

Abstract
Bifidobacteria are early colonizers of the human gut and play central roles in human health and metabolism. To thrive in this competitive niche, these bacteria evolved the capacity to use complex carbohydrates, including mammalian N-glycans. Herein, we elucidated pivotal biochemical steps involved in high-mannose N-glycan utilization by Bifidobacterium longum. After N-glycan release by an endo-β-N-acetylglucosaminidase, the mannosyl arms are trimmed by the cooperative action of three functionally distinct glycoside hydrolase 38 (GH38) α-mannosidases and a specific GH125 α-1,6-mannosidase. High-resolution cryo-electron microscopy structures revealed that bifidobacterial GH38 α-mannosidases form homotetramers, with the N-terminal jelly roll domain contributing to substrate selectivity. Additionally, an α-glucosidase enables the processing of monoglucosylated N-glycans. Notably, the main degradation product, mannose, is isomerized into fructose before phosphorylation, an unconventional metabolic route connecting it to the bifid shunt pathway. These findings shed light on key molecular mechanisms used by bifidobacteria to use high-mannose N-glycans, a perennial carbon and energy source in the intestinal lumen.
The human gastrointestinal tract is home to a complex microbial community, including species from the genus Bifidobacterium, which play central roles in human health and metabolism, such as in the protection against pathogens, production of bioactive molecules (folate and neurotransmitters) and modulation of neuronal development and immunity1–4. Bifidobacteria are more prevalent in infant microbiota, and their persistence throughout the host’s life is a result of their ability to process and uptake complex carbohydrates available in the intestinal lumen5, including resistant starch6, plant polysaccharides7, milk oligosaccharides8 and O- and N-glycans produced by host tissues9. Host glycosylation represents a perennial source of nutrients for human microbiota, and N-glycans are the most commonly found in eukaryotic organisms. N-glycans comprise a group of large oligosaccharides classified into three main classes in mammals: high-mannose, complex and hybrid N-glycans10. These oligosaccharides have a conserved core composed of three moieties of mannose and two moieties of N-acetylglucosamine (GlcNAc) linked to specific asparagine residues of target proteins (Man3GlcNAc2-Asn). However, their arms or antennas, depending on the N-glycan type, can vary greatly in composition and structure, conferring on them a vast range of biological roles from cell adhesion to signaling pathways, among other cellular and metabolic functions11.
Bacterial strategies for N-glycan utilization usually involve elaborate molecular systems known as polysaccharide utilization loci (PULs), which consist of colocalized and co-regulated genes that encode carbohydrate-active enzymes, TonB-dependent transporters and cell surface glycan-binding proteins, allowing the detection, enzymatic breakdown and transport of complex carbohydrates12–15. The N-glycan PULs (N-GULs) from Bacteroidetes, Firmicutes and Pro-teobacteria phyla have been investigated16–18, highlighting the diversity of enzymatic modules that coevolved with the structurally diverse types of N-linked oligosaccharides. For instance, Bacteroides thetaiotaomicron is equipped with distinct N-GULs, such as for mammalian high-mannose N-glycan (HMNG)16, yeast α-mannan16 and complex N-glycan from human immunoglobulins19. For HMNG degradation, Bacteroides thetaiotaomicron encodes a glycoside hydrolase 18 (GH18) endo-β-N-acetylglucosaminidase (ENGase)16,20 that releases the HMNG from its peptide, internalizes HMNG by a SBBP/SusC-D transport system and trims it in the periplasm using three GH92 enzymes, each acting specifically on one of the three types of α-mannosyl linkages16.
In the Gram-positive phylum Actinobacteria, which includes the genus Bifidobacterium, only a few enzymes acting on N-glycan breakdown have been described so far21. They comprise β-mannosidases from the GH family 5_18 (refs.21–23), specialized in the cleavage of the β-1,4 linkages between mannosyl and GlcNAc moieties, as demonstrated by our group23, and ENGases from the GH18 and GH85 families24, which remove the N-glycan from glycoproteins. All these characterized enzymes act on the universal core of N-glycans; but how the ubiquitous and abundant mannosyl extensions are degraded by these bacteria remains elusive. In addition, several genes encoding enzymes putatively involved in α-mannosyl cleavage are present in Actinobacteria, probably offering diverse and cooperative functions for N-glycan breakdown21,25. This gap in knowledge about demannosylation has limited our fundamental understanding of N-glycan processing and metabolism in Actinobacteria and the symbiotic relationship between bifidobacteria and their human hosts.
In this work, using the model organism Bifidobacterium longum, we investigated the molecular mechanisms driving demannosylation of HMNGs, including the downstream metabolic steps for mannose fermentation. Combining biochemical and mutation analyses with mass spectrometry (MS), high-resolution cryo-electron microscopy (cryo-EM) and molecular dynamics analyses, we showed that three GH38 and one GH125 α-mannosidases act cooperatively on the distinct chemical linkages of HMNG mannosyl arms. This system also encodes a GH31 α-glucosidase that enables Bifidobacterium longum to process monoglucosylated HMNGs. Furthermore, we demonstrated that the major N-glycan degradation product, mannose, is connected to the central fermentative pathway bifid shunt by an unconventional metabolic step in which this monosaccharide is isomerized into fructose before phosphorylation. Taken together, these results advance our mechanistic understanding about N-GULs from the phylum Actinobacteria, establishing a biochemical basis for N-glycan degradation in Bifidobacterium species, a key bacterial genus for human health and metabolism.
Results
Bifidobacterium longum is endowed with an N-glycan utilization system
In the Gram-positive phylum Actinobacteria, similar to the PULs described for Gram-negative bacteria, carbohydrate utilization systems involve colocalized and co-regulated genes encompassing distinct carbohydrate-active enzymes and ABC-type membrane transporters. The genome of Bifidobacterium longum NCC2705 contains a predicted N-GUL that encodes ABC transporters, seven GHs and two components of carbohydrate fermentative pathways (Extended Data Fig. 1 and Supplementary Tables 1 and 2).
In this predicted N-GUL, genes encoding a GH85 ENGase (bl1335, protein Bl_Endo85) and a GH5_18 β-mannosidase (bl1333, protein Bl_Man5B) are clustered with five other genes for GH enzymes (Extended Data Fig. 1)25. Four of the genes encode uncharacterized enzymes from the families GH38 (bl1327, bl1328 and bl1329) and GH125 (bl1338), potentially involved in the cleavage of α-mannosides from N-glycans, and the other gene (bl1334) encodes a member of the GH31 family. This family includes the α-subunit (GIIα) of the heterodimeric glucosidase II, which is responsible for the trimming of the two terminal α-1,3-glucosyl residues of Glc2Man9GlcNAc2 during glycoprotein processing in the endoplasmic reticulum26, suggesting a similar role of the bl1334 gene product in the breakdown of glucosylated N-glycans. This gene cluster also includes sequences encoding an ABC transport system, establishing the main prerequisites for a carbohydrate utilization system27. Furthermore, genes encoding a predicted isomerase of the GlcNAc-2-epimerase super-family and a characterized fructokinase28 are also present (Extended Data Fig. 1), which could link N-glycan digestion products to the central carbon metabolism pathway in bifidobacteria, the bifid shunt29.
This gene organization is not restricted to Bifidobacterium longum NCC2705, as it is also found in other species such as Bifidobacterium breve and Bifidobacterium callitrichos (Extended Data Fig. 2a). Variations containing other GH families (GH2, GH3, GH18 or GH20) are present in distinct strains from the same species or in other Bifidobacterium species (Extended Data Fig. 2b). The variations in the N-GUL gene composition in bifidobacteria points to a divergent specialization at the strain level for the use of different types of substrates, such as complex and hybrid types of N-glycans.
Distinct α-mannosidases participate in N-glycan breakdown
The proteins encoded by bl1327–bl1329 genes (named as Bl_Man38A, Bl_Man38B and Bl_Man38C) belong to the GH38 family and share 55–60% sequence identity with one another. These three enzymes exhibit catalytic activity specifically on pNP-α-Man among several synthetic substrates, suggesting that they are α-mannosidases (Supplementary Table 3 and Supplementary Fig. 1). The protein encoded by bl1338 (named as Bl_Man125) does not present activity on any synthetic substrate (Supplementary Table 3), which is not surprising due to the high specificity to α-1,6-mannosyl linkages described for this GH family30. To assess their specificity for native substrates, the activity of the GH38 and GH125 recombinant enzymes on Man9GlcNAc (Extended Data Fig. 1b,c) and Man5GlcNAc (Extended Data Fig. 1d) was examined by liquid chromatography–MS (LC–MS; Fig. 1a,b, Extended Data Figs. 3–5 and Supplementary Figs. 2–4). Zn2+ and Co2+ ions were individually provided in reaction mixtures for GH38 mannosidases, consistent with metal ion dependence displayed by other family members31,32 (Supplementary Figs. 5–7). The three GH38 enzymes showed greater efficiency in the presence of Co2+ ions, with the preference of Bl_Man38A for Man5GlcNAc, releasing mannose and the disaccharide Man-β-1,4-GlcNAc as final products (Fig. 1a,b). Bl_Man38C, however, was more efficient in cleaving Man9GlcNAc, generating mainly Man5GlcNAc, Man4GlcNAc and Man6GlcNAc (Fig. 1a,b). Bl_Man38A and Bl_Man38C combined were able to decompose Man9GlcNAc, demonstrating a complementary role of these two enzymes in N-glycan degradation (Extended Data Fig. 3). Distinct from the other two α-mannosidases, Bl_Man38B was able to cleave all the α-mannosyl linkages from both Man9GlcNAc and Man5GlcNAc substrates (Fig. 1a,b), which is indicative of a generalist activity on HMNGs. Transcriptional studies of Bifidobacterium longum NCC2705 have shown a co-regulation of the expression of Bl_Man38A and Bl_Man38C33, suggesting that bl1328 (encoding Bl_Man38B) can be regulated independently from its neighboring genes, which is in agreement with the functional data. Bl_Man125 alone was not able to degrade either Man9GlcNAc or Man5GlcNAc (Extended Data Fig. 4) but boosted HMNG demannosylation, principally when combined with Bl_Man38B (Extended Data Fig. 5).
Fig. 1 |. Activity of Bl_Man38A, Bl_Man38B and Bl_Man38C on native substrates and disaccharides.
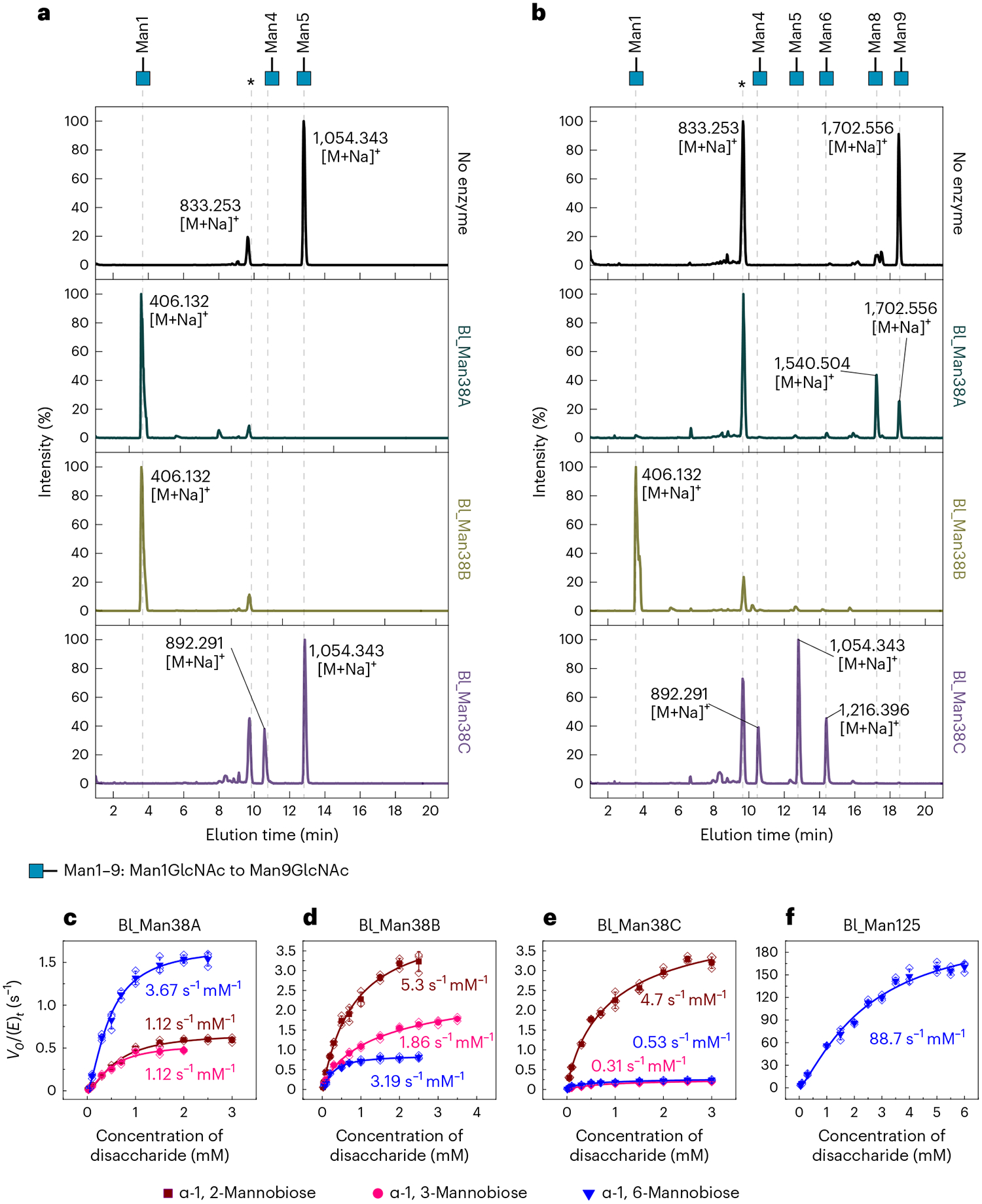
a,b, Assays of Bl_Man38A–Bl_Man38C supplemented with 0.1 mM CoCl2 on Man5GlcNAc (a) or Man9GlcNAc (b) in 60-min reactions. The asterisk (*) indicates internal control (xylohexaose). c–f, Kinetic assays of Bl_Man38A (c), Bl_Man38B (d), Bl_Man38C (e) and Bl_Man125 (f) on the disaccharides α-1,2-mannobiose (dark red), α-1,3-mannobiose (pink) and α-1,6-mannobiose (blue). The reactions containing GH38 α-mannosidases were supplemented with 0.1 mM CoCl2. Results are expressed as mean ± s.d. from three independent experiments. For Bl_Man125, only the kinetics curve for α-1,6-mannobiose is shown because no activity was detected with the other disaccharides. Mass spectra are provided in Supplementary Figs. 2–4. Kinetic parameters for the disaccharide hydrolysis are shown in Supplementary Table 4.
To gain further insights into the functional cooperation between GH38 and GH125 α-mannosidases, regioselectivity was investigated using α-1,2-, α-1,3- and α-1,6-mannobiose as substrates (Fig. 1c–f and Supplementary Table 4). The three GH38 enzymes were able to cleave α-1,2-, α-1,3- and α-1,6-mannobiose, however, with different substrate specificity profiles (Supplementary Table 4). Bl_Man38A showed higher catalytic efficiency for α-1,6-mannobiose than for α-1,2- and α-1,3-mannobiose (Fig. 1c and Supplementary Table 4). α-1,6-Mannosides are the last α-linkages during the N-glycan degradation cascade, and this biochemical profile corroborates with the preference of Bl_Man38A for shorter oligosaccharides generated during N-glycan breakdown. Bl_Man38B was efficient over the three disaccharides, showing higher catalytic efficiency on α-1,2-mannobiose and less activity on α-1,6-mannobiose (Fig. 1d and Supplementary Table 4). The higher efficiency of Bl_Man38B for the more abundant and external α-1,2- and α-1,3-mannosyl linkages in HMNGs is a likely explanation for its capacity to utterly depolymerize the α-mannosyl arms. Bl_Man38C presents a clear preference for α-1,2-mannobiose, with a catalytic efficiency an order of magnitude higher than on the other disaccharides (Fig. 1e and Supplementary Table 4), which is consistent with the specificity for Man9GlcNAc, which has only α-1,2-mannosides available at the cleavable extremities. Bl_Man125 was shown to be strictly an α-1,6-mannosidase with no residual activity on α-1,2- and α-1,3-mannobiose and a catalytic efficiency approximately two orders of magnitude higher for α-1,6-mannobiose than GH38 enzymes, which might explain the cooperative effect with GH38 α-mannosidases, more remarkably with Bl_Man38B.
Together, these results demonstrate complementary roles for Bl_Man38A and Bl_Man38C and reveal Bl_Man38B as a more generalist enzyme that is competent to act at any stage of N-glycan demannosylation and Bl_Man125 as a strict and catalytically efficient α-1,6-mannosidase, which can boost the final steps of N-glycan degradation cascade.
Bifidobacterial GH38 α-mannosidases oligomerize as homotetramers
The activity of the three GH38 α-mannosidases on natural substrates is in accordance with their regioselectivity profiles on disaccharides; however, these results do not explain, for example, why Bl_Man38A is inefficient in breaking down Man9GlcNAc, despite its capacity to cleave all α-1,2-, α-1,3- and α-1,6-mannoside linkages. These mechanistic questions prompted us to unravel the structures of these enzymes to investigate the molecular determinants governing substrate preference and the potential role of remote motifs in this mechanism. Despite obtaining crystals of Bl_Man38A, diffraction experiments yielded only low-resolution data (>5 Å). Considering the molecular weight of these enzymes and their potential to oligomerize, we used single-particle cryo-EM as an alternative for structure determination.
Structures of all three unliganded enzymes were determined at resolutions sufficient to allow clear visualization and interpretation of most side chains (2.8-Å, 3.4-Å and 2.9-Å resolution for Bl_Man38A, Bl_Man38B and Bl_Man38C, respectively; Supplementary Table 5 and Supplementary Figs. 8 and 9), featuring regions of notable higher resolution (up to 2.3 Å; Supplementary Fig. 10). Interestingly, the cryo-EM structures of the three enzymes revealed a tetrameric arrangement in which the active sites are internal to the protein complex (Fig. 2a), and large lateral channels allow the entrance of the substrate (Supplementary Fig. 11). In the tetramers, all the protomers interact with each other mainly through the accessory modules found in these enzymes (Supplementary Fig. 12 and Supplementary Table 6), indicating that domain composition and oligomerization are correlated in the GH38 family. This oligomeric arrangement is also found in yeast α-mannosidases and diverges from other structurally characterized (non-cytosolic mammalian, plant, insect and bacterial) GH38 enzymes that are either monomeric or dimeric (Extended Data Fig. 6 and Supplementary Table 7)34–36. It is worth noting that the mammalian cytosolic α-mannosidase (Man2C1) is also tetrameric, acts specifically on HMNG and is activated by Co2+ ions37; however, its structure remains undetermined. Our results along with phylogenetic analysis (Extended Data Fig. 7) indicate that bifidobacterial, yeast vacuolar35 and mammalian cytosolic37 α-mannosidases establish a subgroup within the GH38 family that shares the same oligomeric state, specific activity on HMNGs and activation by Co2+ ions.
Fig. 2 |. Cryo-EM structure of Bl_Man38A as an archetypal model of the three bifidobacterial α-mannosidases.

a, Tetrameric arrangement of Bl_Man38A colored by chain, except for chain A, which was colored by domains using the color scheme represented in b and c. b, Scheme showing the domain organization. c, Cartoon representation of one protomer of the tetramer colored by domain. d, Conserved metal ion-binding site that composes the −1 subsite complexed with a Zn2+ ion. e, Interactions with a mannose at the −1 subsite of the Bl_Man38AD387A mutant. f, Superposition of residues that compose the −1 subsite for wide-type structures of Bl_Man38A (carbons are in cyan), Bl_Man38B (carbons are in white) and Bl_Man38C (carbons are in dark gray). The corresponding figures for the protomers of Bl_Man38B and Bl_Man38C are presented in Supplementary Figs. 13 and 14.
Bl_Man38A–Bl_Man38C enzymes exhibit a multimodular architecture consisting of six domains (Fig. 2b,c), which include a (β/α)8-barrel catalytic domain, which is conserved across members of the GH38 family (Pfam PF01074.22), two non-conserved N-terminal domains, exhibiting jelly roll and four-helix bundle folds, and three distinct C-terminal domains, an α-mannosidase middle domain with an immunoglobulin/albumin-binding domain-like fold (PF09261.11), a GH38 C-terminal β-sheet domain (PF07748.13) and a C-terminal β-sandwich (PF170677.1). This domain organization is similar to that found in other tetrameric GH38 α-mannosidases35 lacking a β-sheet motif between the two C-terminal domains that is considered pivotal for the dimerization of some bacterial GH38 members, such as from Streptococcus pyogenes36 (Supplementary Fig. 13). Furthermore, the N-terminal domains (jelly roll and four-helix bundle), which are conserved in the tetrameric GH38 members, are not present in other bacterial GH38 members (Supplementary Fig. 13), which have important implications in the mechanism of substrate recognition as discussed later. These analyses indicate that the bifidobacterial α-mannosidases resemble eukaryotic GH38 members with similar function more than other bacterial members so far characterized. The correlation between function and oligomeric state is also supported by the fact that the lysosomal GH38 α-mannosidases from evolutionary distant organisms, such as cattle38 and jack bean39, share the same domain organization and form elongated dimers. This evidence suggests that structural and functional properties in the GH38 family are better correlated with biological function and cellular localization, independent of any phylogenetic correlation at the organism level.
Subsite –1 is a generalist platform for α-mannosyl cleavage
The active site was identified by the presence of the nucleophile and acid/base catalytic residues according to the retaining mechanism of this family and the presence of a divalent metal ion at the −1 subsite37,40 (Fig. 2d and Supplementary Fig. 14). The metal binding site consists of a histidine dyad (His 275/His 276/His 272 and His 597/His 598/His 594 for Bl_Man38A/Bl_Man38B/Bl_Man38C), the catalytic nucleophile (Asp 387/Asp 388/Asp 384) and another conserved aspartic residue (Asp 277/Asp 278/Asp 274). In the unliganded structures, the metal ion was interpreted as being a Zn2+ ion based on comparative activity assays (Fig. 2d and Supplementary Figs. 5–7).
To identify the structural elements related to functional divergence of the three GH38 α-mannosidases, we exhaustively attempted to obtain cryo-EM structures with Man5GlcNAc or Man9GlcNAc using both wild-type enzyme and inactive mutants (Bl_Man38AD387A and Bl_Man38AD487A). Despite all efforts and multiple datasets collected, it was not possible to observe the substrate bound to the active site. However, for the Bl_Man38AD387A mutant, we observed a mannose bound to the active site (Supplementary Fig. 15), allowing us to identify the enzyme–substrate interactions at the −1 subsite (Fig. 2e), which is considered a relevant frame for the catalytic reaction. In this complex, several hydrophobic and polar interactions are involved in saccharide binding, highlighting the pyranosidic ring stacking with Trp 280 and the O2 and O3 mannosyl atoms anchored to the metal ion (2.2 Å). Superposition of the Bl_Man38AD387A–mannose complex with unliganded forms of Bl_Man38A–Bl_Man38C showed that the residues forming the −1 subsite are mostly conserved in the three enzymes (Fig. 2f) and in other members of the GH38 family (Supplementary Fig. 14). This highly energetic binding mode of the mannosyl residue at the −1 subsite based on metal ion, stacking and polar interactions indicates that this subsite is instrumental for the proper binding of the substrate in a catalytic-productive manner, allowing distinct glycosidic bonds between mannosyl residues in N-glycans (α-1,2-, α-1,3- and α-1,6-linkages) to be cleaved by the three enzymes despite the differences in substrate preference.
Structural basis of N-glycan binding in GH38 α-mannosidases
To further define how N-glycans interact with the active-site cleft of these α-mannosidases, we performed docking and molecular dynamics simulations with the preferential substrate for each enzyme: Man5GlcNAc for Bl_Man38A, both Man5GlcNAc and Man9GlcNAc for Bl_Man38B and Man9GlcNAc for Bl_Man38C (Supplementary Fig. 16). The substrates were stabilized at different positions in the simulations for each enzyme; however, the B-arm of N-glycans was accommodated in the −1 subsite for all of them, suggesting that this arm would be the first to be cleaved by these α-mannosidases (Fig. 3).
Fig. 3 |. Structural basis of high-mannose N-glycan recognition by Bl_Man38A–Bl_Man38C.
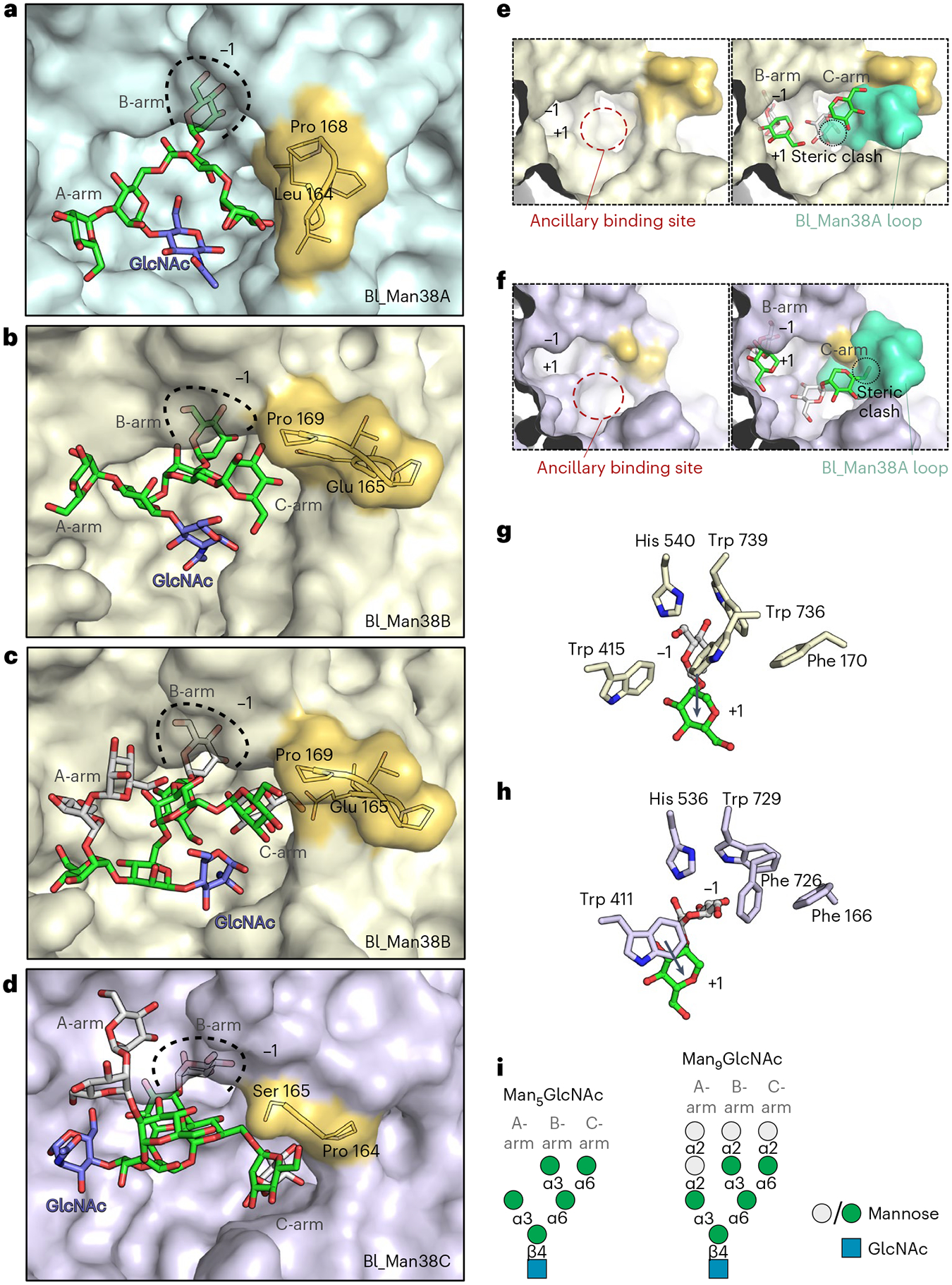
a–d, Representative structures of Bl_Man38A (a), Bl_Man38B (b and c) and Bl_Man38C (d) after docking and energy minimization with Man5GlcNAc (a and b) and Man9GlcNAc (c and d). e,f, Superposition of the hypervariable loop of Bl_Man38A (green) to the active sites of Bl_Man38B (e) and Bl_Man38C (f), showing only the B- and C-arms of Man9GlcNAc (docked and energy-minimized complexes). g,h, Comparison of the putative interactions formed by Trp 736 in Bl_Man38B (g) and its substitution for a phenylalanine residue in Bl_Man38C (Phe 726) (h), showing the shift of orientation of the α-1,2-mannoside in the active site of the enzymes. Only the α-1,2-mannoside of Man9GlcNAc is shown for better visualization. i, Schematic representation of Man5GlcNAc and Man9GlcNAc docked into the structures.
The residues forming the –1 subsite are conserved between Bl_Man38A, Bl_Man38B and Bl_Man38C enzymes, except for a tryptophan substitution by a phenylalanine in Bl_Man38C (Phe 726). The simulations with Man9GlcNAc indicated that this substitution alters the interaction pattern at the –1 subsite, permitting the mannosyl moiety at the +1 subsite to stack with Trp 411 (Fig. 3g,h), which could be associated with regioselectivity. To test this hypothesis, we analyzed the activity of the Bl_Man38CF726W mutant with manno-disaccharides (Supplementary Table 4) and N-glycans (Extended Data Fig. 8). The mutation increased the catalytic efficiency on all disaccharides, retaining the preference for α-1,2-mannobiose; however, a change in the catalytic efficiency between α-1,6- and α-1,3-mannobiose was observed. The Bl_Man38CF726W mutant showed a ~2.5-fold higher catalytic efficiency on α-1,3-mannobiose than on α-1,6-mannobiose (Supplementary Table 4), the opposite observed for the wild-type enzyme. In addition, this mutation decreased the efficiency against α-1,2-linkages on N-glycans (Man9GlcNAc) and increased activity against Man5GlcNAc (Extended Data Fig. 8). These results indicate that Phe 726 contributes to the preference of Bl_Man38C for Man9GlcNAc via a reaccommodating mechanism of N-glycan arms without imposing stereochemical restrictions to the glycosidic bond itself.
The binding mode and conformation of the A-arm varied depending on the enzyme and substrate analyzed (Fig. 3a–d,i). The A-arm was positioned externally to the active site and did not seem to undergo stringent stereochemical impositions due to its length or type of bond. The C-arm extremity of Man9GlcNAc, however, fit into a specific and conserved hydrophobic pocket (Trp 280/Trp 281/Trp 277 and Leu 281/Leu 282/Leu 278 in Bl_Man38A/Bl_Man38B/Bl_Man38C), named as ancillary binding site. At the vicinity of this ancillary binding site, there is a hypervariable loop (164LGVPP168 in Bl_ManA, 165EGPTP169 in Bl_Man38B and 164PS165 in Bl_Man38C) in the three paralogs, which protrudes from the N-terminal jelly roll domain and is only present in tetrameric GH38 α-mannosidases (Supplementary Figs. 17 and 18). Notably, this loop is the most divergent motif in the catalytic interface between the bifidobacterial GH38 enzymes, suggesting that it is involved in N-glycan selectivity.
The hypervariable loop is elongated in Bl_Man38A compared to Bl_Man38C (Supplementary Figs. 17 and 18) or adopts a distinct conformation in relation to Bl_Man38B (Supplementary Figs. 17 and 18), creating steric impediments for the proper accommodation of the Man9GlcNAc C-arm in the Bl_Man38A active site (Fig. 3e,f), which correlates with the low activity of Bl_Man38A against this substrate. To infer whether only this hypervariable loop would determine the specificity of Bl_Man38A for smaller N-glycans, the Bl_Man38AΔLeu 164–Pro 168 mutant was constructed; however, the shortening of this loop aiming to remove the steric impediment did not favor the activity on Man9GlcNAc yet negatively affected the activity on Man5GlcNAc (Extended Data Fig. 9). Therefore, although the hypervariable loop likely represents a steric barrier for Man9GlcNAc binding in Bl_Man38A, other structural elements, including the Trp→Phe substitution in the −1 subsite, might cooperate for a more elaborate mechanism of N-glycan selectivity in these enzymes.
In the Schizosaccharomyces pombe GH38 α-mannosidase35, the hypervariable loop also displays distinct amino acid composition and adopts a different conformation compared to bifidobacterial enzymes (Supplementary Fig. 17), whereas in the Saccharomyces cerevisiae GH38 α-mannosidase34, this loop was not modeled, indicating conformational flexibility. The lack of data regarding regioselectivity and N-glycan preference, associated with the high variability of this loop, make it difficult to infer, by structural analogy, functional aspects of yeast α-mannosidases.
Together, our results highlight that tetrameric GH38 α-mannosidases use a distinct mechanism of substrate recognition compared to monomeric or dimeric GH38 α-mannosidases, attributing a role for the N-terminal jelly roll domain in substrate recognition, in particular to the hypervariable loop, which likely plays a role in substrate binding and might contribute to N-glycan preference.
N-GUL GH31 α-glucosidase cleaves monoglucosylated N-glycans
The Bifidobacterium longum N-GUL also encodes an uncharacterized GH31 enzyme (Bl_Glc31 encoded by bl1334), and, to assess its role in the N-glycan degradation cascade, we constructed a soluble variant that lacks an N-terminal region comprising 51 residues not found in homologous proteins (Supplementary Fig. 19). The activity of the recombinant enzyme was first tested on synthetic substrates (pNP-carbohydrates), indicating that it is an α-glucosidase (named as Bl_Glc31; Supplementary Table 8 and Extended Data Fig. 10a,b). Bl_Glc31 displays sequence similarity (~20–30%) with other characterized bacterial GH31 α-glucosidases, such as Bt_0339 and Bt_3299 from Bacteroides thetaiotaomicron41, Ro-aG1 from Blautia obeum42 and LlGH31 from Lactococcus lactis43. Bacteroides thetaiotaomicron enzymes and Ro-aG1 are involved in starch degradation, whereas LlGH31 was proposed to play a role in the degradation of nigerooligosaccharides43. Therefore, we tested Bl_Glc31 on typical substrates containing α-linked glucosides, maltose and sucrose; however, no activity was detected (Extended Data Fig. 10c–j), which is not surprising considering its colocalization with GH enzymes in an N-GUL.
Given its genomic context and the similarity with the catalytic subunit of glucosidase II (sequence similarity of 31%), we then examined the capacity of Bl_Glc31 to hydrolyze glucosylated N-glycans, in particular the monoglucosylated form of HMNGs (GlcMan9GlcNAc), naturally produced by intestinal epithelial cells (Fig. 4b,c)44. This type of HMNG is abundant in apoptotic vesicles formed from this tissue45 and might serve as a putative source of nutrients for bifidobacteria in their natural environment. MS analysis showed that Bl_Glc31 can specifically cleave GlcMan9GlcNAc, generating only Man9GlcNAc (Fig. 4b,c and Supplementary Fig. 20), which demonstrates biochemically its capacity to remove glucosyl terminations in these types of N-glycans.
Fig. 4 |. Cleavage profile of high-mannose N-glycans by Bl_Endo85 and Bl_Glc31.
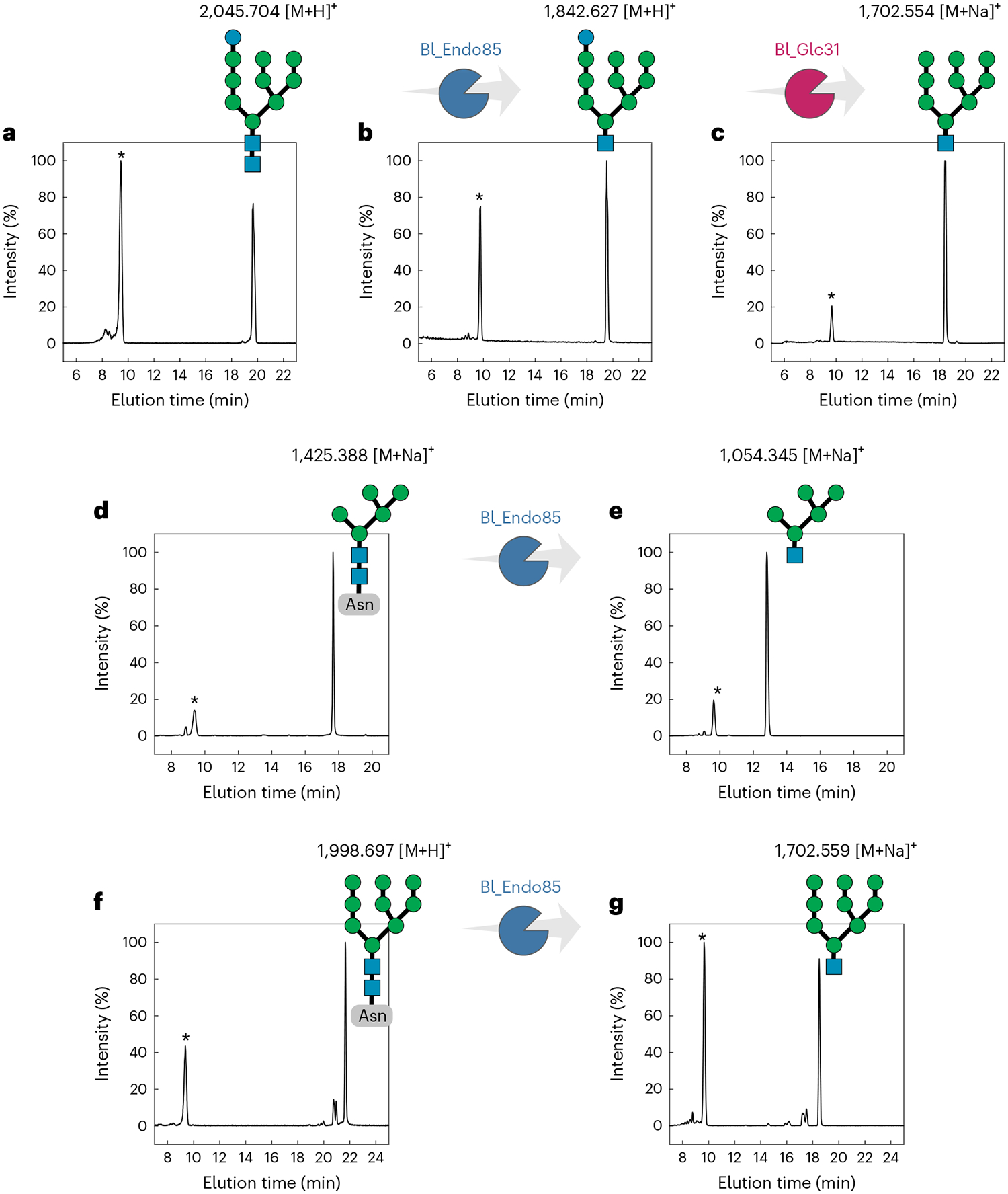
a, Control of GlcMan9GlcNAc2 in which no enzymes were added. b, Reaction of Bl_Endo85 against GlcMan9GlcNAc2, generating GlcMan9GlcNAc. c, Reaction of Bl_Glc31 against GlcMan9GlcNAc, generating Man9GlcNAc. d,f, Controls of Man5GlcNAc2-Asn (d) and Man9GlcNAc2-Asn (f) in which no enzymes were added. e,g, Reactions of Bl_Endo85 against Man5GlcNAc2-Asn (e) and Man9GlcNAc2-Asn (g), generating Man5GlcNAc and Man9GlcNAc, respectively. Asterisks (*) indicate the internal control xylohexaose. m/z values are indicated above the substrates or reaction products, and all the mass spectra are provided in Supplementary Figs. 20 and 21.
As Bl_Glc31 is predicted to be localized in the cytoplasm, it necessitates that Bl_Endo85 would cleave monoglucosylated HMNGs. The ability of Bl_Endo85 to cleave bovine RNAseB N-glycans was previously reported24; however, its activity specifically on Man5GlcNAc2, Man9GlcNAc2 or GlcMan9GlcNAc2 was not demonstrated. Therefore, we assayed Bl_Endo85 on these substrates, showing that this ENGase can act on Man5GlcNAc2 (Fig. 4d,e and Supplementary Fig. 21) and Man9GlcNAc2 (Fig. 4f,g and Supplementary Fig. 21), including the monoglucosylated type (Fig. 4a,b and Supplementary Fig. 20), which supports a role of Bl_Glc31 in the N-glycan degradation cascade. The non-removal of this glucosyl moiety impairs the action of α-mannosidases; as such, this α-glucosidase is mandatory for the utilization of monoglucosylated HMNGs by Bifidobacterium longum. Furthermore, among the bacterial GH31 α-glucosidases, Bl_Glc31 is the first to be described to participate in N-glycan processing, expanding the repertoire of biological functions of bacterial GH31 enzymes.
Unconventional isomerization is key for mannose metabolism
The metabolic pathways of monosaccharides in bifidobacteria are well described; however, the mechanism of mannose metabolism, the major product of HMNG degradation by the N-GUL, remains elusive in these bacteria46. The Bifidobacterium longum NCC2705 genome lacks genes encoding mannose-6-phosphate isomerases, which are instrumental in connecting the phosphorylated mannose in the glycolytic pathway in most organisms. Nevertheless, the gene bl1337 encodes an uncharacterized epimerase that displays 38% sequence identity with YihS, a sulfoquinovose isomerase from Salmonella enterica that is able to convert mannose into fructose without prior phosphorylation47. Because bl1337 is located within the predicted N-GUL in the Bifidobacterium longum NCC2705 genome (Fig. 5a), it was envisaged to play a similar role in the metabolism of N-glycan-derived mannoses.
Fig. 5 |. bl1337 encodes an efficient ManI.
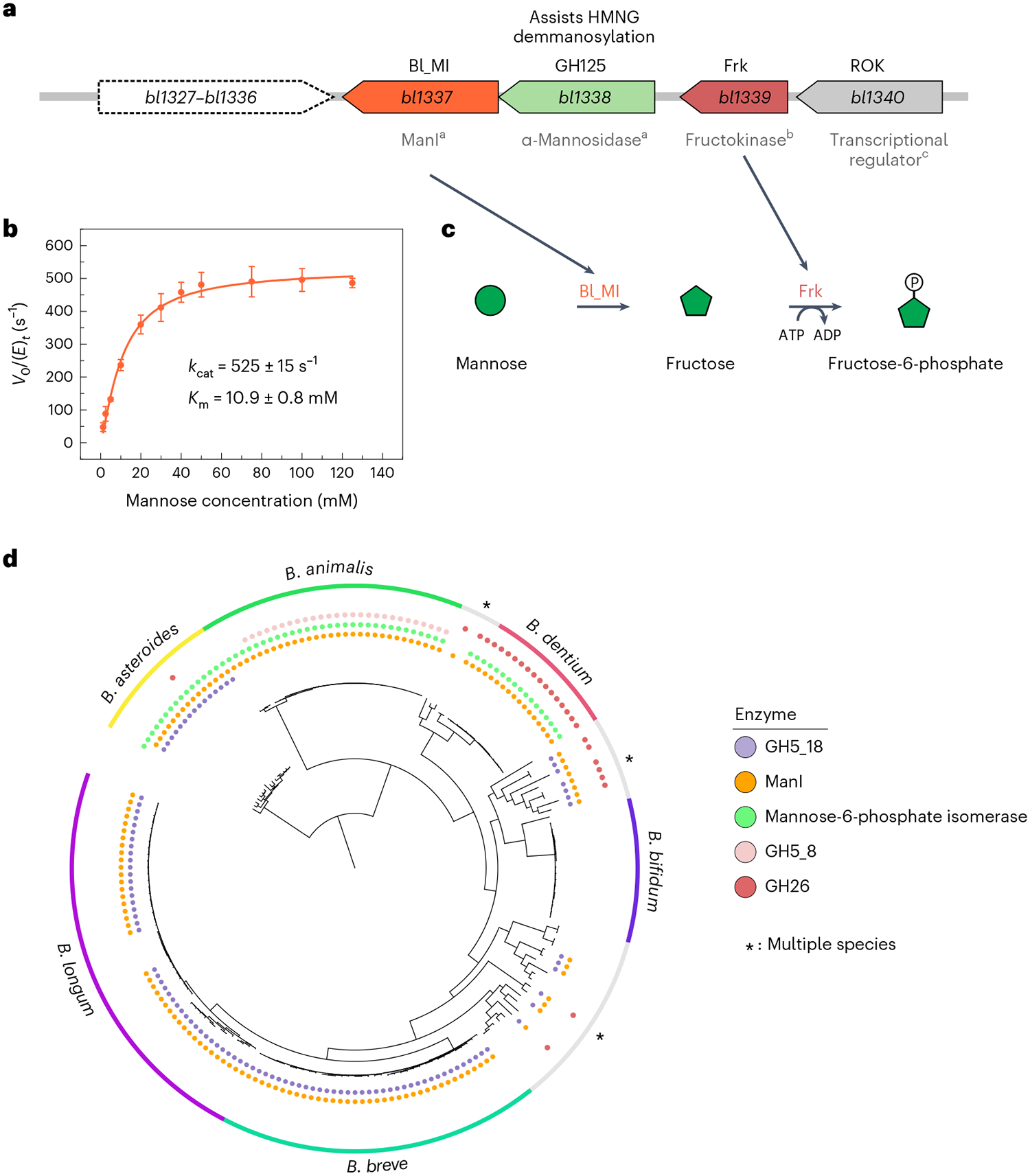
a, Gene cluster including bl1337 (ManI Bl_MI), bl1339 (fructokinase), bl1338 (GH125 α-1,6-mannosidase) and bl1340 (ROK-family transcriptional regulator); a, biochemically characterized in this work; b, characterized in a previous work28; c, activity predicted. b, Substrate saturation curve of Bl_MI including kinetic parameters. Results are expressed as mean ± s.d. from three independent experiments (n = 3). c, Proposed biochemical route for the conversion of mannose to fructose-6-phosphate, a central molecule for the fermentative pathway. d, Phylogenetic tree of Bifidobacterium genomes, indicating the presence of genes encoding ManIs, mannose-6-phosphate isomerases, GH5_18 β-mannosidases (markers for N-glycan degradation) and GH26 and GH5_8 β-mannanases (markers for mannan/glucomannan degradation). Multiple species are detailed in Extended Data Fig. 2.
The gene product of bl1337 was biochemically characterized, demonstrating its ability to isomerize mannose into fructose (Supplementary Fig. 22). The enzyme (named as Bl_MI) displays a kcat of 525 ± 15 s−1 and a K m of 10.9 ± 0.8 mM (kcat /Km = 48 −1s mM−1 ) on mannose at optimum conditions (Fig. 5b and Supplementary Fig. 23), representing the highest catalytic efficiency described so far for the conversion of mannose to fructose (Supplementary Table 9). The fructose generated by Bl_MI could be further converted to fructose-6-phosphate by the fructokinase encoded by bl1339 (ref.28), allowing it to enter the central hexose fermentative pathway of bifidobacteria known as the bifid shunt29 (Fig. 5c).
Evolutionary analysis showed that orthologous genes of bl1337 encoding mannose isomerases (ManIs) are not conserved in all Bifidobacterium species, but they are present in the genomes of most strains that use mannose-containing carbohydrates (Fig. 5d). Bifidobacterium longum, Bifidobacterium breve and Bifidobacterium asteroides contain N-GULs in a genomic region adjacent to the gene encoding ManI. Species that use mannans and glucomannans as mannose sources also encode ManI, exemplified by Bifidobacterium dentium and Bifidobacterium animalis7, which express endo-β-mannanases from families GH26 and GH5_8, respectively. However, for Bifidobacterium asteroides, Bifidobacterium animalis and Bifidobacterium dentium, mannose-6-phosphate isomerases co-occur with ManI, indicating dual metabolic routes for mannose in these species. These analyses show a prevalence of the mannose-to-fructose isomerization route in mannose metabolism in this genus; however, divergent mannose pathways are found at species and strain levels in Bifidobacterium.
Discussion
The successful establishment of a specific bacterial genus in the human gastrointestinal lumen is associated with its ability to obtain carbon and energy from diverse recalcitrant sources in this highly competitive environment. Bifidobacteria are well known for their biochemical capability to process and use milk saccharides; however, their lifelong existence in this niche indicates that alternative molecular strategies for nutrient uptake from distinct glycan sources exist that support their survival.
Herein, we revealed the multienzymatic system in bifidobacteria that allows them to use host HMNGs as an energy and carbon source. Our results (1) illuminate the cooperative action of distinct cytosolic α-mannosidases in N-glycan demannosylation, including structural insights into substrate preference and regioselectivity; (2) unveil the role of a GH31 α-glucosidase for the utilization of monoglucosylated HMNGs and the ability of the Bifidobacterium longum ENGase in releasing this type of HMNG and (3) demonstrate the unconventional bio-catalytic isomerization before phosphorylation as the likely metabolic route connecting mannose to the bifid shunt fermentative pathway in these bacteria. These findings along with previous studies about the GH85 ENGase (Bl_Endo85)24 and the GH5_18 β-mannosidase23 provide a comprehensive understanding of all biochemical steps involved in the elaborate and multienzymatic degradation cascade and utilization of HMNGs by bifidobacteria, as outlined in Fig. 6 (Supplementary Table 10), and poses as a representative model for the phylum Actinobacteria.
Fig. 6 |. Biochemical model for high-mannose N-glycan degradation and metabolism by Bifidocaterium longum.

Schematic representation of a proposed model for high-mannose N-glycan degradation by Bifidobacterium longum NCC2705 based on the biochemical data presented in this work and predicted cellular localization (Supplementary Table 11).
The diverse repertoire of α-mannosidases in Bifidobacterium longum is particularly notable and involves non-trivial cooperation mechanisms between a GH38 generalist α-mannosidase (Bl_Man38B), two complementary GH38 α-mannosidases (Bl_Man38A and Bl_Man38C) and a GH125-specific α-1,6-mannosidase, offering a diversified biochemical toolbox for the processing of N-glycans. This functional aspect along with the sophisticated mechanism of Man1GlcNAc cleavage by a specialist GH5_18 β-mannosidase and the unconventional metabolic fate of mannose highlight the convolution of molecular strategies used by bifidobacteria to use host N-glycans as carbon and energy sources.
It is also remarkable how different this bifidobacterial mechanism of HMNG digestion and metabolism is compared to other human gut or Gram-positive bacteria. For instance, the Gram-negative Bacteroides thetaiotaomicron, also a member of the human gut microbiota, performs N-glycan demannosylation in the perisplasm by a logical and sequential action of three specific GH92 α-mannosidases, and the degradation products follow the classical fermentative pathways16. However, in the Gram-positive bacterium Streptococcus pneumoniae, which belongs to the phylum Firmicutes, the demmanosylation of Man9GlcNAc starts in the extracellular environment by a GH92 α-1,2-mannosidase, generating Man5GlcNAc, which is then cleaved by a GH85 ENGase (EndoD) and internalized17. In the cytoplasm, a putative GH38 α-1,3-mannosidase along with a GH125 α-1,6-mannosidase reduce Man5GlcNAc into Man1GlcNAc. By contrast, N-glycan degradation by the Actinobacteria Bifidobacterium longum starts with the release of the entire N-glycan by a membrane-anchored GH85 ENGase, proceeds with HMNG uptake via ABC transporters and ends in the cytoplasm with complete depolymerization by the action of the multienzymatic system mechanistically described herein (Fig. 6 and Supplementary Table 11). These analyses highlight the diverse molecular strategies present in the bacterial kingdom to cope with HMNG processing, even among Gram-positive phyla or human gut bacteria, shaped through evolution to allow the utilization of host N-glycans as energy and carbon sources in distinct ecological niches and under highly competitive environments.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41589-022-01202-4.
Methods
Phylogenetic analysis
To inspect the genomic organization of N-GULs in Bifidobacterium, a set of 92 single-copy core genes was extracted from each genome, aligned, concatenated and used as input to the FastTree 2.1.10 (ref.48) software to infer an approximate maximum likelihood phylogenetic tree, with a JTT + CAT amino acid substitution model. Local support values were calculated with the Shimodaira–Hasegawa test with 1,000 bootstrap replicates. Homologous genes from Bifidobacterium longum NCC2705 N-glycan utilization loci were annotated using the tool cblaster 1.3.11 (ref.49). The phylogenetic tree was annotated and visualized using itol 6.0 (https://itol.embl.de/).
Molecular cloning and mutagenesis
Escherichia coli codon-optimized open-reading frames encoding Bl_Man38A (bl1327), Bl_Man38B (bl1328), Bl_Man38C (bl1329), Bl_Glc31 (bl1334), Bl_Endo85 (bl1335), Bl_MI (bl1337) and Bl_Man125 (bl1338) were obtained in the pET28a vector from Biomatik or GeneScript (Supplementary Table 12). The mutants Bl_Man38AD387A, Bl_Man38AΔLeu 164–Pro 168, Bl_Man38CF726W, Bl_Man38CW104A and Bl_Glc31ΔMet 1–Arg 51 were constructed using the QuikChange II XL kit (Agilent Technologies).
Protein expression and purification
Proteins were expressed in E. coli BL21 (DE3) cells (Agilent Technologies) in selective LB medium at 20 °C for 16 h. Cells were collected and lysed by sonication on ice. The lysed cells were centrifuged, and the supernatant was applied onto a HiTrap Chelating HP column (GE Healthcare) preequilibrated with 20 mM phosphate buffer (pH 7.4) containing 500 mM NaCl. The proteins were eluted with an imidazole gradient, and fractions containing the enzyme were pooled, concentrated and loaded onto a Superdex 200 16/60 HiLoad column (GE Healthcare) preequilibrated with phosphate buffer (150 mM NaCl and 20 mM sodium phosphate, pH 7.4). Fractions of interest were analyzed by polyacrylamide gel electrophoresis under denaturing conditions and dynamic light scattering (Supplementary Figs. 24–30). Only monodisperse samples were used in the functional and structural experiments.
Source of the carbohydrates
Man9GlcNAc2 and GlcMan9GlcNAc2 were purchased from Dextra Laboratories. Man9GlcNAc2-Asn and Man5GlcNAc2-Asn were prepared by enzymatic digestion of soybean agglutinin isolated from soybean flour with subsequent chromatographic purification following a previously described procedure20. Disaccharides, α-1,2-, α-1,3- and α-1,6-mannobiose, were purchased from Carbosynth. The monosaccharides mannose, fructose, glucose and xylose were purchased from Sigma-Aldrich. Xylobiose, xylotetraose and xylohexaose were purchased from Megazyme. Synthetic substrates p-nitrophenyl-α-d-mannopyranoside, p-nitrophenyl-α-d-glucopyranoside, p-nitrophenyl-α-d-galactopyranoside, p-nitrophenyl-α-d-xylopyranoside, p-nitrophenyl-α-l-arabinofuranoside, p-nitrophenyl-α-l-arabinopyranoside and p-nitrophenyl-α-d-fucopyranoside were purchased from Sigma-Aldrich.
Enzyme assays with synthetic substrates
The activities of Bl_Man38A–Bl_Man38C and Bl_Glc31 were screened against the aforementioned synthetic substrates, and optimum conditions for hydrolysis were determined using their preferential synthetic substrates, p-nitrophenyl-α-d-mannopyranoside and p-nitrophenyl-α-d-glucopyranoside, for Bl_Man38A–Bl_Man38C and Bl_Glc31, respectively (Supplementary Table 13). Reactions with Bl_Man38A–Bl_Man38C were supplemented with 0.1 mM CoCl2. A saturated solution of sodium tetraborate was added to stop the reactions. The p-nitrophenolate released was detected at 400 nm using an Infinite M200 Pro (Tecan), and data were collected using the i-Control 2.0. Enzyme assays consisted of three independent experiments.
Enzyme assays with N-glycans and disaccharides
Bl_Endo85 reactions were performed in 150 mM ammonium acetate (pH 5.0) at 37 °C for 16 h with the following oligosaccharides: Man9GlcNAc2, GlcMan9GlcNAc2, Man9GlcNAc2-Asn and Man5 GlcNAc2-Asn (Supplementary Table 13). Bl_Man38A–Bl_Man38C (0.2 mg ml−1) and Bl_Man125 reactions were performed in 150 mM ammonium acetate (pH 5.5) at 37 °C for 1 h with the oligosaccharides Man9GlcNAc (0.025 mg ml−1) and Man5GlcNAc (0.025 mg ml−1). The reactions with the disaccharides (α-1,2-mannobiose, α-1,3-mannobiose or α-1,6-mannobiose) were performed using 1 mM substrate and 0.8 mg ml−1 enzyme under the same conditions used for the oligosaccharides. In the cooperation assays, Bl_Man38A–Bl_Man38C (0.2 mg ml−1) were combined with Bl_Man125 (0.2 mg ml−1), and the reactions were conducted under the aforementioned conditions for 30 min or 1 h (as indicated in the figure legends). To determine the kinetics parameters of Bl_Man38A–Bl_Man38C, mutants and Bl_Man125 on disaccharides, the reactions were performed at pH 5.5 and 37 °C in a substrate range from 0.1 to 6 mM. The reaction time and enzyme concentration were selected in a linear region of the hydrolysis reaction (Supplementary Table 14). The reactions were stopped with the addition of acetonitrile to a final concentration of 50%. The liquid phase was removed by using a vacuum concentrator, and a solution of 75% acetonitrile (24 μl) and xylohexaose (internal standard; 50 mM, 1 μl) was added to the reactions. Reaction aliquots (10 μl) were analyzed by LC–MS using a Glycan BEH amide column (17 μm × 2.1 mm × 150 mm; Waters Corporation) at 60 °C (liquid phase, 100 mM ammonium formate (pH 4.4) and 100% acetonitrile) in a QExactive Plus (Thermo Fisher Scientific) mass spectrometer coupled to an Ultimate 3000 liquid chromatographer (Thermo Fisher Scientific) operating in ESI (+) at 3.8 kV and 350 °C and auxiliary gas at 400 °C and 15 arbitrary units. The DDA mode (top5) at 70,000 resolution and m/z screening from 150 to 2,100 were used. The reactions with disaccharides were analyzed in an LTQ XL linear ion trap mass spectrometer (Thermo Fisher Scientific), and an ESI (+) ion source was used. The spray voltage was maintained at 3.5 kV, and the ion source temperature and the scan range were set to 275 °C and 190–600 m/z, respectively. MS data were collected and analyzed using the software Xcalibur 4.2.47 (Thermo Fisher Scientific). Xylohexaose was used as an internal standard for both oligosaccharides and disaccharides assays. The internal area of the peaks corresponding to the analytes was adjusted in function of the internal standard. The kinetic parameters were calculated by non-linear regression analysis of the Michaelis–Menten plot using OriginPro 8.1 (OriginLab). Enzyme assays consisted of three independent experiments.
Isomerization assays
The isomerase activity of Bl_MI was analyzed by the carbazole-cysteine method50 and capillary zone electrophoresis (CZE). The reactions were stopped by incubating at 100 °C for 2 min, and the product was detected at 560 nm using an Infinite M200 Pro (Tecan). The pH dependence of enzyme activity was assayed in McIlvaine buffer in a pH range from 4 to 8. The temperature dependence of enzyme activity was assayed in McIlvaine buffer (pH 6.5). To determine the kinetics parameters, the reactions were incubated for 10 min at pH 6.5 and 50 °C in the substrate range from 1 to 120 mM using 0.25 μg ml−1 enzyme (Supplementary Table 14). The kinetic parameters were calculated by non-linear regression analysis of the Michaelis–Menten plot. Enzyme assays consisted of three independent experiments. To analyze the monosaccharides by CZE, product reactions were labeled with 8-aminopyrene-1,3,6-trisulfonic acid (APTS) using 1 M sodium cyanoborohydride and 6.5 mM APTS for 90 min at 60 °C. Samples were resuspended in running buffer (40 mM potassium phosphate, pH 2.5) and analyzed in a P/ACE MDQ instrument (Beckman Coulter) equipped with a laser-induced fluorescence detection module. A capillary of 75 μm internal diameter and 12 cm effective length was used. Electrophoretic conditions were set to 20 kV mA with reverse polarity at 25 °C. The electrophoretic behavior of labeled products was compared to standards. CZE data were collected using the software Karat32 8.0 (Beckman Coulter).
Nuclear magnetic resonance spectroscopy
To confirm the isomerization of mannose into fructose, reactions containing mannose (50 mM) were incubated with 0.00025 mg ml−1 Bl_MI for 10 min or 0.1 mg ml−1 Bl_MI for 30 min at 50 °C in McIlvaine buffer (pH 6.5). The reactions were stopped by incubation at 100 °C for 2 min, and the proteins were removed by filtration using Amicon Ultra centrifugal filters (Millipore) with a 3-kDa pore. Lyophilized samples were dissolved in 0.6 ml of deuterium oxide-containing phosphate buffer (0.1 M, pH 7.4) containing 0.5 mM of 3-(trimethylsilyl)-2,2,3,3-tetradeuteropropionic acid. Spectra were acquired using an Agilent DD2 500-MHz spectrometer (Agilent Technologies) equipped with a triple-resonance probe at 25 °C. 1H-NMR spectra acquisition was performed with 64 scans collected and 32,000 data points over a spectral width of 8,000 Hz. A 1.5-s relaxation delay was incorporated between scans, during which a continual water presaturation radio frequency field was applied to eliminate residual water signal. Two-dimensional (2D) NMR 1H-1H-TOCSY spectra were acquired using a spectral width of 8,000 Hz and 256 increments with 24 transients of 2,000 complex points for each free induction decay. Two-dimensional NMR 1H-13C-HSQC spectra were acquired with a spectral width of 8,000 Hz × 25,133 Hz and 128 increments with 88 transients of 2,000 complex points. In both 2D NMR spectra, as 1H-NMR spectra, a 1.5-s relaxation delay was incorporated between scans with water presaturation radio frequency to eliminate water signal. The reaction products were processed and quantified using the software NMR Suite 8.1 (Chenomx) using 2D 1H-1H-TOCSY/1H-13C-HSQC spectra to confirm the reaction product identity.
Cryo-electron microscopy grid preparation, data collection and processing
Grids were prepared with fresh samples without intermediary steps such as concentration, centrifugation or buffer exchange. The concentrations of the proteins were 1.28, 0.78, 0.34 and 2.2 mg ml−1, and the estimated hydrodynamic radii were 13.5, 14.1, 14.8 and 13.5 nm, respectively, for Bl_Man38A, Bl_Man38B, Bl_Man38C and Bl_Man38AD387A. The complex with mannose was obtained after incubation of Bl_Man38AD387A (18 μM) with 1 mM Man5GlcNAc for 6 h on ice. The grids for cryo-EM were prepared using Vitrobot Mark IV (Thermo Fisher Scientific) operating at 4 °C and 100% humidity. The samples (3 μl) were deposited in Quantifoil R 2/2 mesh 200 (Quantifoil), blotted for 4–5 s and fast vitrified in liquid ethane. Data were collected at the Brazilian Nanotechnology National Laboratory on a transmission electron microscope Titan Krios G3i (Thermo Fisher Scientific) equipped with a Cs corrector operating at 300 kV at a magnification of ×96,000, corresponding to a sampling of 0.67 Å per pixel. Movies were acquired using the software EPU 2.11.1.11REL (Thermo Fisher Scientific) in a Falcon 3EC detector operating in counting mode, with 20 frames per movie, an accumulated dose of 30 e− Å−2 and an applied defocus between 1.0 and 2.0 μm. Collected datasets were submitted to a posteriori camera correction using IMAGIC-4D51. The subsequent processing and three-dimensional (3D) map reconstruction and refinement were performed using cisTEM software52. These steps include movie alignment, CTF determination, particle picking, 2D classification, ab initio 3D reconstruction, 3D refinement and map sharpening. All maps, except Bl_Man38AD387A-MAN were generated without prior information to avoid bias. Fourier shell correlation curves were calculated using the software cisTEM 1.0.0-beta52. Local resolution estimates were calculated using the IMAGIC-4D software with half-maps produced by cisTEM.
Model building, refinement and validation
An ab initio model of Bl_Man38A was built based on the protein sequence and the reconstructed cryo-EM density map using the tool map_to_model from Phenix 1.19.2–4158 (ref.53) and further manually adjusted using the program Coot 0.9.4.1 (ref.54). The Bl_Man38A structure was used to generate a homology model for Bl_Man38B (60% sequence identity) and Bl_Man38C (55% sequence identity). The atomic coordinates were refined using the real_space_refinement from Phenix 1.19.2–4158 (ref.53), and model validation was performed using Molprobity 4.5.1 (ref.55).
Ligand docking and molecular dynamics simulations
Man5GlcNAc and Man9GlcNAc ligands were docked into the cryo-EM atomic coordinates of Bl_Man38A, Bl_Man38B and Bl_Man38C using Autodock Vina 1.1.2 (ref.56). The docking box offset with 30 × 30 × 30 Å3 was centered in the median point between one oxygen of the nucleophile (ASP) and the acid–base (ASH) acid hydrogen. Protonation states were determined at pH 5.5, and visual inspection of the titratable residues in the structures was performed. Molecular dynamics simulations were performed using the Cuda accelerated version of the Amber 20 package (https://ambermd.org/). Initial structures were neutralized, solvated and submitted to two initial steps of energy minimization, restrained and unrestrained. Further, consecutive runs at canonical ensemble (NVT) using heating steps of 100, 200 and 310 K were performed. Production runs consisted of 100-ns simulations with a 2.0-fs timestep along with a shake algorithm to constrain all bonds involving hydrogen atoms. Trajectory analyses were performed using the tool CPPTRAJ 3.0 (ref.57). Structural images were generated using PyMOL 2.2.3 (Schrondiger).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1 |. Genomic organization of the predicted N-glycan utilization loci (N-GUL) from B. longum NCC2705.
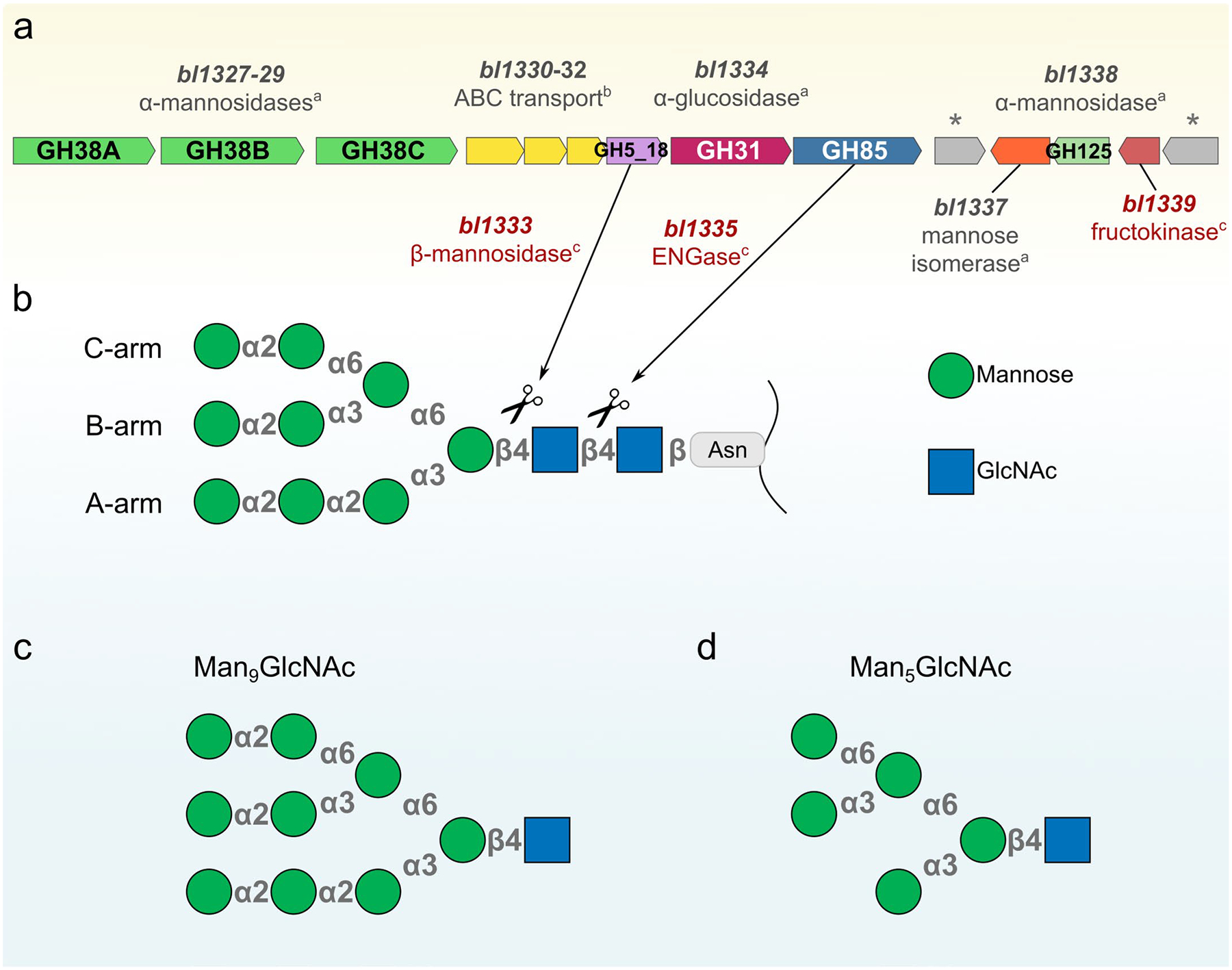
a, Genes that compose N-GUL represented by arrows and their respective gene products. a = biochemically characterized in this work, b = predicted, c = characterized in previous works23,24,28. The symbol * represents transcriptional regulators (bl1336 – encoding a LacI transcriptional regulator and bl1340 – encoding a ROK transcriptional regulator). b, A schematic representation of a high-mannose N-glycan indicating its subunits (geometric forms), glycosidic linkages (gray symbols) and the cleavage sites (scissors) recognized by the GH5 β-mannosidase23 and GH85 ENGase24 enzymes previously characterized. Predictions of uncharacterized enzymes were performed by sequence and hidden-Markov similarities analyses (Supplementary Tables 1, 2). c, Representation of Man9GlcNAc. d, Representation of Man5GlcNAc.
Extended Data
Extended Data Fig. 2 |. Evolutionary analysis of Bifidobacterium strains.

a, Phylogenetic tree of Bifidobacterium genomes. *1: B. ruminantium, B. pseudocatenulatum, B. catenulatum, and B. moukalabense; *2: B. jacchi, B. scardovii, B. samirii, B. biavatii, B. ramosum, B. hapali, B. aerophilum, and B. leontopitheci; *3: B. merycicum, B. callitrichos, B. platyrrhinorum, B. aesculapii, B. parmae, B. stellenboschens, B. eulemuris, B. lemurum, B. scaligerum, B. callitrichidarum, B. myosotis, B. reuteri, B. cebidarum, B. felsineum, B. imperatoris, and B. saguini. b, Examples of N-GUL gene organizations that can be found in intra- and interspecies.
Extended Data
Extended Data Fig. 3 |. Man9GlcNAc breakdown by the action of Bl_Man38A and Bl_Man38C.

a, Negative control of Man9GlcNAc with no enzyme. b, Only Bl_Man38A. c, Only Bl_Man38C. d, Both Bl_Man38A and Bl_Man38C added into the reaction. Note that the combined reaction led to complete depletion of the substrate, forming mainly the final product Man1GlcNAc and residual amounts of Man3GlcNAc.
Extended Data
Extended Data Fig. 4 |. Bl_Man125 displayed no activity over Man5GlcNAc and Man9GlcNAc substrates.

Biochemical profile of Bl_Man125 using Man5GlcNAc (a, b) and Man9GlcNAc (c, d) as putative substrates in 60-min reactions. a, Negative control of Man5GlcNAc with no enzyme. b, Man5GlcNAc with Bl_Man125. c, Negative control of Man9GlcNAc with no enzyme. d, Man9GlcNAc with Bl_Man125.
Extended Data
Extended Data Fig. 5 |. The complementary activity of Bl_Man125 with Bl_Man38A-C enzymes on Man5GlcNAc.

Reactions combining the GH125 α-1,6-mannosidase and the GH38 enzymes using Man5GlcNAc to evaluate a putative booster effect in the degradation of small oligosaccharides. a, Negative control (no enzyme). b, Bl_Man125, c, Bl_Man38A, d, Bl_Man38A and Bl_Man125, e, Bl_Man38B, f, Bl_Man38B and Bl_Man125, g, Bl_Man38C, h, Bl_Man38C and Bl_Man125, i, Bl_Man38A and Bl_Man38C, j, Bl_Man38A, Bl_Man38C and Bl_Man125. Note that a remarkable difference is observed for Bl_Man38B, in which peaks corresponding to Man3GlcNAc and Man2GlcNAc are depleted in the presence of Bl_Man125.
Extended Data
Extended Data Fig. 6 |. The distinct oligomeric states observed in the GH38 family.
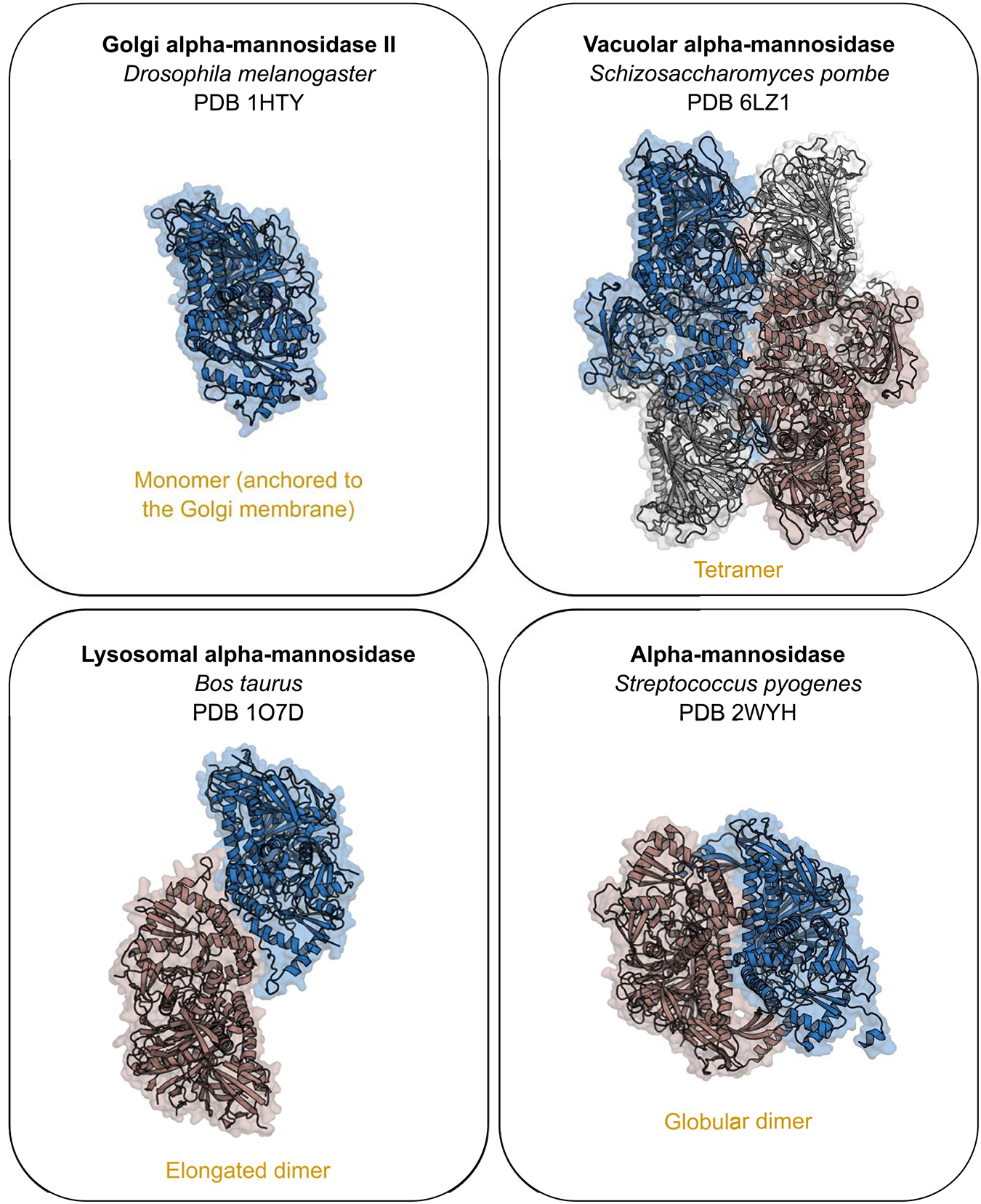
Oligomeric arrangements described for the members of the GH38 family, represented by the structures under the PDB code 1HTY, 6LZ1, 1O7D and 2WYH. Blue protomers were structurally aligned, maintaining the same position and dimension for all the structures. The r.m.s.d. values of the structural comparisons of these enzymes with Bl_Man38A, Bl_Man38B and Bl_Man38C are provided in the Supplementary Table 7.
Extended Data
Extended Data Fig. 7 |. Maximum likelihood phylogeny of characterized members of the GH38 family.

Red circles indicate the sequences containing at least one structure deposited in the Protein Data Bank, with their respective codes assigned. The yellow clade indicates bacterial GH38 α-mannosidases that oligomerizes as globular dimers and is represented by the structure under the PDB code 2WYH. The purple clades correspond to lysosomal and vacuolar (for plants) family members, which oligomerize as elongated dimers as shown for the structure under the PDB code 1O7D. The green clade corresponds to monomeric enzymes that are anchored to Golgi apparatus membrane, represented by the structure under the PDB code 1HTY. The red clades comprise the tetrameric enzymes, grouping cytosolic, vacuolar (for yeasts) and bifidobacterial members in the same phylogenetic branch. Grey clades do not have structural data available to infer the oligomeric state of their members. A GH92 α-mannosidase was used as the outgroup.
Extended Data
Extended Data Fig. 8 |. Functional comparison between the mutant Bl_Man38CF726W and the wild-type Bl_Man38C.

Assays using Man5GlcNAc (a–c) and Man9GlcNAc (d–f) as substrate, in 60-min reactions with no enzyme (a, d), with Bl_Man38C (b, e) and with Bl_Man38CF726W (c, f). *: internal control (xylohexaose). g–i, Kinetic assays with Bl_Man38C and Bl_Man38CF726W on the disaccharides α-1,2-mannobiose (g), α-1,3-mannobiose (h) and α-1,6-mannobiose (i). The reactions were supplemented with 0.1 mM CoCl2. Results are expressed as mean ± SD from three independent experiments (g–i).
Extended Data
Extended Data Fig. 9 |. Functional comparison between the mutant Bl_Man38AΔLeu164-Pro168 and the wild-type Bl_Man38A.

Assays using Man5GlcNAc (a–c) and Man9GlcNAc (d–f) as substrate, with no enzyme (a, d), with Bl_Man38A (b, e) and with Bl_Man38AΔLeu164-Pro168 (c, f). *: internal control (xylohexaose). The reactions were supplemented with 0.1 mM CoCl2.
Extended Data
Extended Data Fig. 10 |. Enzymatic characterization of the α-glucosidase Bl_Glc31.
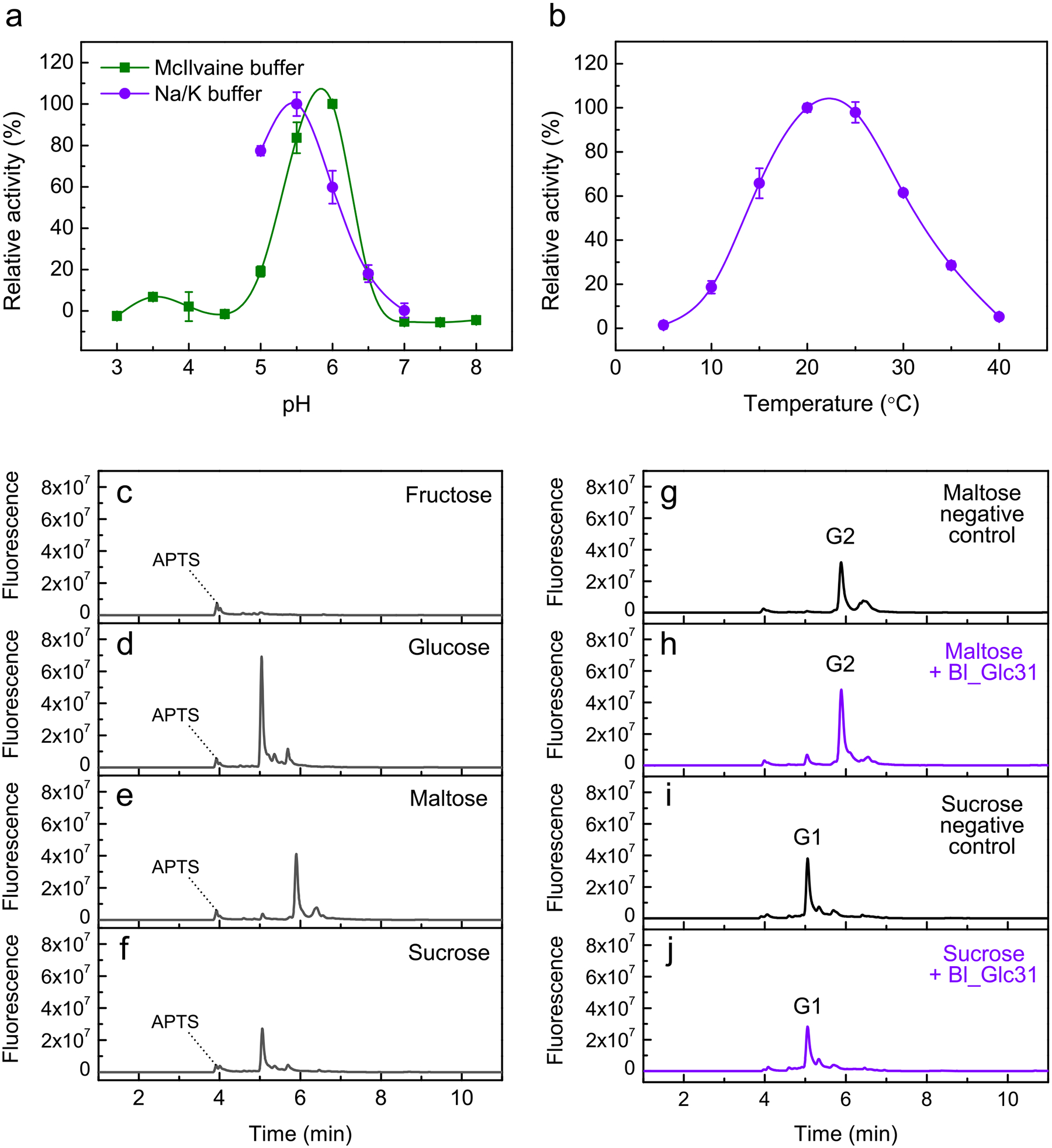
Enzyme activity was analyzed by colorimetric (a, b) and capillary electrophoresis methods (c–j). a, Relative activity of Bl_Glc31 on pNP-α-Glc in function of pH in reactions containing McIlvaine buffer (green) and 200 mM sodium/potassium phosphate (purple) buffers. b, Relative activity of Bl_Glc31 over pNP-α-Glc in 200 mM sodium/potassium phosphate buffer in function of temperature. Data from panels a and b are shown as mean ± SD from three independent experiments (n = 3). c–f, Capillary electrophoresis patterns for fructose (c), glucose (d), maltose (e) and sucrose (f). Note that in the sucrose profile only contaminant glucose is observed, and no sucrose is detected due to its lack of reactivity with APTS. (g–j) Reactions of Bl_Glc31 against maltose and sucrose as putative substrates in optimal conditions of the enzyme (200 mM sodium/potassium phosphate buffer pH 5.5, at 25 °C) for 16 hrs. Due to the long reaction time, negative controls without enzyme were performed to verify possible spontaneous hydrolysis of maltose (g) and sucrose (i). In 4 min of elution, a peak of APTS is seen for all the reactions. G2 = maltose. G1 = glucose.
Supplementary Material
Acknowledgements
We thank R. dos Santos Bezerra for support with some bioinformatics analyses. We thank M. L. Sforca and S. A. Rocco for conducting and analyzing the NMR experiments. We thank L. M. Z. Murakami for support in characterization of isomerase activity. We acknowledge the National Nanotechnology Laboratory (LNNano) for the provision of time (proposal TEM-C2-26053 and TEM-C2-26816) on the transmission electron microscopes JEM 1400 Plus (Jeol), Talos F200C (Thermo Fisher Scientific), Talos Arctica G2 (Thermo Fisher Scientific) and Titan Krios G3i (Thermo Fisher Scientific). We acknowledge the Brazilian Biorenewables National Laboratory (LNBR) for the use of the characterization of macromolecules facility (proposals 26914, 27586 and 20220639) and for the use of the metabolomics facility (proposal 20220817). We acknowledge the Brazilian Biosciences National Laboratory (LNBio) for the use of the NMR facility (proposal 20220793). LNNano, LNBR and LNBio are operated by the Brazilian National Center for Research in Energy and Materials for the Brazilian Ministry for Science, Technology and Innovations (MCTIC). We acknowledge the National Laboratory for Scientific Computing (LNCC/MCTI, Brazil) for providing high-performance computing resources of the SDumont supercomputer (http://sdumont.lncc.br), which have contributed to the research results reported within this paper (proposals 212394 and 221167). This research was supported by grants from Fundação de Amparo à Pesquisa do Estado de São Paulo (grant number 2015/26982-0 to M.T.M., 2017/15340-2 to R.V.P. and 2016/00740-2 to R.L.C.) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq; grant number 306135/2016-7 and 305013/2020-3 to M.T.M.) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES). This work was also supported in part by NIH grant R01AI149297.
Footnotes
Competing interests
The authors declare no competing interests.
Extended data is available for this paper at https://doi.org/10.1038/s41589-022-01202-4.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41589-022-01202-4.
Data availability
Atomic coordinates and structure factors were deposited to the Protein Data Bank (PDB) with the following accession codes: PDB 7UFR/Electron Microscopy Data Bank (EMDB) 26478 (Bl_Man38A), PDB 7UFS/EMDB 26479 (Bl_Man38B), PDB 7UFT/EMDB 26480 (Bl_Man38C) and PDB 7UFU/EMDB 26481 (Bl_Man38AD387A–MAN). Other data generated or analyzed during this study are included in the published article and its Supplementary Information files. Source data are provided with this paper.
References
- 1.Fanning S et al. Bifidobacterial surface-exopolysaccharide facilitates commensal–host interaction through immune modulation and pathogen protection. Proc. Natl Acad. Sci. USA 109, 2108–2113 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andlid TA, D’Aimmo MR & Jastrebova J in The Bifidobacteria and Related Organisms (eds Mattarelli P, Biavati B, Holzapfel WH & Wood BJB) 195–212 (Elsevier, 2018). [Google Scholar]
- 3.Moya-Pérez A, Perez-Villalba A, Benítez-Páez A, Campillo I & Sanz Y Bifidobacterium CECT 7765 modulates early stress-induced immune, neuroendocrine and behavioral alterations in mice. Brain Behav. Immun 65, 43–56 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Luck B et al. Bifidobacteria shape host neural circuits during postnatal development by promoting synapse formation and microglial function. Sci. Rep 10, 7737 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Milani C et al. Genomics of the genus Bifidobacterium reveals species-specific adaptation to the glycan-rich gut environment. Appl. Environ. Microbiol 82, 980–991 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jung DH et al. The presence of resistant starch-degrading amylases in Bifidobacterium adolescentis of the human gut. Int. J. Biol. Macromol 161, 389–397 (2020). [DOI] [PubMed] [Google Scholar]
- 7.la Rosa SL et al. Wood-derived dietary fibers promote beneficial human gut microbiota. mSphere 4, e00554–18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yamada C et al. Molecular insight into evolution of symbiosis between breast-fed infants and a member of the human gut microbiome Bifidobacterium longum. Cell Chem. Biol 24, 515–524 (2017). [DOI] [PubMed] [Google Scholar]
- 9.Katoh T et al. Enzymatic adaptation of Bifidobacterium bifidum to host glycans, viewed from glycoside hydrolases and carbohydrate-binding modules. Microorganisms 8, 481 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Higel F, Seidl A, Sörgel F & Friess W N-Glycosylation heterogeneity and the influence on structure, function and pharmacokinetics of monoclonal antibodies and Fc fusion proteins. Eur. J. Pharm. Biopharm 100, 94–100 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Schjoldager KT, Narimatsu Y, Joshi HJ & Clausen H Global view of human protein glycosylation pathways and functions. Nat. Rev. Mol. Cell Biol 21, 729–749 (2020). [DOI] [PubMed] [Google Scholar]
- 12.Bjursell MK, Martens EC & Gordon JI Functional genomic and metabolic studies of the adaptations of a prominent adult human gut symbiont, Bacteroides thetaiotaomicron, to the suckling period. J. Biol. Chem 281, 36269–36279 (2006). [DOI] [PubMed] [Google Scholar]
- 13.Martens EC, Koropatkin NM, Smith TJ & Gordon JI Complex glycan catabolism by the human gut microbiota: the Bacteroidetes Sus-like paradigm. J. Biol. Chem 284, 24673–24677 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hemsworth GR, Déjean G, Davies GJ & Brumer H Learning from microbial strategies for polysaccharide degradation. Biochem. Soc. Trans 44, 94–108 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Foley MH, Cockburn DW & Koropatkin NM The Sus operon: a model system for starch uptake by the human gut Bacteroidetes. Cell. Mol. Life Sci 73, 2603–2617 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cuskin F et al. Human gut Bacteroidetes can utilize yeast mannan through a selfish mechanism. Nature 517, 165–169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Robb M et al. Molecular characterization of N-glycan degradation and transport in Streptococcus pneumoniae and its contribution to virulence. PLoS Pathog 13, e1006090 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dupoiron S et al. The N-glycan cluster from Xanthomonas campestris pv. campestris: a toolbox for sequential plant N-glycan processing. J. Biol. Chem 290, 6022–6036 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Briliūtė J et al. Complex N-glycan breakdown by gut Bacteroides involves an extensive enzymatic apparatus encoded by multiple co-regulated genetic loci. Nat. Microbiol 4, 1571–1581 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trastoy B et al. Structural basis of mammalian high-mannose N-glycan processing by human gut Bacteroides. Nat. Commun 11, 889 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Higgins MA et al. N-Glycan degradation pathways in gut- and soil-dwelling Actinobacteria share common core genes. ACS Chem. Biol 16, 701–711 (2021). [DOI] [PubMed] [Google Scholar]
- 22.Reichenbach T et al. Structural and biochemical characterization of the Cutibacterium acnes exo-β-1,4-mannosidase that targets the N-glycan core of host glycoproteins. PLoS ONE 13, e0204703 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cordeiro RL et al. N-Glycan utilization by Bifidobacterium gut symbionts involves a specialist β-mannosidase. J. Mol. Biol 431, 732–747 (2019). [DOI] [PubMed] [Google Scholar]
- 24.Garrido D et al. Endo-β-N-acetylglucosaminidases from infant gut-associated bifidobacteria release complex N-glycans from human milk glycoproteins. Mol. Cell Proteomics 11, 775–785 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schell MA et al. The genome sequence of Bifidobacterium longum reflects its adaptation to the human gastrointestinal tract. Proc. Natl Acad. Sci. USA 99, 14422–14427 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trombetta ES, Simons JF & Helenius A Endoplasmic reticulum glucosidase II is composed of a catalytic subunit, conserved from yeast to mammals, and a tightly bound noncatalytic HDEL-containing subunit. J. Biol. Chem 271, 27509–27516 (1996). [DOI] [PubMed] [Google Scholar]
- 27.Parche S et al. Sugar transport systems of Bifidobacterium longum NCC2705. J. Mol. Microbiol. Biotechnol 12, 9–19 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Caescu CI, Vidal O, Krzewinski F, Artenie V & Bouquelet S Bifidobacterium longum requires a fructokinase (Frk; ATP: D-fructose 6-phosphotransferase, EC 2.7.1.4) for fructose catabolism. J. Bacteriol 186, 6515–6525 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fushinobu S Unique sugar metabolic pathways of bifidobacteria. Biosci. Biotechnol. Biochem 74, 2374–2384 (2010). [DOI] [PubMed] [Google Scholar]
- 30.Gregg KJ et al. Analysis of a new family of widely distributed metal-independent α-mannosidases provides unique insight into the processing of N-linked glycans. J. Biol. Chem 286, 15586–15596 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shah N, Kuntz DA & Rose DR Golgi α-mannosidase II cleaves two sugars sequentially in the same catalytic site. Proc. Natl Acad. Sci. USA 105, 9570–9575 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nielsen JW et al. Metal-ion dependent catalytic properties of sulfolobus solfataricus class II α-mannosidase. Biochemistry 51, 8039–8046 (2012). [DOI] [PubMed] [Google Scholar]
- 33.Sonnenburg JL, Chen CTL & Gordon JI Genomic and metabolic studies of the impact of probiotics on a model gut symbiont and host. PLoS Biol 4, 2213–2226 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bertipaglia C et al. Higher-order assemblies of oligomeric cargo receptor complexes form the membrane scaffold of the Cvt vesicle. EMBO Rep 17, 1044–1060 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang J, Wang YY, Du LL & Ye K Cryo-EM structure of fission yeast tetrameric α-mannosidase Ams1. FEBS Open Bio 10, 2437–2451 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Suits MDL et al. Structure and kinetic investigation of Streptococcus pyogenes family GH38 α-mannosidase. PLoS ONE 5, e9006 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Suzuki T et al. Man2C1, an α-mannosidase, is involved in the trimming of free oligosaccharides in the cytosol. Biochem. J 400, 33–41 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heikinheimo P et al. The structure of bovine lysosomal α-mannosidase suggests a novel mechanism for low-pH activation. J. Mol. Biol 327, 631–644 (2003). [DOI] [PubMed] [Google Scholar]
- 39.Howard E et al. Structural basis of outstanding multivalent effects in jack bean α-mannosidase inhibition. Angew. Chem. Int. Ed. Engl 57, 8002–8006 (2018). [DOI] [PubMed] [Google Scholar]
- 40.Yamagishi M, Ishimizu T, Natsuka S & Hase S Co(II)-regulated substrate specificity of cytosolic α-mannosidase. J. Biochem 132, 253–256 (2002). [DOI] [PubMed] [Google Scholar]
- 41.Chaudet MM & Rose DR Suggested alternative starch utilization system from the human gut bacterium Bacteroides thetaiotaomicron. Biochem. Cell Biol 94, 241–246 (2016). [DOI] [PubMed] [Google Scholar]
- 42.Tan K et al. Novel α-glucosidase from human gut microbiome: substrate specificities and their switch. FASEB J 24, 3939–3949 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ikegaya M et al. Structural basis of the strict specificity of a bacterial GH31 α-1,3-glucosidase for nigerooligosaccharides. J. Biol. Chem 298, 101827 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Park D et al. Enterocyte glycosylation is responsive to changes in extracellular conditions: implications for membrane functions. Glycobiology 27, 847–860 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bilyy RO et al. Macrophages discriminate glycosylation patterns of apoptotic cell-derived microparticles. J. Biol. Chem 287, 496–503 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Egan M & Van Sinderen D in The Bifidobacteria and Related Organisms (eds Mattarelli P, Biavati B, Holzapfel WH & Wood BJB) 145–164 (Elsevier, 2018). [Google Scholar]
- 47.Itoh T, Mikami B, Hashimoto W & Murata K Crystal structure of YihS in complex with d-mannose: structural annotation of Escherichia coli and Salmonella enterica yihS-encoded proteins to an aldose–ketose isomerase. J. Mol. Biol 377, 1443–1459 (2008). [DOI] [PubMed] [Google Scholar]
- 48.Price MN, Dehal PS & Arkin AP FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS ONE 5, e9490 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gilchrist CLM et al. cblaster: a remote search tool for rapid identification and visualization of homologous gene clusters. Bioinform. Adv 1, vbab016 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dische Z & Borenfreund E A new spectrophotometric method for the detection and determination of keto sugars and trioses. J. Biol. Chem 192, 583–587 (1951). [PubMed] [Google Scholar]
- 51.van Heel M et al. in International Tables for Crystallography, Volume F, 2nd edition, Crystallography of Biological Macromolecules (eds Arnold E, Himmel DM & Rossmann MG) 624–628 (Wiley, 2012). [Google Scholar]
- 52.Grant T, Rohou A & Grigorieff N CisTEM, user-friendly software for single-particle image processing. eLife 7, e35383 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Adams PD et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Emsley P & Cowtan K Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 55.Chen VB et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr 66, 12–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Trott O & Olson AJ Autodock vina: improving the speed and accuracy of docking. J. Comput. Chem 31, 455–461 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Roe DR & Cheatham TE PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput 9, 3084–3095 (2013). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Atomic coordinates and structure factors were deposited to the Protein Data Bank (PDB) with the following accession codes: PDB 7UFR/Electron Microscopy Data Bank (EMDB) 26478 (Bl_Man38A), PDB 7UFS/EMDB 26479 (Bl_Man38B), PDB 7UFT/EMDB 26480 (Bl_Man38C) and PDB 7UFU/EMDB 26481 (Bl_Man38AD387A–MAN). Other data generated or analyzed during this study are included in the published article and its Supplementary Information files. Source data are provided with this paper.