

Abstract
Treprostinil is a prostacyclin analogue that targets multiple cellular receptors to treat pulmonary arterial hypertension (PAH). In certain scenarios, patients may require aggressive treprostinil titration. Several studies have demonstrated that higher doses of treprostinil lead to greater clinical benefit. Data supports successful transitions from parenteral to oral treprostinil; however, administration routes, transition duration, and transition setting vary in the real‐world. The EXPEDITE clinical trial (NCT03497689) prospectively studied whether rapid parenteral treprostinil induction can be used to achieve high doses of oral treprostinil (total daily dose: ≥12 mg) in prostacyclin naïve PAH patients. Parenteral prostacyclin induction may be more appropriate for patients who need to reach therapeutic dosing more urgently than longer titration durations reported with conventional de novo oral treprostinil initiation. This summary provides strategies utilized in EXPEDITE. Parenteral treprostinil was initiated at 2 ng/kg/min intravenously or subcutaneously; clinicians determined the frequency and dose increment of up‐titration. Two distinct transition schedules from parenteral to oral treprostinil were employed: rapid cross‐titration in an inpatient setting (median: 2 days) or gradual cross‐titration in an outpatient setting (median: 5 days). Patient status was closely monitored after transition; oral treprostinil dose was titrated to clinical effect and tolerability. Factors considered when individualizing dosing strategies included parenteral and oral treprostinil target doses, nursing support, patient education, medication counseling and adverse events management. EXPEDITE demonstrated the time to a therapeutic dose of oral treprostinil is significantly shorter when utilizing a short‐term parenteral induction strategy and may be suitable for patients requiring aggressive titration of oral treprostinil.
Keywords: cross‐titration, prostacyclin, pulmonary arterial hypertension
INTRODUCTION
Pulmonary arterial hypertension (PAH) is a hemodynamic abnormality marked by an elevation in pulmonary arterial pressure and pulmonary vascular resistance (PVR). 1 Elevations in PVR cause an increase in right ventricular (RV) afterload, which may impair RV function and can lead to progressive limiting symptoms and death. 1 Currently approved therapies for the management of PAH target three pathways: nitric oxide, endothelin, and prostacyclin pathways. 2 Treprostinil is a prostacyclin analogue that binds to prostaglandin IP, DP1, and EP2 receptors, which inhibits platelet aggregation and elicits vasodilatory and antiproliferative effects. 3 , 4 , 5 , 6 Treprostinil can be administered subcutaneously (SC), intravenously (IV), orally, or by inhalation. 6 , 7 , 8
Oral treprostinil dose is individualized to clinical effect and tolerability and does not have a labelled maximum dose. 6 Higher doses are associated with greater improvements in clinical parameters (6‐minute walk distance [6MWD], N‐terminal pro‐brain natriuretic peptide [NT‐proBNP], WHO functional class [FC], and decreased risk of hospitalization). 9 , 10 , 11 , 12 , 13 In certain clinical scenarios, patients may require aggressive titration of oral treprostinil. Open‐label and uncontrolled studies have previously demonstrated the ability to aggressively titrate parenteral prostacyclin, 14 , 15 , 16 where doses of 23−139 ng/kg/min have been attained at 12 weeks, 15 , 16 , 17 equivalent to an oral treprostinil total daily dose (TDD) of 11.6−70.1 mg in a 70‐kg patient. 6 Notably, patients with PAH with prior treprostinil exposure tolerate higher doses of oral treprostinil as compared to patients without prior exposure (TDD of 9.0 mg vs. 7.5 mg at 6 months, respectively).
Clinical data supports successful transitions from parenteral to oral treprostinil. 6 , 19 Most notably, an open‐label, multi‐center study in hemodynamically stable patients (n = 33) on long‐term parenteral treprostinil transitioned from parenteral treprostinil (median of 57 ng/min/kg) to oral treprostinil over 5 days in an inpatient setting. Of the 33 patients, 31 patients successfully transitioned and completed the 24‐week study while maintaining their hemodynamic statuses. 20 Real‐world data describe these transitions; however, routes of administration, transition duration, and transition setting vary. 18 , 21 , 22 , 23
Later, a case report in a prostacyclin naïve patient introduced the concept for rapid parenteral induction initiated while inpatient over approximately 1 week before treatment with oral treprostinil. 24 Parenteral induction occurred over 7 days and reached parenteral treprostinil dose of 42 ng/kg/min before transitioning to oral treprostinil dose of 24 mg TDD. A case series of 10 higher risk patients with a mean (SD) baseline REVEAL 2.0 risk score of 10 (2) using the same strategy reported transitioning to a mean (SD) oral treprostinil of 4.7 (1.6) mg TID over 3 days after rapid parenteral induction. 25 Despite previous publications and case reports detailing successful transitions from parenteral treprostinil to oral treprostinil, there is limited guidance detailing how to utilize parenteral treprostinil induction to reach clinically efficacious doses of oral treprostinil.
The EXPEDITE study (NCT03497689) was designed to prospectively evaluate whether rapid parenteral induction over 2−8 weeks can be used to achieve high doses of oral treprostinil. The aim of this report is (1) to introduce a proposed induction strategy and (2) to examine the safety and tolerability of the transition for patients who prefer the convenience of oral prostacyclin therapy or for those who may not be candidates for long‐term parenteral treprostinil therapy. 20 This report utilizes prospectively collected data to summarize the induction and transition strategies utilized in the EXPEDITE study.
INDUCTION AND TRANSITION STRATEGIES
EXPEDITE study design
The EXPEDITE clinical trial was a 16‐week, open‐label, multicenter, uncontrolled study in 35 participants with PAH; 29 participants were included in the per‐protocol population and used for all data presentation herein (Table 1). Study visits included screening, baseline (before the initiation of parenteral treprostinil), and Weeks 2, 4, 8, 12, and 16. Additionally, a post‐transition study visit occurred 1−2 weeks after the transition visit. The parenteral treprostinil induction phase occurred for 2−8 weeks; the remaining time in the study was the cross‐titration and oral treprostinil optimization phases (Figure 1).
Table 1.
Baseline Characteristics of Per‐Protocol Patient Population in the EXPEDITE study.
| Patient characteristics | Total |
|---|---|
| n = 29 | |
| Age—years, median (range) | 56 (35−72) |
| Female sex—no. (%) | 18 (62.1) |
| Weight at baseline—kg, median (range) | 87 (56−152) |
| Race—no. (%) | |
| White | 23 (79.3) |
| Black or African American | 4 (13.8) |
| American Indian or Alaska Native | 1 (3.4) |
| Other | 1 (3.4) |
| Ethnicity—no. (%) | |
| Not Hispanic or Latino | 26 (89.7) |
| Hispanic or Latino | 3 (10.3) |
| Time since PAH diagnosis—weeks, median (range) | 33 (1−274) |
| PAH classification—no. (%) | |
| Idiopathic | 12 (41.4) |
| Associated with connective tissue disease | 9 (31.0) |
| Associated with drug or toxin exposure | 5 (17.2) |
| HIV‐associated | 1 (3.4) |
| Heritable | 1 (3.4) |
| Other | 1 (3.4) |
| PAH background medications—no (%) | |
| None | 6 (20.7) |
| ERA only | 1 (3.4) |
| PDE‐5i/sGCS only | 10 (34.5) |
| ERA + PDE‐5i/sGCS | 12 (41.4) |
| WHO functional class at baseline—no (%) | |
| II | 15 (51.7) |
| III | 14 (48.3) |
| NT‐proBNP—ng/L, median, IQR | 415 (195−1061) |
| 6MWD—m, median, IQR | 363 (288−426) |
| eGFR—no (%) | |
| ≥60 mL/min/1.73m2 | 22 (75.9) |
| <60 mL/min/1.73m2 | 3 (10.3) |
| Not available | 4 (13.8) |
| REVEAL 2.0 risk score—median (IQR) | 7 (5−8) |
| Echocardiographic parameters | |
| TAPSE (mm)—median (IQR) | 17.0 (12.4−21.2) |
| RAA (cm2)—median (IQR) | 20.3 (16.8−28.9) |
| RV/LV ratio, diastole—median (IQR) | 0.8 (0.6−0.9) |
| Pericardial effusion—no (%) | |
| Mild, moderate, or severe | 1 (3.4) |
| Trace or none | 28 (96.6) |
| Hemodynamics (Right Heart Catheterization)a—median (IQR) | |
| mRAP (mmHg) | 7.0 (4.0−10.0) |
| mPAP (mmHg) | 53 (47−58) |
| PAWP (mmHg) | 10 (7−13) |
| PVR (Wood units) | 9.6 (7.5−11.6) |
| Cardiac index (L/min/m2) | 2.4 (2.1−2.7) |
Abbreviations: 6MWD, 6‐minute walk distance; eGFR, estimated glomerular filtration rate; ERA, endothelin receptor antagonists; mPAP, mean pulmonary arterial pressure; mRAP, mean right atrial pressure; NT‐proBNP, N‐terminal pro‐brain natriuretic peptide; PAH, pulmonary arterial hypertension; PAWP, pulmonary artery wedge pressure; PDE‐5i, phosphodiesterase‐5 inhibitor; PVR, pulmonary vascular resistance; RAA, right atrial area; RV/LV ratio, right ventricular to left ventricular ratio; sGCS, soluble guanylate cyclase stimulator; TAPSE, tricuspid annular plane systolic excursion; WHO, World Health Organization.
Baseline hemodynamics were collected in patients up to 180 days screening.
Figure 1.
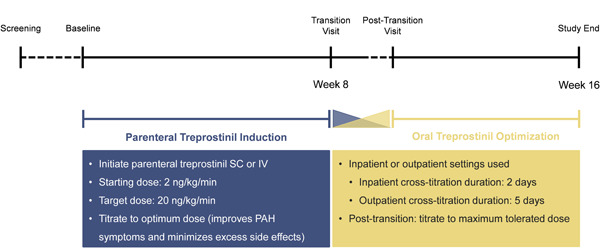
Study Schema. Representative schematic of patients with PAH in the EXPEDITE study who initiated parenteral treprostinil and transitioned to oral treprostinil at Week 8. Transition could occur at Week 2, 4, or 8. The mean (SD) duration of parenteral treprostinil exposure, which includes the parenteral treprostinil induction and the cross‐titration phases, was 55 (13) days. Post‐transition visit occurred 1−2 weeks after the transition visit. The mean (SD) duration of oral treprostinil exposure, which includes the cross‐titration and oral treprostinil optimization phases, was 64 (16) days.
The primary objective of the EXPEDITE study was the percentage of participants who obtained 12 mg TDD (or TDD of 0.171 mg/kg for participants < 70 kg) or higher of oral treprostinil after 16 weeks. This dose goal was based on the hypothesis that parenteral treprostinil induction would allow participants to double the dose of oral treprostinil of 6 mg TDD, which was the dose reached at 16 weeks in randomized placebo‐controlled trials and real‐world evidence studies. 18 , 26 , 27 Secondary objectives included changes in 6MWD, WHO FC, NT‐proBNP, risk, 28 patient‐reported outcomes (treatment satisfaction questionnaire for medication), 29 and emPHasis‐10 questionnaire 30 after 16 weeks and will be discussed in future publications. Adverse events (AE) were collected in the parenteral treprostinil induction, cross‐titration, and oral optimization phases; data analyses were descriptive with no formal statistical testing. Participants were eligible if they had a REVEAL 2.0 risk score of 9 or less and did not receive prostacyclin therapy within 28 days of baseline. As this study focused on the addition of a new therapy rather than the replacement of one therapy by another therapy, participants were eligible regardless of the number of PAH background therapies (0, 1, or 2) they were receiving at baseline. Patients were excluded if they had uncontrolled or severe hypertension.
Thirty‐five participants started parenteral treprostinil and were included in safety analyses. Six participants had major protocol deviations and were excluded from the subsequent dosing analyses. Despite initiating parenteral treprostinil, it was discovered during clinical monitoring that five participants had not actually met eligibility criteria. These five participants should have been considered screen failures, and, subsequently, excluded from the study. One patient voluntarily dislodged their catheter and interrupted parenteral treprostinil infusion for greater than 24 h; they subsequently withdrew consent to participate in the study. The trial protocol was approved by the institutional review board at each participating site. The trial was conducted in accordance with Good Clinical Practice guidelines, and all participants provided written informed consent to participate.
Parenteral treprostinil induction
Following completion of baseline assessments, clinicians were instructed to initiate parenteral treprostinil at 2 ng/kg/min SC or IV (central venous catheter [CVC] or peripherally inserted catheter [PICC]). Clinicians were able to choose whether to initiate therapy in an inpatient or outpatient setting (Figure 1). Investigators chose the frequency and dose increments for up‐titration and were instructed to utilize parenteral therapy to achieve an optimal treprostinil dose; titration was individualized and optimized in each participant to a dose that improved PAH symptomology. There was no maximum parenteral treprostinil dose.
Inpatient initiation of parenteral treprostinil (28 out of 29 participants) was preferred over outpatient initiation and may have facilitated faster titration and closer AE management (Supporting Information: Table S1). IV administration (19 of 29 participants) was more common than SC administration. Six of the 19 participants subsequently transitioned to SC for outpatient administration of treprostinil. Of those who were initiated on IV treprostinil, PICC (18 of 19 participants) was predominantly used, as compared to CVC administration. IV administration may have been more common than SC administration to avoid SC site pain and reaction. Participants in this study were generally titrated more rapidly than parenteral treprostinil patients in real‐world practice (Figure 2). 31
Figure 2.
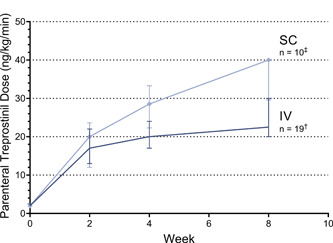
Parenteral Treprostinil Induction. Median (Q1, Q3) parenteral treprostinil dose during EXPEDITE study at Baseline, Week 2, 4, and 8. Clinicians were instructed to initiate parenteral treprostinil at 2 ng/kg/min SC or IV at the Baseline visit in an inpatient or outpatient setting. Clinicians chose the frequency and dose increments for up‐titration with a goal to improve PAH symptomology. In the EXPEDITE study, SC up‐titration of parenteral treprostinil results in higher doses at a faster rate than IV up‐titration. *IV = intravenous, SC = subcutaneous † One patient transitioned at Week 2, four patients transitioned at Week 4, and 14 patients transitioned at Week 8. ‡ One patient transitioned at Week 4, and nine patients transitioned at Week 8. No patients transitioned at Week 2.
The mean (SD) time of parenteral treprostinil exposure was 55 (13) days. The median (range) dose immediately before transition was 24 (6−40) ng/kg/min. Notably, participants initiated on SC treprostinil were able to achieve a higher median dose of 40 ng/kg/min compared to 21 ng/kg/min for participants initiated on IV treprostinil. The higher doses reached by participants on SC treprostinil was likely due to the fact that six of ten participants were enrolled at a single center. This center aimed to reach parenteral treprostinil doses of 40 ng/kg/min at 8 weeks. However, titration was less rapid than previously reported case reports describing inpatient parenteral treprostinil induction over approximately 1 week with rapid transition to oral treprostinil. 24 , 25 In the EXPEDITE study, the maximum parenteral doses achieved during initiation and up‐titration were similar between participants who began transition to oral treprostinil at Week 4 compared to Week 8, suggesting 4 weeks or less may be an adequate time to up‐titrate patients to therapeutic SC or IV doses.
Cross‐titration from parenteral (SC/IV) treprostinil to oral treprostinil
During the transition visit (Week 2, 4, or 8), participants transitioned if they achieved a minimum parenteral treprostinil dose of 20 ng/kg/min. Before transition, 6MWD, WHO FC, NT‐proBNP, and echocardiography parameters were assessed. At the transition visit, the median (IQR) of 6MWD, NT‐proBNP, tricuspid annular plane systolic excursion (TAPSE), and right atrial area for the per‐protocol population were 377 (318, 453) m, 186 (110, 724) ng/L, 18.1 (14.8, 20.9) mm, and 19.3 (16.0, 26.7) cm2, respectively. Of the 29 patients, 76% improved WHO FC, 21% maintained, and 3% worsened at their transition visit. In general, the clinical variables outlined above improved from baseline to transition (Table 1). The investigator used their discretion in determining whether a participant was suitable for transition; no formal guidance on disease stability or required thresholds for clinical parameters before transition were outlined in the protocol. All participants still receiving parenteral treprostinil at Week 8 began transitioning to oral treprostinil regardless of their parenteral treprostinil dose unless deemed unsuitable for transition by their clinician. Transition from parenteral to oral treprostinil could occur over 1−21 days in an inpatient or outpatient setting. Clinicians were instructed to reverse or stop the transition if significant signs or symptoms of PAH or serious safety concerns occurred. Transitioning was conducted via a cross‐tapering method. During transition, oral treprostinil was administered at approximately the same time in which the parenteral treprostinil dose was decreased. The target daily dose of oral treprostinil at the end of transition was calculated using the weight‐based dosing conversion in the oral treprostinil prescribing information 6 (Figure 3, Supporting Information: Table S2).
Figure 3.

Dosing Conversion Steps (with an example). Use the following formula to estimate a target total daily dose of oral treprostinil in mg using a patient's dose of intravenous (IV)/subcutaneous (SC) treprostinil (in ng/kg/min) and weight (in kg)(6).
One participant transitioned at Week 2, five participants transitioned at Week 4, and 23 participants transitioned at Week 8 (Supporting Information: Table S3). Most participants (16 of 29) transitioned in an outpatient setting over a median (range) of 5 (4−14) days with a median (range) parenteral treprostinil dose of 22 (6−40) ng/kg/min at the start of transition. A representative schedule of an outpatient transition is presented in Figure 4. The remaining 13 participants transitioned in an inpatient setting over a median (range) of 2 (1−2) days, with a median (range) parenteral dose of 30 (12−40) ng/kg/min at the start of transition. Figure 5 depicts a representative schedule for participants who transitioned in an inpatient setting. Supporting Information: Table S2 provides example cross‐titration schedules based on patient weight, transition duration, parenteral treprostinil dose, and target oral treprostinil dose.
Figure 4.
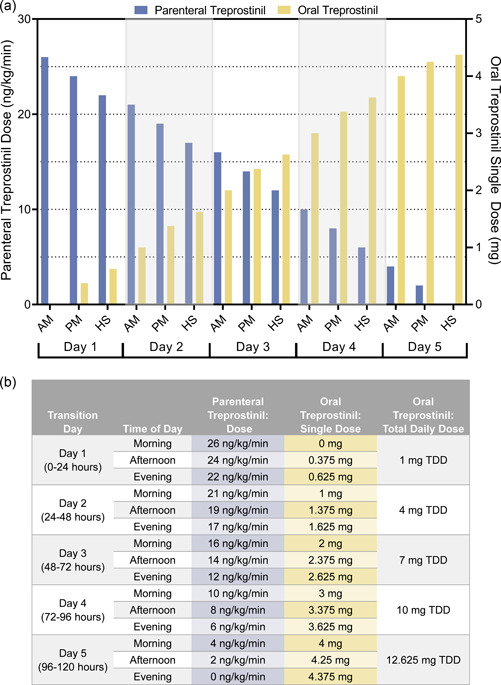
Representative Outpatient Transition Dosing Schedule. For outpatient patients who transitioned from parenteral treprostinil to oral treprostinil, the median parenteral treprostinil dose at transition was 22 ng/kg/min. Outpatient patients gradually discontinued parenteral treprostinil over 5 days while increasing oral treprostinil to the target dosage. Here, the target total daily dose of oral treprostinil is 12.625 mg for a patient weighing 70 kg, based on oral treprostinil total daily dose (mg) = 0.0072 x parenteral treprostinil daily dose (ng/kg/min) x weight (kg). *AM, morning, PM, afternoon, HS, evening, TDD, total daily dose.
Figure 5.

Representative Inpatient Transition Dosing Schedule. For inpatient patients who transitioned from parenteral treprostinil to oral treprostinil, the median parenteral treprostinil dose at transition was 30 ng/kg/min. Inpatient patients gradually discontinued parenteral treprostinil over 1‐2 days while increasing oral treprostinil to the target dosage. Here, the target dose of oral treprostinil is 6 mg TDD for a patient weighing 70 kg, based on oral treprostinil total daily dose (mg) = 0.0072 x parenteral treprostinil daily dose (ng/kg/min) x weight (kg). *AM, morning; PM, afternoon; HS, evening, TDD, total daily dose.
Oral treprostinil optimization
At the post transition visit 1−2 weeks after initiating transition, participants returned to the study site clinic to assess AEs, WHO FC, 6MWD, concomitant medications, and vital signs. The mean (SD) TDD at the post‐transition visit was 16.6 (8.1) mg. Clinicians were encouraged to continue oral treprostinil titration in an outpatient setting by increasing the dose by 0.125 mg TID every 3−4 days as tolerated with the goal of achieving a maximum tolerated dose by Week 16. From the post‐transition visit to Week 16, many participants (13 of 28) continued to up‐titrate oral treprostinil after their post‐transition visit, while nine participants maintained and six participants decreased their oral treprostinil dose. At the end of the EXPEDITE study, 79.3% of participants achieved an oral treprostinil dose of at least 12 mg TDD at Week 16; the mean (SD) TDD was 16.4 (7.5) mg at Week 16. The mean (SD) oral treprostinil exposure time was 64 (16) days throughout the entire study.
Safety and adverse event management
In the EXPEDITE study, 28 of the 29 participants remained on oral treprostinil through study end at Week 16, and one discontinued early due to death. Notably, no participants switched back to parenteral treprostinil after receiving oral treprostinil during the EXPEDITE study. In the safety population (n = 35), the most common AEs were headache (86%), nausea (71%), diarrhea (60%), flushing (57%), jaw pain (51%), extremity pain (43%), and vomiting (43%). The number of participants experiencing prostanoid‐related AEs was similar when participants received parenteral treprostinil versus when receiving oral treprostinil, except for an increase in flushing and decrease in headache while receiving oral treprostinil (Table 2). While participants were on parenteral treprostinil, 69%, 55%, and 34% of participants in the per‐protocol population (n = 29) received ondansetron, acetaminophen, and loperamide for prostacyclin‐related nausea/vomiting, headache, and diarrhea, respectively. When on oral treprostinil, 48%, 38%, and 38% of participants used ondansetron, acetaminophen, and loperamide, respectively. Overall, the use of all concomitant medications decreased after transitioning from parenteral to oral treprostinil, likely due to the majority of titration occurring during the parenteral treprostinil induction phase. The concomitant medications used in this study were the same as the three most commonly used in the ADAPT registry and consistent with expert recommendations from two independent Delphi consensus statements on the AE management for oral treprostinil. 32 , 33 , 34
Table 2.
Prostanoid‐related Adverse Events. The number and associated percentage of patients who experienced an AE during each phase of the study.
| Parenteral Treprostinil Phase | Oral Treprostinil Phase | ||
|---|---|---|---|
| (Before Transition) | Transition Phase | (After Transition) | |
| n = 35 | n = 33 | n = 32 | |
| Adverse events, n (%) | |||
| Headache | 28 (80) | 19 (58) | 19 (59) |
| Vomiting | 13 (37) | 5 (15) | 9 (28) |
| Nausea | 24 (69) | 21 (64) | 21 (66) |
| Extremity pain | 12 (34) | 10 (30) | 11 (34) |
| Diarrhea | 18 (51) | 17 (52) | 17 (53) |
| Jaw pain | 16 (46) | 15 (46) | 16 (50) |
| Flushing | 10 (29) | 10 (30) | 17 (53) |
Compared to data from the 16‐week FREEDOM‐C and FREEDOM‐C2 studies, 26 , 27 AEs during the oral treprostinil phase were qualitatively similar despite higher doses reached during EXPEDITE. Ten participants experienced at least one serious adverse event (SAE) with a total of 16 SAEs among the 10 participants. For the one participant who died, the cause of death was worsening right heart failure. This participant with connective tissue disease‐associated PAH was enrolled with a REVEAL 2.0 risk score of 9 (6MWD 370 m, WHO FC II, NT‐proBNP 2361 ng/L, and TAPSE 7.7 mm). The participant began transitioning from a parenteral treprostinil dose of 21 ng/kg/min at Week 4, with a corresponding decline in clinical parameters (6MWD 294 m, WHO FC III, NT‐proBNP 2661 ng/kg/min, and TAPSE 9.5 mm). The participant ultimately perished from right heart failure approximately three months after transitioning to oral treprostinil. The Investigator deemed the death unrelated to oral treprostinil or the transition. However, the long‐term safety of this induction strategy has yet to be established. Two participants discontinued oral treprostinil early due to treatment‐emergent AEs (sepsis and nausea) considered unrelated to study drugs.
CONCLUSION
The EXPEDITE study supports utilizing parenteral treprostinil induction therapy to rapidly reach therapeutic doses of oral treprostinil in prostacyclin naïve patients. The time to reach a therapeutic dose of oral treprostinil following parenteral treprostinil induction was shorter compared to historical de novo oral treprostinil starts. 18 Historically, higher doses of oral treprostinil, in particular doses greater than 9 mg TDD, have been associated with greater clinical benefits. 9 , 10 , 11 , 12
IV administration, predominantly via a PICC line, in an inpatient setting was preferred for parenteral treprostinil induction, though outpatient SC initiation and up‐titration was also used. Similar parenteral treprostinil doses were achieved regardless of duration of induction. Before transition, clinical status was assessed to determine if transition to oral treprostinil is appropriate. Two distinct transition strategies were employed: rapid cross‐titration in an inpatient setting or gradual cross‐titration in an outpatient setting. Patient status was closely monitored after transition, and oral treprostinil dose was titrated to clinical effect and tolerability. Further analyses on efficacy, safety, and quality of life after utilizing the parenteral treprostinil induction strategy will be discussed in future publications.
De novo initiations of oral treprostinil may be appropriate in patients who have adequate time to titrate therapy or those unwilling or unable to manage an infusion pump. Right‐heart imaging should be considered when choosing the appropriate dosing strategy. 35 Other factors to consider when planning individualized dosing strategies are the target doses for parenteral and oral treprostinil, nursing support, patient education and medication counseling, and AE management. A shorter (e.g., 1 week) inpatient admission with planned discharge on oral treprostinil may be considered to quickly attain the 3 mg (9 mg TDD) target dose that has been previously associated with better outcomes. 10 There are several limitations to our study: this was an open‐label study, the sample size was relatively small, the study was of a relatively short duration, and there was no control arm. This open‐label study did not employ randomization, so measures to minimize bias and procedures relating to randomization could not be used. This treatment strategy may be limited to motivated patients and experienced multidisciplinary health care teams. Finally, the long‐term safety and outcomes associated with this treatment approach need to be further explored. The EXPEDITE study induction strategy may be appropriate for patients requiring an aggressive titration of oral treprostinil.
AUTHOR CONTRIBUTIONS
This study was sponsored, designed, and executed by United Therapeutics. Benjamin Wu, Stephanie Hwang, Scott Seaman, and Meredith Broderick led data analyses, wrote the initial draft of the manuscript, and revised the manuscript throughout its development. John F. Kingrey, Chad E. Miller, Veronica Franco, Jimmy S. Smith, Ronald Zolty, Ronald J. Oudiz, Jean M. Elwing, Jessica H. Huston, Lana Melendres‐Groves, Ashwin Ravichandran, and Vijay Balasubramanian actively recruited, treated participants in the study, interpreted data, and revised the manuscript. Franck F. Rahaghi provided data interpretation and reviewed and revised the manuscript. All authors approved the decision to submit the manuscript for publication.
CONFLICT OF INTEREST STATEMENT
J. F. Kingrey is part of the speaker bureau for United Therapeutics, Bayer pharmaceuticals, and Janssen Pharmaceuticals, attends advisory boards for United Therapeutics, Bayer, and Janssen, consults for United Therapeutics, Bayer, Janssen, and conducts research for United Therapeutics, Janssen, Acceleron, Gossamer Bio. C. E. Miller is part of the speaker bureau for United Therapeutics, Janssen, and Bayer, attends advisory boards for United Therapeutics and Janssen, consults for United Therapeutics, and conducts clinical research for United Therapeutics, Janssen, Bayer, and Insmed. J. S. Smith serves as a speaker for and has received research funds from Bayer and United Therapeutics. V. Franco has served on advisory boards for United Therapeutics and has supported research for United Therapeutics, Merck, Acceleron, Johnson and Johnson, Respira, Aerovate, Cereno, and Abbott. R. Zolty has received payments for consulting on behalf of United Therapeutics, Bayer, and Janssen/Johnson and Johnson. R. J. Oudiz has received grants from Acceleron/Merck, Gossamer Bio, Insmed, Janssen, and United Therapeutics and honoraria/speaker fees from Merck, Janssen, and United Therapeutics. J. M. Elwing consults for United Therapeutics, Altavant, Aerovate, Bayer, Gossamer Bio, Liquida, and Merck and has received research support and grants from Janssen, United Therapeutics, Liquidia, Phase Bio, Gossamer Bio, Bayer, Acceleron, Altavant, Aerovate, Tenax, and Pharmosa. J. H. Huston has conducted clinical trials for Acceleron, CardiolRx, and Aadi Bioscience. L. Melendres‐Groves has received honoraria and/or fees for consultancy and advisory committees for United Therapeutics Corporation outside of submitted works. A. Ravichandran reports personal fees from United Therapeutics, Bayer, and Actelion outside the submitted work. V. Balasubramanian serves as a consultant for United Therapeutics. B. Wu, S. Hwang, S. Seaman, and M. Broderick are paid employees of United Therapeutics. F. F. Rahaghi has consulted for United Therapeutics, Janssen PH, Merck, and Altavant; served as a speaker for United Therapeutics, Janssen PH, and Bayer; and received research funding from Janssen, Bayer, Merck, Gossamer, and Bellerophon.
ETHICS STATEMENT
The trial protocol was approved by the institutional review board at each participating site. The trial was conducted in accordance with Good Clinical Practice guidelines, and all participants provided written informed consent to participate.
Supporting information
Supporting information.
ACKNOWLEDGMENTS
The authors would like to thank Eric Shen for clinical conduct and oversight as well as Dana Cella for statistical support. This study was funded by United Therapeutics Corporation. Guarantor John F. Kingrey.
Kingrey JF, Miller CE, Franco V, Smith JS, Zolty R, Oudiz RJ, Elwing JM, Huston JH, Melendres‐Groves L, Ravichandran A, Balasubramanian V, Wu B, Hwang S, Seaman S, Broderick M, Rahaghi FF. Implementing the EXPEDITE parenteral induction protocol: Rapid parenteral treprostinil titration and transition to oral treprostinil. Pulm Circ. 2023;13:e12255. 10.1002/pul2.12255
REFERENCES
- 1. Humbert M, Lau EMT, Montani D, Jaïs X, Sitbon O, Simonneau G. Advances in therapeutic interventions for patients with pulmonary arterial hypertension. Circulation. 2014;130(24):2189–208. [DOI] [PubMed] [Google Scholar]
- 2. Hassoun PM. Pulmonary arterial hypertension. N Engl J Med. 2021;385(25):2361–76. [DOI] [PubMed] [Google Scholar]
- 3. Clapp LH, Gurung R. The mechanistic basis of prostacyclin and its stable analogues in pulmonary arterial hypertension: role of membrane versus nuclear receptors. Prostaglandins Other Lipid Mediat. 2015;120:56–71. [DOI] [PubMed] [Google Scholar]
- 4. Benyahia C, Boukais K, Gomez I, Silverstein A, Clapp L, Fabre A, Danel C, Leséche G, Longrois D, Norel X. A comparative study of PGI2 mimetics used clinically on the vasorelaxation of human pulmonary arteries and veins, role of the DP‐receptor. Prostaglandins Other Lipid Mediat. 2013;Dec 107:48–55. [DOI] [PubMed] [Google Scholar]
- 5. Whittle BJ, Silverstein AM, Mottola DM, Clapp LH. Binding and activity of the prostacyclin receptor (IP) agonists, treprostinil and iloprost, at human prostanoid receptors: treprostinil is a potent DP1 and EP2 agonist. Biochem Pharmacol. 2012;84(1):68–75. [DOI] [PubMed] [Google Scholar]
- 6. Orenitram (treprostinil) extended release tablets, US package insert, United Therapeutics Corporation; 2019. [Google Scholar]
- 7. Remodulin (treprostinil), US package insert, United Therapeutics Corporation; 2002. [Google Scholar]
- 8. Tyvaso (treprostinil), US package insert, United Therapeutics Corporation; 2009. [Google Scholar]
- 9. Ramani G, Cassady S, Shen E, Broderick M, Wasik A, Sui Q, Nelsen A. Novel dose–response analyses of treprostinil in pulmonary arterial hypertension and its effects on six‐minute walk distance and hospitalizations. Pulm Circ. 2020;10(3):2045894020923956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. White RJ, Grünig E, Jerjes‐Sanchez C, Bohns Meyer GM, Pulido T, Sepulveda P, Wang KY, Deng CQ, Grover R, Solum D, Ousmanou A, Tapson VF. Dose‐response relationship of oral treprostinil for secondary endpoints in the FREEDOM‐EV study. Euro Respir J. 2019;54(suppl 63):PA5462. [Google Scholar]
- 11. White RJ, Rao Y. Novel analysis of the oral treprostinil combination therapy trial data. Am J Respir Crit Care Med. 2016;193(12):1434–6. [DOI] [PubMed] [Google Scholar]
- 12. Kumar P, Arneson C, Laliberte K, Nelsen AC. Dose‐response relationship of oral treprostinil diolamine (TRE) in patients with pulmonary arterial hypertension (PAH). Am J Respir Crit Care Med. 2013;187:A3271. [Google Scholar]
- 13. White RJ, Jerjes‐Sanchez C, Bohns Meyer GM, Pulido T, Sepulveda P, Wang KY, Grünig E, Hiremath S, Yu Z, Gangcheng Z, Yip WLJ, Zhang S, Khan A, Deng CQ, Grover R, Tapson VF; FREEDOM‐EV Investigators . Combination Therapy with Oral Treprostinil for Pulmonary Arterial Hypertension. A Double‐Blind Placebo‐controlled Clinical Trial. Am J Respir Crit Care Med. 2020;201(6):707–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sitbon O, Vonk Noordegraaf A. Epoprostenol and pulmonary arterial hypertension: 20 years of clinical experience. Eur Respir Rev. 2017;26(143):160055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Grünig E, Benjamin N, Lange TJ, Krueger U, Klose H, Neurohr C, Wilkens H, Halank M, Seyfarth HJ, Held M, Traube A, Pernow M, Grover ER, Egenlauf B, Gerhardt F, Viethen T, Rosenkranz S. Safety, tolerability and clinical effects of a rapid dose titration of subcutaneous treprostinil therapy in pulmonary arterial hypertension: a prospective multi‐centre trial. Respiration. 2016;92(6):362–70. [DOI] [PubMed] [Google Scholar]
- 16. Hansen L, Rischard F, Knoper S. Rapid inpatient titration of intravenous treprostinil for severe pulmonary arterial hypertension. J Heart Lung Transplant. 2014;33(4):S309. [Google Scholar]
- 17. Sitbon O, Manes A, Jais X, Pallazini M, Humbert M, Presotto L, Paillette L, Zaccardelli D, Davis G, Jeffs R, Simonneau G, Galie N. Rapid switch from intravenous epoprostenol to intravenous treprostinil in patients with pulmonary arterial hypertension. J Cardiovasc Pharmacol. 2007;49(1):1–5. [DOI] [PubMed] [Google Scholar]
- 18. Balasubramanian VP, Messick CR, Broderick M, Nelsen AC. Dosing characteristics of oral treprostinil in real‐world clinical practice. Pulm Circ. 2018; 8(2):1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ivy DD, Feinstein JA, Yung D, Mullen MP, Kirkpatrick EC, Hirsch R, Austin ED, Fineman J, Truong U, Solum D, Deng CQ, Hopper RK. Oral treprostinil in transition or as add‐on therapy in pediatric pulmonary arterial hypertension. Pulm Circ. 2019;9(3):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chakinala MM, Feldman JP, Rischard F, Mathier M, Broderick M, Leedom N, Laliberte K, White RJ. Transition from parenteral to oral treprostinil in pulmonary arterial hypertension. J Heart Lung Transplant. 2017;Feb 36(2):193–201. [DOI] [PubMed] [Google Scholar]
- 21. Sahay S, Ravichandran A, Parikh K, Gordon K, Broderick M, Lee D, Swisher J. Real‐world transitions from parenteral, inhaled, and oral prostacyclin‐class therapies to oral treprostinil: Interim data from the ADAPT registry. Am J Respir Crit Care Med. 2020;201:A3811. [Google Scholar]
- 22. Maestas T, Hansen LM, Vanderpool RR, Desai AA, Airhart S, Knapp SM, Cohen A, Feldman J, Rischard FP. Right ventricular afterload predicts long‐term transition from parenteral to oral treprostinil in pulmonary arterial hypertension. Pulm Circ. 2018;8(4):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jarrett H, Jonnalagadda AK, Liu SD, Bagnola AJ, Lewis D, Shlobin OA, Barnett CF. Rapid transition from parenteral to oral treprostinil in pulmonary hypertension is feasible and safe: a retrospective cohort study. J Heart Lung Transplant. 2018;37(4):S494. [Google Scholar]
- 24. Gleason JB, Dolan J, Piran P, Rahaghi FF. The rapid initiation, titration, and transition from intravenous to oral treprostinil in a patient with severe pulmonary arterial hypertension. Case Rep Pulmonol. 2015;e498981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chin JA, Kaur J, Mehta JP, Rahaghi FF. Rapid initiation, titration, and transition from IV to oral treprostinil: A retrospective study. Am J Respir Crit Care Med. 2020;201:A3823. [Google Scholar]
- 26. Tapson VF, Torres F, Kermeen F, Keogh AM, Allen RP, Frantz RP, Badesch DB, Frost AE, Shapiro SM, Laliberte K, Sigman J, Arneson C, Galiè N. Oral treprostinil for the treatment of pulmonary arterial hypertension in patients on background endothelin receptor antagonist and/or phosphodiesterase type 5 inhibitor therapy (The FREEDOM‐C Study). Chest. 2012;142(6):1383–90. [DOI] [PubMed] [Google Scholar]
- 27. Tapson VF, Jing ZC, Xu KF, Pan L, Feldman J, Kiely DG, Kotlyar E, McSwain CS, Laliberte K, Arneson C, Rubin LJ. Oral treprostinil for the treatment of pulmonary arterial hypertension in patients receiving background endothelin receptor antagonist and phosphodiesterase type 5 inhibitor therapy (The FREEDOM‐C2 Study). Chest. 2013;144(3):952–8. [DOI] [PubMed] [Google Scholar]
- 28. Galiè N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, Simonneau G, Peacock A, Noordegraaf AV, Beghetti M, Ghofrani A, Gomez Sanchez MA, Hansmann G, Klepetko W, Lancellotti P, Matucci M, McDonagh T, Pierard LA, Trindade PT, Zompatori M, Hoeper M, ESC Scientific Document Group . 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension: the joint task force for the diagnosis and treatment of pulmonary hypertension of the european society of cardiology (ESC) and the european respiratory society (ERS): endorsed by: association for european paediatric and congenital cardiology (AEPC), international society for heart and lung transplantation (ISHLT). Eur Heart J. 2016;37(1):67–119. [DOI] [PubMed] [Google Scholar]
- 29. Atkinson MJ, Sinha A, Hass SL, Colman SS, Kumar RN, Brod M, Rowland CR. Validation of a general measure of treatment satisfaction, the treatment satisfaction questionnaire for medication (TSQM), using a national panel study of chronic disease. Health Qual Life Outcomes. 2004;2:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yorke J, Corris P, Gaine S, Gibbs JSR, Kiely DG, Harries C, Pollock V, Armstrong I. emPHasis‐10: development of a health‐related quality of life measure in pulmonary hypertension. Eur Respir J. 2014;43(4):1106–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Balasubramanian VP, Safdar Z, Sketch MR, Broderick M, Nelsen AC, Lee D, Melendres‐Groves L. Real‐world dosing characteristics and utilization of parenteral treprostinil in the outpatient setting. Pulm Circ. 2022;12:e12016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kingrey J, Swisher J, Ravichandran A, Gordon J, Gordon K, Broderick M, Carrell G, Lee D, Sahay S. Interim data from the ADAPT registry: Patient‐Reported Real‐World tolerability and management of adverse events with oral treprostinil. Chest. 2020;158(4):A2169–A2170. [Google Scholar]
- 33. Rahaghi FF, Feldman JP, Allen RP, Tapson V, Safdar Z, Balasubramanian VP, Shapiro S, Mathier MA, Elwing JM, Chakinala MM, White RJ. Recommendations for the use of oral treprostinil in clinical practice: a Delphi consensus project pulmonary circulation. Pulm Circ. 2017;7(1):167–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Brewer J, Anguiano RH, Awdish RL, Coons J, Kimber A, Morrison M, Paulus S, Schmit A, Sexparth F, Swetz K, Verlinden N. An expert panel delphi consensus statement on appropriate patient selection and management of adverse events in patients receiving oral treprostinil for the treatment of pulmonary arterial hypertension. Am J Respir Crit Care Med. 2021;203:A3618. [Google Scholar]
- 35. Mercurio V, Hassan HJ, Naranjo M, Cuomo A, Mazurek JA, Forfia PR, Balasubramanian A, Simpson CE, Damico RL, Kolb TM, Mathai SC, Hsu S, Mukherjee M, Hassoun PM. Risk stratification of patients with pulmonary arterial hypertension: the role of echocardiography. J Clin Med. 2022;11(14):4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.