

Abstract
The RNA decapping scavenger protein, DcpS, has recently been identified as a dependency in acute myeloid leukemia. The potent DcpS inhibitor RG3039 attenuates AML cell viability, and shRNA knockdown of DcpS is also antiproliferative. Importantly, DcpS was found to be non-essential in normal human hematopoietic cells, which opens a therapeutic window for AML treatment by DcpS modulation. Considering this strong DcpS dependence in AML cell lines, we explored PROTAC-mediated degradation as an alternative strategy to modulate DcpS activity. Herein, we report the development of JCS-1, a PROTAC exhibiting effective degradation of DcpS at nanomolar concentrations. JCS-1 non-covalently binds DcpS with an RG3039-based warhead and recruits the E3 ligase VHL, which induces potent, rapid, and sustained DcpS degradation in several AML cell lines. JCS-1 serves as a chemical biology tool to interrogate DcpS degradation and associated changes in RNA processes in different cellular contexts, which may be an attractive strategy for the treatment of AML and other DcpS-dependent genetic disorders.
Introduction:
The 5’ mRNA cap structure enhances translational efficiency and protects the mRNA transcript from premature degradation.1,2,3 The scavenger mRNA-decapping enzyme DcpS is a histidine triad (HIT) pyrophosphatase that, along with Dcp2, is responsible for the final hydrolysis of the cap structure during the 5’ to 3’ mRNA degradation process in eukaryotes.4,5,6,7,8,9,10,11,12 Furthermore, DcpS has multiple functional roles in RNA regulation,17,18,19 such as participating in micro-RNA (miRNA) turnover,13,14 pre-mRNA splicing,15 and the 5’ to 3’ mRNA decay pathway.16
Although the interplay of the many functions of DcpS with other RNA processing machinery is not fully understood, DcpS has been implicated in several diseases, the first being spinal muscular atrophy (SMA).20 SMA is caused by deletion or mutation of both copies of the survival motor neuron 1 (SMN1) gene, which encodes SMN, a protein necessary for proper motor neuron development and function. The severity of SMA symptoms is dependent on the variable copy number of the related gene SMN2; patients with higher levels of SMN2 expression have less severe symptoms.21 C5-substituted quinazolines were found to be potent inducers of SMN2 expression but the molecular target was not known.22,23 In subsequent protein microarray array studies using a radio-labelled C5-substituted analog, this class of compounds was found to bind and inhibit DcpS.20,24 Candidate C5-quinazoline inhibitor RG3039 was advanced as a potential SMA therapy, where it was shown to improve survival and motor function in SMA mice.23,25 RG3039 showed promising results during a phase 1 clinical trial, but its development as an SMA therapy was later terminated due to an inability to increase SMN1 protein levels in humans.24 DcpS mutations have also revealed a crucial role of mRNA processing in intellectual development, as autosomal recessive variants have been identified as drivers of intellectual disability during development.26,27
In addition to SMA and developmental intellectual disability, DcpS was recently identified as a driver of acute myeloid leukemia (AML).1 This unexpected finding illustrates the knowledge gap in understanding the role of aberrant mRNA processing in different disease states. Given the diverse and often untreatable nature of this class of blood cancers, identifying novel therapeutic interventions for AMLs is of great interest.28 Genome-wide CRISPR-Cas9 knockout screening using AML cell lines revealed that DcpS is an essential gene for AML cell survival.1,29 DcpS inhibition with RG3039 imparts potent antileukemic activity in both cellular and human AML xenograft models1. Further, shRNA silencing of DcpS demonstrated antileukemic activity, corroborating that the activity of RG3039 in AML cells was due to DcpS inhibition.1 Hematopoietic cells containing bi-allelic DcpS loss-of-function mutations were phenotypically normal, indicating DcpS is dispensable in normal blood cells but necessary in AML.1 Therefore, we became interested in generating a molecular degrader of DcpS to interrogate the impact of sustained DcpS degradation, rather than inhibition or gene silencing, on AML progression.
PROteolysis TArgeting Chimeras (PROTACs) have emerged as a new and promising modality in drug discovery. These heterobifunctional molecules simultaneously engage a protein of interest (POI) and an E3 ligase (e.g. cereblon (CRBN) or Von-Hippel Lindau (VHL)), forming a ternary complex and enabling the E3 ligase to ubiquitinate the POI on proximal lysine residues.30,31,32,33 The ubiquitinated POI is subsequently recognized and degraded by the 26S proteasome. Major advantages of target degradation are the elimination of non-enzymatic (ex. scaffolding) roles that are not typically attenuated by traditional small molecule inhibitors and the ability to observe pharmacological effects at lower drug concentrations due to the catalytic nature of the degrader molecule30,31,32,33. We generated PROTACs based on RG3039 as the DcpS binding warhead, resulting in the discovery of a VHL-recruiting DcpS degrader JCS-1. Our PROTAC induces rapid and sustained degradation of DcpS via a PROTAC mechanism in multiple AML cell lines, leading to decreased proliferation. JCS-1 is a valuable chemical tool to interrogate the impact of DcpS degradation on mRNA biology in many cellular contexts and may serve as a blueprint for the development of clinical candidate DcpS degraders.
Results:
RG3039-based VHL-recruiting PROTACs degrade endogenous DcpS in MOLM-14 cells
Considering its positive Phase 1 trial data, we chose RG3039 (Figure 1A) as the DcpS recruiting element to design DcpS-targeting PROTACs. Using reported co-crystal structures, the observed binding poses of analogs and docking of RG3039 onto DcpS revealed that the dichlorobenzene motif does not form significant interactions with DcpS (PDB: 1ST0, 3BL7, and 3BL9).34 We decided to eliminate the halogens in favor of a meta-phenol vector to enable facile linker attachment during PROTAC synthesis. To confirm that the removal of the halogens and addition of linkers did not disrupt binding, engagement of DcpS was determined in MOLM-14 lysates using the CEllular Thermal Shift Assay (CETSA).35 VHL-recruiting PROTACs ranging in linker length from 6 to 18 atoms were able to bind and stabilize DcpS upon heating lysates to 68°C at 0.1–1 μM concentrations (SI Figure 1).
Figure 1: JCS-1 degrades endogenous DcpS in AML cell lines:

A) Chemical structures of RG3039 (DcpS inhibitor), JCS-1 (active PROTAC), and JCS-2 (inactive epimeric control PROTAC). B) JCS −1 degrades DcpS in a dose-dependent manner in MOLM-14 cells. Cells were treated for 24 hours. Blots show lysates from independent wells harvested side-by-side on the same day. Quantified data represents the mean ± SD from three independent biological replicates. One-way ANOVA, *** p < 0.005; **** p < 0.001
Confident that our PROTACs would engage DcpS in cells, we synthesized a library of >20 DcpS-targeting PROTACs recruiting VHL or CRBN (SI Table 1). Either the amide linked (left-hand (lh)) or phenol linked (right-hand (rh)) VHL ligand, developed in our laboratory,36 was used to recruit VHL, while a 4-hydroxy thalidomide derivative was used to recruit CRBN. Interestingly, only JCS-1, which linked des-chloro RG3039 to the phenol VHL ligand via a 4 PEG (17 atom) linker was able to induce significant DcpS degradation in multiple AML cell lines. The observed degradation was efficacious, with maximal degradation (Dmax) of ~98% and DC50 (the concentration at which half the Dmax is reached) of 87 nM in MOLM-14 AML cells (Figure 1B). We further tested a small panel of AML cell lines and JCS-1 was able to efficiently induce DcpS degradation with DC50 values ranging from 13 nM to 87 nM and Dmax >90% in all cell lines (SI Figure 2).
To ensure that the 17 atom, 5 PEG linker was the optimal linker length for inducing DcpS degradation, PROTACs were synthesized with linker lengths of 15, 16, 18, or 19 atoms (SI Table 1). Remarkably, potent degradation was observed for the 17-linker atom PROTAC JCS-1, with modest degradation observed for the 16 and 18 atom derivatives and no degradation observed for the PROTACs with 15 or 19 atom linkers. These data illustrate the stringent requirements for ternary complex formation and subsequent ubiquitination and degradation in this system.
Characterization of DcpS degradation by JCS-1 in MOLM-14 cells:
PROTAC targets are marked for degradation via ubiquitination, which is dependent on the ternary complex formation between the target, the PROTAC, and the E3 ligase.37 The hydroxyproline moiety of the VHL ligand is crucial for binding to the E3 ligase, and inversion of the stereochemistry of the 4-hydroxyproline abrogates VHL binding.36,38 We generated JCS-2 (Figure 1A) as an inactive epimeric control PROTAC that cannot bind to VHL to form the necessary ternary complex needed for ubiquitination. As expected, the inverted hydroxyproline epimeric control PROTAC JCS-2 did not induce DcpS degradation (Figure 2). Additionally, we disrupted VHL binding by competing with excess VHL ligand, which inhibited ternary complex formation and subsequent DcpS degradation. Neddylation of CUL2, a VHL adaptor protein, and functional proteasomes are both required for target degradation via the canonical PROTAC mechanism. Degradation by JCS-1 was inhibited by the neddylation inhibitor MLN4924 (MLN) and proteasome inhibitor epoxomicin, providing further evidence that degradation by JCS-1 follows the predicted PROTAC mechanism.39 Finally, free DcpS inhibitor RG3039 does not induce DcpS degradation, nor does a combination of un-linked RG3039 and VHL ligand, the free components of the PROTAC. (Figure 2). Together, these data clearly demonstrate that JCS-1 induces the degradation of DcpS via a bona fide PROTAC mechanism.
Figure 2: DcpS degradation by JCS-1 in MOLM-14 cells is via PROTAC mechanism:
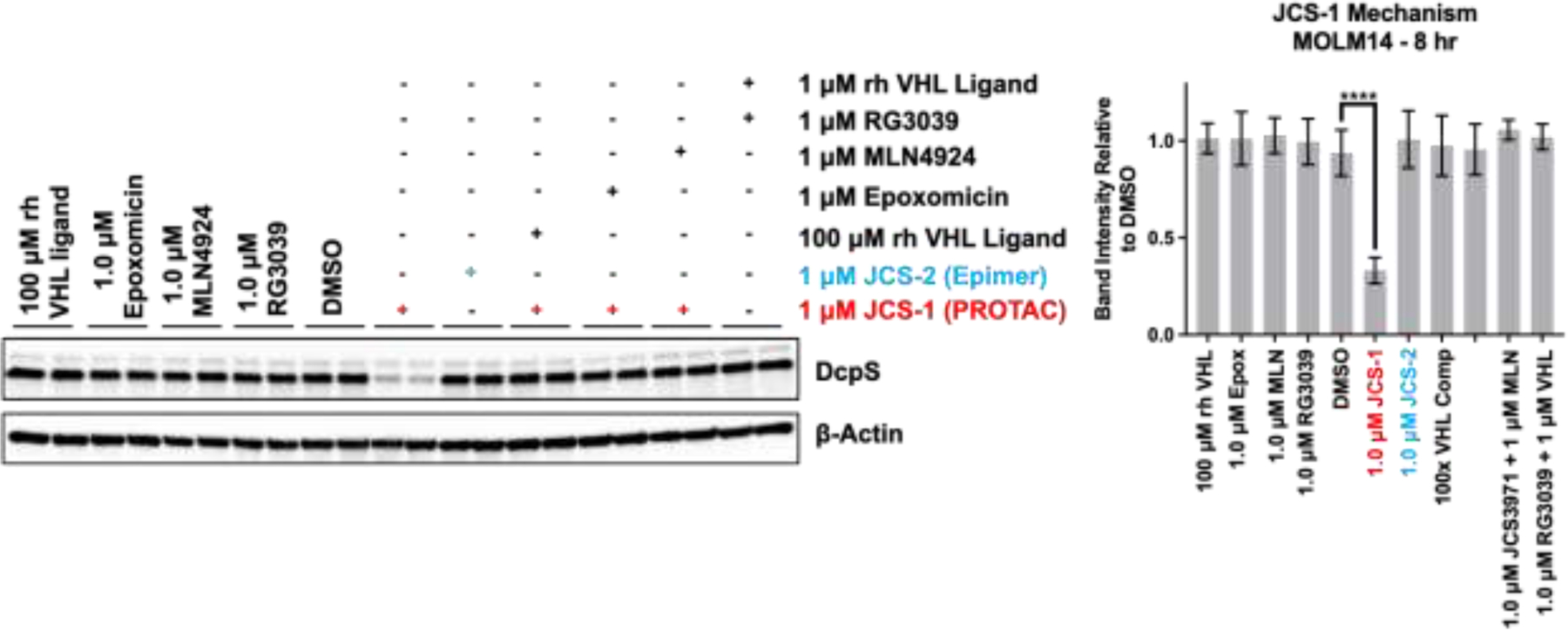
Inactive epimer JCS-2 does not induce degradation, and degradation by JCS-1 is prevented by VHL ligand competition, proteasome inhibition by epoxomicin (Epox), and neddylation inhibition by MLN4924 (MLN). Inhibitor RG3039 alone or combined with the VHL ligand also do not induce degradation. Blots show lysates from independent wells harvested side-by-side on the same day. Quantitation to the right. Quantified data represents the mean ± SD from three independent biological replicates. One-way ANOVA, **** p < 0.001
Next, we interrogated the kinetics of DcpS degradation in MOLM-14 cells. A 24 hour time course analysis revealed that at 1 μM JCS-1, degradation occurs as rapidly as 2 hours after treatment, with maximal degradation observed between 12 and 24 hours (Figure 3A). Similar data was observed in time courses performed with MV411 cells (SI Figure 3A). We also carried out an extended time courses to determine how quickly DcpS is resynthesized after PROTAC depletion. We found degradation to be highly sustained by a single treatment of 1 μM JCS-1 (SI Figure 3B), so we conducted a washout experiment to determine whether the slow recovery of DcpS levels is due to slow DcpS resynthesis or residual PROTAC activity (Figure 3B). MOLM-14 cells were treated for 24 hours with 1 μM JCS-1 after which treatment media was exchanged with fresh media containing either DMSO or excess VHL ligand (10 μM) to compete away residual PROTAC binding to VHL. Interestingly, the media washout with DMSO alone was not sufficient to recover DcpS levels even after over 72 hours, indicating that the residual PROTAC that remains after washout is yet able to potently degrade newly synthesized DcpS faster than the cell can produce it. However, when competing the residual PROTAC activity with free VHL ligand, DcpS levels began to recover within 24 hours. These data indicate that DcpS resynthesis is slow and that the PROTAC is likely to act catalytically, as expected for a noncovalent degrader.
Figure 3: DcpS degradation by JCS-1 in MOLM-14 cells is rapid and sustained:
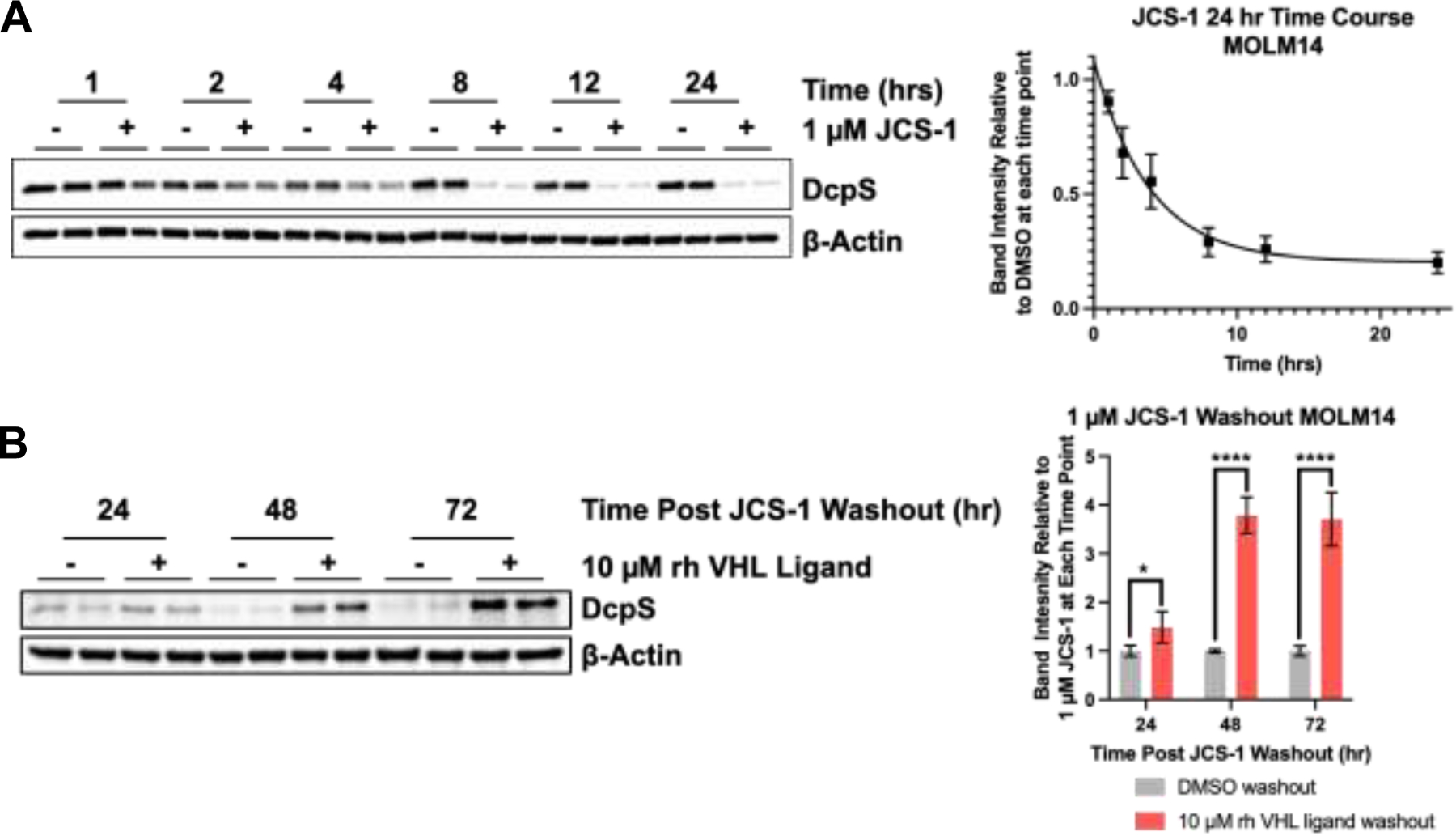
A) 24 hour time course for DcpS degradation by 1 μM JCS-1 in MOLM-14 cells. B) DcpS recovery following washout after 24 hour initial treatment with 1 μM JCS-1 with or without 10 μM VHL-ligand to compete against residual PROTAC action following washout. Quantified data represents the mean ± SD from three independent biological replicates. One-way ANOVA, * p < 0.05, **** p < 0.001
DcpS degradation by JCS-1 inhibits MOLM-14 cells proliferation and viability
We next sought to determine the effect of DcpS degradation on AML cell proliferation and viability. To this end, we conducted 12 day cell counting and 5 day MTS experiments in MOLM14 cells. Cells were treated with either the epimer, JCS-2, or the PROTAC, JCS-1, to understand how AML cell viability is affected when DcpS is inhibited versus degraded, respectively. The epimer was used to interrogate the effect of inhibition rather than RG3039 because the epimer has identical physicochemical properties (e.g., solubility and permeability) to the PROTAC.
To assess the effects on AML proliferation, cells were treated every 3 days with either DMSO, 1 μM JCS-1, or 1 μM JCS-2 and counted on the day of treatment for a total of 12 days. A significant decrease in cell number was observed at 6, 9, and 12 days for the PROTAC-treated cells compared to those treated with DMSO (Figure 4A). Epimer treatment significantly decreased cell number by day 6, however it was to a lesser extent than the PROTAC treated cells. Moreover, by 9 days epimer-treated cells had similar cell counts as DMSO-treated cells. Using the MTS assay, we found that the PROTAC was ~3x more potent than the epimer with IC50 values of 1.1 ± 0.1 and 3.6 ± 0.6 μM respectively after 5 days of compound treatment (Figure 4B). Similar sensitivity to the PROTAC was seen in MV411 cells, but not OCI-AML3 cells or the T-ALL cell line MOLT4 (SI Figure 4). These data demonstrate that PROTAC-mediated degradation of DcpS is more antiproliferative than inhibition by a physiochemically equivalent inhibitor in MOLM14 and MV411 AML cells. This difference suggests that JCS-1 induced DcpS degradation also prevents scaffolding roles of DcpS, e.g. binding to the spliceosome or nuclear pore complexes1, and that these other activities of DcpS may contribute to AML progression.
Figure 4: Effects of degradation vs inhibition on MOLM14 cell proliferation and viability.
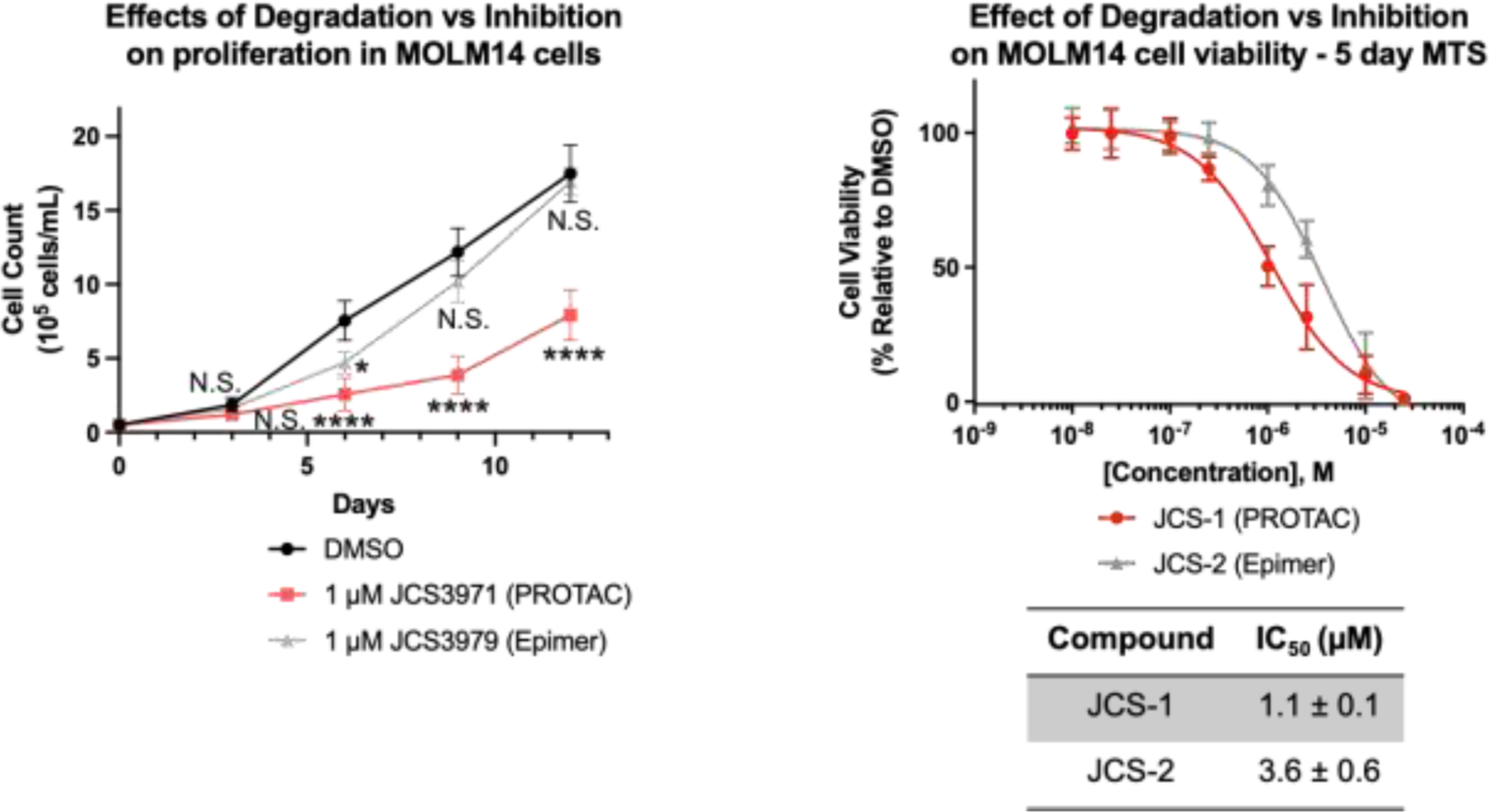
(A) Cells were seeded at a density of 0.5 × 105 cells/mL and treated with compounds every 3 days for 12 days. Significant reduction in cell proliferation by the PROTAC was observed between days 3 and 12. Proliferation was reduced by the epimer at day 6, however recovered to DMSO-treated levels by day 9. (B) MTS assay monitoring cell viability over 96 hours. The PROTAC is ~3x more potent than the epimer. Quantitation for A) and B) represents mean ± SD from three independent biological replicates performed in technical triplicate. Two-way ANOVA, Not significant (N.S.), * p < 0.05, **** p <0.0001.
Degradation and inhibition of DcpS have differential impacts on the mRNA pool of MOLM-14 cells
To explore the differential impacts of DcpS degradation and inhibition on global mRNA expression and stability, we performed a TimeLapse-seq experiment on MOLM-14 cells treated with 1 μM JCS-1, JCS-2, or RG3039 for 6 or 24 hours.40 During the last 2 hours of treatment, cells were fed 100 μM 4-thiouridine (s4U), which is recognized as uridine by the RNA machinery and incorporated into newly synthesized RNA. Upon isolation of the metabolically labeled RNA, TimeLapse chemistry converts 4-thiouridine into cytidine analogs via recoding of the hydrogen-bonding patterns (SI Fig 5).41 Sequencing then allows for the profiling of ‘new’ RNA with U-to-C mutational marks. By comparing changes in total RNA counts and TimeLapse induced mutation rates relative to control conditions, we were able to delineate the impacts of DcpS degradation and inhibition on the transcriptional dynamics of specific genes. Quality control data from the TimeLapse-seq experiment (SI Figure 6) demonstrates that biological replicates were positively associated and clustered closely to each other in principle component analysis (PCA). We chose the 6 hour time point for further analysis because the gene signatures after 24 hour PROTAC treatment were generally correlated with cell stress and cytotoxicity.
TimeLapse-seq analysis of JCS-1-treated MOLM-14 cells revealed modest changes in overall mRNA expression relative to DMSO-treated controls as observed in previous studies (Figure 5A).1 Of the 8629 detected genes, 407 were differentially expressed (padj < 0.05). We observed a comparable amount of genes upregulated and downregulated after 6 hours of treatment, suggesting DcpS degradation did not skew overall mRNA transcript levels. Notably, while the majority of transcripts were unaffected by JCS-1 treatment, numerous genes implicated in AML pathogenesis and cell viability were significantly differentially expressed (padj < 0.05, | L2FC (log 2 fold change) > 0.6). BCL2, NRAS, and CDK6, which have all been posited as potential AML therapeutic targets, were downregulated in JCS-1 treated samples relative to DMSO treated controls.42,43,44 Given that DcpS has at least one known function independent of its catalytic decapping activity14 (i.e. control of cytoplasmic miRNA turnover), we next investigated the effect of DcpS degradation versus inhibition on the mRNA pool of MOLM-14 cells (Figure 5B). Specifically, we were interested to see whether treatment with JCS-1 might also elicit changes in mRNA expression patterns not observed under JCS-2 treatment. Interestingly, only a small subset of all identified genes exhibited significant differential expression between PROTAC and epimer treated samples (padj < 0.05, |L2FC| > 0.6). Using quantitative real-time quantitative PCR (RT-qPCR), we validated several hit transcripts from the Time LapSeq dataset. Both SNORD99 and DDX24 expression were significantly increased in JCS-1, but not in JCS-2 or RG3039 treated cells compared to DMSO respectively (SI Figure 7). A similar trend, although not significant, was observed for SNORD73A. SNORDs are non-coding, small nucleolar RNAs that facilitate posttranscriptional modification of RNA species.45 Although there is no known role for SNORD99 or SNORD73A in AML progression, a similar RNA species, SNORD42A, has been shown to promote AML progression through site specific methylation of 18S ribosomal RNA.46 Our data suggests that SNORD99 and SNORD73A could be modifying another, yet undiscovered, RNA species important for AML progression. From these experiments, we were able to elucidate AML-relevant genes downregulated specifically by DcpS degradation, which may explain the stronger antiproliferative effect of the PROTAC compared to the epimer. However, our TimeLapse-seq data overall suggests that the majority of total mRNA changes were driven by suppression of DcpS’s decapping function. We remain interested in further interrogating other biological roles of DcpS that might affect specific transcript levels independently of its catalytic activity.
Figure 5: TimeLapse-seq enables profiling of differential gene expression kinetics upon DcpS degradation and inhibition.
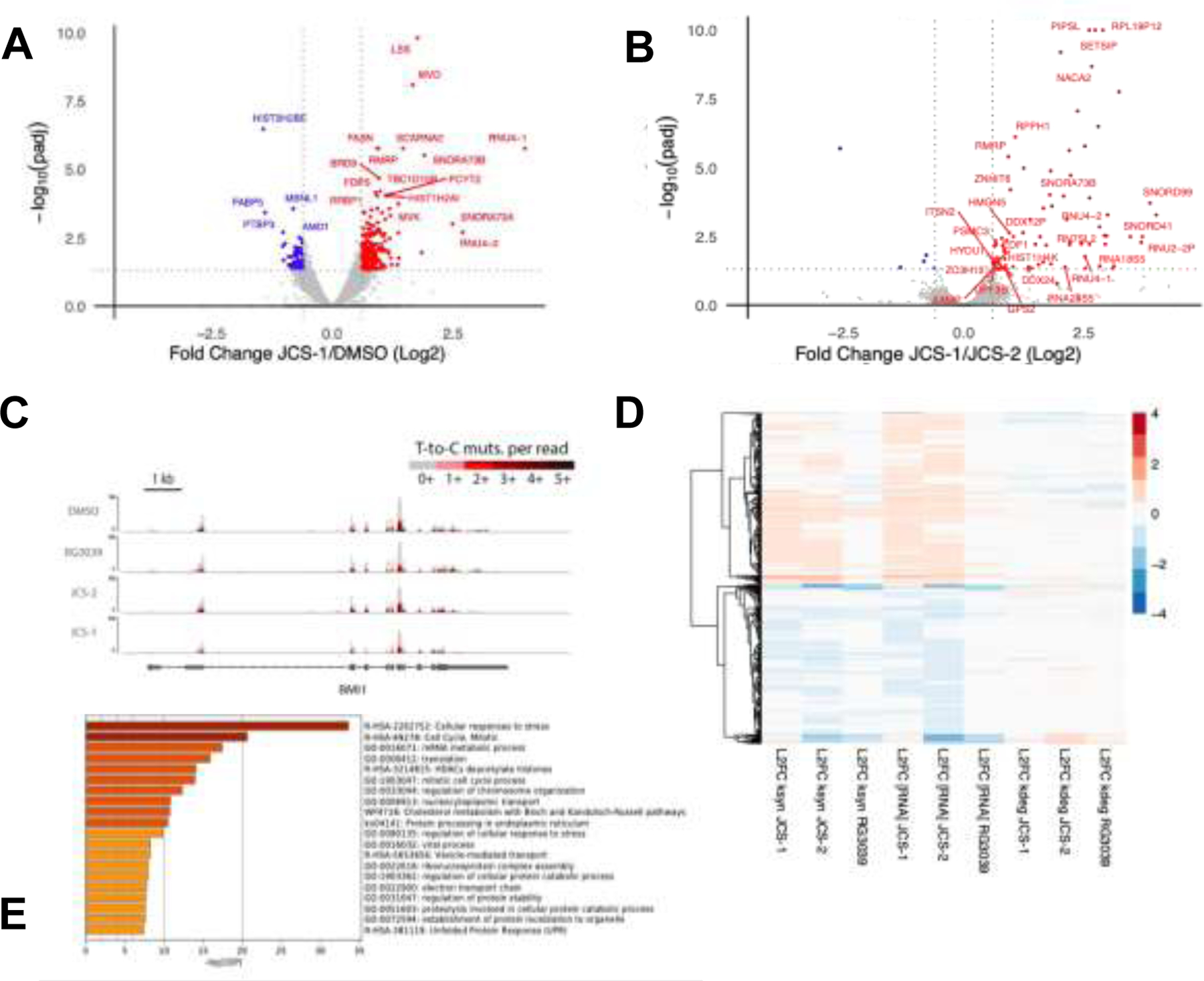
(A) Volcano plot describing log2 fold changes in total RNA in MOLM-14 cells treated with JCS-1 (1 μM, 6 hr) versus DMSO. Points in red represent transcripts upregulated in JCS-1 treated samples with padj < 0.05 and log2 fold change > 0.6. Points in blue represent transcripts downregulated in JCS-1 treated samples with padj < 0.05 and log2 fold change < −0.6. All log2 fold change and padj values were calculated according to DESeq2. Data points represent two biological replicates. Additional data point labels are assigned in SI Excel Sheet 1. (B) Volcano plot describing log2 fold changes in total RNA in MOLM-14 cells treated with JCS-1 (1 μM, 6 hr) versus JCS-2 (1 μM, 6 hr). Red and blue colored points represent differentially expressed genes across treatments with padj < 0.05 and |log2 fold change| > 0.6. Transcripts that were not significantly differentially expressed in JCS-1 treated samples relative to DMSO treated samples (see Fig. 5.A) were excluded from labeling. Data points represent two biological replicates. (C) TimeLapse-seq tracks of BMI1 in MOLM-14 cells treated with DMSO, RG3039, JCS-2, or JCS-1. BMI1, an AML implicated proto-oncogene, was identified as significantly downregulated in JCS-1 treatments. Increased T-to-C mutational content indicates higher s4U incorporation corresponding to newly synthesized RNA. (D) TimeLapse-seq profiling estimates decay- and synthesis-driven changes in total RNA. The heat map shown describes the log2 fold differences in total RNA and ksyn and kdeg rates for transcripts in JCS-1, JCS-2, and RG3039 treated samples relative to DMSO treated controls. Rows represent 8275 selected genes that exhibited significant differential expression (padj < 0.05) in either of the JCS-1, JCS-2, or RG3039 treatments relative to DMSO. Color of the displayed rows indicate the sign and magnitude of log2 fold change. Transcripts with similar expression patterns were clustered together in the dendrogram on the left. (E) Metascape gene enrichment analysis of MOLM-14 cells treated with JCS-1 (1 μM, 6 hr). Gene signatures were identified by filtering for differentially expressed transcripts with padj < 0.10.
TimeLapse-seq mutational profiles of individual genes were used to probe the effects of DcpS degradation and inhibition on mRNA stability. As seen in the sample TimeLapse tracks for the known oncogene BMI (Figure 5C), U-to-C mutations distinguish newly synthesized RNA from old RNA. As demonstrated by Schofield et al.,40 this added layer of temporal information can be used to estimate the RNA decay and synthesis rates (kdeg and ksyn) of all identified transcripts (Figure 5D). We observed that both DcpS degradation and inhibition elicited changes in total RNA that were primarily synthesis driven. Unlike the decapping enzyme Dcp2, which has been shown to act on a broad set of target substrates41,45, DcpS does not appear to serve a more general role as a direct regulator of mRNA stability.
Finally, we performed Gene Set Enrichment Analysis (GSEA) and Metascape Analysis (http://metascape.org)47 on JCS-1 treated MOLM-14 cells to investigate the potential mechanisms by which DcpS degradation decreases cell proliferation and viability (Figure 5E). As expected, JCS-1 treatment resulted in the differential expression of genes related to the cellular stress response and had broad effects on fundamental biological processes such as mitosis and cholesterol synthesis. Interestingly, under both analyses, transcripts downregulated by JCS-1 treatment showed enrichment for gene signatures related to nucleocytoplasmic carrier activity, mRNA metabolic processing, and ribonucleoprotein complex assembly. Indeed, previous immunoprecipitation (IP) experiments have also directly linked DcpS to components of the pre-mRNA metabolic pathway, including the nuclear pore complex and spliceosome1. Additionally, GSEA Analysis highlighted enrichment of the Interleukin-4 (IL-4) response (FDR=0.05) in transcripts upregulated by JCS-1 treatment. IL-4 has been shown to restrict AML cell growth and enrichment of this gene expression signature suggests skewing towards type II immune response upon DcpS degradation.48 Ultimately, treatment of MOLM-14 cells with JCS-1 induced broad anti-proliferative cellular effects, and the AML-specific dependency on DcpS is likely mediated via various processes.
Discussion:
Herein we report the development of a DcpS degrading PROTAC that could serve as a useful chemical biology tool to further probe DcpS biology in AML or other disease states. Our PROTAC JCS-1 couples a modified version of the DcpS inhibitor, RG3039, to the right-handed orientation of the VHL ligand developed in our laboratory.38 JCS-1 mediated recruitment of VHL to DcpS induces rapid (Figure 3A) and highly sustained (Figure 3B) degradation of endogenous DcpS in several AML cell lines including MOLM-14 (Figure 1), MV411 (SI Figure 2A) and OCI-AML3 (SI Figure 2B) with DC50 values ranging from 13 to 89 nM. We observed modest attenuation of MOLM-14 and MV411 cell growth using JCS-1 when comparing to the physicochemical equivalent, non-degrading epimer JCS-2 (Figure 4 and SI Figure 4A). To further interrogate the impact of DcpS degradation on the homeostasis of the mRNA pool of MOLM-14 cells we used TimeLapse-Seq to identify AML-relevant transcripts that were specifically altered by PROTAC treatment (Figure 5 and SI Figure 7).41,49
Our PROTAC is not as antiproliferative in AML cells as would be expected when considering previous work by the Maeda group that demonstrated a strong DcpS-dependency for AML.1 The inhibitor RG3039, which the authors employed alongside shRNA-mediated knockdown and CRISPR/Cas9-mediated knockout to interrogate DcpS dependency, has been demonstrated to accumulate in the lysosome.50 It remains possible that the localization of the inhibitor, or other off target effects, cause RG3039 to be a more potent inhibitor of AML proliferation when compared to our PROTAC.
Our TimeLapse-Seq experiments demonstrate modest changes in overall mRNA expression upon degradation and inhibition of DcpS with JCS-1 and JCS-2, respectively. Previously performed RNA-seq experiments carried out on AML cell lines treated with the DcpS inhibitor RG3039 also saw subtle effects on the mRNA pool.1 Nonetheless, our sequencing results reveal perturbation of key transcripts and pathways that may be responsible for the MOLM-14 dependency on DcpS. Additional insights provided by the TimeLapse mutational profiling suggests that observed changes in total RNA levels are driven primarily by RNA synthesis rather than RNA decay. Ultimately, this suggests that the mRNA decapping activity of DcpS does not have direct widespread effects on mRNA expression or stability. Rather, our findings agree with previous reports that DcpS may play a significant biological role via its interaction with pre-mRNA processing machineries, specifically regulating first-intron splicing of pre-mRNA through direct interaction with the spliceosome1,15.
The interplay of the multiple roles of DcpS in mRNA homeostasis are not fully understood. While the enzymatic decapping activity of DcpS is well characterized, more recently identified roles in miRNA turnover, splicing, and RNA export are still being researched1,15. When these distinct, but highly related functions are better characterized, it may allow for a better understanding of the role DcpS plays in disease. Our tool compound JCS-1 can be used to study how sustained DcpS degradation effects both the enzymatic and scaffolding roles of DcpS in DcpS-driven disease states, such as SMA, AML, along with other cancer types that may be discovered to have DcpS dependencies.52
Supplementary Material
Acknowledgements:
We would like to thank John Hines for his careful reading and editing of this manuscript. C. M. C. is funded by the NIH (R35CA197589) and is supported by an American Cancer Research Professorship. T.M. is funded by grant AMED #18063889 and a Grant-in-Aid for Scientific Research 20H05699. M. J. B. acknowledges support from the NIH (F31CA232477 and 5T32GM067543).
Declaration of Competing Interests:
C.M.C. is a shareholder in Arvinas, Inc. and in Halda, LLC, for which he consults and receives laboratory research support.
References:
- 1.Yamauchi T et al. Genome-wide CRISPR-Cas9 Screen Identifies Leukemia-Specific Dependence on a Pre-mRNA Metabolic Pathway Regulated by DCPS. Cancer Cell 33, 386–400.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu H, Rodgers ND, Jiao X & Kiledjian M The scavenger mRNA decapping enzyme DcpS is a member of the HIT family of pyrophosphatases. EMBO J. 21, 4699–4708 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arribas-Layton Marcos 1, 3, Wu Donghui 2, 3, Lykke-Andersen Jens 1, 4, and Song Haiwei 2. Structural and functional control of the eukaryotic mRNA decapping machinery. Biochim Biophys Acta. 1829, 580–589 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu SW, Rajagopal V, Patel SS & Kiledjian M Mechanistic and kinetic analysis of the dcps scavenger decapping enzyme. J. Biol. Chem 283, 16427–16436 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gu M et al. Insights into the structure, mechanism, and regulation of scavenger mRNA decapping activity. Mol. Cell 14, 67–80 (2004). [DOI] [PubMed] [Google Scholar]
- 6.Siwaszek A, Ukleja M & Dziembowski A Proteins involved in the degradation of cytoplasmic mRNA in the major eukaryotic model systems. RNA Biol. 11, 1122–1136 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Song Man-Gen, Li You, and K. M Multiple mRNA Decapping Enzymes in Mammalian Cells. Mol. Cell 40, 423–432 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiao Xinfu 1, Xiang Song 2, Oh ChanSeok 1, Martin Charles E. 1, Tong Liang 2, and K. M Identification of a quality control mechanism for mRNA 5’-end capping. Nature 467, 608–611 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Milac AL, Bojarska E & Wypijewska del Nogal A Decapping Scavenger (DcpS) enzyme: Advances in its structure, activity and roles in the cap-dependent mRNA metabolism. Biochim. Biophys. Acta - Gene Regul. Mech 1839, 452–462 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Neu A, Neu U, Fuchs AL, Schlager B & Sprangers R An excess of catalytically required motions inhibits the scavenger decapping enzyme. Nat. Chem. Biol 11, 697–704 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taverniti V & Seraphin B Elimination of cap structures generated by mRNA decay involves the new scavenger mRNA decapping enzyme Aph1/FHIT together with DcpS. Nucleic Acids Res. 43, 482–492 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fuchs AL, Wurm JP, Neu A & Sprangers R Molecular basis of the selective processing of short mRNA substrates by the DcpS mRNA decapping enzyme. Proc. Natl. Acad. Sci. U. S. A 117, 19237–19244 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou M, Bail S, Plasterer HL, Rusche J & Kiledjian M DcpS is a transcript-specific modulator of RNA in mammalian cells. Rna 21, 1306–1312 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meziane O et al. The human decapping scavenger enzyme DcpS modulates microRNA turnover. Sci. Rep 5, 1–8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shen V, Liu H, Liu SW, Jiao X & Kiledjian M DcpS scavenger decapping enzyme can modulate pre-mRNA splicing. Rna 14, 1132–1142 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Van Dijk E, Hir H. Le & Séraphin B DcpS can act in the 5′−3′ mRNA decay pathway in addition to the 3′−5′ pathway. Proc. Natl. Acad. Sci. U. S. A 100, 12081–12086 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Y, Song M & Kiledjian M Differential utilization of decapping enzymes in mammalian mRNA decay pathways. Rna 17, 419–428 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xiangyue Wu and Brewer G, The Regulation of mRNA Stability in Mammalian Cells: 2.0 Xiangyue. Gene 500, 10–21 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Braun KA & Young ET Coupling mRNA Synthesis and Decay. Mol. Cell. Biol 34, 4078–4087 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Singh J et al. DcpS as a therapeutic target for spinal muscular atrophy. ACS Chem. Biol 3, 711–722 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.MacKenzie D et al. Human Growth Hormone Increases SMN Expression and Survival in Severe Spinal Muscular Atrophy Mouse Model. J. Neuromuscul. Dis 1, 65–74 [PubMed] [Google Scholar]
- 22.Gentillon C, Connell AJ, Kirk RW & Butchbach MER The effects of C5-substituted 2,4-diaminoquinazolines on selected transcript expression in spinal muscular atrophy cells. PLoS One 12, 1–22 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gogliotti RG et al. The dcpS inhibitor RG3039 improves survival, function and motor unit pathologies in two SMA mouse models. Hum. Mol. Genet 22, 4084–4101 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cherry JJ et al. In vitro and in vivo effects of 2,4 diaminoquinazoline inhibitors of the decapping scavenger enzyme DcpS: Context-specific modulation of SMN transcript levels. PLoS ONE 12, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Van Meerbeke JP et al. The DcpS inhibitor RG3039 improves motor functionin SMA mice. Hum. Mol. Genet 22, 4074–4083 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ahmed I et al. Mutations in DCPS and EDC3 in autosomal recessive intellectual disability indicate a crucial role for mRNA decapping in neurodevelopment. Hum. Mol. Genet 24, 3172–3180 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ng CKL et al. Loss of the scavenger mRNA decapping enzyme DCPS causes syndromic intellectual disability with neuromuscular defects. Hum. Mol. Genet 24, 3163–3171 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ferrara F & Schiffer CA Acute myeloid leukaemia in adults. Lancet 381, 484–495 (2013). [DOI] [PubMed] [Google Scholar]
- 29.Shalem O, et al. Genome-Scale CRISPR-Cas9 Knockout Screening in Human Cells. 343, 84–88 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burslem GM & Crews CM Small-Molecule Modulation of Protein Homeostasis. Chem. Rev 117, 11269–11301 (2017). [DOI] [PubMed] [Google Scholar]
- 31.Bond MJ & Crews CM Proteolysis targeting chimeras (PROTACs) come of age: Entering the third decade of targeted protein degradation. RSC Chem. Biol 2, 725–742 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Samarasinghe KTG & Crews CM Targeted protein degradation: A promise for undruggable proteins. Cell Chem. Biol 28, 934–951 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alabi SB & Crews CM Major advances in targeted protein degradation: PROTACs, LYTACs, and MADTACs. J. Biol. Chem 296, 100647 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Han GW et al. Crystal structure of an Apo mRNA decapping enzyme (DcpS) from Mouse at 1.83 Å resolution. Proteins Struct. Funct. Genet 60, 797–802 (2005). [DOI] [PubMed] [Google Scholar]
- 35.Jafari R et al. The cellular thermal shift assay for evaluating drug target interactions in cells. Nat. Protoc 9, 2100–2122 (2014). [DOI] [PubMed] [Google Scholar]
- 36.Buckley DL et al. Targeting the von Hippel-Lindau E3 ubiquitin ligase using small molecules to disrupt the VHL/HIF-1α interaction. J. Am. Chem. Soc 134, 4465–4468 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chamberlain PP & Hamann LG Development of targeted protein degradation therapeutics. Nat. Chem. Biol 15, 937–944 (2019). [DOI] [PubMed] [Google Scholar]
- 38.Buckley DL et al. Small-molecule inhibitors of the interaction between the E3 ligase VHL and HIF1α. Angew. Chemie - Int. Ed 51, 11463–11467 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meng L et al. Epoxomicin, a potent and selective proteasome inhibitor, exhibits in vivo antiinflammatory activity. Proc. Natl. Acad. Sci. U. S. A 96, 10403–10408 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schofield JA, Duffy EE, Kiefer L, Sullivan MC & Simon MD TimeLapse-seq: Adding a temporal dimension to RNA sequencing through nucleoside recoding. Nat. Methods 15, 221–225 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luo Y et al. Discovery of cellular substrates of human RNA-decapping enzyme DCP2 using a stapled bicyclic peptide inhibitor. Cell Chem. Biol 28, 463–474.e7 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wei Y et al. Targeting Bcl-2 Proteins in Acute Myeloid Leukemia. Front. Oncol 10, 1–11 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang S et al. Mutational spectrum and prognosis in NRAS-mutated acute myeloid leukemia. Sci. Rep 10, 1–9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu W et al. CDK6 Is a Potential Prognostic Biomarker in Acute Myeloid Leukemia. Front. Genet 11, 1–11 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liang J et al. Small Nucleolar RNAs: Insight Into Their Function in Cancer. Front. Oncol 9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pauli C et al. Site-specific methylation of 18S ribosomal RNA by SNORD42A is required for acute myeloid leukemia cell proliferation. Blood. 135(23), 2059–2070 (2020) [DOI] [PubMed] [Google Scholar]
- 47.Zhou Y et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun 10, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Platanias LC Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol 5, 375–386 (2005). [DOI] [PubMed] [Google Scholar]
- 49.Luo Y, Schofield JA, Simon MD & Slavoff SA Global Profiling of Cellular Substrates of Human Dcp2. Biochemistry 59, 4176–4188 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gopalsamy A et al. Design of Potent mRNA Decapping Scavenger Enzyme (DcpS) Inhibitors with Improved Physicochemical Properties To Investigate the Mechanism of Therapeutic Benefit in Spinal Muscular Atrophy (SMA). J. Med. Chem 60, 3094–3108 (2017). [DOI] [PubMed] [Google Scholar]
- 51.Menduti G, Rasà DM, Stanga S & Boido M Drug Screening and Drug Repositioning as Promising Therapeutic Approaches for Spinal Muscular Atrophy Treatment. Front. Pharmacol 11, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.