

Abstract
Genetic analyses have linked BTBD9 to restless legs syndrome (RLS) and sleep regulation. Btbd9 knockout mice show RLS-like motor restlessness. Previously, we found hyperactivity of cerebellar Purkinje cells (PCs) in Btbd9 knockout mice, which may contribute to the motor restlessness observed. However, underlying mechanisms for PC hyperactivity in Btbd9 knockout mice are unknown. Here, we used dissociated PC recording, brain slice recording and western blot to address this question. Our dissociated recording shows that knockout PCs had increased TEA-sensitive, Ca2+-dependent K+ currents. Applying antagonist to large conductance Ca2+-activated K+ (BK) channels further isolated the increased current as BK current. Consistently, we found increased amplitude of afterhyperpolarization and elevated BK protein levels in the knockout mice. Dissociated recording also shows a decrease in TEA-insensitive, Ca2+-dependent K+ currents. The result is consistent with reduced amplitude of tail currents, mainly composed of small conductance Ca2+-activated K+ (SK) currents, in slice recording. Our results suggest that BK and SK channels may be responsible for the hyperactivity of knockout PCs. Recently, BTBD9 protein was shown to associate with SYNGAP1 protein. We found a decreased cerebellar level of SYNGAP1 in Btbd9 knockout mice. However, Syngap1 heterozygous knockout mice showed nocturnal, instead of diurnal, motor restlessness. Our results suggest that SYNGAP1 deficiency may not contribute directly to the RLS-like motor restlessness observed in Btbd9 knockout mice. Finally, we found that PC-specific Btbd9 knockout mice exhibited deficits in motor coordination and balance similar to Btbd9 knockout mice, suggesting that the motor effect of BTBD9 in PCs is cell-autonomous.
Keywords: Btbd9, restless legs syndrome, cerebellar Purkinje cells, BK channels, SK channels, SYNGAP1
Introduction
BTBD9 is a gene ubiquitously expressed (Fagerberg et al., 2014). The protein encoded by the gene contains a BTB/POZ domain, which is responsible for protein-protein interaction (Stogios et al., 2005). Polymorphisms in BTBD9 are associated with restless legs syndrome (RLS) (Winkelmann et al., 2007; Jimenez-Jimenez et al., 2018; Akcimen et al., 2020), sleep duration, sleep timing, and daytime sleepiness (Jones et al., 2019; Wang et al., 2019). Restless legs syndrome is considered a sleep disorder. It affects up to 10% of the general population (Gossard et al., 2021), causing patients to feel the urge to move, which may or may not be accompanied by uncomfortable sensations (Trenkwalder et al., 2018). The symptoms of RLS usually happen in the evening or during periods of rest, leading to a sleep disturbance (Gossard et al., 2021). Although the pathophysiology of RLS is mostly unclear, gene silencing of the BTBD9 homolog in flies leads to increased motor activity, decreased dopamine levels, and fragmented sleep patterns (Freeman et al., 2012). Similarly, Btbd9 systemic knockout (Btbd9 KO) mice also show motor restlessness, thermal hypersensitivity, dysregulated iron homeostasis, and a disruption in sleep structure (DeAndrade et al., 2012a). Two independent studies also observed sleep deficits in different lines of Btbd9 KO mice (Muramatsu et al., 2019; Gao et al., 2022). These studies indicate a role of BTBD9 in the generation of RLS-like phenotypes, and Btbd9 KO mice are useful in understanding the function of BTBD9 in motor control, sleep regulation, thermosensation, and iron homeostasis (DeAndrade and Li, 2015; Allen et al., 2017).
Using Btbd9 KO mice, we have found that Btbd9 knockout throughout the whole body leads to decreased neural activity in the cerebellum, more non-tonic Purkinje cells (PCs), and increased excitability of tonic PCs (Lyu et al., 2020a). The cerebellum is important for maintaining balance and fine-tuning and coordination of movements. Consistent with the changes in PCs, Btbd9 KO showed deficiency in rotarod and beam walking tests, both of which indicate that Btbd9 KO also has impaired motor balance and coordination (Lyu et al., 2020b). Although it is hard to rule out the influence of brain circuits and development, the results suggest a function of Btbd9 protein in PC activity, which may contribute to the behavioral output of Btbd9 KO mice. We next investigated the RLS-like behaviors, including circadian-dependent hyperactivity and thermosensation, in PC-specific Btbd9 KO mice (Btbd9 pKO). We found that Btbd9 knockout only in cerebellar PCs causes increased locomotion during the rest phase, but not during the active phase, and is insufficient for developing a sensory deficit (Lyu et al., 2020a). Our results confirmed that Btbd9 deficiency in PCs is responsible for generating motor-aspect of RLS-like phenotypes. However, what underlies the changes in PC activity, and whether Btbd9 pKO can result in deficits in motor balance and coordination is unknown.
BK channel regulates action potential (AP) repolarization and afterpotentials of PCs (Niday and Bean, 2021). Ex vivo, both BK channel blocker and BK activator increased the firing frequency of the PCs (Sausbier et al., 2004; Liu et al., 2022). In vivo, gain or loss of function of the BK channel has been reported to be associated with motor impairment. BK KO mice showed deficits in motor coordination, with significantly smaller afterhyperpolarization (AHP) amplitude and slightly longer mean AP 90–10% decay time of PCs (Sausbier et al., 2004). PC-specific BK KO mice displayed motor alterations similar to those observed in global BK KO mice (Chen et al., 2010). With motor impairment as well, the C9orf72 KO mouse line also showed hyperactivity of PCs accompanied by a significant increase of BK protein in the cerebellum (Liu et al., 2022). Similarly, Dyt1 knock-in mice (Liu et al., 2020) and PKCγ knockout mice (Watanave et al., 2022) displayed motor defects, larger BK current, and increased cerebellar BK protein.
SK channel also regulates AHP and the activity of PCs (Womack and Khodakhah, 2003). Applying the SK blocker decreased the amplitude of the AHP and increased the firing rate of PCs without affecting the shape of AP (Womack and Khodakhah, 2003). Intraperitoneal (IP) injection of SK channel inhibitor in wildtype (WT) mice induces an irregular pattern of PC activity. In contrast, IP injection of SK channel-positive modulator decreases the spontaneous firing rate of cerebellar PCs (Egorova et al., 2016). Mice with SK2 deletion specifically in PCs displayed a significant increase in the firing of PCs (Grasselli et al., 2020). However, although systemic SK KO mice showed motor impairment, the deficits are not apparent in mice with SK2 deletion specifically in PCs (Grasselli et al., 2020).
Btbd9 is known to interact with Syngap1 (Li et al., 2017; Wilkinson et al., 2017). SYNGAP1 is part of the postsynaptic density protein complex. It accelerates GTP-to-GDP conversion and thus directly inactivates small GTPase proteins within the Ras superfamily (Weldon et al., 2018). Loss of function of SYNGAP1 leads to reduced cognitive function, altered excitatory-inhibitory balance, and changes in brain morphology and innate behavior (Kilinc et al., 2018). Mutations in SYNGAP1 have been linked to autism (Berryer et al., 2013). Children with autism have been reported to have a high prevalence of RLS symptoms (Russell et al., 2017). A retrospective study indicates all individuals with SYNGAP1 mutations had impairment in gross motor function (Jimenez-Gomez et al., 2019). It is unclear whether loss of Btbd9 can decrease SYNGAP1 level and if a deficiency in SYNGAP1 will lead to RLS-like behavior in mice.
We hypothesize that BTBD9 deficiency leads to altered K+ channel activity and decreased level of SYNGAP1, which are responsible for the increased excitability observed in Btbd9 KO PCs and motor restlessness and incoordination. To test the hypothesis, we first isolated and compared different K+ currents in cerebellar PCs of Btbd9 KO and WT mice using dissociated neuron recording, followed by brain slice recording to compare tail currents, and western blot to quantify protein levels of ion channels and SYNGAP1 in the cerebellum. We further analyzed action AP and AHP based on the whole-cell patch-clamp recordings performed with PCs before (Lyu et al., 2020a). To explore the contribution of SYNGAP1 to the behavioral phenotype of Btbd9 KO mice, we reanalyzed the home-cage activity data of Syngap1−/+ mice (Nakajima et al., 2019). Finally, we tested motor coordination and balance of Btbd9 pKO mice to determine if motor phenotypes observed in Btbd9 KO (Lyu et al., 2020b) are cell autonomous.
Experimental Procedures
Mice
The experiments were conducted in accordance with national legislation and associated guidelines, and the procedures were approved by the Institutional Animal Care and Use Committee of the University of Florida. Experiments were conducted with males to minimize the variations caused by estrous cycles in females. The generation of Btbd9 KO mice and Btbd9 pKO mice has been described before (Lyu et al., 2020a).
Dissociated PC recording
Experiments were conducted according to protocols described previously (Raman and Bean, 1999; Tian et al., 2014; Liu et al., 2020). It should be noted that healthy dissociated PCs can only be isolated from mice younger than 21 d of age. Investigators who performed the recordings were blind to the genotypes. Eight KO cells and 5 WT cells were recorded from 3 Btbd9 KO pups and 3 WT littermates. Briefly, mice (13–18 d) were sacrificed, and the cerebellum was rapidly removed to a Tyrode’s (TYR) Solution (in mM): 150 NaCl, 4 KCl, 2 CaCl2, 2 MgCl2, 10 HEPES, and 10 glucose (buffered to pH 7.4 with NaOH). The vermal layer of the cerebellum was removed and minced in an ice-cold, oxygenated dissociation solution containing (in mM): 82 Na2SO4, 30 K2SO4, 5 MgCl2, 10 HEPES, 10 glucose, and 0.001% phenol red (pH 7.4). The minced tissue was subsequently incubated in the dissociation solution with 3 mg/ml protease XXIII (pH 7.4) at 35 °C for 7 min while oxygen was blown over the surface of the solution. Subsequently, the tissue was washed in warm, oxygenated dissociation solution containing 1 mg/ml bovine serum albumin and 1 mg/ml trypsin inhibitor and then maintained in TYR solution while oxygen was blown over the surface of the fluid. Occasionally, one-fifth of the tissue was withdrawn and triturated with a fire-polished Pasteur pipette to liberate individual neurons, after which 300 μl of the solution was transferred to poly-D-lysine coated cover glass.
The acutely dissociated PCs were allowed to settle on poly-D-lysine coated cover glass for 5 min before transferring the cover glass to the recording chamber. After washing out tissue debris with TYR solution, PCs with a pear-like shape and smooth surface were selected. Recording pipettes (5–10 MΩ) were filled with an intracellular solution containing (in mM): 122 K-gluconate, 9 NaCl, 1.8 MgCl2, 0.9 EGTA, 9 HEPES, 14 Tris-creatine phosphate, 4 MgATP, and 0.3 Tris-GTP (pH 7.2). The control external solution was TYR solution, which was applied to the recorded cell during the gigaseal formation. After adding 300 nM tetrodotoxin (TTX) to TYR solution to block all major sodium (Na+) channels, the solution was referred to as “Ca ECS” (current a, Fig. 1B). A “Co ECS” solution was made by replacing CaCl2 in Ca ECS with 2mM CoCl2, which reversibly blocks Ca2+ currents through all types of Ca2+ channels (currents b, Fig. 1D). To block certain K+ channels, we added tetraethylammonium (TEA; 1mM) to Ca ECS and Co ECS to make “CaTEA ECS” (currents c, Fig. 1E) and “CoTEA ECS” (currents d, Fig. 1F), respectively.
Figure 1.
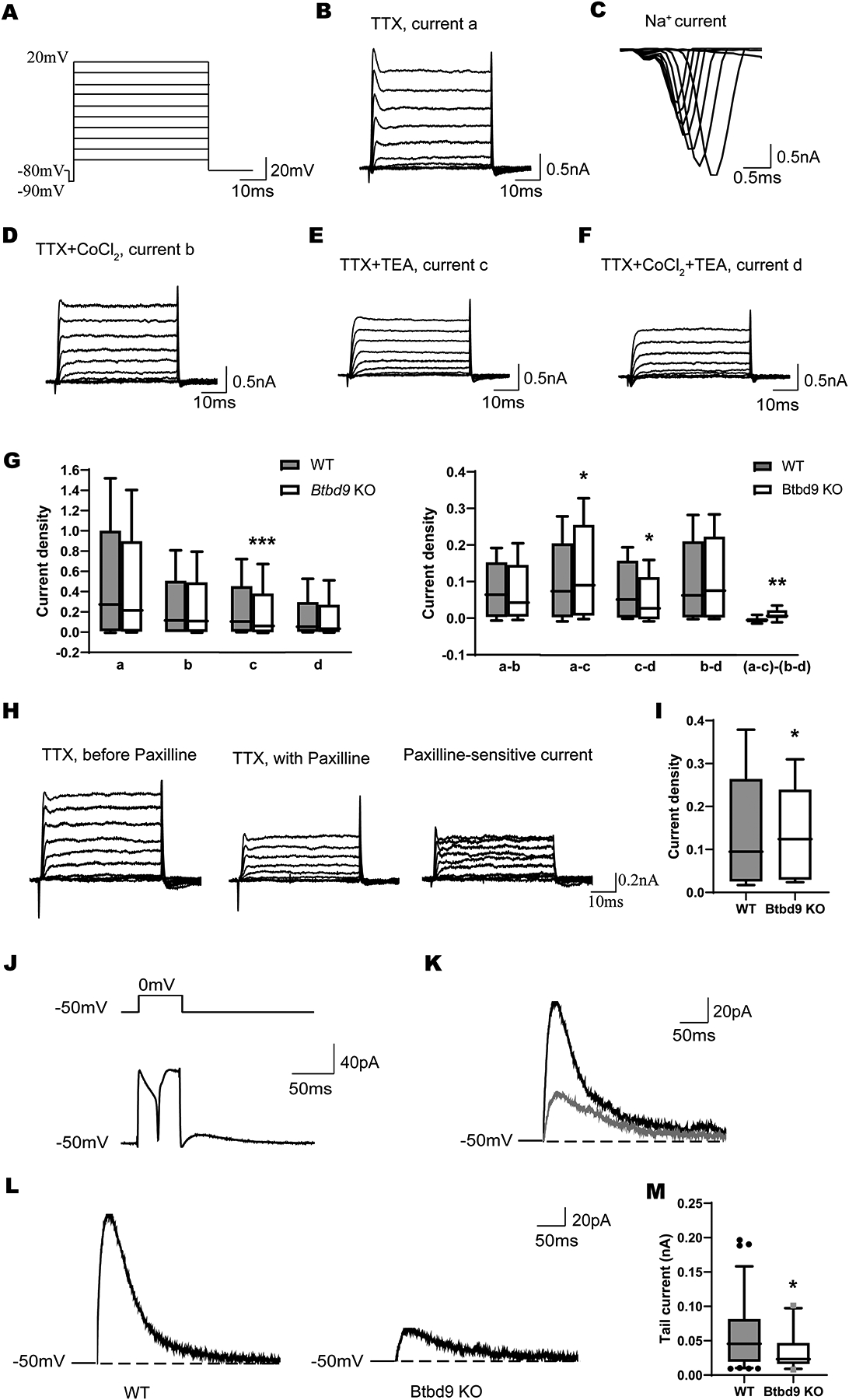
Dissociated PC recording. (A) Voltage steps were used for whole-cell voltage-clamp recording of acutely dissociated PCs. Representative traces from a WT neuron bathed in: (B) Ca ECS solution containing 300 nM TTX, (D) Co ECS solution containing 2 mM Co2+ and 300nM TTX, (E) CaTEA ECS solution containing 2 mM Ca2+, 300 nM TTX, and 1 mM TEA, (F) CoTEA ECS solution containing 2 mM Co2+, 300 nM TTX, and 1 mM TEA. (G) Densities of currents c and c-d were significantly decreased in the Btbd9 KO PCs, while densities of currents a-c and (a-c)-(b-d) were significantly increased in the Btbd9 KO PCs, compared with the WT PCs (KO, n=3 mice and 8 cells; WT, n=3 mice and 5 cells). (H) Representative traces from a WT neuron before (left) and after (middle) the application of Paxilline. (I) Densities of Paxilline-sensitive currents significantly increased in the Btbd9 KO PCs (KO, n=3 mice and 7 cells; WT, n=5 mice and 12 cells). (J) Voltage step (top) to evoke the calcium spike and tail current (bottom). (K) Tail current (black) was reduced after 100 nM apamin (gray). (L) Representative tail current traces in Btbd9 KO and WT mice. (M) significant reduction of the tail current in KO mice (KO, n=1 mouse and 4 cells; WT, n=2 mice and 8 cells). a, total K+ currents; b, Ca2+-independent K+ currents; c, TEA-insensitive K+ currents; d, TEA-insensitive, Ca2+-independent K+ currents; a-b, Ca2+-dependent K+ currents; a-c, TEA-sensitive K+ currents; c-d, TEA-insensitive, Ca2+-dependent K+ currents; b-d, TEA-sensitive, Ca2+-independent K+ currents; (a-c)-(c-d), TEA-sensitive, Ca2+-dependent K+ currents. Data in G and I are presented as box-and-whiskers plots, with the “box” depicting the median and the 25th and 75th quartiles and the “whisker” showing the 5th and 95th percentile. ***, p ≤ 0.001, **, p ≤ 0.01, *, p ≤ 0.05.
The change of different ECS was achieved by turning a 3-way stopcock with a gravity-driven perfusion system. Whole-cell recordings of dissociated PCs were performed at room temperature (23 ± 1 °C) using the Axopatch 1D Amplifier (Molecular Devices). Data were acquired using pCLAMP 10 software, with P/N subtraction of the leak currents built into the software. Signals were filtered at 5 kHz, and digitized at 10 kHz with a DigiData 1440 (Molecular Devices, Union City, CA). After establishing the whole-cell configuration, the cell was voltage-clamped at −80 mV (without correcting liquid junction potential).
Fast and slow capacitances were removed using capacitance compensation. The patch-clamp amplifier also compensated for series resistance. For isolation of K+ currents, voltage step protocols were repeated when the cell was exposed to Ca ECS, Co ECS, CaTEA ECS, and CoTEA ECS sequentially. The duration for each voltage step was 50 ms. For the quantification of ionic currents of interest, three identical recordings from the last three ECS solutions were averaged. Data were analyzed using Clampfit 10.3 software.
For each cell, subtractions were normalized with cell capacitance to obtain current densities. Then the value was normalized to the maximal current obtained with TTX or Ca ECS. Different types of K+ currents were analyzed separately. Each cell had 10 normalized data points, corresponding to voltage steps from −70mV to 20mV, which were included in the statistical analysis. Voltage steps and age were included as continuous variables.
Seven KO cells and twelve WT cells were recorded from the second batch of animals of 3 Btbd9 KO pups and 5 WT littermates (14–20 d). All procedures were the same as described. Cells were recorded in Ca ECS solutions for 5 min with or without 1 μM Paxilline (Fig. 1H).
Voltage clamp recording to determine Purkinje cell SK current
Experiments were conducted as described by others (Tian et al., 2014). The Investigator who performed the electrophysiological recordings was blind to the genotypes. A total of 4 mice were used. Eight cells from 2 WT mice and four cells from 1 Btbd9 KO littermate at 21 to 22 days of age, which generated 105 instances of calcium spike-induced tail currents, were used to quantify tail current amplitude. Animal ID, cell ID, traces, and voltage steps were all used to fit a multiple-level nested model (SAS GENMOD). The number of calcium spikes, the peak of the last calcium spike to the end of the voltage step, and the voltage step were used as covariates.
To confirm the major component of tail current is SK current, apamin, a blocker for SK channel, was used with 1 WT mouse at 20 days of age in an additional experiment. Mice were anesthetized by the inhalation of isoflurane, decapitated, and the brains were rapidly removed. Three hundred μm-thick cerebellar sagittal brain slices were cut in ice-cold, oxygenated cutting saline (in mM): (180 sucrose, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 10 D-glucose, 1 CaCl2, 10 MgCl2, and 10 glucose) using a Vibratome (Leica VT 1000s) and the slices will be recovered in a holding chamber for 60 min at 35°C with artificial cerebrospinal fluid (ACSF). Final concentrations of ACSF (in mM): 126 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 1 MgCl2, 2 CaCl2, and 10 glucose. The slices were then incubated at room temperature. The patch electrode was filled with an intracellular solution containing 135 mM potassium gluconate, 10 mM KCl, 1 mM MgCl2, 2 mM Na2-ATP, 0.4 mM Na3-GTP, 10 mM HEPES (pH 7.2–7.3 with KOH). Each slice was transferred to a submerged recording chamber with the continuous flow (2 ml/min) of ACSF.
Calcium spikes and invoked SK current were recorded by the voltage clamp at whole-cell recording mode with IR-DIC and an Axopatch 1D amplifier (Axon Instruments, Foster City, CA). The slice was perfused with ACSF with 1μM TTX and 1mM TEA to block voltage-gated Na+ channels, and most of the voltage-gated K+ channels, PCs were held at −50 mV before stepping to potentials ranging from 0 to 90 mV every 30 s for a period of 100–300 ms to invoke Ca2+ spike complexes. At the end of the voltage step, the membrane potential was stepped back to −50 mV to allow Ca2+- activated tail current generation. (Fig 1J).
Western blot
Western blot was performed as previously described (Yokoi et al., 2015b; Yokoi et al., 2015a; Yokoi et al., 2020). The individual cerebellum was dissected from 5 Btbd9 KO mice and 5 WTs with an average age of 3 mo (63–147 d) and homogenized in 200 μl of ice-cold lysis buffer (Tris/HCl 50mM, pH=7.4; NaCl 175mM; EDTA 5mM, pH=8.0) containing protease inhibitor cocktail (Roche). Twenty-two μl of ice-cold 10% Triton X-100 was added to the homogenate. The mixtures were incubated for 30 min on ice and centrifuged at 10,000 × g for 15 min at 4 °C. The supernatant was used as protein samples for the western blot. The protein concentration of the supernatant was measured by protein assay reagent (Bio-Rad). An aliquot of the supernatant was mixed with 2 × loading buffer containing 2-mercaptoethanol, boiled for 5 min, chilled on ice, and spun down. The proteins were separated on a 10% SDS-PAGE gel and transferred to Millipore Immobilon-FL transfer membranes (PVDF). The PVDF membranes were washed in 0.1M PBS for 5 min and blocked with LICOR Odyssey blocking buffer for 1 h. The membranes were cropped around the position of the bands of protein size markers (Precision Plus Protein™ All Blue Standards, Bio-Rad, #1610373) indicated by the manufacturers. For small conductance Ca2+-activated K+ (SK) channels, the membrane was cut between 37 KDa and 50 KDa, and between 50 KDa and 75 KDa. We cut the membranes between 100 KDa and 250 KDa to probe large-conductance Ca2+-activated K+ (BK) channels, 50 KDa to 100 KDa to probe Kv1.2 channels, 150 KDa to 250 KDa to probe P/Q channels, slightly above 100 KDa to below 250 KD to probe SYNGAP1, or slightly above 25 KDa to little above 37 KDa to probe glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The membranes were incubated overnight at 4 °C with rabbit polyclonal anti-KCNN2 (KCa2.2, SK2) antibody (Alomone lab, #APC-028) at 1:4000 dilution, mouse monoclonal anti-Slo1 (BK) antibody, clone L6/60 (Sigma-Aldrich, MABN70) at 1:4000 dilution, mouse monoclonal anti-KCNA2 antibody (for Kv1.2; Sigma-Aldrich, SAB5200059) at 1:4000 dilution, rabbit polyclonal anti- Ca2+ channel antibody (α1A subunit; P/Q-type of voltage-gated Ca2+ channel; Sigma-Aldrich, C1353) at 1:1000 dilution, rabbit polyclonal anti-SYNGAP1 antibody (Proteintech, 19739–1-AP) at 1:2000 dilution, or goat GAPDH antibody (Santa Cruz, sc-20357) at 1:2000 dilution in the blocking buffer. The membranes were washed with 0.1M PBS containing 0.1% Tween 20 for 4 times at 5 min each, then treated for 1 h with LI-COR IRDye 800CW donkey anti-goat IgG (H+L), LI-COR IRDye 800CW donkey anti-mouse IgG (H+L), or LI-COR IRDye 680RD donkey anti-rabbit IgG (H+L) at 1:15,556 dilution. After washing 4 times with 0.1M PBS containing 0.1% Tween 20 for 5 min each and 0.1M PBS 3 times for 5 min each, the membranes were dried, and the signals were detected and quantified by an LI-COR Odyssey imaging system.
Rotarod and beam walking
Eleven Btbd9 pKO mice and 15 control littermates with an average age of 7 mo (176–262 d) were tested as described previously (Dang et al., 2005; Lyu et al., 2020b). The rotarod apparatus started at an initial speed of 4 rpm and gradually accelerated at a rate of 0.2 rpm/s. The latency to fall was measured with a cutoff time of 3 min. Mice were tested for three trials each day for 2 d. The trials on the same day were performed approximately 1 h apart.
Animals were allowed to rest for a week before the beam walking test. Animals were trained to traverse a medium square beam (14 mm wide) in 3 consecutive trials each day for 2 d. After training was completed, the experiment commenced with recordings of the number of hind paw slips for each of the two trials per beam. On the first test day, animals were made to cross the medium square and round beam (17 mm diameter), and on the second day, a small round beam (10 mm diameter) and an additional small square beam (7 mm wide). It happens that the animals cannot finish the trial and drop down from the beam. In this case, it was assigned the largest number observed during the whole beam walking experiment.
Statistical analysis
AP and AHP data were derived from the whole-cell patch-clamp recording experiment conducted before (Lyu et al., 2020a) using the Mini Analysis Program (KO, n=6 mice and 42 cells; WT, n=6 mice and 43 cells). Analysis of AP and AHP was performed as described by others (Sausbier et al., 2004). The home-cage activity of Syngap1−/+ mice was published earlier (Nakajima et al., 2019) and used for sleep-like behavior analysis (Syngap1−/+, n=19; WT, n=14). All experimental data were tested for normality using the SPSS statistical package (version 26). Isolated currents obtained from the dissociated PC recording, tail currents, western blot of SYNGAP1, rise time and decay time of AP, AP area, AP half-width, AHP amplitude, and behavioral data were not normally distributed and analyzed by the generalized linear model (GENMOD, SAS University Edition). We did not use the nonparametric test because the method does not have a nesting function and does not allow us to consider “age” as a covariate. The amplitude of AP was normally distributed and therefore analyzed by mixed-model ANOVA (JMP Pro 15). Western blot data of SK, BK, Kv1.2, and P/Q channels were analyzed with Student’s t-test. Age was used as a continuous variable in both SAS GENMOD and ANOVA. Data in the text are presented as “mean ± standard error of the mean (SEM)” unless specifically marked.
Results
Decreased SK but increased BK current density of Btbd9 KO PCs
We first used the voltage clamp technique to compare ionic currents in Btbd9 KO PCs. Due to PC’s extensive dendritic materials and space clamp issues, it is extremely challenging to perform voltage clamp of PCs in adult brain slices. Acutely dissociated PCs can be used instead to directly record different types of K+ currents (Raman and Bean, 1999; Tian et al., 2014). However, healthy dissociated PCs can only be isolated from mice younger than 21 days of age. Dissociated PCs were identified by their large diameter and characteristic pear-like shape. Currents were recorded in response to voltage steps under five different conditions. The first recording assay was conducted without the interference of any drugs. Then TTX (300 nM) was introduced to block voltage-gated Na+ channels (currents a, Fig. 1B). Na+ currents were obtained by subtracting currents a from the whole currents (Fig. 1C). Next, CoCl2 (2 mM) was added to replace CaCl2, blocking Ca2+ channels and Ca2+-activated K+ channels (currents b, Fig. 1D). TEA (1 mM) was used to block TEA-sensitive K+ channels (currents c, Fig. 1E). Finally, TTX, CoCl2, and TEA were used all together to reveal the TEA-insensitive, Ca2+-independent K+ channels (currents d, Fig. 1F). Ca2+-dependent K+ currents were obtained by subtracting currents b from currents a (a-b). TEA-sensitive K+ currents were obtained by subtracting currents c from currents a (a-c). Subtraction of currents d from currents c resulted in TEA-insensitive, Ca2+-dependent K+ currents (c-d), which mainly contain small conductance Ca2+-activated K+ (SK) currents (Ishii et al., 1997). Subtraction of currents d from currents b resulted in TEA-sensitive, Ca2+-independent K+ currents (b-d). Subtraction between TEA-sensitive K+ currents, (a-c), and TEA-sensitive, Ca2+-independent K+ currents, (b-d), led to TEA-sensitive, Ca2+-dependent K+ currents [(a-c)-(b-d)], which is mostly contributed by BK channels (Benton et al., 2013). There was a significant 42% decrease in TEA-insensitive K+ currents (Fig. 1G, c, KO, 0.034 ± 0.003, WT, 0.059 ± 0.007, p < 0.001, GENMOD with a gamma distribution), which was attributed to a significant 38% decrease in SK current density (Fig. 1G, c–d, KO, 0.021 ± 0.004, WT, 0.034 ± 0.003, p = 0.033, GENMOD with a gamma distribution). There was a 23% increase in TEA-sensitive current (Fig. 1G, a–c, KO, 0.050 ± 0.005, WT, 0.040 ± 0.001, p = 0.034, GENMOD with a gamma distribution). Further analysis showed a significant 57% increase in BK current density of the Btbd9 KO PCs (Fig. 1G, (a–c)–(b–d), KO, 0.022 ± 0.004, WT, 0.014 ± 0.001, p = 0.008, GENMOD with a gamma distribution). These results together suggest PCs of juvenile KO mice have already shown functional changes in SK and BK channels, which are important for the neural development of firing patterns and frequency (Kshatri et al., 2018).
To further confirm the increased current density observed from the BK channel in KO mice, recordings were performed in solutions with TTX first, then were repeated by adding Paxilline, a BK channel blocker (Fig. 1H). Subtractions between the two recordings resulted in Paxilline-sensitive currents (Fig. 1H), which were significantly increased in the KO mice (15%, Fig. 1I, KO, 0.110 ± 0.016, WT, 0.083 ± 0.001, p = 0.0498, GENMOD with a gamma distribution).
To further confirm the decreased current density observed from the SK channel in KO mice, we used voltage steps to evoke tail currents in brain slice recording (Fig. 1J, the current after the end of the voltage step). Most of the tail current was responsive to apamin, an SK channel blocker, indicating SK current is the main component of the tail current (Fig. 1K). Btbd9 KO mice had a significant reduction in the tail current amplitude (18%, Fig. 1L, M, KO, 0.039 ± 0.003, WT, 0.048 ± 0.001, p = 0.024, GENMOD with a gamma distribution), suggesting a decreased SK current.
Increased cerebellar level of BK channels in Btbd9 KO mice
To explore if there exist ion channel changes in adult mice, we analyzed the related protein levels in the cerebellum using western blot. Previously, we have found that tonic PCs in Btbd9 KO have increased spontaneous firing frequency and enhanced response to current steps (Lyu et al., 2020a). The firing of PCs is regulated by the TEA-insensitive, Ca2+-dependent K+ channels, including SK channels (Tian et al., 2014); the TEA-sensitive, Ca2+-dependent K+ channels comprise BK channels (Womack and Khodakhah, 2002); and the Ca2+-independent K+ currents contain Kv1.2 channels (Xie et al., 2010; Williams et al., 2012). In addition, it has been shown that P/Q-type Ca2+ channels solely provide Ca2+ for both SK and BK channels (Womack et al., 2004). Hence, P/Q-type Ca2+ channels were included in our analysis. Although the expression levels of SK, Kv1.2, and P/Q channels were not statistically different between Btbd9 KO and WT mice (Fig. 2A, KO, 0.318 ± 0.032, WT, 0.255 ± 0.027, p = 0.165; C, KO, 0.807 ± 0.137, WT, 0.795 ± 0.139, p = 0.950; D, KO, 0.089 ± 0.025, WT, 0.096 ± 0.015, p = 0.801, all Student’s t-test), BK channels showed a significant increase (47%) in Btbd9 KO compared to WT mice (Fig. 2B, KO, 0.637 ± 0.069, WT, 0.434 ± 0.046, p = 0.040, Student’s t-test). In summary, the western blot data suggest that the adult Btbd9 KO has increased BK channel protein in their cerebellum, which may contribute to the increased excitability in KO PCs.
Figure 2.
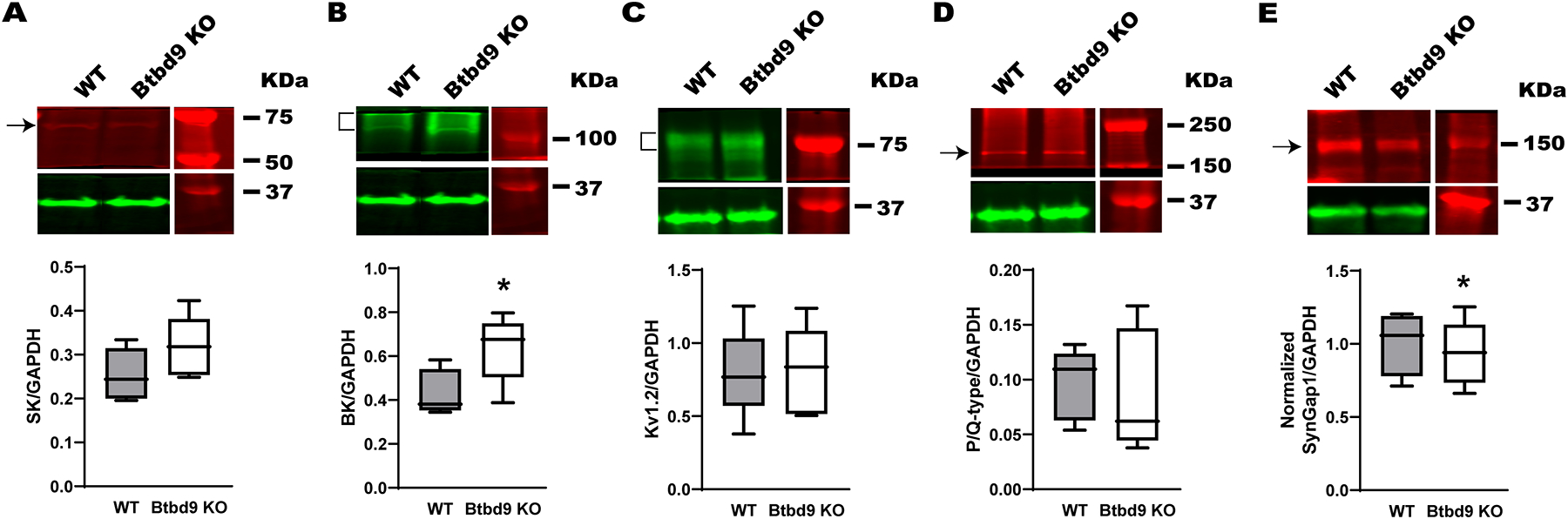
Western blot of cerebellar tissues from the systematic Btbd9 KO mice (n=5) and their WT littermates (n=5). While the SK channel (A), P/Q, and Kv1.2 channels (C, D) were not different between the Btbd9 KO mice and the WTs, Btbd9 KO mice had increased levels of the BK channel (B). (E) Btbd9 KO mice had decreased levels of SYNGAP1. Target protein bands were normalized to the GAPDH at the bottom of each figure. Blots were cropped to show the representative bands. The experiment was done in duplicate. Data are presented as box-and-whiskers plots, with the “box” depicting the median and the 25th and 75th quartiles and the “whisker” showing the 5th and 95th percentile. *, p ≤ 0.05.
Increased AHP amplitude of Btbd9 KO PCs
BK channels regulate the shape of AP and AHP (Sah and Faber, 2002). Increased levels of BK channels would be expected to cause increased AHP amplitude and smaller AP half-width of neurons (Haghdoost-Yazdi et al., 2008). We have previously published our brain slice recording results from PCs containing information about AP and AHP (Lyu et al., 2020a). Further analysis showed that the AHP amplitude was significantly larger in Btbd9 KO compared to WT PCs (Fig. 3H, KO, 12.606 ± 0.418, WT, 11.098 ± 0.584, p = 0.047, GENMOD with a gamma distribution). However, the half-width of AP was not significantly decreased in Btbd9 KO PCs (Fig. 3G, KO, 0.673 ± 0.022, WT, 0.627 ± 0.016, p = 0.089, GENMOD with a gamma distribution). It should be noted that knockout of BK channels in mice only has a modest effect on the AP itself, with the main impact of decreased AHP amplitude (Sausbier et al., 2004). Our analysis also showed that the amplitude (Fig. 3C, KO, 15.375 ± 0.474, WT, 15.160 ± 0.469, p = 0.751, mixed model ANOVA) and decay time (Fig. 3E, KO, 0.473 ± 0.017, WT, 0.442 ± 0.017, p = 0.215, GENMOD with a gamma distribution) of AP were not statistically different between Btbd9 KO and WT PCs. However, Btbd9 KO PCs had an increased rise time of AP (Fig. 3D, KO, 2.374 ± 0.017, WT, 2.275 ± 0.023, p = 0.001, GENMOD with a gamma distribution) and increased area of AP (Fig. 3F, KO, 12.979 ± 0.547, WT, 11.318 ± 0.367, p = 0.007, GENMOD with a gamma distribution). The results indicate potential changes in other voltage-gated ion channels responsible for the AP generation.
Figure 3.
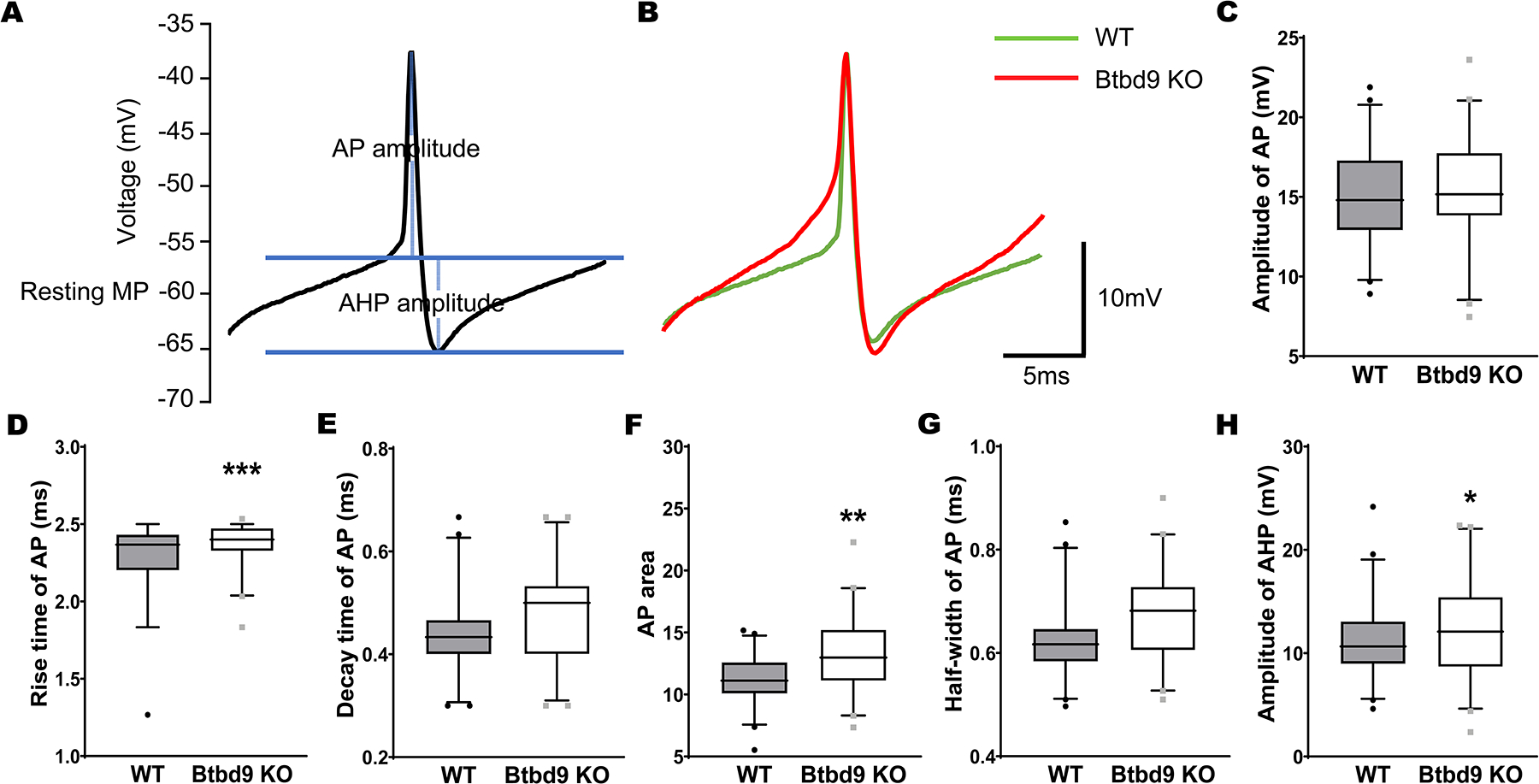
Comparison of AP and AHP from the whole-cell recording of PCs of the systemic Btbd9 KO mice (n=6 mice and 42 cells) and their WT littermates (n=6 mice and 43 cells). (A) Diagram showing how AP and AHP were measured. The y-axis shows the voltages without adjusting the junction potential (−14.5 mV). (B) Representative traces of AP and AHP from WT and KO mice. (C-G) AP parameters. The rise time and area of AP were significantly increased in Btbd9 KO PCs compared with that of the WTs. (H) The amplitude of AHP was significantly increased in Btbd9 KO PCs compared with that of the WTs. Data in C-H are presented as box-and-whiskers plots, with the “box” depicting the median and the 25th and 75th quartiles and the “whisker” showing the 5th and 95th percentile. ***, p ≤ 0.001, **, p ≤ 0.01, *, p ≤ 0.05.
Decreased cerebellar level of SYNGAP1 in Btbd9 KO mice
Previous studies found putative in vitro interaction between BTBD9 and SYNGAP1 (Li et al., 2017; Wilkinson et al., 2017). Here, we validated such an interaction in the Btbd9 KO cerebellum. Western blot analysis indicates a decreased level of SYNGAP1 protein in the Btbd9 KO cerebellum (Fig. 2E, p = 0.020, GENMOD with a gamma distribution), supporting such an interaction.
The increased home-cage activity of Syngap1−/+ mice during the dark phase
To further understand if the decreased cerebellar level of SYNGAP1 is associated with RLS-like phenotypes observed in the Btbd9 KO mice (DeAndrade et al., 2012a), we made a home-cage activity comparison between Syngap1−/+ mice and their control littermates based on the data collected before (Nakajima et al., 2019). As described, Syngap1−/+ mice and WTs were housed individually in their home cage, and their locomotor activity was monitored for 7 days by video cameras connected to a computer. We only included the last 4 days’ data for analyses as we did before (Sausbier et al., 2004; Lyu et al., 2019; Lyu et al., 2020b). The activity levels of the first 3 days were not stable (Nakajima et al., 2019), and the mice are considered to be accommodating to the environment.
We divided the activity level of each mouse into the light phase (7:00 to 18:00) and dark phase (19:00 to 6:00) and summed the activity level for each mouse during each phase. Each mouse had 4 days’ and 4 nights’ data (coded as ‘Period’ in analyses). Day and night activity levels were separately compared between two genotypes with the period as a within-subject and a covariate using the GENMOD with a gamma distribution. We also summed each mouse’s activity level during each hour. The activity levels of each hour were separately compared between two genotypes with the period as a within-subject and a covariate using the GENMOD with a gamma distribution. A principal feature of RLS is a desire to move, especially during the rest phase (Trenkwalder et al., 2018). Here, opposite to the Btbd9 KO mice, Syngap1−/+ mice showed an increased home-cage activity level during the dark phase (Fig. 4B, left panel, Syngap1−/+, 19592.459 ± 1769.898, WT, 10855.205 ± 1064.515, p < 0.001, GENMOD with a gamma distribution), which is usually the active phase of rodents, but not the light phase (Fig. 4A, left panel, Syngap1−/+, 4837.726 ± 262.666, WT, 4804.460 ± 345.548, p = 0.939, GENMOD with a gamma distribution) when rodents usually are sleeping. We then computed the probability of waking by coding the activity level as 1 if the mouse had movements during a minute and coded the activity level as 0 if the mouse did not have any movements during that minute. The probability of day and night were separately compared between two genotypes with period, hour, and minute as within-subjects using the GENMOD with a binomial distribution. Syngap1−/+ mice had an increased likelihood of waking in the active phase (Fig. 4D, Syngap1−/+, 0.961 ± 0.006, WT, 0.897 ± 0.011, p < 0.001, GENMOD with a binomial distribution), but not the rest phase (Fig. 4C, Syngap1−/+, 0.678 ± 0.006, WT, 0.702 ± 0.015, p = 0.148, GENMOD with a binomial distribution). The results suggest that the reduction of SYNGAP1 protein itself in Btbd9 KO mice may not contribute directly to RLS-like phenotypes.
Figure 4.
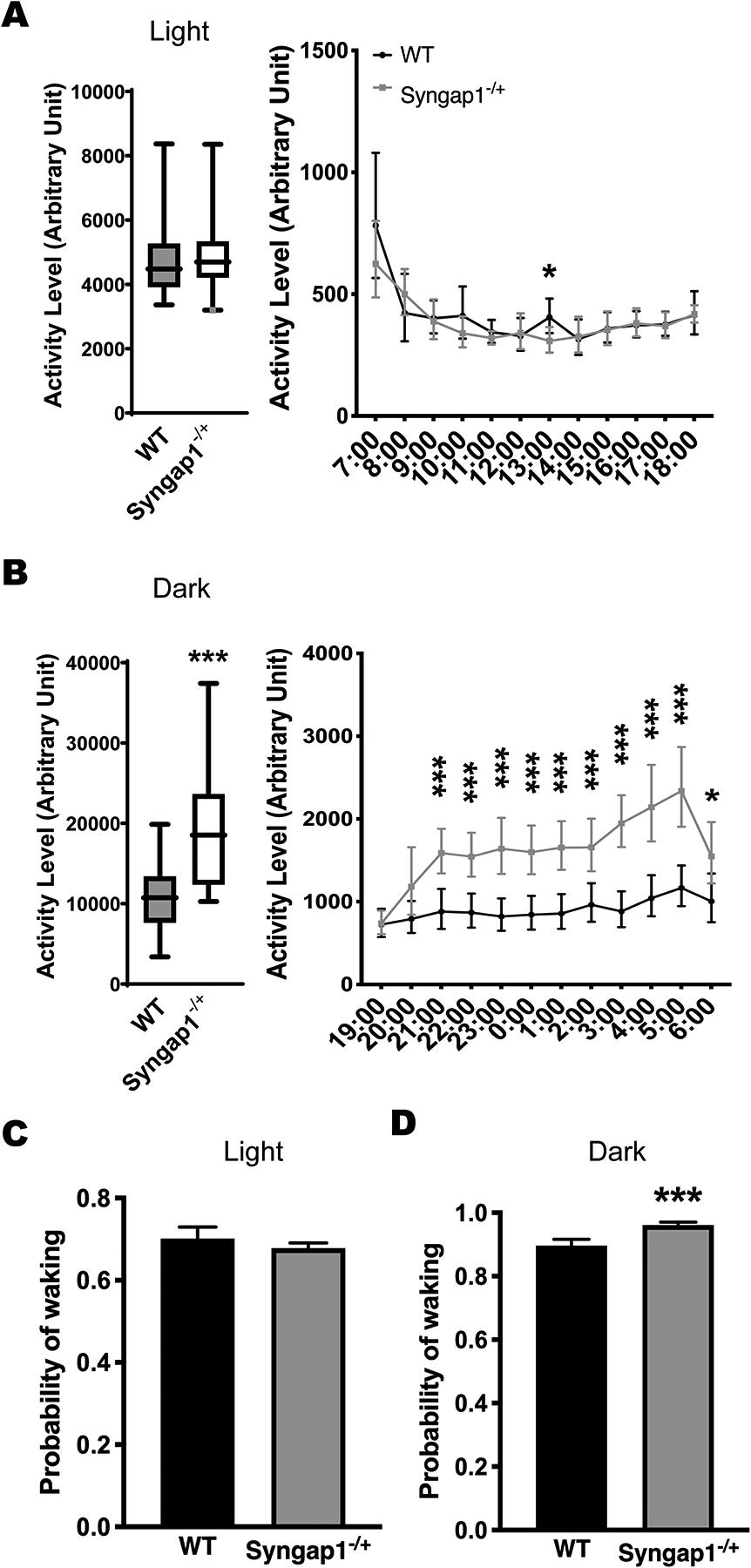
The home-cage activity of Syngap1−/+ (n=19) compared with the WT mice (n=14). (A) Syngap1−/+ mice did not have activity changes during the light phase compared with the WT mice. (B) Syngap1−/+ mice showed hyperactivity during the dark phase. Data are presented as box-and-whiskers plots, with the “box” depicting the median and the 25th and 75th quartiles and the “whisker” showing the 5th and 95th percentile. The hourly activity levels are presented next to the corresponding box-and-whiskers plots. Significant p values are marked above each time point. (C, D) Syngap1−/+ mice showed an increased probability of waking during the dark phase but not during the light phase. Histograms represent means plus 95% CIs. ***, p ≤ 0.001, *, p ≤ 0.05.
Motor coordination and balance deficits of Btbd9 pKO mice
Btbd9 KO mice have decreased latency to fall in the rotarod test and increased number of slips in the beam walking test (Lyu et al., 2020b) besides RSL-like phenotypes (DeAndrade et al., 2012a). The rotarod test assesses gross motor ability to maintain balance and coordination when the animals are challenged by an accelerating rotating rod. In contrast, the beam walking test assesses fine motor balance and coordination skills (Dang et al., 2005). Btbd9 pKO mice showed increased locomotor activity levels and the probability of waking, especially during the rest phase (Lyu et al., 2020a), which is consistent with what we found in Btbd9 KO mice. Here, we investigated whether Btbd9 pKO mice had similar motor coordination and balance deficits as Btbd9 KO. We used the same experimental setup to find that pKO mice showed a motor deficit in the rotarod (Fig. 5A, right panel, pKO, 81.997 ± 4.328, Control, 98.197 ± 6.387, p = 0.032, GENMOD with a gamma distribution) but not in the beam walking test (Fig. 5B, pKO, 1.133 ± 0.356, Control, 1.102 ± 0.294, p = 0.938, GENMOD with a negative binomial distribution). Therefore, PC-specific BTBD9 deficiency in mice is sufficient to cause RLS-like motor restlessness (Lyu et al., 2020a) and can lead to motor coordination and balance deficits.
Figure 5.
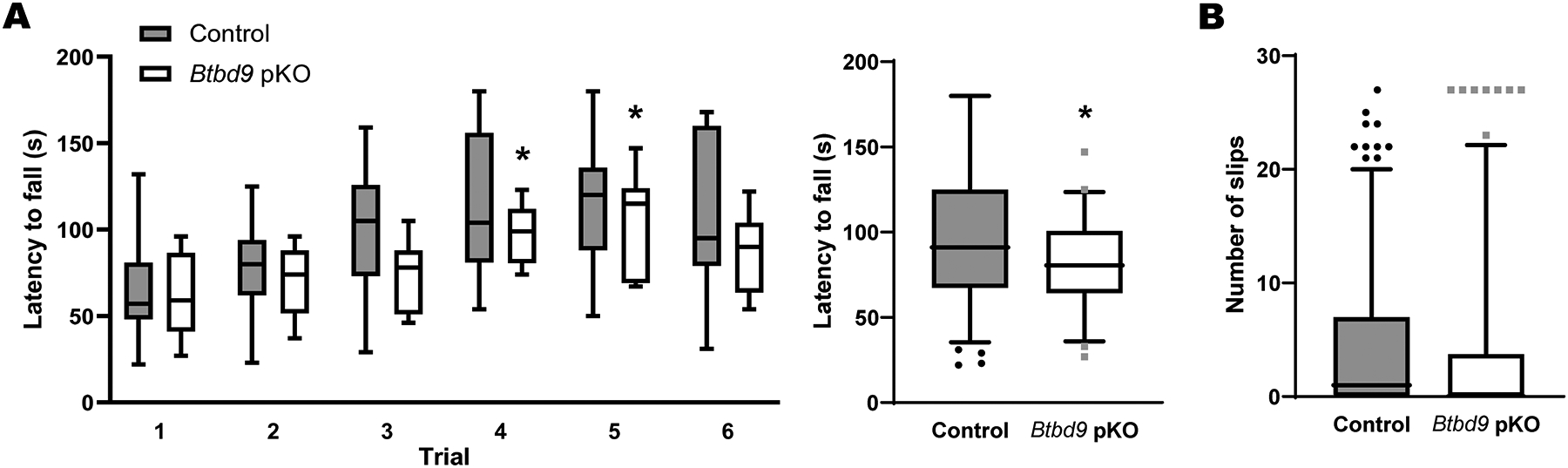
Rotarod and beam walking tests for Btbd9 pKO mice (n=11) and their control littermates (n=15). (A) Btbd9 pKO mice showed decreased latency to fall in trials 3 and 4 (left), and when 6 trials were considered as repeated measurements (right). (B) Btbd9 pKO mice had a similar number of slips in the beam walking test compared with controls. Data are presented as box-and-whiskers plots, with the “box” depicting the median and the 25th and 75th quartiles and the “whisker” showing the 5th and 95th percentile. *, p ≤ 0.05.
Discussion
Our previous study showed that Btbd9 KO mice had increased neural activity in cerebellar PCs (Lyu et al., 2020a). Furthermore, Btbd9 pKO mice resemble the diurnal motor restlessness of Btbd9 KO mice (Lyu et al., 2020a). Based on these findings, we proposed that a lack of BTBD9 protein influences the function of the cerebellum, which may contribute to RLS-like circadian-dependent hyperactivity. However, the underlying mechanisms for the observed activity changes in PCs and RLS-like behavior are unclear. In this study, we used three lines of mutated mice, which are Btbd9 KO, Btbd9 pKO, and Syngap1−/+. To answer this question, we performed dissociated neuron recording, brain slice recording, western blot, and behavioral tests, including home-cage activity (Nakajima et al., 2019), rotarod and beam walking. As summarized in Figure 6, our results demonstrate that Btbd9 KO had an increased BK but decreased SK activity level in PCs at the early postnatal stage. Moreover, adult Btbd9 KO had an increased protein level of BK channels, consistent with an increased amplitude of AHP observed from KO PCs. Although the protein level of SYNGAP1 decreased in the cerebellum of Btbd9 KO, Syngap1−/+ mice did not have RLS-like diurnal motor restlessness. Finally, PC-specific BTBD9 deficiency is sufficient for mice to develop motor coordination and balance deficits.
Figure 6.
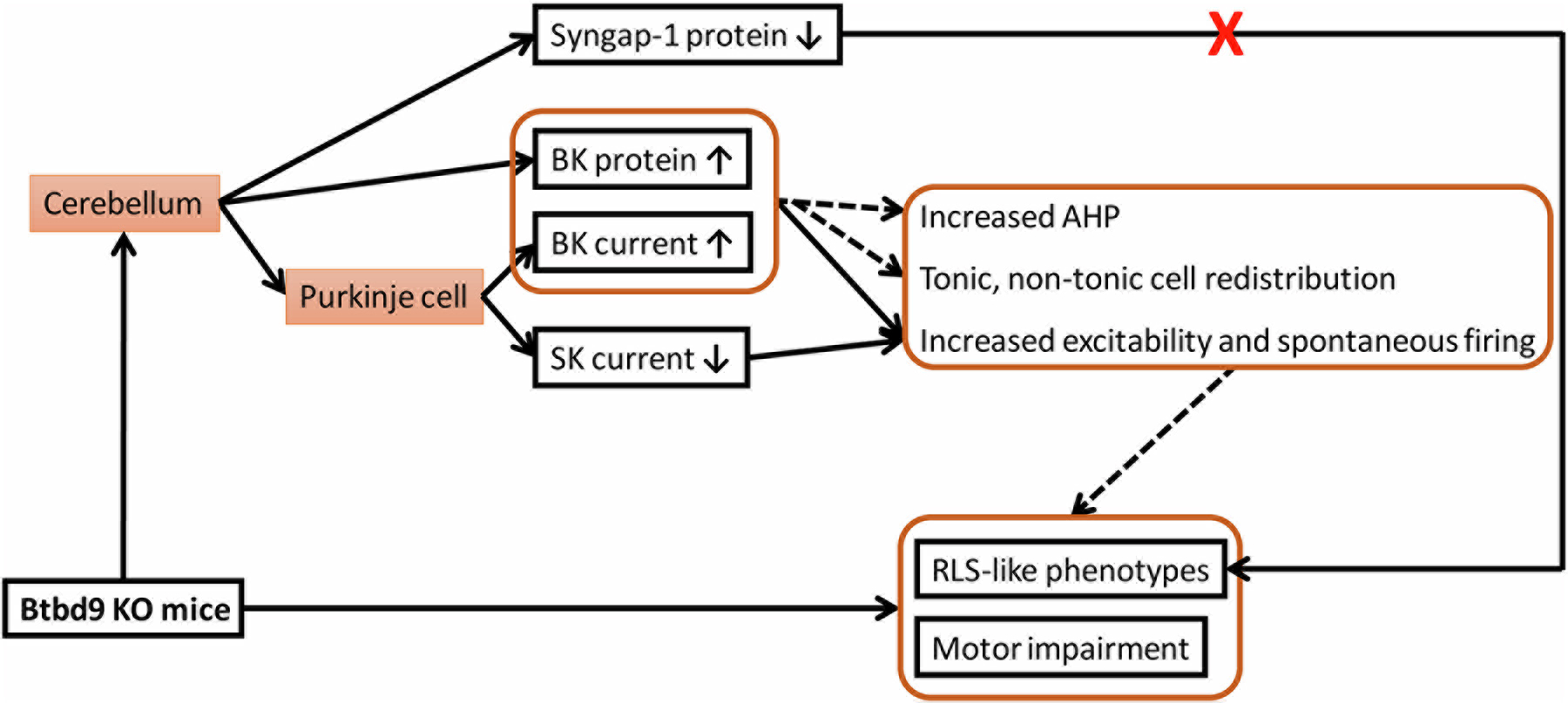
Summary of the findings. A solid line indicates a validated relationship, and a dotted line represents a possible contributing factor.
Dissociated neuron recording indicated an increased current density of BK channels in juvenile Btbd9 KO PCs, and western blot showed an increased protein level of BK channels in the adult Btbd9 KO cerebellum. The two findings are consistent and supported by the recently published hyperexcitability and cell-type redistribution of KO PCs (Lyu et al., 2020a), a larger AHP of KO PCs, and increased firing frequency with a BK agonist (Liu et al., 2022). First, BK channels have been found to contribute to the fast AHP following spontaneous APs (Sah and Faber, 2002). Specifically, the strong activation of BK channels enhances the amplitude of AHP (Haghdoost-Yazdi et al., 2008). In addition, BK KO mice had reduced spontaneous activity of the PCs (Sausbier et al., 2004). Although we did not have evidence showing that a pharmacological blockage of BK channels in KO PCs would rescue the deficit, we were able to show that pharmacological stimulation of BK channels in WT PCs leads to increased firing frequency, as observed in KO PCs (Liu et al., 2022). It can be inferred that the activity increase of BK channels would be more likely to result in increased spontaneous activity of the PCs, which is precisely our case. However, another study showed that inhibition of BK channels in PCs leads to increased firing frequency in rats with ages less than 16 d (Womack et al., 2009). It should be noted that the effectiveness of iberiotoxin, a BK channel blocker, disappears when animals are older than 20 d (Womack et al., 2009). The mice we used in the whole-cell patch-clamp recording were around 8 mo of age. Therefore, the effect of blocking the BK channel in increasing firing frequency may have been greatly diminished. Moreover, blocking the BK channel acutely with the drugs is different from chronic manipulations like gene knockout. Secondly, BK channels are also important for the transition between tonic and non-tonic firing (Gu et al., 2007). In hippocampal neurons (Brenner et al., 2005; Wang et al., 2016), adrenal chromaffin cells (Martinez-Espinosa et al., 2014), and neocortical pyramidal neurons (Shruti et al., 2008), enhanced BK current is linked with paradoxical increases in excitability and predisposition to non-tonic firing. Furthermore, BK currents are involved in the circadian regulation of the suprachiasmatic nucleus and generate bidirectional effects on neuronal firing under distinct conditions during day and night (Montgomery and Meredith, 2012). Finally, BK channels are associated with motor coordination in mice. Mouse models with motor impairment, including C9orf72 KO mice (Liu et al., 2022), Dyt1 knock-in mice (Liu et al., 2020), and PKCγ KO mice (Watanave et al., 2022), show a significant increase in BK protein in the cerebellum.
RLS-associated genes, CRBN and SUN1 (Tilch et al., 2020), have been found to be able to increase the BK channel activity and influence the function of the cerebellum, respectively. The deficiency of SUN1, which is enriched in PCs, causes cerebellar ataxia in mice (Wang et al., 2015). CRL4ACRBN, an E3 ubiquitin ligase, targets the BK channel for polyubiquitination. Inactivation of CRL4ACRBN leads to markedly increased BK channel activity (Liu et al., 2014). Additionally, Crbn KO animals have been found to show enhanced BK channel activity (Choi et al., 2018). Our results showed that Btbd9 deficiency led to increased BK channel activity. These findings demonstrate the importance of BK channels and the cerebellum in the pathogenesis of RLS.
The BACK domain of BTBD9 has been implied to help maintain substrate orientation in Cullin3-based E3 ligase complexes (Stogios and Prive, 2004; Stogios et al., 2005). The BTBD9 ortholog dBTBD9 has been found to colocalize with Cullin-3 in Drosophila (Freeman et al., 2012). BTBD9 is thought to work with Cullin-3 to regulate levels of IRP2, which is degraded by over-expressing BTBD9 in HEK cells (Freeman et al., 2012). Cullin-RING and BTBD9 complexes have been identified to interact with tumor necrosis factor α-induced protein 1 (TNFAIP1) and promote its polyubiquitination and degradation (Li et al., 2020). Therefore, loss-of-function in BTBD9 may affect the normal ubiquitylation and proteolysis process. The increased protein level of BK channels may be caused by the same loss of function of BTBD9 in protein ubiquitylation. Interestingly, Dynamin-1 was found to be significantly elevated in the systemic Btbd9 KO mice (DeAndrade et al., 2012b; Lyu et al., 2020c). Dynamin-1 may represent another potential ubiquitylation target that is regulated by BTBD9 protein.
There was a decreased current density of SK channels detected by dissociated recording and a reduced tail current detected by brain slice recording in juvenile Btbd9 KO PCs. However, we could not find protein level changes for the SK channel in adult KO mice. It has been found that blockage of the SK channel increased the firing frequency of PCs in mice (Womack and Khodakhah, 2003). Moreover, PC-specific SK2 deletion in mice leads to an increase in the firing of PCs (Grasselli et al., 2020). A reduction in the SK channel also enhances the coupling of dendritic EPSPs to spike output (Ohtsuki and Hansel, 2018), facilitating enhanced activity levels and excitability of PCs. Therefore, decreased levels of SK current may be another contributor to the increased excitability and firing of tonic PCs observed in Btbd9 KO mice. However, SK channel may not be responsible for the increased AHP amplitude of KO PCs and the motor impairment found in KO mice. It has been reported that SK channel blockage in PCs leads to a decrease in AHP amplitude (Womack and Khodakhah, 2003). PC-specific SK2 KO mice do not have significant impairment in locomotion and do not show ataxia-like tremors (Grasselli et al., 2020).
Large-scale protein interactome analysis has revealed that BTBD9 protein interacts with SYNGAP1, a synaptic Ras GTPase activating protein abundant in the neurons’ postsynaptic density (Li et al., 2017; Wilkinson et al., 2017). Our western blot analysis showed a decreased level of SYNGAP1 protein in the cerebellum of Btbd9 KO mice, which is consistent with the proposed interaction in vivo. However, Syngap1−/+ mice failed to show RLS-like phenotypes, indicating that decreased SYNGAP1 level may not be directly related to the diurnal motor restlessness observed in the Btbd9 KO mice. On the other hand, one of the primary neuronal functions of the SYNGAP1 protein is to regulate the balance between excitation and inhibition. Loss of SYNGAP1 can lead to epilepsy or seizures (Berryer et al., 2013; Mignot et al., 2016). SYNGAP1 protein has been mostly studied in the context of the postsynaptic density of excitatory neurons; however, a recent study also revealed the role of SYNGAP1 protein in the GABAergic inhibitory neurons (Berryer et al., 2016). According to Allen Brain Atlas, SYNGAP1 is highly expressed in the PCs and surrounding cells. A reduced cerebellar level of SYNGAP1 protein might lead to increased excitability of PCs either in a cell-autonomous manner or through cell-cell interactions with other cells.
Btbd9 pKO mice showed a motor deficit in the rotarod test, similar to Btbd9 KO mice (Lyu et al., 2020b). However, Btbd9 KO mice showed an increased number of slips in the beam walking test (Lyu et al., 2020b), which is not the case for the pKO mice. This is reasonable because, in addition to the cerebellum, Btbd9 KO mice have BTBD9 deficiency in other brain regions, like the striatum and the cerebral cortex, both of which are important for motor controls. BTBD9 deficiency, specifically in the cerebral cortex, has been shown to be sufficient to cause motor deficits in the beam walking test (Lyu et al., 2020b). This study extends our mechanistic understanding of the BTBD9 function in the cerebellum and behavioral output of the Btbd9 KO mice. Future studies will test the PC activity in Btbd9 pKO mice, the function of BK channel blockers in relieving RLS-like phenotypes in rodents, and their potential in clinical settings.
Highlights.
Increased BK but decreased SK currents, and enhanced AHP in PCs of Btbd9 KO mice.
Increased BK channel protein levels in the cerebellum of Btbd9 KO mice.
SYNGAP1 protein level decreased in Btbd9 KO mice due to its interaction with BTBD9.
Syngap1−/+ mice did not have restless legs syndrome-like diurnal motor restlessness.
Btbd9 PC-specific KO developed motor coordination and balance deficits as Btbd9 KO.
Acknowledgments:
We thank Tom Otis, Indira Raman, and Bruce Bean for teaching the Purkinje cell patch-clamp recording in brain slices and preparation of dissociated PCs, Dr. Jinbin Tian from Michael Zhu lab for the voltage-clamp recording of dissociated PCs and tail current in brain slices, and Drs. Noboru H. Komiyama (The University of Edinburgh), Seth G. N. Grant (The University of Edinburgh), Tsuyoshi Miyakawa (Fujita Health University), and Ryuichi Nakajima (Fujita Health University) for sharing home-cage activity data of Syngap1−/+ mice.
Funding:
This work was supported by the National Institute of Health [R21NS065273, R01NS082244, and R21NS111498], and the Restless Legs Syndrome Foundation.
Abbreviations
- AHP
afterhyperpolarization
- AP
action potential
- Btbd9 KO
Btbd9 knockout mice
- Btbd9 pKO
Btbd9 Purkinje cell-specific knockout mice
- KO
knockout
- PC
Purkinje cell
- RLS
restless legs syndrome
- WT
wildtype
Footnotes
Conflict of interest:
none
References:
- Akcimen F, Ross JP, Sarayloo F, Liao C, De Barros Oliveira R, Ruskey JA, Bourassa CV, Dion PA, Xiong L, Gan-Or Z, Rouleau GA (2020) Genetic and epidemiological characterization of restless legs syndrome in Quebec. Sleep 43. [DOI] [PubMed] [Google Scholar]
- Allen RP, Donelson NC, Jones BC, Li Y, Manconi M, Rye DB, Sanyal S, Winkelmann J (2017) Animal models of RLS phenotypes. Sleep Med 31:23–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benton MD, Lewis AH, Bant JS, Raman IM (2013) Iberiotoxin-sensitive and -insensitive BK currents in Purkinje neuron somata. J Neurophysiol 109:2528–2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berryer MH, Chattopadhyaya B, Xing P, Riebe I, Bosoi C, Sanon N, Antoine-Bertrand J, Levesque M, Avoli M, Hamdan FF, Carmant L, Lamarche-Vane N, Lacaille JC, Michaud JL, Di Cristo G (2016) Decrease of SYNGAP1 in GABAergic cells impairs inhibitory synapse connectivity, synaptic inhibition and cognitive function. Nat Commun 7:13340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berryer MH et al. (2013) Mutations in SYNGAP1 cause intellectual disability, autism, and a specific form of epilepsy by inducing haploinsufficiency. Hum Mutat 34:385–394. [DOI] [PubMed] [Google Scholar]
- Brenner R, Chen QH, Vilaythong A, Toney GM, Noebels JL, Aldrich RW (2005) BK channel beta4 subunit reduces dentate gyrus excitability and protects against temporal lobe seizures. Nat Neurosci 8:1752–1759. [DOI] [PubMed] [Google Scholar]
- Chen X, Kovalchuk Y, Adelsberger H, Henning HA, Sausbier M, Wietzorrek G, Ruth P, Yarom Y, Konnerth A (2010) Disruption of the olivo-cerebellar circuit by Purkinje neuron-specific ablation of BK channels. Proc Natl Acad Sci U S A 107:12323–12328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi TY, Lee SH, Kim YJ, Bae JR, Lee KM, Jo Y, Kim SJ, Lee AR, Choi S, Choi LM, Bang S, Song MR, Chung J, Lee KJ, Kim SH, Park CS, Choi SY (2018) Cereblon Maintains Synaptic and Cognitive Function by Regulating BK Channel. J Neurosci 38:3571–3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang MT, Yokoi F, McNaught KS, Jengelley TA, Jackson T, Li J, Li Y (2005) Generation and characterization of Dyt1 DeltaGAG knock-in mouse as a model for early-onset dystonia. Exp Neurol 196:452–463. [DOI] [PubMed] [Google Scholar]
- DeAndrade MP, Li Y (2015) Chapter 80 - Btbd9 Knockout Mice as a Model of Restless Legs Syndrome. In: Movement Disorders (Second Edition) pp 1191–1205: Academic Press. [Google Scholar]
- DeAndrade MP, Johnson RL Jr., Unger EL, Zhang L, van Groen T, Gamble KL, Li Y (2012a) Motor restlessness, sleep disturbances, thermal sensory alterations and elevated serum iron levels in Btbd9 mutant mice. Hum Mol Genet 21:3984–3992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeAndrade MP, Zhang L, Doroodchi A, Yokoi F, Cheetham CC, Chen HX, Roper SN, Sweatt JD, Li Y (2012b) Enhanced hippocampal long-term potentiation and fear memory in Btbd9 mutant mice. PLoS One 7:e35518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egorova PA, Zakharova OA, Vlasova OL, Bezprozvanny IB (2016) In vivo analysis of cerebellar Purkinje cell activity in SCA2 transgenic mouse model. J Neurophysiol 115:2840–2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagerberg L et al. (2014) Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol Cell Proteomics 13:397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman A, Pranski E, Miller RD, Radmard S, Bernhard D, Jinnah HA, Betarbet R, Rye DB, Sanyal S (2012) Sleep fragmentation and motor restlessness in a Drosophila model of Restless Legs Syndrome. Curr Biol 22:1142–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, Wang A, Zhao Y, Zhang X, Yuan X, Li N, Xu C, Wang S, Zhu Y, Zhu J, Guan J, Liu F, Yin S (2022) Integrative Proteome and Ubiquitinome Analyses Reveal the Substrates of BTBD9 and Its Underlying Mechanism in Sleep Regulation. ACS Omega 7:11839–11852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gossard TR, Trotti LM, Videnovic A, St Louis EK (2021) Restless Legs Syndrome: Contemporary Diagnosis and Treatment. Neurotherapeutics 18:140–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grasselli G, Boele HJ, Titley HK, Bradford N, van Beers L, Jay L, Beekhof GC, Busch SE, De Zeeuw CI, Schonewille M, Hansel C (2020) SK2 channels in cerebellar Purkinje cells contribute to excitability modulation in motor-learning-specific memory traces. PLoS Biol 18:e3000596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu N, Vervaeke K, Storm JF (2007) BK potassium channels facilitate high-frequency firing and cause early spike frequency adaptation in rat CA1 hippocampal pyramidal cells. J Physiol 580:859–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghdoost-Yazdi H, Janahmadi M, Behzadi G (2008) Iberiotoxin-sensitive large conductance Ca2+ -dependent K+ (BK) channels regulate the spike configuration in the burst firing of cerebellar Purkinje neurons. Brain Res 1212:1–8. [DOI] [PubMed] [Google Scholar]
- Ishii TM, Maylie J, Adelman JP (1997) Determinants of apamin and d-tubocurarine block in SK potassium channels. J Biol Chem 272:23195–23200. [DOI] [PubMed] [Google Scholar]
- Jimenez-Gomez A, Niu S, Andujar-Perez F, McQuade EA, Balasa A, Huss D, Coorg R, Quach M, Vinson S, Risen S, Holder JL Jr. (2019) Phenotypic characterization of individuals with SYNGAP1 pathogenic variants reveals a potential correlation between posterior dominant rhythm and developmental progression. J Neurodev Disord 11:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Jimenez FJ, Alonso-Navarro H, Garcia-Martin E, Agundez JAG (2018) Genetics of restless legs syndrome: An update. Sleep Med Rev 39:108–121. [DOI] [PubMed] [Google Scholar]
- Jones SE et al. (2019) Genetic studies of accelerometer-based sleep measures yield new insights into human sleep behaviour. Nat Commun 10:1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilinc M, Creson T, Rojas C, Aceti M, Ellegood J, Vaissiere T, Lerch JP, Rumbaugh G (2018) Species-conserved SYNGAP1 phenotypes associated with neurodevelopmental disorders. Mol Cell Neurosci 91:140–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kshatri AS, Gonzalez-Hernandez A, Giraldez T (2018) Physiological Roles and Therapeutic Potential of Ca(2+) Activated Potassium Channels in the Nervous System. Front Mol Neurosci 11:258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Zhang W, Yang H, Howrigan DP, Wilkinson B, Souaiaia T, Evgrafov OV, Genovese G, Clementel VA, Tudor JC, Abel T, Knowles JA, Neale BM, Wang K, Sun F, Coba MP (2017) Spatiotemporal profile of postsynaptic interactomes integrates components of complex brain disorders. Nat Neurosci 20:1150–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Zhang W, Liu Y, Liu X, Cai L, Kang J, Zhang Y, Chen W, Dong C, Zhang Y, Wang M, Wei W, Jia L (2020) The CRL3(BTBD9) E3 ubiquitin ligase complex targets TNFAIP1 for degradation to suppress cancer cell migration. Signal Transduct Target Ther 5:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Ye J, Zou X, Xu Z, Feng Y, Zou X, Chen Z, Li Y, Cang Y (2014) CRL4A(CRBN) E3 ubiquitin ligase restricts BK channel activity and prevents epileptogenesis. Nat Commun 5:3924. [DOI] [PubMed] [Google Scholar]
- Liu Y, Xing H, Wilkes BJ, Yokoi F, Chen H, Vaillancourt DE, Li Y (2020) The abnormal firing of Purkinje cells in the knockin mouse model of DYT1 dystonia. Brain Res Bull 165:14–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Xing H, Ernst AF, Liu C, Maugee C, Yokoi F, Lakshmana M, Li Y (2022) Hyperactivity of Purkinje cell and motor deficits in C9orf72 knockout mice. Mol Cell Neurosci 121:103756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyu S, Xing H, DeAndrade MP, Perez PD, Yokoi F, Febo M, Walters AS, Li Y (2020a) The Role of BTBD9 in the Cerebellum, Sleep-like Behaviors and the Restless Legs Syndrome. Neuroscience 440:85–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyu S, Xing H, DeAndrade MP, Liu Y, Perez PD, Yokoi F, Febo M, Walters AS, Li Y (2019) The Role of BTBD9 in Striatum and Restless Legs Syndrome. eNeuro 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyu S, Xing H, DeAndrade MP, Perez PD, Zhang K, Liu Y, Yokoi F, Febo M, Li Y (2020b) The role of BTBD9 in the cerebral cortex and the pathogenesis of restless legs syndrome. Exp Neurol 323:113111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyu S, Doroodchi A, Xing H, Sheng Y, DeAndrade MP, Yang Y, Johnson TL, Clemens S, Yokoi F, Miller MA, Xiao R, Li Y (2020c) BTBD9 and dopaminergic dysfunction in the pathogenesis of restless legs syndrome. Brain Struct Funct 225:1743–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Espinosa PL, Yang C, Gonzalez-Perez V, Xia XM, Lingle CJ (2014) Knockout of the BK beta2 subunit abolishes inactivation of BK currents in mouse adrenal chromaffin cells and results in slow-wave burst activity. J Gen Physiol 144:275–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignot C et al. (2016) Genetic and neurodevelopmental spectrum of SYNGAP1-associated intellectual disability and epilepsy. J Med Genet 53:511–522. [DOI] [PubMed] [Google Scholar]
- Montgomery JR, Meredith AL (2012) Genetic activation of BK currents in vivo generates bidirectional effects on neuronal excitability. Proc Natl Acad Sci U S A 109:18997–19002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muramatsu K, Chikahisa S, Shimizu N, Sei H, Inoue Y (2019) Rotigotine suppresses sleep-related muscle activity augmented by injection of dialysis patients’ sera in a mouse model of restless legs syndrome. Sci Rep 9:16344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima R, Takao K, Hattori S, Shoji H, Komiyama NH, Grant SGN, Miyakawa T (2019) Comprehensive behavioral analysis of heterozygous Syngap1 knockout mice. Neuropsychopharmacol Rep 39:223–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niday Z, Bean BP (2021) BK Channel Regulation of Afterpotentials and Burst Firing in Cerebellar Purkinje Neurons. J Neurosci 41:2854–2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtsuki G, Hansel C (2018) Synaptic Potential and Plasticity of an SK2 Channel Gate Regulate Spike Burst Activity in Cerebellar Purkinje Cells. iScience 1:49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Bean BP (1999) Ionic currents underlying spontaneous action potentials in isolated cerebellar Purkinje neurons. J Neurosci 19:1663–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell M, Baldwin CM, McClain D, Matthews N, Smith C, Quan SF (2017) Symptoms of Restless Legs Syndrome in Biological Caregivers of Children with Autism Spectrum Disorders. J Clin Sleep Med 13:105–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah P, Faber ES (2002) Channels underlying neuronal calcium-activated potassium currents. Prog Neurobiol 66:345–353. [DOI] [PubMed] [Google Scholar]
- Sausbier M, Hu H, Arntz C, Feil S, Kamm S, Adelsberger H, Sausbier U, Sailer CA, Feil R, Hofmann F, Korth M, Shipston MJ, Knaus HG, Wolfer DP, Pedroarena CM, Storm JF, Ruth P (2004) Cerebellar ataxia and Purkinje cell dysfunction caused by Ca2+-activated K+ channel deficiency. Proc Natl Acad Sci U S A 101:9474–9478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shruti S, Clem RL, Barth AL (2008) A seizure-induced gain-of-function in BK channels is associated with elevated firing activity in neocortical pyramidal neurons. Neurobiol Dis 30:323–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stogios PJ, Prive GG (2004) The BACK domain in BTB-kelch proteins. Trends Biochem Sci 29:634–637. [DOI] [PubMed] [Google Scholar]
- Stogios PJ, Downs GS, Jauhal JJ, Nandra SK, Prive GG (2005) Sequence and structural analysis of BTB domain proteins. Genome Biol 6:R82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian J, Tep C, Benedick A, Saidi N, Ryu JC, Kim ML, Sadasivan S, Oberdick J, Smeyne R, Zhu MX, Yoon SO (2014) p75 regulates Purkinje cell firing by modulating SK channel activity through Rac1. J Biol Chem 289:31458–31472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilch E et al. (2020) Identification of Restless Legs Syndrome Genes by Mutational Load Analysis. Ann Neurol 87:184–193. [DOI] [PubMed] [Google Scholar]
- Trenkwalder C, Allen R, Hogl B, Clemens S, Patton S, Schormair B, Winkelmann J (2018) Comorbidities, treatment, and pathophysiology in restless legs syndrome. Lancet Neurol 17:994–1005. [DOI] [PubMed] [Google Scholar]
- Wang B, Bugay V, Ling L, Chuang HH, Jaffe DB, Brenner R (2016) Knockout of the BK beta4-subunit promotes a functional coupling of BK channels and ryanodine receptors that mediate a fAHP-induced increase in excitability. J Neurophysiol 116:456–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H et al. (2019) Genome-wide association analysis of self-reported daytime sleepiness identifies 42 loci that suggest biological subtypes. Nat Commun 10:3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JY, Yu IS, Huang CC, Chen CY, Wang WP, Lin SW, Jeang KT, Chi YH (2015) Sun1 deficiency leads to cerebellar ataxia in mice. Dis Model Mech 8:957–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanave M, Takahashi N, Hosoi N, Konno A, Yamamoto H, Yasui H, Kawachi M, Horii T, Matsuzaki Y, Hatada I, Hirai H (2022) Protein kinase Cgamma in cerebellar Purkinje cells regulates Ca(2+)-activated large-conductance K(+) channels and motor coordination. Proc Natl Acad Sci U S A 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weldon M, Kilinc M, Lloyd Holder J Jr., Rumbaugh G (2018) The first international conference on SYNGAP1-related brain disorders: a stakeholder meeting of families, researchers, clinicians, and regulators. J Neurodev Disord 10:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson B, Li J, Coba MP (2017) Synaptic GAP and GEF Complexes Cluster Proteins Essential for GTP Signaling. Sci Rep 7:5272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams MR, Fuchs JR, Green JT, Morielli AD (2012) Cellular mechanisms and behavioral consequences of Kv1.2 regulation in the rat cerebellum. J Neurosci 32:9228–9237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkelmann J et al. (2007) Genome-wide association study of restless legs syndrome identifies common variants in three genomic regions. Nat Genet 39:1000–1006. [DOI] [PubMed] [Google Scholar]
- Womack MD, Khodakhah K (2002) Characterization of large conductance Ca2+-activated K+ channels in cerebellar Purkinje neurons. Eur J Neurosci 16:1214–1222. [DOI] [PubMed] [Google Scholar]
- Womack MD, Khodakhah K (2003) Somatic and dendritic small-conductance calcium-activated potassium channels regulate the output of cerebellar Purkinje neurons. J Neurosci 23:2600–2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Womack MD, Chevez C, Khodakhah K (2004) Calcium-activated potassium channels are selectively coupled to P/Q-type calcium channels in cerebellar Purkinje neurons. J Neurosci 24:8818–8822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Womack MD, Hoang C, Khodakhah K (2009) Large conductance calcium-activated potassium channels affect both spontaneous firing and intracellular calcium concentration in cerebellar Purkinje neurons. Neuroscience 162:989–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie G, Harrison J, Clapcote SJ, Huang Y, Zhang JY, Wang LY, Roder JC (2010) A new Kv1.2 channelopathy underlying cerebellar ataxia. J Biol Chem 285:32160–32173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoi F, Dang MT, Liu J, Gandre JR, Kwon K, Yuen R, Li Y (2015a) Decreased dopamine receptor 1 activity and impaired motor-skill transfer in Dyt1 DeltaGAG heterozygous knock-in mice. Behav Brain Res 279:202–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoi F, Chen HX, Dang MT, Cheetham CC, Campbell SL, Roper SN, Sweatt JD, Li Y (2015b) Behavioral and electrophysiological characterization of Dyt1 heterozygous knockout mice. PLoS One 10:e0120916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoi F, Oleas J, Xing H, Liu Y, Dexter KM, Misztal C, Gerard M, Efimenko I, Lynch P, Villanueva M, Alsina R, Krishnaswamy S, Vaillancourt DE, Li Y (2020) Decreased number of striatal cholinergic interneurons and motor deficits in dopamine receptor 2-expressing-cell-specific Dyt1 conditional knockout mice. Neurobiol Dis 134:104638. [DOI] [PMC free article] [PubMed] [Google Scholar]