

Abstract

Single-site catalysts (SSCs) have attracted significant research interest due to their high metal atom utilization. Platinum single sites trapped in the defects of carbon substrates (trapped Pt-SSCs) have been proposed as efficient and stable electrocatalysts for the hydrogen evolution reaction (HER). However, the correlation between Pt bonding environment, its evolution during operation, and catalytic activity is still unclear. Here, a trapped Pt-SSC is synthesized by pyrolysis of H2PtCl6 chemisorbed on a polyaniline substrate. In situ heated scanning transmission electron microscopy and temperature-dependent X-ray photoelectron spectroscopy clarify the thermally induced structural evolution of Pt during pyrolysis. The results show that the nitrogen in polyaniline coordinates with Pt ions and atomically disperses them before pyrolysis and traps Pt sites at pyridinic N defects generated during the substrate graphitization. Operando X-ray absorption spectroscopy confirms that the trapped Pt-SSC is stable at the HER working potentials but with inferior electrocatalytic activity compared with metallic Pt nanoparticles. First principle calculations suggest that the inferior activity of trapped Pt-SSCs is due to their unfavorable hydrogen chemisorption energy relative to metallic Pt(111) surfaces. These results further the understanding of the structure–property relationship in trapped Pt-SSCs and motivate a detailed techno-economic analysis to evaluate their commercial applicability.
Keywords: platinum single-site catalysts, in situ characterization, Pt-SSC formation mechanism, hydrogen evolution reaction, DFT calculations
Introduction
Proton exchange membrane (PEM) water electrolysis is one of the most promising technologies to generate green hydrogen from renewable energy sources.1 However, it relies on rare and expensive noble metal electrocatalysts, which undermines its economic viability.2,3 Single-site catalysts (SSCs), a new frontier in heterogeneous catalysis where isolated metal sites are supported on a substrate,4−9 have been proposed to alleviate this problem by maximizing noble metal utilization without compromising activity.10,11
Metallic Pt nanoparticles on a carbon support are the state-of-the-art hydrogen evolution reaction (HER) electrocatalyst in PEM electrolyzers.12,13 However, the current required mass loading of Pt nanoparticles may constrain large-scale (terawatt) deployment of PEM water electrolyzers.3 Highly dispersed Pt-SSCs have the potential to reduce the mass loading of Pt by maximizing atom utilization.14,15 Pt-SSCs trapped in defective carbon substrates have been reported to display higher mass activities for the HER compared to Pt nanoparticles as they fully expose the metal sites.16−19 However, their intrinsic catalytic activity relative to metallic nanoparticles is still a topic of debate in the field due to a lack of a detailed investigation into their structure–property relationship.12,20,21 Specifically, several reports have shown that Pt-SSCs on carbon substrates have higher mass activity than commercial Pt nanoparticles.17,18,22 On the contrary, other studies20,23 have suggested that Pt nanoparticles may be more active than oxidized Pt-SSCs for the HER. Thus, it is important to conduct a deep and fair evaluation of both Pt-SSCs and Pt nanoparticles for HER.
This fundamental interrogation requires a well-defined trapped Pt-SSCs structure that can then be used to investigate the bonding environment of Pt, its evolution under working conditions, and correlate it to catalytic activity and stability.23−25 One-pot pyrolysis, whereby a mixture of metal precursors and a carbon matrix are pyrolyzed at high temperatures (700–1000 °C), is commonly used to synthesize trapped SSCs.26−30 However, due to the complexity of the precursor mixture and the dynamic structural evolution of single sites induced by heating,31,32 a trial and error approach is commonly used to refine the synthesis conditions. A fundamental mechanistic understanding of the formation mechanism of Pt-SSCs is essential for the development of novel and reproducible synthesis methodologies.
Here we synthesize a Pt-SSC trapped in an N-doped graphene substrate by pyrolyzing H2PtCl6 chemisorbed on polyaniline (PANI).33 The formation mechanism of the trapped Pt-SSC is investigated by in situ heated scanning transmission electron microscopy (STEM) and temperature-dependent X-ray photoelectron spectroscopy (XPS) to probe the physical and chemical structures of Pt sites at various temperatures (25–800 °C). We show that N in PANI can coordinate with Pt-ions in the complex and trap Pt in the defects during substrate graphitization. After defining the bonding environment of the Pt-SSC with extensive in situ and ex situ characterization techniques, we investigate its evolution under working conditions using operando X-ray absorption spectroscopy (XAS). We show that the Pt-SSC structure is stable and that Pt remains in its (+2) oxidation state at the HER working potential. Finally, we compare the catalytic activity of our model Pt-SSC to that of Pt nanoparticles, with the model Pt-SSC demonstrating inferior catalytic properties. First principle calculations suggest that the inferior activity of the trapped Pt-SSC is due to its unfavorable hydrogen chemisorption energy relative to metallic Pt(111) surfaces.
Results and Discussion
Pt Single-Sites on Polyaniline
A Pt-polyaniline (PANI) composite with well-dispersed Pt sites (Figure 1a) was synthesized via a wet impregnation method from H2PtCl6 and PANI (details in the experimental section). PANI is a commonly used substrate for the synthesis of SSCs because of its ability to coordinate with metal-ion complexes34,35 and hinder their aggregation.36−38 High-resolution annular dark-field (ADF) STEM images collected after the impregnation process (Figure 1b–d and Supplementary Figure S1) display Pt sites atomically dispersed on the polyaniline substrate with loading up to 10 wt%. The density of isolated Pt sites in 2 wt%, 5 wt% and 10 wt% samples are ∼3, ∼7 and ∼14 nm–2, respectively (as determined by calculating the number of bright dots in the STEM images which are representative of each sample).
Figure 1.

(a) Schematic of the synthesis process of Pt-PANI and trapped Pt-SSC. High-resolution ADF-STEM images of 2 wt% (b), 5 wt% (c) and 10 wt% (d) Pt-PANI shows Pt individually anchored on the PANI substrate. (e) FT-IR spectra of PANI and 10 wt% Pt single sites on PANI. (f) FT-EXAFS spectra of Pt(Py)2Cl2 (blue), PtCl2 (yellow), meso-tetraphenylporphine-Pt(II) (PtTPP) (green), and Pt-PANI (red). (g) XPS of Pt 4f for Pt-PANI.
The interaction between 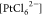 and PANI was investigated by Fourier-transform
infrared spectroscopy (FT-IR) and XAS. As shown in the FT-IR spectra
of the 10 wt% Pt-PANI sample (Figure 1e), in addition to the characteristic peaks of PANI,
new peaks corresponding to Pt-Cl and Pt-N vibrations are present at
334/370 cm1 and 920/1003 cm1, respectively.39 These new vibrations indicate that Pt-ions are
anchored on the PANI substrate by interactions with N; the remaining
Cl ligands are from the precursor. To provide further information
on the bonding environment of Pt, EXAFS was performed at the Pt L3-edge and compared with reference Pt structures. As shown
in Figure 1f, both
the Fourier-transformed (FT)-EXAFS spectra of the Pt-PANI (red) and
Pt(Py)2Cl2 (blue) feature two peaks at 1.93
and 1.50 Å, which are in agreement with the positions of the
Pt-Cl bond in PtCl2 (yellow) at 1.93 Å and Pt-N bond
in meso-tetraphenylporphine-Pt(II) (PtTPP, green) at 1.56 Å,
respectively. The EXAFS data provide further evidence that Pt single
sites bond with N in the PANI substrate and remaining Cl ligands.
and PANI was investigated by Fourier-transform
infrared spectroscopy (FT-IR) and XAS. As shown in the FT-IR spectra
of the 10 wt% Pt-PANI sample (Figure 1e), in addition to the characteristic peaks of PANI,
new peaks corresponding to Pt-Cl and Pt-N vibrations are present at
334/370 cm1 and 920/1003 cm1, respectively.39 These new vibrations indicate that Pt-ions are
anchored on the PANI substrate by interactions with N; the remaining
Cl ligands are from the precursor. To provide further information
on the bonding environment of Pt, EXAFS was performed at the Pt L3-edge and compared with reference Pt structures. As shown
in Figure 1f, both
the Fourier-transformed (FT)-EXAFS spectra of the Pt-PANI (red) and
Pt(Py)2Cl2 (blue) feature two peaks at 1.93
and 1.50 Å, which are in agreement with the positions of the
Pt-Cl bond in PtCl2 (yellow) at 1.93 Å and Pt-N bond
in meso-tetraphenylporphine-Pt(II) (PtTPP, green) at 1.56 Å,
respectively. The EXAFS data provide further evidence that Pt single
sites bond with N in the PANI substrate and remaining Cl ligands.
The oxidation state of Pt in the Pt-PANI sample is further clarified by XPS and XANES. A Pt 4f7/2 peak with a binding energy of 72.9 eV, corresponding to an oxidation state of + 2,33 is present in the Pt core level XPS spectrum of Pt-PANI (Figure 1g). The XANES of Pt-L3 in Pt-PANI and Pt(Py)2Cl2 have similar edge-line position and white-line intensity (Figure S4), which also indicates the oxidation state of Pt in the Pt-PANI sample is + 2. Pt is partially reduced from a + 4 to a + 2 oxidation state as it transitions from the H2PtCl6 precursor to become anchored on the PANI substrate. The presence of metal-bonded Cl peaks at 198.3 eV from Pt-Cl in the Cl 2p spectra can also prove the successful anchoring of Pt sites (Supplementary Figure S5). Combining the STEM, FT-IR, EXAFS and XPS data, we confirm that Pt-ions are well dispersed throughout the sample due to their coordination to the aniline N in PANI.
Trapped Pt-SSC on N-Doped Graphene
Pt-PANI samples with various Pt loadings (10 wt%, 5 wt%, 2 wt% and 1 wt%) were pyrolyzed at 800 °C under N2 for 1.5 h and charactrized by ex situ STEM (Figure 2a–h). Pt nanoparticles can be identified in the pyrolyzed Pt-PANI samples with Pt loadings of 10 wt% (Figure 2a,e), 5 wt% (Figure 2b,f), and 2 wt% (Figure 2c,g), mainly at the edge of the substrates. More specifically, the 10 wt% Pt-PANI sample is covered with 3–5 nm nanoparticles remain after pyrolysis (a), while there are ∼150 and ∼42 nanoparticles by counts in the b (5 wt%) and c (2 wt%) samples, respectively. At the lowest Pt loading of 1 wt%, almost all the Pt forms a trapped single-site (Figure 2d,h). Therefore, we selected the 1 wt% Pt-PANI sample as a model for further investigation (see discussion in Supplementary Figure S2).
Figure 2.

Low- (a–d) and high-resolution (e–h) STEM images of Pt-PANI with various loadings after pyrolysis in N2 at 800 °C for 1.5 h, which are representative of each sample; 10 wt% (a, e), 5 wt% (b, f), 2 wt% (c, g) and 1 wt% (d, h). The red circles highlight the formed nanoparticles, and the yellow rectangles outline the areas where high-resolution STEM images were recorded (e–h). (i) Raman spectra of the PANI substrate before and after pyrolysis. CPMAS NMR spectra of 15N (j) and FT-EXAFS (k) of 1 wt% Pt-PANI before and after pyrolysis in N2 at 800 °C for 1.5 h.
Raman spectroscopy and cross-polarization magic-angle spinning nuclear magnetic resonance spectroscopy (CPMAS NMR) were used to confirm the structure of the PANI after pyrolysis. The ex situ Raman spectra of PANI before and after pyrolysis (Figure 2i) show that all the characteristic PANI peaks vanish after pyrolysis, while two new peaks at 1360 and 1580 cm–1 appear which are from the D and G bands of graphene. This indicates the formation of a defective graphene substrate.40 Furthermore, the new peaks formed after pyrolysis in the 15N CPMAS NMR spectra (Figure 2j), ranging from −50 to −200 ppm, demonstrate the formation of pyridinic N and graphitic N.41 The Raman and NMR data together suggest that the PANI substrate becomes N-doped graphene after pyrolysis.
Figure 2k shows the Pt L3-edge EXAFS spectra of the Pt-SSC before and after pyrolysis. After pyrolysis, the Pt-Cl peak at 1.93 Å disappears, leaving the peak at 1.58 Å (Pt-N) as the main peak. The absence of a significant Pt-Pt peak at 2.73 Å means that the Pt sites remain isolated and bonded to N as trapped Pt-SSCs.
Formation Mechanism of Trapped Pt-SSC
In situ heating STEM was used to track the spacial evolution of the Pt sites in the 10 wt% Pt-PANI sample with increasing temperature to form trapped Pt-SSC. The majority of the Pt sites in the bulk remain isolated even when the temperature reaches 800 °C (Figure 3a–d and Supplementary Figure S11). On the contrary, some of the Pt sites along the edge (Figure 3e–h and Supplementary Figures S12 and S13) start to aggregate at 500 °C and further transform into nanoparticles at 800 °C (Figure 2a,e). The formation of Pt nanoparticles confirms that Pt sites tend to aggregate at high temperatures, especially with relatively high Pt loadings.
Figure 3.

In situ heating STEM images showing the spacial evolution of Pt sites at the bulk (a–d) and edge (e–h) in a sample with 10 wt% Pt on PANI with increasing temperatures (RT, 350 °C, 500 °C, and 800 °C) under vacuum. (i–k) Temperature-dependent XPS data of 1 wt% Pt-PANI under vacuum to show how the bonding environment of Pt evolves during pyrolysis. Pt 4f spectra (i), N 1s spectra (j), and Cl 2p spectra (k). (l) XANES of the trapped Pt-SSC synthesized by pyrolysis, compared with the Pt TPP complex and Pt foil. The inset is a schematic of the trapped Pt-SSC structure; black for carbon, blue for N, and gray for Pt.
Temperature-dependent XPS was then performed to track the changes in the bonding environment of Pt in a 1 wt% Pt-PANI sample (10 wt% in the Supplementary Figure S14) core level for Pt 4f, N 1s, Cl 2p, C 1s, and O 1s were recorded. As Figure 3i shows, the Pt 4f peaks shift to lower binding energies with increasing temperature; the Pt 4f7/2 shifts from 72.8 eV (Pt(+2)) at room temperature (RT) to 72.0 eV at 700 °C. This suggests a change in Pt’s chemical environment as the pyrolysis temperature increases, possibly toward more metallic Pt. Further, the Pt 4f peaks exhibit a more asymmetric peak shape at 700 °C, which is indicative of this more metallic Pt bonding environment. More importantly, as the temperature increases to 700 °C, the N 1s peak gradually splits into two peaks: a graphitic N peak at 400.9 eV and a pyridinic N peak at 398.3 eV (Figure 3j).33,42 This significant change indicates the graphitization of the PANI substrate at high temperatures, which is also supported by the shifts in the C-N peak at C 1s (Supplementary Figure S15). In comparison, the Cl spectra show that peaks from Cl bonded to Pt sites at 198.3 eV and organic Cl peaks at 200 eV from PANI at RT disappear by the time the samples reach 300 °C (Figure 3k). Oxygen peaks also disappear at this temperature (Supplementary Figure S15).
Overall, as can be seen in survey scans (Supplementary Figure S16), although the intensity of the N peaks has dropped, the disappearance of the Cl and O peaks indicates that only the remaining N can continuously bond with the Pt to trap it at high temperatures. Based on the different electronic density of N sites, it is more likely that pyridinic N, rather than graphitic N, donates electrons to bond with Pt sites.43 By comparing the XANES data of the trapped Pt-SSC with PtTPP and Pt foil (Figure 3l), we can confirm that the Pt sites in trapped Pt-SSC have a similar oxidation state and bonding environment as Pt(II) in the PtTPP complex, which is consistent with our XPS results (Figure 3i). Therefore, we fit the EXAFS data in Figure 2k with calculated DFT models (see discussion in Supplementary Figure S7 and Table S2), we propose the following atomic structure for trapped Pt-SSC: one Pt site bonds with 3 N and 1 C on the N-doped graphene (Pt-N3-C structure of the inset in Figure 3l).
In summary, in the Pt-PANI samples, Pt sites are atomically bonded with N and Cl, yielding a formal oxidation state of +2. During the pyrolysis process, they are then gradually donated electrons by the pyridinic N sites and finally become trapped in the pyridinic N defects formed during the substrate graphitization. Pt-sites in Pt-PANI samples with higher Pt loading are prone to reduction to their metallic state and aggregation into nanoparticles, especially at edge areas (Figure 3h), when the surrounding N sites are evaporated at high temperatures.
HER Activity of Trapped Pt-SSC
After clarifying the structure of the trapped Pt-SSC (1 wt% Pt-PANI after pyrolysis), its catalytic activity for the HER was evaluated using a customized three-electrode setup and compared with other trapped Pt-SSC samples (5 and 10 wt% Pt-PANI after pyrolysis) and commercial 20 wt% Pt/C (1 μgPt cm–2). The three-electrode setup is specially designed to minimize the mass transfer limitations in the HER (Supplementary Figure S17 and Supplementary Figure S18), which is critical for an accurate evaluation of the HER catalytic activity. Their activities were analyzed using cyclic voltammetry (CV) at a scan rate of 10 mV s–1 in 0.5 M H2SO4 saturated with pure H2. Representative structures of the trapped Pt samples (1 wt%, 5 wt% and 10 wt% Pt-PANI after ex situ pyrolysis) and commercial 20 wt% Pt/C are shown as STEM images in Figure 2 and Supplementary Figure S24. As previously discussed, the trapped 1 wt% Pt sample (trapped Pt-SSC) is a single-site structure, the trapped 10 wt% Pt sample is composed of well-dispersed nanoparticles and single sites, and the 20 wt% Pt/C sample is covered with nanoparticles with an average size of 2–3 nm (Supplementary Figure S24).
From the CV curves shown in Figure 4a, the trapped 1 wt% Pt sample (trapped Pt-SSC, blue line) has a much higher onset overpotential than the other samples (the overpotential to reach a current density of 1 mA cm–2); the 1 wt% sample requires 483.0 ± 13.7 mV compared with only 15.0 ± 0.5 mV for the Pt/C (yellow). Electrochemical active surface area (ECSA) specific current density and Pt-mass specific current both show the same activity trend (Supplementary Figure S20). The HER activities of trapped Pt samples increase significantly with Pt loading (from blue 1 wt% to green 5 wt% to red 10 wt%). The trapped 10 wt% Pt sample, which contains vastly more nanoparticles, shows superior activity, needing only 76.0 ± 8.6 mV vs RHE to reach a current density of 1 mA cm–2, as seen in Figure 4a. This is 407 mV and 290 mV lower than the trapped 1 wt% and 5 wt% samples, respectively.
Figure 4.
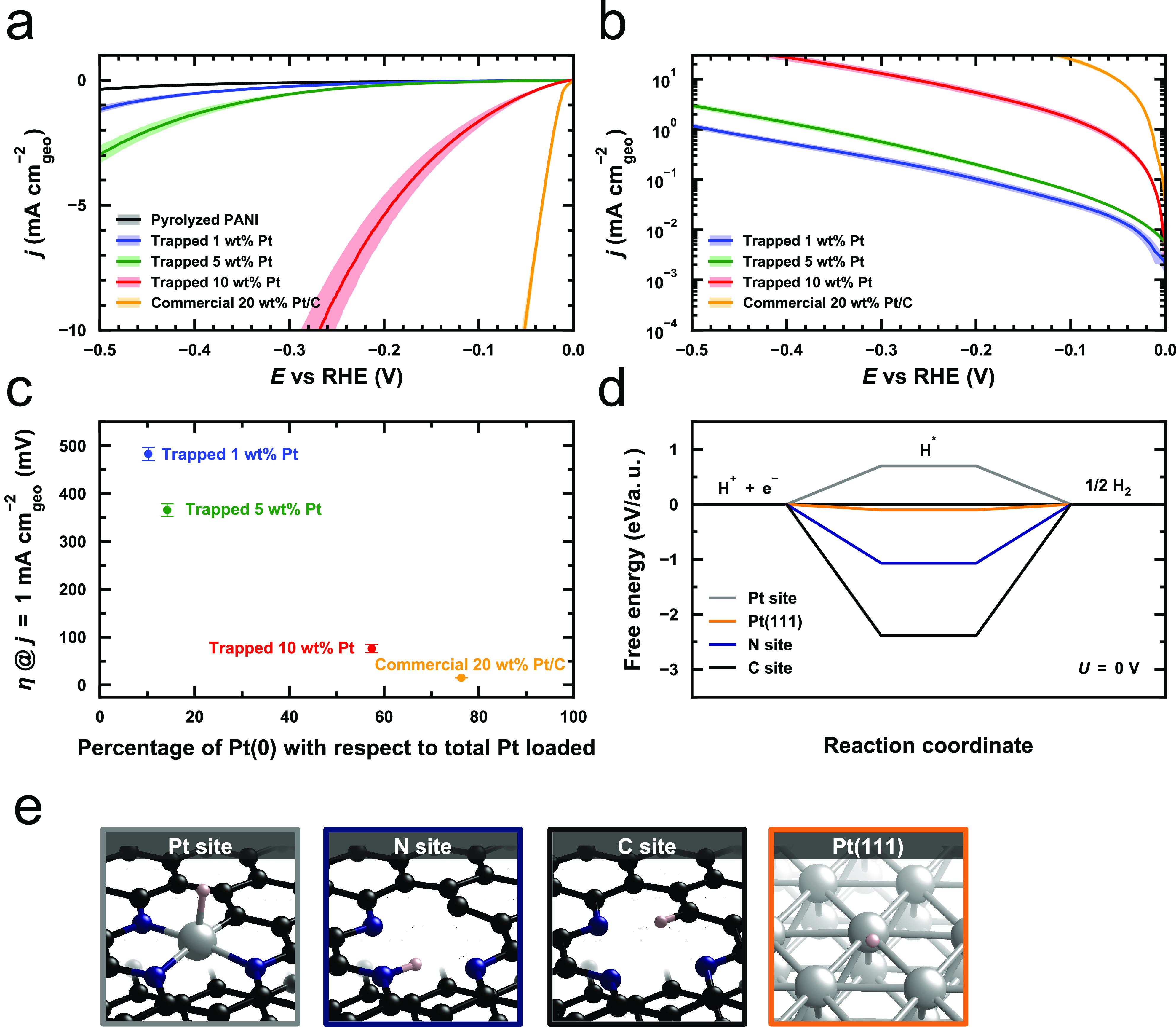
(a) HER polarization curves for trapped Pt-SSC samples synthesized by one-pot pyrolysis and commercial 20 wt% Pt/C in 0.5 M H2SO4 electrolyte, showing the overpotential needed to reach a specific geometric current density. Solid curves represent the averaged current response obtained from a minimum of three separate measurements, and the shaded regions show the range of standard errors relative to the mean values. (b) The areal current density on a logarithm-10 scale derived from the average CV curves for Pt samples in (a). (c) The correlation between the onset overpotentials (η at the current density of 1 mA cm–2) and the percentage of Pt(0) with respect to total Pt loaded in an ex situ pyrolyzed Pt samples. (d) Free energy diagram for the hydrogen evolution at the reaction potential U = 0 V. The data are calculated at 1 bar of H2 and pH = 0 at 298 K. (e) Relaxed structures of hydrogen adsorbed on Pt site, N site, C site, and the FCC site on Pt(111) surface. Pink for hydrogen, black for carbon, blue for N, and metallic for Pt.
Furthermore, Figure 4b and the corresponding Tafel analysis (Supplementary Figure S21) show a comparison of the HER kinetics by calculating the base-10 logarithm of the geometric current density in Figure 4a. In Pt single site-dominated samples (trapped 1 wt% and 5 wt% Pt) are much slower than in Pt nanoparticle-dominated samples (trapped 10 wt% Pt and 20 wt% Pt/C). This indicates that the trapped Pt-SSCs may have a different reaction mechanism for the HER compared to that of the metallic Pt nanoparticles.44 Correlation between the oxidation state of Pt (Supplementary Figure S25) and the HER activity is demonstrated in Figure 4c. The more reduced Pt structure with a higher number of nanoparticles has a lower onset overpotential for the HER, which is consistent with metallic Pt being more active than the oxidized trapped Pt-SSCs trapped on the N-doped graphene.20 Combining STEM, XPS and HER data, we conclude that the model trapped Pt-SSCs for the HER show inferior HER activity than metallic nanoparticles. This is evidenced by comparing the HER performance of 1 wt% trapped Pt-SSC, an oxidized Pt single-site sample, with the commercial 20 wt% Pt/C, a metallic Pt-nanoparticle structure (further discussion in Supplementary Figure S22).
The origin of the low HER activity of the trapped Pt-SSCs can be probed using a first-principles approach (Figure 4d). We calculate the hydrogen chemisorption free energy using density-functional theory (DFT) at an applied external potential of U = 0 V and pH = 0 on the Pt, C, and N sites of the Pt-N3-C model shown in Figure 4e. The hydrogen chemisorption free energy is an indicator of the HER activity for Pt group metals, with values closest to 0 eV showing the highest HER activity.45 A 0.70 eV hydrogen chemisorption energy for the Pt site is observed (gray line and gray frame in Figure 4d and e). This value is further from the thermo-neutral value than that of the Pt(111) surface (−0.10 eV), implying that the occurrence of the HER may be less favorable on the single Pt site than on the metallic Pt(111) surface.23,46 Strong hydrogen adsorption on the N (−1.07 eV, the blue line in Figure 4d and e) and C site (−2.39 eV, black line in Figure 4d and e) can also be observed, suggesting that these two sites are unlikely to catalyze the HER. The poorer activity of the Pt-SSC can be attributed to the lower electron density available for protons at the Pt site relative to that in Pt metal or intermediate nanoclusters. These observations are in agreement with our experimentally measured activities.
Structure of Trapped Pt-SSC at Working Potentials
Finally, operando XAS was employed to investigate the structure of the trapped Pt-SSCs under working potentials for the HER. Operando XAS spectra of the Pt L3-edge were recorded alongside the current when various potentials were applied to the sample (from 0.50 V (OCV) to −0.30 V vs RHE). The setup of the operando experiment is shown in Supplementary Figure S30. The chronoamperometry (CA) curves recorded at 7 potentials are shown in Figure 5a, revealing that hydrogen starts to evolve significantly below −0.1 V vs RHE. From the FT-EXAFS spectra of the Pt L3-edge at all potentials (Supplementary Figure S28), the peak at 1.50–1.60 Å, representing the Pt-N bond, is dominant throughout the experiment. No Pt-Pt peak is observed, but some distortion of the Pt-N peak with decreasing potential is apparent.
Figure 5.
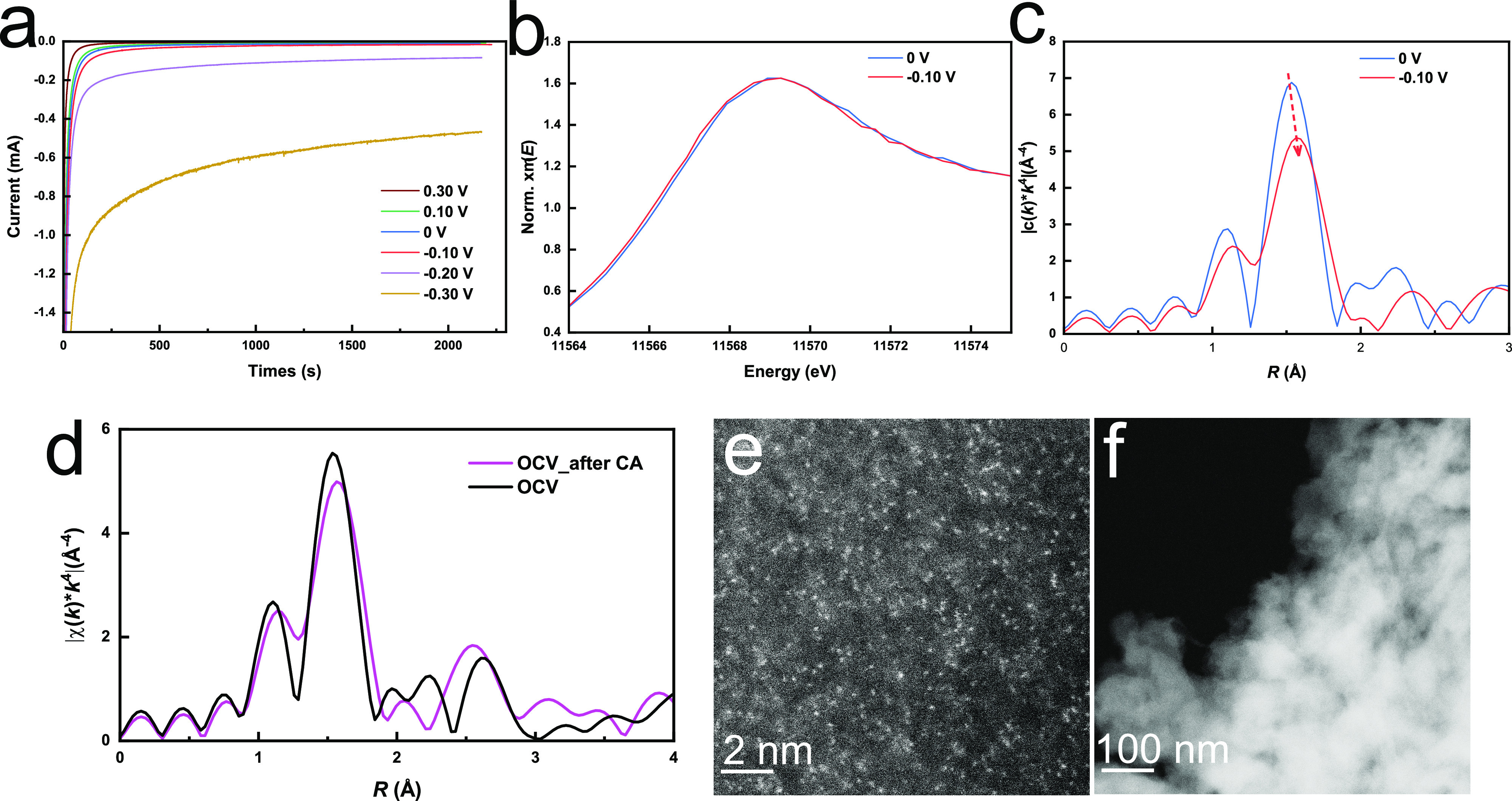
(a) Chronoamperometry curves of trapped Pt-SSCs at different applied potentials. (b) and (c) XANES and FT-EXAFS spectra of the Pt L3-edge for the trapped Pt-SSCs highlighted at 0 and −0.10 V. (d) FT-EXAFS spectra of the Pt-L3 edge for trapped Pt-SSCs at the OCV before and after CA tests. (e) and (f) Atomic STEM images of trapped Pt-SSC after 2 h of CA test at −0.2 V vs RHE with a scale bar of 2 and 100 nm, respectively, showing no Pt nanoparticle can be observed after the standard stability test.
From OCV to 0 V vs RHE, there is no shift in the absorption edge and the white-line intensity of the XANES spectra (Supplementary Figure S28), indicating no significant change in the oxidation state of Pt above 0 V. A slight reduction of Pt is found from 0 to −0.1 V with the white line shifting negatively by 0.1 eV (Figure 5b) when the HER occurs. An apparent distortion in the Pt-N peak in the FT-EXAFS spectra can also be observed at −0.1 V in Figure 5c. Fitting the operando EXAFS data with the proposed Pt-N3-C structure (Supplementary Table S3) suggests that this corresponds with a shortening of the Pt-N bond by ∼ 0.01 Å. The slight reduction of Pt and the decrease in the Pt-N bond length indicate that the trapped Pt-SSCs have been reduced slightly to initiate the reaction.
At the working potentials for the HER (−0.1 V, −0.2 V, and −0.3 V vs RHE), the oxidation state of Pt remains stable, as shown by the XANES spectra in Supplementary Figure S28c, and lack of observed Pt-Pt peak at 2.70 Å (Supplementary Figure S28a). The data show that Pt remains stable in a single-site structure under HER working conditions. Even when the potential returns to the OCV after the CA tests, the Pt-N bonding is retained (Figure 5d and Supplementary Figure S29). The stability of the trapped Pt-SSCs is further proven by the ADF-STEM images of the used catalyst. As shown in Figure 5e, f, no Pt aggregates are recorded after performing the CA test for 2 h at −0.2 V vs RHE. Overall, the data show that the trapped Pt-SSCs retain a stable structure under an HER working potential, with no aggregation occurring during normal operation.
Conclusions
In this work, model-trapped Pt-SSCs on N-doped graphene were prepared by pyrolyzing atomically dispersed Pt sites on a PANI substrate. In situ characterization experiments (STEM and XPS) were combined to clarify the formation mechanism of trapped Pt-SSCs. We found that N sites on the substrate could persistently bond with Pt sites during pyrolysis and trap Pt as isolated sites in pyridinic N defects after graphitization of the substrate. By combining in situ characterization data with complementary ex situ characterization results (XAS, Raman, and CPMAS NMR), the atomic structure of the trapped Pt-SSCs was identified: Pt(+2) single sites bond with three pyridinic Ns and one carbon on the N-doped graphene substrate. The local structure of Pt sites of trapped Pt-SSC is an analogue of Pt macrocyclic Pt(II) complexes.
Next, the intrinsic activity of the model trapped Pt-SSCs for the HER was clarified. HER polarization curves show that an additional 468 mV is required for the trapped 1 wt% Pt-SSC to reach the current density of 1 mA cm–2 compared with commercial 20 wt% Pt/C, a metallic Pt-nanoparticle structure. By correlating the activity and oxidation state of our Pt catalysts and commercial Pt nanoparticles, we confirmed that a metallic Pt structure is more active for the HER because of its lower hydrogen chemisorption energy. We believe this is due to Pt-SSCs having a lower electron density relative to metallic Pt nanoparticles, which is attributed to their oxidized state in this bonding environment. The operando XAS experiment shows a trapped Pt-SSC is indeed stable at the HER working potentials. While metallic Pt single-site structures that are stable under the HER conditions have not been well-demonstrated to date, our study suggests that a detailed techno-economic analysis is needed to evaluate if the benefits of the reduced metallic loading and long-term stability of trapped Pt single-sites compared to state-of-the-art Pt nanoparticles outweigh their lower catalytic activity and more complex synthetic procedure.
Acknowledgments
The authors would like to acknowledge the financial support of the Jiangsu Industrial Technology Research Institute (JITRI), the Henry Royce Institute (through UK Engineering and Physical Sciences Research Council grant EP/R010145/1) and EPSRC (EP/T001038/1) for capital equipment. R.S.W. acknowledges support from a UKRI Future Leaders Fellowship (MR/V024558/1). We acknowledge Diamond Light Source for time on B18 Beamline under Proposal SP-30237. We are grateful to the David Cockayne Centre for Electron Microscopy for the use of their electron microscopes.
Data Availability Statement
The authors declare that all data supporting the findings of this study are included within the paper and its Supporting Information files. Source data are available from the corresponding author upon reasonable request.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.3c01513.
Experimental method and materials; XANES and XPS spectra of Pt-PANI; additional temperature-dependent XPS spectra of 1 wt% Pt-PANI; size distribution of Pt single sites and Pt nanoparticles of trapped 1 wt% Pt-SSC; ex situ and operando EXAFS analysis of trapped 1 wt% Pt-SSC, including photograph of the operando XAS setup; discussions on the protocol of HER testing and the catalytic and DFT calculation results (PDF)
Author Contributions
⊥ P.T. and P.-Y.H. contributed equally to this paper. P.T. and M.P. conceived and designed the experiments. P.T. prepared all the samples and performed the in situ and ex-situ aberration-corrected STEM characterization and analyzed the data under the supervision of J.H.W. J.E.S. performed the XPS measurements under the supervision of R.S.W. and contributed to XPS data interpretation. P.T. did the FT-IR experiment and explained the data. P.-Y.H. collected the Raman data and analyzed it with P.T. D.G., P.T., and P.-Y.H. conducted the operando XAFS experiments at Diamond Light Source under the proposal of SP_30237. P.T. analyzed the operando XAS data. P.-Y.H. and P.T. performed the electrochemical tests. H.G. conducted the CPMAS NMR experiment and analyzed the data. P.-Y.H. conducted the DFT calculations. P.T. and C.W. wrote the manuscript with input from all authors. M.P. supervised the design of the project and gave in-depth scientific input.
The authors declare no competing financial interest.
Supplementary Material
References
- Turner J. A. Sustainable hydrogen production. Science 2004, 305, 972–974. 10.1126/science.1103197. [DOI] [PubMed] [Google Scholar]
- Kibsgaard J.; Chorkendorff I. Considerations for the scaling-up of water splitting catalysts. Nature Energy 2019, 4, 430–433. 10.1038/s41560-019-0407-1. [DOI] [Google Scholar]
- Hubert M. A.; King L. A.; Jaramillo T. F. Evaluating the Case for Reduced Precious Metal Catalysts in Proton Exchange Membrane Electrolyzers. ACS Energy Letters 2022, 7, 17–23. 10.1021/acsenergylett.1c01869. [DOI] [Google Scholar]
- Yang X.-F.; Wang A.; Qiao B.; Li J.; Liu J.; Zhang T. Single-atom catalysts: a new frontier in heterogeneous catalysis. Accounts of chemical research 2013, 46, 1740–1748. 10.1021/ar300361m. [DOI] [PubMed] [Google Scholar]
- Cui X.; Li W.; Ryabchuk P.; Junge K.; Beller M. Bridging homogeneous and heterogeneous catalysis by heterogeneous single-metal-site catalysts. Nature Catalysis 2018, 1, 385–397. 10.1038/s41929-018-0090-9. [DOI] [Google Scholar]
- Wang A.; Li J.; Zhang T. Heterogeneous single-atom catalysis. Nature Reviews Chemistry 2018, 2, 65–81. 10.1038/s41570-018-0010-1. [DOI] [Google Scholar]
- Fei H.; Dong J.; Chen D.; Hu T.; Duan X.; Shakir I.; Huang Y.; Duan X. Single atom electrocatalysts supported on graphene or graphene-like carbons. Chemical Society Reviews 2019, 48, 5207–5241. 10.1039/C9CS00422J. [DOI] [PubMed] [Google Scholar]
- Resasco J.; Yang F.; Mou T.; Wang B.; Christopher P.; Resasco D. E. Relationship between Atomic Scale Structure and Reactivity of Pt Catalysts: Hydrodeoxygenation of m-Cresol over Isolated Pt Cations and Clusters. ACS Catalysis 2020, 10, 595–603. 10.1021/acscatal.9b04330. [DOI] [Google Scholar]
- Dong C.; Gao Z.; Li Y.; Peng M.; Wang M.; Xu Y.; Li C.; Xu M.; Deng Y.; Qin X.; Huang F.; Wei X.; Wang Y.-G.; Liu H.; Zhou W.; Ma D. Fully exposed palladium cluster catalysts enable hydrogen production from nitrogen heterocycles. Nature Catalysis 2022, 5, 485–493. 10.1038/s41929-022-00769-4. [DOI] [Google Scholar]
- Zhu C.; Shi Q.; Feng S.; Du D.; Lin Y. Single-atom catalysts for electrochemical water splitting. ACS Energy Letters 2018, 3, 1713–1721. 10.1021/acsenergylett.8b00640. [DOI] [Google Scholar]
- Ding S.; Hulsey M. J.; Perez-Ramirez J.; Yan N. Transforming energy with single-atom catalysts. Joule 2019, 3, 2897–2929. 10.1016/j.joule.2019.09.015. [DOI] [Google Scholar]
- Hansen J. N.; Prats H.; Toudahl K. K.; Secher N. M.; Chan K.; Kibsgaard J.; Chorkendorff I. Is There Anything Better than Pt for HER?. ACS Energy Letters 2021, 6, 1175–1180. 10.1021/acsenergylett.1c00246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalitis C. M.; Kramer D.; Sharman J.; Wright E.; Kucernak A. R. Pt Nano-Particle Performance for PEFC Reactions at Low Catalyst Loading and High Reactant Mass Transport. ECS Transactions 2013, 58, 39–47. 10.1149/05801.0039ecst. [DOI] [Google Scholar]
- Zhang L.; Doyle-Davis K.; Sun X. Pt-Based electrocatalysts with high atom utilization efficiency: from nanostructures to single atoms. Energy & Environmental Science 2019, 12, 492–517. 10.1039/C8EE02939C. [DOI] [Google Scholar]
- Pu Z.; Amiinu I. S.; Cheng R.; Wang P.; Zhang C.; Mu S.; Zhao W.; Su F.; Zhang G.; Liao S.; Sun S. Single-Atom Catalysts for Electrochemical Hydrogen Evolution Reaction: Recent Advances and Future Perspectives. Nano-Micro Letters 2020, 12, 21. 10.1007/s40820-019-0349-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng N.; Stambula S.; Wang D.; Banis M. N.; Liu J.; Riese A.; Xiao B.; Li R.; Sham T.-K.; Liu L.-M.; Botton G. A.; Sun X. Platinum single-atom and cluster catalysis of the hydrogen evolution reaction. Nat. Commun. 2016, 7, 13638. 10.1038/ncomms13638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.; Chen Y.; Zhou L.; Chen C.; Han Z.; Zhang B.; Wu Q.; Yang L.; Du L.; Bu Y.; Wang P.; Wang X.; Yang H.; Hu Z. The simplest construction of single-site catalysts by the synergism of micropore trapping and nitrogen anchoring. Nat. Commun. 2019, 10, 1657. 10.1038/s41467-019-09596-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D.; Li X.; Chen S.; Yan H.; Wang C.; Wu C.; Haleem Y. A.; Duan S.; Lu J.; Ge B.; Ajayan P. M.; Luo Y.; Jiang J.; Song L. Atomically dispersed platinum supported on curved carbon supports for efficient electrocatalytic hydrogen evolution. Nature Energy 2019, 4, 512–518. 10.1038/s41560-019-0402-6. [DOI] [Google Scholar]
- Yang Q.; Liu H.; Yuan P.; Jia Y.; Zhuang L.; Zhang H.; Yan X.; Liu G.; Zhao Y.; Liu J.; Wei S.; Song L.; Wu Q.; Ge B.; Zhang L.; Wang K.; Wang X.; Chang C.-R.; Yao X. Single Carbon Vacancy Traps Atomic Platinum for Hydrogen Evolution Catalysis. J. Am. Chem. Soc. 2022, 144, 2171–2178. 10.1021/jacs.1c10814. [DOI] [PubMed] [Google Scholar]
- Wang J.; Tan H. Y.; Kuo T. R.; Lin S. C.; Hsu C. S.; Zhu Y.; Chu Y. C.; Chen T. L.; Lee J. F.; Chen H. M. In Situ Identifying the Dynamic Structure behind Activity of Atomically Dispersed Platinum Catalyst toward Hydrogen Evolution Reaction. Small 2021, 17, 2005713. 10.1002/smll.202005713. [DOI] [PubMed] [Google Scholar]
- Ou H.; Wang D.; Li Y. How to select effective electrocatalysts: Nano or single atom?. Nano Select 2021, 2, 492–511. 10.1002/nano.202000239. [DOI] [Google Scholar]
- Li J.; Banis M. N.; Ren Z.; Adair K. R.; Doyle-Davis K.; Meira D. M.; Finfrock Y. Z.; Zhang L.; Kong F.; Sham T. K.; Li R.; Luo J.; Sun X. Unveiling the Nature of Pt Single-Atom Catalyst during Electrocatalytic Hydrogen Evolution and Oxygen Reduction Reactions. Small 2021, 17, 2007245. 10.1002/smll.202007245. [DOI] [PubMed] [Google Scholar]
- Fang S.; Zhu X.; Liu X.; Gu J.; Liu W.; Wang D.; Zhang W.; Lin Y.; Lu J.; Wei S.; Li Y.; Yao T. Uncovering near-free platinum single-atom dynamics during electrochemical hydrogen evolution reaction. Nat. Commun. 2020, 11, 1–8. 10.1038/s41467-020-14848-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Yang X.; Zhang J.; Huang Y.; Liu B. In Situ/Operando Techniques for Characterization of Single-Atom Catalysts. ACS Catalysis 2019, 9, 2521–2531. 10.1021/acscatal.8b04937. [DOI] [Google Scholar]
- Speck F. D.; Paul M. T.; Ruiz-Zepeda F.; Gatalo M.; Kim H.; Kwon H. C.; Mayrhofer K. J.; Choi M.; Choi C. H.; Hodnik N.; Cherevko S. Atomistic Insights into the Stability of Pt Single-Atom Electrocatalysts. J. Am. Chem. Soc. 2020, 142, 15496–15504. 10.1021/jacs.0c07138. [DOI] [PubMed] [Google Scholar]
- Fei H.; Dong J.; Feng Y.; Allen C. S.; Wan C.; Volosskiy B.; Li M.; Zhao Z.; Wang Y.; Sun H.; An P.; Chen W.; Guo Z.; Lee C.; Chen D.; Shakir I.; Liu M.; Hu T.; Li Y.; Kirkland A. I.; Duan X.; Huang Y. General synthesis and definitive structural identification of MN4C4 single-atom catalysts with tunable electrocatalytic activities. Nature Catalysis 2018, 1, 63–72. 10.1038/s41929-017-0008-y. [DOI] [Google Scholar]
- Xia C.; Qiu Y.; Xia Y.; Zhu P.; King G.; Zhang X.; Wu Z.; Kim J. Y. T.; Cullen D. A.; Zheng D.; Li P.; Shakouri M.; Heredia E.; Cui P.; Alshareef H. N.; Hu Y.; Wang H. General synthesis of single-atom catalysts with high metal loading using graphene quantum dots. Nature Chemistry 2021, 13, 887–894. 10.1038/s41557-021-00734-x. [DOI] [PubMed] [Google Scholar]
- Hai X.; Xi S.; Mitchell S.; Harrath K.; Xu H.; Akl D. F.; Kong D.; Li J.; Li Z.; Sun T.; Yang H.; Cui Y.; Su C.; Zhao X.; Li J.; Pérez-Ramírez J.; Lu J. Scalable two-step annealing method for preparing ultra-high-density single-atom catalyst libraries. Nature Nanotechnology 2022, 17, 174–181. 10.1038/s41565-021-01022-y. [DOI] [PubMed] [Google Scholar]
- Ju W.; Bagger A.; Hao G. P.; Varela A. S.; Sinev I.; Bon V.; Cuenya B. R.; Kaskel S.; Rossmeisl J.; Strasser P. Understanding activity and selectivity of metal-nitrogen-doped carbon catalysts for electrochemical reduction of CO2. Nature Communications 2017, 8, 1–9. 10.1038/s41467-017-01035-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y.; Lu B.; Chen S. Carbon-Supported Single Atom Catalysts for Electrochemical Energy Conversion and Storage. Adv. Mater. 2018, 30, 1801995. 10.1002/adma.201801995. [DOI] [PubMed] [Google Scholar]
- Zhao S.; Cheng Y.; Veder J.-P.; Johannessen B.; Saunders M.; Zhang L.; Liu C.; Chisholm M. F.; De Marco R.; Liu J.; Yang S.-Z.; Jiang S. P. One-Pot Pyrolysis Method to Fabricate Carbon Nanotube Supported Ni Single-Atom Catalysts with Ultrahigh Loading. ACS Applied Energy Materials 2018, 1, 5286–5297. 10.1021/acsaem.8b00903. [DOI] [Google Scholar]
- Coperet C. Single-sites and nanoparticles at tailored interfaces prepared via surface organometallic chemistry from thermolytic molecular precursors. Accounts of chemical research 2019, 52, 1697–1708. 10.1021/acs.accounts.9b00138. [DOI] [PubMed] [Google Scholar]
- Kaiser S. K.; Fako E.; Manzocchi G.; Krumeich F.; Hauert R.; Clark A. H.; Safonova O. V.; López N.; Pérez-Ramírez J. Nanostructuring unlocks high performance of platinum single-atom catalysts for stable vinyl chloride production. Nature Catalysis 2020, 3, 376–385. 10.1038/s41929-020-0431-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G.; Johnston C. M.; Mack N. H.; Artyushkova K.; Ferrandon M.; Nelson M.; Lezama-Pacheco J. S.; Conradson S. D.; More K. L.; Myers D. J.; Zelenay P. Synthesis–structure–performance correlation for polyaniline–Me–C non-precious metal cathode catalysts for oxygen reduction in fuel cells. J. Mater. Chem. 2011, 21, 11392–11405. 10.1039/c0jm03613g. [DOI] [Google Scholar]
- Chung H. T.; Cullen D. A.; Higgins D.; Sneed B. T.; Holby E. F.; More K. L.; Zelenay P. Direct atomic-level insight into the active sites of a high-performance PGM-free ORR catalyst. Science 2017, 357, 479–484. 10.1126/science.aan2255. [DOI] [PubMed] [Google Scholar]
- Djara R.; Holade Y.; Merzouki A.; Masquelez N.; Cot D.; Rebière B.; Petit E.; Huguet P.; Canaff C.; Morisset S.; Napporn T.; et al. Insights from the physicochemical and electrochemical screening of the potentiality of the chemically synthesized polyaniline. J. Electrochem. Soc. 2020, 167, 066503. 10.1149/1945-7111/ab7d40. [DOI] [Google Scholar]
- Ding S.; Guo Y.; Hülsey M. J.; Zhang B.; Asakura H.; Liu L.; Han Y.; Gao M.; Hasegawa J.-Y.; Qiao B.; Zhang T.; Yan N. Electrostatic Stabilization of Single-Atom Catalysts by Ionic Liquids. Chem 2019, 5, 3207–3219. 10.1016/j.chempr.2019.10.007. [DOI] [Google Scholar]
- Huang W.-Y.; Chang M.-Y.; Wang Y.-Z.; Huang Y.-C.; Ho K.-S.; Hsieh T.-H.; Kuo Y.-C. Polyaniline Based Pt-Electrocatalyst for a Proton Exchanged Membrane Fuel Cell. Polymers 2020, 12, 617. 10.3390/polym12030617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernooij R. R.; Joshi T.; Shaili E.; Kubeil M.; Appadoo D. R.; Izgorodina E. I.; Graham B.; Sadler P. J.; Wood B. R.; Spiccia L. Comprehensive vibrational spectroscopic investigation of trans, trans, trans-[Pt(N3)3(OH)2(py)2], a Pt (IV) diazido anticancer prodrug candidate. Inorganic chemistry 2016, 55, 5983–5992. 10.1021/acs.inorgchem.6b00476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaniyoor A.; Ramaprabhu S. A Raman spectroscopic investigation of graphite oxide derived graphene. AIP Advances 2012, 2, 032183. 10.1063/1.4756995. [DOI] [Google Scholar]
- Kim G.; Lee J.; Liu T.; Grey C. P. Characterizing Nitrogen Sites in Nitrogen-Doped Reduced Graphene Oxide: A Combined Solid-State 15N NMR, XPS, and DFT Approach. Journal of Physical Chemistry C 2021, 125, 10558–10564. 10.1021/acs.jpcc.1c02669. [DOI] [Google Scholar]
- Han L.; Cheng H.; Liu W.; Li H.; Ou P.; Lin R.; Wang H. T.; Pao C. W.; Head A. R.; Wang C. H.; Tong X.; Sun C. J.; Pong W. F.; Luo J.; Zheng J. C.; Xin H. L. A single-atom library for guided monometallic and concentration-complex multimetallic designs. Nat. Mater. 2022, 21, 681–688. 10.1038/s41563-022-01252-y. [DOI] [PubMed] [Google Scholar]
- Lin Y. C.; Teng P. Y.; Yeh C. H.; Koshino M.; Chiu P. W.; Suenaga K. Structural and Chemical Dynamics of Pyridinic-Nitrogen Defects in Graphene. Nano Letters 2015, 15, 7408–7413. 10.1021/acs.nanolett.5b02831. [DOI] [PubMed] [Google Scholar]
- Prats H.; Chan K. The determination of the HOR/HER reaction mechanism from experimental kinetic data. Phys. Chem. Chem. Phys. 2021, 23, 27150–27158. 10.1039/D1CP04134G. [DOI] [PubMed] [Google Scholar]
- Norskov J. K.; Bligaard T.; Logadottir A.; Kitchin J.; Chen J. G.; Pandelov S.; Stimming U. Trends in the exchange current for hydrogen evolution. Journal of The Electrochemical Society 2005, 152, J23. 10.1149/1.1856988. [DOI] [Google Scholar]
- Hossain M. D.; Liu Z.; Zhuang M.; Yan X.; Xu G.-L.; Gadre C. A.; Tyagi A.; Abidi I. H.; Sun C.-J.; Wong H.; Guda A.; Hao Y.; Pan X.; Amine K.; Luo Z. Rational Design of Graphene-Supported Single Atom Catalysts for Hydrogen Evolution Reaction. Advanced Energy Materials 2019, 9, 1803689. 10.1002/aenm.201803689. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all data supporting the findings of this study are included within the paper and its Supporting Information files. Source data are available from the corresponding author upon reasonable request.