

Abstract
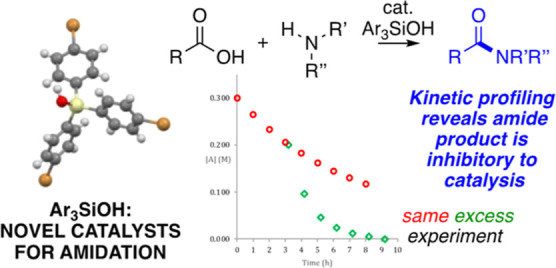
Triarylsilanols have been reported as the first silicon-centered molecular catalysts for direct amidation of carboxylic acids with amines as identified after a screen of silanols, silanediols, disiloxanediols, and incompletely condensed silsesquioxanes as potential homogeneous catalysts. Subsequent synthesis and testing of various electronically differentiated triarylsilanols have identified tris(p-haloaryl)silanols as more active than the parent triarylsilanol, where the bromide congener is found to be the most active. Catalyst decomposition can be observed by NMR methods, but RPKA methods reveal that product inhibition is operative, where tertiary amides are more inhibitory than secondary amides. Studies using an authentically synthesized triaryl silylester as a putative intermediate in the catalytic system enable a plausible mechanism to be proposed as supported by computationals.
Introduction
The catalytic amidation of carboxylic acids1 thereby avoiding stoichiometric quantities of activating agent2 is the focus of much current research.3 Significant progress has been made with the development of boron-based catalysts,4−7 oxophilic metal catalysts,8 and other catalytic systems.9 However, while there are several reports of successful silicon-based reagents10 and silica gels11 for direct amidation, there are no examples of silicon-centered molecular catalysts for direct amidations.12 Furthermore, while various proposals for the mechanistic operation of catalytic amidations have been moot,4−9 only limited direct experimental evidence for presumed activated intermediates has been garnered.13 Herein, we report on the discovery, synthesis, and use of various electronically differentiated triarylsilanols as the first silicon-centered molecular catalysts for direct amidation. Kinetic profiling reveals that these reactions are subject to product inhibition and also sheds light on the reactivity differences between different types of acid–amine combinations. Moreover, we also report on the preparation and utilization of catalytically competent intermediates including an expected on-cycle triarylsilyl ester and an unexpected off-cycle silanaminium salt.
Results and Discussion
On the basis that silica gels have been successfully employed for direct amidation of carboxylic acids (vide supra), we considered that silanols A, silanediols B, disiloxanediols C, and incompletely condensed silsesquioxanes D as molecular analogues of silica gel may also be proficient as homogeneous catalysts (Figure 1a).14 Accordingly, an initial screen of such species at 10 mol % loading using a model amidation of aliphatic carboxylic acid 1a with primary amine 2a in the ideal 1:1 stoichiometry to give amide 3a (Figure 1b) for 1 h in refluxing toluene was conducted (Figure 1c).15 In the event, once the background conversion (11%) with no added catalyst had been taken into account for this acid–amine combination, the 25% conversion obtained with triphenylsilanol 4 revealed it to be a lead catalyst for further study.16 Pleasingly, and in contrast to the other putative silanol, silanediol, and disiloxanediols catalysts that were screened, no irreversible condensation of triphenylsilanol 4 to catalytically unreactive hexaphenyldisiloxane [(Ph3Si)2O] was observed under these conditions.17,18
Figure 1.
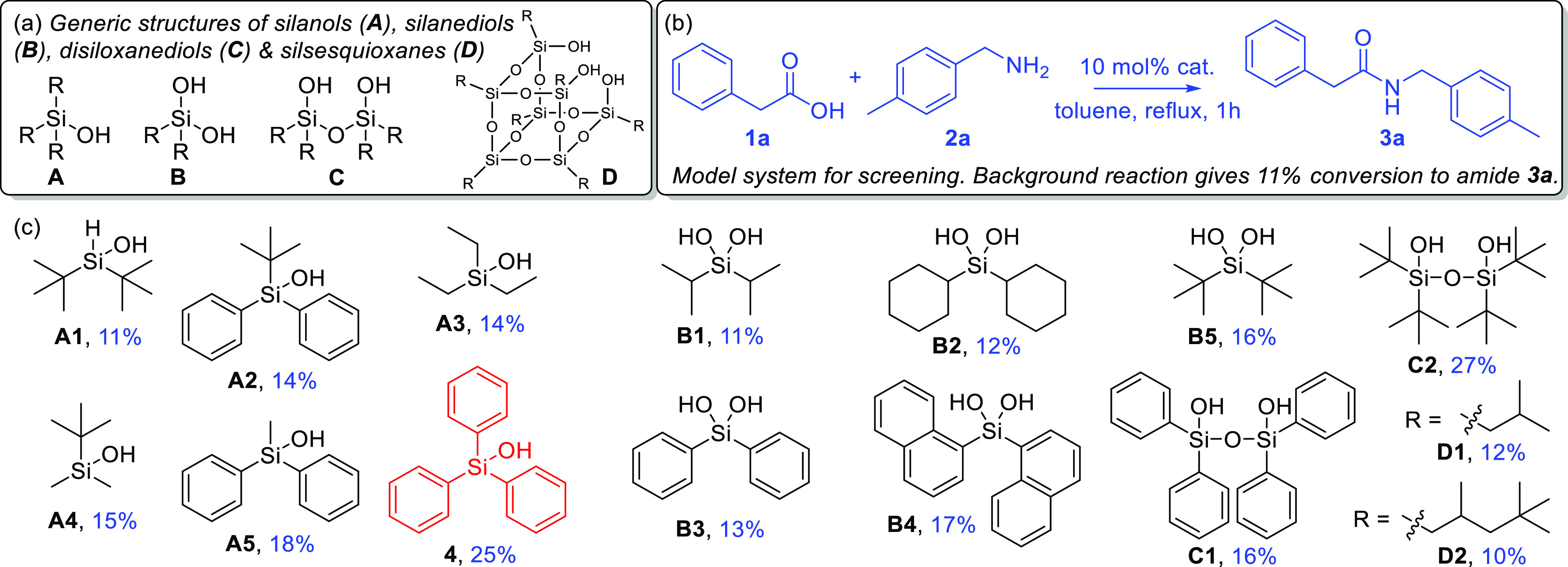
(a) Generic structures of silica molecular analogues A–D, (b) the model amidation reaction to give amide 3a, and (c) a screen of potential catalysts with percentage conversions for the model amidation.
As the next stage of the investigation, a series of known (5a–j)19,20 and new (5k) electronically differentiated triaryl silanes were prepared via metalation of the appropriate aryl bromide followed by reaction with trichlorosilane (Figure 2a). Subsequent hydrolysis provided known (6a, 6c, 6e, 6f, and 6h)21 and new (6b, 6d, 6g, and 6i–k) silanols as potential amidation catalysts.22 Each silanol was then screened against the model amidation reaction (cf., Figure 1b) for its catalytic activity (Figure 2b). In addition, an assessment of catalyst integrity post-amidation was performed by 1H NMR analysis compared to an internal standard. Here, the concern is that condensation to the corresponding (assumed) catalytically inactive disiloxane (R3SiOSiR3)23 and/or other processes that cleave the Si–aryl bond(s) may occur, although this analysis did not allow the distinction between these two different possibilities.
Figure 2.

(a) Preparative route to silanes 5a–k and silanols 6a–k, (b) a screen of the catalytic activity of silanols 6a–k in the model amidation reaction (cf., Figure 1b) including an assessment of catalyst integrity post-amidation, and (c) X-ray crystal structures of novel silanols 6d and 6k.
Inspection of the results for silanol catalysts 6a–k shows that under the specified conditions of the model reaction (10 mol % catalyst, 1h reaction time), increased conversions to amide 3a (6j, 6k: 46%; cf., 4: 25%) can be obtained. Further inspection reveals a trend for increased activity for those catalysts bearing electron-withdrawing groups (and reduced activity for those with electron donating groups). Indeed, a Hammett plot of substituent constant24 vs log(kR/kH)25 for silanols 6b–j (Figure 3a) yields a value of ρ = +0.82,26 implicating the buildup of negative charge in the rate determining step of this catalytic amidation but also signifying that the aryl groups are somewhat remote from the location of the charge. This implies that additional and/or more strongly electron-withdrawing groups should further improve the catalytic activity. Unfortunately, while triphenylsilanol 4 itself (σp = 0) and alkyl substituted silanols 6c–d (σp ca. −0.2) did not display any detectable catalyst decomposition,27 inspection of the catalyst integrity data for electron-deficient silanols 6e–j shows that the onset of catalyst decomposition under these conditions has already begun, ranging from slight-to-moderate (0.1 ≥ σp/m ≥ 0.3) to severe (0.4 ≥ σp/m ≥ 0.6) (Figure 3b). This trend is continued for strongly electron-deficient tris(trifluorophenyl)silanol 6k (not shown in Figure 3b), which, while giving the maximum observed conversion in this catalyst screen, resulted in complete catalyst decomposition. This, therefore, effectively marks the limit of electron-withdrawing substituents that can be introduced into these silanols for use in such direct amidation reactions. Moreover, inspection of the results for strongly electron-rich substituents (σp < −0.2) show that non-negligible amounts of catalyst decomposition are observable here too, as exemplified by p-methoxy silanol 6b. These results suggest that more than one pathway of catalyst deactivation is in operation. This is consistent with previous studies where it is known that electron donating groups enhance the aryl–silicon bond cleavage of triarylsilanes under acidic conditions, whereas strongly electron-deficient triarylsilanes rapidly decompose under the action of base.28
Figure 3.

(a, left) Hammett plot for catalytic amidation using catalysts 6b–j and 4, and (b, right) plot of substituent constants (σp/m) vs catalyst integrity post-amidation. The red box denotes the catalysts selected for further study.
On the basis of their improved performance compared to parent silanol 4, counterbalanced by only slight-to-moderate loss of catalyst integrity in the range (0.1 ≥ σp/m ≥ 0.3), the tris(p-haloaryl)silanols 6e (p-F), 6f (p-Cl) and 6h (p-Br) were selected for further study, where—of those selected—silanol 6h had been found to provide the highest conversion to amide 3a after 1h reaction time (cf., Figure 2). A comparison of conversion versus time profiles at 10 mol % (Figure 4a) and 30 mol % (Figure 4b) loading for these different catalysts for the model amidation reveals two main features. First, bromide congener 6h displays superior catalytic activity at both loadings and over the entire course of the reaction. Notably, at 30 mol % loading of p-bromo silanol 6h, the initial rate is ca. 10 times faster than the background reaction, and quantitative conversion to amide 3a was observed after 6h. Based on this finding, we selected 30 mol % catalyst loading as our standard loading for further studies (vide infra). Second, we note that the increased loadings give increased initial rates with all catalysts, although the responses are not linear and is indicative of complex kinetic behavior (vide infra).
Figure 4.
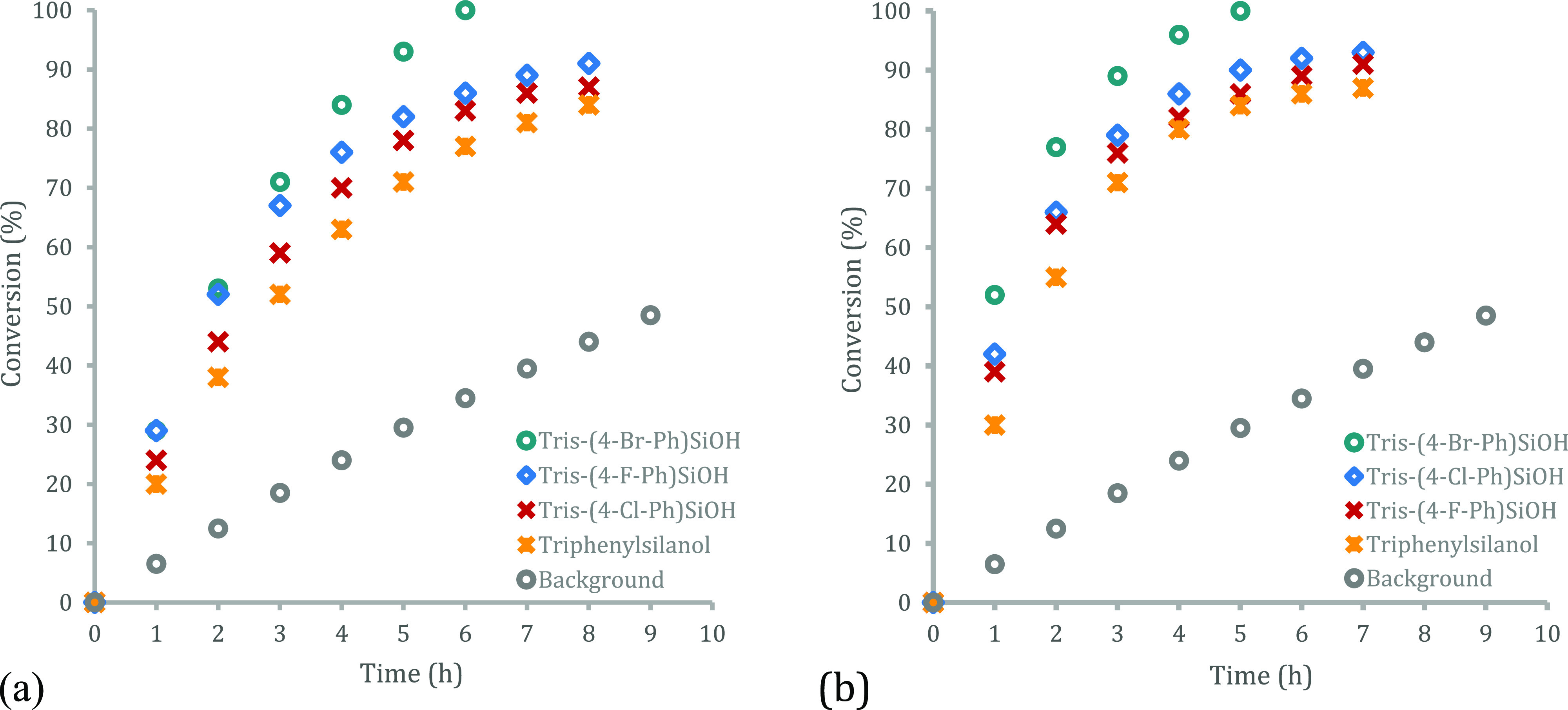
Conversion vs time plots for the model amidation reaction (cf., Figure 1b) with triarylsilanol catalysts 4, 6e, 6f, and 6h at (a, left) 10 mol %, (b, right) and 30 mol % catalyst loadings.
We next elected to explore preparative amidation reactions using 30 mol % catalyst 6h. Phenylacetic acid and benzoic acid were chosen as representative alkyl and aryl carboxylic acids to undergo attempted catalytic amidation with bromosilanol 6h, where aromatic carboxylic acids are known to be more difficult to amidate compared to aliphatic ones.10j 4-Methylbenzylamine, morpholine, and N-methylbenzylamine were chosen as representative primary, cyclic, and acyclic secondary amines, which are increasingly resistant to amidation. Aniline was also selected as an aromatic amine with reduced nucleophilicity. These acid–amine combinations allow benchmarking against catalytic amidation reactions in the literature.
There are very few reports on high-yielding catalytic amidation reactions of aromatic carboxylic acids in the ideal 1:1 acid/amine stoichiometry in the literature,4−9 and perhaps unsurprisingly, catalytic quantities of bromosilanol 6h failed to provide any quantities of amides 3f–3h, which are among the most difficult combinations of acid and amine (Figure 5). Pleasingly, some activity was found for the formation of aromatic amide 3e albeit in low conversion after 24 h. Aliphatic amide 3a was formed in quantitative yield in a reaction time of 6 h, albeit where the background reaction is itself complete in 24 h. This result demonstrates the need to run background reactions when reporting direct amidation reactions. For tertiary amides 3b and 3c, some catalytic activity was observed over the background reaction rate, but the acceleration was only modest, and increasing the concentration did not ameliorate the situation. In these cases, after evaporative removal of the toluene solvent as the first step in the workup procedure, quantities of a white solid in a yellow oil was observed which, when isolated, proved to be hexa(4-bromophenyl)disiloxane,29 characterized for the first time by crystallography (see the Supporting Information), thereby implicating catalyst deactivation by condensation. In contrast, for anilide 3d, the catalyst activity over the background reaction was more marked compared with what might have been expected, with the more nucleophilic secondary amines giving rise to amides 3b and 3c. With these admittedly disappointing results in hand, we sought to conduct additional kinetic experiments to interrogate the system further.30,31
Figure 5.

Substrate scope of catalytic amidation with bromosilanol 6h at 0.2 or 1.0 M concentration in both acid and amine with isolated yields after acid–base workup and dry column vacuum chromatography. The figures in parentheses are isolated yield of a background reaction without catalyst, where the amide products were pure after acid–base workup. Amide 3a precipitated from the reaction mixture on cooling and could be collected by filtration. Amide 3d was triturated with n-hexane to remove unreacted aniline.
Accordingly, a “same excess” experiment32 using catalyst 6h for the model system (cf., Figure 1a) and time-adjusted analysis33 showed that the reaction profiles did not overlay (Figure 6a), in line with our expectations regarding the previously observed loss of catalyst integrity as determined by NMR spectroscopy (cf., Figures 2 and 3) and by the observation of disiloxane formation when using secondary amines. However, a second same excess experiment with added product amide 3a gave plots that nearly overlaid (Figure 6b), showing instead that for this acid–amine combination, amide product inhibition is the dominant factor at play rather than catalyst decomposition, and these findings are also consistent with the determined catalyst order.31 A further reaction with water added instead (Figure 6c) shows that it also contributes to the inhibition (although we expect it is lost as it is generated in refluxing toluene). A fourth experiment (Figure 6d) with both added water and amide gave plots that almost perfectly overlaid. The very slight divergence of this latter plot can therefore be attributed to the catalyst decomposition.
Figure 6.

“Same excess” concentration vs time plots for the model amidation reaction (cf., Figure 1b) (a, top left) red circles t0 = 0.3 M [1a], 0.3 M [2a], and 0.06 M [6h] and time-adjusted green diamonds t3.2 = 0.2 M [1a], 0.2 M [2a], and 0.06 M [6h]; (b, top right) blue triangles with added 0.1 M [3a]; (c, bottom left) brown triangles with added 0.1 M [H2O]; and (d, bottom right) green triangles with added 0.1 M [3a] amide and 0.1 M [H2O].
With this system being established, it was realized that it could be exploited to examine any inhibitory effect of different amides without the need to establish new conditions (Figure 7). Accordingly, each of anilide 3d and tertiary amides 3b and 3c were individually added in further same excess experiments. Inspection of the plots reveal that anilide 3d more strongly inhibits the reaction than amide product 3a itself (Figure 7b). Tertiary amides 3b and 3c (Figure 7d,c respectively) inhibit the reaction even more strongly. The experiments, therefore, demonstrate that while it is to be expected that tertiary amides are inherently harder to form than secondary amides, to their reinforcing detriment, tertiary amide products inhibit the reaction more strongly than secondary amides. These results, therefore, go some way to rationalize the marked differences in reactivity in the catalytic formation of secondary amide 3a versus tertiary amides 3b and 3c (cf., Figure 5).
Figure 7.

“Same excess” concentration vs time plots for the model amidation reaction (cf., Figure 1b) (a, top left) red circles: t0 = 0.3 M [1a], 0.3 M [2a], and 0.06 M [6h]; time-adjusted blue diamonds: t3.2 = 0.2 M [1a], 0.2 M [2a], 0.06 M [6h], and 0.1 M [3a]; (b, top right) orange triangles with added 0.1 M [3d] amide; (c, bottom left) orange triangles with added 0.1 M [3c]; and (d, bottom right) orange triangles with added 0.1 M [3b] amide and 0.1 M [H2O].
Turning now to the question of catalyst decomposition quantification, 1H NMR methods had been used in the initial screening of arylsilanols (cf., Figure 2) to quantify catalyst integrity but suffered from overlapping resonances of catalyst, disiloxane, substrates, and products in the aromatic region of the spectrum. In an effort to overcome this difficulty, a HPLC method was developed for ex situ monitoring of catalyst integrity. Catalyst integrity monitoring experiments (0.06 M 6h in toluene) at reflux, on its own, and with the individual components (0.2 M in acid, amine, or amide) of the model reaction (cf., Figure 1b) showed that the silanol was thermally stable, acid and amide were individually tolerated, and the catalyst concentration did not decrease at all, but the use of primary amine 2a showed ca. 20% catalyst depletion to 0.050 M after 8 h at reflux (see the Supporting Information for data). These experiments show that silanol 6h is sensitive to base-mediated decomposition. For secondary amine N-methylbenzylamine, the catalyst concentration reduced to 0.042 M (ca. 30% catalyst depletion), consistent with the increased basicity of secondary alkyl amines over primary alkyl amines. In contrast, for the same experiment employing aniline, 97% catalyst integrity (0.058 M) was observed at the end of the same period, consistent with its reduced basicity.
Thus, in amidation reactions catalyzed by silanol 6h, it is to be expected that secondary alkyl amines will contribute to more extensive catalyst decomposition than primary alkyl amines. Thus, a second, detrimentally reinforcing—and perhaps now dominant— factor for these amine types is expected to be at play and further explains the poor catalytic conversions of secondary amines into tertiary amides 3b and 3c (cf., Figure 5). In the case of aniline, catalyst decomposition is expected to be less important and rationalizes the modest catalytic activity observed despite aniline’s lower nucleophilicity. In the event, ex situ HPLC monitoring of catalytic amidation runs of phenylacetic acid with 4-methylbenzylamine, morpholine, N-methylbenzylamine, and aniline to give amides 3a–3d with 39, 49, 52, and 13% catalyst depletion, respectively, after 8 h refluxing in toluene (Figure 8). Evidently, under the conditions of the amidation, more severe catalyst depletion occurs than with each individual amine alone. We infer that acid and amine must act in conjunction to effect catalyst decomposition, and the more basic the amine, the more severe the depletion. Catalyst decomposition in the case of secondary amines is further accentuated by prolonged high concentrations of acid and base since the conversion to tertiary amide products is low.
Figure 8.

Plot of [6h] vs time in catalytic amidation reactions of phenyl acetic acid with 4-methylbenzylamine, N-methylbenzylamine, morpholine, and aniline as monitored by HPLC methods.
To elucidate the mechanism of these triarylsilanol-catalyzed direct amidation reactions, silyl ester 7—as a putative intermediate34 in the catalytic cycle—was prepared by the modification of a known procedure from triphenylsilane35 (Scheme 1a). Attempted formation of amide 3a via the stoichiometric reaction of silyl ester 7 with amine 2a in toluene at room temperature led unexpectedly instead to the immediate formation of a white precipitate, which we formulate as silanaminium carboxylate salt 8.36 In support of this formulation, a precipitate that behaved identically was obtained when silyl amine 9 was combined in a stoichiometric fashion with carboxylic acid 1a. To our surprise, redissolution of salt 8 in rigorously dried chloroform-d gave a clean 1:1 mixture of ester 7 and amine 2a. This mixture evolved over several days at room temperature to produce a mixture of amide 3a, silanol 4, disiloxane 10, and ammonium carboxylate salt 11. Here, we invoke productive attack of amine 2a on ester 7 to give amide 3a, thereby releasing free silanol 4. Amine-mediated condensation of silanol 4 with a second equivalent of ester evidently allows the formation of disiloxane 10, thereby concomitantly producing the ammonium carboxylate salt 11. In accord with these observations, attempts to crystallize the silanaminium salt 8 from toluene, chloroform, or dichloromethane provided only ammonium carboxylate salt 11. In contrast, the mixing of adamantane-1-carboxylic acid with silyl amine 9 allowed for the crystallization (CH2Cl2/hexane) of adamantyl silyl ester 12. Inspection of the residual solution showed that no amide formation had occurred, showing that all the other downstream species must arise from the initial nucleophilic attack on silyl ester 7 (itself immediately regenerated on dissolution of the silanaminium salt 8), where attack of 12 is prohibited under these conditions for steric reasons. These experiments, therefore, implicate silyl ester 7 as the key intermediate in the formation of amide 3a, which is also evidently energetically accessible from silanol 4 under the conditions of the catalytic reactions and supported by computations.37 We invoke a mechanistic scenario that allows for any Lewis basic lone pair in the system to attack at the acyl carbon or at the silicon center of silyl ester 7 (Scheme 1b). Thus, attack at the acyl carbon by amine 2a irreversibly gives product 3a, whereas attack at Si gives silyl amine 9; attack by water at either C or Si return silanol 4 and carboxylic acid 1a; attack by silanol 4 at Si irreversibly gives disiloxane 10, while attack at C is a degenerate exchange. Computations also helped to identify the possible mechanisms for amide inhibition as a Lewis base by interaction with the Si center; two of these are shown in Scheme 1b as amide-silyl ester dispersion complex 13 and silyl imidate 14.37,38 The high calculated free energy of silyl imidate 14 does not support an inhibitory mechanism, whereas dispersion complex 13 is a better candidate. Finally, the use of catalytic quantities (10 mol %) of either silyl ester 7, silanaminium salt 8 or silyl amine 9 were all found to provide identical levels of conversion in the model reaction as triphenylsilanol 4 itself (cf., Figure 1b) and is consistent with our mechanistic proposal.39
Scheme 1. (a, Top) Preparation of Silyl Ester 7 and Silyl Amine 9 and Chemistry of the Resulting Silanaminium Carboxylate Salt 8 and (b, Bottom) Plausible Catalytic Cycle.
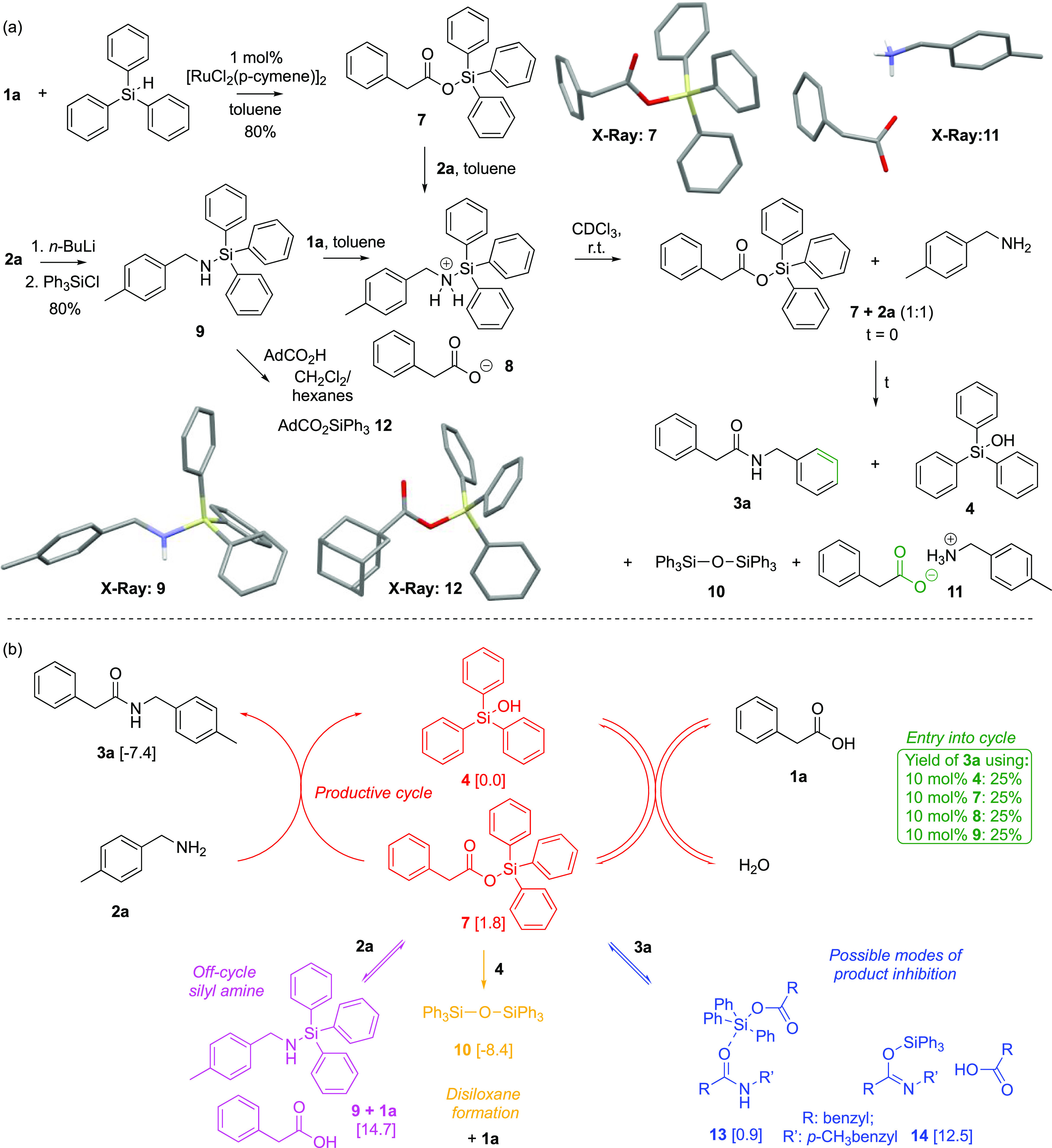
Numbers in square brackets are computed relative free energies ΔG384 in kcal/mol @ 4 atm. standard state (∼0.13 M) for discrete species involved in the catalytic cycle using the B3LYP+GD3+BJ/Def-TZVPP/SCRF = toluene density functional procedure (see ref (37)).
Conclusions
In conclusion, we have discovered that triarylsilanols act as the first silicon-centered catalysts for direct amidation of carboxylic acids with amines. Synthesis and screening of a range of electronically differentiated triaryl silanols identified tris(p-haloaryl)silanols as more active catalysts than the parent triphenylsilanol, where the bromide congener was found to be the most active. Although catalyst degradation had been observed by inspection of the final product mixtures by NMR spectroscopy, same excess experiments instead identify amide product inhibition as the dominant detrimental effect in these catalytic runs, and is testament to the power of RPKA methods. Given that most catalytic direct amidation reactions involve Lewis acidic catalysts, Lewis basic amide product inhibition may be widespread.13b Tertiary amide products were found to be more inhibitory than secondary amide products, thereby detrimentally reinforcing the inherent lower reactivity of secondary amines compared to primary amines in direct amidation reactions. As observed above, we expect this effect to be universal in other direct amidation reactions catalyzed by Lewis acidic catalysts. In addition, the silanol catalyst was found to be subject to condensation to catalytically inactive disiloxane under the reaction conditions, the extent of which correlated with the basicity of the amine employed. Since secondary alkyl amines are more basic than primary alkyl amines, this further detrimentally impacts on the use of secondary amines to form tertiary amide products. Studies using an authentically synthesized triaryl silylester as a putative intermediate in the catalytic system, and supported by calculations, enable a plausible mechanism of amidation to be proposed whereby the Lewis acidic silicon and acyl carbon centers of the silyl ester are able to interact with any Lewis basic species, including the amide product.
Experimental Section
General Considerations
Unless otherwise noted, reagents were purchased from commercial sources and used as received. Di-t-butylhydridosilanol (A1),40 dicyclohexanesilanediol (B2),41 1,1,3,3-tetra-t-butyldisiloxane-1,3-diol (C2),42 tris(4-N,N-dimethylaminophenyl)silane (5a),19a tris(4-chlorophenyl)silane (5f),19d tris(3-chlorophenyl)silane (5g),19e tris(3-(trifluoromethyl)phenyl)silane (5i),19g and tetrakis(trimethylsilyl)methane were available as previously synthesized legacy chemicals in the Lickiss Group, Imperial College London. t-Butyldimethylsilanol (A4), diphenylsilanediol (B3), and 1,1,3,3-tetraphenyldisiloxane-1,3-diol (C1), incompletely condensed silsesquioxanes D1 and D2 and triphenylsilanol (4) were commercially available and used as received. 4-Methylbenzylamine was distilled under reduced pressure from CaH2 and stored in Schlenk tubes over activated 4 Å molecular sieves.
All reactions were carried out in oven-dried glassware, under an inert atmosphere of nitrogen, unless otherwise stated. Reaction temperatures other than ambient temperature were achieved using DrySyn heating blocks, dry ice/acetone bath (−78 °C), and ice/water bath (0 °C). Volatiles were removed in vacuo by rotary evaporation. Kieselgel-60 F254 pre-coated aluminum-backed plates were used for analytical thin layer chromatography and visualized using UV light (254 nm) or chemical staining with basic aqueous potassium permanganate solution.
1H NMR (400 MHz), 13C{1H} NMR (101MHz), 19F/19F{1H} NMR (377 MHz), and 29Si{1H} NMR (80 MHz) spectra were recorded at 298 K on a Bruker AV400 spectrometer. Chemical shifts (δ) are reported in ppm relative to solvent signals (δ = 7.26 and 77.16 ppm for CDCl3). Coupling constants (J) are quoted in Hz. Abbreviations used for multiplicity are as follows: s—singlet, d—doublet, dd—doublet of doublets, ddd—doublet of doublet of doublets, ddt—doublet of doublet of triplets, t—triplet, tt—triplet of triplets, app.t—apparent triplet, q—quartet, app. quint—apparent quintet, br—broad, m—multiplet. Structural assignments were made with additional information from gCOSY, gHSQC, and gHMBC experiments. Melting points were measured using a Lambda Photometrics MPA 100 OptiMelt melting point apparatus. High-resolution mass spectroscopy (HRMS) spectra were recorded by the Imperial College Department of Chemistry Mass Spectrometry Service. X-ray crystal structures were determined using an Agilent Xcalibur 3E diffractometer; for details, see the Supporting Information.
Procedures for the Synthesis of Molecular Silica Analogues A2, A3, A5, B1, B4, and B5
The appropriate silane (10.0 mmol) was added dropwise to a vigorously stirred solution of triethylamine (2.8 mL, 20.1 mmol) in Et2O (30.0 mL) and H2O (1.0 mL). After 45 min, the mixture was transferred to a separating funnel containing an aqueous solution of HCl (1.0 M, 21.0 mL, 21.0 mmol). The aqueous layer was extracted with Et2O (3 × 10.0 mL), the combined organic portions were washed with brine, dried over MgSO4, filtered, and the volatiles were evaporated.
t-Butyldiphenylsilanol (A2)43
t-Butyldiphenylsilanol was prepared using t-butyl(chloro)diphenylsilane (2.6 mL, 10.0 mmol). Then, it was purified by silica plug and eluted with hexane, to remove non-polar components, and then with THF to yield t-butyldiphenylsilanol (A2) (2.36 g, 9.2 mmol, 92%) as a white solid; mp 59.4–60.3 °C. ATR–FTIR 3256 cm–1; 1H NMR (400 MHz, CDCl3): δ 7.73–7.71 (m, 4H), 7.44–7.36 (m, 6H), 1.88 (br s, 1H), 1.07 (s, 9H); 13C{1H} NMR (101 MHz, CDCl3): δ 135.3, 134.9, 129.8, 127.9, 26.7, 19.2; 29Si{1H} NMR (80 MHz, CDCl3): δ −5.6; HRMS (EI+) m/z: [M]+• calcd for C16H20OSi, 256.1283; found, 256.1282.
Triethylsilanol (A3)44
Triethylsilanol (A3) was prepared using triethylchlorosilane (1.7 mL, 10.0 mmol). Then, it was purified by silica plug and eluted with hexane, to remove non-polar components, and then with Et2O to yield triethylsilanol (A3) (1.16 g, 8.8 mmol, 88%) as a colorless oil. ATR–FTIR 3293 cm–1; 1H NMR (400 MHz, CDCl3): δ 2.70 (s, 1H), 0.94 (t, J = 8.0 Hz, 9H), 0.56 (q, J = 8.0 Hz, 6H); 13C{1H} NMR (101 MHz, CDCl3): δ 6.7, 5.9; 29Si{1H} NMR (80 MHz, CDCl3): δ 19.0; HRMS (EI+) m/z: [M]+• calcd for C6H16OSi, 132.0970; found, 132.0970.
Methyldiphenylsilanol (A5)45
Methyldiphenylsilanol (A5) was prepared using chloro(methyl)diphenylsilane (2.1 mL, 10.0 mmol). Then, it was purified by silica plug and eluted with hexane, to remove non-polar components, and then with Et2O to yield methyldiphenylsilanol (A5) (2.1 g, 9.8 mmol, 98%) as a colorless oil. ATR–FTIR 3278 cm–1; 1H NMR (400 MHz, CDCl3): δ 7.65–7.63 (m, 4H), 7.49–7.45 (m, 2H), 7.43–7.39 (m, 4H), 3.28 (br s, 1H), 0.67 (s, 3H); 13C{1H} NMR (101 MHz, CDCl3): δ 137.1, 134.1, 129.9, 128.0, −1.2; 29Si{1H} NMR (80 MHz, CDCl3): δ −2.8; HRMS (EI+) m/z: [M]+• calcd for C13H14OSi, 214.0814; found, 214.0813.
Di-i-propylsilanediol (B1)46
Di-i-propylsilanediol (B1) was prepared using diisopropyldichlorosilane (1.8 mL, 10.0 mmol), followed by recrystallization from (hexane/CH2Cl2, 8:2) to yield di-i-propylsilanediol (B1) (1.29 g, 8.6 mmol, 86%) as colorless needles; mp 113.2–114.4 °C. ATR–FTIR 3235 cm–1; 1H NMR (400 MHz, CDCl3): δ 2.05 (s, 2H), 0.99 (d, J = 6.5 Hz, 12H), 1.02–0.93 (m, 2H); 13C{1H} NMR (101 MHz, CDCl3): δ 17.0, 12.7; 29Si{1H} NMR (80 MHz, CDCl3): δ −3.7; HRMS (EI+) m/z: [M]+• calcd for C6H16O2Si, 148.0920; found, 148.0923.
Procedure for the Synthesis of Di(naphthalen-1-yl)silanediol (B4)14a
To a solution of 1-bromonaphthalene (7.0 mL, 50.0 mmol) in Et2O (300 mL) at −78 °C was added n-BuLi (2.43 M, 41.15 mL, 100 mmol) at a rate of 3 mL/min. The resulting pale yellow-orange solution was stirred at −78 °C for 1 h, allowed to warm to room temperature and stirred for 2 h. Subsequently, the solution was re-cooled to −78 °C and transferred dropwise by cannula to a solution of silicon tetrachloride (2.86 mL, 25 mmol) in Et2O (200 mL) at −78 °C with immediate precipitation of a white solid. After complete transfer, the mixture was allowed to warm to room temperature and stirred for 48 h. The solvent was removed in vacuo to afford a yellow oil containing a white precipitate, and a saturated solution of aqueous NaHCO3 was added to neutralize the mixture to pH 7. The mixture was extracted with Et2O (3 × 25 mL), the combined organics were washed with brine, dried over MgSO4, filtered, and evaporated. Trituration (hexane/Et2O, 8:2) afforded di(naphthalen-1-yl)silanediol (B4) (6.59 g, 23.3 mmol, 93%) as a white solid; mp 155.2–156.2 °C. ATR–FTIR 3565, 3353 cm–1; 1H NMR (400 MHz, methanol-d4): δ 8.41 (d, J = 8.3 Hz, 2H), 7.97 (dd, J = 6.8, 1.3 Hz, 2H), 7.90 (dd, J = 8.3, 1.1 Hz, 2H), 7.82 (dd, J = 7.7, 1.8 Hz, 2H), 7.46–7.33 (m, 6H), 4.90 (s, 2H); 13C{1H} NMR (101 MHz, methanol-d4): δ 138.4, 136.3, 136.2, 134.8, 131.6, 130.2, 129.6, 126.7, 126.4, 126.0; 29Si{1H} NMR (80 MHz, methanol-d4): δ −29.2; HRMS (ES–, TOF) m/z: [M – H]− calcd for C20H15O2Si, 315.0841; found, 315.0837.
Procedure for the Synthesis of Di-t-butylsilanediol (B5)47
A solution of di-t-butylhyridosilanol (A1) (96 mg, 0.6 mmol) in THF (5 mL) was added dropwise over 10 min to an aqueous solution of NaOH (7.7 M, 248 μL, 1.9 mmol) with vigorous stirring and open to air to allow the escape of the hydrogen generated. Once the addition was complete, the mixture was monitored by TLC (hexane/Et2O, 9:1) until complete disappearance of the starting silane was observed. The solvent was removed in vacuo, and aqueous HCl solution (1.0 M, 2 mL, 2 mmol) was added to the resulting emulsion. Et2O (5 mL) was added, and the aqueous phase was extracted with Et2O (2 × 5 mL). The combined organics were washed with brine, dried over MgSO4, filtered, and evaporated to yield a white solid. The solid was purified by silica plug and eluted with hexane, to remove non-polar components, and then with Et2O to give di-t-butylsilanediol (B5) (103 mg, 0.58 mmol, 97%) as colorless crystals; mp 151.3–152.3 °C. ATR–FTIR 3293 cm–1; 1H NMR (400 MHz, CDCl3): δ 2.17 (br s, 2H), 1.04 (s, 18H); 13C{1H} NMR (101 MHz, CDCl3): δ 27.4, 19.9; 29Si{1H} NMR (80 MHz, CDCl3): δ −7.1; HRMS (ES–, TOF) m/z: [M – H + HCO2H]− calcd for C9H21O4Si, 221.1209; found, 221.1202.
General Procedure for the Molecular Silica Analogue and Silanol Screening (Figures 1 and 2)
Phenylacetic acid 1a (136 mg, 1.0 mmol), 4-methylbenzylamine 2a (130 μL, 1.0 mmol), molecular silicon analogues A1–5, B1–5, C1–2, D1–2 or triarylsilanols 4, 6a–k (0.1 mmol), and toluene (5.0 mL) were placed in a 10 mL round-bottom flask equipped with a stirrer bar. The mixture was purged under a flow of nitrogen for 5 min before bringing to reflux for 1 h. The reaction mixture was allowed to cool, and the solvent was removed in vacuo. CDCl3 was added until a homogeneous solution was observed at which point decamethylcyclopentasiloxane (10 μL, 0.0258 mmol) was added as an internal standard. A 1H NMR spectrum was recorded, and conversions were calculated by integration against the internal standard. Reactions were conducted in triplicate, and the average conversion is reported. For the use of silanols 4, 6a–k catalyst integrity was also determined by integration against the internal standard.
General Procedures A1–A3 and B1–B3 for the Formation of Triarylsilanes and Silanols, Respectively
General Procedure A1
A solution of aryl bromide (18 mmol) in Et2O (10 mL) was added over 5 min to magnesium turnings (437 mg, 18 mmol) that had been vigorously pre-stirred for 1 h. The reaction mixture was stirred as described below, and the resulting solution was cooled to −78 °C and trichlorosilane (0.45 mL, 4.5 mmol) was added. After stirring as described below, the solvent was removed in vacuo and aqueous HCl solution (0.1 M, 10 mL, 1 mmol) was added. The aqueous phase was extracted with Et2O (3 × 5 mL), and the combined organics were washed with brine, dried over MgSO4, filtered, and evaporated.
General Procedure A2
To a stirred solution of arylbromide (18 mmol) in Et2O (25 mL) was added a solution of n-BuLi in hexane (2.27 M, 8.13 mL, 18.5 mmol) at −78 °C. After allowing the reaction mixture to stir at 0 °C for 1 h, then room temperature for 1.5 h, it was cooled to −78 °C, and trichlorosilane (0.45 mL, 4.5 mmol) was added dropwise. The reaction mixture was stirred at −78 °C for 2 h, allowed to warm to room temperature for 2 h, and aqueous HCl solution (0.1 M, 10 mL, 1 mmol) was added. The layers were separated, the aqueous phase was extracted with Et2O (3 × 5 mL), and the combined organics were washed with brine, dried over MgSO4, filtered, and evaporated.
General Procedure A3
To a stirred solution of arylbromide (10 mmol) in anhydrous Et2O (30 mL) held at −78 °C was added a solution of iPrMgCl in THF (1.61 M, 6.06 mL, 10 mmol) over 30 min. After stirring at −78 °C for 30 min, trichlorosilane (0.30 mL, 3 mmol) was added dropwise and the reaction mixture was allowed to warm to room temperature overnight. The precipitate formed was removed by filtration, and the solvent was removed in vacuo.
General Procedure B1
A solution of the silane in CH3CN was added to a stirred solution of [Ru(p-cymene)Cl2]2 (1.8 mg, 0.01 mmol) in CH3CN (1 mL) and H2O (138 μL). The solution was brought to 80 °C, stirred as detailed below, and the solvent was evaporated.
General Procedure B2
A solution of silane (2.0 mmol) in THF (10 mL) was added dropwise over 10 min to an aqueous solution of NaOH (7.7 M, 600 μL, 4.62 mmol) while vigorously stirring in the open air to allow the escape of generated H2. Once TLC monitoring showed the reaction was complete, the solvent was removed in vacuo and aqueous HCl solution (1.0 M, 5 mL, 5 mmol) was added to the resulting emulsion. Et2O (10 mL) was added, the layers were separated, and the aqueous phase was further extracted with Et2O (3 × 5 mL). The combined organics were washed with brine, dried over MgSO4, filtered, and evaporated.
General Procedure B3
10 wt % Pd/C (100 mg, 0.1 mmol) and H2O (100 μL, 5.5 mmol) were added sequentially to a solution of silane (1.0 mmol) in THF (5 mL) at room temperature with immediate evolution of gas. The mixture was stirred for 45 min, passed through a plug of silica, eluting with CH2Cl2, and the solvent was evaporated.
Tris(4-methoxyphenyl)silane (5b)19b
Tris(4-methoxyphenyl)silane (5b) was prepared according to general procedure A1 with stirring for 2 h at room temperature for Grignard formation and 4 h at reflux for aryl Grignard addition. Purification by column chromatography (hexane/Et2O, 1:1), gave tris(4-methoxyphenyl)silane (5b) (400 mg, 1.13 mmol, 25%) as a colorless oil, that over weeks solidified into a white solid; mp 75.2–76.1 °C. ATR–FTIR 2114 cm–1; 1H NMR (400 MHz, CDCl3): δ 7.48 (d, J = 8.6 Hz, 6H), 6.92 (d, J = 8.6 Hz, 6H), 5.41 (s, 1JSi–H = 196.5 Hz 1H), 3.82 (s, 9H); 13C{1H} NMR (101 MHz, CDCl3): δ 161.1, 137.4, 125.0, 114.0, 55.2; 29Si{1H} NMR (80 MHz, CDCl3): δ −19.3. A mass spectrum molecular ion could not be observed by electron or chemical ionization techniques.
Tri-p-tolylsilane (5c)19c
Tri-p-tolylsilane (5c) was prepared according to general procedure A1 with stirring for 5 h at room temperature for Grignard formation and 4 h at reflux for aryl Grignard addition. Purification by column chromatography (hexane) gave tri-p-tolylsilane (5c) (542 mg, 1.8 mmol, 40%) as a white solid; mp 81.6–83.0 °C. ATR–FTIR 2114 cm–1; 1H NMR (400 MHz, CDCl3): δ 7.51 (d, J = 7.7 Hz, 6H), 7.23 (d, J = 7.7 Hz, 6H), 5.47 (s, 1JSi–H = 197.6 Hz, 1H), 2.40 (s, 9H); 13C{1H} NMR (101 MHz, CDCl3): δ 139.8, 135.9, 130.3, 129.0, 21.7; 29Si{1H} NMR (80 MHz, CDCl3): δ −18.6.
Tris(4-(t-butyl)phenyl)silane (5d)19d
Tris(4-(t-butyl)phenyl)silane (5d) was prepared according to general procedure A2 followed by recrystallization from hexane to give tris(4-(t-butyl)phenyl)silane (5d) (1.54 g, 3.6 mmol, 80%) as a colorless solid; mp 168.4–169.7 °C. 1H NMR (400 MHz, CDCl3): δ 7.55–7.52 (m, 6H), 7.41–7.38 (m, 6H), 5.42 (s, 1JSi–H = 197.1 Hz, 1H), 1.32 (s, 27H); 13C{1H} NMR (101 MHz, CDCl3): δ 152.7, 135.8, 130.4, 125.1, 34.9, 31.4; 29Si{1H} NMR (80 MHz, CDCl3): δ −19.2; HRMS (EI+) m/z: [M]+• calcd for C30H40Si, 428.2899; found, 428.2916.
Tris(4-fluorophenyl)silane (5e)19c
Tris(4-fluorophenyl)silane (5e) was prepared according to general procedure A1 with stirring at 0 °C, followed by reflux for 1 h to complete Grignard formation and 16 h at room temperature for aryl Grignard addition. Purification by column chromatography (hexane) gave tris(4-fluorophenyl)silane (5e) (1.13 g, 3.6 mmol, 80%) as a colorless oil that over time crystallized as colorless crystals; mp 42.3–43.9 °C. ATR–FTIR 2129 cm–1; 1H NMR (400 MHz, CDCl3): δ 7.67–7.62 (m, 6H), 7.21–7.16 (m, 6H), 5.61 (s, 1JSi–H = 201.8 Hz, 1H); 13C{1H} NMR (101 MHz, CDCl3): δ 164.5 (d, 1JC–F = 250.3 Hz, para), 137.9 (d, 3JC–F = 7.7 Hz, ortho), 128.6 (d, 4JC–F = 3.6 Hz, ipso), 115.7 (d, 2JC–F = 20.0 Hz, meta); 19F NMR (377 MHz, CDCl3): δ −109.6 (tt, 3JF–H = 9.2 Hz, 4JF–H = 5.6 Hz); 29Si{1H} NMR (80 MHz, CDCl3): δ −18.8; HRMS (EI+) m/z: [M – H]+ calcd for C18H12SiF3, 313.0660; found, 313.0670.
Tris(4-bromophenyl)silane (5h)19f
Tris(4-bromophenyl)silane (5h) was prepared according to general procedure A2 using 1,4-dibromobenzene (4.72 g, 20.0 mmol), n-BuLi (2.5 M in hexane, 8.00 mL, 20.0 mmol), and trichlorosilane (0.61 mL, 6.0 mmol). Recrystallization from hexane gave tris(4-bromophenyl)silane (5h) (2.68 g, 5.4 mmol, 90%) as white crystalline needles; mp 109.9–110.5 °C. 1H NMR (400 MHz, CDCl3): δ 7.53 (d, J = 8.2 Hz, 6H), 7.37 (d, J = 8.2 Hz, 6H), 5.38 (s, 1JSi–H = 204.1 Hz, 1H); 13C{1H} NMR (101 MHz, CDCl3): δ 137.3, 131.7, 131.1, 125.5; 29Si{1H} NMR (80 MHz, CDCl3): δ −18.4; HRMS (EI+) m/z: [M]+• calcd for C18H1279Br3Si, 492.8258; found, 492.8271.
Tris(4-(trifluoromethyl)phenyl)silane (5j)19d
Tris(4-(trifluoromethyl)phenyl)silane (5j) was prepared according to general procedure A1, using THF (9 mL) as the reaction solvent, with stirring at room temperature for 1 h followed by reflux for 1 h to complete Grignard formation and 16 h at room temperature for aryl Grignard addition. Purification by column chromatography (hexane) gave tris(4-(trifluoromethyl)phenyl)silane (5j) (1.17 g, 2.52 mmol, 56%) as a colorless solid; mp 74.5–75.3 °C. ATR–FTIR 2162 cm–1; 1H NMR (400 MHz, CDCl3): δ 7.71–7.67 (m, 12H), 5.60 (s, 1JSi–H = 206.5 Hz, 1H); 13C{1H} NMR (101 MHz, CDCl3): δ 136.4, 136.2, 132.7 (q, 2JC–F = 32.4 Hz), 125.1 (q, 3JC–F = 3.7 Hz), 124.1 (q, 1JC–F = 272.5 Hz); 19F{1H} NMR (377 MHz, CDCl3): δ −63.2; 29Si{1H} NMR (80 MHz, CDCl3): δ −18.6; HRMS (EI+) m/z: [M]+• calcd for C21H13F9Si, 464.0643; found, 464.0629.
Tris(3,4,5-trifluorophenyl)silane (5k)
Tris(3,4,5-trifluorophenyl)silane (5k) was prepared according to general procedure A3, followed by recrystallization from hexane to give tris(3,4,5-trifluorophenyl)silane (5k) (773 mg, 1.83 mmol, 61%) as colorless crystals; mp 80–82 °C. 1H NMR (400 MHz, CDCl3): δ 7.10–7.06 (app. t, JH–F = 6.8 Hz, 6H), 5.39 (s, 1JSi–H = 215 Hz, 1H); 13C{1H} NMR (101 MHz, CDCl3): δ 151.9 (ddd, 1JC–F = 256.5 Hz, 2JC–F = 9.9 Hz, 3JC–F = 2.6 Hz, meta), 141.8 (dt, 1JC–F = 257.6 Hz, 2JC–F = 14.9 Hz, para), 126.7 (q, JC–F = 4.1 Hz, ipso), 119.4 (dd, 2JC–F = 14.3 Hz, 3JC–F = 5.5 Hz, ortho); 19F{1H} NMR (377 MHz, CDCl3): δ −132.3 (d, JF–F = 20.5 Hz, meta), −155.4 (t, JF–F = 20.5 Hz, para); 29Si{1H} NMR (80 MHz, CDCl3): δ −16.9; HRMS (EI+) m/z: [M – H]+ calcd for C18H6SiF9, 421.0095; found, 421.0103.
Tris(4-N,N-dimethylaminophenyl)silanol (6a)19a
Tris(4-N,N-dimethylaminophenyl)silanol (6a) was prepared according to general procedure B1 using silane 5a (599 mg, 1.54 mmol) in CH3CN (2.5 mL) for 24 h. Purification by column chromatography (hexane/EtOAc, 9:1) gave tris-(4-N,N-dimethylaminophenyl)silanol (6a) (475 mg, 1.174 mmol, 76%) as a white solid; mp 183.0–183.7 °C. 1H NMR (400 MHz, CDCl3): δ 7.54 (d, J = 8.7 Hz, 6H), 6.75 (d, J = 8.7 Hz, 6H), 2.99 (br s, 18H), 2.39 (s, 1H); 13C{1H} NMR (101 MHz, CDCl3): δ 151.4, 136.4, 122.2, 111.8, 40.3; 29Si{1H} NMR (80 MHz, CDCl3): δ −10.6; HRMS (ES+, TOF) m/z: [M + H]+ calcd for C24H32N3OSi, 406.2315; found, 406.2316.
Tris(4-methoxyphenyl)silanol (6b)
Tris(4-methoxyphenyl)silanol (6b) was prepared according to general procedure B1 using an added solution of silane 5b (175 mg, 0.5 mmol) in CH3CN (1.5 mL) for 5 h. Purification by silica plug, followed by eluting with hexane to remove non-polar components and then with CH2Cl2 gave tris(4-methoxyphenyl)silanol (6b) (124 mg, 0.375 mmol, 71%) as a colorless oil. ATR–FTIR 3349 cm–1; 1H NMR (400 MHz, CDCl3): δ 7.54 (d, J = 8.5 Hz, 6H), 6.92 (d, J = 8.5 Hz, 6H), 3.82 (s, 9H), 2.38 (s, 1H); 13C{1H} NMR (101 MHz, CDCl3): δ 161.3, 136.7, 127.0, 113.8, 55.2; 29Si{1H} NMR (80 MHz, CDCl3): δ −11.7; HRMS (ES–, TOF) m/z: [M – H]− calcd for C21H21O4Si, 365.1209; found, 365.1213.
Tris-p-tolylsilanol (6c)21b
Tris-p-tolylsilanol (6c) was prepared according to general procedure B2 using silane 5c on a 1.5 mmol scale. Purification by silica plug, followed by eluting with hexane, to remove non-polar components, and then with Et2O gave tri-p-tolylsilanol (6c) (449 mg, 1.41 mmol, 94%) as a white solid; mp 100.7–101.2 °C. 1H NMR (400 MHz, CDCl3): δ 7.54 (d, J = 7.7 Hz, 6H), 7.22 (d, J = 7.7 Hz, 6H), 2.58 (s, 1H), 2.40 (s, 9H); 13C{1H} NMR (101 MHz, CDCl3): δ 140.1, 135.2, 132.1, 128.8, 21.7; 29Si{1H} NMR (80 MHz, CDCl3): δ −11.8; HRMS (EI+) m/z: [M]+• calcd for C21H22OSi, 318.1440; found, 318.1447.
Tris(4-(t-butyl)phenyl)silanol (6d)
Tris(4-(t-butyl)phenyl)silanol (6d) was prepared according to general procedure B3 using silane 5d to give tris(4-(t-butyl)phenyl)silanol (6d) (444 mg, 1.0 mmol, 100%) as a white solid; mp 226–227 °C. ATR–FTIR 3662 cm–1; 1H NMR (400 MHz, CDCl3): δ 7.60 (d, J = 8.3 Hz, 6H), 7.41 (d, J = 8.3 Hz, 6H), 2.39 (s, 1H), 1.33 (s, 27H); 13C{1H} NMR (101 MHz, CDCl3): δ 153.1, 135.0, 132.2, 125.0, 34.9, 31.4; 29Si{1H} NMR (80 MHz, CDCl3): δ −12.4; HRMS (APCI–) m/z: [M – H]− calcd for C30H39OSi, 443.2765; found, 443.2753.
Tris(4-fluorophenyl)silanol (6e)21c
Tris(4-fluorophenyl)silanol (6e) was prepared according to general procedure B2 using silane 5e on a 0.6 mmol scale. Purification by silica plug, followed by eluting with hexane, to remove non-polar components, and then with Et2O gave tris(4-fluorophenyl)silanol (6e) (190 mg, 0.58 mmol, 96%) as a white solid; mp 88.2–89.7 °C. ATR–FTIR 3207 cm–1; 1H NMR (400 MHz, CDCl3): δ 7.56 (ddt, J = 8.4, 6.1, 2.3 Hz, 6H), 7.12–7.07 (m, 6H), 2.72 (s, 1H); 13C{1H} NMR (101 MHz, CDCl3): δ 164.6 (d, 1JC–F = 250.6 Hz, para), 137.2 (d, 3JC–F = 7.8 Hz, ortho), 130.5 (d, 4JC–F = 4.3 Hz, ipso), 115.5 (d, 2JC–F = 19.9 Hz, meta); 19F{1H} NMR (377 MHz, CDCl3): δ −109.4; 29Si{1H} NMR (80 MHz, CDCl3): δ −12.86; HRMS (ES–, TOF) m/z: [M – H]− calcd for C18H12OSiF3, 329.0610; found, 329.0613.
Tris(4-chlorophenyl)silanol (6f)21d
Tris(4-chlorophenyl)silanol (6f) was prepared according to general procedure B2 using silane 5f. Purification by silica plug and eluting with hexane, removing non-polar components, and then Et2O gave tris(4-chlorophenyl)silanol (6f) (713 mg, 1.88 mmol, 94%) as a white solid; mp 119.9–120.7 °C. ATR–FTIR 3144 cm–1; 1H NMR (400 MHz, CDCl3): δ 7.51–7.48 (m, 6H), 7.40–7.37 (m, 6H), 2.64 (s, 1H); 13C{1H} NMR (101 MHz, CDCl3): δ 137.2, 136.3, 132.6, 128.6; 29Si{1H} NMR (80 MHz, CDCl3): δ −12.9.
Tris(3-chlorophenyl)silanol (6g)
Tris(3-chlorophenyl)silanol (6g) was prepared according to general procedure B2 using silane 5g on a 1.9 mmol scale. Purification by silica plug, followed by eluting with hexane, to remove non-polar components, and then with Et2O gave tris(3-chlorophenyl)silanol (6g) (728 mg, 1.9 mmol, 100%) as a colorless oil. ATR–FTIR 3262 cm–1; 1H NMR (400 MHz, CDCl3): δ 7.55–7.54 (m, 3H), 7.46–7.42 (m, 6H), 7.35–7.31 (m, 3H), 3.34 (s, 1H); 13C{1H} NMR (101 MHz, CDCl3): δ 136.4, 134.8, 134.6, 132.9, 130.9, 129.8; 29Si{1H} NMR (80 MHz, CDCl3): δ −14.6; HRMS (EI+) m/z: [M]+• calcd for C18H13OSi35Cl3, 377.9801; found, 377.9816.
Tris(4-bromophenyl)silanol (6h)21c
Tris(4-bromophenyl)silanol (6h) was prepared according to general procedure B2 using silane 5h. Purification by silica plug, followed by eluting with hexane, to remove non-polar components, and then with Et2O gave tris(4-bromophenyl)silanol (6h) (995 mg, 1.9 mmol, 97%) as a white solid; mp 125.9–126.5 °C. ATR–FTIR 3155 cm–1; 1H NMR (400 MHz, CDCl3): δ 7.54 (d, J = 8.4 Hz, 6H), 7.42 (d, J = 8.4 Hz, 6H), 2.56 (s, 1H); 13C{1H} NMR (101 MHz, CDCl3): δ 136.5, 133.0, 131.5, 125.9; 29Si{1H} NMR (80 MHz, CDCl3): δ −12.6; HRMS (ES–, TOF) m/z: [M – H]− calcd for C18H12OSi79Br3, 508.8208; found, 508.8219.
Tris(3-(trifluoromethyl)phenyl)silanol (6i)
Tris(3-(trifluoromethyl)phenyl)silanol (6i) was prepared according to general procedure B2 using silane 5i on a 0.6 mmol scale. Purification by silica plug, followed by eluting with hexane, to remove non-polar components, and then with Et2O gave tris(3-(trifluoromethyl)phenyl)silanol (6i) (273 mg, 0.57 mmol, 95%) as a white solid; mp 82–83 °C. ATR–FTIR 3202 cm–1; 1H NMR (400 MHz, CDCl3): δ 7.89–7.88 (m, 3H), 7.77–7.74 (m, 6H), 7.58–7.54 (m, 3H), 2.83 (s, 1H); 13C{1H} NMR (101 MHz, CDCl3): δ 138.3, 135.0, 131.3 (q, J = 3.6 Hz), 131.0 (q, J = 32.1 Hz), 128.8, 127.7 (q, J = 3.7 Hz), 124.2 (q, J = 272.5 Hz); 19F{1H} NMR (377 MHz, CDCl3): δ −62.9; 29Si{1H} NMR (80 MHz, CDCl3): δ −14.6; HRMS (ES–, TOF) m/z: [M – H]− calcd for C21H12OF9Si, 479.0514; found, 479.0506.
Tris(4-(trifluoromethyl)phenyl)silanol (6j)
Tris(4-(trifluoromethyl)phenyl)silanol (6j) was prepared according to general procedure B2 using silane 5j on a 1.5 mmol scale. Purification by silica plug, followed by eluting with hexane, to remove non-polar components, and then with Et2O gave tris(4-(trifluoromethyl)phenyl)silanol (6j) (595 mg, 1.24 mmol, 85%) as a white solid; mp 101.8–102.9 °C. ATR–FTIR 3194 cm–1; 1H NMR (400 MHz, CDCl3): δ 7.74 (d, J = 7.8 Hz, 6H), 7.68 (d, J = 7.8 Hz, 6H), 3.19 (s. 1H); 13C{1H} NMR (101 MHz, CDCl3): δ 138.3, 135.3, 132.8 (q, J = 32.3 Hz), 125.0 (q, J = 3.8 Hz), 124.0 (q, J = 272.5 Hz); 19F{1H} NMR (377 MHz, CDCl3): δ −63.2; 29Si{1H} NMR (79 MHz, CDCl3): δ −14.5; HRMS (ES–, TOF) m/z: [M – H]− calcd for C21H12OSiF9, 479.0514; found, 479.0504.
Tris(3,4,5-trifluorophenyl)silanol (6k)
Tris(3,4,5-trifluorophenyl)silanol (6k) was prepared according to general procedure B2 using silane 5k on a 0.6 mmol scale to give tris-(3,4,5-trifluorophenyl)silanol (6k) (269 mg, 0.6 mmol, 99%) as a white solid; mp 266–268 °C. ATR–FTIR 3222 cm–1; 1H NMR (400 MHz, CDCl3): δ 7.14 (app. t, JH–F = 6.9 Hz, 6H), 2.98 (s, 1H); 13C{1H} NMR (101 MHz, CDCl3): δ 151.8 (ddd, 1JC–F = 256.8 Hz, 2JC–F = 10.4 Hz, 3JC–F = 2.6 Hz, meta), 142.0 (dt, 1JC–F = 257.5 Hz, 2JC–F = 15.1 Hz, para), 127.8 (q, J = 3.4 Hz, ipso), 118.7 (dd, 2JC–F = 13.9 Hz, 3JC–F = 5.4 Hz, ortho); 19F NMR (377 MHz, CDCl3): δ −132.4 (dd, 3JF–F = 20.0, 3JF–H = 6.3 Hz, meta), −155.2 (tt, 3JF–F = 20.0, 4JF–H = 6.9 Hz, para); 29Si{1H} NMR (80 MHz, CDCl3): δ −15.9; HRMS (ES–, TOF) m/z: [M – H]− calcd for C18H6OSiF9, 437.0044; found, 437.0032.
General Procedure for Conversion versus Time Plots with Catalytic Quantities of Silanols 4, 6e–f, and 6h (Figure 4)
Phenylacetic acid (0.41 g, 3.0 mmol), 4-methylbenzylamine (0.38 mL, 3.0 mmol), triarylsilanols 4, 6e, 6f, 6h (10 or 30 mol %), tetrakis(trimethylsilyl)methane (30 mg, 0.098 mmol) as internal standard, and toluene (15 mL) were placed within a 100 mL two neck round-bottom flask. The mixture was purged under a flow of nitrogen for 5 min before bringing to reflux. After the elapsed time, an aliquot from the reaction mixture was taken and transferred to a glass vial. Chloroform-d was added until a homogeneous solution was observed at which point the 1H NMR spectra were recorded. Conversions were calculated by integration against the internal standard. Reactions were conducted in triplicate, and the average conversions are reported.
General Procedure for Silanol 6h Catalyzed Preparation of Amides (Figure 5)
Carboxylic acid (5.0 mmol), amine (5.0 mmol), tris(4-bromophenyl)silanol 6h (0.76 g, 1.5 mmol, 30 mol %), and toluene (25 mL) were charged into a 1000 mL round-bottom flask. This mixture was purged under a flow of nitrogen for 5 min after which the mixture was heated to reflux for 24 h. The solution was then allowed to cool and concentrated in vacuo. The residue was diluted with EtOAc (20 mL), and the resulting solution was washed with aqueous NaOH (25 mL, 0.4 M) and aqueous HCl (25 mL, 1.0 M) solutions. The organic layer was dried over MgSO4, filtered, and evaporated. The residue was purified by dry column vacuum chromatography (2–8% EtOAc in petroleum ether) to yield amide products.
N-(4-Methylbenzyl)-2-phenylacetamide (3a)10j
Following the general procedure, the product precipitated from the reaction upon cooling. Collection by filtration and washing with toluene gave amide 3a (0.72 g, 100%) as a white solid; mp 139.0–140.0 °C (lit.10j mp 139.0–139.3 °C); 1H NMR (400 MHz, CDCl3): δ 7.37–7.32 (m, 2H), 7.31–7.27 (m, 3H), 7.11–7.06 (m, 4H), 5.64 (br s, 1H), 4.37 (d, J = 5.7 Hz, 2H), 3.63 (s, 2H), 2.31 (s, 3H); 13C{1H} NMR (101 MHz, CDCl3): δ 170.9, 137.2, 135.1, 134.9, 129.6, 129.4, 129.1, 127.6, 127.5, 44.0, 43.5, 21.2.
1-Morpholino-2-phenylethan-1-one (3b)10j
Following the general procedure gave amide 3b (0.21 g, 1.0 mmol, 20%) as a pale yellow crystalline solid; mp 65.8–66.8 °C (lit.10j mp 65–67 °C); 1H NMR (400 MHz, CDCl3): δ 7.37–7.34 (m, 2H), 7.30–7.27 (m, 3H), 3.76 (s, 2H), 3.67 (s, 4H), 3.52–3.49 (m, 2H), 3.47–3.45 (m, 2H); 13C{1H} NMR (101 MHz, CDCl3): δ 169.7, 134.8, 128.8, 128.6, 126.9, 66.8, 66.5, 46.6, 42.2, 40.9.
N-Benzyl-N-methyl-2-phenylacetamide (3c)10j
Following the general procedure gave amide 3c (0.12 g, 0.5 mmol, 10%) as a colorless oil; 1H NMR (400 MHz, CDCl3): δ major rotamer 7.40–7.10 (m, 10H), 4.65 (s, 2H), 3.82 (s, 2H), 2.93 (s, 3H); minor rotamer 7.40–7.10 (m, 10H), 4.56 (s, 2H), 3.79 (s, 2H), 2.99 (s, 3H); 13C{1H} NMR (101 MHz, CDCl3): δ 171.5, 171.1, 137.3, 136.5, 135.1, 134.9, 128.9, 128.8, 128.7, 128.6, 128.0, 127.6, 127.3, 126.8, 126.4, 53.6, 50.9, 41.2, 40.9, 35.2, 34.0.
N,2-Diphenylacetamide (3d)10j
Following the general procedure followed by trituration with n-hexane gave amide 3d (0.35 g, 1.6 mmol, 30%) as a white solid; mp 113.4–114.8 °C (lit.10j mp 113–115 °C); 1H NMR (400 MHz, CDCl3): δ 7.43–7.39 (m, 4H), 7.36–7.33 (m, 3H), 7.30–7.28 (m, 2H), 7.12 (br s, 1H), 7.07 (t, J = 7.4 Hz, 1H), 3.74 (s, 2H); 13C{1H} NMR (101 MHz, CDCl3): δ 169.2, 137.7, 134.6, 129.6, 129.4, 129.0, 127.8, 124.6, 119.9, 45.0.
N-(4-Methylbenzyl)benzamide (3e)10j
Following the general procedure gave amide 13 (90 mg, 0.4 mmol, 8%) as a white solid; mp 140.3–141 °C (lit.10j mp 140–141 °C); 1H NMR (400 MHz, CDCl3): δ 7.80 (dd, J = 7.1, 1.7 Hz, 2H), 7.55–7.49 (m, 1H), 7.48–7.41 (m, 2H), 7.30–7.25 (m, 2H), 7.19 (d, J = 7.8 Hz, 2H), 6.40 (br s, 1H), 4.63 (d, J = 5.6 Hz, 2H), 2.37 (s, 3H); 13C{1H} NMR (101 MHz, CDCl3): δ 167.4, 137.5, 135.3, 134.6, 131.5, 129.5, 128.6, 128.0, 127.1, 44.0, 21.2.
General Procedure for Silanol 6h Catalyzed Concentration versus Time Plots (Figures 6 and 7)
Phenylacetic acid (0.41g, 3.0 mmol, 1 equiv), 4-methylbenzylamine (0.38 mL, 3.0 mmol, 1 equiv), tris(4-bromophenyl)silanol 6h (0.46 g, 0.9 mmol, 30 mol %), tetrakis(trimethylsilyl)methane (30 mg, 0.098 mmol) as internal standard, and toluene (15 mL, 0.2 M) were charged into a 100 mL two-neck round-bottomed flask. N-(4-Methylbenzyl)-2-phenylacetamide 3a (0.36 g, 1.5 mmol, 0.1 M) and/or H2O (27 μL, 1.5 mmol, 0.1 M) or amides 3b–d (1.5 mmol, 0.1 M) were added, and the mixture was purged under a flow of N2 for 5 min before bringing to reflux. After the elapsed time, an aliquot from the reaction mixture was taken and transferred to a glass vial. Chloroform-d was added until a homogeneous solution was observed at which point 1H NMR spectra were recorded. Concentrations were calculated by integration against the internal standard. Reactions were conducted in triplicate, and the average concentrations are reported.
General Procedure for Determining Catalyst 6h Integrity (Figure 8)
Phenylacetic acid (0.41 g, 3.0 mmol, 1 equiv), amine (3.0 mmol, 1 equiv), tris(4-bromophenyl)silanol 6h (0.46 g, 0.9 mmol, 30 mol %), durene (30 mg, 0.2 mmol) as internal standard, and toluene (15 mL, 0.2 M) were charged into a 100 mL two-neck round-bottom flask. The mixture was purged under a flow of N2 for 5 min before bringing to reflux for the required time. After the elapsed time, an aliquot (100 μL) was taken and added to iPrOH (1000 μL) to give a homogeneous solution. Samples were analyzed on a reverse phase 25 cm × 4.6 mm, 5 μm SUPELCOSIL LC-18 column @ 222/230 nm, 80:20 MeCN/H2O, flow rate 2.0 mL/min. Catalyst integrity was determined by integration against the internal standard and by reference to a pre-determined calibration curve.
Triphenylsilyl 2-Phenylacetate 7
A stirred solution of triphenylsilane (2.63 g, 10.1 mmol), phenylacetic acid (1.37 g, 10.1 mmol), and [Ru(p-cymene)Cl2]2 (62 mg, 0.10 mmol) in toluene (15 mL) was held at 50 °C for 18 h. The solution turned from pale orange to deep red/brown over this time. The solvent was evaporated, and the solid residue was dissolved in Et2O (50 mL) and passed through a column of dry activated carbon. The solvent was evaporated, and recrystallization from hexane gave triphenylsilyl 2-phenylacetate (7) (3.18 g, 8.08 mmol, 80%) as colorless blocks; mp 91.2–92.0 °C. 1H NMR (400 MHz, CDCl3): δ 7.61–7.59 (m, 6H), 7.49–7.44 (m, 3H), 7.39–7.36 (m, 6H), 7.34–7.29 (m, 5H), 3.79 (s, 2H); 13C{1H} NMR (101 MHz, CDCl3): δ 171.2, 135.8, 134.2, 132.1, 130.6, 129.6, 128.7, 128.0, 127.2, 43.3; 29Si{1H} NMR (80 MHz, CDCl3): δ −9.9; HRMS (EI+) m/z: [M]+• calcd for C26H22O2Si, 394.1389; found, 394.1395.
N-(4-Methylbenzyl)(triphenylsilyl)amine 9
To a solution 4-methylbenzylamine (113 μL, 0.87 mmol) in hexane (10 mL) at −78 °C was added dropwise a solution of n-BuLi in hexane (2.35 M, 370 μL, 0.87 mL) over a 5 min period to form a dull pink solution. Triphenylchlorosilane (258 mg, 0.87 mmol) as a solution in Et2O (10 mL) was added dropwise, forming a pale-yellow solution with a white precipitate. The reaction mixture was stirred for 2 h at room temperature, the solvent was evaporated, and the resulting solid was triturated with boiling hexane. The product crystallized from the triturate to give N-(4-methylbenzyl)(triphenylsilyl)amine (9) (263 mg, 0.70 mmol, 80%) as a colorless crystalline solid; mp 85 °C. 1H NMR (400 MHz, CDCl3): δ 7.71–7.68 (m, 6H), 7.48–7.44 (m, 3H), 7.43–7.39 (m, 6H), 7.23 (d, J = 7.8 Hz, 2H), 7.14 (d, J = 7.8 Hz, 2H), 4.11 (d, J = 7.5 Hz, 2H), 2.37 (s, 3H), 1.55 (t, J = 7.5 Hz, 1H); 13C NMR (101 MHz, CDCl3): δ 140.6, 136.2, 135.7, 135.1, 129.8, 129.1, 128.0, 127.2, 46.5, 21.2; 29Si NMR (80 MHz, CDCl3): δ −16.0; HRMS (EI+) m/z: [M]+• calcd for C26H25NSi, 379.1756; found, 379.1760.
Acknowledgments
We thank The Pharmacat Consortium and Pfizer for a studentship (to B.C.R), the Punjab Educational Endowment Fund (PEEF) for financial support (to R.Q.), and Jian Kwan for the initial preparation and crystallization of ester 7. We thank the EPSRC for the grant EP/T030534/1 (to H.S.R.).
Data Availability Statement
The data underlying this study are available in the published article and its online Supporting Information as well as openly available as part of the PhD thesis of B. C. Rowley in the Imperial College London Institutional Repository (Spiral) at https://doi.org/10.25560/78224. Computational data are openly available at the Imperial College research data repository at DOI: 10.14469/hpc/12480.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.3c00585.
Copies of NMR spectra for new compounds, catalyst integrity and VTNA plots, and X-ray crystallographic data (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Allen C. L.; Williams J. M. Metal-Catalysed Approaches to Amide Bond Formation. Chem. Soc. Rev. 2011, 40, 3405–3415. 10.1039/C0CS00196A. [DOI] [PubMed] [Google Scholar]; b Pattabiraman V. R.; Bode J. W. Rethinking Amide Bond Synthesis. Nature 2011, 480, 471–479. 10.1038/nature10702. [DOI] [PubMed] [Google Scholar]; c Lundberg H.; Tinnis F.; Selander N.; Adolfsson H. Catalytic Amide Formation from Non-Activated Carboxylic Acids and Amines. Chem. Soc. Rev. 2014, 43, 2714–2742. 10.1039/C3CS60345H. [DOI] [PubMed] [Google Scholar]; d Sabatini M. T.; Boulton L.; Sneddon H. F.; Sheppard T. D. A Green Chemistry Perspective on Catalytic Amide Bond Formation. Nat. Catal. 2019, 2, 10–17. 10.1038/s41929-018-0211-5. [DOI] [Google Scholar]; e Wang X. Challenges and Outlook for Catalytic Direct Amidation Reactions. Nat. Catal. 2019, 2, 98–102. 10.1038/s41929-018-0215-1. [DOI] [Google Scholar]; f Todorovic M.; Perrin D. M. Recent Developments in Catalytic Amide Bond Formation. Pept. Sci. 2020, 112, e24210 10.1002/pep2.24210. [DOI] [Google Scholar]; g Taussat A.; de Figueiredo R. M.; Campagne J.-M. Direct Catalytic Amidations from Carboxylic Acid and Ester Derivatives: A Review. Catalysts 2023, 13, 366. 10.3390/catal13020366. [DOI] [Google Scholar]
- a Constable D. J. C.; Dunn P. J.; Hayler J. D.; Humphrey G. R.; Leazer J. L. Jr.; Linderman R. J.; Lorenz K.; Manley J.; Pearlman B. A.; Wells A.; Zaks A.; Zhang T. Y. Key Green Chemistry Research Areas—A Perspective From Pharmaceutical Manufacturers. Green Chem. 2007, 9, 411–420. 10.1039/B703488C. [DOI] [Google Scholar]; b Bryan M. C.; Dunn P. J.; Entwistle D.; Gallou F.; Koenig S. G.; Hayler J. D.; Hickey M. R.; Hughes S.; Kopach M. E.; Moine G.; Richardson P.; Roschangar F.; Steven A.; Weiberth F. J. Key Green Chemistry Research Areas From A Pharmaceutical Manufacturers’ Perspective Revisited. Green Chem. 2018, 20, 5082–5103. 10.1039/C8GC01276H. [DOI] [Google Scholar]
- For selected recent reviews see:; a Lanigan R. M.; Sheppard T. D. Recent Developments in Amide Synthesis: Direct Amidation of Carboxylic Acids and Transamidation Reactions. Eur. J. Org. Chem. 2013, 2013, 7453–7465. 10.1002/ejoc.201300573. [DOI] [Google Scholar]; b de Figueiredo R. M.; Suppo J.-S.; Campagne J.-M. Nonclassical Routes for Amide Bond Formation. Chem. Rev. 2016, 116, 12029–12122. 10.1021/acs.chemrev.6b00237. [DOI] [PubMed] [Google Scholar]; c Ojeda-Porras A.; Gamba-Sánchez D. Recent Developments in Amide Synthesis using Nonactivated Starting Materials. J. Org. Chem. 2016, 81, 11548–11555. 10.1021/acs.joc.6b02358. [DOI] [PubMed] [Google Scholar]; d Muramatsu W.; Hattori T.; Yamamoto H. Game Change from Reagent-To Substrate-Controlled Peptide Synthesis. Bull. Chem. Soc. Jpn. 2020, 93, 759–767. 10.1246/bcsj.20200057. [DOI] [Google Scholar]; e Santos A. S.; Silva A. M.; Marques M. M. B. Sustainable Amidation Reactions–Recent Advances. Eur. J. Org. Chem. 2020, 2020, 2501–2516. 10.1002/ejoc.202000106. [DOI] [Google Scholar]; f Massolo E.; Pirola M.; Benaglia M. Amide bond formation strategies: Latest Advances on a Dateless Transformation. Eur. J. Org. Chem. 2020, 2020, 4641–4651. 10.1002/ejoc.202000080. [DOI] [Google Scholar]; g Pedrood K.; Bahadorikhalili S.; Lotfi V.; Larijani B.; Mahdavi M. Catalytic and Non-Catalytic Amidation of Carboxylic Acid Substrates. Mol. Divers. 2021, 26, 1311–1344. 10.1007/s11030-021-10252-0. [DOI] [PubMed] [Google Scholar]; h Muramatsu W.; Hattori T.; Yamamoto H. Amide Bond Formation: Beyond the Dilemma between Activation and Racemisation. Chem. Commun. 2021, 57, 6346–6359. 10.1039/D1CC01795K. [DOI] [PubMed] [Google Scholar]
- Aryl and alkyl boronic and borinic acids, representative examples:; a Ishihara K.; Ohara S.; Yamamoto H. 3, 4, 5-Trifluorobenzeneboronic Acid as an Extremely Active Amidation Catalyst. J. Org. Chem. 1996, 61, 4196–4197. 10.1021/jo9606564. [DOI] [PubMed] [Google Scholar]; b Maki T.; Ishihara K.; Yamamoto H. New Boron (III)-Catalyzed Amide and Ester Condensation Reactions. Tetrahedron 2007, 63, 8645–8657. 10.1016/j.tet.2007.03.157. [DOI] [Google Scholar]; c Arnold K.; Batsanov A. S.; Davies B.; Whiting A. Synthesis, Evaluation and Application of Novel Bifunctional N,N-di-isopropylbenzylamineboronic Acid Catalysts for Direct Amide Formation between Carboxylic Acids and Amines. Green Chem. 2008, 10, 124–134. 10.1039/B712008G. [DOI] [Google Scholar]; d Al-Zoubi R. M.; Marion O.; Hall D. G. Direct and Waste-Free Amidations and Cycloadditions by Organocatalytic Activation of Carboxylic Acids at Room Temperature. Angew. Chem., Int. Ed. 2008, 47, 2876–2879. 10.1002/anie.200705468. [DOI] [PubMed] [Google Scholar]; e Gernigon N.; Al-Zoubi R. M.; Hall D. G. Direct Amidation of Carboxylic Acids Catalyzed by ortho-iodo Arylboronic Acids: Catalyst Optimization, Scope, and Preliminary Mechanistic Study Supporting a Peculiar Halogen Acceleration Effect. J. Org. Chem. 2012, 77, 8386–8400. 10.1021/jo3013258. [DOI] [PubMed] [Google Scholar]; f Gernigon N.; Zheng H.; Hall D. G. Solid-Supported ortho-iodoarylboronic Acid Catalyst for Direct Amidation of Carboxylic Acids. Tetrahedron Lett. 2013, 54, 4475–4478. 10.1016/j.tetlet.2013.06.043. [DOI] [Google Scholar]; g Yamashita R.; Sakakura A.; Ishihara K. Primary Alkylboronic Acids as Highly Active Catalysts for the Dehydrative Amide Condensation of α-Hydroxycarboxylic Acids. Org. Lett. 2013, 15, 3654–3657. 10.1021/ol401537f. [DOI] [PubMed] [Google Scholar]; h Fatemi S.; Gernigon N.; Hall D. G. A Multigram-Scale Lower E-Factor Procedure for MIBA-Catalyzed Direct Amidation and its Application to the Coupling of Alpha And Beta Aminoacids. Green Chem. 2015, 17, 4016–4028. 10.1039/C5GC00659G. [DOI] [Google Scholar]; i Tam E. K. W.; Liu L. Y.; Liu L. Y.; Chen A. 2-Furanylboronic Acid as an Effective Catalyst for the Direct Amidation of Carboxylic Acids at Room Temperature. Eur. J. Org. Chem. 2015, 2015, 1100–1107. 10.1002/ejoc.201403468. [DOI] [Google Scholar]; j Mohy El Dine T.; Erb W.; Berhault Y.; Rouden J.; Blanchet J. Catalytic Chemical Amide Synthesis at Room Temperature: One more Step toward Peptide Synthesis. J. Org. Chem. 2015, 80, 4532–4544. 10.1021/acs.joc.5b00378. [DOI] [PubMed] [Google Scholar]; k El Dine T. M.; Rouden J.; Blanchet J. Borinic acid Catalysed Peptide Synthesis. Chem. Commun. 2015, 51, 16084–16087. 10.1039/C5CC06177F. [DOI] [PubMed] [Google Scholar]; l Ishihara K.; Lu Y. Boronic Acid–DMAPO Cooperative Catalysis for Dehydrative Condensation between Carboxylic Acids and Amines. Chem. Sci. 2016, 7, 1276–1280. 10.1039/C5SC03761A. [DOI] [PMC free article] [PubMed] [Google Scholar]; m Lu Y.; Wang K.; Ishihara K. Design of Boronic Acid–Base Complexes as Reusable Homogeneous Catalysts in Dehydrative Condensations between Carboxylic Acids and Amines. Asian J. Org. Chem. 2017, 6, 1191–1194. 10.1002/ajoc.201700194. [DOI] [Google Scholar]; n Wang K.; Lu Y.; Ishihara K. The ortho-Substituent on 2,4-bis(trifluoromethyl)phenylboronic Acid Catalyzed Dehydrative Condensation between Carboxylic Acids and Amines. Chem. Commun. 2018, 54, 5410–5413. 10.1039/C8CC02558D. [DOI] [PubMed] [Google Scholar]; o Du Y.; Barber T.; Lim S. E.; Rzepa H. S.; Baxendale I. R.; Whiting A. A Solid-Supported Arylboronic Acid Catalyst for Direct Amidation. Chem. Commun. 2019, 55, 2916–2919. 10.1039/C8CC09913H. [DOI] [PubMed] [Google Scholar]; p Al-Zoubi R. M.; Al-Jammal W. K.; McDonald R. Regioselective Synthesis of ortho-iodobiphenylboronic Acid Derivatives: A Superior Catalyst for Carboxylic Acid Activation. New J. Chem. 2020, 44, 3612–3623. 10.1039/C9NJ05708K. [DOI] [Google Scholar]; q Zhou J.; Paladino M.; Hall D. G. Direct Boronic Acid Promoted Amidation of Carboxylic Acids with Poorly Nucleophilic Amines. Eur. J. Org. Chem. 2022, 2022, e202201050 10.1002/ejoc.202201050. [DOI] [Google Scholar]; r Pan B.; Huang D.-M.; Sun H.-T.; Song S. N.; Su X. B. Heterocyclic Boron Acid Catalyzed Dehydrative Amidation of Aliphatic/Aromatic Carboxylic Acids with Amines. J. Org. Chem. 2023, 88, 2832–2840. 10.1021/acs.joc.2c02515. [DOI] [PubMed] [Google Scholar]
- Boric acid, representative examples:; a Tang P. Boric Acid Catalyzed Amide Formation from Carboxylic Acids and Amines: N-Benzyl-4-phenylbutyramide. Org. Synth. 2005, 81, 262–272. 10.1002/0471264229.os081.28. [DOI] [Google Scholar]; b Anderson J. E.; Davis R.; Fitzgerald R. N.; Haberman J. M. Selective Phenol Alkylation: An Improved Preparation of Efaproxiral. Synth. Commun. 2006, 36, 2129–2133. 10.1080/00397910600636527. [DOI] [Google Scholar]; c Mylavarapu R. K.; Gcm K.; Kolla N.; Veeramalla R.; Koilkonda P.; Bhattacharya A.; Bandichhor R. Boric acid Catalyzed Amidation in the Synthesis of Active Pharmaceutical Ingredients. Org. Process Res. Dev. 2007, 11, 1065–1068. 10.1021/op700098w. [DOI] [Google Scholar]; d Janvier M.; Moebs-Sanchez S.; Popowycz F. Bio-Based Amides from Renewable Isosorbide by a Direct and Atom-Economic Boric Acid Amidation Methodology. Eur. J. Org. Chem. 2016, 2016, 2308–2318. 10.1002/ejoc.201600186. [DOI] [Google Scholar]; e Tang P.; Yuan Q.; Yun F.; Cheng C.; Zhang J.; Li J.; Liu X.; Xie R. Boric Acid Catalyzed Direct Amidation between Amino-Azaarenes and Carboxylic Acids. Synthesis 2016, 49, 1583–1596. 10.1055/s-0036-1588126. [DOI] [Google Scholar]
- Boronates, recent representative examples:; a Sabatini M. T.; Boulton L. T.; Sheppard T. D. Borate Esters: Simple Catalysts for the Sustainable Synthesis of Complex Amides. Sci. Adv. 2017, 3, e1701028 10.1126/sciadv.1701028. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Sabatini M. T.; Karaluka V.; Lanigan R. M.; Boulton L. T.; Badland M.; Sheppard T. D. Protecting-Group-Free Amidation of Amino Acids Using Lewis Acid Catalysts. Chem.—Eur. J. 2018, 24, 7033–7043. 10.1002/chem.201800372. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Ghorpade S. A.; Sawant D. N.; Sekar N. Triphenyl Borate Catalyzed Synthesis of Amides from Carboxylic Acids and Amines. Tetrahedron 2018, 74, 6954–6958. 10.1016/j.tet.2018.10.030. [DOI] [Google Scholar]; d Coomber C. E.; Laserna V.; Martin L. T.; Smith P. D.; Hailes H. C.; Porter M. J.; Sheppard T. D. Catalytic Direct Amidations in tert-Butyl Acetate Using B(OCH2CF3)3. Org. Biomol. Chem. 2019, 17, 6465–6469. 10.1039/C9OB01012B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- B3NO2 heterocycles, diborons, diboronic acids and anhydrides, representative examples:; a Noda H.; Furutachi M.; Asada Y.; Shibasaki M.; Kumagai N. Unique Physicochemical and Catalytic Properties Dictated by the B3NO2 Ring System. Nat. Chem. 2017, 9, 571–577. 10.1038/nchem.2708. [DOI] [PubMed] [Google Scholar]; b Liu Z.; Noda H.; Shibasaki M.; Kumagai N. Catalytic Oligopeptide Synthesis. Org. Lett. 2018, 20, 612–615. 10.1021/acs.orglett.7b03735. [DOI] [PubMed] [Google Scholar]; c Sawant D. N.; Bagal D. B.; Ogawa S.; Selvam K.; Saito S. Diboron-Catalyzed Dehydrative Amidation of Aromatic Carboxylic Acids with Amines. Org. Lett. 2018, 20, 4397–4400. 10.1021/acs.orglett.8b01480. [DOI] [PubMed] [Google Scholar]; d Opie C. R.; Noda H.; Shibasaki M.; Kumagai N. All Non-Carbon B3NO2 Exotic Heterocycles: Synthesis, Dynamics, and Catalysis. Chem.—Eur. J. 2019, 25, 4648–4653. 10.1002/chem.201900715. [DOI] [PubMed] [Google Scholar]; e Shimada N.; Hirata M.; Koshizuka M.; Ohse N.; Kaito R.; Makino K. Diboronic Acid Anhydrides as Effective Catalysts for the Hydroxy-Directed Dehydrative Amidation of Carboxylic Acids. Org. Lett. 2019, 21, 4303–4308. 10.1021/acs.orglett.9b01484. [DOI] [PubMed] [Google Scholar]; f Michigami K.; Sakaguchi T.; Takemoto Y. Catalytic Dehydrative Peptide Synthesis with gem-Diboronic Acids. ACS Catal. 2019, 10, 683–688. 10.1021/acscatal.9b03894. [DOI] [Google Scholar]; g Koshizuka M.; Makino K.; Shimada N. Diboronic Acid Anhydride-Catalyzed Direct Peptide Bond Formation enabled by Hydroxy-Directed Dehydrative Condensation. Org. Lett. 2020, 22, 8658–8664. 10.1021/acs.orglett.0c03252. [DOI] [PubMed] [Google Scholar]
- a Sugi Y.; Terada Y.; Ieda N.; Komura K. Multivalent Metal Salts as Versatile Catalysts for the Amidation of Long-Chain Aliphatic Acids with Aliphatic Amines. Synthesis 2008, 2008, 2318–2320. 10.1055/s-2008-1067168. [DOI] [Google Scholar]; b Allen C. L.; Chhatwal A. R.; Williams J. M. Direct Amide Formation from Unactivated Carboxylic Acids and Amines. Chem. Commun. 2012, 48, 666–668. 10.1039/C1CC15210F. [DOI] [PubMed] [Google Scholar]; c Adolfsson H.; Lundberg H.; Tinnis F. Titanium(IV) Isopropoxide as an Efficient Catalyst for Direct Amidation of Nonactivated Carboxylic Acids. Synlett 2012, 23, 2201–2204. 10.1055/s-0032-1316993. [DOI] [Google Scholar]; d Lundberg H.; Tinnis F.; Adolfsson H. Direct Amide Coupling of Non-activated Carboxylic Acids and Amines Catalysed by Zirconium(IV) Chloride. Chem.—Eur. J. 2012, 18, 3822–3826. 10.1002/chem.201104055. [DOI] [PubMed] [Google Scholar]; e Tinnis F.; Lundberg H.; Adolfsson H. Direct Catalytic Formation of Primary and Tertiary Amides from Non-Activated Carboxylic Acids, Employing Carbamates as Amine Source. Adv. Synth. Catal. 2012, 354, 2531–2536. 10.1002/adsc.201200436. [DOI] [Google Scholar]; f Lundberg H.; Adolfsson H. Hafnium-Catalyzed Direct Amide Formation at Room Temperature. ACS Catal. 2015, 5, 3271–3277. 10.1021/acscatal.5b00385. [DOI] [Google Scholar]; g Wang Z.; Bao X.; Xu M.; Deng Z.; Han Y.; Wang N. Direct Formation of Amides from Carboxylic Acids and Amines Catalyzed by Niobium(V) Oxalate Hydrate. ChemistrySelect 2018, 3, 2599–2603. 10.1002/slct.201800204. [DOI] [Google Scholar]; h Cheng L.; Ge X.; Huang L. Direct amidation of Non-Activated Phenylacetic Acid and Benzylamine Derivatives Catalysed by NiCl2. R. Soc. Open Sci. 2018, 5, 171870. 10.1098/rsos.171870. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Li N.; Wang L.; Zhang L.; Zhao W.; Qiao J.; Xu X.; Liang Z. Air-stable Bis(pentamethylcyclopentadienyl) Zirconium Perfluorooctanesulfonate as an Efficient and Recyclable Catalyst for the Synthesis of N-substituted Amides. ChemCatChem 2018, 10, 3532–3538. 10.1002/cctc.201800590. [DOI] [Google Scholar]; j De Azambuja F.; Parac-Vogt T. N. Water-tolerant and Atom Economical Amide Bond Formation by Metal-Substituted Polyoxometalate Catalysts. ACS Catal. 2019, 9, 10245–10252. 10.1021/acscatal.9b03415. [DOI] [Google Scholar]; k Lundberg H.; Tinnis F.; Adolfsson H. Zirconium Catalyzed Amide Formation without Water Scavenging. Appl. Organomet. Chem. 2019, 33, e5062 10.1002/aoc.5062. [DOI] [Google Scholar]; l Muramatsu W.; Yamamoto H. Tantalum-Catalyzed Amidation of Amino Acid Homologues. J. Am. Chem. Soc. 2019, 141, 18926–18931. 10.1021/jacs.9b08415. [DOI] [PubMed] [Google Scholar]; m Muramatsu W.; Hattori T.; Yamamoto H. Substrate-Directed Lewis-Acid Catalysis for Peptide Synthesis. J. Am. Chem. Soc. 2019, 141, 12288–12295. 10.1021/jacs.9b03850. [DOI] [PubMed] [Google Scholar]; n Wang H.; Dong W.; Hou Z.; Cheng L.; Li X.; Huang L. Direct Amidation of Non-Activated Carboxylic Acid and Amine Derivatives Catalyzed by TiCp2Cl2. Appl. Organomet. Chem. 2020, 34, e5568 10.1002/aoc.5568. [DOI] [Google Scholar]; o Ali M. A.; Nath A.; Jannat M.; Islam M. M. Direct Synthesis of Diamides from Dicarboxylic Acids with Amines Using Nb2O5 as a Lewis Acid Catalyst and Molecular Docking Studies as Anticancer Agents. ACS Omega 2021, 6, 25002–25009. 10.1021/acsomega.1c04069. [DOI] [PMC free article] [PubMed] [Google Scholar]; p Zhang Y.; de Azambuja F.; Parac-Vogt T. N. Zirconium Oxo Clusters as Discrete Molecular Catalysts for the Direct Amide Bond Formation. Catal. Sci. Technol. 2022, 12, 3190–3201. 10.1039/D2CY00421F. [DOI] [Google Scholar]; q Wang A.; Xie Y.; Wang J.; Shi D.; Yu H. Atom-Economic Amide Synthesis by Using an Iron-substituted Polyoxometalate Catalyst. Chem. Commun. 2022, 58, 1127–1130. 10.1039/D1CC05417A. [DOI] [PubMed] [Google Scholar]
- For recent representative examples see:; a Lenstra D. C.; Rutjes F. P.; Mecinović J. Triphenylphosphine-Catalysed Amide Bond Formation between Carboxylic Acids and Amines. Chem. Commun. 2014, 50, 5763–5766. 10.1039/C4CC01861C. [DOI] [PubMed] [Google Scholar]; b Krause T.; Baader S.; Erb B.; Gooßen L. J. Atom-Economic Catalytic Amide Synthesis from Amines and Carboxylic Acids Activated in situ with Acetylenes. Nat. Commun. 2016, 7, 11732–11737. 10.1038/ncomms11732. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Ganesan S.; Kothandapani J.; Ganesan A. Nano-Magnetic Sulfonic Acid Catalyzed Facile Synthesis of Diverse Amide Derivatives. Synthesis 2016, 49, 685–692. 10.1055/s-0036-1588319. [DOI] [Google Scholar]; d Philpott H. K.; Thomas P. J.; Tew D.; Fuerst D. E.; Lovelock S. L. A Versatile Biosynthetic Approach to Amide Bond Formation. Green Chem. 2018, 20, 3426–3431. 10.1039/C8GC01697F. [DOI] [Google Scholar]; e Manova D.; Gallier F.; Tak-Tak L.; Yotava L.; Lubin-Germain N. Lipase-Catalyzed Amidation of Carboxylic Acid and Amines. Tetrahedron Lett. 2018, 59, 2086–2090. 10.1016/j.tetlet.2018.04.049. [DOI] [Google Scholar]; f Potadar S. M.; Mali A. S.; Waghmode K. T.; Chaturbhuj G. U. Repurposing n-Butyl Stannoic Acid as Highly Efficient Catalyst for Direct Amidation of Carboxylic Acids with Amines. Tetrahedron Lett. 2018, 59, 4582–4586. 10.1016/j.tetlet.2018.11.036. [DOI] [Google Scholar]; g Srivastava V.; Singh P. K.; Singh P. P. Visible Light Photoredox Catalysed Amidation of Carboxylic Acids with Amines. Tetrahedron Lett. 2019, 60, 40–43. 10.1016/j.tetlet.2018.11.050. [DOI] [Google Scholar]; h Handoko; Satishkumar S.; Panigrahi N. R.; Arora P. S. Rational Design of an Organocatalyst for Peptide Bond Formation. J. Am. Chem. Soc. 2019, 141, 15977–15985. 10.1021/jacs.9b07742. [DOI] [PubMed] [Google Scholar]; i Zarecki A. P.; Kolanowski J. L.; Markiewicz W. T. Microwave-Assisted Catalytic Method for a Green Synthesis of Amides Directly from Amines and Carboxylic Acids. Molecules 2020, 25, 1761. 10.3390/molecules25081761. [DOI] [PMC free article] [PubMed] [Google Scholar]; j Li Z.; Liu L.; Xu K.; Huang T.; Li X.; Song B.; Chen T. Palladium-Catalyzed N-Acylation of Tertiary Amines by Carboxylic Acids: A Method for the Synthesis of Amides. Org. Lett. 2020, 22, 5517–5521. 10.1021/acs.orglett.0c01869. [DOI] [PubMed] [Google Scholar]; k Saito S.; Movahed F. S.; Sawant D. N.; Bagal D. B. Tris(o-phenylenedioxy)cyclotriphosphazene as a Promoter for the Formation of Amide Bonds between Aromatic Acids and Amines. Synthesis 2020, 52, 3253–3262. 10.1055/s-0040-1707174. [DOI] [Google Scholar]; l Wang J.; Hou H.; Hu Y.; Lin J.; Wu M.; Zheng Z.; Xu X. Visible-Light-Induced Direct Construction of Amide Bond from Carboxylic Acids with Amines in Aqueous Solution. Tetrahedron Lett. 2021, 65, 152801. 10.1016/j.tetlet.2020.152801. [DOI] [Google Scholar]; m de Azambuja F.; Loosen A.; Conic D.; van den Besselaar M.; Harvey J. N.; Parac-Vogt T. N. En Route to a Heterogeneous Catalytic Direct Peptide Bond Formation by Zr-Based Metal–Organic Framework Catalysts. ACS Catal. 2021, 11, 7647–7658. 10.1021/acscatal.1c01782. [DOI] [Google Scholar]; n Handoko; Panigrahi N. R.; Arora P. S. Two-Component Redox Organocatalyst for Peptide Bond Formation. J. Am. Chem. Soc. 2022, 144, 3637–3643. 10.1021/jacs.1c12798. [DOI] [PubMed] [Google Scholar]; o Zhang L.; Jiang J.; Li L.; Chen Q.; Zhang L.; Sun H.; Li C. Sustainable Synthesis of Amides from Carboxylic Acids and Equivalent Amounts of Amines Using a Reusable Brønsted Acidic Ionic Liquid as a Catalyst and a Solvent. ACS Sustain. Chem. Eng. 2022, 10, 8433–8442. 10.1021/acssuschemeng.2c01434. [DOI] [Google Scholar]; p Su J.; Mo J. N.; Chen X.; Umanzor A.; Zhang Z.; Houk K. N.; Zhao J. Generation of Oxyphosphonium Ions by Photoredox/Cobaloxime Catalysis for Scalable Amide and Peptide Synthesis in Batch and Continuous-Flow. Angew. Chem., Int. Ed. 2022, 61, e202112668 10.1002/anie.202112668. [DOI] [PubMed] [Google Scholar]
- For representative examples see:; a Chan T. H.; Wong L. T. L. Silicon Tetrachloride as a Coupling Reagent for Amide Formation. J. Org. Chem. 1969, 34, 2766–2767. 10.1021/jo01261a064. [DOI] [Google Scholar]; b Chou W.-C.; Chou M.-C.; Lu Y.-Y.; Chen S.-F. HMDS-Promoted in situ Amidation Reactions of Carboxylic Acids and Amines. Tetrahedron Lett. 1999, 40, 3419–3422. 10.1016/S0040-4039(99)00505-5. [DOI] [Google Scholar]; c van Leeuwen S. H.; Quaedflieg P. J. L. M.; Broxterman Q. B.; Liskamp R. M. J. Synthesis of Amides from Unprotected Amino Acids by a Simultaneous Protection–Activation Strategy using Dichlorodialkyl Silanes. Tetrahedron Lett. 2002, 43, 9203–9207. 10.1016/S0040-4039(02)02275-X. [DOI] [Google Scholar]; d Tozawa T.; Yamane Y.; Mukaiyama T. A Convenient Method for Preparations of 1-Acylimidazoles and Carboxamides by using Novel Imidazolylsilane Derivatives. Chem. Lett. 2005, 34, 734–735. 10.1246/cl.2005.734. [DOI] [Google Scholar]; e Tozawa T.; Yamane Y.; Mukaiyama T. An Effective Method for the Synthesis of Carboxamides by using Tetrakis(pyridine-2-yloxy)silane as a Mild Coupling Reagent. Chem. Lett. 2005, 34, 1334–1335. 10.1246/cl.2005.1334. [DOI] [Google Scholar]; f Tozawa T.; Yamane Y.; Mukaiyama T. An Efficient Method for the Preparation of Carboxamides by Dehydration Condensation using Tetrakis(1,1,1,3,3,3-hexafluoro-2-propoxy)silane. Chem. Lett. 2005, 34, 1586–1587. 10.1246/cl.2005.1586. [DOI] [Google Scholar]; g Ruan Z.; Lawrence R. M.; Cooper C. B. Phenylsilane as an Active Amidation Reagent for the Preparation of Carboxamides and Peptides. Tetrahedron Lett. 2006, 47, 7649–7651. 10.1016/j.tetlet.2006.08.024. [DOI] [Google Scholar]; h Aspin S. J.; Taillemaud S.; Cyr P.; Charette A. B. 9-Silafluorenyl Dichlorides as Chemically Ligating Coupling Agents and Their Application in Peptide Synthesis. Angew. Chem., Int. Ed. 2016, 55, 13833–13837. 10.1002/anie.201606120. [DOI] [PubMed] [Google Scholar]; i Sayes M.; Charette A. B. Diphenylsilane as a Coupling Reagent for Amide Bond Formation. Green Chem. 2017, 19, 5060–5064. 10.1039/C7GC02643A. [DOI] [Google Scholar]; j Braddock D. C.; Lickiss P. D.; Rowley B. C.; Pugh D.; Purnomo T.; Santhakumar G.; Fussell S. J. Tetramethyl Orthosilicate (TMOS) as a Reagent for Direct Amidation of Carboxylic Acids. Org. Lett. 2018, 20, 950–953. 10.1021/acs.orglett.7b03841. [DOI] [PubMed] [Google Scholar]; k Morisset E.; Chardon A.; Rouden J.; Blanchet J. Phenysilane and Silicon Tetraacetate: Versatile Promotors for Amide Synthesis. Eur. J. Org. Chem. 2020, 2020, 388–392. 10.1002/ejoc.201901660. [DOI] [Google Scholar]; l D’Amaral M. C.; Jamkhou N.; Adler M. J. Efficient and Accessible Silane-Mediated Direct Amide Coupling of Carboxylic Acids and Amines. Green Chem. 2021, 23, 288–295. 10.1039/D0GC02833A. [DOI] [Google Scholar]; m Muramatsu W.; Yamamoto H. Peptide Bond Formation of Amino Acids by Transient Masking with Silylating Reagents. J. Am. Chem. Soc. 2021, 143, 6792–6797. 10.1021/jacs.1c02600. [DOI] [PubMed] [Google Scholar]; n Braddock D. C.; Davies J. J.; Lickiss P. D. Methyltrimethoxysilane (MTM) as a Reagent for Direct Amidation of Carboxylic Acids. Org. Lett. 2022, 24, 1175–1179. 10.1021/acs.orglett.1c04265. [DOI] [PMC free article] [PubMed] [Google Scholar]; o Lainer T.; Czerny F.; Haas M. Solvent-Free Amide Bond Formation Using a Variety of Methoxysilanes as Coupling Agent. Org. Biomol. Chem. 2022, 20, 3717–3720. 10.1039/D2OB00589A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For representative examples see:; a Comerford J. W.; Clark J. H.; Macquarrie D. J.; Breeden S. W. Clean, Reusable and Low Cost Heterogeneous Catalyst for Amide Synthesis. Chem. Commun. 2009, 2562–2564. 10.1039/B901581G. [DOI] [PubMed] [Google Scholar]; b Yang X.-D.; Zeng X.-H.; Zhao Y.-H.; Wang X.-Q.; Pan Z.-Q.; Li L.; Zhang H.-B. Silica Gel-Mediated Amide Bond Formation: An Environmentally Benign Method for Liquid-Phase Synthesis and Cytotoxic Activities of Amides. J. Comb. Chem. 2010, 12, 307–310. 10.1021/cc900135f. [DOI] [PubMed] [Google Scholar]; c Komura K.; Nakano Y.; Koketsu M. Mesoporous Silica MCM-41 as a Highly Active, Recoverable and Reusable Catalyst for Direct Amidation of Fatty Acids and Long-Chain Amines. Green Chem. 2011, 13, 828–831. 10.1039/C0GC00673D. [DOI] [Google Scholar]; d Ojeda-Porras A.; Hernández-Santana A.; Gamba-Sánchez D. Direct Amidation of Carboxylic Acids with Amines under Microwave Irradiation using Silica Gel as a Solid Support. Green Chem. 2015, 17, 3157–3163. 10.1039/C5GC00189G. [DOI] [Google Scholar]; e Komura K.; Tamura M.; Murase D. Direct Amide Synthesis from Equimolar Amounts of Carboxylic Acid and Amine Catalyzed by Mesoporous Silica SBA-15. Synthesis 2015, 47, 769–776. 10.1055/s-0034-1379966. [DOI] [Google Scholar]; f Tamura M.; Murase D.; Komura K. Mesoporous Silica Catalyzed the Direct Amidation of Palmitic Acid and Hexylamine and Unique Dependence of Reaction Rate on Pore Size with p6mm Topological Catalyst. Chem. Lett. 2016, 45, 451–453. 10.1246/cl.160049. [DOI] [Google Scholar]; g Zakharova M. V.; Kleitz F.; Fontaine F.-G. Lewis Acidity Quantification and Catalytic Activity of Ti, Zr and Al-Supported Mesoporous Silica. Dalton Trans. 2017, 46, 3864–3876. 10.1039/C7DT00035A. [DOI] [PubMed] [Google Scholar]
- For the use of a catalytic silicon entity used in conjunction with a stoichiometric silicon reagent see:; a Muramatsu W.; Manthena C.; Nakashima E.; Yamamoto H. Peptide Bond-Forming Reaction via Amino Acid Silyl Esters: New Catalytic Reactivity of an Aminosilane. ACS Catal. 2020, 10, 9594–9603. 10.1021/acscatal.0c02512. [DOI] [Google Scholar]; b Muramatsu W.; Yamamoto H. Organocatalytic Activation of Inert Hydrosilane for Peptide Bond Formation. Org. Lett. 2022, 24, 7194–7199. 10.1021/acs.orglett.2c02947. [DOI] [PubMed] [Google Scholar]; For a review on stoichiometric silicon reagents for direct amidation see:; c Davies J. J.; Braddock D. C.; Lickiss P. D. Silicon Compounds as Stoichiometric Coupling Reagents for Direct Amidation. Org. Biomol. Chem. 2021, 19, 6746–6760. 10.1039/D1OB01003D. [DOI] [PubMed] [Google Scholar]
- For representative experimental and computational investigations see:; a Charville H.; Jackson D.; Hodges G.; Whiting A. The Thermal and Boron-Catalysed Direct Amide Formation Reactions: Mechanistically Understudied yet Important Processes. Chem. Commun. 2010, 46, 1813–1823. 10.1039/B923093A. [DOI] [PubMed] [Google Scholar]; b Lundberg H.; Tinnis F.; Zhang J.; Algarra A. G.; Himo F.; Adolfsson H. Mechanistic Elucidation of Zirconium-Catalyzed Direct Amidation. J. Am. Chem. Soc. 2017, 139, 2286–2295. 10.1021/jacs.6b10973. [DOI] [PubMed] [Google Scholar]; c Jiang Y.-Y.; Zhu L.; Liang Y.; Man X.; Bi S. Mechanism of Amide Bond Formation from Carboxylic Acids and Amines Promoted by 9-Silafluorenyl Dichloride Derivatives. J. Org. Chem. 2017, 82, 9087–9096. 10.1021/acs.joc.7b01637. [DOI] [PubMed] [Google Scholar]; d Jiang Y.-Y.; Hu B.; Xu Z.-Y.; Zhang R.-X.; Liu T.-T.; Bi S. Boron Ester-Catalyzed Amidation of Carboxylic Acids with Amines: Mechanistic Rationale by Computational Study. Chem.—Asian J. 2018, 13, 2685–2690. 10.1002/asia.201800797. [DOI] [PubMed] [Google Scholar]; e Arkhipenko S.; Sabatini M. T.; Batsanov A. S.; Karaluka V.; Sheppard T. D.; Rzepa H. S.; Whiting A. Mechanistic Insights into Boron-Catalysed Direct Amidation Reactions. Chem. Sci. 2018, 9, 1058–1072. 10.1039/C7SC03595K. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Rimola A.; Fabbiani M.; Sodupe M.; Ugliengo P.; Martra G. How Does Silica Catalyze the Amide Bond Formation under Dry Conditions? Role of Specific Surface Silanol Pairs. ACS Catal. 2018, 8, 2558–2568. 10.1021/acscatal.7b03961. [DOI] [Google Scholar]; g Noda H.; Asada Y.; Shibasaki M.; Kumagai N. Neighboring Protonation Unveils Lewis Acidity in the B3NO2 Heterocycle. J. Am. Chem. Soc. 2019, 141, 1546–1554. 10.1021/jacs.8b10336. [DOI] [PubMed] [Google Scholar]; h Hu B.; Jiang Y.-Y.; Liu P.; Zhang R.-X.; Zhang Q.; Liu T.-T.; Bi S. The Mechanism and Structure–Activity Relationship of Amide Bond Formation by Silane Derivatives: A Computational Study. Org. Biomol. Chem. 2019, 17, 9232–9242. 10.1039/C9OB01605H. [DOI] [PubMed] [Google Scholar]
- For the use of silanols A, silanediols B and siloxanediols C as (hydrogen-bonding) catalysts for other processes see:; a Schafer A. G.; Wieting J. M.; Mattson A. E. Silanediols: A New Class of Hydrogen Bond Donor Catalysts. Org. Lett. 2011, 13, 5228–5231. 10.1021/ol2021115. [DOI] [PubMed] [Google Scholar]; b Tran N. T.; Min T.; Franz A. K. Silanediol Hydrogen-Bonding Activation of Carbonyl Compounds. Chem.—Eur. J. 2011, 17, 9897–9900. 10.1002/chem.201101492. [DOI] [PubMed] [Google Scholar]; c Beemelmanns C.; Husmann R.; Whelligan D. K.; Özçubukçu S.; Bolm C. Planar-Chiral Bis-silanols and Diols as H-Bonding Asymmetric Organocatalysts. Eur. J. Org. Chem. 2012, 2012, 3373–3376. 10.1002/ejoc.201200548. [DOI] [Google Scholar]; d Min T.; Fettinger J. C.; Franz A. K. Enantiocontrol with a Hydrogen-bond Directing Pyrrolidinylsilanol Catalyst. ACS Catal. 2012, 2, 1661–1666. 10.1021/cs300290j. [DOI] [Google Scholar]; e Tran N. T.; Wilson S. O.; Franz A. K. Cooperative Hydrogen-Bonding Effects in Silanediol Catalysis. Org. Lett. 2012, 14, 186–189. 10.1021/ol202971m. [DOI] [PubMed] [Google Scholar]; f Schafer A. G.; Wieting J. M.; Fisher T. J.; Mattson A. E. Chiral Silanediols in Anion-Binding Catalysis. Angew. Chem., Int. Ed. 2013, 52, 11321. 10.1002/anie.201305496. [DOI] [PubMed] [Google Scholar]; g Hardman-Baldwin A. M.; Mattson A. E. Silanediol-Catalyzed Carbon Dioxide Fixation. ChemSusChem 2014, 7, 3275–3278. 10.1002/cssc.201402783. [DOI] [PubMed] [Google Scholar]; h Wieting J. M.; Fisher T. J.; Schafer A. G.; Visco M. D.; Gallucci J. C.; Mattson A. E. Preparation and Catalytic Activity of BINOL-Derived Silanediols. Eur. J. Org. Chem. 2015, 2015, 525–533. 10.1002/ejoc.201403441. [DOI] [Google Scholar]; i Diemoz K. M.; Wilson S. O.; Franz A. K. Synthesis of Structurally Varied 1,3-Disiloxanediols and their Activity as Anion-Binding Catalysts. Chem.—Eur. J. 2016, 22, 18349–18353. 10.1002/chem.201604103. [DOI] [PubMed] [Google Scholar]; j Diemoz K. M.; Hein J. E.; Wilson S. O.; Fettinger J. C.; Franz A. K. Reaction Progress Kinetics Analysis of 1,3-Disiloxanediols as Hydrogen-Bonding Catalysts. J. Org. Chem. 2017, 82, 6738–6747. 10.1021/acs.joc.7b00875. [DOI] [PubMed] [Google Scholar]; k Velásquez-Hernández M. d. J.; Torres-Huerta A.; Hernández-Balderas U.; Martínez-Otero D.; Núñez-Pineda A.; Jancik V. Novel Route to Silanetriols and Silanediols Based On Acetoxysilylalkoxides. Polyhedron 2017, 122, 161–171. 10.1016/j.poly.2016.10.051. [DOI] [Google Scholar]; l Hurkes N.; Belaj F.; Koe J. R.; Pietschnig R. S. Synthesis, structure and catalytic properties of bis[2-(trifluoromethyl)phenyl]silanediol: Silanediol catalysis. Appl. Organomet. Chem. 2018, 32, e4427 10.1002/aoc.4427. [DOI] [Google Scholar]; m Jagannathan J. R.; Diemoz K. M.; Targos K.; Fettinger J. C.; Franz A. K. Kinetic and Binding Studies Reveal Cooperativity and Off-Cycle Competition for H-Bonding Catalysis with Silsesquioxane Silanols. Chem.—Eur. J. 2019, 25, 14953–14958. 10.1002/chem.201903693. [DOI] [PubMed] [Google Scholar]; n Fox F.; Neudörfl J. M.; Goldfuss B. Silanediol versus Chlorosilanol: Hydrolyses and Hydrogen-Bonding Catalyses with Fenchole-Based Silanes. Beilstein J. Org. Chem. 2019, 15, 167–186. 10.3762/bjoc.15.17. [DOI] [PMC free article] [PubMed] [Google Scholar]; o Pérez-Pérez J.; Hernández-Balderas U.; Martínez-Otero D.; Jancik V. Bifunctional Silanol-based HBD Catalysts for CO2 Fixation into Cyclic Carbonates. New J. Chem. 2019, 43, 18525–18533. 10.1039/C9NJ04840E. [DOI] [Google Scholar]; p Chen G.; Zhang Y.; Xu J.; Liu X.; Liu K.; Tong M.; Long Z. Imidazolium-Based Ionic Porous Hybrid Polymers with POSS-Derived Silanols for Efficient Heterogeneous Catalytic CO2 Conversion under Mild Conditions. J. Chem. Eng. 2020, 381, 122765. 10.1016/j.cej.2019.122765. [DOI] [Google Scholar]
- All reactions were conducted in triplicate or more. Conversions were determined by comparison with an internal standard. The error in conversions was determined to be ±1%. The acid and amine in these reactions form an insoluble salt in toluene at room temperature. As the reaction temperature approaches reflux, the salt dissolves.
- Triphenyl carbinol was examined as a potential catalyst under the standard conditions (15% conversion to amide 3), showing that the use of the silanol is beneficial. The use of 2,2,2-trifluoroethanol (pKa: 12.5, DMSO) and p-nitrophenol (pKa: 10.8, DMSO) as protic additives with pKa values similar triphenylsilanol (10.8, THF) did not induce significant catalytic activity over the background reaction.
- As expected, a control experiment using hexaphenyldisiloxane as a potential catalyst showed no change to the background conversion.
- Individual control experiments for 18 h in refluxing toluene with hydrocinnamic acid and benzylamine as representative acid and amine components were also conducted. Triphenylsilanol proved to be entirely inert to the presence of the acid, whereas up to ca. 20% of the corresponding disiloxane was observed with the added amine after 1 h. Concomitant steady and substantial depletion (ca. 60%, 18 h) of triphenylsilanol was also observed in the latter experiment.
- 5a:; a Gilman H.; Plunkett M. A.; Dunn G. E. Some Organosilicon Compounds Containing the p-Dimethylaminophenyl Group. J. Am. Chem. Soc. 1951, 73, 1686–1688. 10.1021/ja01148a079. [DOI] [Google Scholar]; 5b, 5c, 5f & 5j:; b Benkeser R. A.; Riel F. The Reactions of Some Triarylsilanes with Methyllithium and Phenylisopropylpotassium. J. Am. Chem. Soc. 1951, 73, 3472–3474. 10.1021/ja01151a142. [DOI] [Google Scholar]; 5b, 5c, 5e:; c Prince P. D.; Bearpark M. J.; McGrady G. S.; Steed J. W. Hypervalent hydridosilicates: Synthesis, Structure and Hydride Bridging. Dalton Trans. 2008, 271–282. 10.1039/B713427D. [DOI] [PubMed] [Google Scholar]; ; 5b–f, 5h & 5j:; d Akhani R. K.; Moore M. I.; Pribyl J. G.; Wiskur S. L.; Wiskur S. L. Linear Free-Energy Relationship and Rate Study on a Silylation-Based Kinetic Resolution: Mechanistic Insights. J. Org. Chem. 2014, 79, 2384–2396. 10.1021/jo402569h. [DOI] [PubMed] [Google Scholar]; ; 5g:; e Ali M.; Eaborn C.; Walton D. R. M. Organosilicon Compounds: LI. Further Studies of the Base-Catalysed Solvolysis of N-(triorganosilyl) anilines. J. Organomet. Chem. 1974, 78, 83–92. 10.1016/S0022-328X(00)86449-9. [DOI] [Google Scholar]; ; 5h:; f Wander M.; Hausoul P. J. C.; Sliedregt L. A. J. M.; van Steen B. J.; van Koten G.; Klein Gebbink R. J. M. Synthesis of Polyaryl Rigid-Core Carbosilane Dendrimers for Supported Organic Synthesis. Organometallics 2009, 28, 4406–4415. 10.1021/om900265v. [DOI] [Google Scholar]; ; 5i:; g Gilman H.; Goodman J. J. Some Monomeric Organosilicon Compounds of High Thermal Stability. J. Org. Chem. 1957, 22, 45–47. 10.1021/jo01352a011. [DOI] [Google Scholar]
- Silanes 5a, 5f, 5g & 5i were available to us in-house and did not require repreparation.
- 6a:; a Reference (19a); 6c:; b Steele A. R.; Kipping F. S.; Dickinson R.; Chapman A. C. Some Derivatives of Tri-p-tolylsilicane. J. Chem. Soc. 1929, 357–358. 10.1039/JR9290000357. [DOI] [Google Scholar]; ; 6e, 6f & 6h; c Pauling H.; Andrews D. A.; Hindley N. C. The Rearrangement of α-Acetylenic Alcohols to α, β-Unsaturated Carbonyl Compounds by Silylvanadate Catalysts. Helv. Chim. Acta 1976, 59, 1233–1243. 10.1002/hlca.19760590426. [DOI] [Google Scholar]; ; 6f:; d Gilman H.; Miller L. S. The Preparation of Some Chlorinated Aryl Silanes for the Examination of Their Insecticidal Properties. J. Am. Chem. Soc. 1951, 73, 968–970. 10.1021/ja01147a023. [DOI] [Google Scholar]
- The first X-ray crystal structures for tris(4-halophenyl)silanes 5e & 5f and tris(4-halophenyl)silanols 6e, 6f & 6h have been obtained as part of this work (see Supporting Information).
- Indeed, attempted recrystallisation of 6e and 6i from hexane led instead to crystallisation as their respective disiloxanes ((4-FC6H4)3Si)2O and ((3-CF3C6H4)3Si)2O which were characterised by X-ray crystallography (see Supporting Information).
- The log(kR/kH) values were obtained by first subtracting the known background conversion from the observed conversion to give a corrected catalytic conversion for each silanol. At low conversions (as in these cases) it is approximated that the kR/kH ≈ %corrected conversion (R)/%corrected conversion (H).
- Hansch C.; Leo A.; Taft R. W. A Survey of Hammett Substituent Constants and Resonance and Field Parameters. Chem. Rev. 1991, 91, 165–195. 10.1021/cr00002a004. [DOI] [Google Scholar]
- Here, dimethylamino silanol 6a is an evident outlier (not plotted), which we attribute to protonation of the amine centres under the reaction conditions thereby changing the electronics of the properties of this catalyst. In this respect, we note the σp value of the −NMe3+ substituent is 0.82. Unfortunately, the catalyst integrity for 6a could not be determined due to overlapping signals in the 1H NMR spectrum.
- One should also note that a consequence of catalyst decomposition is to reduce the true value of ρ: a Hammett plot of non-decomposing catalysts 6c–e & 4 provided ρ = +1.71.
- a Eaborn C. Cleavages of Aryl-Silicon and Related Bonds by Electrophiles. J. Organomet. Chem. 1975, 100, 43–57. 10.1016/S0022-328X(00)88933-0. [DOI] [Google Scholar]; b Brook M. A.Cleavage of Si-C Bonds. In Silicon in Organic, Organometallic and Polymer Chemistry; Wiley, 2000; pp 552–607. [Google Scholar]; c Shirai S.; Goto Y.; Mizoshita N.; Ohashi M.; Tani T.; Shimada T.; Hyodo S.; Inagaki S. Theoretical Studies on Si–C Bond Cleavage in Organosilane Precursors during Polycondensation to Organosilica Hybrids. J. Phys. Chem. A 2010, 114, 6047–6054. 10.1021/jp101242g. [DOI] [PubMed] [Google Scholar]; d Reich H. J.Mechanism of C-Si Bond Cleavage using Lewis Bases (n → σ*). In Lewis Base Catalysis in Organic Synthesis; Vedejs E., Denmark S. E., Eds.; John Wiley & Sons, 2016; Vol. 1, Chapter 8, pp 233–280. [Google Scholar]
- Delmas L. C.; Horton P. N.; White A. J. P.; Coles S. J.; Lickiss P. D.; Davies R. P. Siloxane-Based Linkers in The Construction of Hydrogen Bonded Assemblies and Porous 3D MOFs. Chem. Commun. 2017, 53, 12524–12527. 10.1039/C7CC06022J. [DOI] [PubMed] [Google Scholar]
- Preliminary kinetic experiments with either an excess of acid 1a or amine 2a in 0.3 vs 0.2 and 0.06 M catalyst 6h unexpectedly resulted in slower rates of reaction than the ideal 1:1 acid/amine at both 0.2 and 0.3 M concentrations. In turn this meant that different excess experiments (see ref (32)) gave rise to negative orders in acid and amine (see Supporting Information for detail).
- Experiments to determine the order in catalyst with runs at [6h] = 0.06 M vs 0.03 M with [1a] = [2a] = 0.2 M gave a catalyst order of 2.6 (see Supporting Information for detail). A catalyst order >1 is consistent with product inhibition operating:Alamillo-Ferrer C.; Hutchinson G.; Burés J. Mechanistic interpretation of orders in catalyst greater than one. Nat. Rev. Chem. 2022, 7, 26–34. 10.1038/s41570-022-00447-w. [DOI] [PubMed] [Google Scholar]
- Blackmond D. G. Reaction Progress Kinetic Analysis: A Powerful Methodology for Mechanistic Studies of Complex Catalytic Reactions. Angew. Chem., Int. Ed. 2005, 44, 4302–4320. 10.1002/anie.200462544. [DOI] [PubMed] [Google Scholar]
- Baxter R. D.; Sale D.; Engle K. M.; Yu J.-Q.; Blackmond D. G. Mechanistic Rationalization of Unusual Kinetics in Pd-Catalyzed C–H Olefination. J. Am. Chem. Soc. 2012, 134, 4600–4606. 10.1021/ja207634t. [DOI] [PubMed] [Google Scholar]
- D‘Amaral M. C.; Andrews K. G.; Denton R.; Adler M. J. Silyl Esters as Reactive Intermediates in Organic Synthesis. Synthesis 2023, 10.1055/a-2083-8591. [DOI] [Google Scholar]
- Ojima Y.; Yamaguchi K.; Mizuno N. An Efficient Solvent-Free Route to Silyl Esters and Silyl Ethers. Adv. Synth. Catal. 2009, 351, 1405–1411. 10.1002/adsc.200900230. [DOI] [Google Scholar]
- Silanaminium carboxylate salt 8, mp 68–70 °C, is difficult to otherwise characterize as it immediately reverts to silyl ester 7 and amine 2a on dissolution in solvent. We attribute its initial formation in toluene solution to a kinetic attack of the amine on the ester at silicon (driven by its precipitation). Redissolution results in the thermodynamic preference for O-silylation.
- For all managed computational research data, including input and output files see:Braddock D. C.; Rowley B. C.; Lickiss P. D.; Fussell S. F.; Qamar R.; Pugh D.; Rzepa H. S.; White J. P. On the Use of Triarylsilanols as Catalysts for Direct Amidation of Carboxylic Acids. Imperial College Research Data Repository 2023, 10.14469/hpc/12480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapid and reversible formation of a pentacoordinate amide-tris(pentafluorophenyl)silyl fluoride complex has been observed by NMR methods in CDCl3 on mixing tris(pentafluorophenyl)silyl fluoride with an amide. A tetrahedral cationic imidate was observed with the corresponding silyl triflate:Dilma A. D.; Levin V. V.; Korlyukov A. A.; Belyakov P. A.; Struchkova M. I.; Antipin M. Yu.; Tartakovsky V. A. Complexation of tris(pentafluophenyl)silanes with neutral Lewis bases. J. Organomet. Chem. 2008, 693, 1005–1019. 10.1016/j.jorganchem.2007.12.022. [DOI] [Google Scholar]
- The catalytic cycle could also be entered using 10 mol % Ph3SiH (or 10 mol % Ph3SiCl) giving identical conversions to amide 3a (25%), which again implicates the intermediacy of silyl ester 7. For the formation of a silyl ester from PhSiH3 and a carboxylic acid see:Andrews K. G.; Denton R. M. A More Critical Role for Silicon in the Catalytic Staudinger Amidation: Silanes as Non-innocent Reductants. Chem. Commun. 2017, 53, 7982–7985. 10.1039/C7CC03076B. [DOI] [PubMed] [Google Scholar]
- Davies A. G.; Neville A. G. A Study by Electron Spin Resonance Spectroscopy of the di-t-Butylsilanone Radical Anion. J. Organomet. Chem. 1992, 436, 255–263. 10.1016/0022-328X(92)85058-5. [DOI] [Google Scholar]
- Cusa N. W.; Kipping F. 308. Organic Derivatives of Silicon. Part XLVII. Cyclohexylphenyl and Cyclohexyl Derivatives. J. Chem. Soc. 1932, 2205–2209. 10.1039/jr9320002205. [DOI] [Google Scholar]
- Graalmann O.; Klingebiel U.; Clegg W.; Haase M.; Sheldrick G. M. Stufenweise Synthese ketten-und ringförmiger Siloxane—Kristallstrukturen. Chem. Ber. 1984, 117, 2988–2997. 10.1002/cber.19841170916. [DOI] [Google Scholar]
- Sieburth S. M.; Mu W. Silanol synthesis: reaction of hexaphenylcyclotrisiloxane with organometallic reagents. J. Org. Chem. 1993, 58, 7584–7586. 10.1021/jo00078a047. [DOI] [Google Scholar]
- Denmark S. E.; Butler C. R. Vinylation of Aromatic Halides using Inexpensive Organosilicon Reagents. Illustration of Design of Experiment Protocols. J. Am. Chem. Soc. 2008, 130, 3690–3704. 10.1021/ja7100888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubiel S.; Griffith G.; Long C.; Baker G.; Smith R. E. Determination of Reactive Components in Silicone Foams. Anal. Chem. 1983, 55, 1533–1537. 10.1021/ac00260a020. [DOI] [Google Scholar]
- Eaborn C. 537. Organosilicon Compounds. Part III. Some Sterically Hindered Compounds. J. Chem. Soc. 1952, 2840–2846. 10.1039/JR9520002840. [DOI] [Google Scholar]
- Doyle M. P.; West C. T. Silane Reductions in Acidic Media. IV. Reductions of alkyl-substituted Cyclohexanones by mono-di-and trialkylsilanes. Stereochemistry of Alcohol and Ether Formation. J. Org. Chem. 1975, 40, 3821–3829. 10.1021/jo00914a002. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this study are available in the published article and its online Supporting Information as well as openly available as part of the PhD thesis of B. C. Rowley in the Imperial College London Institutional Repository (Spiral) at https://doi.org/10.25560/78224. Computational data are openly available at the Imperial College research data repository at DOI: 10.14469/hpc/12480.