

Abstract

The efficient transformation of hydroxyproline “doubly customizable units” into rigid hexahydropyrimidine units takes place in good global yields and generates compounds of pharmaceutical interest. In particular, the process can readily provide access to peptidomimetics and peptides with reversed sequences or with valuable turns.
Introduction
The recent introduction of hydroxyproline “doubly customizable units” allows the generation of a variety of modified amines and peptides in good yields and very few steps (Scheme 1, conversion 1 → 3).1,2 A first modification (conversion 1 → [4] → 2) is carried out using an oxidative radical decarboxylation, which generates acyliminium intermediate 4. This ion reacts with carbon nucleophiles to afford 2-alkyl pyrrolidines in good yield and excellent 2R or 2S purity due to the control provided by the stereogenic center at C-4.1a After deprotecting the 4-hydroxy group, compound 2 can undergo a second oxidative radical scission. The resulting products 5 have an N,O-acetal and an α-lateral chain with a terminal carbonyl group. Both chains can be functionalized independently,1,3 and thus, we have reported the formation of acyclic products 3 with three different, tailor-made substituents.1a
Scheme 1. Previous Results and Current Work on Doubly Customizable Units.

However, the possibility of using the newly formed chains to create other rigid, cyclic rings with new functionalities and conformational properties was very attractive. When the Nu group in compound 5 was an α- or β-ester (e.g., Nu = CO2Me or CH2CO2Me), treatment with a primary amine under reductive amination conditions provided an α- or β-amino lactam such as 6a.1b,3d However, the formation of a different heterocycle, using both the aldehyde and N,O-group, while Nu remained unaffected, had not been achieved. This transformation is not trivial, since the aldehyde group was the first to react, and the resulting linear chain extended away from the relatively bulky N-acetoxymethyl group. Therefore, non-cyclic products 3 were obtained.1,3 In this paper, we report a change in the reductive amination–cyclization conditions that allows the transformation of aldehydes 5 into hexahydropyrimidines 6b, which in addition to their pharmacological utility, can be valuable units to modulate a peptide secondary structure.
Hexahydropyrimidines are valuable components of different drugs4,5 and have been usually prepared from a diamine derivative and an aldehyde,4b,4c,5a,6 although cycloadditions from imines have also been reported.7 This methodology works well for the preparation of simple hexahydropyrimidines but offers little selectivity when several reactive amine or amide groups are present in the molecule, particularly in complex ones such as peptides. Unlike the classical approach, the present methodology uses a dicarbonyl compound (substrate 5) that reacts with an amine (as commented in Schemes 1 and 2). Since the carbonyl groups are selectively generated from the “customizable” residue before the reductive amination, the process allows a mild, selective introduction of the heterocycle in complex molecules.
Scheme 2. Use of In Situ-Formed Hexahydropyrimidines for Modulation of the Peptide Structure.

In addition to the synthesis of hexahydropyrimidines, we were interested in their potential to modulate a peptide secondary structure. As shown in Scheme 2, the precursor dicarbonyl compound 5 can react with one or two amino groups (placed on the same or different residues) to create peptides with reversed sequences (conversion I → II) or to generate turns (conversion III → IV).
In fact, linear α,α-diamines have been introduced by Chorev and Goodman8 and others9,10 to obtain partial retro- and retroinverso peptides with promising bioactivities and superior resistance to proteases.9,10 However, using cyclic α,α-diamines (II) such as hexahydropyrimidines introduces extra rigidity in the system, which is valuable for a better control of biological interactions.
With respect to the formation of peptide turns (IV), our in situ-formed nitrogen heterocycles would serve as “turn templates”. The literature reports other natural and synthetic turn templates (such as morpholino-, piperazine-, indolizidine-, bicyclic lactam-, and dibenzofuran-containing amino acids, among others),11−13 but unlike them, the hexahydropyrimidines would be formed during peptide ligation, allowing in situ turn generation at specific positions. Since the starting aldehydes 5 can have S or R chains, the conformation of the molecule can be modulated. In the present paper, we will discuss the preliminary results and feasibility of this versatile strategy.
Results and Discussion
The formation of the dicarbonyl compounds 5 from commercial or readily available l-hydroxyproline (Hyp) derivatives 1 is summarized in Scheme 3. The first transformation of the “doubly customizable” Hyp unit afforded 2-alkyl-4-hydroxypyrrolidines 2a–2d in good yields (Scheme 3) via a sequential oxidative radical decarboxylation–alkylation, according to our reported methodology.1a Similarly, the oxidative radical scission of compounds 2a–2d to give aldehydes 5a–5d as pure enantiomers also proceeded efficiently.
Scheme 3. Oxidative Radical Scission of Substrates 2a–2d to Give Pure Enantiomers 5a–5d.

Once the dicarbonyl substrates 5a–5c were available, their reductive amination was studied using alanine methyl ester as a model amine (Scheme 4). When the reaction was carried out with NaBH(OAc)3 in dichloroethane, the expected amination took place to give products 7–8. However, when the conditions were modified, and NaBH4 in methanol was introduced, the reductive amination was followed by an intramolecular cyclization. Under these conditions, the N-acetoxymethyl group was likely converted into an imino moiety, which underwent addition of the nearby amino group to give the hexahydropyrimidines 9–11.
Scheme 4. Reductive Amination of Scission Products 5a–5c.

Particularly interesting is the α,β- peptide 11, which presents not only the conformational constraints due to the dihydropyrimidine ring but also those induced by the α,α-dimethylated β-amino acid. In previous works, we observed that this moiety favors the formation of unusually expanded β-turns and γ- and δ-turns depending on its stereochemistry.14 Therefore, the following studies were devoted to the formation of hybrid peptides15 by reaction of substrate 5c and its isomer 5d with different amino acids and small peptides.
Thus, aldehyde 5c was reacted with derivatives of glycine, serine, and phenylalanine to give the hexahydropyrimidines 12–14 (Scheme 5), while the aldehyde isomer 5d was treated with glycine methyl ester to afford product 15. The formation of 12 and its epimer 15 took place in similar yield (65 and 62%, respectively), and all products 12–15 were easily purified. In these hexahydropyrimidine derivatives, two important structural changes are achieved: an increased system rigidity and also a reversal of the N → C direction, as will be commented later.
Scheme 5. Synthesis of Hexahydropyrimidines with Different Reversed Sequences.

The treatment of aldehydes 5c and 5d with peptides gave products with different flexibility (Scheme 6, compounds 16–23) depending on the reductive amination conditions. Thus, peptides where all the side chains were linear (products 16, 17, 20, and 21) were obtained with the less polar system (triacetoxyborohydride in DCE and Et3N). However, peptides with a rigid hexahydropyrimidine core (products 18, 19, 22, and 23) were obtained with sodium borohydride in methanol. These heterocyclic compounds were obtained in 50–68% yield and were easily purified; again, the stereochemistry of the starting aldehyde 5c or 5d did not influence the reaction yields.
Scheme 6. Synthesis of Hexahydropyrimidines in Peptides.

The different results obtained under both reaction conditions deserve some discussion. As commented in Introduction, we had only achieved cyclic products when compounds related to 5c and 5d were treated with a primary amine (such as benzylamine) under reductive amination conditions, giving amino lactams.1b However, when an α-substituted primary amine was used (such as α-methylbenzylamine), the cyclization did not take place due to steric hindrance.1b The amines derived from amino acids and peptides are also α-substituted primary amines and do not cyclize. Moreover, due to their superior bulkiness, they are likely away from the α,α-dimethyl ester chain and closer to the N-acetoxymethyl group. Under the more polar conditions, the cleavage of the acetoxymethyl group is favored with formation of an intermediate imine, which is intramolecularly trapped by the adjacent amine groups from the new amino acid or peptide chains, resulting in the hexahydropyrimidines. On the contrary, under the previous, less polar conditions, the acetoxy group remained intact and an acyclic compound was formed.
Interestingly, as happened with the α,α-diamines of partially modified retro- and retroinverso peptides,8−10 a reversal of the N → C direction was achieved (shown for compound 18, Figure 1). The 3D representation of an energy-minimized conformation for compound 18 (Figure 1) presents the reversed sequences, with the chains in different spatial orientations.16
Figure 1.
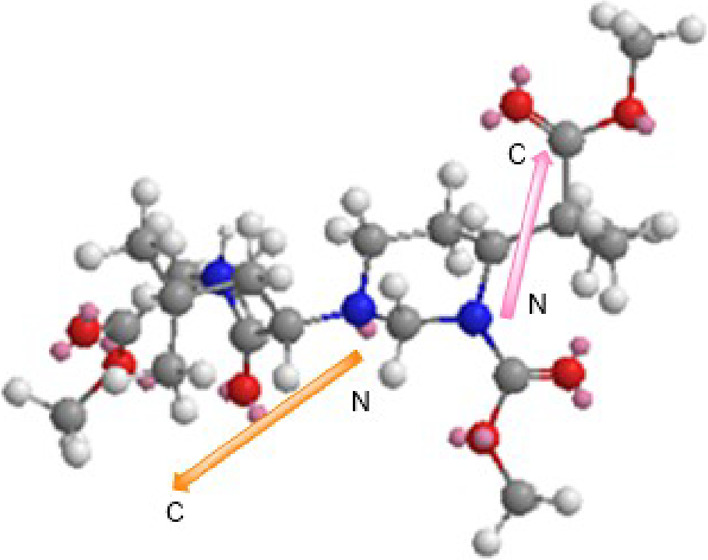
3D representation of the minimized conformation for compound 18, showing reversed sequences.
Finally, the reaction of 5c with tripeptide 25 under the reductive amination conditions was studied (Scheme 7). In this case, two products were obtained in 66% global yield, the valuable bicyclic hexahydropyrimidine 26 (36%), together with imidazolidine 27 (30%). The formation of compound 27 suggests that the formation of the imidazolidine ring takes place first, instead of the expected reductive amination. Then, the N,O-acetal is cleaved to an imine, followed by either addition of the ornithine α-amino group (to give 26)17 or addition of the solvent (or sodium methoxide formed in situ) to give the N-methoxymethyl derivative 27.18
Scheme 7. Turn-Inducing Hexahydropyrimidine 26.

The stereochemistry of compound 26 was determined with NOESY experiments (Figure 2) and by comparison of experimental JH,H with theoretical ones for both isomers.19 The 3D representation of the energy-minimized structure whose JH,H matched the experimental ones (Figure 2) shows both the β-amino acid and the α-peptide backbones in close proximity due to the system rigidity. Therefore, the bicyclic unit formed during peptide ligation could be effective for creating turns.
Figure 2.

3D representation of the minimized conformation for compound 26 with NOESY correlations shown by arrows (strong interactions in orange and weak interactions in green). The rigid bicyclic unit is formed during peptide ligation and forces the peptide backbone to form a turn as shown.
The stereochemistry of the monocyclic compound 27 could not be determined with NOESY experiments. No correlations were observed between the aminal proton and CHNOrn, but this result is not conclusive for opposite spatial orientation. Future work with other models is planned to solve this point. In any case, the possibility of obtaining systems with different rigidity levels is quite interesting for structural and biomedical studies. We are currently optimizing the cyclization conditions, using less nucleophilic solvents, and studying the reaction in larger peptide units, where new interactions and hydrogen bonds are expected. However, the current protocol already represents a promising tool to create conformationally constrained peptides and generate turn templates in situ, thus modulating the bioactivity of the peptides.
Conclusions
The efficient conversion of hydroxyproline “doubly customizable units” into functionalized hexahydropyrimidines, heterocycles found in compounds of pharmaceutical interest, is presented herein. This work also introduces their use to provide access to peptidomimetics and peptides with reversed sequences or with valuable turns.
The hydroxyproline unit can be cleaved at two points, and a one-pot decarboxylation–alkylation process provided 2-substituted 4-hydroxypyrrolidines, which were used as substrates for the scission of the pyrrolidine ring at C4-C5. This strategy was developed in our preliminary paper to give compounds with three new linear chains or amino lactams, but in this work, the conditions were modified to attach the C-α and N1-substituents so that a hexahydropyrimidine ring was formed. This transformation was not trivial, since under most conditions, the newly formed chains extended away from each other, hindering the formation of cyclic compounds. Under the current new conditions, we achieved the generation of the rigid hexahydropyrimidine core, which proved valuable to create peptides with reversed sequence directions.
In addition, a further extension of this methodology created a hexahydropyrimidine-containing bicycle, which could be used as a “turn template” in peptides. Since the template was formed in situ during peptide ligation, this methodology would allow an easy access to peptides containing turns at selected positions while clipping two peptide chains.
Experimental Section
General Methods
Commercially available reagents and solvents were of analytical grade or were purified by standard procedures prior to use. All reactions involving air- or moisture-sensitive materials were carried out under a nitrogen atmosphere. Melting points were determined with a hot-stage apparatus and were uncorrected. Optical rotations were measured at the sodium line at ambient temperature (26 °C) in CHCl3 solutions. The NMR spectra were determined at 500 or 400 MHz for 1H and 125.7 or 100.6 MHz for 13C, at 26 °C or 70 °C, as stated for each case. Sometimes, due to slower rotamer interconversion at 26 °C, two (or more) sets of signals are visible at room temperature, while only one set of signals (rotamer average) is seen at 70 °C due to faster rotamer interconversion. For some compounds, the 1H NMR spectra show some signals as broad bands (br b) due to equilibria between rotamers.
Structural assignments were made with additional information from 2D experiments, such as COSY, HSQC, NOESY, and/or HMBC experiments.
The oxidative radical scissions were carried out at 26 °C under irradiation with a commercial (DIY shops) cool white LED light (14 W, 1100 lumens, 400–750 nm, with peaks at 432 nm/blue and 556 nm/green), placed 20 cm away from the standard (borosilicate) reaction flask. The LED light can be replaced by a commercial 80 W tungsten filament lamp, which provides light in a continuous spectrum of 340–1400 nm, which is perceived as white light, and which offers similar results.1a In both cases, the reaction should be carried out until the disappearance of the starting material.
1H NMR spectra are reported as follows (s = singlet, d = doublet, t = triplet, dd = doublet of doublets, ddd = doublet of doublet of doublets, q = quartet, m = multiplet, br = broad, br b = broad band, and br s = broad singlet; coupling constant(s) in Hz). The mass spectra were carried out using electrospray ionization techniques (ESI) or electronic impact (EI); the latter was determined at 70 eV using an ion trap mass analyzer. Merck silica gel 60 PF254 and 60 (0.063–0.2 mm) were used for preparative thin layer chromatography and column chromatography, respectively. The reagent for TLC analysis was KMnO4 in NaOH/K2CO3 aqueous solution, and TLC was heated until the development of color.
General Procedure for the Scission of the Pyrrolidine C4–C5 Bond
To a solution of the 2-alkyl-2-hydroxypyrrolidine (0.2 mmol) in dry dichloromethane (4 mL) were added iodine (25 mg, 0.1 mmol) and DIB (129 mg, 0.4 mmol). The resulting mixture was stirred for 3 h at 26 °C under irradiation with visible light (cool white LED lamp). Then, the reaction mixture was poured into 10% aqueous Na2S2O3 (10 mL) and extracted with CH2Cl2. The organic layer was dried over sodium sulfate, filtered, and concentrated under vacuum. The residue was purified by chromatography on silica gel (hexanes/ethyl acetate) to give the scission products 5a–5d.
N-(Acetoxymethyl)-N-(methoxycarbonyl)-(3S)-aminohexanal (5a)
Obtained from 2-propyl-4-hydroxypyrrolidine 2a (37 mg, 0.2 mmol) according to the general pyrrolidine scission procedure. After work-up and solvent evaporation, the residue was purified by radial chromatography (hexanes/EtOAc, 80:20), yielding aldehyde 5a (30.5 mg, 63%) as a colorless viscous oil, whose characterization data were already reported.1a
(3R)-[N-(Acetoxymethyl)-N-(methoxycarbonyl)amino]-5-(oxo)-5-(phenyl)pentanal (5b)
Obtained from compound 2b (53 mg, 0.2 mmol) according to the general pyrrolidine scission procedure. After work-up and solvent evaporation, the residue was purified by radial chromatography (hexanes/EtOAc, 80:20), yielding aldehyde 5b (48 mg, 74%) as a colorless viscous oil, whose characterization data were already reported.1a
Methyl (3R)-[N-(Acetoxymethyl)-N-(methoxycarbonyl)amino]-2,2-dimethyl-5-oxopentanoate (5c)
Obtained from compound 2c (49 mg, 0.2 mmol) according to the general pyrrolidine scission procedure. After work-up and solvent evaporation, the residue was purified by radial chromatography (hexanes/EtOAc, 80:20), yielding aldehyde 5c (39 mg, 65%) as a colorless viscous oil, whose characterization data were already reported.1a
Methyl (3S)-[N-(Acetoxymethyl)-N-(methoxycarbonyl)amino]-2,2-dimethyl-5-oxopentanoate (5d)
Obtained from compound 2d (49 mg, 0.2 mmol) according to the general pyrrolidine scission procedure. After work-up and solvent evaporation, the residue was purified by radial chromatography (hexanes/EtOAc, 70:30), yielding the aldehyde 5d (38 mg, 63%) as a colorless viscous oil, whose characterization data were already reported.1a
General Reductive Amination Procedure to Prepare Acyclic Compounds Such as Products 7 and 8
A solution of aldehyde 5 (0.15 mmol) in dry dichloroethane (3 mL) was treated with the corresponding amine (0.21 mmol) and Et3N (28 μL, 20 mg, 0.2 mmol). The resulting mixture was stirred for 30 min, and then NaBH(OAc)3 (51 mg, 0.24 mmol) was added. The stirring continued for 4 h, and then the mixture was poured into saturated aqueous NaHCO3 and extracted with CH2Cl2. The organic layer was dried over sodium sulfate, filtered, and concentrated under vacuum. The residue was purified by chromatography on silica gel (hexanes/ethyl acetate) to give the reductive amination products, such as compounds 7 or 8.
N-[(3S)-[N-(Acetoxymethyl)-N-(methoxycarbonyl)amino]hexyl]-L-alanine Methyl Ester (7)
Obtained from aldehyde 5a (37 mg, 0.15 mmol) according to the general procedures for the reductive amination using H-Ala-OMe hydrochloride as a reagent (29.3 mg, 0.21 mmol). After work-up and solvent evaporation, the residue was purified by rotatory chromatography (hexanes/EtOAc, 40:60), yielding diamine 7 (30.1 mg, 60%) as a syrup. [α]D: +11 (c 0.07, CHCl3). IR (CHCl3) νmax 3478, 1697, 1685, 1521, 1215, 1045, 928 cm–1. 1H NMR (400 MHz, 26 °C, CDCl3) rotamer mixture δH 5.40–5.20 (2H, m), 4.13/4.02 (1H/1H, br s/br s), 3.73 (3H, s), 3.71 (3H, s), 3.35–3.25 (2H, m), 2.60–2.55 (1H, m), 2.50–2.40 (1H, m), 2.05 (3H, s), 1.70–1.62 (3H, m), 1.50–1.37 (1H, m), 1.33–1.20 (2H, m), 1.27 (3H, d, J = 6.8 Hz), 0.88 (3H, t, J = 7.2 Hz). 13C{1H} NMR (125.7 MHz, 26 °C, CDCl3) rotamer mixture δc 176.1 (C), 170.6 (C, CO), 156.6 (C), 69.2 (CH2), 56.7 (CH), 54.8/54.6 (CH), 53.1/52.7 (CH3), 51.8/51.6 (CH3), 44.9/44.7 (CH2), 36.0/35.6 (CH2), 34.5/34.2 (CH2), 21.0 (CH3), 19.6/19.4 (CH2), 19.2/19.0 (CH3), 13.89/13.84 (CH3). HRMS (ESI-TOF) calcd for C15H28N2O6Na [M + Na]+, 355.1845; found, 355.1858. Anal. calcd for C15H28N2O6: C, 54.20; H, 8.49; N, 8.43. Found: C, 54.37; H, 8.31; N, 8.62.
Methyl N-[N-(Acetoxymethyl)-N-(methoxycarbonyl)-2,2-dimethyl-1-methyl-D-β-norvalin-5-yl]-L-alanine (8)
Obtained from aldehyde 5c (45.5 mg, 0.15 mmol) according to the general procedures for the reductive amination using H-Ala-OMe hydrochloride as a reagent (29.3 mg, 0.21 mmol). After work-up and solvent evaporation, the residue was purified by rotatory chromatography (hexanes/EtOAc, 40:60), yielding diamine 8 (39.2 mg, 67%) as a syrup. [α]D: +8 (c 0.39, CHCl3). IR (CHCl3) νmax 3438, 1727, 1699, 1213, 1047 cm–1. 1H NMR (500 MHz, 26 °C, CDCl3) rotamer mixture δH 5.43–5.23 (2H, m), 4.60–4.40 (1H, m), 3.71 (3H, s), 3.68 (3H, s), 3.64 (3H, s), 3.32–3.23 (1H, m), 2.67–2.58 (1H, m), 2.43–2.30 (1H, m), 2.04 (3H, s), 1.90–1.80 (1H, m), 1.76–1.62 (1H, br b), 1.25 (3H, d, J = 6.5 Hz), 1.20 (3H, s), 1.16 (3H, s). 13C{1H} NMR (125.7 MHz, 26 °C, CDCl3) rotamer mixture δc 176.5 (C), 175.9 (C), 170.4 (C), 157.5/157.3 (C), 69.5/69.1 (CH2), 59.9/59.6 (CH), 56.6 (CH), 53.4 (CH3), 52.0 (CH3), 51.8 (CH3), 47.1 (C), 45.2/44.9 (CH2), 29.3/29.1 (CH2), 24.2 (CH3), 22.1/21.7 (CH3), 21.0 (CH3), 19.0 (CH3). HRMS (ESI-TOF) calcd for C17H30N2O8Na [M + Na]+, 413.1900; found, 413.1904. Anal. calcd for C17H30N2O8: C, 52.30; H, 7.75; N, 7.18. Found: C, 52.38; H, 7.86; N, 7.13.
General Procedure for Reductive Amination–Cyclization of Aldehydes 5a–5d to Prepare Hexahydropyrimidines
A solution of the aldehyde (0.15 mmol) in dry methanol (3 mL) was treated with the corresponding amine (1.4 equiv, 0.21 mmol) and Et3N (28 μL, 20 mg, 0.20 mmol). The resulting mixture was stirred for 1 h, and then NaBH4 (7.4 mg, 0.195 mmol) was added. The stirring continued for 72 h, and then the reaction mixture was poured into saturated aqueous NaHCO3 and extracted with CH2Cl2. The organic layer was dried over sodium sulfate, filtered, and evaporated under vacuum. The residue was purified by chromatography on silica gel (hexanes/ethyl acetate mixtures) to give the hexahydropyrimidines.
Methyl 2S-(1-Methoxycarbonyl-6S-(propyl)hexahydropyrimidin-3-yl)propanoate (9)
Obtained from aldehyde 5a (37 mg, 0.15 mmol) according to the general procedures for the reductive amination–cyclization using H-Ala-OMe hydrochloride as a reagent (29.3 mg, 0.21 mmol). After work-up and solvent evaporation, the residue was purified by rotatory chromatography (hexanes/EtOAc, 50:50), yielding compound 9 (24.8 mg, 61%) as a syrup. [α]D: +7 (c 0.29, CHCl3). IR (CHCl3) νmax 1715, 1683, 1450, 1296, 1029 cm–1. 1H NMR (400 MHz, CDCl3, 26 °C) rotamer mixture δH 4.75–4.67 (1H, m), 4.30–4.20 (1H, m), 3.78–3.67 (1H, m), 3.72 (3H, s), 3.68 (3H, s), 3.47 (1H, dt, J = 6.8, 7.2 Hz), 2.89–2.82 (1H, m), 2.76 (1H, dd, J = 12.4, 11.2 Hz), 1.97–1.86 (1H, m), 1.75–1.65 (1H, m), 1.46–1.36 (2H, m), 1.32 (3H, d, J = 7.2 Hz), 1.31–1.22 (2H, m), 0.92 (3H, t, J = 7.4 Hz). 13C{1H} NMR (100.6 MHz, CDCl3, 26 °C) rotamer mixture δc 173.5 (C), 155.9 (C), 59.0 (CH2 + CH), 52.5 (CH3), 51.6 (CH3), 49.6 (CH), 42.0 (CH2), 31.9 (CH2), 27.0 (CH2), 19.2 (CH2), 15.6 (CH3), 13.9 (CH3). HRMS (ESI-TOF) calcd for C13H24N2O4Na [M + Na]+, 295.1634; found, 295.1622. Anal. calcd for C13H24N2O4: C, 57.33; H, 8.88; N, 10.29. Found: C, 57.68; H, 8.49; N, 10.08.
Methyl 2S-(1-Methoxycarbonyl-6R-(2-oxo-2-phenylethyl)hexahydropyrimidin-3-yl)propanoate (10)
Obtained from aldehyde 5b (48.2 mg, 0.15 mmol) according to the general procedures for the reductive amination–cyclization using H-Ala-OMe hydrochloride as a reagent (29.3 mg, 0.21 mmol). After work-up and solvent evaporation, the residue was purified by rotatory chromatography (hexanes/EtOAc, 50:50), yielding compound 10 (31.2 mg, 60%) as a syrup. [α]D: −9 (c 0.15, CHCl3). IR (CHCl3) νmax 1733, 1697, 1449, 1230, 1045 cm–1. 1H NMR (500 MHz, CD3CN, 70 °C) δH 7.97 (2H, d, J = 7.1 Hz), 7.62 (1H, dd, J = 7.5, 7.3 Hz), 7.51 (2H, dd, J = 7.9, 7.5 Hz), 4.79–4.74 (1H, m), 4.67 (1H, d, J = 11.7 Hz), 3.93 (1H, d, J = 11.9 Hz), 3.69 (3H, s), 3.56 (3H, s), 3.47 (1H, q, J = 7.1 Hz), 3.38 (1H, dd, J = 15.8, 8.0 Hz), 3.24 (1H, dd, J = 15.7, 6.3 Hz), 2.89–2.84 (1H, m), 2.80 (1H, ddd, J = 12.5, 12.5, 3.0 Hz), 1.98–1.90 (1H, m), 1.55 (1H, dq, J = 13.7, 2.8 Hz), 1.27 (3H, d, J = 7.0 Hz). 13C{1H} NMR (125.7 MHz, CD3CN, 70 °C) δc 199.6 (C), 174.5 (C), 156.7 (C), 138.6 (C), 134.3 (CH), 129.9 (2 × CH), 129.3 (2 × CH), 60.7 (CH), 60.5 (CH2), 53.2 (CH3), 52.2 (CH3), 48.8 (CH), 43.5 (CH2), 40.6 (CH2), 28.4 (CH2), 16.0 (CH3). HRMS (ESI-TOF) calcd for C18H24N2O5Na [M + Na]+, 371.1583; found, 371.1585. Anal. calcd for C18H24N2O5: C, 62.05; H, 6.94; N, 8.04. Found: C, 62.17; H, 6.65; N, 7.85.
Methyl 2S-(1-Methoxycarbonyl-6R-(1-methoxy-2-methyl-1-oxopropan-2-yl)hexahydropyrimidin-3-yl)propanoate (11)
Obtained from aldehyde 5c (49.5 mg, 0.15 mmol) according to the general procedures for the reductive amination–cyclization using H-Ala-OMe hydrochloride as a reagent (29.3 mg, 0.21 mmol). After work-up and solvent evaporation, the residue was purified by rotatory chromatography (hexanes/EtOAc, 50:50), yielding compound 11 (32.1 mg, 65%) as a syrup. [α]D: +16 (c 0.37, CHCl3). IR (CHCl3) νmax 3438, 1723, 1698, 1449, 1218, 1047 cm–1. 1H NMR (500 MHz, CD3CN, 70 °C) δH 4.68 (1H, d, J = 12.6 Hz), 4.26 (1H, t, J = 6.6 Hz), 3.93 (1H, d, J = 12.6 Hz), 3.67 (3H, s), 3.65 (3H, s), 3.64 (3H, s), 3.40 (1H, q, J = 7.1 Hz), 2.79 (1H, ddd, J = 12.0, 8.0, 4.4 Hz), 2.70–2.64 (1H, m), 1.88–1.83 (2H, m), 1.23 (1H, d, J = 7.0 Hz), 1.20 (3H, s), 1.19 (3H, s). 13C{1H} NMR (125.7 MHz, CD3CN, 25 °C) δc 178.0 (C), 174.8 (C), 158.5 (C), 61.1 (CH), 60.9 (CH2), 58.1 (CH), 53.4 (CH3), 52.7 (CH3), 52.2 (CH3), 48.3 (C), 44.7 (CH2), 24.6 (CH3), 24.3 (CH2), 23.2 (CH3), 16.3 (CH3). HRMS (ESI-TOF) calcd for C15H26N2O6Na [M + Na]+, 353.1689; found, 353.1689. Anal. calcd for C15H26N2O6: C, 54.53; H, 7.93; N, 8.48. Found: C, 54.72; H, 7.76; N, 8.57.
Methyl 2-(1-Methoxycarbonyl-6R-(1-methoxy-2-methyl-1-oxopropan-2-yl)hexahydropyrimidin-3-yl)acetate (12)
Obtained from aldehyde 5c (49.5 mg, 0.15 mmol) according to the general procedures for the reductive amination–cyclization using H-Gly-OMe hydrochloride as a reagent (26.4 mg, 0.21 mmol). After work-up and solvent evaporation, the residue was purified by rotatory chromatography (hexanes/EtOAc, 50:50), yielding compound 12 (30.8 mg, 65%) as a syrup. [α]D: +39 (c 0.17, CHCl3). IR (CHCl3) νmax 1733, 1697, 1449, 1230, 1045 cm–1. 1H NMR (500 MHz, CD3CN, 70 °C) δH 4.71 (1H, d, J = 13.0 Hz), 4.29 (1H, t, J = 6.5 Hz), 3.96 (1H, d, J = 13.0 Hz), 3.66 (3H, s), 3.65 (3H, s), 3.64 (3H, s), 3.36 (1H, d, J = 16.7 Hz), 3.25 (1H, d, J = 16.7 Hz), 2.92 (1H, ddd, J = 12.0, 9.5, 4.0 Hz), 2.57 (1H, ddd, J = 11.9, 7.0, 4.5 Hz), 1.97–1.89 (1H, m), 1.80–1.72 (1H, m), 1.21 (3H, s), 1.20 (3H, s). 13C{1H}NMR (125.7 MHz, CD3CN, 25 °C) δc 178.2 (C), 172.3 (C), 158.8 (C), 62.2 (CH2), 58.0 (CH), 56.3 (CH2), 53.8 (CH3), 53.0 (CH3), 52.6 (CH3), 49.1 (CH2), 48.4 (C), 24.6 (CH3), 23.2 (CH2), 23.1 (CH3). HRMS (ESI-TOF) calcd for C14H24N2O6Na [M + Na]+, 339.1532; found, 339.1529. Anal. calcd for C14H24N2O6: C, 53.15; H, 7.65; N, 8.86. Found: C, 52.89; H, 7.80; N, 8.56.
Methyl 3-Benzyloxy-2S-(1-methoxycarbonyl-6R-(1-methoxy-2-methyl-1-oxopropan-2-yl)hexahydropyrimidin-3-yl)propanoate (13)
Obtained from aldehyde 5c (49.5 mg, 0.15 mmol) according to the general procedures for the reductive amination–cyclization using H-Ser(OBn)-OMe hydrochloride as a reagent (49.0 mg, 0.21 mmol). After work-up and solvent evaporation, the residue was purified by rotatory chromatography (hexanes/EtOAc, 50:50), yielding compound 13 (32.5 mg, 50%) as a syrup. [α]D: +8 (c 0.64, CHCl3). IR (CHCl3) νmax 1723, 1517, 1421, 1218, 1054 cm–1. 1H NMR (500 MHz, CD3CN, 25 °C) rotamer mixture δH 7.40–7.33 (5H, m), 5.14 (2H, s), 4.66 (1H, d, J = 11.2 Hz), 4.23 (1H, t, J = 5.9 Hz), 4.00 (1H, br dd, J = 10.3, 2.0 Hz), 3.73 (1H, dd, J = 11.3, 5.8 Hz), 3.68–3.64 (1H, m), 3.60 (9H, s), 3.43 (1H, t, J = 6.3 Hz), 2.80–2.75 (1H, m), 2.73–2.67 (1H, m), 1.87–1.76 (2H, m), 1.19/1.14 (3H/3H, s/s), 1.16/1.12 (3H/3H, s/s). 13C{1H} NMR (125.7 MHz, CD3CN, 25 °C) rotamer mixture δc 178.0 (C), 172.2 (C), 158.6 (C), 137.7 (C), 129.9 (2 × CH), 129.5/129.4 (2 × CH), 129.3 (CH), 68.2/67.8 (CH), 67.6/67.4 (CH2), 61.6 (CH2), 61.3 (CH2), 58.1 (CH), 53.6/53.5 (CH3), 52.8/52.7 (CH3), 52.7/52.6 (CH3), 48.4 (C), 45.0 (CH2), 24.65/24.57 (CH2), 23.5/23.3 (CH3), 22.4 (CH3). HRMS (ESI-TOF) calcd for C21H29N2O7 [M + 2H – Me]+, 423.2158; found, 423.2123. Anal. calcd for C22H32N2O7: C, 60.54; H, 7.39; N, 6.42. Found: C, 60.77; H, 7.16; N, 6.30.
Methyl 2S-(1-Methoxycarbonyl-6R-(1-methoxy-2-methyl-1-oxopropan-2-yl)hexahydropyrimidin-3-yl)-3-phenylpropanoate (14)
Obtained from aldehyde 5c (49.5 mg, 0.15 mmol) according to the general procedures for the reductive amination–cyclization using H-Phe-OBn hydrochloride as a reagent (61.3 mg, 0.21 mmol). After work-up and solvent evaporation, the residue was purified by rotatory chromatography (hexanes/EtOAc, 50:50), yielding compound 14 (38.3 mg, 53%) as a syrup. [α]D: +5 (c 0.72, CHCl3). IR (CHCl3) νmax 1723, 1702, 1449, 1221, 1047 cm–1. 1H NMR (500 MHz, CD3CN, 70 °C) δH 7.37–7.18 (10H, m), 5.06 (2H, s), 4.70 (1H, d, J = 12.1 Hz), 4.26 (1H, t, J = 6.6 Hz), 3.95 (1H, d, J = 12.2 Hz), 3.68–3.62 (1H, m), 3.62 (3H, s), 3.59 (3H, s), 3.03 (1H, dd, J = 13.8, 8.2 Hz), 2.94 (1H, dd, J = 13.9, 6.9 Hz), 2.86–2.82 (1H, m), 1.83 (2H, q, J = 6.2 Hz), 1.17 (3H, s), 1.15 (3H, s). 13C{1H} NMR (125.7 MHz, CD3CN, 70 °C) δc 178.0 (C), 172.8 (C), 158.5 (C), 139.6 (C), 137.6 (C), 130.5 (2 × CH), 129.8 (2 × CH), 129.6 (2 × CH), 129.5 (2 × CH), 129.4 (CH), 127.6 (CH), 68.0 (CH), 67.3 (CH2), 61.7 (CH2), 57.9 (CH), 53.5 (CH3), 52.7 (CH3), 48.4 (C), 44.8 (CH2), 36.9 (CH2), 24.8 (CH2), 24.6 (CH3), 23.3 (CH3). HRMS (ESI-TOF) calcd for C27H34N2O6Na [M + Na]+, 505.2315; found, 505.2315. Anal. calcd for C27H34N2O6: C, 67.20; H, 7.10; N, 5.81. Found: C, 67.04; H, 7.47; N, 5.83.
Methyl 2-(1-Methoxycarbonyl-6S-(1-methoxy-2-methyl-1-oxopropan-2-yl)hexahydropyrimidin-3-yl)acetate (15)
Obtained from aldehyde 5d (49.5 mg, 0.15 mmol) according to the general procedures for the reductive amination–cyclization using H-Gly-OMe hydrochloride as a reagent (26.4 mg, 0.21 mmol). After work-up and solvent evaporation, the residue was purified by rotatory chromatography (hexanes/EtOAc, 50:50), yielding tetrahydropyrimidine 15 (29.0 mg, 62%) as a syrup. [α]D: −41 (c 0.15, CHCl3). IR (CHCl3) νmax 1733, 1697, 1449, 1230, 1045 cm–1. 1H NMR (500 MHz, CD3CN, 70 °C) δH 4.71 (1H, d, J = 13.0 Hz), 4.29 (1H, dd, J = 7.0, 6.0 Hz), 3.96 (1H, d, J = 13.0 Hz), 3.66 (3H, s), 3.65 (3H, s), 3.64 (3H, s), 3.36 (1H, d, J = 16.6 Hz), 3.25 (1H, d, J = 16.6 Hz), 2.92 (1H, ddd, J = 12.5, 9.4, 3.6 Hz), 2.57 (1H, ddd, J = 11.5, 6.8, 4.5 Hz), 1.97–1.89 (1H, m), 1.80–1.74 (1H, m), 1.21 (3H, s), 1.20 (3H, s). 13C{1H} NMR (125.7 MHz, CD3CN, 25 °C) δc 178.2 (C), 172.3 (C), 158.8 (C), 62.2 (CH2), 58.0 (CH), 56.3 (CH2), 53.8 (CH3), 53.0 (CH3), 52.6 (CH3), 49.1 (CH2), 48.4 (C), 24.6 (CH3), 23.2 (CH2), 23.1 (CH3). HRMS (ESI-TOF) calcd for C14H24N2O6Na [M + Na]+, 339.1532; found, 339.1530. Anal. calcd for C14H24N2O6: C, 53.15; H, 7.65; N, 8.86. Found: C, 53.40; H, 7.37; N, 8.74.
Methyl N-[N-(Acetoxymethyl)-N-(methoxycarbonyl)-2,2-dimethyl-1-methyl-D-β-norvalin-5-yl]-L-leucinyl-L-alaninate (16)
Obtained from aldehyde 5c (45.5 mg, 0.15 mmol) according to the general procedure for the reductive amination using H-Leu-Ala-OMe hydrochloride (53 mg, 0.21 mmol) as a reagent. After work-up and solvent evaporation, the residue was purified by rotatory chromatography (hexanes/EtOAc, 40:60), yielding diamine 16 (60.9 mg, 81%) as a syrup. [α]D: +14 (c 0.22, CHCl3). IR (CHCl3) νmax 3433, 1723, 1523, 1226, 1046 cm–1. 1H NMR (500 MHz, 70 °C, CD3CN) δH 7.55–7.40 (1H, br b), 5.38 (1H, d, J = 11.0 Hz), 5.23 (1H, d, J = 11.5 Hz), 4.50–4.37 (2H, m), 3.70 (3H, s), 3.69 (3H, s), 3.65 (3H, s), 2.99 (1H, dd, J = 8.5, 5.5 Hz), 2.60–2.47 (2H, m), 1.99 (3H, s), 1.90–1.79 (1H, m), 1.75–1.68 (2H, m), 1.47–1.41 (1H, m), 1.39–1.34 (1H, m), 1.37 (3H, d, J = 6.9 Hz), 1.22 (3H, s), 1.19 (3H, s), 0.94 (3H, d, J = 6.5 Hz), 0.91 (3H, d, J = 6.5 Hz). 13C{1H} NMR (100.6 MHz, 26 °C, CDCl3) rotamer mixture δc 177.6 (C), 174.8/174.5 (C/C), 173.5 (C), 170.2 (C), 157.6/157.3 (C/C), 69.4/69.2 (CH2), 61.7 (CH), 59.4/59.2 (CH), 53.5 (CH3), 52.3 (CH3), 52.0 (CH3), 47.2 (CH), 46.9 (C), 45.9/45.6 (CH2), 42.9 (CH2), 29.3/28.7 (CH), 24.9 (CH2), 24.1 (CH3), 23.3 (CH3), 22.1 (CH3), 21.7 (CH3), 20.9 (CH3), 18.3 (CH3). HRMS (ESI-TOF) calcd for C23H41N3O9Na [M + Na]+, 526.2740; found, 526.2743. Anal. calcd for C23H41N3O9: C, 54.86; H, 8.21; N, 8.34. Found: C, 55.17; H, 8.22; N, 8.46.
Methyl N-[N-(Acetoxymethyl)-N-(methoxycarbonyl)-2,2-dimethyl-1-methyl-D-β-norvalin-5-yl]-L-leucinyl-L-leucinate (17)
Obtained from aldehyde 5c (45.5 mg, 0.15 mmol) according to the general procedure for the reductive amination using H-Leu-Leu-OMe hydrochloride (61.5 mg, 0.21 mmol) as a reagent. After work-up and solvent evaporation, the residue was purified by rotatory chromatography (hexanes/EtOAc, 40:60), yielding diamine 17 (70.8 mg, 87%) as a syrup. [α]D: +17 (c 0.17, CHCl3). IR (CHCl3) νmax 3488, 1733, 1719, 1702, 1217, 1050 cm–1. 1H NMR (400 MHz, 70 °C, CD3CN) δH 7.50–7.32 (1H, br b), 5.41 (1H, d, J = 11.2 Hz), 5.28 (1H, d, J = 11.2 Hz), 4.52–4.45 (2H, m), 3.73 (3H, s), 3.71 (3H, s), 3.68 (3H, s), 3.03 (1H, dd, J = 8.2, 5.8 Hz), 2.63–2.50 (2H, m), 2.01 (3H, s), 1.90–1.80 (1H, m), 1.79–1.68 (3H, m), 1.69–1.64 (2H, m), 1.51–1.35 (2H, m), 1.25 (3H, s), 1.22 (3H, s), 0.98 (3H, d, J = 6.0 Hz), 0.97 (3H, d, J = 6.8 Hz), 0.95 (3H, d, J = 6.4 Hz), 0.94 (3H, d, J = 6.0 Hz). 13C{1H} NMR (100.6 MHz, 70 °C, CD3CN) δc 177.8 (C), 176.1 (C), 174.6 (C), 171.2 (C), 158.7 (C), 71.2 (CH2), 63.1 (CH), 61.8 (CH), 54.0 (CH3), 52.8 (CH3), 52.7 (CH3), 51.6 (CH), 48.4 (C), 47.1 (CH2), 44.5 (CH2), 42.1 (CH2), 30.2 (CH2), 26.12 (CH), 26.06 (CH), 24.7 (CH3), 23.7 (CH3), 23.5 (CH3), 23.0 (CH3), 22.7 (CH3), 22.3 (CH3), 21.4 (CH3). HRMS (ESI-TOF) calcd for C26H47N3O9Na [M + Na]+, 568.3210; found, 568.3209. Anal. calcd for C26H47N3O9: C, 57.23; H, 8.68; N, 7.70. Found: C, 57.29; H, 8.59; N, 7.80.
Methyl [2S-(1-Methoxycarbonyl-6R-(1-methoxy-2-methyl-1-oxopropan-2-yl)hexahydropyrimidin-3-yl)-4-methylpentanoyl]-L-alanine (18)
Obtained from aldehyde 5c (49.5 mg, 0.15 mmol) according to the general procedures for the reductive amination–cyclization using H-Leu-Ala-OMe as a reagent (45.0 mg, 0.21 mmol). After work-up and solvent evaporation, the residue was purified by rotatory chromatography (hexanes/EtOAc, 40:60), yielding hexahydropyrimidine 18 (40.3 mg, 60%) as a syrup. [α]D: +21 (c 0.48, CHCl3). IR (CHCl3) νmax 3438, 1720, 1705, 1513, 1218, 1049 cm–1. 1H NMR (500 MHz, CD3CN, 70 °C) δH 7.07 (1H, br s), 4.72 (1H, d, J = 12.4 Hz), 4.41 (1H, quin, J = 7.3 Hz), 4.28 (1H, dd, J = 5.4, 7.1 Hz), 3.82 (1H, d, J = 12.4 Hz), 3.69 (3H, s), 3.66 (3H, s), 3.64 (3H, s), 3.10 (1H, dd, J = 5.8, 8.5 Hz), 2.83–2.77 (1H, m), 2.71–2.65 (1H, m), 1.89–1.82 (2H, m), 1.68–1.60 (1H, m), 1.54–1.42 (2H, m), 1.36 (3H, d, J = 7.2 Hz), 1.21 (3H, s), 1.20 (3H, s), 0.92 (3H, d, J = 6.2 Hz), 0.91 (3H, d, J = 6.2 Hz). 13C{1H} NMR (125.7 MHz, CD3CN, 25 °C) δc 178.1 (C), 174.5 (C), 173.4 (C), 158.5 (C), 65.7 (CH), 61.1 (CH2), 57.9 (CH), 53.5 (CH3), 52.9 (CH3), 52.7 (CH3), 49.0 (CH), 48.3 (C), 46.1 (CH2), 39.6 (CH2), 26.4 (CH), 24.8 (CH3), 24.6 (CH2), 23.8 (CH3), 23.4 (CH3), 22.8 (CH3), 18.3 (CH3). HRMS (ESI-TOF) calcd for C21H37N3O7Na [M + Na]+, 466.2529; found, 466.2529. Anal. calcd for C21H37N3O7: C, 56.87; H, 8.41; N, 9.47. Found: C, 57.08; H, 8.51; N, 9.58.
Methyl [2S-(1-Methoxycarbonyl-6R-(1-methoxy-2-methyl-1-oxopropan-2-yl)hexahydropyrimidin-3-yl)-4-methylpentanoyl]-L-leucine (19)
Obtained from aldehyde 5c (49.5 mg, 0.15 mmol) according to the general procedures for the reductive amination–cyclization using H-Leu-Leu-OMe as a reagent (54.2 mg, 0.21 mmol). After work-up and solvent evaporation, the residue was purified by rotatory chromatography (hexanes/EtOAc, 40:60), yielding compound 19 (43.6 mg, 60%) as a syrup. [α]D: +22 (c 0.45, CHCl3). IR (CHCl3) νmax 3434, 1723, 1520, 1449, 1211, 1054 cm–1. 1H NMR (500 MHz, CD3CN, 70 °C) δH 6.94 (1H, d, J = 7.4 Hz), 4.73 (1H, d, J = 12.4 Hz), 4.43 (1H, td, J = 8.1, 6.8 Hz), 4.28 (1H, dd, J = 7.2, 5.4 Hz), 3.83 (1H, d, J = 12.4 Hz), 3.67 (3H, s), 3.66 (3H, s), 3.64 (3H, s), 3.10 (1H, dd, J = 8.0, 6.6 Hz), 2.79 (1H, ddd, J = 12.4, 9.3, 3.9 Hz), 2.70–2.65 (1H, m), 1.89–1.81 (2H, m), 1.73–1.64 (1H, m), 1.65–1.59 (3H, m), 1.48–1.45 (2H, m), 1.21 (3H, s), 1.20 (3H, s), 0.95 (3H, d, J = 6.5 Hz), 0.93 (6H, d, J = 6.5 Hz), 0.90 (3H, d, J = 6.5 Hz). 13C{1H} NMR (125.7 MHz, CD3CN, 70 °C) δc 178.0 (C), 174.4 (C), 173.8 (C), 158.6 (C), 65.9 (CH), 61.3 (CH2), 57.9 (CH), 53.5 (CH3), 52.8 (CH3), 52.7 (CH3), 51.7 (CH), 48.3 (C), 46.0 (CH2), 41.8 (CH2), 40.1 (CH2), 26.4 (CH), 26.0 (CH), 24.7 (CH3), 24.6 (CH2), 23.8 (CH3), 23.42 (CH3), 23.36 (CH3), 22.8 (CH3), 22.2 (CH3). HRMS (ESI-TOF) calcd for C24H43N3O7Na [M + Na]+, 508.2999; found, 508.2999. Anal. calcd for C24H43N3O7: C, 59.36; H, 8.93; N, 8.65. Found: C, 59.46; H, 8.73; N, 9.63.
N-[N-(Acetoxymethyl)-N-(methoxycarbonyl)-2,2-dimethyl-1-methyl-L-β-norvalin-5-yl]-L-leucinyl-L-alanine Methyl Ester (20)
Obtained from aldehyde 5d (45.5 mg, 0.15 mmol) according to the general procedure for the reductive amination using H-Leu-Ala-OMe hydrochloride (53 mg, 0.21 mmol) as a reagent. After work-up and solvent evaporation, the residue was purified by rotatory chromatography (hexanes/EtOAc, 40:60), yielding diamine 20 (61.7 mg, 82%) as a syrup. [α]D: −5 (c 0.42, CHCl3). IR (CHCl3) νmax 3434, 1704, 1570, 1450, 1295, 1088 cm–1. 1H NMR (400 MHz, 70 °C, CD3CN) δH 7.42–7.32 (1H, br b), 5.38 (1H, d, J = 10.8 Hz), 5.27 (1H, d, J = 11.2 Hz), 4.50–4.37 (2H, m), 3.69 (6H, s), 3.65 (3H, s), 3.02 (1H, dd, J = 7.9, 5.7 Hz), 2.55 (2H, dd, J = 7.6, 6.0 Hz), 1.99 (3H, s), 1.92–1.83 (1H, m), 1.77–1.66 (2H, m), 1.49–1.44 (1H, m), 1.40–1.33 (1H, m), 1.37 (3H, d, J = 7.2 Hz), 1.22 (3H, s), 1.19 (3H, s), 0.95 (3H, d, J = 6.4 Hz), 0.92 (3H, d, J = 6.8 Hz). 13C{1H} NMR (100.6 MHz, 70 °C, CD3CN) δc 177.8 (C), 175.9 (C), 174.6 (C), 171.3 (C), 164.3 (C), 71.3 (CH2), 62.6 (CH), 61.6 (CH), 54.0 (CH3), 52.9 (CH3), 52.7 (CH3), 49.0 (CH), 48.5 (C), 46.8 (CH2), 44.5 (CH2), 30.1 (CH2), 26.1 (CH), 24.7 (CH3), 23.7 (CH3), 23.0 (CH3), 22.7 (CH3), 21.4 (CH3), 18.3 (CH3). HRMS (ESI-TOF) calcd for C23H41N3O9Na [M + Na]+, 526.2741; found, 526.2737. Anal. calcd for C23H41N3O9: C, 54.86; H, 8.21; N, 8.34. Found: C, 54.83; H, 8.31; N, 8.20.
N-[N-(Acetoxymethyl)-N-(methoxycarbonyl)-2,2-dimethyl-1-methyl-L-β-norvalin-5-yl]-L-leucinyl-L-leucine Methyl Ester (21)
Obtained from aldehyde 5d (45.5 mg, 0.15 mmol) according to the general procedure for the reductive amination using H-Leu-Leu-OMe hydrochloride as a reagent (61.5 mg, 0.21 mmol). After work-up and solvent evaporation, the residue was purified by rotatory chromatography (hexanes/EtOAc, 40:60), yielding diamine 21 (69.0 mg, 84%) as a syrup. [α]D: −8 (c 0.48, CHCl3). IR (CHCl3) νmax 3436, 1704, 1570, 1450, 1295, 1088 cm–1. 1H NMR (400 MHz, 70 °C, CD3CN) δH 7.30–7.24 (1H, m), 5.38 (1H, d, J = 11.2 Hz), 5.27 (1H, d, J = 11.2 Hz), 4.50–4.40 (2H, m), 3.69 (3H, s), 3.68 (3H, s), 3.65 (3H, s), 3.03 (1H, dd, J = 7.8, 5.9 Hz), 2.60–2.52 (2H, m), 1.99 (3H, s), 1.90–1.80 (1H, br b), 1.78–1.62 (6H, m), 1.51–1.44 (1H, m), 1.41–1.31 (1H, m), 1.22 (3H, s), 1.19 (3H, s), 0.95 (3H, d, J = 6.4 Hz), 0.94 (3H, d, J = 6.8 Hz), 0.92 (6H, d, J = 6.4 Hz). 13C{1H} NMR (100.6 MHz, 70 °C, CD3CN) δc 177.8 (C), 176.1 (C), 174.5 (C), 171.3 (C), 158.7 (C), 71.2 (CH2), 62.7 (CH), 61.7 (CH), 54.0 (CH3), 52.8 (CH3), 52.7 (CH3), 51.7 (CH), 48.5 (C), 46.8 (CH2), 44.6 (CH2), 42.0 (CH2), 30.1 (CH2), 26.2 (2 × CH), 24.7 (CH3), 23.7 (CH3), 23.5 (CH3), 23.1 (CH3), 22.7 (CH3), 22.3 (CH3), 21.4 (CH3). HRMS (ESI-TOF) calcd for C26H47N3O9Na [M + Na]+, 568.3210; found, 568.3215. Anal. calcd for C26H47N3O9: C, 57.23; H, 8.68; N, 7.70. Found: C, 57.30; H, 8.63; N, 7.72.
Methyl [2S-(1-Methoxycarbonyl-6R-(1-methoxy-2-methyl-1-oxopropan-2-yl)hexahydropyrimidin-3-yl)-4-methylpentanoyl]-L-alanine (22)
Obtained from aldehyde 5d (49.5 mg, 0.15 mmol) according to the general procedures for the reductive amination–cyclization using H-Leu-Ala-OMe as a reagent (45.0 mg, 0.21 mmol). After work-up and solvent evaporation, the residue was purified by rotatory chromatography (hexanes/EtOAc, 40:60), yielding compound 22 (45.0 mg, 68%) as a syrup. [α]D: −21 (c 0.52, CHCl3). IR (CHCl3) νmax 3438, 1718, 1700, 1447, 1214, 1047 cm–1. 1H NMR (500 MHz, CD3CN, 70 °C) δH 6.94 (1H, d, J = 5.8 Hz), 4.82 (1H, d, J = 12.8 Hz), 4.38 (1H, quint, J = 7.2 Hz), 4.28 (1H, dd, J = 7.6, 4.7 Hz), 3.89 (1H, d, J = 12.8 Hz), 3.68 (3H, s), 3.66 (3H, s), 3.64 (3H, s), 3.09 (1H, dd, J = 8.4, 6.1 Hz), 2.80 (1H, ddd, J = 12.0, 9.8, 3.6 Hz), 2.62 (1H, dt, J = 12.0, 6.0 Hz), 1.95–1.89 (1H, m), 1.83–1.78 (1H, m), 1.68–1.62 (1H, m), 1.58–1.46 (2H, m), 1.35 (3H, d, J = 7.3 Hz), 1.21 (3H, s), 1.20 (3H, s), 0.93 (3H, d, J = 6.5 Hz), 0.91 (3H, d, J = 6.6 Hz). 13C{1H} NMR (125.7 MHz, CD3CN, 70 °C) δc 178.1 (C), 174.4 (C), 173.4 (C), 158.3 (C), 65.4 (CH), 59.3 (CH2), 57.8 (CH), 53.4 (CH3), 52.8 (CH3), 52.7 (CH3), 49.1 (CH), 48.3 (C), 47.3 (CH2), 39.7 (CH2), 26.1 (CH), 24.8 (CH3), 24.3 (CH2), 23.7 (CH3), 23.4 (CH3), 22.9 (CH3), 18.3 (CH3). HRMS (ESI-TOF) calcd for C21H37N3O7Na [M + Na]+, 466.2529; found, 466.2526. Anal. calcd for C21H37N3O7: C, 56.87; H, 8.41; N, 9.47. Found: C, 56.76; H, 8.62; N, 9.24.
Methyl [2S-(1-Methoxycarbonyl-6R-(1-methoxy-2-methyl-1-oxopropan-2-yl)hexahydropyrimidin-3-yl)-4-methylpentanoyl]-L-leucine (23)
Obtained from aldehyde 5d (49.5 mg, 0.15 mmol) according to the general procedures for the reductive amination–cyclization using H-Leu-Leu-OMe as a reagent (54.0 mg, 0.21 mmol). After work-up and solvent evaporation, the residue was purified by rotatory chromatography (hexanes/EtOAc, 40:60), yielding compound 23 (47.8 mg, 65%) as a syrup. [α]D: −37 (c 0.37, CHCl3). IR (CHCl3) νmax 3435, 1721, 1702, 1518, 1424, 1213, 1043 cm–1. 1H NMR (500 MHz, CD3CN, 70 °C) δH 6.87 (1H, d, J = 7.3 Hz), 4.84 (1H, d, J = 12.8 Hz), 4.42 (1H, q, J = 7.8 Hz), 4.29 (1H, dd, J = 7.6, 4.7 Hz), 3.91 (1H, d, J = 12.8 Hz), 3.67 (3H, s), 3.66 (3H, s), 3.64 (3H, s), 3.11 (1H, dd, J = 8.7, 5.8 Hz), 2.83 (1H, ddd, J = 12.1, 9.7, 3.5 Hz), 2.58 (1H, dt, J = 11.9, 5.9 Hz), 1.99–1.89 (1H, m), 1.83–1.78 (1H, m), 1.71–1.48 (6H, m), 1.21 (3H, s), 1.20 (3H, s), 0.94 (3H, d, J = 6.5 Hz), 0.93 (3H, d, J = 6.4 Hz), 0.92 (3H, d, J = 6.3 Hz), 0.90 (3H, d, J = 6.4 Hz). 13C{1H} NMR (125.7 MHz, CD3CN, 70 °C) δc 178.0 (C), 174.3 (C), 173.7 (C), 158.3 (C), 65.5 (CH), 59.1 (CH2), 57.8 (CH), 53.4 (CH3), 52.8 (CH3), 52.7 (CH3), 51.8 (CH), 48.4 (C), 47.6 (CH2), 41.8 (CH2), 39.9 (CH2), 26.1 (CH), 26.0 (CH), 24.8 (CH2), 24.2 (CH3), 23.8 (CH3), 23.43 (CH3), 23.42 (CH3), 22.8 (CH3), 22.1 (CH3). HRMS (ESI-TOF) calcd for C24H43N3O7Na [M + Na]+, 508.2999; found, 508.3001. Anal. calcd for C24H43N3O7: C, 59.36; H, 8.93; N, 8.65. Found: C, 59.50; H, 9.09; N, 8.36.
N2-(tert-Butoxycarbonyl)-N5-(benzyloxycarbonyl)-L-ornithyl-L-phenylalanine Methyl Ester (24)
HBTU (1.71 g, 4.50 mmol) and DIPEA (2.15 mL, 1.60 g, 12.27 mmol) were added to a solution of commercial Boc-Orn(Cbz)-OH (1.50 g, 4.09 mmol) and l-phenylalanine methyl ester hydrochloride (883 mg, 4.09 mmol) in dry CH2Cl2 (15 mL) at 0 °C. The reaction mixture was stirred at room temperature for 2 h, then poured into water, and washed with saturated aqueous NaHCO3 solution (3 × 20 mL) and 5% HCl (3 × 20 mL). The organic layer was dried and evaporated as usual, and the residue was purified by column chromatography (hexanes/EtOAc, 6:4), giving dipeptide 24 (1.82 mg, 84%) as a crystalline solid: mp 124–126 °C (from EtOAc/pentane); [α]D: −37 (c 0.37, CHCl3). IR (CHCl3) νmax 3442, 1716, 1685, 1517, 1218, 1051 cm–1. 1H NMR (500 MHz, CD3CN, 70 °C) δH 7.38–7.20 (10H, m), 6.85 (1H, d, J = 6.7 Hz), 5.55–5.45 (1H, br b), 5.30–5.40 (1H, br b), 5.08 (1H, d, J = 12.9 Hz), 5.06 (1H, d, J = 13.0 Hz), 4.68 (1H, ddd, J = 7.8, 7.7, 5.8 Hz), 4.04–3.98 (1H, m), 3.66 (3H, s), 3.18–3.06 (3H, m), 3.01 (1H, dd, J = 14.0, 7.7 Hz), 1.74–1.66 (1H, m), 1.52–1.45 (3H, m), 1.42 (9H, s). 13C{1H} NMR (125.7 MHz, CD3CN, 70 °C) δc 173.4 (C), 173.1 (C), 158.0 (C), 156.9 (C), 138.9 (C), 138.2 (C), 130.6 (2 × CH), 129.8 (2 × CH), 129.7 (2 × CH), 129.1 (CH), 129.0 (2 × CH), 128.1 (CH), 80.5 (C), 67.3 (CH2), 55.7 (CH), 54.9 (CH), 53.0 (CH3), 41.5 (CH2), 38.8 (CH2), 30.8 (CH2), 29.0 (3 × CH3), 27.3 (CH2). HRMS (ESI-TOF/QTOF) calcd for C28H37N3O7Na [M + Na]+, 550.2529; found, 550.2549. Anal. calcd for C28H37N3O7: C, 63.74; H, 7.07; N, 7.96. Found: C, 63.52; H, 7.06; N, 7.58.
N2-(tert-Butoxycarbonyl)-N5-(benzyloxycarbonyl)-L-ornithyl-L-phenylanyl-L-leucine Methyl Ester (25)
To a solution of dipeptide 24 (1.82 g, 3.45 mmol) in MeOH (15 mL) at 0 °C was added 2 N KOH (7 mL). The reaction mixture was allowed to reach room temperature and stirred for 2 h. Then, it was acidified to pH 2–3 with 5% HCl, poured into water, and extracted with EtOAc. The organic layer was dried and evaporated as usual, affording the corresponding acid that was dissolved in CH2Cl2 and treated with H-Leu-OMe hydrochloride (627 mg, 3.45 mmol), HBTU (1.44 g, 3.80 mmol), and DIPEA (1.82 mL, 10.4 mmol). After stirring for 2 h, the reaction mixture was poured into water and washed with saturated aqueous NaHCO3 solution (3 × 20 mL) and 5% HCl (3 × 20 mL). The organic layer was dried and evaporated as usual, and the residue was purified by column chromatography (hexanes/EtOAc, 1:1), giving tripeptide 25 (1.74 mg, 79%) as a crystalline solid: mp 137–139 °C (from EtOAc/pentane); [α]D: −21 (c 0.47, CHCl3). IR (CHCl3) νmax 3441, 3348, 1708, 1521, 1421, 1213, 1046 cm–1. 1H NMR (500 MHz, CD3CN, 70 °C) δH 7.39–7.21 (10H, m), 6.84 (1H, d, J = 7.4 Hz), 6.79 (1H, d, J = 7.3 Hz), 5.55–5.45 (1H, br b), 5.43–5.30 (1H, br b), 5.08 (2H, s), 4.60 (1H, dt, J = 8.1, 5.5 Hz), 4.43 (1H, dt, J = 8.4, 5.5 Hz), 3.97 (1H, dt, J = 7.7, 5.5 Hz), 3.66 (3H, s), 3.16–3.05 (3H, m), 2.96 (1H, dd, J = 14.1, 7.9 Hz), 1.71–1.44 (7H, m), 1.42 (9H, s), 0.92 (3H, d, J = 6.4 Hz), 0.90 (3H, d, J = 6.2 Hz). 13C{1H} NMR (125.7 MHz, CD3CN, 70 °C) δc 174.0 (C), 173.4 (C), 172.1 (C), 157.9 (C), 157.0 (C), 139.0 (C), 138.7 (C), 130.7 (2 × CH), 129.8 (2 × CH), 129.7 (2 × CH), 129.1 (CH), 129.0 (2 × CH), 127.9 (CH), 80.6 (C), 67.3 (CH2), 56.0 (CH), 55.4 (CH), 52.9 (CH3), 52.4 (CH), 42.0 (CH2), 41.5 (CH2), 38.9 (CH2), 30.6 (CH2), 29.0 (3 × CH3), 27.3 (CH2), 25.9 (CH), 23.3 (CH3), 22.4 (CH3). HRMS (ESI-TOF) calcd for C34H48N4O8Na [M + Na]+, 663.3370; found, 663.3381. Anal. calcd for C34H48N4O8: C, 63.73; H, 7.55; N, 8.74. Found: C, 63.34; H, 7.53; N, 8.49.
Methyl (2S-((3S,7R,8aR)-3-(3-[(Benzyloxycarbonyl)amino]propyl)-7-(1-methoxy-2-methyl-1-oxopropan-2-yl)-2-oxohexahydroimidazo[1,2-c]pyrimidin-1(5H)-yl)-3-phenylpropanoyl)-L-leucine (26) and Methyl [(2S)-[(4S)-4-(3-(Benzyloxycarbonylamino)prop-1-yl)-2-[4-methoxy-2R-(N-methoxycarbonyl-N-methoxymethylamino)-3,3-dimethyl-4-oxobutyl]-5-oxo-imidazolidin-1-yl]-3-phenylpropanoyl]-L-leucine (27)
A solution of peptide 25 (134.6 mg, 0.21 mmol) in a 1:1 mixture of TFA:DCM (1 mL) was stirred at 0° for 1 h. Then, the solvent was evaporated under vacuum and the residue was dissolved in methanol and added to a solution of aldehyde 5c (49.5 mg, 0.15 mmol) and Et3N, which was later treated with NaBH4 according to the general reductive amination protocol. After work-up and solvent evaporation, the residue was purified by rotatory chromatography (hexanes/EtOAc, 40:60), yielding compounds 26 (41.1 mg, 36%) and 27 (35.3 mg, 30%) as colorless oils.
Compound 26
[α]D: −26 (c 0.59, CHCl3). IR (CHCl3) νmax 3445, 1712, 1696, 1518, 1444, 1213, 1045 cm–1. 1H NMR (400 MHz, CD3CN, 70 °C) δH 7.40–7.23 (10H, m), 6.97 (1H, d, J = 7.6 Hz), 5.58–5.44 (1H, br b), 5.10 (2H, s), 4.81 (1H, d, J = 11.0 Hz), 4.51–4.42 (2H, m), 4.38 (1H, td, J = 11.8, 2.5 Hz), 4.25 (1H, dd, J = 12.2, 6.6 Hz), 3.70 (3H, s), 3.67 (6H, s), 3.59 (1H, d, J = 11.0 Hz), 3.39–3.25 (2H, m), 3.22–3.18 (1H, m), 3.13–3.07 (2H, m), 1.83 (1H, ddd, J = 13.5, 6.7, 2.7 Hz), 1.74–1.34 (6H, m), 1.26–1.15 (2H, m), 1.12 (3H, s), 1.08 (3H, s), 0.98 (3H, d, J = 6.3 Hz), 0.96 (3H, d, J = 6.2 Hz). 13C{1H} NMR (100.6 MHz, CD3CN, 70 °C) δc 177.1 (C), 174.5 (C), 174.0 (C), 170.9 (C), 158.7 (C), 157.7 (C), 139.1 (C), 138.9 (C), 130.4 (2 × CH), 129.9 (2 × CH), 129.8 (2 × CH), 129.1 (CH), 129.0 (2 × CH), 128.1 (CH), 73.0 (CH), 67.2 (CH2), 62.5 (CH), 61.5 (CH2), 58.9 (CH), 58.0 (CH), 53.8 (CH3), 53.0 (CH3), 52.7 (CH3), 52.6 (CH), 48.2 (C), 42.13 (CH2), 42.06 (CH2), 34.5 (CH2), 29.3 (CH2), 27.8 (CH2), 26.6 (CH2), 26.1 (CH), 23.3 (CH3), 22.7 (CH3), 22.6 (CH3), 22.0 (CH3). HRMS (ESI-TOF) calcd for C40H55N5O10Na [M + Na]+, 788.3847; found, 788.3843; calcd for C40H56N5O10 [M + H]+, 766.4027; found, 766.4026.
Compound 27
[α]D: +6 (c 0.34, CHCl3). IR (CHCl3) νmax 3446, 1710, 1700, 1680, 1520, 1419, 1219, 1048 cm–1. 1H NMR (500 MHz, CD3CN, 70 °C) δH 7.65 (1H, d, J = 6.5 Hz), 7.39–7.20 (10H, m), 5.60–5.45 (1H, br b), 5.07 (2H, s), 4.60–4.40 (4H, m), 4.48–4.41 (1H, td, J = 8.4, 5.7 Hz), 3.98 (1H, br dd, J = 10.2, 5.7 Hz), 3.83–3.76 (1H, m), 3.67 (3H, s), 3.66 (3H, s), 3.60 (3H, s), 3.53 (1H, dd, J = 13.7, 10.4 Hz), 3.45 (1H, dd, J = 7.6, 4.6 Hz), 3.28 (1H, dd, J = 13.7, 5.7 Hz), 3.22 (3H, br s), 3.16 (2H, q, J = 6.7 Hz), 1.77–1.55 (7H, m), 1.53–1.45 (1H, m), 1.34 (2H, br t, J = 13.1, 12.9 Hz), 1.15 (3H, s), 1.13 (3H, s), 0.95 (3H, d, J = 6.6 Hz), 0.94 (3H, d, J = 6.5 Hz). 13C{1H} NMR (125.7 MHz, CD3CN, 70 °C) δc 177.7 (C), 177.4 (C), 174.2 (C), 171.7 (C), 158.7 (C), 157.7 (C), 139.3 (C), 139.0 (C), 130.5 (2 × CH), 129.8 (2 × CH), 129.7 (2 × CH), 129.1 (CH), 129.0 (2 × CH), 128.0 (CH), 74.6 (CH), 67.2 (CH2), 62.9 (CH), 60.1 (CH), 56.8 (CH3), 55.2 (CH), 53.7 (CH3), 53.0 (CH3), 52.7 (CH3), 52.4 (CH), 48.3 (C), 42.1 (CH2), 42.0 (CH2), 36.9 (CH2), 35.7 (CH2), 32.2 (CH2), 27.3 (CH2), 26.1 (CH), 24.6 (CH3), 23.5 (CH3), 23.4 (CH3), 22.5 (CH3). HRMS (ESI-TOF) calcd for C41H59N5O11Na [M + Na]+, 820.4109; found, 820.4106; calcd for C40H56N5O10 [M + H]+, 766.4027; found, 766.4023.
Acknowledgments
This work was mainly financed by project RETOS-SELECTFIGHT (PID2020-116688RB-C21) of the Plan Estatal I + D, Ministry of Science, Spain (with FEDER funds). We also thank project ProID2020010134 (Programa de Subvenciones a la Realización I + D Ma Carmen Betancourt y Molina from ACIISI-Gobierno de Canarias with FEDER funds). D.H. also acknowledges her contract TRANSALUDAGRO financed by Cabildo de Tenerife, Program TF INNOVA 2016-21 (with MEDI & FDCAN Funds). M.P. thanks predoctoral grants from Gobierno de Canarias (Convocatoria Tesis 2020) and Ministerio de Ciencia, Innovación y Universidades, Spain (FPU grants). M.P. is currently a student of the Ph.D. program “Ciencias Médicas y Farmacéuticas, Desarrollo y Calidad de Vida” of the University of La Laguna. Finally, we also acknowledge support of the publication fee by CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URIC).
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.3c00673.
1H and 13C NMR of compounds 7–23, COSY of 21, and HSQC of compounds 17, 18, 21, and 23; 1H/13C NMR, COSY, HSQC, HMBC, and NOESY of “turn-inducing” compounds 26 and 27 and 1H/13C NMR of their precursors 24 and 25; MM2 calculations of minimum-energy conformations of compounds 26 and epi-26 and key angles for coupling constant calculations (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Hernández D.; Carro C.; Boto A. “Doubly Customizable” Unit for the Generation of Structural Diversity: From Pure Enantiomeric Amines to Peptide Derivatives. J. Org. Chem. 2021, 86, 2796–2809. 10.1021/acs.joc.0c02751. [DOI] [PubMed] [Google Scholar]; b Hernández D.; Carro C.; Boto A. Structural Diversity by Using Amino Acid “Customizable Units:” Conversion of Hydroxyproline (Hyp) into Nitrogen Heterocycles. Amino Acids 2022, 54, 955–966. 10.1007/s00726-022-03159-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a review, see:; Boto A.; González C. C.; Hernández D.; Romero-Estudillo I.; Saavedra C. J. Site-selective modification of peptide backbones. Org. Chem. Front. 2021, 8, 6720–6759. 10.1039/D1QO00892G. [DOI] [Google Scholar]
- a Saavedra C. J.; Carro C.; Hernández D.; Boto A. Conversion of “Customizable Units” into N-Alkyl Amino Acids and Generation of N-Alkyl Peptides. J. Org. Chem. 2019, 84, 8392–8410. 10.1021/acs.joc.9b00114. [DOI] [PubMed] [Google Scholar]; b Saavedra C. J.; Cuevas F.; Romero-Estudillo I.; Boto A. Synthesis of Diketopiperazine Scaffolds with tailored N- and α-chains by Selective Modification of Customizable Units. Adv. Synth. Catal. 2020, 362, 3158–3169. 10.1002/adsc.202000470. [DOI] [Google Scholar]; c Cuevas F.; Saavedra C. J.; Romero-Estudillo I.; Boto A.; Ordóñez M.; Vergara I. Structural diversity using Hyp “customizable units:” proof of-concept synthesis of sansalvamide-related antitumoral peptides. Eur. J. Org. Chem. 2021, 933–943. 10.1002/ejoc.202001427. [DOI] [Google Scholar]; d Romero-Estudillo I.; Boto A. Domino process achieves site-selective peptide modification with high optical purity. Applications to chain diversification and peptide ligation. J. Org. Chem. 2015, 80, 9379–9391. 10.1021/acs.joc.5b00932. [DOI] [PubMed] [Google Scholar]
- a For the antimicrobial hexetidine, see: https://pubchem.ncbi.nlm.nih.gov/compound/Hexetidine,accessed on 09-01-2023.; b Pikul S.; McDow-Dunham K.; Almstead N. G.; De B.; Natchus M. G.; Taiwo Y. O.; Williams L. E.; Hynd B. A.; Hsieh L. C.; Janusz M. J.; Gu F.; Mieling G. E. Heterocycle-Based MMP Inhibitors with P2’ Substituents. Bioorg. Med. Chem. Lett. 2001, 11, 1009–1013. 10.1016/S0960-894X(01)00137-8. [DOI] [PubMed] [Google Scholar]; c Bender S. L.; Melnick M. J.. Metalloproteinase inhibitors, pharmaceutical compositions containing them and their pharmaceutical uses. Patent US5753653A (1998).
- Some antibiotic natural products were also reported as hydropyrimidines, but the structure was corrected later:; a Wu G.; Liu S.; Wang T.; Jiang Z.; Lv K.; Wang Y.; Sun C. Total Synthesis of Originally Proposed and Revised Structure of Hetiamacin A. Org. Lett. 2018, 12, 3566–3569. [DOI] [PubMed] [Google Scholar]; b Liu S. W.; Jin J.; Chen C.; Liu J.-M.; Li J.-Y.; Wang F.-F.; Jiang Z.-K.; Hu J. H.; Gao Z.-X.; Yao F.; You X.-F.; Si S.-Y.; Sun C.-H. PJS, a novel isocoumarin with hexahydropyrimidine ring from Bacillus subtilis. J. Antibiot. 2013, 66, 281–284. 10.1038/ja.2012.118. [DOI] [PubMed] [Google Scholar]
- Other syntheses from diamines and aldehydes:; a Lindahl F.; Hoang H. N.; Fairlie D. P.; Cooper M. A. Facile synthesis of mono- and bis-methylated Fmoc-Dap, -Dab and -Orn amino acids. Chem. Commun. 2015, 51, 4496–4498. [DOI] [PubMed] [Google Scholar]; b Naphthoquinone derivatives compound and a composition containing the same for antibacterial activity. Patent KR102381397, 2022. (Kyung Hee University).
- For cycloadditions:; Chang T.-C.; Pradipta A. R.; Tanaka K. Enantioselective synthesis of cyclic and linear diamines by imine cycloadditions. Chirality 2020, 32, 1160–1168. 10.1002/chir.23265. [DOI] [PubMed] [Google Scholar]
- a Chorev M.; Goodman M. A Dozen Years of Retro-Inverso Peptidomimetics. Acc. Chem. Res. 1993, 26, 266–273. 10.1021/ar00029a007. [DOI] [Google Scholar]; b Chorev M. The Partial Retro–Inverso Modification: A Road Traveled Together. Biopolymers 2005, 80, 67–84. 10.1002/bip.20219. [DOI] [PubMed] [Google Scholar]
- a Fletcher M. D.; Campbell M. M. Partially Modified Retro-Inverso Peptides: Development, Synthesis, and Conformational Behavior. Chem. Rev. 1998, 98, 763–796. 10.1021/cr970468t. [DOI] [PubMed] [Google Scholar]; b Frank H. G.; Knorr K.; Haberl U.; Rybka A.. Isosteric Transformation. Patent WO2005/090389 (2005).
- For some recent reviews on retropeptides, see:; a Doti N.; Mardirossian M.; Sandomenico A.; Ruvo M.; Caporale A. Recent Applications of Retro-Inverso Peptides. Int. J. Mol. Sci. 2021, 22, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Rai J. Peptide and protein mimetics by retro and retroinverso analogs. Chem. Biol. Drug Des. 2019, 93, 724–736. 10.1111/cbdd.13472. [DOI] [PubMed] [Google Scholar]; c Preston G. W. Different directions for retro-inverso peptides. J Pep Sci. 2022, 28, e3384. [DOI] [PubMed] [Google Scholar]
- For some reviews on cyclic and polycyclic turn templates, see:; a Venkatraman J.; Shankaramma S. C.; Balaram P. Design of Folded Peptides. Chem. Rev. 2001, 101, 3131–3152. 10.1021/cr000053z. [DOI] [PubMed] [Google Scholar]; b Vasudev P. G.; Chatterjee S.; Shamala N.; Balaram P. Structural Chemistry of Peptides Containing Backbone Expanded Amino Acid Residues: Conformational Features of β, γ, and Hybrid Peptides. Chem. Rev. 2011, 111, 657–687. 10.1021/cr100100x. [DOI] [PubMed] [Google Scholar]; c Jwad R.; Weissberger D.; Hunter L. Strategies for fine-tuning the conformation of cyclic peptides. Chem. Rev. 2020, 120, 9743–9789. 10.1021/acs.chemrev.0c00013. [DOI] [PubMed] [Google Scholar]
- For other examples of β-turn cyclic templates, see:; a Martínez L.; Martorell G.; Sampedro A.; Ballester P.; Costa A.; Rotger C. Hydrogen Bonded Squaramide-Based Foldable Module Induces Both β- and α-Turns in Hairpin Structures of α-Peptides in Water. Org. Lett. 2015, 17, 2980–2983. [DOI] [PubMed] [Google Scholar]; b Fuller A. A.; Du D.; Liu F.; Davoren J. E.; Bhabha G.; Kroon G.; Case D. A.; Dyson H. J.; Powers E. T.; Wipf P.; Gruebele M.; Kelly J. W. Evaluating β-turn mimics as β-sheet folding nucleators. PNAS 2009, 106, 11067–11072. 10.1073/pnas.0813012106. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Kim K.; Germanas J. P. Peptides Constrained to Type VI β-Turns. 1. Evidence for an Exceptionally Stable Intramolecular Hydrogen Bond. J. Org. Chem. 1997, 62, 2847–2852. 10.1021/jo961180r. [DOI] [PubMed] [Google Scholar]; d Kim K.; Germanas J. P. Peptides Constrained to Type VI β-Turns. 2. Antiparallel β-Ladder Formation. J. Org. Chem. 1997, 62, 2853–2860. [DOI] [PubMed] [Google Scholar]
- For other examples of γ- and other turn cyclic templates, see:; a Yuan Z.-Q.; Blomberg D.; Sethson I.; Brickmann K.; Ekholm K.; Johansson B.; Nilsson A.; Kihlberg J. Synthesis and Pharmacological Evaluation of an Analogue of the Peptide Hormone Oxytocin That Contains a Mimetic of an Inverse γ-Turn. J. Med. Chem. 2002, 45, 2512–2519. [DOI] [PubMed] [Google Scholar]; b Farahani M. D.; Honarparvar B.; Albericio F.; Maguire G. E. M.; Govender T.; Arvidsson P. I.; Kruger H. G. Proline N-oxides: modulators of the 3D conformation of linear peptides through “NO-turns.”. Org. Biomol. Chem. 2014, 12, 4479–4490. 10.1039/c4ob00433g. [DOI] [PubMed] [Google Scholar]; c Balo R.; Jiménez A.; Reza D.; Estévez R. J.; Estévez J. C. Peptides Incorporating 3,4-Dihydroxyprolines: Synthesis and Structural Study. Chem. Proc. 2022, 8, 72. [Google Scholar]
- a Saavedra C. J.; Hernández R.; Boto A.; Álvarez E. Catalytic, One-pot synthesis of β-amino acids from α-amino acids. Preparation of α,β-peptide derivatives. J. Org. Chem. 2009, 74, 4655–4665. [DOI] [PubMed] [Google Scholar]; b Saavedra C. J.; Boto A.; Hernández R.; Miranda J. I.; Aizpurua J. M. Conformation and chiral effects in α,β,α-tripeptide. J. Org. Chem. 2012, 77, 5907–5913. [DOI] [PubMed] [Google Scholar]
- a See also:Chatterjee S.; Roy R. S.; Balaram P. Expanding the polypeptide backbone: hydrogen-bonded conformations in hybrid polypeptides containing the higher homologues of α-amino acids. J. R. Soc., Interface 2007, 4, 587–606. 10.1098/rsif.2006.0203. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Chatterjee S.; Vasudev P. G.; Raghothama S.; Ramakrishnan C.; Shamala N.; Balaram P. Expanding the Peptide β-Turn in α,γ Hybrid Sequences: 12 Atom Hydrogen Bonded Helical and Hairpin Turns. J. Am. Chem. Soc. 2009, 131, 5956–5965. 10.1021/ja900618h. [DOI] [PubMed] [Google Scholar]
- The 3D representation corresponds to an MM2-minimized conformation for compound 18, calculated with Chem3D Pro (64bit), version 22.0.0.22, from PerkinElmer.
- An interesting report of the trapping of imine intermediates by amides to create cyclic peptides (CyClick chemistry) was recently reported. The reaction is exclusively intramolecular:; Adebomi V.; Cohen R. D.; Wills R.; Chavers H. A. H.; Martin G. E.; Raj M. CyClick Chemistry for the Synthesis of Cyclick Peptides. Angew. Chem., Int. Ed. 2019, 58, 19073–19080. [DOI] [PubMed] [Google Scholar]
- Alternatively, compound 26 may come from 27 or its epimer, with the protic solvent promoting substitution of the methoxy group by the amino group.
- The 3D representation corresponds to an MM2-minimized conformation for compound 26. A minimized conformation was also calculated for its 8a-epimer epi-26 to determine torsion angles and coupling constants, as commented in the Supporting Information. The minimized conformations were calculated with Chem3D Pro (64bit), version 22.0.0.22, from PerkinElmer. For 7-H, the experimental coupling constant values were J = 12.2, 6.6 Hz; for 8a-H, they were J = 11.8, 2, 5, and 2.5 Hz; and for 8-Ha, they were J = 13.5 (geminal with 8-Hb), 6.7, and 2.7 Hz. The proton 8-Hb was overlapped with other signals, and its J could not be calculated. The theoretical values for compound 26 were then calculated using Jmax = 12 Hz; for 7-H, they were J = 12 and 5.1 Hz, and for 8a-H, they were J = 9.7 and 2.1 Hz. The theoretical values for epi-26 (the epimer at 8a-C) were also calculated using Jmax = 12 Hz; for 7-H, they were J = 11.7 and 3.5Hz, and for 8a-H, they were J = 11.9 and 4.8 Hz.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.