

Abstract
Also known as electrospray, electrohydrodynamic atomization has been used extensively in the last 15 years to develop polymer-based particles for drug delivery in cell and animal models. More recently, novel core-shell, multi-axial, and other electrospray particles have been developed from an array of polymers for a variety of biomedical applications. This review focuses on electrospray as a novel method of particle fabrication for drug delivery, specifically highlighting the applications of these particle systems in cell culture and animal models while also discussing polymers used for particle fabrication. Applications of electrospray particles to treat glioma, ovarian cancer, and breast cancer are reviewed. Additionally, delivery of antibiotics, gene therapy, and bacterial cells formulated in electrospray particles is discussed. Finally, vaccines as well as drug eluting particles for differentiation of stem cells and tissue engineering are highlighted. The article concludes with a discussion of where the future of electrospray technology can go to strengthen its foothold in the biomedical field.
Keywords: Polymeric particles, Cancer, Antimicrobial, Gene delivery, Cell delivery, Vaccines, Tissue engineering
1. Introduction
Electrospray (ES), also known as electrohydrodynamic atomization, has emerged as an advanced technology to generate particles for drug delivery. ES is akin to electrospinning, but is usually performed with more dilute polymer solutions while using the same experimental setup: a high-voltage power source, a glass syringe, a syringe pump, and a grounded conductive surface [1] (Fig. 1a). Moreover, ES has the versatility to produce particles comprised of polymers [2,3], microbubbles [4], polyplexes [5], amphiphilic polymers [6,7], silica [8], and lipids [9,10]. These particles have been used for the encapsulation of small molecules, proteins, theranostic elements, nucleotides, cells, and other therapeutics [2,7]. In addition, polymeric particles have been shown to encapsulate multiple elements as well as incorporate multiple polymers to form di-, tri-, tetra-, and additional layered polymeric particles [2].
Fig. 1.
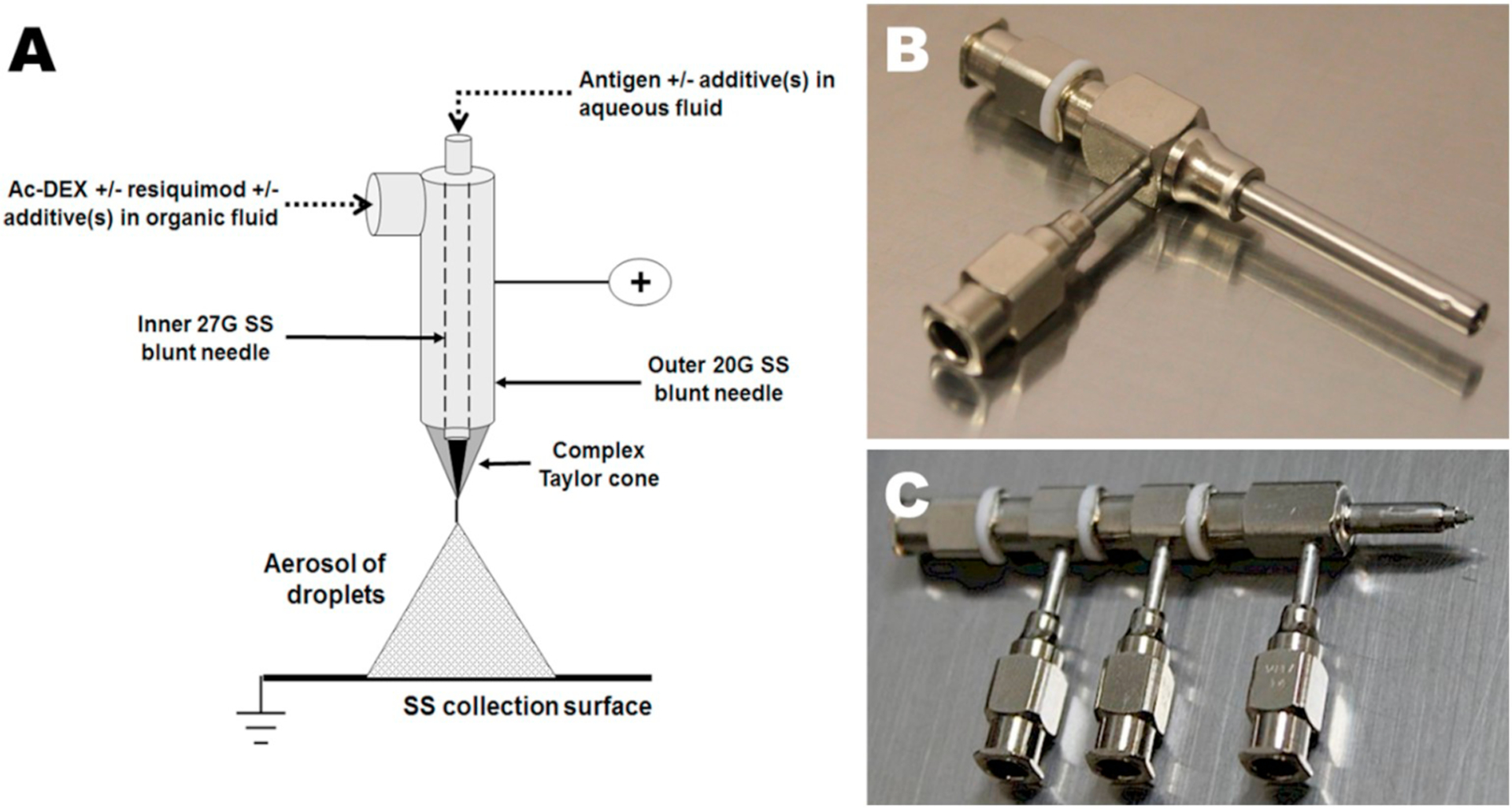
(A) Co-axial ES apparatus for generation of acetalated dextran (Ac-DEX) microparticles encapsulating the adjuvant resiquimod and a protein antigen in a single microparticle [11]. (B) Co-axial needle [12] and (C) Tetra-axial needle [12]. The Taylor cone would be created at the right-hand tip, with Luer-Lock style imports at the left and facing down.
Typically, ES involves the generation of an aerosol spray of the cargo(s) and encapsulant matrix(es). Using a syringe pump at a low flow rate of hundreds of microliters per hour, a volatile solution with the matrix(es) and encapsulate(s) dissolved is sprayed through a needle onto or into a solid or liquid substrate. Applying positive or negative charge to the needle or collection substrate in order to create a difference in electrical potential on the order of tens of kilovolts (kV) results in the formation of a Taylor Cone at the needle orifice. In a monoaxial (single needle) spray, all of the sprayed constituents mix throughout the process; however, in a multi-axial spray (Fig. 1), the individual solutions (e.g. protein in aqueous phase and polymer in an organic solvent) do not mix until they leave their respective channels in the syringe needle and enter the Taylor Cone (Fig. 1a). This limited mixing can facilitate the formation of core-shell particles wherein an inner core particle is formed with an outer layer around it [13]. At the tip of the Taylor Cone the solution is ejected in a jet toward the oppositely charged collection substrate. At relatively high polymer concentrations, polymer chain entanglement results in high viscosity of the solution, such that the Taylor Cone jet forms a continuous fiber travelling to the collection substrate, known as electrospinning. At lower solution viscosities, aerosol droplets are ejected due to repulsion forces that are an effect of like charges overcoming the cohesive forces of surface tension and viscosity, resulting in an electrospray. Maintenance of a stable spray requires balancing the solution properties with the flow rate, along with a number of other parameters (Fig. 2). After the droplets are aerosolized, they travel to a grounded or oppositely charged conductive plate or liquid where they collect. As the droplets move toward the substrate, the volatile solvent carrier evaporates, increasing the droplet charge density and leading the droplets to split into even smaller droplets, which increases the overall droplet surface area and leads to rapid evaporation of solvent over a relatively short distance. The effect of this evaporation on the particle morphology can vary according to the ratio of the rate of evaporation to the rate of solute diffusion within the solvent, which can be represented as the Peclet number [14]. A low rate of diffusion of the solutes through the droplet, corresponding to a higher Peclet number, results in accumulation of solutes at the surface of the droplet as the solvent evaporates, leading to the formation of a hollow droplet which eventually crumples or collapses into a toroid or wrinkled particle shape. Higher rates of diffusion, corresponding to a lower Peclet number, allow solutes to reequilibrate to a more energetically favorable homogenous spherical distribution, resulting in the formation of more smooth particles [15]. At times, an increase in the temperature of the air surrounding the spray, via heat lamp or other system, is used to fully evaporate the solvent. If the solvent is not fully evaporated at time of impact on the grounded plate, the particles will aggregate and/or flatten. Temperature, distance to plate, flow rates, viscosity, charge, salt concentration of solutions, solvent used, and numerous other variables can be finetuned to optimize particle production and vary morphology. These factors can lead to complex and novel particles that can have several benefits.
Fig. 2.
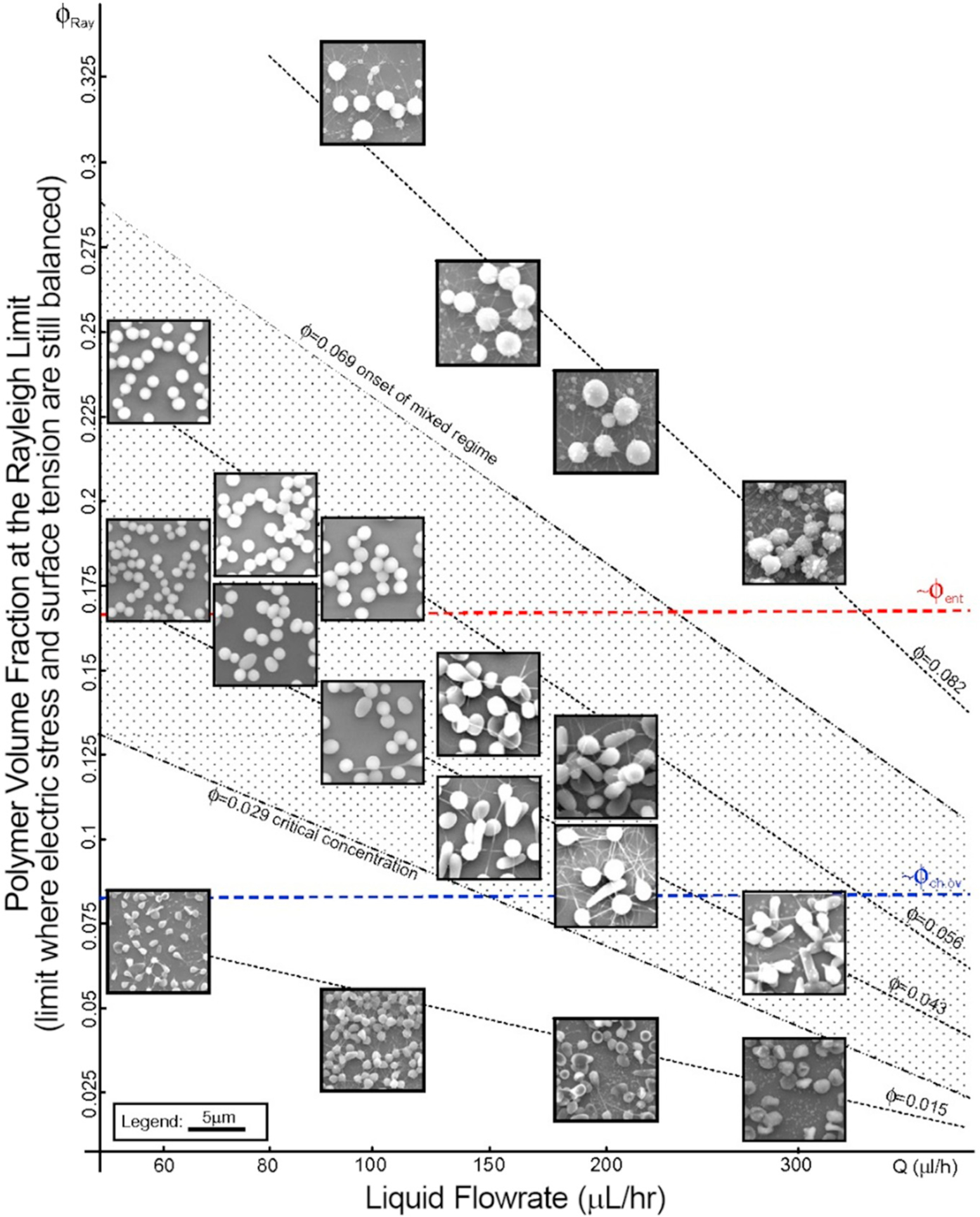
Figure modified from Almería et al. [16] to illustrate polydispersity of PLGA ES particles as a function of liquid flowrate (Q (μl/h)) and polymer volume fraction at the Rayleigh limit (ϕ). The Rayleigh limit is the point at which the electric stress and surface tension become unbalanced and the Taylor cone, or the flow in general, becomes unstable. Entanglement volume fraction, ϕent, is the volume fraction of polymer within the droplet that is large enough to form an entangled network of polymer chains. The overlap volume fraction, ϕch.ov, is the polymer volume fraction where the average distance between chains is equivalent to their radius in solution.
No matter how the particles are formed, or the cargo incorporated, there are several benefits to the generation of nano/microparticles using ES, especially in comparison to other common polymeric particle fabrication techniques such as emulsion, coacervation, or even microfluidic devices. The generation of particles via ES can lead to a less polydisperse size population than other methods, while minimizing batch-to-batch variation [9,10,13]. Additionally, compounds with a broader range of partition coefficients can generally be accommodated with ES since mono- or multi-axial generation of particles facilitate the use of a range of solvents and aqueous solutions individually or cosprayed. In addition, emulsification prior to spraying has been reported, which could further expand the number and types of cargoes that can be used [17]. Better solubility of the cargo in addition to using a continuous phase of air, rather than water or another solvent, leads to another advantage of ES: high encapsulation efficiencies (EEs) [18]. Whereas compounds can partition into the continuous aqueous phase during standard emulsion methods, in ES the spray aerosol is generated into air, limiting the ability of the cargo to diffuse away. Having a high EE facilitates facile encapsulation of multiple agents at a controlled amount (e.g. ratio to each other) that is often difficult to achieve with other methods where the inclusion of multiple agents may affect the EE of individual agents [11,19]. Another benefit is that the scalability of ES is improved compared to emulsion and coacervation processes because it is a continuous rather than batch method that scales linearly with the number of ES heads [20,21]. This scalable production has been applied in materials coatings and can be advantageous when developing low cost therapies and vaccines. The importance of production cost is illustrated by the limited advancement of poly(lactic-co-glycolic acid) (PLGA) tetanus vaccines by the World Health Organization (WHO), in part because production costs were too significant [22]. Another aspect of the limited advancement of PLGA tetanus vaccines was the denaturation of the protein antigen during the emulsion process, not only due to prolonged contact with the solvent, but also the high-speed shearing during emulsion. Proper protein tertiary and perhaps even quaternary structure is important in both protein therapeutics and for B cell activation in vaccines [23]. ES has been shown to be less harsh on proteins compared to methods like emulsion because it uses low shear forces and minimal contact time with solvents, on the order of microseconds in the Taylor Cone during a co-axial spray [11,24]. As such, ES has also been shown not to denature DNA during encapsulation [25]. Finally, when ES is used with multi-axial needles (Fig. 1) to develop immiscible layers of polymer, it can create core-shell or multilayer particles (Fig. 3). This can facilitate an additional controlled release feature of the particle, further dictating the kinetic release rates of the encapsulates [13]. These aforementioned features of ES make it a very advantageous particle generation method for a wide range of uses.
Fig. 3.
Fluorescent micrograph of coumarin-6 encapsulated in the core of a core-shell co-axial ES microparticle comprised of poly(D, l-lactide) and poly(D,l-lactide-co-glycolide) [26].
ES has been used for a vast variety of applications including material coatings [27], solar cells [28], tissue engineering [29], microfluidics [30], microelectromechanical systems (MEMs) [31], and drug delivery [13]. Since its first application in or around the mid-2000s for drug encapsulation [32], studies are beginning to move from developing the technique to applying it to relevant in vitro and in vivo systems. Several reviews exist regarding configurations and optimization of ES parameters, with most focusing on development of the particle and/or in vitro release of cargo [1,2,13,29,33–36]. This review focuses on the reports of these particles being applied to cells and/or animal models and reports on the outcomes of these studies as they relate to advancements in the field of drug delivery, vaccine delivery, and tissue engineering.
2. Matrices used in electrospray
A variety of polymers have been applied to ES systems (Table 1), and this illustrates the broad variety of conditions that can be used to spray and form particles. When choosing a polymer there are several desirable physiochemical properties such as electrical conductivity, viscosity, and solubility that can be finely tuned to get the desired Taylor cone stability or particle size. Furthermore, if multi-axial ES is used, the miscibility of the solvents and polymers used will dictate whether core-shell or mixed phase particles are formed. There are several types of polymers reported here that are used in cell and animal models. These include polyesters, stimuli responsive polymers, natural polymers, and other non-degradable polymers.
Table 1.
Summary of matrices used in electrospray for drug delivery.
| Polymer class | Polymer | Degradation mechanism | Degradation rate | Degradation by-products | Ref |
|---|---|---|---|---|---|
| Polyesters | Poly(lactic acid) (PLA) | Hydrolysis of ester bonds | Months | Lactic acid | [37–39] |
| Poly(glycolic acid) (PGA) | Glycolic acid | [37–39] | |||
| Poly(lactic-co-glycolic acid) (PLGA) | Lactic acid and glycolic acid | [37–39] | |||
| Polycaprolactone (PCL) | Caproic acid | [37,40] | |||
| Stimuli responsive | 2-(40-N-dimethylamino-4-nitro-[1,10-biphenyl]-3-yl)butane-l,4-diyl dicarbonyl | Light responsive | Minutes when exposed to visible light (λex = 400–500 nm) | Photocleaved nitro-alkene derivative; secondary products not yet identified | [41] |
| Acetalated Dextran (Ac-DEX; Ace-DEX) | Hydrolysis of acetal groups | Tunable, ranging from hours to months based on reaction time and molecular weight | Dextran and exceedingly low levels of acetone and a short chain alcohol | [3,42–48] | |
| Eudagrits (methacrylic acid and methyl methacrylate co-polymers) | Not biodegradable | – | – | [49] | |
| Natural polymers | Alginate | Not biodegradable | – | – | [50] |
| Cellulose Derivatives | Hydrolysis and cellulases | Varies | Glucose; other degradation products vary by derivatives | [51–53] | |
| Gelatin | Hydrolysis and enzymatic degradation | Weeks or months | Prolylhydroxyproline and glycine | [54–57] | |
| Gliadin | Proteases | Slow: can be resistant to digestion | Glutamine and proline residues | [58,59] | |
| Fibroin | Not biodegradable | – | – | [60] | |
| Maize arabinoxylans | Enzymatic degradation by gut resident microbes | De-branching occurs on the order of days | Arabinose and xylose | [61–64] | |
| Chitosan | Enzymatic degradation | Varies based on degree of deacetylation and molecular weight | Glucosamine and chito-oligosaccharides | [65–67] | |
| Starches from yams | Hydrolysis and enzymatic degradation | Hours | Amylose and Amylopectin | [65,68] | |
| Cationic polymer and lipid | Poyethylenimine (PEI) | Non-biodegradable | – | – | [69–72] |
| 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP) | Hydrolysis of ester linkage | Unsaturated diacyl tails and amine head group | [70,73,74] | ||
| Other non-degradable | Ecoflex® | Non-biodegradable | – | – | [75] |
| polymers | Polyethylene glycol (PEG) | – | – | [76–78] | |
| Polyvinyl alcohol (PVA) | – | – | [79,80] |
2.1. Polyesters
The most used polymers for ES are by far poly(lactic-co-glycolic acid) (PLGA) and poly(lactic acid) (PLA). These are both polyesters, along with another commonly used polymer, polycaprolactone (PCL) and poly(glycolic acid) (PGA). Polyesters are biodegradable via hydrolyzable ester bonds. The ratio of glycolic acid and lactic acid in PLGA can be changed to somewhat alter the degradation rate, but generally 50:50 or 75:25 (PLA:PGA) polymers are used, with degradation rates on the order of months. With PLGA, the hydrolytic by-products are lactic acid and glycolic acid, with PCL yielding caproic acid. These acidic by-products have been shown to result in low pH locally [37], and in a particle form, PLGA has been shown to have pH ~3 inside particles [38,39]. Despite this, PLGA/PLA is a constituent of over a dozen FDA approved therapies, and because it has a valid drug master file, it is a common go-to polymer for drug delivery applications. Similarly, PCL has been approved in drug delivery and other devices.
2.2. Stimuli responsive
In this review, two primary stimuli are used to trigger the breakdown of ES particles: light and pH. Stimuli responsive materials can allow for release of encapsulate at a desired location or cell in the body, which can mitigate off-site toxicity as well as optimize therapy delivery. Carling et al. [41] formed a new light sensitive polymer called 2-(40-N-dimethylamino-4-nitro-[1,10-biphenyl]-3-yl)butane-1,4-diyl dicarbonyl for stimuli responsive delivery. As designed, the polymer degrades rapidly when exposed to visible light (λex = 400–500 nm). This polymer was developed to degrade when visible light was focused at an intensity of ~0.21 W/cm2 in the area of the delivered particles.
Acetalated dextran (Ac-DEX) has been used extensively by our lab and a few others for vaccine and drug delivery, including formulation via ES [3]. Ac-DEX is formed from dextran, an FDA approved polymer which is used as a plasma expander. The polymer is acid-labile and degrades more rapidly at lower pH, as observed intracellularly, near inflammation, or in tumors. The by-products of Ac-DEX degradation are primarily dextran, as well as exceedingly low levels of acetone and a short chain alcohol (ethanol or methanol) [42], well below what is produced naturally in the body or would be toxic to even pregnant women [43,44]. A unique feature of Ac-DEX is its finely tunable degradation rate (from hours to months) [81] that can be adjusted with reaction time and dextran molecular weight [42,46]. Additionally, early studies with the material indicate that Ac-DEX stabilizes proteins outside the cold-chain [47]. Ac-DEX has yet to be in any FDA approved products, but has illustrated little to no toxicity in animal models [3,48].
Eudragits are another class of pH responsive polymers. They are a co-polymer comprised of methacrylic acid - methyl methacrylate, typically used for oral (PO) delivery [49]. They are non-degradable, nonabsorbable, and non-toxic polymers. The L series polymers are anionic and dissolve around pH > 6 for enteric coating and delivery to the small intestine. Eudragit RL series polymers have quaternary ammonium groups and although they are water insoluble, they are often applied for film coating because they afford sustained release via polymer swelling in aqueous systems.
2.3. Natural polymers
Besides polyesters, natural polymers are the most commonly used type of polymers applied in ES. Natural polymers can sometimes have distinct characteristics and easy and inexpensive sourcing compared to synthetic polymers, but they can also have known allergic responses that can limit their use.
Alginate is a natural anionic polymer derived from brown algae [50]. It can be easily cross-linked in the presence of divalent cations such as Ca2+. Unless modified to become degradable, alginate is generally non-degradable in mammals. Particles made from alginate can be easily fabricated by dripping a solution of the polymer into a Ca2+ aqueous solution. Because of the ease of fabrication, alginate is used commercially in food (e.g. gummies, boba tea) as well as for oral delivery capsules.
Cellulose derivatives are used for ES and are usually modified to make them more soluble in organic solvents. Ethyl (hydroxyethyl) cellulose (EHEC) is a cellulose based polymer where ethyl and hydroxyethyl groups are linked to anhydroglucose units by ether linkages. Carboxymethylcellulose (CMC) has carboxymethyl on many of the hydroxyl groups of the glucopyranose monomers [51]. Both polymers degrade as cellulose degrades. They are both also used commercially as a thickening agent and to deliver drugs orally.
Gliadin is a wheat storage protein [58]. It is the water-soluble portion of gluten, which can limit its use to individuals who are not gluten sensitive. To that point, gliadin is the main immunogen of Celiac disease. Gliadin is often a contaminant detected in pharmaceutical preparations and is typically purified for formulation use.
Gelatin is a water soluble, denatured version of collagen. It is used as a gelling agent in many things including food and pharmaceuticals. As a protein, it is degradable in the body. It is often used to enhance cell adhesion for electrospinning [54] since it possesses binding groups for integrin binding of most cells.
Fibroin is an insoluble protein that is contained in spider silk [60]. Its primary structure consists of repeat sequences of amino acids (GlySer-Gly-Ala-Gly-Ala)n, to form anti-parallel beta sheets. These consecutive beta-sheets give the material a high stiffness and strength. Some bacteria are able to degrade fibroin, while mammalian species cannot.
Maize arabinoxylans are the main non-cellulose polysaccharides in corn. They can form gels in aqueous environments. Bran is the primary by-product of corn milling and mostly comprised of cellulose. Corn wax is the solid that is formed from the chilling of corn oil during the refining process and can be used for soap stock [61]. As polysaccharides, the structures are likely degradable, although this is not well understood.
Chitosan is a polysaccharide that can be isolated from various shellfish such as shrimp and crab. Since it is isolated from shellfish, there is concern that it could cause anaphylaxis in those who have shellfish allergies. A unique property of chitosan is that it is mucoadhesive and is commonly used for PO delivery, as it will attach to the mucous layer of the gastrointestinal tract. It is degradable and considered non-toxic but has not been FDA approved for use as a drug delivery polymer.
Yams are a very common plant across the globe. The starches from a yam have been shown to be viscous and stable in acidic conditions. They can be substituted for modified starches in processed foods and have been applied in the food industry. Their application in pharmaceutical has not been well reported [65].
2.4. Cationic polymers and lipids
Polyethylenimine (PEI) is a polymer comprised of repeat amine groups. It can be linear or branched. Abundant amines make the polymer highly cationic. When mixed with DNA or other negatively charged species, it can form polyplexes where the oppositely charged species intermix. At high levels PEI can be cytotoxic because the cations can complex to the cell’s DNA [69].
1,2-dioleoyl-3-trimethylammonium-propane (DOTAP) is a cationic phospholipid. It can be used in liposomes and other lipid complexes to provide a positive charge. Additionally, it can serve as an amphiphilic lipid to reduce surface tension and stabilize the surface of the particle.
2.5. Other non-degradable polymers
Ecoflex® is a rubbery material that is commonly used to make masks and prosthetics. It is approved for dermal contact but has not been approved for use in the body. The polymer is a non-degradable platinum-catalyzed silicone. Its ability to stretch and deform can afford it unique properties when formulated into particles.
Polyethylene glycol (PEG) is one of the most commonly used polymers for biomedical applications since it is highly thermodynamically favorable for it to interact with water. Due to this, it has anti-fouling properties that resist protein and cell adhesion, leading to reduced clearance by the reticuloendothelial system. This leads to long circulation times when therapeutics that are PEGylated (covalently bound to PEG) are given intravenously. Moreover, steric protection of protein therapeutics can occur, leading to reduced absorption or degradation of these therapies. The first PEGylated therapy was PEG covalently attached to interferon, and since then there have been more than a dozen PEGylated therapies including conjugates to proteins, peptides, aptamers, and even small molecules. Liposomal doxorubicin (Doxil®) is a PEGylated liposome and the most commercially applied liposomal therapy. PEG is not degradable, and often is used at molecular weights which will clear the kidney (< 40 kDa) because it will, over time, disassociate from the conjugate. There are reports of immune responses to PEG, which can complicate its use in humans [76].
Polyvinyl alcohol (PVA) is a common additive in polyester emulsions. It is a non-degradable material at realistic timescales in the body and is also water soluble. It is used as a thickening agent in drug formulations like eye drops. The addition of PVA to emulsions or ES can result in a more viscous solution, which could result in a more stable emulsion or Taylor cone.
3. Applications
3.1. Drug, protein, cell and gene delivery
3.2. Cancer drug delivery
The most common application of ES particles is for delivery of chemotherapeutics to treat cancer (Table 2). Since most ES particles are too large to exhibit the enhanced permeability and retention (EPR) effect [95], and intravenously (IV) injected particles primarily accumulate in the liver and spleen, direct injection into the tumor or peritumoral injection would be preferred. Intratumoral (IT) injections can be clinically relevant [96] for melanoma and other accessible cancers, and as cancer imaging techniques improve [97], this method of administration becomes more and more of an option for treatments.
Table 2.
Electrospray technologies developed for cancer drug delivery.
| Polymer | Cargo | Axial | Particle size range | Cell model | Animal model | Ref |
|---|---|---|---|---|---|---|
| PCL | Taxol | Mono | 1–15 μm | C6 Glioma | − | [82] |
| PLGA | Paclitaxel | Mono | 200 nm-10 μm | C6 Glioma | – | [83] |
| PLGA | Paclitaxel | Mono | 13–17 μm | – | C6 Glioma cells subQ (BALB/c nude mice) | [84] |
| PLGA+Alginate | Paclitaxel | Mono (two-step) | 10–15 μm (post-first step) 1.6–1.7 mm (post-second step) | C6 Glioma | C6 Glioma cells subQ (BALB/c nude mice) | [85] |
| EHEC | Fe3O4 NPs | Mono | 1–10 μm | GL261 Glioma | – | [86] |
| PLA PLGA | PaclitaxelSuramin | Co (miscible) | 10–20 μm | U87 Glioma | U87 Glioma cells subQ (BALB/c nude mice) | [87] |
| Gliadin or Gliadin-Gelatin | Cyclophosphamide | Mono | 200–400 nm | MCF7 Breast | – | [88] |
| PLGA+PCL or PVP | Combretastatin A4 Doxorubicin | Co (miscible) | 400–500 nm | B16-F10 Melanoma HUVECs | – | [89] |
| Silk Fibroin | Cisplatin | Mono | 59–75 nm | A549 Lung L929 Fibroblasts | – | [90] |
| PLGA+Albumin | (5,10,15, 20-tetrakis(mesohydroxyphenyl)porphyrin) (mTHPP), Dasatinib | Mono | 80–90 nm | U87MG Glioma | No disease (Wistar rats) | [91] |
| PLA | Gambogic-acid | Mono | 70–7500 nm | – | H22 cells orthotopic | [92] |
| Silk Fibroin PVA | Doxorubicin | Co (miscible) | 300–1800 nm | MDA-MB-231 Breast | MDA-MB-231 cells subQ (BALB/c nude mice) | [79] |
| Ecoflex®PEG | Docetaxel | Mono | 360 nm | SKOV-3 Ovarian | SKOV-3 cells subQ (B6 nude) | [75] |
| PLGA | Paclitaxel Etoposide | Co (miscible) | 1–4 μm | Saos-2 Osteosarcoma | – | [93] |
| Ac-DEX | cGAMP | Co (semimiscible) | 1–3 μm | BMDCs | B16F10 in C57BL/6 mice Orthotopic E077 in C57BL/6 and 6(Cg)- Tmem173tm1.2Camb/J (Tmem173−/−) mice | [94] |
3.2.1. Glioma treatment
Cancers affecting glial cells in the brain, known as gliomas, are the most studied cancers for application of ES particles (Table 2). Since surgical resection is the standard of care for brain cancer [98], interstitial therapy, where therapeutic agents are administered to the tumor resection cavity, can be easily applied. The FDA approved Carmustineloaded Gliadel® wafers are based on this practice. This therapy has significant room for improvement since Carmustine has low tumoricidal activity and is rapidly cleared from the brain upon release from the polymer wafer [99–101]. In 2005, Ding et al. [82] first reported the application of PCL particles encapsulating taxol, prepared via monoaxial ES, for the treatment of rat C6 Glioma cells in vitro. They noted that cellular uptake of particles was approximately 60% after 2 h of incubation and varied slightly with different processing parameters, including where the particles were collected during post spray processing (e.g. filter, pre-filter). Xie et al. [83] then expanded on this work in the following year by preparing paclitaxel-loaded PLGA ES particles 200 nm to 10 μm in size. These particles illustrated an increase in the ratio of G2/M (interphase to mitosis) in C6 cells, mostly as the dose of paclitaxel increased in the particles. Moreover, C6 viability decreased with drug loaded particle treatment in a time but not dose dependent manner, when given in vitro. Later, Ranganath et al. [85] (Fig. 4) took ES paclitaxel PLGA microparticles suspended in a sodium alginate solution and electrosprayed them a second time directly into a CaCl2 solution. They then encapsulated the PLGA microparticles into alginate hydrogel beads approximately 1.6 mm in size. When C6 glioma cells were cultured with the microparticle-hydrogel composite, greater apoptosis was observed for the encapsulated group at 6 days, compared to the soluble drug control; however, it was not significant versus the soluble drug at 2 and 4 days. The 50% lethal dose (LD50) of the formulation was significantly changed when the gelation time of the alginate beads (time in the CaCl2 solution) was altered, with increased gelation time leading to reduced cytotoxicity. Using another glioma cell line, GL261, Hayashi et al. [86] report cytotoxicity of red blood cell-like ES EHEC particles on the order of 1 to 10 μm in size, encapsulating superparamagnetic magnetite nanoparticles. In a very limited study, the cytotoxicity of the composite microparticles was reported to be the same as the control. In another study, Nie et al. [87] used co-axial ES to co-encapsulate paclitaxel and suramin in PLGA particles approximately 10 to 20 μm in size. When evaluating the co-therapy particles against a human glioma cell line, U87, they note an ~20% reduction in proliferation and viability after 3 days. Also, higher levels of apoptosis were observed with the co-therapy particles compared to free compound, but only after 6 days. Ramachandran et al. [91] also used U87 cells and treated them with PLGA ES particles encapsulating the photo-labile porphyrin 5,10,15, 20-tetrakis(meso-hydroxyphenyl)porphyrin (mTHPP) coated in albumin with dasatinib. When applied to U87 cells, a dose and time dependence for uptake was reported. When laser assisted activation of mTHPP was used, they noted an increase in reactive oxygen species (ROS) production by the glioma cells. Additionally, they noted inhibition of cell migration as a result of dasatinib. With these studies, they illustrated activity on U87 cells of both porphyrin and the tyrosine kinase inhibitor. In general, these studies illustrate that ES particles can release chemotherapeutics to lyse glioma cells in culture, particularly at longer time points, indicative of a controlled release system.
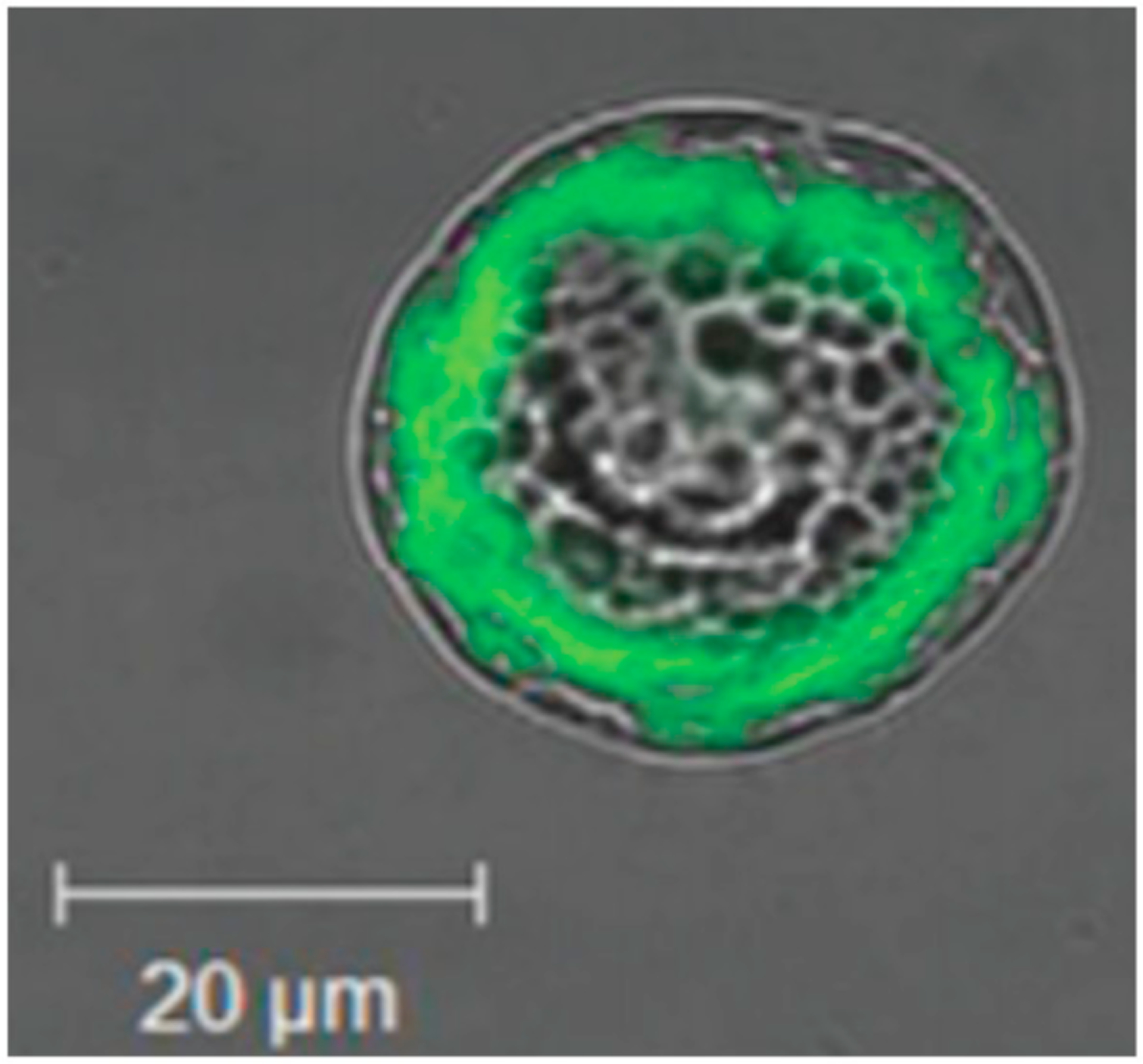
Fig. 4.

Ranganath et al.’s [85] encapsulation of ES paclitaxel PLGA particles in alginate (A-C), and then in vitro evaluation for apoptosis when cultured with C6 glioma cells (D) and when used peritumorally to treat a C6 subcutaneous tumor (E). Micrograph B is of the PLGA paclitaxel particles, and micrograph C is of the alginate particles, with the arrows indicating PLGA particles.
Although promising in vitro studies have been reported for the treatment of glioma cells with ES particles, evaluation in an animal model can advance pre-clinical studies toward the eventual translation of these therapies. Naraharisetti et al. [84] encapsulated both paclitaxel and etanidazole into 15 μm particles using monoaxial ES. When these particles were given in peritumoral implants for the treatment of a subcutaneous C6 tumor in BALB/c nude mice, cell death was observed histologically at the tumor edge. Decreased tumor volume was observed in the co-therapy particle group compared to soluble therapies. In comparison to other particle types, ES and spray dried particles displayed the same tumor inhibition with a four times lower dose of paclitaxel compared to disc shaped particles. Using the same animal model, Ranganath et al. [85] (Fig. 4) expand on their in vitro studies to show that after 21 days, tumor volume was reduced by paclitaxel loaded PLGA particles prepared via monoaxial ES, compared to untreated and soluble drug controls. They performed an IT incision to deliver 2 mg of particles and used valid sham controls for this method. Nie et al. [87] used a similar model with U87 cells in place of C6 cells to continue their in vitro work. Co-encapsulating paclitaxel and suramin, they found that localization of one drug versus the other in the core or shell of the particles did not result in significant differences in GBM growth. Additionally, statistical differences were not noted between individually delivered drugs and co-encapsulated compounds for tumor growth inhibition. However, histology showed fewer tumor cells and reduced proliferation with co-encapsulated particles. These studies illustrate the ability of ES particles to treat glioma cancer burdens in vivo, and significantly reduce the tumor burden in comparison to soluble drug controls. Moreover, ES affords the co-encapsulation of multiple drug therapies for enhanced treatment of glioma.
3.2.2. Other cancers
Several other studies have reported ES particles encapsulating chemotherapeutics for the treatment of other cancers such as breast, skin, lung, liver, or ovarian. Two studies have evaluated ES particles for the treatment of breast cancer. Using doxorubicin encapsulated in 300–1800 nm co-axial electrosprayed silk and PVA particles, Cao et al. [79] observed a dose and time response in cytotoxicity when MDA-MB-231 breast cancer cells were cultured with the particles. The cytotoxicity observed upon treatment with the doxorubicin particles was greater than treatment with soluble drug at the same concentration. Additionally, apoptosis increased in a dose and time responsive fashion with the particles and further increased when ultrasound was introduced to the culture. Applying MCF7 breast cancer cells in vitro, Gulfam et al. [88] used monoaxial ES to encapsulate cyclophosphamide into 200–400 nm gliadin and gelatin-gliadin nanoparticles. When cells were cultured with the particles, B-cell lymphoma 2 (BCL-2), a regulator of cell death, was downregulated in cells treated with soluble drug or particle-delivered drug. Moreover, they report there were no significant differences in BrdU incorporation between the soluble and encapsulated drug after 24 h, indicating no significant differences in cell proliferation. Looking at melanoma, Cao et al. [89] loaded combretastatin A4 and doxorubicin into composite PLGA, PCL, and PVP particles using a co-axial ES system. They evaluated the drug activity in vitro against B16-F10 melanoma cells and human umbilical vein endothelial cells (HUVECs). They noted that the co-encapsulated particles induced apoptosis in both cell types. Applying silk fibroin particles approximately 60 nm in size and encapsulating cisplatin, Qu et al. [90] evaluated lung cancer cells. They observed an increase in platinum uptake in lung cancer A549 cells over non-cancer L929 cells when cultured with particles containing the platinum-based cisplatin. The particles were able to induce cytotoxicity at an increased rate versus soluble drug or empty carrier. Yin et al. [92] used ES to encapsulate gambogic acid into PLA microparticles to evaluate in vivo for the treatment of a hepatocellular carcinoma using an H22 cell orthotopic model. In a distribution study with intravenous administration, they found that particles smaller than 185 nm accumulated the most in the liver, whereas those around 350 nm in size collected most extensively in the spleen and those approximately 7 μm in size had the highest accumulation in the lungs. This distribution correlated with the ratio of gambogic acid in a specific tissue to the ratio in all the tissues evaluated (i.e., heart, liver, spleen, lung, kidney). Survival time of mice inoculated with liver cancer was improved with the drug loaded particle group over the soluble drug group. Zhang et al. [93] used co-axial ES to encapsulate both paclitaxel and etoposide into core-shell PLGA particles. They report that the encapsulated drugs increased cytotoxicity when applied to osteosarcoma cells, in comparison to free drug alone. Varshosaz et al. [75] (Fig. 5) evaluated ovarian cancer SKOV-3 cells both in vitro and in vivo using monoaxial ES particles comprised of Ecoflex® and PEG to encapsulate docetaxel. They noted that in vitro, particles significantly decreased cell viability versus free drug at the same concentration. This correlated with particle uptake, in that rhodamine B labeled particles were taken up at an increased rate compared to free dye, as measured with flow cytometry. When applied in vivo to a B6 nude mouse model with subcutaneously introduced SKOV-3 ovarian cancer cells, the tumor size was significantly reduced in the particle treated group compared to the saline or Taxotere control without significant difference in body weights between groups. These results in an in vivo ovarian cancer model indicate that chemotherapeutic-loaded ES particles can reduce in vivo tumor burden, advancing the promising application of ES particles for delivery of chemotherapeutics. Watkins-Schulz et al. [94] used the stimulator of interferon genes (STING) agonist, cyclic guanosine monophosphate–adenosine monophosphate (cGAMP), as immunotherapy for cancer and illustrated innate immune signaling activity in bone-marrow derived dendritic cells (BMDCs). Production of inflammatory cytokines interferon-beta (IFN-β), tumor necrosis factor (TNF), and interlukin-6 (IL-6) all were significantly greater when cGAMP was encapsulated in electrosprayed Ac-DEX microparticles than when given in soluble form. cGAMP microparticles were injected via various routes in a subcutaneous B16F10 mouse model of melanoma, with IT injection illustrating the greatest reduction in tumor burden. Using this route of administration, tumor burden was significantly reduced in both melanoma and breast cancer (e077), in a manner that was dependent on natural killer (NK) and CD8+ T cells. When a STING knock-out mouse (6(Cg)-Tmem173tm1.2Camb/J (Tmem173−/−)) was used, cancer growth was not significantly changed, indicating that STING is required for cGAMP microparticle efficacy. All of these studies collectively expand on the application of ES particles to deliver chemotherapeutics to impact the growth and viability of several cancer types.
Fig. 5.
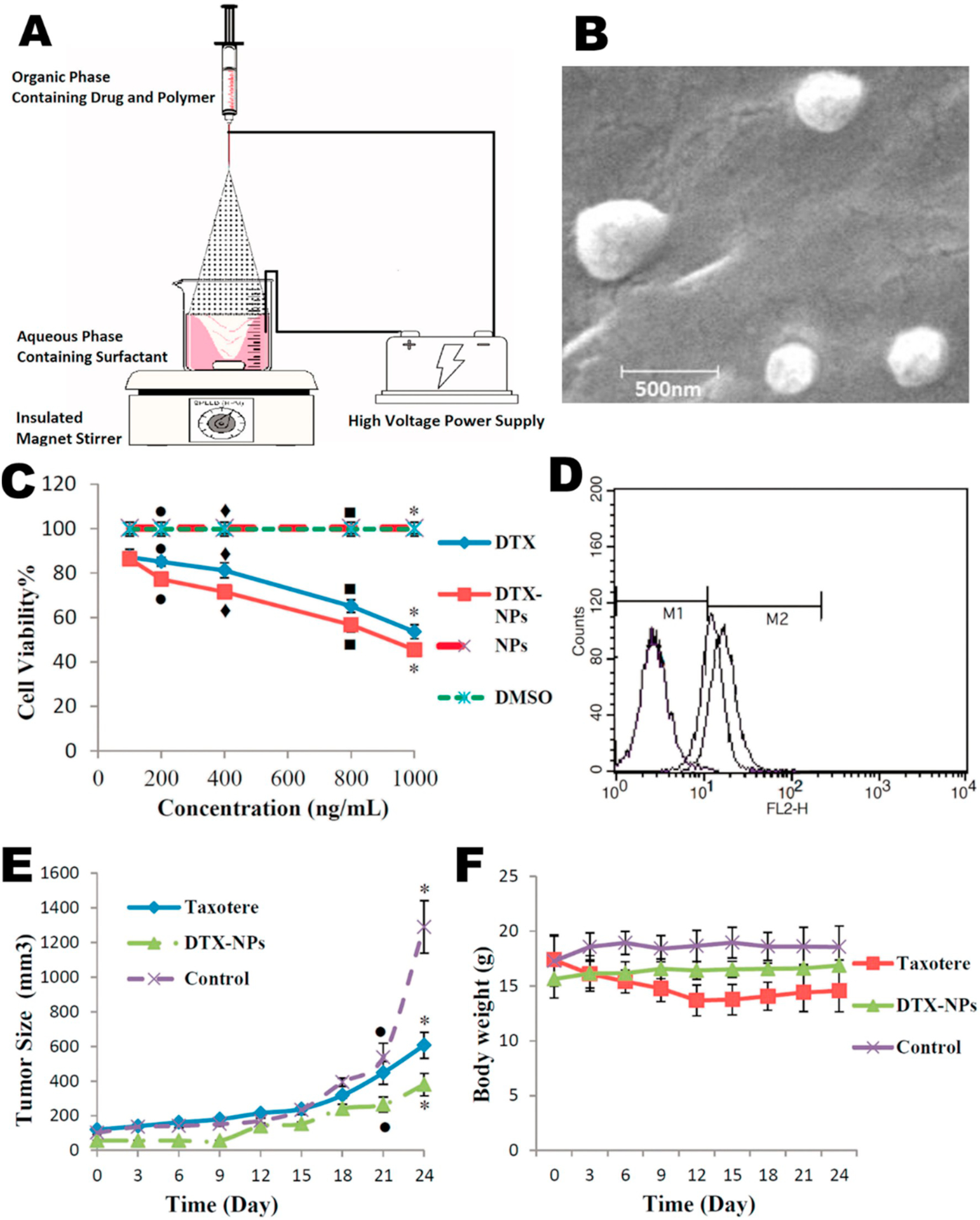
Varshosaz et al. [75] applied Ecoflex® and PEG particles encapsulating docetaxel for the in vitro and in vivo treatment of ovarian cancer. (A) They prepared their particles by spraying into an aqueous phase containing a surfactant to stabilize and prevent coalescence of sprayed particles. Note the necessity of insulating the magnetic stirrer from the high voltage of the spray apparatus. (B) SEM micrograph of particles formed. (C) Presents cell viability in vitro where DTX refers to docetaxel, and NP nanoparticles.(D) The uptake of particles labeled with rhodamine B, as measured by flow cytometry, where M1 indicates autofluorescence of blank cells and the M2 region indicates greater fluorescence due to particle uptake. Each of the three particle groups in the M2 region were treated with rhodamine-loaded particles while the group in the M1 region was treated with unloaded particles. (E) Size of subcutaneous ovarian tumor with treatment of drug (Taxotere)-loaded particle, drug alone or with no treatment. (F) Body weight of each treatment group in E.
3.3. Antimicrobial delivery
The controlled release of antimicrobials can lead to reduced treatment frequency, which can result in reduced emergence of drug resistance and better treatment of biofilms [104]. To this end, several anti-microbial compounds have been encapsulated in ES particles and have demonstrated efficacy both in vitro and in vivo (Table 3). Duong et al. [18] loaded the toll-like receptor 7/8 agonist resiquimod into Ac-DEX microparticles using monoaxial ES. Particles were made with different drug loadings. When cultured with RAW macrophages in vitro, less resiquimod was needed for similar cytokine production when the drug was encapsulated in Ac-DEX particles, but this result was only observed when particles had a higher drug loading. Viability and proliferation of cells was equivalent for all particle groups except at high concentrations of particle and with the lower drug loaded particles. Using an in vivo visceral Leishmania donovani model of infection, mice were treated with either blank or drug-loaded microparticles via IV injection on day 14, post infection. Parasite burden in the bone marrow of infected mice at day 28 was significantly reduced by treatment with resiquimod loaded microparticles compared to treatment with empty microparticles. Parasite burden in the liver and spleen were numerically reduced but were only significantly lower than the PBS control in the liver. In another study evaluating the toxicity of tetracycline hydrochloride encapsulated in monoaxial CMC alginate particles, Lai et al. [102] applied particles 500 nm in size to 3T3 fibroblasts and human embryonic kidney (HEK293) cells. They illustrated that cytotoxicity remained near 100% at a concentration up to 200 μg/mL at 5 and 24 h. Mai et al. [103] (Fig. 6) encapsulated curcumin in PLA monoaxial ES particles, forming 1–20 μm particles. They evaluated antibacterial efficacy in Escherichia coli (E. coli) and Staphylococcus aureus (S. aureus) using a disc approach on bacterial lawns on agar. They illustrated that discs comprised of the antibiotic loaded particles limited the growth of either bacteria. They also noted low levels of hemolysis and cytotoxicity in rat adrenal medulla (PC12) and human embryonic kidney (293 T) cells, and these low levels were observed to have a dose response. When human dermal fibroblasts were grown on a layer of particles, a growth rate similar to tissue culture plastic control was observed at 1, 3, and 5 days. These studies are a start to applying an ES platform to an area of increasing interest, antimicrobials. As drug resistance to conventional antimicrobials increases, ES particles could be a valid controlled release system that should be explored to decrease dosing frequency.
Table 3.
Electrospray technologies for delivery of antimicrobial therapies.
| Polymer | Cargo | Axial | Particle size range | Cell model | Animal model | Ref |
|---|---|---|---|---|---|---|
| Ac-DEX | Resiquimod | Mono | 1–2 μm | RAW 264.7 Macrophages | Visceral Leishmaniasis (BALB/C mice) | [18] |
| CMC Alginate | Tetracycline hydrochloride | Mono | 400–600 nm | 3T3 Fibroblasts HEK293 | – | [102] |
| PLA | Curcumin | Mono | 1–20 μm |
E. coli
S. aureus HEK293T cells PC12 cells Human dermal fibroblasts |
− | [103] |
Fig. 6.
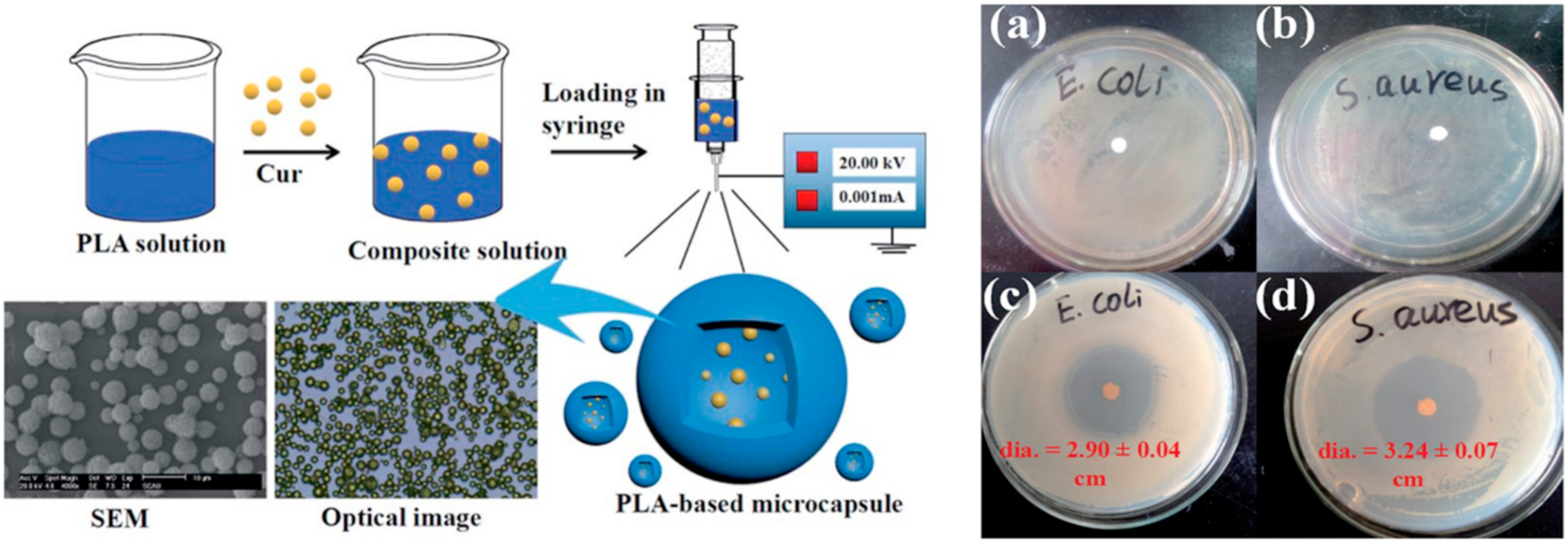
Fabrication by Mai et al. [103] of PLA particles encapsulating curcumin (left) with scale bar equal to 10 μm. Incubation of control particles (a–b) and curcumin loaded particles (c–d) with two different bacteria (right), demonstrating the effective inhibition of bacterial growth by the curcumin-loaded particles.
3.4. Oral delivery
Oral delivery of therapeutics is always challenging due to significant barriers which can include extreme pH levels, intermittent fluid flow, high amounts of metabolizing enzymes, and a discontinuous mucosal layer across the entire gastrointestinal tract [109]. Platforms that can effectively deliver biologics and small molecules which do not obey Lipinski’s rules through the oral route are desired, as current approaches result in very poor bioavailability. Several ES particles have been applied for oral delivery (Table 4). For example, Hao et al. [105] encapsulated proton pump inhibitor omeprazole into Eudragit L 100–55 monoaxial ES particles. At the highest drug to polymer ratio (0.8), cytotoxicity in Caco-2 intestinal epithelial cells was significantly lower at 24 h compared to untreated controls. Later at 36 and 48 h, particles with lower drug to polymer ratios (0.4 and 0.6) also had reduced cytotoxicity compared to controls. In a separate work, Hao et al. [106] evaluated the cytotoxicity of PLGA particles encapsulating the anti-ulcer medication metronidazole with GES-1 (gastric epithelial) cells. These formulations were non-toxic to cells up to 800 μg/mL at 24 h. Cytotoxicity decreased very slightly with 2–4% w/w loaded particles at the 36 and 48 h time points. In a third study by Hao et al. [107], aspirin was encapsulated via co-axial ES into Eudragit L 100–55 and Eudragit RS particles. Cytotoxicity of Caco-2 cells when incubated with aspirin-loaded particles was significantly reduced after 24 h at a particle concentration of 100 μg/mL and greater. For all evaluated drug concentrations, cytotoxicity was significantly reduced at 36 and 48 h. A dose and time dependent response was observed with cytotoxicity measurements, reporting levels as low as 65% viability for 800 μg/mL at 48 h. When Caco-2 cells were imaged for uptake by epifluorescence, there was notable uptake at 1 and 2 h. In another application of ES particles for oral delivery, Paz-Samaniego et al. [108] (Fig. 7) used corn-based bran and wax arabinoxylans to encapsulate bacteria and insulin via tetra-axial ES. Using the SIMulator Gastro-Intestinal (Simgi®)—a computer-controlled model of a human GI tract that consists of five successive reactors, simulating the stomach, small intestine, and three parts of the large intestine—they report that 24% of insulin was released before the colon compartment, with the remainder released in the colon. Bacteria deposition was noted in the colon compartment, but earlier deposition was not discussed. Overall, oral delivery of small molecule drugs, proteins, or cells has been reported with ES particles, illustrating the promise of this technique in developing platforms for oral drug delivery.
Table 4.
Electrospray particle systems for oral delivery.
| Polymer | Cargo | Axial | Particle size range | Cell model | Ref |
|---|---|---|---|---|---|
| Eudragit L 100–55 | Omeprazole | Mono | 200–350 nm | Caco-2 cells | [105] |
| PLGA | Metronidazole | Mono | 1–8 μm | GES-1 cells | [106] |
| Eudragit L 100–55 Eudragit RS | Aspirin | Co (miscible) | 280–435 nm | Caco-2 cells | [107] |
| Maize Bran Arabinoxylans Maize Wax Arabinoxylans |
Bifidobacterium longum B. adolescents Insulin |
Tetra (miscible) | 2–3 μm | Simgi® | [108] |
Fig. 7.
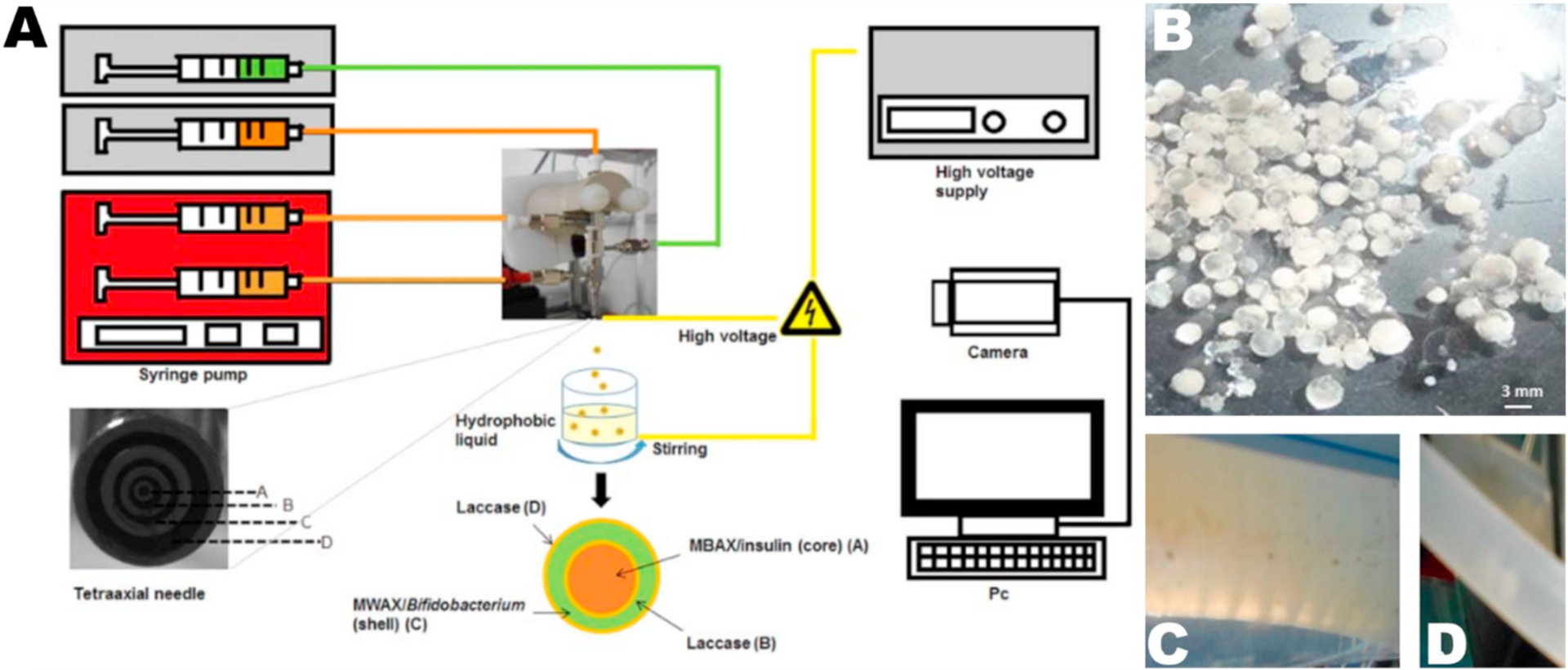
Paz-Samaniego et al. [108] generated ES particles from corn-based arabinoxylans to encapsulate bacteria and insulin via tetra-axial spraying (A). (B) Photograph of particles encapsulating two bacteria and insulin. Particles encapsulating insulin and bacteria in the stomach (C) and small intestine (D) compartments of the GI tract simulator Simgi®.
3.5. Gene delivery
For gene delivery, PEI has been used in two ES applications, one that formed PEI-DNA polyplexes during spraying and another that induced transfection as part of the process. Zeles-Hahn et al. [70] sprayed PEI and the cationic lipid DOTAP with plasmid DNA encoding luciferase. They reported a significant increase in cytotoxicity when 1:10 DNA to PEI polyplexes were applied to human derived tracheal epithelial cells, but such an increase was not observed with a 1:4 polyplex. Both lactate dehydrogenase (LDH) release and interleukin-8 (IL-8) were measured, demonstrating both cytotoxicity and cytokine production increase in < 10 h. Lee et al. [71] electrosprayed PEI with plasmid encoding green fluorescent protein (GFP) and 3T3 fibroblasts, all in a single spray, as a form of aerosol electroporation. They concluded that transfection increased when different media was sprayed and when a range of voltages were applied to the spray needle. The viability of the cells decreased with increasing electrical potential difference between the spray needle and substrate. Together, these studies demonstrate promise for the future applications of ES in gene transfection (Table 5).
Table 5.
Electrospray technologies for gene delivery.
3.6. Treatment of inflammation and oxidative stress
ES particles have been used for a variety of other drug delivery applications including treatment of inflammation and oxidative stress. In a study using a photosensitive polymer, 2-(40-N-dimethylamino-4-nitro-[1,10-biphenyl]-3-yl)butane-1,4-diyl dicarbonyl, Carling et al. [41] (Fig. 8) evaluated the inhibition of inflammation by encapsulating dexamethasone using monoaxial ES to form particles approximately 1 μm in size. Using a carrageenan-induced hind paw inflammation model and treating with particles, they report that application of light to rapidly degrade the particles resulted in significantly reduced footpad thickness, indicating reduced inflammation, compared to dexamethasone alone or drug in the particle without light exposure. Stubelius et al. [110] used ES to encapsulate dexamethasone in Ac-DEX particles and evaluated them in cells, a dermal air pouch model, and an animal model for gout. They report reduced IL-1β levels in mouse bone marrow derived macrophages and CXCL1 levels in epithelial cells when cells were stimulated with lipopolysaccharide (LPS) and monosodium urate (MSU) crystals and then incubated with particles. Upon treatment with dexamethasone-loaded Ac-DEX particles, pro-inflammatory cytokine levels were significantly reduced compared to untreated PBS controls but not compared to dexamethasone-loaded PLGA particles or the free drug. Prophylactic treatment of an MSU mouse model of gout lead to significantly reduced tissue infiltration of the MSU crystals compared to untreated control, free dexamethasone, and PLGA-encapsulated dexamethasone. Prophylactic treatment was also evaluated in a skin air-pouch model to determine the inflammatory response of the formulation. Similar to the gout model, the Ac-DEX-encapsulated dexamethasone group significantly outperformed the other groups in the reduction of pro-inflammatory cytokines and immune cell infiltrates.
Fig. 8.
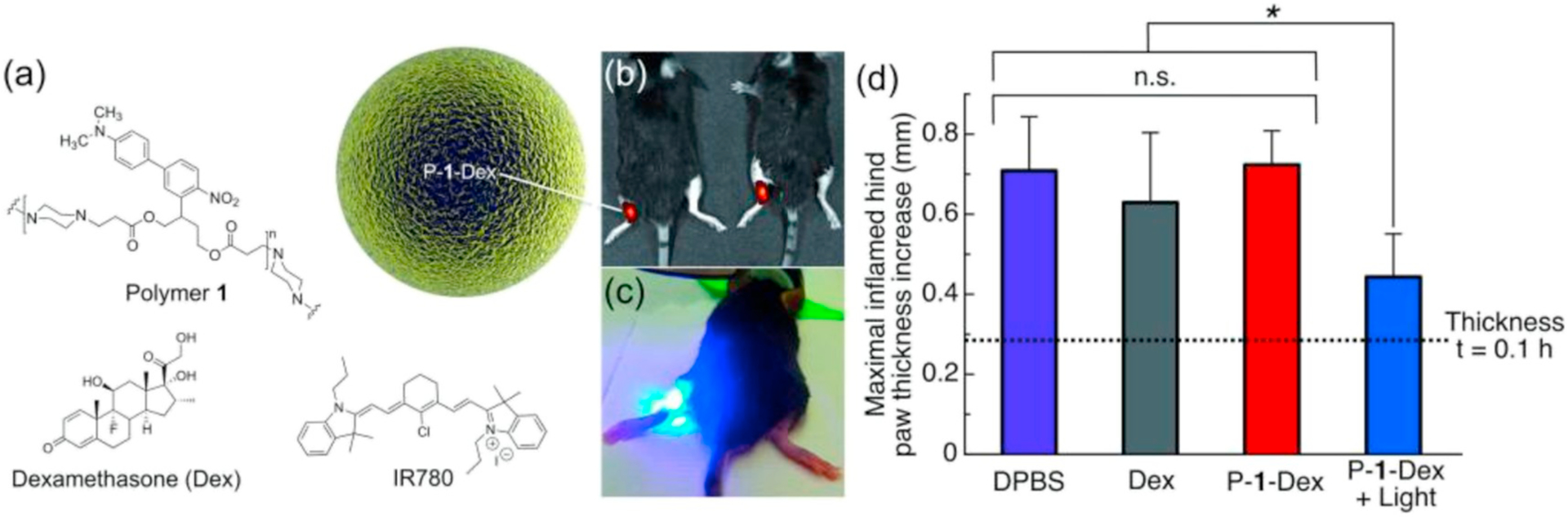
Carling et al. [41] applied 2-(40-N-dimethylamino-4-nitro-[1,10-biphenyl]-3-yl)butane-1,4-diyl dicarbonyl (Polymer 1) to encapsulate dexamethasone in ES particles (a). IR780 is a near IR fluorescent probe. (b) Illustrates location of injection on the mouse, with (c) showing application of light. In (d), Carrageenan-induced hind paw inflammation was induced and treated with either saline control (DPBS), dexamethasone (Dex) and dexamethasone encapsulated in Polymer 1, with or without subsequent near IR light stimulation (P-1-Dex and P-1-Dex + Light, respectively). Significant reduction in paw thickness increase was observed with the light-triggered release of dexamethasone from the particles.
Looking at oxidative stress, Xing et al. [111] used a tri-axial ES to make porous PCL particles loaded with antioxidant Ganoderma lucidum polysaccharide (GLP) in a solid core-porous shell structure intended for pulmonary delivery. Though the particles seem large with an average size of 13.37 μm, the theoretical mass mean aerodynamic diameter (MMADt) was estimated to be 4.66 μm. This MMADt paired with the particle porosity made the formulation promising for aerosolization and lung delivery [111]. These particles were administered to an H2O2-induced oxidative stress in vitro model with MRC-5 human lung fibroblasts. After 48 h treatment with particles, reactive oxygen species (ROS) staining indicated a reduction in ROS, and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) indicated an increase in cell proliferation and viability. Furthermore, Sudan Red G loaded particles were delivered via a dry powder insufflator, in vivo to determine biodistribution upon inhalation by pathogen-free KunMing mice. Fluorescent intensity measured in vivo demonstrated delivery to the lung tissue and a retention time > 48 h. Overall, these applications of ES for stimulus-sensitive delivery and pulmonary delivery demonstrate the promise of the ES method for control of oxidative stress (Table 6).
Table 6.
Electrospray technologies for treatment of inflammation and oxidative stress.
| Polymer | Cargo | Axial | Particle size range | Cell model | Animal model | Ref |
|---|---|---|---|---|---|---|
| 2-(40-N-dimethylamino-4-nitro- [1,1O-biphenyl] −3-yl)butane-l ,4-diyl dicarbonyl | Dexamethasone | Mono | 1 μm | RAW 264.7 macrophages | Carrageenan-induced hind paw inflammation (C57BL/6 mice) | [41] |
| Ac-DEX PLGA | Dexamethasone | Mono | 3.3 μm | Cl 66 Endothelial cells C57B1/6 Bone-marrow derived macrophages | Dermal air pouch (inflammation model) Monosodium urate crystal (Gout) model | [110] |
| PCL | Ganoderma lucidum polysaccharide | Tri | 13.37 μm | MRC-5 human lung fibroblasts | Pathogen-free KunMing mice | [111] |
3.7. Vaccine delivery
The application of ES for production of particulate vaccines takes advantage of the reduced denaturation of encapsulated proteins and peptides compared to other formulation methods, such as emulsion. Reduced denaturation is desirable for subunit vaccines that use proteins as antigens. In the evaluation of vaccines, only innate signaling can be evaluated in vitro with leukocytes, so mouse or higher species models must be used to fully elucidate the protection afforded by vaccines. Suksamran et al. [65] report development of an orally delivered vaccine comprised of alginate or yam starch encapsulating the model protein ovalbumin and coated with a cationically modified chitosan through electrostatic interactions. They noted that when the particles were applied to Caco-2 cells, cell viability was essentially 100% in particle concentrations up to about 1 mg/mL, and cytotoxicity increased to near 50% at > 10 mg/mL of particles. They then evaluated adhesion of the particles in an ex vivo porcine intestine model. Adhesion to the porcine intestine after 30 min was significantly greater for cation-coated particles relative to uncoated particles, and a fairly constant percent adhesion was observed in all groups out to the last time point at 2.5 h. Particles were then evaluated in BALB/c mice for antibody titer generation following oral vaccination on day zero with another orally administered booster dose on day fourteen. Significantly greater total IgG and IgA titers were observed from mice vaccinated with chitosancoated, OVA-loaded alginate yam core particles compared to individual polymer formulations and formulations without yam protein. Doavi et al. [112] used a monoaxial electrospray strategy to encapsulate a synthetic E. coli antigen, rEIT, into chitosan or trimethylated chitosan (TMC) particles. They administered the antigen encapsulated in either vehicle intranasally to BALB/c mice, with boosts on days 28 and 42 following initial vaccination. Comparable levels of anti-rEIT IgG in serum was observed when immunizing with ES formulations or a positive control consisting of rEIT injected intraperitoneally with Freud’s adjuvant. In addition, they saw increased anti-rEIT IgA titers in serum, fecal, and eyewash fluids compared to both the soluble controls and the intraperitoneal positive control. Subsequently, they observed significantly reduced adhesion of E. coli O157:H7 to cultured Caco-2 monolayers in the presence of sera from the intranasally immunized mice relative to unimmunized mice. Bacterial shedding from the intestines was also significantly reduced compared to soluble controls in both groups immunized with ES particles. Gallovic et al. [11] generated an anthrax vaccine using Ac-DEX to encapsulate recombinant protective antigen (rPA) and resiquimod either together or in separate particles using co-axial ES (Fig. 9). Vaccinating on day 0 and 21 subcutaneously in BALB/c mice, we showed that vaccination with ES particles significantly increased the amount of total IgG, IgG2a isotype, and neutralizing antibodies compared to particles made through emulsion. Also, ES particles protected significantly better after a lethal aerosol anthrax challenge. Moreover, we showed that ES particles protected better than rPA with alum or FDA approved Biothrax. In a lethal challenge, we reported that, surprisingly, co-encapsulated particles (rPA and resiquimod) provided significantly reduced protection than when adjuvant and antigen were separately delivered, despite having similar total IgG titers. Junkins et al. [48] also applied Ac-DEX using a co-axial ES system to develop an influenza vaccine using hemagglutinin (HA) and STING agonist cGAMP. In this study, cGAMP was encapsulated in Ac-DEX particles and HA was delivered in soluble form. When culturing BMDCs isolated from C57Bl/6 mice with cGAMP particles, ES Ac-DEX particles had significantly greater induction of IFN-γ and IL-6 than unloaded particles, ES PLGA particles, emulsion Ac-DEX particles, liposomes, or transfection agent co-administered with cGAMP. We also reported that no significant differences in IFN-γ secretion were observed when treating BMDCs with cGAMP particles before or after sterilization with gamma-radiation. Vaccinating mice via intramuscular (IM) injection on day 0 and 21, we reported there was greater total IgG, IgG2a isotype, soluble cytokines, and cells producing IFN-γ and IL-2 after HA re-stimulation compared to controls. These differences became significantly greater as the concentration of cGAMP decreased. Additionally, mouse toxicity studies were performed, and across six different parameters, cGAMP-loaded particles at the highest loading did not illustrate any signs of toxicity. Furtmann et al. [80] used monoaxial ES to encapsulate Cytomegalovirus (CMV) pp65 IE-1 peptide. They then administered the particles to peripheral blood mononuclear cells (PBMCs). They noted that cytotoxicity was not decreased with increasing concentration of peptide loaded particles, and particles did not significantly change cytotoxicity. Additionally, monocytes primarily took up the particles in contrast to B cells, T cells, and NK cells, which is logical for 200 nm sized particles as they can only effectively be engulfed by phagocytic cells [113]. TNF-α and IFN-γ positive T cells were near equal with those treated with soluble peptide after antigen re-stimulation. Although, they reported significantly increased T cell proliferation with particle treated PMBCs compared to those treated with soluble peptide. Increased peptide tetramer staining over soluble and blank particle control was also reported after particle treatment. Chen et al. [45] used Ac-DEX and encapsulated ovalbumin and murabutide, separately, using co-axial ES to explore the effect of particle degradation on vaccine responses. When particles encapsulating the NOD-2 agonist murabutide were applied to JAWSII dendritic cells, we found that significantly higher production of cytokines over soluble, blank, or media control was observed when the adjuvant was delivered with ES particles. Furthermore, cell viability was near 100% for up to 400 ng/mL of murabutide. To understand the effects of degradation individually on adjuvant (murabutide) and antigen (ovalbumin), particles encapsulating these compounds were co-delivered with soluble ovalbumin or monophosphoryl lipid A (MPL), respectively. After IM vaccination on day 0 and 21 with particles of three different Ac-DEX degradation rates for both adjuvant and antigen delivery, total sera IgG and isotypes were shown to be uniquely dependent on the degradation rate of Ac-DEX polymer. Moreover, cellular responses were influenced by degradation, in a manner unique in some cases from what was observed with humoral responses. These studies support the idea that encapsulation with ES is desirable for protein delivery, as a denatured protein would not elicit a strong humoral or protective response for many of these evaluated systems. As the application of ES for vaccine development grows, other benefits like scalability and dry powder formulation will likely also contribute to the success of this platform (Table 7).
Fig. 9.
Gallovic et al. [11] ES of Ac-DEX to encapsulate rPA (PA) and resiquimod (Resi). Micrograph of (A) emulsion (EM) and (B) ES particles. (C) Circular dichroism of rPA from different particles and blank particles spiked with protein ideally stored. (D) Total serum IgG after vaccination with experimental groups. (E) Toxin neutralization of serum antibodies isolated from mice after vaccination. (F) Survival after vaccination and lethal challenge with different experimental groups.
Table 7.
Electrospray technologies for vaccine delivery.
| Polymer | Cargo | Axial | Particle size range | Cell model | Animal model | Ref |
|---|---|---|---|---|---|---|
| Alginate n-(4-n,n-dimethylaminocinnamyl) chitosan Yam starch | Ovalbumin | Mono | 0.75–3μim | Caco-2 cells ex vivo Porcine Intestine | BALB/c mice | [65] |
| Chitosan Trimethylated Chitosan | Recombinant synthetic antigen EIT | Mono | 300–600 nm | Caco-2 cells | BALB/c mice | [112] |
| Ac-DEX | rPA Resiquimod |
Co (immiscible) | 1–3 μm | – | BALB/c mice | [11] |
| Ac-DEX PLGA | cGAMP | Co (semi-miscible) | 1–3 μm | BMDCs | C57BL/6 mice | [48] |
| Ac-DEX PLGA | cGAMP Resiquimod |
Co (semi-miscible) | [19] | |||
| PLGA PVA | Cytomegalovirus (CMV) peptides pp65 & IE-1 | Mono | 150–800 nm | PBMCs | – | [80] |
| Ac-DEX | Ovalbumin Murabutide |
Co (semi-miscible) | 500–600 nm | JAWSII dendritic cells | C57BL/6 mice | [45] |
3.8. Drug release for tissue engineering
The range of tissue engineering applications of ES particles is vast and includes drug releasing particles as well as using blank particles to create microstructures, such as pillars of particles or particle laden nanofibers [29]. These surfaces create structure for new tissue growth, differentiation and repair. To accomplish this, even a variety of matrix material, outside the realm of polymers have been used. For example, Link et al. [124] report the electrospray of decellularized extracellular matrix to form nanoparticles and promote lung repair. Several reviews discuss the application of electrospray particles strictly for tissue engineering [29,125,126]. In noting the studies that generate drug eluting particles applied in vitro or in vivo, two primary applications are found: stem cell differentiation and encapsulation or bone tissue repair Table 8.
Table 8.
Electrospray technologies for tissue engineering.
| Polymer | Cargo | Axial | Particle size range | Cell model | Animal model | Ref |
|---|---|---|---|---|---|---|
| PLGA Gelatin | Insulin-like growth factor | Co (immiscible) | 5 μm | MSCs SMCs |
– | [114] |
| PLA PLGA | PDGF Simvastatin |
Co (semi-miscible) | 15–20 μm | – | Osteotomy repair (Sprague-Dawley rats) | [115] |
| PLGA | Simvastatin | Mono | 1.6–8 μm | MG-63 osteoblasts | – | [116] |
| PLA PLGA | PDGF Simvastatin |
Co (semi-miscible) | 18–21 μm | – | Osteotomy repair (Sprague-Dawley rats) | [117] |
| Alginate CMC | R1 Embryonic stem cells | Co (miscible) | 285–345 μm | – | – | [118] |
| PLGA | BSA SDF-1α |
Co (semi-miscible) | 4 μm | MSCs | – | [119] |
| PCL Silk fibroin | Ascorbic acid Dexamethasone | Mono | 0.7–8 μm | ADSCs | – | [120] |
| PLA PLGA | VEGF BMP-2 |
Co (semi-miscible) | 0.5–1.5 μm | MECs BMSCs |
Calvarial bone defects (Sprague-Dawley rats) | [121] |
| PLGA | 3T3 fibroblasts | Co (immiscible) | ~10 μm | – | – | [122] |
| PLGA | BMP-2 | Co (semi-miscible | 439 nm | – | Titanium screw implant model (Sprague-Dawley rats) | [123] |
ES has been used to generate particle laden scaffolds or scaffold components for stem cell differentiation, and it is also used to encapsulate cells for tissue engineering applications. Wang et al. [114] (Fig. 10) fabricated PLGA and gelatin particles to encapsulate insulin-like growth factor (IGF). These particles were then sprayed on to polyurethane urea scaffolds that were simultaneously being electrospun. When mesenchymal stem cells (MSCs) were seeded on the particle laden scaffolds, the IGF released from lower molecular weight PLGA lead to increased proliferation of rat aortic smooth muscle cells (SMCs). MSC viability on the scaffolds was the greatest with the low molecular weight particles incorporated in the scaffold; however, those grown on high molecular weight particle scaffolds exhibited similar viability. Under hypoxic conditions, the MSCs had significantly lower viability when grown on a scaffold without IGF incorporated in the particles compared to being grown on a scaffold with IGF loaded in the ES particles. In another approach, focusing on the encapsulation of cells, Zhao et al. [118] electrosprayed R1 Embryonic stem cells in either alginate or carboxymethyl cellulose (CMC) to form either gel (alginate) or liquid CMC containing particles. They showed that the gel core had decreased pluripotency markers and increased differentiation markers for cardiac cells, whereas the liquid core particles displayed the opposite trends. However, in culture, the cells in the liquid core displayed significantly more beating foci, indicative of cardiomyocyte activity. Additionally, the gel core particles still displayed increased foci compared to tissue culture control. Zamani et al. [119] encapsulated Stromal derived factor-1α (SDF-1α) with and without bovine serum albumin (BSA) in ES PLGA co-axial particles. When the particles were applied to MSCs in vitro, a significant increase in number of migrating cells was observed in response to particles containing both BSA and SDF-1α compared to particles without BSA and other controls. Moreover, MSCs proliferated at an increased rate when cultured with extract released from the BSA stabilized particles. Gandhimathi et al. [120] used monoaxial ES to encapsulate ascorbic acid and dexamethasone into PCL and silk fibroin, forming particles 0.72 to 4 μm in size. When the co-therapy particles were incubated with human adipose derived stem cells (ADSC), cells grown on the particles had increased growth across all timepoints compared to tissue culture controls, individual and combined polymer blanks, and polymers with ascorbic acid. Quantification of alkaline phosphatase (ALP) activity showed that osteogenic differentiation was highest in the co-therapy particulate group, as well as the particle group without dexamethasone. This mirrored the highest mineral deposition observed. Evaluating the encapsulation of cells via ES, Esfahani et al. [122] encapsulated 3T3 fibroblasts into rigid PLGA microparticles using co-axial ES. They illustrated encapsulation of GFPexpressing fibroblasts using flow cytometry and microscopy. In the same year, Chang et al. [123] loaded bone morphogenetic protein-2 (BMP-2) into PLGA particles and then embedded them in a cryogel scaffold. The scaffold was placed under the head of a screw as it was implanted into the mandible of a rat. The bone to volume ratio around the screw increased for all groups, compared to PBS control, but the deposition of new bone was greatest for the particle-containing group. These studies indicate that systems with ES particles can influence stem cell differentiation into a variety of different tissues.
Fig. 10.
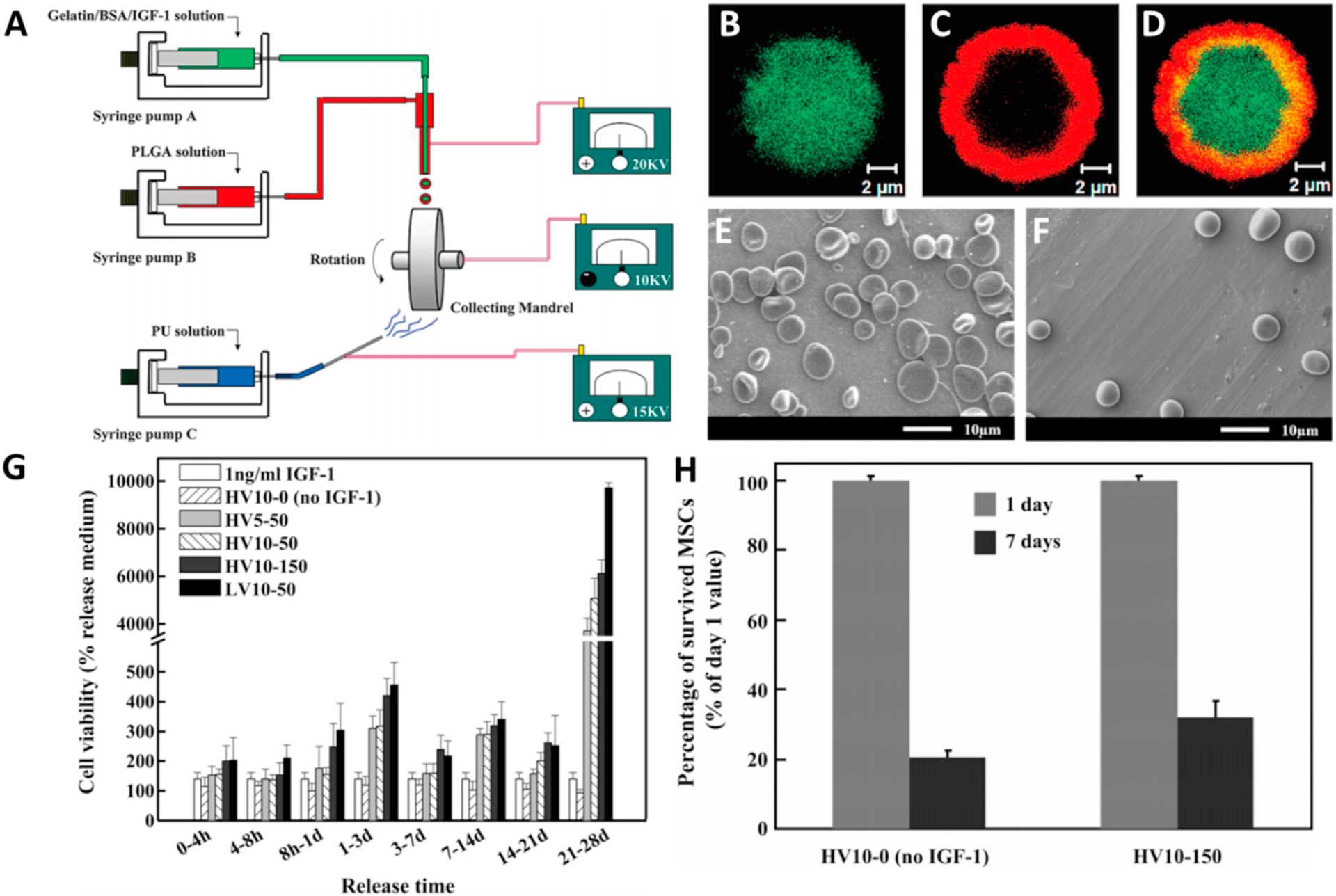
Wang et al. [114] combined spraying and aligned spinning apparatus to create particle-laden electrosprayed scaffolds (A). (B–D) Rhodamine-B loaded PLGA (red) shell and FITC-BSA (green) loaded core. Scanning electron micrograph of (E) 5% and (F) 10% IGF-1 loaded PLGA particles. (G) Bioactivity of IGF-1 released on SMCs. HV stands for high molecular weight, and LV low molecular weight. The numbers refer to loading of IGF-1 in μg/mL. (H) The percentage of surviving MSCs under hypoxic conditions and nutrient starvation conditions.
For bone tissue repair a variety of ES platforms have been evaluated. Chang et al. [115] have used PLA and PLGA and co-axial ES to encapsulate platelet-derived growth factor (PDGF) and simvastatin, used to stimulate osteoblastic activity [127]. Osteotomies (i.e. holes were drilled in the bone) were performed on Sprague-Dawley rats, where a section of bone was removed from the maxillae. Looking at osteotomy repair and bone regeneration, they showed that simvastatin-in-core and PDGF-in-shell particles had significantly greater percent of bone fill compared to controls, individual drug groups, or the PDGF-in-core and simvastatin-in-shell group. Significantly fewer inflammatory cells were observed in the co-delivered groups. However, tartrate-resistant acid phosphatase positive (TRAP+; a marker of osteoclasts) cells and apoptotic cells (except for the PDGF-in-core/simvastatin-in-shell particles) in all drug treatment groups were similar by day 14. They continued this work, using the same particle system and animal model, osteotomy repair in Sprague-Dawley rats [123]. They then report significantly greater osteogenesis in the osteotomy with core-shell PDGF and simvastatin particles at day 14, but by day 28 bone growth was similar in all drug groups, including individually delivered agents. TRAP+ cells significantly increased, compared to BSA core/shell control, after treatment with simvastatin-in-core with BSA-in-shell or simvastatin-in-core and PDGF-in-shell after 28 days. Nath et al. [116] used simvastatin alone in PLGA monoaxial ES particles. When these particles were cultured with MG-63 osteoblast cells the cytotoxicity of simvastatin loaded particles was reduced compared to PLGA alone, especially at higher drug loading. Expression of osteogenic factors, determined by PCR, significantly increased with simvastatin dose and time up to 14 days compared to the PLGA control for all recorded time points. Wang et al. [121] (Fig. 11) encapsulated vascular endothelial growth factor (VEGF) and BMP-2 into 500 nm PLGA and PLA co-axial ES particles. Using microvascular epithelial cells (MECs) and the osteogenic differentiation of bone marrow stem cells (BMSCs) they showed that VEGF induces MEC proliferation and gene expression of ALP, and osteopontin (OPN) and BMP-2 were significantly increased with BMP-2 treatment after 14 days. Using a rat calvarial bone defect model, they showed that treatment with co-therapy particles lead to significant increases in bone volume at 2, 4, and 8 weeks compared to soluble agent, even co-delivered soluble agent. Additionally, CD-31 presence at 4 and 8 weeks was significantly greater compared to soluble controls, combination soluble controls, and blank particles, indicating greater vascularization upon co-therapy particle treatment. Increased osteocalcin (OCN) for co-therapy particles versus other groups was illustrated via histology. The application of ES particles for bone tissue engineering has illustrated strong repair, bone growth, and expression of repair-associated factors. These promising results should encourage further exploration of the applications of ES therapies for bone growth and repair. ES is starting to emerge as a platform for tissue regeneration and these results are promising in the field of tissue regeneration and drug delivery.
Fig. 11.
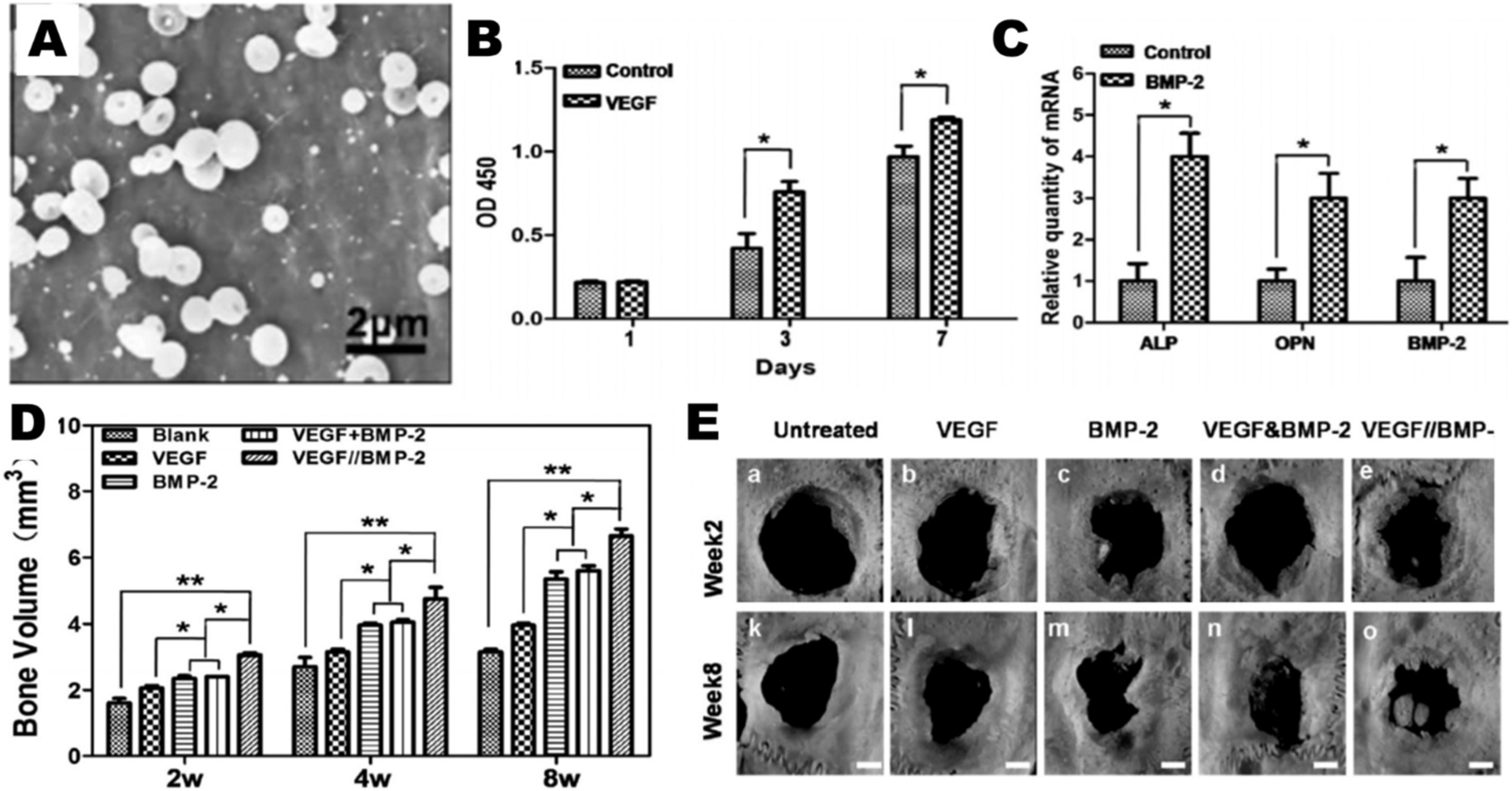
Wang et al. [121] used PLGA and PLA to particles to encapsulate VEGF and BMP-2. (A) Scanning electron micrograph of ES particles. (B) Proliferation of MECs when cultured with VEGF loaded particles. (C) RT-PCR was used to report gene expression profiles of ALP, OPN, and BMP-2 on day 14 Following treatment with BMP-2 loaded particles. (D, E) Bone volume measured by 3D μ-CT of rat calvarial bone defects treated with different groups, and images of osteotomies in each group at 2 and 8 weeks after treatment.
4. State of the art and future perspectives
As demonstrated by the works highlighted here, ES technology is promising for various biomedical applications, including cancer treatment, vaccine development, oral drug delivery, and drug eluting tissue engineering platforms. ES also promises advantages over other particle fabrication techniques when it comes to manufacturing. Additionally, ES demonstrates improved encapsulation efficiency over other techniques. Despite these advantages, there is a need for improved methods to evaluate particle cytotoxicity in cell culture and for more rigorous animal models in the evaluation of ES drug-loaded particles.
With regard to manufacturing, the scalability and size of the ES system could advance its application in cell and animal models. The scalability of ES is a significant advantage of the platform. Although others have studied multi-plexing of ES systems where multiple spray nozzles operate in parallel [20,21], this has not been reported to be applied to in vitro and in vivo models. Comparison of multi-plexed versus single-head systems in vivo would advance the scalability potential of the platform. To display a bioequivalence of sorts between the two methods would lead to better acceptance of ES for the fabrication of particles en masse. In addition to scalability, a smaller size of particles would lead to a better embrace of ES particles by the biomedical community. A majority of the studies above report particles with diameters > 200 nm and into the micron range. To target non-phagocytic cells and for better intravenous delivery of particles, which would be especially helpful for the delivery of chemotherapeutic or theranostic agents in cancer treatment, sizes 100 nm or below are needed. Generation of particles in this size range and in a multi-plexed model would have significant impact on ES for biomedical applications.
In selection of cargos, a focus should be placed on molecules that need to be encapsulated for maximum efficacy. Also, molecules that have poor encapsulation with other methods—often due to partitioning into an aqueous continuous phase present during processing—increases the rationale for using ES over other methods. The use of dexamethasone by several of the aforementioned groups is one example of this, as emulsion-based encapsulation of dexamethasone can lead to low loading of the drug [128], whereas ES can lead to higher encapsulation efficiencies [41,120]. The vaccine adjuvants resiquimod [18,129] and cGAMP [48] are also examples where encapsulation efficiencies are poor with emulsion but increase with ES. In the cases of cGAMP and the adjuvant murabutide [45], the cell membrane partition coefficients of the adjuvants are exceedingly low, and therefore copious amounts of soluble drug are needed to permeate the cell’s membrane (clinical trial #NCT02675439). Using ES to form particles that release their cargo intracellularly, the barrier of the cell membrane is overcome. By identifying therapeutics that would partition into an aqueous phase easily, one can rationally maximize the benefits of ES formulation.
There are several areas of improvement for evaluating particle cytotoxicity, including assay selection, controls, selectivity of formulation, and concentration range. With regards to assays, many of the proposed studies focus on cytotoxicity measurements using a MTT based assay. With MTT-based assays, the number of mitochondria is measured, which can be an indicator of cell viability as well as proliferation. If the particles are of a small enough size, taking into consideration the polydispersity of the sample, they may be taken up by cells, slowing the cells’ proliferation rate but not significantly affecting their viability. Earlier, our group did a study comparing viability as measured by an LDH assay and MTT assay [81]. We noted that there was significantly reduced viability as measured by MTT, but the LDH assay revealed very little toxicity. It was then hypothesized that engulfment of particles was likely the cause of the decreased MTT, which of course was reflected in a dose response fashion as observed with the MTT but not LDH assay (data not shown in manuscript). Although we did not conduct additional assays, we could have used a terminal deoxynucleotidyl transferase (TdT) dUTP Nick-End Labeling (TUNEL), or fluorescent live/dead assay to better evaluate cell viability. Several of the aforementioned papers used an LDH assay; however, other assays could be used to support the data found with the LDH assay. Additionally, when doing a cytotoxicity evaluation, drug-loaded and blank particles should both be used. Without the use of both blank and loaded particles, it is difficult to discern if observed cytotoxicity is the result of the carrier or the drug. To save samples, blank particles can just be used at the highest concentration. Finally, for chemotherapeutic studies, a selectivity of cytotoxicity in cancer versus non-cancer cells is preferred. Qu et al. [90] demonstrated this effect in their study, and it added to the understanding that with the ES formulation, drug is primarily affecting cancer cells over non-cancer cells. Also, to illustrate greater difference between soluble and encapsulated agents, a greater range of concentration is sometimes warranted, wherein low or high concentrations might lead to significant differences in either therapeutic effect or cytotoxicity. Controlled release systems can take longer to result in noticeable differences, so changes in concentration and time can make a significant difference in evaluating particles. Taken together, careful consideration of these aspects can improve the accuracy, quality and rigor of the cytotoxicity evaluation reported with ES or other particle systems.
Finally, the ES field will advance when more rigorous animal models are used for evaluation. For instance, orthotopic or spontaneous tumor models are considered more relevant than subcutaneous models. Orthotopic glioma models [130] in mice or rats would better mimic clinical standards for the disease and allow therapies to be more translatable. Similarly, more rigorous orthotopic models of triple negative breast cancer [131] would be the next step beyond subcutaneous models. Although these are challenging models, they can be accessed through collaboration and offer the potential to expand translational application of ES therapies.
5. Conclusion
As ES matures as a platform for generation of particle-based drug release systems, it is important to apply these systems in relevant in vitro and in vivo models. The work presented here on delivery of drugs, cells, genes, and vaccines, as well as tissue engineering, highlights the wonderful potential of this technology. As the application of ES progresses in the biomedical field, a greater impact of the work can also be realized through further optimization of the particles generated by the method and better choices of cargos, assays, and animal models.
Acknowledgements
NIH R01AI14133301 Micro-particle delivery of a potent intracellular adjuvant for a Universal Flu Vaccine *** NIH R41AI14079501 Advancing formulation of sting agonist for Universal Flu Vaccine **** NIH R01AI13752502 Biomaterials to study tolerance immune induction kinetics.
References
- [1].Nikolaou M, Krasia-Christoforou T, Eur. J. Pharm. Sci 113 (2018) 29–40. [DOI] [PubMed] [Google Scholar]
- [2].Qi S, Craig D, Adv. Drug Deliv. Rev 100 (2016) 67–84. [DOI] [PubMed] [Google Scholar]
- [3].Bachelder EM, Pino EN, Ainslie KM, Chem. Rev 117 (2017) 1915–1926. [DOI] [PubMed] [Google Scholar]
- [4].Farook U, Stride E, Edirisinghe MJ, Moaleji R, Med. Biol. Eng. Comput 45 (2007) 781–789. [DOI] [PubMed] [Google Scholar]
- [5].Wu Y, Duong A, Lee LJ, Wyslouzil BE, Electrospray Production of Nanoparticles for Drug/Nucleic Acid Delivery, Intech Europe, Rijeka, 2012. [Google Scholar]
- [6].Souva MS, Nabar GM, Winter JO, Wyslouzil BE, J. Colloid Interface Sci 512 (2018) 411–418. [DOI] [PubMed] [Google Scholar]
- [7].Duong AD, Ruan G, Mahajan K, Winter JO, Wyslouzil BE, Langmuir 30 (2014) 3939–3948. [DOI] [PubMed] [Google Scholar]
- [8].Sayed E, Karavasili C, Ruparelia K, Haj-Ahmad R, Charalambopoulou G, Steriotis T, Giasafaki D, Cox P, Singh N, Giassafaki LN, Mpenekou A, Markopoulou CK, Vizirianakis IS, Chang MW, Fatouros DG, Ahmad Z, J. Control. Release 278 (2018) 142–155. [DOI] [PubMed] [Google Scholar]
- [9].Collier MA, Bachelder EM, Ainslie KM, Pharm. Res 34 (2017) 419–426. [DOI] [PubMed] [Google Scholar]
- [10].Duong AD, Collier MA, Bachelder EM, Wyslouzil BE, Ainslie KM, Mol. Pharm 13 (2016) 92–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Gallovic MD, Schully KL, Bell MG, Elberson MA, Palmer JR, Darko CA, Bachelder EM, Wyslouzil BE, Keane-Myers AM, Ainslie KM, Adv. Healthc. Mater 5 (20) (2016) 2617–2627. [DOI] [PubMed] [Google Scholar]
- [12].Corp R-HI., 1961.
- [13].Davoodi P, Feng F, Xu Q, Yan WC, Tong YW, Srinivasan MP, Sharma VK, Wang CH, J. Control. Release 205 (2015) 70–82. [DOI] [PubMed] [Google Scholar]
- [14].Leong KH, J. Aerosol Sci 18 (1987) 511–524. [Google Scholar]
- [15].Yao J, Lim LK, Xie J, Hua J, Wang CH, J. Aerosol Sci 39 (2008) 987–1002. [Google Scholar]
- [16].Almeria B, Deng W, Fahmy TM, Gomez A, J. Colloid Interface Sci 343 (2010) 125–133. [DOI] [PubMed] [Google Scholar]
- [17].Wang Y, Yang X, Liu W, Zhang F, Cai Q, Deng X, J. Microencapsul 30 (2013) 490–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Duong AD, Sharma S, Peine KJ, Gupta G, Satoskar AR, Bachelder EM, Wyslouzil BE, Ainslie KM, Mol. Pharm 10 (2013) 1045–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Collier MA, Junkins RD, Gallovic MD, Johnson BM, Johnson MM, Macintyre AN, Sempowski GD, Bachelder EM, Ting JP, Ainslie KM, Mol. Pharm 15 (11) (2018) 4933–4946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Parhizkar M, Reardon PJT, Knowles JC, Browning RJ, Stride E, Pedley RB, Grego T, Edirisinghe M, Mater. Des 126 (2017) 73–84. [Google Scholar]
- [21].Lojewski B, Yang W, Duan H, Xu C, Deng W, Aerosol Sci. Technol 47 (2013) 146–152. [Google Scholar]
- [22].Johansen P, Martinez Gomez JM, Gander B, Expert Rev. Vaccines 6 (2007) 471–474. [DOI] [PubMed] [Google Scholar]
- [23].Paul WE, Fundamental Immunology, 4th ed., Raven Press, New York, 1994. [Google Scholar]
- [24].Xie J, Ng WJ, Lee LY, Wang CH, J. Colloid Interface Sci 317 (2008) 469–476. [DOI] [PubMed] [Google Scholar]
- [25].Davies LA, Hannavy K, Davies N, Pirrie A, Coffee RA, Hyde SC, Gill DR, Pharm. Res 22 (2005) 1294–1304. [DOI] [PubMed] [Google Scholar]
- [26].Chang PC, Chung MC, Lei C, Chong LY, Wang CH, J. Biomed. Mater. Res. A 100 (2012) 2970–2978. [DOI] [PubMed] [Google Scholar]
- [27].Jaworek A, Sobczyk AT, Krupa A, J. Aerosol Sci 125 (2018) 57–92. [Google Scholar]
- [28].Sengupta D, Das P, Mondal B, Mukherjee K, Renew. Sust. Energ. Rev 60 (2016) 356–376. [Google Scholar]
- [29].Jayaraman P, Gandhimathi C, Venugopal JR, Becker DL, Ramakrishna S, Srinivasan DK, Adv. Drug Deliv. Rev 94 (2015) 77–95. [DOI] [PubMed] [Google Scholar]
- [30].Kameoka J, Orth R, Ilic B, Czaplewski D, Wachs T, Craighead HG, Anal. Chem 74 (2002) 5897–5901. [DOI] [PubMed] [Google Scholar]
- [31].Chiarot PR, Sullivan P, Mrad RB, J. Microelectromech. Syst 20 (2011) 1241–1249. [Google Scholar]
- [32].Marijnissen J, Ciach T, Müller M, Winkels T, KB G, Schmidt-Ott A, Luding S, Experiment and simulation of electrospray particle-flows for controlled release of drugs, in: Hoogendoorn SP, Luding S, Bovy PHL, Schreckenberg M, Wolf DE (Eds.), Traffic and Granular Flow’03, Springer, Berlin, Heidelberg, 2005. [Google Scholar]
- [33].Boda SK, Li X, Xie J, J. Aerosol Sci 125 (2018) 164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Bock N, Dargaville TR, Woodruff MA, Prog. Polym. Sci 37 (2012) 1510–1551. [Google Scholar]
- [35].Mehta P, Haj-Ahmad R, Rasekh M, Arshad MS, Smith A, van der Merwe SM, Li X, Chang M-W, Ahmad Z, Drug Discov. Today 22 (2017) 157–165. [DOI] [PubMed] [Google Scholar]
- [36].Enayati M, Chang M-W, Bragman F, Edirisinghe M, Stride E, Colloids Surf. A Physicochem. Eng. Asp 382 (2011) 154–164. [Google Scholar]
- [37].Lu L, Peter SJ, Lyman MD, Lai HL, Leite SM, Tamada JA, Uyama S, Vacanti JP, Langer R, Mikos AG, Biomaterials 21 (2000) 1837–1845. [DOI] [PubMed] [Google Scholar]
- [38].Li L, Schwendeman SP, J. Control. Release 101 (2005) 163–173. [DOI] [PubMed] [Google Scholar]
- [39].Ding AG, Schwendeman SP, Pharm. Res 25 (2008) 2041–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Woodruff MA, D.W H, Prog. Polym. Sci 35 (2010) 1217–1256. [Google Scholar]
- [41].Carling CJ, Viger ML, Huu VA, Garcia AV, Almutairi A, Chem. Sci 6 (2015) 335–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Broaders KE, Cohen JA, Beaudette TT, Bachelder EM, Frechet JM, Proc. Natl. Acad. Sci. U. S. A 106 (2009) 5497–5502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Reichard GA Jr., Haff AC, Skutches CL, Paul P, Holroyde CP, Owen OE, J. Clin. Invest 63 (1979) 619–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].EPA, in: EPA (Ed.), www.epa.gov, 2005.
- [45].Chen N, Johnson MM, Collier MA, Gallovic MD, Bachelder EM, Ainslie KM, J. Control. Release 273 (2018) 147–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kauffman KJ, Do C, Sharma S, Gallovic MD, Bachelder EM, Ainslie KM, ACS Appl. Mater. Interfaces 4 (2012) 4149–4155. [DOI] [PubMed] [Google Scholar]
- [47].Kanthamneni N, Sharma S, Meenach SA, Billet B, Zhao JC, Bachelder EM, Ainslie KM, Int. J. Pharm 431 (2012) 101–110. [DOI] [PubMed] [Google Scholar]
- [48].Junkins R, Gallovic M, Johnson B, Collier M, Watkins-Schulz R, Cheng N, David C, McGee C, Sempowski G, Shterev I, McKinnone K, Bachelder E, Ainslie K, Ting J, Control J. Release 21 (2017) 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Thakral S, Thakral NK, Majumdar DK, Expert Opin. Drug Deliv 10 (2013) 131–149. [DOI] [PubMed] [Google Scholar]
- [50].Lee KY, Mooney DJ, Prog. Polym. Sci 37 (2012) 106–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Hollabaugh CB, Burt LH, Walsh AP, Industrial & Engineering Chemistry 37 (1945) 943–947. [Google Scholar]
- [52].Chassard C, Delmas E, Robert C, Bernalier-Donadille A, FEMS Microbiol. Ecol 74 (2010) 205–213. [DOI] [PubMed] [Google Scholar]
- [53].Shokri J, Adibkia K, Application of cellulose and cellulose derivatives in pharmaceutical industries, in: Godbout T.v.d.V.a.L.(Ed.), Cellulose-medical, Pharmaceutical and Electronic Applications, IntechOpen, 2013. [Google Scholar]
- [54].Stankus JJ, Guan J, Fujimoto K, Wagner WR, Biomaterials 27 (2006) 735–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Ullm S, Kruger A, Tondera C, Gebauer TP, Neffe AT, Lendlein A, Jung F, Pietzsch J, Biomaterials 35 (2014) 9755–9766. [DOI] [PubMed] [Google Scholar]
- [56].Zhu S, Huang M, Feng G, Miao Y, Wu H, Zeng M, Lo YM, Food Science & Nutrition 6 (2018) 1023–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Kuijpers AJ, van Wachem PB, van Luyn MJ, Plantinga JA, Engbers GH, Krijgsveld J, Zaat SA, Dankert J, Feijen J, J. Biomed. Mater. Res 51 (2000) 136–145. [DOI] [PubMed] [Google Scholar]
- [58].Barak S, Mudgil D, Khatkar BS, Crit. Rev. Food Sci. Nutr 55 (2015) 357–368. [DOI] [PubMed] [Google Scholar]
- [59].Helmerhorst EJ, Zamakhchari M, Schuppan D, Oppenheim FG, PLoS One 5 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Qi Y, Wang H, Wei K, Yang Y, Zheng RY, Kim IS, Zhang KQ, Int. J. Mol. Sci (2017) 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Shriner RL, Nabenhauer FP, Anderson RJ, J. Am. Chem. Soc 49 (1927) 1290–1294. [Google Scholar]
- [62].Van Laere KMJ, Hartemink R, Bosveld M, Schols HA, Voragen AGJ, Agric J. Food Chem. 48 (2000) 1644–1652. [DOI] [PubMed] [Google Scholar]
- [63].Koukiekolo R, Cho H-Y, Kosugi A, Inui M, Yukawa H, Doi RH, Appl. Environ. Microbiol 71 (2005) 3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Rose DJ, Patterson JA, Hamaker BA, Agric J. Food Chem. 58 (2010) 493–499. [DOI] [PubMed] [Google Scholar]
- [65].Suksamran T, Ngawhirunpat T, Rojanarata T, Sajomsang W, Pitaksuteepong T, Opanasopit P, Int. J. Pharm 448 (2013) 19–27. [DOI] [PubMed] [Google Scholar]
- [66].Kean T, Thanou M, Adv. Drug Deliv. Rev 62 (2010) 3–11. [DOI] [PubMed] [Google Scholar]
- [67].Bellich B, D’Agostino I, Semeraro, Gamini A, Cesaro A, Marine Drugs 14 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Qiao D, Tu W, Liao A, Li N, Zhang B, Jiang F, Zhong L, Zhao S, Zhang L, Lin Q, Carbohydr. Polym 1 (2019) 199–207. [DOI] [PubMed] [Google Scholar]
- [69].Vancha AR, Govindaraju S, Parsa KV, Jasti M, Gonzalez-Garcia M, Ballestero RP, BMC Biotechnol. 4 (2004) 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Zeles-Hahn MG, Lentz YK, Anchordoquy TJ, Lengsfeld CS, J. Electrost 69 (2011) 67–77. [Google Scholar]
- [71].Lee MC, Seonwoo H, Garg P, Jang KJ, Pandey S, Kim HB, Park SB, Ku JB, Kim JH, Lim KT, Chung JH, RSC Adv. 8 (2018) 6452–6459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Wen Y, Pan S, Luo X, Zhang X, Zhang W, Feng M, Bioconjug. Chem 20 (2009) 322–332. [DOI] [PubMed] [Google Scholar]
- [73].Zhong Z, Ji Q, Zhang JA, J. Pharm. Biomed. Anal 51 (2010) 947–951. [DOI] [PubMed] [Google Scholar]
- [74].Simoes S, Filipe A, Faneca H, Mano M, Penacho N, Duzgunes N, Pedroso de Lima M, Expert Opinion on Drug Delivery 2 (2005) 237–254. [DOI] [PubMed] [Google Scholar]
- [75].Varshosaz J, Ghassami E, Noorbakhsh A, Jahanian-Najafabadi A, Minaiyan M, Behzadi R, Drug Dev. Ind. Pharm 44 (2018) 1012–1022. [DOI] [PubMed] [Google Scholar]
- [76].McSweeney MD, Versfeld ZC, Carpenter DM, Lai SK, Clin. Transl. Sci 11 (2018) 162–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Rudmann DG, Alston JT, Hanson JC, Heidel S, Toxicol. Pathol 41 (7) (2013) 970–983. [DOI] [PubMed] [Google Scholar]
- [78].Webster R, Didier E, Harris P, Siegel N, Stadler J, Tilbury L, Smith D, Drug Metab. Dispos 35 (2007) 9–16. [DOI] [PubMed] [Google Scholar]
- [79].Cao Y, Liu F, Chen Y, Yu T, Lou D, Guo Y, Li P, Wang Z, Ran H, Sci. Rep 7 (2017) 11913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Furtmann B, Tang J, Kramer S, Eickner T, Luderer F, Fricker G, Gomez A, Heemskerk B, Jahn PS, J. Pharm. Sci 106 (2017) 3316–3327. [DOI] [PubMed] [Google Scholar]
- [81].Chen N, Collier MA, Gallovic MD, Collins GC, Sanchez CC, Fernandes EQ, Bachelder EM, Ainslie KM, Int. J. Pharm 512 (2016) 147–157. [DOI] [PubMed] [Google Scholar]
- [82].Ding L, Lee T, Wang CH, J. Control. Release 102 (2005) 395–413. [DOI] [PubMed] [Google Scholar]
- [83].Xie J, Marijnissen JC, Wang CH, Biomaterials 27 (2006) 3321–3332. [DOI] [PubMed] [Google Scholar]
- [84].Kumar Naraharisetti P, Yung Sheng Ong B, Wei Xie J, Kam Yiu Lee T, Wang CH, Sahinidis NV, Biomaterials 28 (2007) 886–894. [DOI] [PubMed] [Google Scholar]
- [85].Ranganath SH, Kee I, Krantz WB, Chow PK, Wang CH, Pharm. Res 26 (2009) 2101–2114. [DOI] [PubMed] [Google Scholar]
- [86].Hayashi K, Ono K, Suzuki H, Sawada M, Moriya M, Sakamoto W, Yogo T, Small 6 (2010) 2384–2391. [DOI] [PubMed] [Google Scholar]
- [87].Nie H, Fu Y, Wang CH, Biomaterials 31 (2010) 8732–8740. [DOI] [PubMed] [Google Scholar]
- [88].Gulfam M, Kim JE, Lee JM, Ku B, Chung BH, Chung BG, Langmuir 28 (2012) 8216–8223. [DOI] [PubMed] [Google Scholar]
- [89].Cao Y, Wang B, Wang Y, Lou D, RSC Adv. 4 (2014) 30430–30439. [Google Scholar]
- [90].Qu J, Liu Y, Yu Y, Li J, Luo J, Li M, Mater. Sci. Eng. C Mater. Biol. Appl 44 (2014) 166–174. [DOI] [PubMed] [Google Scholar]
- [91].Ramachandran R, Malarvizhi GL, Chandran P, Gupta N, Menon D, Panikar D, Nair S, Koyakutty M, J. Biomed. Nanotechnol 10 (2014) 1401–1415. [DOI] [PubMed] [Google Scholar]
- [92].Yin D, Yang Y, Cai H, Wang F, Peng D, He L, Mol. Pharm 11 (2014) 4107–4117. [DOI] [PubMed] [Google Scholar]
- [93].Zhang M, Tang Y, Zhu Z, Zhao H, Yao J, Sun D, J. Biomater. Sci. Polym. Ed 29 (2018) 1949–1963. [DOI] [PubMed] [Google Scholar]
- [94].Watkins-Schulz R, Tiet P, Gallovic MD, Junkins RD, Batty C, Bachelder EM, Ainslie KM, Ting JPY, Biomaterials 205 (2019) 94–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Danhier F, Control J. Release 244 (2016) 108–121. [DOI] [PubMed] [Google Scholar]
- [96].Marabelle A, Tselikas L, de Baere T, Houot R, Ann. Oncol 28 (2017) xii33–xii43. [DOI] [PubMed] [Google Scholar]
- [97].Solorio L, Patel RB, Wu H, Krupka T, Exner AA, Ther. Deliv 1 (2010) 307–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Adamson C, Kanu OO, Mehta AI, Di C, Lin N, Mattox AK, Bigner DD, Expert Opin. Investig. Drugs 18 (2009) 1061–1083. [DOI] [PubMed] [Google Scholar]
- [99].Grossman SA, Reinhard C, Colvin OM, Chasin M, Brundrett R, Tamargo RJ, Brem H, J. Neurosurg 76 (1992) 640–647. [DOI] [PubMed] [Google Scholar]
- [100].Arifin DY, Lee KY, Wang CH, J. Control. Release 137 (2009) 203–210. [DOI] [PubMed] [Google Scholar]
- [101].Arifin DY, Lee KY, Wang CH, Smith KA, Pharm. Res 26 (2009) 2289–2302. [DOI] [PubMed] [Google Scholar]
- [102].Lai W-F, Susha AS, Rogach AL, Wang G, Huang M, Hu W, Wong W-T, RSC Adv. 7 (2017) 44482–44491. [Google Scholar]
- [103].Mai Z, Chen J, He T, Hu Y, Dong X, Zhang H, Huang W, Ko F, Zhou W, RSC Adv. 7 (2017) 1724–1734. [Google Scholar]
- [104].Bayramov DF, Neff JA, Adv. Drug Deliv. Rev 112 (2017) 48–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Hao S, Wang Y, Wang B, Deng J, Liu X, Liu J, Mater. Sci. Eng. C Mater. Biol. Appl 33 (2013) 4562–4567. [DOI] [PubMed] [Google Scholar]
- [106].Hao S, Wang Y, Wang B, Deng J, Zhu L, Cao Y, Mater. Sci. Eng. C Mater. Biol. Appl 39 (2014) 113–119. [DOI] [PubMed] [Google Scholar]
- [107].Hao S, Wang B, Wang Y, Xu Y, J. Nanopart. Res 16 (2014) 2204. [Google Scholar]
- [108].Paz-Samaniego R, Rascón-Chu A, Brown-Bojorquez F, Carvajal-Millan E, Pedroza-Montero M, Silva-Campa E, Sotelo-Cruz N, López-Franco YL, Lizardi-Mendoza J, J. Appl. Polym. Sci 135 (2018) 46411. [Google Scholar]
- [109].Ainslie KM, Lowe RD, Beaudette TT, Petty L, Bachelder EM, Desai TA, Small 5 (2009) 2857–2863. [DOI] [PubMed] [Google Scholar]
- [110].Stubelius A, Sheng W, Lee S, Olejniczak J, Guma M, Almutairi A, Small 14 (2018) e1800703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Xing Z, Zhang C, Zhao C, Ahmad Z, Li JS, Chang MW, Eur. J. Pharm. Sci 125 (2018) 64–73. [DOI] [PubMed] [Google Scholar]
- [112].Doavi T, Mousavi SL, Kamali M, Amani J, Ramandi MF, Iran. Biomed. J (2016) 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Manolova V, Flace A, Bauer M, Schwarz K, Saudan P, Bachmann MF, Eur. J. Immunol 38 (2008) 1404–1413. [DOI] [PubMed] [Google Scholar]
- [114].Wang F, Li Z, Tamama K, Sen CK, Guan J, Biomacromolecules 10 (2009) 2609–2618. [DOI] [PubMed] [Google Scholar]
- [115].Chang PC, Lim LP, Chong LY, Dovban AS, Chien LY, Chung MC, Lei C, Kao MJ, Chen CH, Chiang HC, Kuo YP, Wang CH, J. Dent. Res 91 (2012) 618–624. [DOI] [PubMed] [Google Scholar]
- [116].Nath SD, Son S, Sadiasa A, Min YK, Lee BT, Int. J. Pharm 443 (2013) 87–94. [DOI] [PubMed] [Google Scholar]
- [117].Chang PC, Chong LY, Dovban AS, Lim LP, Lim JC, Kuo MY, Wang CH, Tissue Eng Part A 20 (2014) 356–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Zhao S, Agarwal P, Rao W, Huang H, Zhang R, Liu Z, Yu J, Weisleder N, Zhang W, He X, Integr. Biol. (Camb) 6 (2014) 874–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Zamani M, Prabhakaran MP, Thian ES, Ramakrishna S, J. Colloid Interface Sci 451 (2015) 144–152. [DOI] [PubMed] [Google Scholar]
- [120].Gandhimathi C, Edwin NXH, Jayaraman P, Venugopal JR, Ramakrishna S, Kumar SD, Journal of Drug Metabolism & Toxicology (2015) 1–7. [Google Scholar]
- [121].Wang Y, Wei Y, Zhang X, Xu M, Liu F, Ma Q, Cai Q, Deng X, Chem. Eng. J 273 (2015) 490–501. [Google Scholar]
- [122].Esfahani RR, Jun H, Rahmani S, Miller A, Lahann J, ACS Omega 2 (2017) 2839–2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Chang HC, Yang C, Feng F, Lin FH, Wang CH, Chang PC, J. Formos. Med. Assoc 116 (2017) 973–981. [DOI] [PubMed] [Google Scholar]
- [124].Link PA, Ritchie AM, Cotman GM, Valentine MS, Dereski BS, Heise RL, Tissue Eng J. Regen. Med 12 (2018) 2331–2336. [DOI] [PubMed] [Google Scholar]
- [125].Zong H, Xia X, Liang Y, Dai S, Alsaedi A, Hayat T, Kong F, Pan JH, Mater. Sci. Eng. C Mater. Biol. Appl 92 (2018) 1075–1091. [DOI] [PubMed] [Google Scholar]
- [126].Soares RMD, Siqueira NM, Prabhakaram MP, Ramakrishna S, Mater. Sci. Eng. C Mater. Biol. Appl 92 (2018) 969–982. [DOI] [PubMed] [Google Scholar]
- [127].Park JB, Med. Oral Patol. Oral Cir. Bucal 14 (2009) e485–e488. [PubMed] [Google Scholar]
- [128].Peine KJ, Guerau-de-Arellano M, Lee P, Kanthamneni N, Severin M, Probst GD, Peng HY, Yang YH, Vangundy Z, Papenfuss TL, LovettRacke AE, Bachelder EM, Ainslie KM, Mol. Pharm 11 (2014) 828–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Schully KL, Sharma S, Peine KJ, Pesce J, Elberson MA, Fonseca ME, Prouty AM, Bell MG, Borteh H, Gallovic M, Bachelder EM, Keane-Myers A, Ainslie KM, Pharm. Res 30 (2013) 1349–1361. [DOI] [PubMed] [Google Scholar]
- [130].Graham-Gurysh E, Moore KM, Satterlee AB, Sheets KT, Lin FC, Bachelder EM, Miller CR, Hingtgen SD, Ainslie KM, Mol. Pharm 15 (2018) 1309–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Cheng N, Watkins-Schulz R, Junkins RD, David CN, Johnson BM, Montgomery SA, Peine KJ, Darr DB, Yuan H, McKinnon KP, Liu Q, Miao L, Huang L, Bachelder EM, Ainslie KM, Ting JP, JCI Insight (2018) 3. [DOI] [PMC free article] [PubMed] [Google Scholar]