated (red circles), upregulated (green circles) and not deregulated genes (black circles) in TSA-treated cells compared to vehicle-treated cells according to two cut-off thresholds based on adjusted p value (Padj) and Log2 fold change (Log2FC).
(B and C) Ingenuity Pathway analysis (IPA) of downregulated (B) and upregulated (C) genes in Min6 cells on TSA treatment.
(D and E) Pie chart displaying proportion of TSA-dependent downregulated (D) and upregulated (E) genes in Min6 cells associated with a specific functional genomic region.
(F and G) Metascape analysis of genes with an active promoter that are downregulated (F) and upregulated (G) on TSA treatment.
See also Tables S1, S2, S3 and Figures S5 and S6.
Regarding upregulated genes, 46% were associated with silent functional genomic regions (bivalent promoter, 18%, heterochromatin, 23% and poised enhancers, 5%) whereas genes associated with active functional genomic regions were less represented (active promoter, 1% and active enhancers, 3%) (Figure 6E). Again, 48% of the downregulated genes were not associated with previously defined functional genomic regions. This analysis however highlighted the direct link between altered epigenomic profile and increased gene expression in response to TSA treatment because previous data demonstrated that silent regions showed either an enrichment in H3K9ac/H3K27ac associated with a depletion in H3K27me3 (i.e., bivalent promoter and poised enhancers; Figures 5D and 5E, respectively) or only depletion in H3K27me3 (i.e., heterochromatin; Figure S4B). These observations were exemplified through a series of selected β-cell (Pdx1 and Mafa) and α-cell markers (Arx and Mafb) demonstrating that β-cell genes were depleted in H3K9ac/H3K27ac marks, whereas α-cell genes were slightly increased in H3K9ac/H3K27ac marks but depleted in H3K27me3 mark (Figure S5C).
Consistent with the analysis of the effects of TSA on H3K9ac, H3K27ac and H3K27me3 levels within the conserved functional genomic regions in Min6 cells, these results showed a direct functional repercussion of the epigenomic alteration of these regions on the regulation of gene expression. This was all the more likely because the genes associated with inactive promoters whose H3K9ac, H3K27ac and H3K27me3 profile were marginally altered in response to TSA treatment were not enriched neither in downregulated genes (2.17%, Figure 6D) nor in upregulated genes (2.09%, Figure 6E). Gene ontology analysis using Metascape revealed that most of the downregulated genes on TSA treatment were associated with chromatin organization, RNA splicing and insulin secretion (Figure 6F). Conversely, upregulated genes were enriched in pathways that are unrelated to β-cell function, such as fat cell differentiation or lipid biosynthetic process (Figure 6G). With the aim to link TSA-induced epigenomic remodeling to alteration of pancreatic β-cell functionality, H3K9ac and H3K27ac levels were monitored within TSS-centered promoters of the genes involved in insulin secretion (GOBP_Insulin_secretion gene set, n = 583 transcripts) both in vehicle- and TSA-treated Min6 cells. Considering that not all promoters of those genes were TSA sensitive, a k-means clustering was applied to discriminate the potentially remodeled promoters from the non-affected ones. This analysis indeed showed that only a part of these genes harbored a significant decrease in the levels of H3K9ac and H3K27ac (Cluster 1, 118 genes, Figure S6A and Table S2), whereas the genes belonging to cluster 2 displayed minor changes in these two H3 acetylation marks (Cluster 2, 169 genes, Figure S6A and Table S3) in response to TSA treatment. The functional impact of this TSA-induced epigenomic remodeling within promoter of cluster 1-associated genes was next evaluated by performing a gene set enrichment analysis. This showed that those genes were significantly enriched in the vehicle-treated cells (Figure S6B) suggesting that the alteration of β-cell functionality could be consecutive of TSA-induced epigenomic remodeling in regulatory regions especially promoters of some key insulin secretion-associated genes, such as Kcnj11 or Abcc8 (Figure S6C), associated with a decrease of their expression levels (Figure S6D). Altogether, these data suggest that TSA treatment functionally affects cell fate through an epigenome-wide remodeling associated with transcriptome changes that alter β-cell identity and function.
The transcriptome of FACS-sorted TSA-treated mouse β cells identifies partially conserved genomic mode of action of TSA-mediated HDAC inhibition
Having demonstrated a direct link between the epigenome alteration and modulation of gene expression in Min6 cells in response to HDAC inhibition, we next sought to determine whether the transcriptome as well as the associated-chromatin features could be conserved in mouse β cells. Pancreatic islets of Langerhans isolated from RIPCre-tdTomato mice expressing the red fluorescent protein td-Tomato under the control of the β-cell specific Cre recombinase were treated by either vehicle (DMSO 0.1%) or TSA (0.5 μM, 16 h) and then subjected to FACS analysis to sort Tomato positive cells, corresponding to a β-cell enriched population. RNA-seq experiment was performed on this β-cell enriched population (Figure 7A). To compare these data with those obtained in Min6 cells, an adjusted p value and Log2FC cut-off thresholds (Padj<0.05 and −1>Log2FC > 1) were applied, respectively. In these conditions, 1,182 genes were significantly downregulated and 1331 genes were significantly upregulated on TSA treatment (Figure 7B, Table S4). IPA further confirmed that downregulated genes from TSA-treated sorted β-cells were mostly associated with insulin secretion and MODY signaling pathways (Figure 7C), whereas upregulated genes were found to be involved in the regulation of axonal guidance signaling, molecular mechanisms of cancer or WNT/β-catenin signaling (Figure 7D).
Figure 7.

Transcriptome of FACS-sorted β-cell from ex vivo TSA-treated mouse Langerhans islets is partly shared with Min6 cells
(A) Schematic representation of β-cell preparation from mouse Langerhans islets.
(B) Volcano plot displaying downregulated (red circles), upregulated (green circles) and not deregulated genes (black circles) in β-cell from TSA-treated mouse Langerhans islets compared to β-cell from vehicle-treated mouse Langerhans islets according to two cut-off thresholds based on adjusted p value (Padj) and Log2 fold change (Log2FC).
(C and D) Top 5 canonical pathways identified by IPA of downregulated (C) and upregulated (D) genes in TSA-treated mouse sorted-β cells.
(E) Venn diagram identifying common TSA-dependent up- and downregulated genes between Min6 cells and β-cell from mouse Langerhans islets.
(F and G) GO analysis of common TSA-dependent downregulated (F) and upregulated (G) genes. Gene ontology analysis was performed using Metascape by filtering output only on GO Biological process. The 5 more enriched GO Biological processes are displayed as -Log10(q) (i.e., -Log10(Padj)).
(H and I) Pie chart displaying proportion of TSA-dependent downregulated (H) and upregulated (I) genes in β-cell from mouse Langerhans islets associated with a specific functional genomic region.
See also Table S4.
To focus our analysis on conserved transcriptomic pattern between FACS-sorted β cells and Min6 cells, we next intersected the related dataset in a Venn diagram (Figure 7E). Among upregulated genes defined in FACS-sorted TSA-treated β-cells, 53% (694 genes) and 2% (26 genes) were shared with upregulated and downregulated genes detected in Min6 cells, respectively. Of note, 46% of upregulated genes were specific to FACS-sorted TSA-treated β-cell. Downregulated genes in TSA-treated β-cell sorted were also mostly conserved compared to downregulated genes in TSA-treated Min6 cells because 53% were shared whereas only 5.6% were upregulated in Min6 cells. TSA treatment also induced a β-cell sorted specific transcriptomic signature because 41% of genes were not overlapping neither with downregulated nor upregulated genes in Min6 cells. This analysis suggested that HDAC inhibition through TSA treatment induced a transcriptomic signature resulting at least partly from conserved molecular mechanisms between ex vivo TSA-treated β-cell and the Min6 β-cell model.
To go further in the functional characterization of these conserved deregulated genes on TSA treatment, a gene ontology analysis using IPA was conducted on both conserved down- and upregulated genes. Conserved downregulated genes were associated with biological processes mostly related to β-cell function (e.g., regulation of hormone levels and endocrine pancreas development, Figure 7F) whereas conserved upregulated genes were rather associated with biological processes unrelated to β-cell function (e.g., axon development and fat cell differentiation, Figure 7G). Altogether, this analysis suggested that the alteration of β-cell function on HDAC inhibition could be conserved and related to selective downregulation of β-cell specific genes. To ascertain this hypothesis, we conducted a Gene Set Enrichment Analysis (GSEA) to monitored β-cell specific gene set enrichment within conserved downregulated genes (Figure S5B). As expected, β-cell specific genes were mostly enriched in vehicle-compared to TSA-treated β-cell-sorted, thus supporting our gene ontology analysis.
With the aim to connect our transcriptomic data with functional genomic data from mouse β cells, we next sought to determine the functional genomic regions to which conserved deregulated genes were associated. By using the previously defined conserved functional genomic regions (Figure 2B), we were able to demonstrate that most of the conserved downregulated genes were associated with active regions (active promoter, 42% and active enhancers, 13%) (Figure 7H) whereas upregulated genes were rather associated with silent regions (bivalent promoter, 18%, poised enhancers, 6% and heterochromatin, 19%) (Figure 7I). Of note, these results were close to results obtained in Min6 cells (Figures 6D and 6E) suggesting that a part of genomic regions functionally affected on TSA treatment were conserved between β-cell-sorted and Min6 cells.
Transcriptome-wide analysis of EndoC-βH1 and human islets on HDAC inhibition identifies conserved TSA-sensitive genes related to β-cell identity
Given that HDAC inhibition led to an important redistribution of histone acetylation at genome-wide level leading to a drastic modulation of gene expression at transcriptome-wide level both in Min6 and mouse β-cell sorted cells, we next asked whether TSA treatment could also reprogram the transcriptome of human β cells. Consequently, EndoC-βH1 cells and human pancreatic islets were independently treated either with vehicle (DMSO 0.1%) or TSA (0.5 μM) during 16 h and the resulting transcriptome was recorded through RNA-seq experiments as depicted in Figures 8A and 8B, respectively.
Figure 8.

TSA treatment of the human β cell line EndoCβH1 and ex vivo human islets of Langerhans affects insulin secretion related pathways
(A and B) Schematic representation of EndoC-βH1 and human islets treatment by TSA and subsequent RNA-seq analysis.
(C) Volcano plot displaying downregulated (red circles), upregulated (green circles) and not deregulated genes (black circles) in EndoC-βH1 control and TSA-treated cells.
(D and E) Top 5 canonical pathways identified by IPA of downregulated (D) and upregulated (E) genes in TSA-treated EndoC-βH1 cells.
(F) Volcano plot displaying downregulated (red circles), upregulated (green circles) and not deregulated genes (black circles) in TSA-treated human islet compared to untreated controls.
(G and H) Top 5 canonical pathways identified by IPA of downregulated (D) and upregulated (E) genes in TSA-treated human pancreatic islets.
In EndoC-βH1 cells, by applying an adjusted p value cut-off threshold at 0.05 (Padj<0.05), 4717 genes were significantly downregulated and 5443 genes were significantly upregulated (Table S5). As in Min6 cells, we applied a second cut-off threshold based on Log2 fold change (Log2FC) to select only the most deregulated genes. Using these two cut-off thresholds, 2793 genes were downregulated (Padj<0.05, Log2FC < −1), 3243 genes were upregulated (Padj<0.05, Log2FC > 1) on TSA treatment (Figure 8C and Table S5). These results showed that the transcriptome of EndoC-βH1 cell line was highly sensitive to and profoundly affected by TSA treatment, as observed in the Min6 cell line and FACS-sorted β cells. Pathway analysis of downregulated genes suggested that TSA affects pathways controlling cell cycle and cell proliferation, but also insulin secretion (Log(pVal) = 1.8; Z score = −5.38; Figure 8D). Amongst genes that are downregulated and identified as β-cell identity genes,18 ABCC8, MAFA, GLP1R and PDX1 were found to be negatively affected. Conversely, TSA-mediated upregulated genes were associated with canonical pathways that were unrelated to β-cell functions, such as fibrosis, axonal guidance or osteoarthritis pathways (Figure 8E). In non-diabetic human islets, 1099 genes were significantly downregulated and 854 genes were significantly upregulated, after applying cut-off thresholds (Padj<0.05, −1>Log2FC > 1, Figure 8F and Table S6). Among the canonical pathways that were affected in downregulated genes, phagosome formation, G-protein coupled receptor signaling and CREB signaling in neurons were the most represented (Figure 8G). Again, insulin secretion signaling pathway was found to be affected by TSA treatment (Log(pVal) = 1.64; Z score = −3.15). For upregulated genes, IPA revealed that genes involved in pathways unrelated to pancreatic islet function were enriched following TSA treatment, such as axonal guidance signaling, synaptogenesis or semaphorin signaling pathways (Figure 8H). Although we did not measure the GSIS of EndoC-βH1 and human islets in response to TSA treatment, these results suggest that TSA induces a profound transcriptomic reprogramming in human β-cell line.
Discussion
Here we show that blocking HDAC activity with TSA profoundly impairs insulin secretion. This functional effect is associated with an epigenomic reprogramming, associated with loss of β-cell identity genes, in mouse as well as in human β cells or islets. Although the contributions of HDACs to endocrine pancreas development are well circumscribed,19,20 our results demonstrate that they also contribute to the maintenance of mature β-cell identity, plasticity and function.
It has been recently suggested that enhancement of ROS (reactive oxygen species) production on TSA treatment in Min6 cells could have negative effects of HDACi on β-cell function.21 An increase of IRS2 (Insulin Receptor Substrate) expression in response to TSA treatment in Min6 cells has been reported to be causal of the impairment of β-cell function on HDAC inhibition.22 Our transcriptomic analyses reveal that TSA treatment decreases rather than increases IRS2 expression (Table S1) and mostly affects the expression of a large set of genes involved in various biological process, suggesting that the effect of HDACi on β-cell function is probably more complex than anticipated. Indeed, among deregulated genes in response to TSA treatment, most of β-cell specific genes are downregulated whereas a series of α-cell specific genes as well as genes unrelated to endocrine functions are upregulated. In accordance with previous results from Kubicek et al. observed in TSA-treated murine β-TC6 cells,15 these results suggest that global HDAC inhibition leads to an alternative transcriptional program leading to a switch from β-cell identity toward an undefined alternative identity partly overlapping with α-cell identity. Also in line with the analysis restricted to some β-cell markers from Yamoto E.,21 our data confirm that β-cell identity is effectively directly impaired by HDAC inhibition.
In this context, we demonstrate here that this alternative transcriptional program is likely consecutive to a drastic alteration of epigenome on HDAC inhibition in β-cell, especially through modification of histone acetylation profile within promoters. Whereas the definition of H3K9ac-enriched promoters at genome-wide level is consistent with global increase of histone acetylation level, it is much more counterintuitive to identify H3K9ac-depleted promoters on HDAC inhibition. Indeed, we found most of the β-cell specific genes promoters surprisingly H3K9ac-depleted in response to HDAC inhibition and correlated with a decrease of gene expression, because inhibition of HDAC deacetylase activity should trigger increased acetylation of histone proteins, including H3. As TSA is an unspecific class I and class II HDAC inhibitor, this experimental evidence suggests that one or several class I and/or class II HDAC enzymes could play a direct active role in the regulation of expression of genes involved in the β-cell identity. In accordance with this hypothesis, class I (i.e., HDAC1, 2 and 3) and class IIb (i.e., HDAC6) HDAC enzymes were shown to be bound within promoter of transcriptionally active gene promoters in human CD4+ T cells.23 However, we did not demonstrate any involvement of neither HDAC1 nor HDAC6 in the direct regulation of transcriptionally active genes in β cell (data not shown) suggesting that (1) conversely to observations from CD4+ T cells, HDAC1 and HDAC6 are not directly involved in the regulation of β-cell specific genes, (2) other HDAC could be implicated in this process such as HDAC2 and/or HDAC3.13,23 Alternatively, depletion of acetylation in promoter of transcriptionally active genes on HDAC inhibition in β cell could be consecutive to the loss of a HAT enzyme binding within promoter of these genes which has been recently proposed for CBP/p300 as an alternative mechanistic model explaining promoter histone deacetylation in response to HDAC inhibition in endothelial cells.24
We also provided evidences that HDAC inhibition also leads to enhancer remodeling. Enhancers are cell specific non-coding elements of the genome involved in long-distance cell-specific regulation of gene expression directly related to cell identity, especially β-cell identity.25 It has been recently reported that the HDAC inhibition using largazole induces a H3K27ac-depletion at enhancers in HCT116 cells26 suggesting a role of HDAC in enhancer activity. Although it remains to experimentally demonstrate that HDACs directly bind to β-cell enhancers, our results emphasize a crucial role of HDAC enzymes in enhancer activity to maintain an appropriate transcriptional pattern. At a mechanistic level, the level of enhancer activity is especially directly related to the amount of local production of enhancers RNA (eRNA)27 playing a direct role in the regulation of enhancer target genes expression28 In addition, as eRNA are transcribed from active enhancers, they exhibit tissue and lineage specificity, and serve as markers of cell state and function.29 Consequently, we hypothesize that remodeling of β-cell enhancers on HDAC inhibition could lead to an alteration of the β-cell specific eRNA expression profile, thus partly contributing to affect specific gene expression profile as recently pointed out in human BT474 cells.30 To date, eRNAs are not well characterized in β cells and the link between HDAC and eRNA needs to be further investigated to better define β-cell identity as well as improve molecular knowledge on β-cell function. In addition, whether HDACi-dependent reprogramming of enhancer epigenome contributes to altered binding of transcription factors remains to be investigated. Although these data need to be experimentally confirmed, our in silico analysis of upstream regulators of TSA-treated Min6 cells RNA-seq data using IPA suggest that the transcriptional activity of several β-cell specific transcription factors, such as HNF-1a or Pdx-1, may be decreased on TSA treatment. Conversely, the activity of ubiquitous, non-β-cell specific transcription factors, such as Sox2, ESR1, AHR or Gli-1, may be increased in TSA treated Min6 cells. Whether their altered transcriptional activity is associated with epigenomic enhancer reprogramming remains to be defined.
In summary, based on a pharmacological approach using the pan-HDAC inhibitor TSA, we provide comprehensive information indicating that HDAC inhibition negatively acts on β-cell transcriptional program through epigenome-wide remodeling, leading to an alteration of β-cell functional properties. These results also indicate that β-cell identity is likely under the control of HDAC activity acting both on gene expression as well as enhancer activity. The key role of acetyl-coA generation in the control of acetylation dependent-cellular metabolism has been previously demonstrated.31,32 Controlling HDAC activity may be considered as one of the mechanisms linking metabolism to gene expression and epigenome regulation, where HDACs could represent a rheostat that “senses” the metabolic condition of the cells, and translate it into an appropriate response. This is of particular interest for metabolic organs, such as liver, muscle and pancreas, which are continuously subjected to changes in nutrient conditions and should rapidly adapt to this environment. In this context, a molecular link between metabolism and β-cell chromatin state is crucial to maintain proper cellular function.33 In addition, other inhibitors of HDAC, such as butyrate, have been shown to exert negative effects on β-cell function and identity,34,35 suggesting that blocking HDAC activity, as proposed in anticancer therapies, may potentially have metabolic side effects. Other investigations are currently in progress to better define the role of each HDAC in this process.
Limitations of the study
A first limitation of this study is the use of a pan-HDAC inhibitor, which does not inhibit specific HDAC enzymes. The recent observations that Hdac3-β-cell specific deficient mice have improved glucose tolerance,13 and that blocking HDAC3 protects β-cells from apoptosis,9 suggest that specific HDAC may play key roles in pancreatic β cells. Conversely, TSA might operate through a broader inhibition of HDAC functions, rather than affecting specific HDAC enzymes, leading to a global negative effect on β-cell function. Further studies using specific HDAC inhibitors, such as the HDAC3 specific inhibitor RGFP966, may help to fully understand the precise contribution of these enzymes to β-cell biology.36
Second, although this study connects the negative effects of HDACi on β-cell function to the global remodeling of epigenome (i.e., histone acetylation) at genome-wide level, we cannot rule out that alteration of acetylome at the proteome-wide level on HDAC inhibition is related to the observed phenotype. Indeed, acetylation is a largely widespread conserved post-translational modification also affecting non-histone proteins/enzymes to regulate their functions.37 As acetylome recently defined in rat islet is enriched in metabolic pathways of β cells related to nutrient sensing,38 we propose that HDAC inhibition using hydroxamic acid such as TSA leads to a global protein deacetylation involved in these pathways impairing protein/enzyme functions concomitantly to epigenome remodeling inducing in turn β-cell failure. Nevertheless, given the specificity of action of TSA on class I and II HDAC, this implies that these two classes of enzymes play a role on both nuclear and cytoplasmic non-histone protein deacetylation process, which has not been yet reported in β cell whereas Sirt3 belonging to class III HDAC takes part to this process in rat Ins1 cells.38
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-IgG | Santa Cruz | sc2025; RRID:AB_737182 |
| Rabbit anti-H3K4me1 | Active Motif | 39297; RRID:AB_2615075 |
| Mouse anti-H3K4me3 | Active Motif | 61379; RRID:AB_2793611 |
| Mouse anti-H3K27me3 | Active Motif | 61017; RRID:AB_2614987 |
| Mouse anti-H3K27ac | Active Motif | 39685; RRID:AB_2793305 |
| Rabbit anti-H3K9ac | Abcam | ab4441; RRID:AB_2118292 |
| Rabbit anti-H3 | Abcam | ab1791; RRID:AB_302613 |
| Goat anti-rabbit HRP-conjugated | Sigma | A9169; RRID:AB_258434 |
| Rabbit anti-mouse HRP-conjugated | Sigma | A9044; RRID:AB_258431 |
| Biological samples | ||
| Human islets | ECIT Islet for Basic Research program | https://ecit.dri-sanraffaele.org |
| Chemicals, peptides, and recombinant proteins | ||
| Trichostatin A | Sigma | T1952-200UL |
| Critical commercial assays | ||
| BCA assay | Pierce | 23227 |
| ECL kit | Pierce | 34076 |
| Mouse insulin kit | Mercodia | 10-1247-01 |
| Human insulin kit | Mercodia | 10-1113-01 |
| RNeasy plus mini kit | Qiagen | 74134 |
| RNeasy plus micro kit | Qiagen | 74034 |
| TruSeq Stranded mRNA library prep | Illumina | 20020594 |
| Nextflex rapid DNA seq kit 2.0 | Perkin Elmer | NOVA-5188-01 |
| Transcriptor universal cDNA master | Roche | 05893151001 |
| Transcriptase inverse SuperScript™ III | Invitrogen | 18080093 |
| FastStart SYBR Green Master | Roche | FSSGMMRO |
| Deposited data | ||
| Raw and analyzed data | This paper | GEO: GSE234144 and GSE234148 |
| The Polycomb-dependent epigenome controls β-cell dysfunction, dedifferentiation and diabetes | Lu TT et al.17 | GEO: GSE110648 |
| Experimental models: Cell lines | ||
| Min6 | AddexBio | C0018008; RRID:CVCL_0431 |
| EndoC-βH1 | Human Cell Design | https://www.humancelldesign.com/human-beta-cells-endoc-bh1/; RRID:CVCL_L909 |
| Experimental models: Organisms/strains | ||
| C57Bl6/J | Charles River Laboratories | 000664 |
| LSL-Td-Tomato | The Jackson Laboratory | 007905 |
| Rip-cre | Herrera, P.L. et al.39 | RRID: MGI:6195021 |
| Oligonucleotides | ||
| Cyclo_Mm_F | ATGGCACTGGCGGCAGGTCC | |
| Cyclo_Mm_R | TTGCCATTCCTGGACCCAAA | |
| Pdx1_Mm_F | ATTGTGCGGTGACCTCGGGC | |
| Pdx1_Mm_R | GATGCTGGAGGGCTGTGGCG | |
| Mafa_Mm_F | TCCGACTGAAACAGAAGCGG | |
| Mafa_Mm_R | CTCTGGAGCTGGCACTTCTC | |
| Foxo1_Mm_F | TGCCCAACCAAAGCTTCCCACA | |
| Foxo1_Mm_R | TGGACTGCTCCTCAGTTCCTGCT | |
| Ins1_Mm_F | GCCAAACAGCAAAGTCCAGG | |
| Ins1_Mm_R | GTTGAAACAATGACCTGCTTGC | |
| Software and algorithms | ||
| GSEA | Subramanian et al.40; Mootha et al.41 | http://www.broadinstitute.org/gsea/; RRID:SCR_003199 |
| Metascape | Zhou et al.42 | http://metascape.org/gp/index.html#/main/step1; RRID:SCR_016620 |
| GREAT | McLean et al.16 | http://great.stanford.edu/public/html/splash.php; RRID:SCR_005807 |
| Ingenuity Pathway Analysis | Qiagen | http://www.ingenuity.com/products/pathways_analysis.html; RRID:SCR_008653 |
| Bcl2fastq conversion software | Illumina |
https://support.illumina.com/sequencing/sequencing_software/bcl2fastq-conversion-software.html; version 2.19.1 and version 2.20; RRID:SCR_015058 |
| Cutadapt | http://code.google.com/p/cutadapt/ | version 1.7.1; RRID:SCR_011841 |
| STAR | http://code.google.com/p/rna-star/ | version 2.5.2b; RRID:SCR_004463 |
| RSEM | http://deweylab.biostat.wisc.edu/rsem/ | version 1.3; RRID:SCR_013027 |
| DEseq2 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html | RRID:SCR_015687 |
| Trim Galore | http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ | version 0.4.5; RRID:SCR_011847 |
| Bowtie2 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml | version 2.3.5.1; RRID:SCR_016368 |
| MACS2 | https://github.com/macs3-project/MACS | RRID:SCR_013291 |
| GraphPad Prism 9.0 | GraphPad | RRID:SCR_002798 |
Resource availability
Lead contact
Further information and request for reagents and resources should be directed to and will be fulfilled by the Lead Contact, Dr Jean Sébastien Annicotte (jean-sebastien.annicotte@inserm.fr)
Materials availability
All unique/stable reagents generated in this study are available from the lead contact with a completed Materials Transfer Agreement.
Experimental model and study participant details
Mice were maintained according to European Union guidelines for the use of laboratory animals. In vivo experiments were performed in compliance with the French ethical guidelines for studies on experimental animals (animal house agreement no. A59-35015, Authorization for Animal Experimentation, project approval by our local ethical committee no. APAFIS#2915-201511300923025v4). C57bl6J (Charles River Laboratories) and Rip-CreLox-STOP-Lox-Tomato (obtained after breeding of Rip-Cre mice39 with LSL-Td-Tomato (Jax, #stock number 007905)) mice were maintained under 12 h light/dark cycle and were fed ad libitum. All experiments were performed on 12-week-old male mice. The influence of sexual dimorphism was not interrogated in this study.
Method details
Cell culture and chemicals
Chemicals, unless stated otherwise, were purchased from Sigma-Aldrich. Min6 cells (Addexbio) were maintained in DMEM high Glucose glutaMAX medium (Gibco, 31966-021) supplemented with 15% SVF, 0.1% β-mercaptoethanol and antibiotics (penicillin/streptomycin). Trichostatin A (Sigma, T1952-200UL) was used at different time and concentrations, as indicated. EndoC-βH1 cells were purchased from Human Cell Design and cultured according supplier recommendations.
Human pancreatic tissue was harvested from human, non-diabetic, adult donors. Isolation and pancreatic islet culture were performed using established isolation procedures as described.43 Human islets were shipped and maintained in culture medium at 37°C in a humidified chamber with 5% CO2 in glucose free RPMI 1640 supplemented with 100 U ml−1 penicillin, 100 U ml−1 streptomycin and 11 mM glucose. Human islets were treated either with vehicle (DMSO 0.1%) or TSA (0.5 μM) during 16 h and snap-frozen for further processing.
Dot blot and western blot experiments
Min6 cells were cultured in 24-well plates (2.105 cells/well) and were resuspended in 100 μL of RIPA buffer (Thermo Scientific, 89901), then incubated at 4°C during 30 min under agitation. After centrifugation (15000 g, 20 min, 4°C), supernatant was harvested and protein concentration was measured using BCA assay (Pierce, 23227). Samples were denatured (95°C, 5 min) and 1 μg of total protein were dotted on nitrocellulose membrane using a dot blotter (Cleaver Scientific Ltd, CSL-D96). After drying (15 min, RT), membranes were saturated in saturation buffer (5% free fatty acids milk in TBS 1X, 2 h, room temperature), then incubated with primary antibody diluted in saturation buffer (16 h, 4°C). After incubation with HRP-coupled secondary antibody diluted in saturation buffer (1 h, room temperature), membranes were revelated using ECL kit (Pierce, 34076) on Chemidoc XRS+ imager (Biorad). Western blot was performed as previously described.44 Min6 cells were washed with PBS 1X and lysed with lysis buffer (50 mM Tris-HCl pH 8, 137 mM NaCl, 10% glycerol, 1% NP-40) supplemented with Protease Inhibitor cocktail and phosphatase inhibitors. Immunoblotting experiments were performed using 20 μg of total proteins and loaded on Precast SDS gel (Biorad), proteins were then transferred on nitrocellulose membrane and were further incubated in saturation buffer (TBS-Tween 0.05%-BSA 5%). Membranes were incubated 16 h at 4°C with primary antibodies as indicated in saturation buffer. After washing, membranes were incubated with the secondary antibody conjugated with horseradish peroxidase in saturation buffer (1 h, room temperature) and membranes were subsequently revealed using ECL kit (Pierce, 34076) on Chemidoc XRS+ imager (Biorad).
Dot blot and western blot images were processed and analyzed using ImageJ software. The level of acetylation was normalized to the level of histone H3.
Mouse pancreatic islet isolation and cell sorting
Mouse pancreatic islets were prepared as described previously.45 Briefly, pancreata were digested using type V collagenase (Sigma, ref C9263-1G) for 10 min at 37°C. After digestion, pancreatic islets were separated in a density gradient medium (Histopaque) and purified by handpicking under a binocular magnifier. Pancreatic islets were cultured for 24 h before further processing. For cell sorting experiments, mouse pancreatic islets isolated from RipCre-tdTomato mice were trypsinized for 7 min with 1 mM of trypsine (Gibco). Dissociated pancreatic cells were directly run into an Influx sorter (Becton Dickinson) equipped with a 86 μm nozzle and tuned at a pressure of 24.7 psi and a frequency of 48.25 kHz. Sample fluid pressure was adjusted to reach and event rate of 10 000 events/second. β cells and other pancreatic cells were selected as Tomato + (Tom+, β cells) and Tomato – (Tom-, non-β cells) and sorted in purity mode (phase mask 16/16) directly in RNeasy kit Lysis buffer for further processing.
Glucose stimulated insulin secretion (GSIS)
Min6 cells cultured in 96-well plates (2.104 cells/well) were glucose-starved in 200 μL of starvation buffer (Krebs Ringer buffer (KRB) supplemented with BSA 0.5%, 1 h, 37°C, 5% CO2). After incubation, the starvation buffer was discarded and cells were incubated in 200 μL of 2.8 mM glucose-concentrated starvation buffer (1 h, 37°C, 5% CO2). After 2.8 mM glucose samples recovering, cells were incubated in 200 μL of 20 mM glucose-concentrated starvation buffer (1 h, 37°C, 5% CO2). After 20 mM glucose samples recovering, the intracellular insulin content was recovered in 100 μL of lysis buffer (Ethanol 75%, HCl 1.5%). Insulin concentration was measured using mouse or human Insulin ELISA kits according the manufacturer’s instructions (Mercodia).
RNA extraction and RT-qPCR
Total RNA was extracted from Min6 cells using RNeasy Plus mini kit and from pancreatic islets using RNeasy Plus micro kit (Qiagen, ref. 74034) following the manufacturer’s instruction. Total RNA sample concentrations were determined using a Nanodrop spectrophotometer (Implen). 100 ng of RNA was used for reverse transcription (RT) using Transcriptor universal cDNA master mix (Roche) for Min6 cells or superscript III enzyme (Invitrogen) for mouse pancreatic islets according to the manufacturer’s instructions. Gene expression was measured through quantitative real-time PCR (qPCR) using FastStart SYBR Green master mix (Roche) according to the manufacturer’s recommendations in a LightCycler 480 II device (Roche). Mouse RT-qPCR results were normalized to endogenous cyclophilin reference mRNA levels. The results are expressed as the relative mRNA level of a specific gene expression using the formula 2−ΔΔCt.
RNA sequencing (RNA-seq)
RNA quality was verified using RNA 6000 nanochips (Agilent, #5067-1511) on the Agilent 2100 bioanalyzer (Agilent, #G2939A). 500 ng of purified RNA with RNA integrity number (RIN) ≥8 was subsequently used for library preparation with the TruSeq Stranded mRNA library Prep (Illumina, #20020594) and sequenced on the Illumina NextSeq500 system using a paired-end 2 × 75 bp protocol. Raw sequencing data are available under the accession number GSE234148.
Chromatin immunoprecipitation sequencing (ChIP-seq)
20.106 Min6 cells were treated with formaldehyde at a final concentration of 1% to cross link DNA and protein complexes during 10 min. The reaction was stopped by the addition of Glycine 0.125 M during 5 min. Cells were lysed and DNA-protein complexes were sheared using the Bioruptor Pico (Diagenode, #B01060010) for 8 min. The sheared chromatin was followed by immunoprecipitation with either the non-specific antibody IgG (Santa Cruz, #sc2025), H3K4me1 (Active motif, #39297), H3K4me3 (Active motif, #61379), H3K27me3 (Active motif, #61017), H3K27ac (Active motif, #39685) or H3K9ac (Abcam, #ab4441). 1 ng of eluted and purified DNA was used to prepare libraries with the Nextflex rapid DNA seq kit 2.0 (Perkin Elmer, #NOVA-5188-01) on the Illumina NextSeq500 system using a single read 100 bp protocol. Raw sequencing data are available under the accession number GSE234144.
Bioinformatic analysis
RNA-seq
The demultiplexing of sequence data (from BCL files generated by Illumina sequencing systems to standard FASTQ file formats) was performed using bcl2fastq Conversion Software (Illumina; version 2.19.1). Trimming of residuals adapters and low-quality reads was performed using Cutadapt software (version 1.7.1). Subsequently, sequence reads from FASTQ files were mapped to the mouse genome (mm10) using STAR Aligner (version 2.5.2b). On average, 38M reads were generated per sample, and 93.5% +/− 0.9% of them were accurately mapped. The counting of the different genes and isoforms was performed using RSEM (version 1.3). Finally, differential expression was performed using DESeq2 package.
ChIP-seq
The demultiplexing of sequence data (from BCL files generated by Illumina sequencing systems to standard FASTQ file formats) was performed using bcl2fastq Conversion Software (Illumina; version 2.20). Trimming of residuals adapters and low-quality reads was performed using TrimGalore (version 0.4.5). Subsequently, sequence reads from FASTQ files were mapped to the mouse genome (mm10) using Bowtie2 Aligner (version 2.3.5.1). Finally peak-calling was performed using MACS2 software (version 2.2.7.1).
Pathway analysis
RNA-seq data and integrated ChIP-seq/RNA-seq data were uploaded to Metascape website42 or Ingenuity Pathway Analysis software (Qiagen) or GSEA40,41 as specified. For RNA-seq data, an adjusted p value <0.05, LogFC>1 and LogFC<−1 were set as thresholds as indicated and pathway analyses were performed using the core analysis function, including canonical pathways, upstream regulators, diseases, biological functions and molecular networks.
Quantification and statistical analysis
Data are presented as mean ± s.e.m. Statistical analyses were performed using a two-tailed unpaired Student’s t test, one-way analysis of variance (ANOVA) followed by Dunnett’s post hoc test or two-way ANOVA with Tukey’s post hoc tests comparing all groups to each other, using GraphPad Prism 9.0 software. Differences were considered statistically significant at p < 0.05 (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 and ∗∗∗∗p < 0.0001).
Acknowledgments
The authors thank the members of the INSERM U1283-EGID and INSERM UMR1167-RID-AGE for helpful discussions. We are grateful to Yannick Campion for his support and his administrative contribution during this project. Human islets were provided through the JDRF award 31-2008-416 (ECIT Islet for Basic Research program). The authors thank “France Génomique” consortium (ANR-10-INBS-009), UMS2014-US41 and the Experimental Resources platform from Université de Lille. This work was supported by the Agence Nationale de la Recherche (ANR) grants (EGID ANR-10-LABX-46; LIGAN-PM Equipex 2010 ANR-10-EQPX-07-01; ANR-16-IDEX-0004 ULNE; ANR BETAPLASTICITY ANR-17-CE14-0034), EFSD, Inserm, CNRS, Université de Lille, Institut Pasteur de Lille (CTRL Melodie), Fondation pour la Recherche Médicale (FDT202106013015, EQU202103012732), I-SITE ULNE (EpiRNAdiab Sustain grant), Conseil Régional Hauts de France, Métropole Européenne de Lille, Société d’Accélération du Transfert de Technologie Nord and Société Francophone du Diabète.
Authors contributions
Conceptualization, F.O. and J.S.A.; Methodology, F.O., M.M., Am.B., and J.S.A.; Investigation, F.O., M.M., M.D., B.T., L.B., C.B., E.D., S.A., Al.B., E.B., O.M.C., L.P., G.P., L.R., C.C., F.B., and E.C.; Resources, C.G., J.E., D.D., J.K.C., F.P., B.S., P.F., and Am.B.; Data curation, F.O., M.M., M.D., L.B., S.A., and Al.B.; Writing – original draft, F.O., M.M., and J.S.A.; Writing – Review and Editing, J.E., P.F., and Am.B.; Visualization, F.O., M.M., and J.S.A.; Supervision, F.O., Am.B., and J.S.A.; Funding acquisition, J.S.A.
Declaration of interests
The authors declare that there is no conflict of interests regarding the publication of this article.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: June 30, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.107231.
Contributor Information
Frédérik Oger, Email: Frederik.oger@univ-lille.fr.
Jean-Sébastien Annicotte, Email: jean-sebastien.annicotte@inserm.fr.
Supplemental information
Data and code availability
-
•
This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table. All original data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Huising M.O. Paracrine regulation of insulin secretion. Diabetologia. 2020;63:2057–2063. doi: 10.1007/s00125-020-05213-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jennings R.E., Scharfmann R., Staels W. Transcription factors that shape the mammalian pancreas. Diabetologia. 2020;63:1974–1980. doi: 10.1007/s00125-020-05161-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rabhi N., Hannou S.A., Froguel P., Annicotte J.-S. Cofactors as metabolic sensors driving cell adaptation in physiology and disease. Front. Endocrinol. 2017;8:304. doi: 10.3389/fendo.2017.00304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Ruijter A.J.M., van Gennip A.H., Caron H.N., Kemp S., van Kuilenburg A.B.P. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem. J. 2003;370:737–749. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mouchiroud L., Eichner L.J., Shaw R.J., Auwerx J. Transcriptional coregulators: fine-tuning metabolism. Cell Metabol. 2014;20:26–40. doi: 10.1016/j.cmet.2014.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Z., Song J., Milne T.A., Wang G.G., Li H., Allis C.D., Patel D.J. Pro isomerization in MLL1 PHD3-bromo cassette connects H3K4me readout to CyP33 and HDAC-mediated repression. Cell. 2010;141:1183–1194. doi: 10.1016/j.cell.2010.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McClure J.J., Li X., Chou C.J. Advances and challenges of HDAC inhibitors in cancer therapeutics. Adv. Cancer Res. 2018;138:183–211. doi: 10.1016/bs.acr.2018.02.006. [DOI] [PubMed] [Google Scholar]
- 8.Larsen L., Tonnesen M., Ronn S.G., Størling J., Jørgensen S., Mascagni P., Dinarello C.A., Billestrup N., Mandrup-Poulsen T. Inhibition of histone deacetylases prevents cytokine-induced toxicity in beta cells. Diabetologia. 2007;50:779–789. doi: 10.1007/s00125-006-0562-3. [DOI] [PubMed] [Google Scholar]
- 9.Chou D.H.C., Holson E.B., Wagner F.F., Tang A.J., Maglathlin R.L., Lewis T.A., Schreiber S.L., Wagner B.K. Inhibition of histone deacetylase 3 protects beta cells from cytokine-induced apoptosis. Chem. Biol. 2012;19:669–673. doi: 10.1016/j.chembiol.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lindeløv Vestergaard A., Heiner Bang-Berthelsen C., Fløyel T., Lucien Stahl J., Christen L., Taheri Sotudeh F., de Hemmer Horskjær P., Stensgaard Frederiksen K., Greek Kofod F., Bruun C., et al. MicroRNAs and histone deacetylase inhibition-mediated protection against inflammatory beta-cell damage. PLoS One. 2018;13:e0203713. doi: 10.1371/journal.pone.0203713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khan S., Jena G. Valproic acid improves glucose homeostasis by increasing beta-cell proliferation, function, and reducing its apoptosis through HDAC inhibition in juvenile diabetic rat. J. Biochem. Mol. Toxicol. 2016;30:438–446. doi: 10.1002/jbt.21807. [DOI] [PubMed] [Google Scholar]
- 12.Lundh M., Galbo T., Poulsen S.S., Mandrup-Poulsen T. Histone deacetylase 3 inhibition improves glycaemia and insulin secretion in obese diabetic rats. Diabetes Obes. Metabol. 2015;17:703–707. doi: 10.1111/dom.12470. [DOI] [PubMed] [Google Scholar]
- 13.Remsberg J.R., Ediger B.N., Ho W.Y., Damle M., Li Z., Teng C., Lanzillotta C., Stoffers D.A., Lazar M.A. Deletion of histone deacetylase 3 in adult beta cells improves glucose tolerance via increased insulin secretion. Mol. Metabol. 2017;6:30–37. doi: 10.1016/j.molmet.2016.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ye J. Improving insulin sensitivity with HDAC inhibitor. Diabetes. 2013;62:685–687. doi: 10.2337/db12-1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kubicek S., Gilbert J.C., Fomina-Yadlin D., Gitlin A.D., Yuan Y., Wagner F.F., Holson E.B., Luo T., Lewis T.A., Taylor B., et al. Chromatin-targeting small molecules cause class-specific transcriptional changes in pancreatic endocrine cells. Proc. Natl. Acad. Sci. USA. 2012;109:5364–5369. doi: 10.1073/pnas.1201079109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McLean C.Y., Bristor D., Hiller M., Clarke S.L., Schaar B.T., Lowe C.B., Wenger A.M., Bejerano G. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 2010;28:495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu T.T.H., Heyne S., Dror E., Casas E., Leonhardt L., Boenke T., Yang C.H., Sagar L., Arrigoni L., Dalgaard K., et al. The polycomb-dependent epigenome controls beta cell dysfunction, dedifferentiation, and diabetes. Cell Metabol. 2018;27:1294–1308.e7. doi: 10.1016/j.cmet.2018.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Gurp L., Fodoulian L., Oropeza D., Furuyama K., Bru-Tari E., Vu A.N., Kaddis J.S., Rodríguez I., Thorel F., Herrera P.L. Generation of human islet cell type-specific identity genesets. Nat. Commun. 2022;13:2020. doi: 10.1038/s41467-022-29588-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haumaitre C., Lenoir O., Scharfmann R. Histone deacetylase inhibitors modify pancreatic cell fate determination and amplify endocrine progenitors. Mol. Cell Biol. 2008;28:6373–6383. doi: 10.1128/MCB.00413-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lenoir O., Flosseau K., Ma F.X., Blondeau B., Mai A., Bassel-Duby R., Ravassard P., Olson E.N., Haumaitre C., Scharfmann R. Specific control of pancreatic endocrine beta- and delta-cell mass by class IIa histone deacetylases HDAC4, HDAC5, and HDAC9. Diabetes. 2011;60:2861–2871. doi: 10.2337/db11-0440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamato E. High dose of histone deacetylase inhibitors affects insulin secretory mechanism of pancreatic beta cell line. Endocr. Regul. 2018;52:21–26. doi: 10.2478/enr-2018-0004. [DOI] [PubMed] [Google Scholar]
- 22.Kawada Y., Asahara S.I., Sugiura Y., Sato A., Furubayashi A., Kawamura M., Bartolome A., Terashi-Suzuki E., Takai T., Kanno A., et al. Histone deacetylase regulates insulin signaling via two pathways in pancreatic beta cells. PLoS One. 2017;12:e0184435. doi: 10.1371/journal.pone.0184435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Z., Zang C., Cui K., Schones D.E., Barski A., Peng W., Zhao K. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell. 2009;138:1019–1031. doi: 10.1016/j.cell.2009.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rafehi H., Balcerczyk A., Lunke S., Kaspi A., Ziemann M., Kn H., Okabe J., Khurana I., Ooi J., Khan A.W., et al. Vascular histone deacetylation by pharmacological HDAC inhibition. Genome Res. 2014;24:1271–1284. doi: 10.1101/gr.168781.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pasquali L., Gaulton K.J., Rodríguez-Seguí S.A., Mularoni L., Miguel-Escalada I., Akerman İ., Tena J.J., Morán I., Gómez-Marín C., van de Bunt M., et al. Pancreatic islet enhancer clusters enriched in type 2 diabetes risk-associated variants. Nat. Genet. 2014;46:136–143. doi: 10.1038/ng.2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sanchez G.J., Richmond P.A., Bunker E.N., Karman S.S., Azofeifa J., Garnett A.T., Xu Q., Wheeler G.E., Toomey C.M., Zhang Q., et al. Genome-wide dose-dependent inhibition of histone deacetylases studies reveal their roles in enhancer remodeling and suppression of oncogenic super-enhancers. Nucleic Acids Res. 2018;46:1756–1776. doi: 10.1093/nar/gkx1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Danko C.G., Hyland S.L., Core L.J., Martins A.L., Waters C.T., Lee H.W., Cheung V.G., Kraus W.L., Lis J.T., Siepel A. Identification of active transcriptional regulatory elements from GRO-seq data. Nat. Methods. 2015;12:433–438. doi: 10.1038/nmeth.3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Melo C.A., Drost J., Wijchers P.J., van de Werken H., de Wit E., Oude Vrielink J.A.F., Elkon R., Melo S.A., Léveillé N., Kalluri R., et al. eRNAs are required for p53-dependent enhancer activity and gene transcription. Mol. Cell. 2013;49:524–535. doi: 10.1016/j.molcel.2012.11.021. [DOI] [PubMed] [Google Scholar]
- 29.Arnold P.R., Wells A.D., Li X.C. Diversity and emerging roles of enhancer RNA in regulation of gene expression and cell fate. Front. Cell Dev. Biol. 2019;7:377. doi: 10.3389/fcell.2019.00377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Greer C.B., Tanaka Y., Kim Y.J., Xie P., Zhang M.Q., Park I.H., Kim T.H. Histone deacetylases positively regulate transcription through the elongation machinery. Cell Rep. 2015;13:1444–1455. doi: 10.1016/j.celrep.2015.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wellen K.E., Hatzivassiliou G., Sachdeva U.M., Bui T.V., Cross J.R., Thompson C.B. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324:1076–1080. doi: 10.1126/science.1164097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wellen K.E., Thompson C.B. Cellular metabolic stress: considering how cells respond to nutrient excess. Mol. Cell. 2010;40:323–332. doi: 10.1016/j.molcel.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vanderkruk B., Hoffman B.G. Metabolism as a central regulator of beta-cell chromatin state. FEBS J. 2021;288:3683–3693. doi: 10.1111/febs.15562. [DOI] [PubMed] [Google Scholar]
- 34.Bridgeman S., Ellison G., Newsholme P., Mamotte C. The HDAC inhibitor butyrate impairs beta cell function and activates the disallowed gene hexokinase I. Int. J. Mol. Sci. 2021;22:13330. doi: 10.3390/ijms222413330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu H.K., Green B.D., Flatt P.R., McClenaghan N.H., McCluskey J.T. Effects of long-term exposure to nicotinamide and sodium butyrate on growth, viability, and the function of clonal insulin secreting cells. Endocr. Res. 2004;30:61–68. doi: 10.1081/erc-120028485. [DOI] [PubMed] [Google Scholar]
- 36.Xu W.S., Parmigiani R.B., Marks P.A. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene. 2007;26:5541–5552. doi: 10.1038/sj.onc.1210620. [DOI] [PubMed] [Google Scholar]
- 37.Zhao S., Xu W., Jiang W., Yu W., Lin Y., Zhang T., Yao J., Zhou L., Zeng Y., Li H., et al. Regulation of cellular metabolism by protein lysine acetylation. Science. 2010;327:1000–1004. doi: 10.1126/science.1179689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang Y., Zhou F., Bai M., Liu Y., Zhang L., Zhu Q., Bi Y., Ning G., Zhou L., Wang X. The pivotal role of protein acetylation in linking glucose and fatty acid metabolism to beta-cell function. Cell Death Dis. 2019;10:66. doi: 10.1038/s41419-019-1349-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Herrera P.L. Adult insulin- and glucagon-producing cells differentiate from two independent cell lineages. Development. 2000;127:2317–2322. doi: 10.1242/dev.127.11.2317. [DOI] [PubMed] [Google Scholar]
- 40.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mootha V.K., Lindgren C.M., Eriksson K.F., Subramanian A., Sihag S., Lehar J., Puigserver P., Carlsson E., Ridderstråle M., Laurila E., et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 42.Zhou Y., Zhou B., Pache L., Chang M., Khodabakhshi A.H., Tanaseichuk O., Benner C., Chanda S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019;10:1523. doi: 10.1038/s41467-019-09234-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kerr-Conte J., Vandewalle B., Moerman E., Lukowiak B., Gmyr V., Arnalsteen L., Caiazzo R., Sterkers A., Hubert T., Vantyghem M.C., Pattou F. Upgrading pretransplant human islet culture technology requires human serum combined with media renewal. Transplantation. 2010;89:1154–1160. doi: 10.1097/TP.0b013e3181d154ac. [DOI] [PubMed] [Google Scholar]
- 44.Rabhi N., Hannou S.A., Gromada X., Salas E., Yao X., Oger F., Carney C., Lopez-Mejia I.C., Durand E., Rabearivelo I., et al. Cdkn2a deficiency promotes adipose tissue browning. Mol. Metabol. 2018;8:65–76. doi: 10.1016/j.molmet.2017.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bourouh C., Courty E., Rolland L., Pasquetti G., Gromada X., Rabhi N., Carney C., Moreno M., Boutry R., Caron E., et al. The transcription factor E2F1 controls the GLP-1 receptor pathway in pancreatic beta cells. Cell Rep. 2022;40:111170. doi: 10.1016/j.celrep.2022.111170. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table. All original data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.