

Abstract
Intravitreal anti-vascular endothelial growth factor (VEGF) drugs have revolutionized the treatment of neovascular age-related macular degeneration (NVAMD). However, many patients suffer from incomplete response to anti-VEGF therapy (IRT), which is defined as (1) persistent (plasma) fluid exudation; (2) unresolved or new hemorrhage; (3) progressive lesion fibrosis; and/or (4) suboptimal vision recovery. The first three of these collectively comprise the problem of persistent disease activity (PDA) in spite of anti-VEGF therapy. Meanwhile, the problem of suboptimal vision recovery (SVR) is defined as a failure to achieve excellent functional visual acuity of 20/40 or better in spite of sufficient anti-VEGF treatment. Thus, incomplete response to anti-VEGF therapy, and specifically PDA and SVR, represent significant clinical unmet needs.
In this review, we will explore PDA and SVR in NVAMD, characterizing the clinical manifestations and exploring the pathobiology of each. We will demonstrate that PDA occurs most frequently in NVAMD patients who develop high-flow CNV lesions with arteriolarization, in contrast to patients with capillary CNV who are highly responsive to anti-VEGF therapy. We will review investigations of experimental CNV and demonstrate that both types of CNV can be modeled in mice. We will present and consider a provocative hypothesis: formation of arteriolar CNV occurs via a distinct pathobiology, termed neovascular remodeling (NVR), wherein blood-derived macrophages infiltrate the incipient CNV lesion, recruit bone marrow-derived mesenchymal precursor cells (MPCs) from the circulation, and activate MPCs to become vascular smooth muscle cells (VSMCs) and myofibroblasts, driving the development of high-flow CNV with arteriolarization and perivascular fibrosis. In considering SVR, we will discuss the concept that limited or poor vision in spite of anti-VEGF may not be caused simply by photoreceptor degeneration but instead may be associated with photoreceptor synaptic dysfunction in the neurosensory retina overlying CNV, triggered by infiltrating blood-derived macrophages and mediated by Müller cell activation Finally, for each of PDA and SVR, we will discuss current approaches to disease management and treatment and consider novel avenues for potential future therapies.
Keywords: Neovascular age-related macular degeneration, Choroidal neovascularization, Anti-VEGF, Anti-VEGF resistance, Persistent disease activity, Suboptimal vision recovery, Macrophage, Monocyte, Mesenchymal precursor cell, Neovascular remodeling, Photoreceptor synaptic dysfunction, Müller cell
1. Introduction
Neovascular age-related macular degeneration (NVAMD) remains the leading cause of severe vision loss in the elderly (Congdon et al., 2004; Pennington and DeAngelis, 2016). NVAMD affects 5% of individuals age 70 or older, and over 15 million people worldwide are affected by the disease, with this number forecasted to double by the year 2050 with the growth of the elderly population (National Eye Institute, 2020). NVAMD is characterized by the onset, formation, and growth of pathologic macular neovascularization (MNV), manifest as either neovascularization originating from the subjacent choriocapillaris into Bruch’s membrane and subretinal space, choroidal neovascularization (CNV), or neovascularization originating from the retinal circulation, retinal angiomatous proliferation (RAP) or Type 3 MNV. In this review, the consensus clinical terminology of MNV (Spaide et al., 2020) will be used when broadly referring to NVAMD disease; specific terminologies, especially CNV, will be used to refer to specific types of neovascularization.
Intravitreal drugs directed against vascular endothelial growth factor (VEGF-A, or VEGF), including ranibizumab, bevacizumab, aflibercept, and brolucizumab, have revolutionized the treatment of NVAMD. Clinical trial data for these drugs as a class have consistently demonstrated that regular, ongoing treatment, administered by monthly, “treat-and-extend” or pro re nata (PRN, or “as needed”) regimens, can produce, on average, significant improvement in vision (i.e., defined as gain of three or more lines of visual acuity) for approximately 30–35% of NVAMD patients (Brown et al., 2006; Rosenfeld et al., 2006; Martin et al., 2011; Heier et al., 2012; Dugel et al., 2020a).
Because of the efficacy of anti-VEGF treatment, it is frequently assumed that, for the majority of patients with NVAMD, these drugs stabilize disease progression, restore vision and prevent progressive vision loss. In fact, this is not the case. Many patients suffer from incomplete response to anti-VEGF therapy, which is defined as (1) persistent (plasma) fluid exudation, as evident by optical coherence tomography (OCT) and/or fluorescein angiography (FA); (2) unresolved or new hemorrhage; (3) progressive lesion fibrosis; and/or (4) suboptimal vision recovery. The first three of these, unresolved fluid, hemorrhage, and progressive fibrosis, collectively comprise the problem of persistent disease activity (PDA) in spite of anti-VEGF therapy. PDA affects up to 50% of NVAMD patients even after sustained treatment for 1 year, with specific rates varying according to specific choice and dosage of anti-VEGF agent (Martin et al. 2011, 2012; Heier et al., 2012). PDA is associated with both increased treatment burden (frequent monthly injections) and with increased risk of long-term vision loss (Ying et al., 2014). Meanwhile, the problem of suboptimal vision recovery (SVR) is defined as a failure to achieve excellent functional visual acuity of 20/40 or better in spite of anti-VEGF treatment, affecting over 60% of NVAMD patients even after sustained treatment for 1 year (Brown et al., 2006; Rosenfeld et al., 2006; Martin et al., 2011; Heier et al., 2012; Dugel et al., 2020a). Since the broad goals of modern-day treatment for not only NVAMD but for all ocular diseases are to achieve sustainable excellent vision (defined as visual acuity of 20/40 or better) for as many patients as possible while either restoring ocular health or maintaining disease quiescence (defined as free of fluid exudation, hemorrhage, and progressive fibrosis), incomplete response to anti-VEGF therapy, and specifically PDA and SVR, represent significant clinical unmet needs.
In this review, we will delve into the clinical problems of PDA and SVR in NVAMD, defining and characterizing the clinical manifestations and exploring the pathobiology of each. We will demonstrate that PDA occurs most frequently in NVAMD patients who develop Arteriolar CNV lesions, in contrast to patients with Capillary CNV who are highly responsive to anti-VEGF therapy. We will review investigations of experimental CNV, demonstrating that both types of CNV can be modeled in mice, and that formation of Arteriolar CNV occurs via a distinct pathobiology, termed neovascular remodeling (NVR): blood- derived macrophages infiltrate the incipient CNV lesion, recruit bone marrow-derived mesenchymal precursor cells (MPCs) from the circulation, and activate MPCs to become vascular smooth muscle cells (VSMCs) and myofibroblasts, driving the development of CNV with arteriolarization and perivascular fibrosis. In considering SVR, we will discuss the concept that limited or poor vision in spite of anti-VEGF may not be caused solely by photoreceptor degeneration and loss but instead may be associated with (potentially reversible) photoreceptor synaptic dysfunction. We will review investigations of experimental CNV in mice demonstrating that (1) both Müller cell activation and photoreceptor synaptic dysfunction develop in neurosensory retina overlying CNV, leading to physiologic vision loss; (2) Müller cell activation and synaptic dysfunction are associated with recruitment of blood-derived macrophages from retinal vessels into the retina overlying CNV, and these phenomena are progressive, extending laterally within the retina along with the leading edge of the growing CNV; 3) targeting retina-infiltrating macrophages may prevent synaptic dysfunction and physiologic vision loss. Finally, for each of PDA and SVR, we will discuss current approaches to disease management and treatment and consider novel avenues for therapy, highlighting potential novel targets and mechanisms of action for the next generation of NVAMD therapies.
2. Persistent disease activity (PDA) in NVAMD
2.1. Clinical manifestations
PDA in spite of anti-VEGF therapy, which is also known by other terms, such as anti-VEGF resistant NVAMD or refractory or recalcitrant disease, is defined as (1) persistent intraretinal, subretinal, or sub-retinal pigment epithelium (RPE) fluid; (2) persistent or new hemorrhage; and/or (3) progressive lesion fibrosis, assessed after initial loading dose (i.e., three monthly doses) or after a period of sustained treatment (e.g. 1 year) (Fig. 1). For the purposes of this review, we will also include patients with apparent quiescence with monthly anti-VEGF loading doses, but who then exhibit recurrent or worsening disease activity on attempted extension to longer (e.g., every 6, 8, or 10 week) treatment intervals. This is particularly relevant because real-world clinical outcomes are inferior as compared to clinical trial outcomes due to the problem of undertreatment, in which patients receive less frequent therapy in practice than they would otherwise receive in a protocol-driven clinical trial (Lad et al., 2014); this has been extensively reviewed previously (Mehta et al., 2018). Further, as we will discuss in this and the following Sections, this group of patients with PDA upon extension shares phenotypic overlap and similar risk for vision loss as patients with PDA on monthly therapy, if they are not maintained on long-term monthly injections.
Fig. 1.

Manifestations of persistent disease activity (PDA) after a period of sustained treatment with intravitreal anti-VEGF (vascular endothelial growth factor) medicines (i.e., after initial loading dose, 6 months, 1 year, etc.). (D) Demonstrates an example by optical coherence tomography (OCT) of worsening subretinal fluid, new focus of intraretinal fluid, and enlarging shallow spongiform pigment epithelial detachment (PED), post-anti-VEGF therapy, as compared to baseline (A). (E) Demonstrates an example by fluorescein angiography (FA) of macular neovascularization (MNV) enlargement and growth and persistent leakage, post-anti-VEGF therapy, as compared to baseline (B). (F) Demonstrates an example by color fundus photography of persistent hemorrhage and progressive fibrosis extending into the fovea, post-anti-VEGF therapy, as compared to baseline (C).
Persistent fluid in NVAMD can manifest in different compartments of the retina, as evident by optical coherence tomography (OCT): within the retina (intraretinal fluid), between the neurosensory retina and the RPE (subretinal fluid), or subjacent to the RPE (sub-RPE fluid, also known as serous pigment epithelium detachment (PED)). Not all types of persistent fluid are equivalent. For example, persistent intraretinal fluid is associated with long-term vision loss, but persistent subretinal fluid does not necessarily correlate with vision in studies of patients receiving regular (i.e., monthly) anti-VEGF treatment (discussed in Section 2.1) (Ying et al. 2014, 2018; Sharma et al., 2016; Guymer et al., 2019).
Meanwhile, the management of serous PEDs is controversial (Khanani et al., 2018; Cheong et al., 2020). While patients with large vascularized serous PEDs (e.g., those > 400 μm in height) were excluded from some clinical trials of anti-VEGF drugs, subsequent studies have demonstrated visual gains for NVAMD patients with PEDs following anti-VEGF treatment, though presence or size of PED itself does not necessarily correlate with visual acuity (Sarraf et al., 2016; Clemens et al., 2020). Another consideration is RPE tear, a complication that can cause vision loss in patients with serous PEDs, especially when fovea-involving (Ersoz et al., 2017). The spontaneous rate of RPE tear in the natural history of PED has been reported between 3 and 12.5% (Casswell et al., 1985; Chuang and Bird, 1988; Hartnett et al., 1992; Pauleikhoff et al., 2002), while the rate of RPE tear with anti-VEGF therapy has been reported between 2.8% and 24% (Chiang et al., 2008; Lommatzsch et al., 2009; Smith et al., 2009; Guber et al., 2013). Due to risks of RPE tear, some investigators advocate observation without treatment of isolated PEDs or PRN management of persistent PEDs, with anti-VEGF administered for recurrent subretinal or intraretinal fluid (Ersoz et al., 2017). However, in post-hoc analysis of the aflibercept VIEW2 study, eyes with PED suffered loss of visual acuity gains on switch from fixed-interval dosing to flexible PRN dosing, with vision loss associated with development of intraretinal fluid (Schmidt-Erfurth et al., 2015). Thus, while consensus is lacking, management of serous PEDs in NVAMD must balance the risks of undertreatment with the potential risks of RPE tear. In general, active fluid in NVAMD represents exudation from leaking aberrant neovessels, which requires ongoing follow-up and treatment to prevent vision loss (Martin et al., 2011; Maguire et al., 2016).
Persistent fluid is surprisingly frequent, with rate of occurrence dependent on both treatment regimen and specific choice of anti-VEGF drug. In the Comparison of AMD Treatment Trial (CATT), which compared ranibizumab vs. bevacizumab and monthly vs. PRN treatment regimens for each drug, rates of persistent retinal fluid were expectedly higher for rigorous PRN treatment than for monthly treatment: at 1 year, persistent fluid by OCT was present in 71.2% and 79% for ranibizumab PRN and bevacizumab PRN, respectively, while rates for ranibizumab monthly and bevacizumab monthly were 53.2% and 70.9%, respectively (Martin et al., 2011). At 2 years, rates of persistent fluid leakage by were similarly higher for bevacizumab as compared to ranibizumab, when comparing the same treatment strategy (Martin et al., 2012).
Similar data on OCT fluid and FA leakage were observed in the U.K.-based IVAN study comparing ranibizumab and bevacizumab (Chakravarthy et al., 2012). In the LUCAS study comparing treat-and-extend bevacizumab vs. ranibizumab, the rates of persistent fluid by OCT were 53% for bevacizumab and 35% for ranibizumab; moreover, 33% of patients receiving ranibizumab and 47% of patients receiving bevacizumab could not be extended beyond 4 week treatment interval due to fluid leakage on attempted extension (Berg et al. 2015, 2016). In the Phase 3 VIEW1/VIEW2 studies of aflibercept, rates of persistent fluid by OCT at 1 year were approximately 20% for every 4 week dosing and approximately 28% for every 8 week dosing (Heier et al., 2012). In the Phase 3 HAWK and HARRIER studies of brolucizumab as compared to aflibercept, rates for brolucizumab 6 mg (dosed q8/q12 weeks) were 31.2% and 25.8% at 48 weeks in each study, respectively, while rates of OCT retinal fluid for aflibercept 2 mg every 8 weeks were 44.6% and 43.9% in each study, respectively (Dugel et al., 2020a). While these rates varied across studies, these data collectively suggest that persistent fluid in spite of monthly anti-VEGF therapy ranges between 25 and 50%, with higher rates for bevacizumab and perhaps lower rates for ranibizumab, aflibercept, and brolucizumab. These rates of persistent fluid increase to approximately 40–65% when including patients who are unable to extend beyond four-week treatment interval due to recurrent fluid (Berg et al. 2015, 2016).
Hemorrhage from neovascularization into retinal tissue, especially subretinal hemorrhage, is a disease manifestation of NVAMD that can lead to irreversible vision loss (Avery et al., 1996) (Fig. 1C). While the precise mechanisms of tissue damage and vision loss are not definitively established, they may include direct toxic effects of iron on photoreceptors and RPE, physical separation of the photoreceptors from the RPE causing indirect damage to both cell types, and fibrin-blood clot damage to tissue architecture (Glatt and Machemer, 1982; Toth et al., 1991; He et al., 2007). Regular treatment with anti-VEGF therapy reduces the risk of new and recurrent hemorrhage in NVAMD, presumably by stabilizing tight junctions between endothelial cells and improving structural integrity of neovessels (Altaweel et al., 2015). However, as demonstrated in the ranibizumab ANCHOR and MARINA studies, the rate of hemorrhage in spite of ongoing monthly anti-VEGF therapy was not insignificant, at 8.0–8.8%, suggesting that other mechanisms beyond VEGF-mediated neovessel permeability may also promote hemorrhage (Brown et al., 2006; Rosenfeld et al., 2006). Notably, rates of hemorrhage increase in the setting of less intensive anti-VEGF treatment regimens. The PIER study evaluated the efficacy of quarterly (every three month dosing) with ranibizumab 0.5 mg or ranibizumab 0.3 mg following loading dose with three initial monthly injections in each treatment arm, as compared to sham treatment (Regillo et al., 2008). Rates of hemorrhage for the quarterly-ranibizumab treatment arms of PIER (23.7% for the 0.3 mg arm and 28.3% for the 0.5 mg arm) were nearly three times the rate seen with monthly treatment in ANCHOR and MARINA and were not different than the rate for the sham control group of PIER (22.4%) (Barbazetto et al., 2010).
Progressive fibrosis, or aberrant deposition of connective tissue typically in the subretinal or subRPE space, is evident as scar formation by clinical exam (Fig. 1C) and dense hyperreflective thickening by OCT and can be evident as progressive lesion growth by FA (Fig. 1B). Progressive fibrosis also occurs with surprising frequency in NVAMD. Fibrotic scars developed in 24.7% of all eyes treated in the CATT study, regardless of treatment regimen, with increased risk of scar associated with baseline characteristics of Type 2 MNV (i.e., predominantly classic CNV) leakage pattern, larger lesion size, increased foveal retinal thickness, subretinal fluid, and presence of subretinal hyperreflective material (SHRM) on OCT (Daniel et al., 2014). Also, in CATT, mean growth in lesion size by FA was +1.6, +1.9, and +3.0 mm2 for bevacizumab monthly, ranibizumab PRN, and bevacizumab PRN, respectively, indicating that a high percentage of eyes continued to remain active with ongoing fibrovascular tissue growth, reflective of the high rate of observed scar formation (Martin et al., 2012).
To assess the rate of PDA among NVAMD patients in a real-world clinical practice and to better understand the extent or relative severity of PDA, we performed analyses of response to anti-VEGF therapy among NVAMD patients at the Duke Center for Macular Diseases. We developed the Duke Disease Activity Severity Scale to comprehensively assess intraretinal fluid, subretinal fluid, and subRPE fluid by OCT; lesion activity by FA, and hemorrhage by color fundus photography (Table 1). We categorically graded each metric of PDA as mild, moderate, or severe and defined the presence of moderate-to-severe PDA or progressive disease (worsening in one or more metrics) as clinically significant. In retrospective analyses of NVAMD patients treated with bevacizumab or ranibizumab using treat-and-extend, we found that the rates of moderate to severe PDA/progressive disease were 25% among patients with Type 2 MNV, 41% among patients with Type 1 MNV, and 61% among patients with serous pigment epithelium detachment (PED), following 1 year of treatment (Lad et al., 2012; Mettu et al., 2012a; Serrano et al., 2012). We have also performed a prospective, open-label study of treatment response among newly diagnosed, previously treatment-naïve NVAMD patients, the PERSIST study (ClinicalTrials.gov NCT02367365), and we observed that the rates of moderate to severe PDA/progressive disease was 24.5% among all NVAMD patients receiving aflibercept on a treat-and-extend basis (Mettu et al., 2016). These findings suggest that the prevalence of PDA in spite of treat-and-extend anti-VEGF therapy in a real-world NVAMD clinical practice largely mirror those observed in prospective clinical trials of intravitreal anti-VEGF agents.
Table 1.
Duke Disease Activity Severity Scale. Different manifestations of disease activity were categorically assessed as none, mild, moderate, and severe, based on specific measurements for each manifestation of disease. Persistent disease activity (PDA) was deemed to be clinically significant if any single manifestation of disease activity was graded as moderate or severe or if there was evidence of progressive disease in any metric of disease activity.
| Degree of Activity | OCT |
FA |
CFP |
||
|---|---|---|---|---|---|
| Serous PED (SPED) | Intraretinal Fluid (IRF) | Subretinal Fluid (SRF) | CNV Activity | Hemorrhage | |
|
| |||||
| None (0) | Flat or Trace sub-RPE fluid (<25 μm) | None or Microcysts | None or Trace SRF (<10 μm) | Uniform stain without leakage | None |
| Mild (1) | Small SPED (25–199 μm) | Mild IRF (RT < 250 μm) | Mild SRF (10–49 μm) | Progressive staining or trace leakage | Trace or dot hemorrhage |
| Moderate (2) | Medium SPED (200–399 μm) | Moderate IRF (RT 250–349 μm) | Moderate SRF (50–99 μm) | Active small area leakage (<0.5 DA) | Small hemorrhage (<0.5 DA) |
| Severe (3) | Large SPED (≥400 μm) | Severe IRF (RT ≥ 350 μm) | Severe SRF (≥100 μm) | Active large area leakage (≥0.5 DA) | Large hemorrhage (≥0.5 DA) |
| Progressive Disease | Evidence of disease worsening in any metric of disease activity, including evidence of fibrosis, lesion growth or worsened hemorrhage | ||||
2.2. Relationship between disease activity and vision
2.2.1. Effects of PDA on vision outcomes
Intuitively, PDA should be associated with worse long-term vision outcomes, and there is considerable evidence to support this concept. In the PIER study (described above), mean visual acuity gains achieved by month 3, following loading dose with three initial monthly ranibizumab injections, were not sustained following switch to quarterly treatment (Regillo et al., 2008). In subgroup analysis of the PIER study, patients with PDA by OCT and FA at month 3, after ranibizumab loading dose, had a cumulative mean net loss of −1.8 letters at month 12. In comparison, patients with quiescent disease at month 3 had a cumulative mean net gain of +10.2 letters at month 12 (Brown et al., 2013a). Similarly, in the VIEW1 and VIEW2 phase 3 studies of aflibercept, patients in one study group were switched to every 8 week treatment with aflibercept following loading dose with three monthly aflibercept, while patients in another study group received every month treatment for the duration of the study. Patients manifesting PDA at month 3 following loading dose had less robust gains in visual acuity upon switch to every 8 week treatment, as compared to patients with PDA following loading dose who continued to receive every 4 week treatment (Jaffe et al., 2016). Collectively, these data indicate that PDA has significant and negative consequences for vision, particularly in the setting of less frequent dosing regimens.
Consistent with these findings, the HORIZON and SEVEN-UP studies, ranibizumab trial extension studies, found that a switch to treatment at physician discretion without a regular or protocol-defined treatment regimen led to progressive loss of mean visual acuity gains in the five years following the clinical trial period, decreasing from mean + 11.2 letters after 2 years of monthly therapy to mean − 8.2 letters at year 7 (five years later), in association with a high rate of persistent fluid (67.8%), hemorrhage (24.1%), and macular fibrosis (61.4%) (Singer et al., 2012; Rofagha et al., 2013). Similar findings of progressive loss of vision gains and PDA in the setting of undertreatment were also observed in long-term follow-up studies of patients in the CATT and IVAN studies (Evans et al., 2020; Maguire et al., 2016).
In analyses of the ANCHOR and MARINA Phase 3 ranibizumab studies, increased total MNV lesion area at month 24 was associated with 3-line loss in visual acuity (Rosenfeld et al., 2011). Meanwhile, several manifestations of PDA, including lesion growth, progressive fibrosis, hemorrhage, and intraretinal fluid, were associated with sustained significant vision loss in the CATT study (Ying et al., 2014). These data highlight that certain manifestations of PDA are directly associated with worse vision outcomes.
Not all manifestations of PDA influence visual outcomes equally, however (as noted in Section 2.1). Persistent subretinal fluid was not associated with vision loss in the CATT study (Ying et al. 2014, 2018). In fact, a number of eyes with persistent subretinal fluid sustained better visual outcomes over the course of the study, raising the possibility that subretinal fluid might somehow be beneficial to vision and prompting some investigators to question whether the presence of subretinal fluid should be an indication for anti-VEGF treatment (Sharma et al., 2016; Arnold et al., 2016). However, findings of the CATT study do not support the conclusion that persistent subretinal fluid should be tolerated without treatment. Since the presence of subretinal fluid was a criterion for treatment in the PRN study groups, all patients with persistent subretinal fluid (whether in PRN or fixed monthly treatment groups) continued to receive ongoing monthly treatment in CATT. Subsequently, the FLUID study has compared tolerating subretinal fluid in a treat-and-extend pattern (allowing for longer interval between treatments even if a small amount of subretinal fluid was present), vs. more aggressive treatment without extension of treatment interval when subretinal fluid was present (Guymer et al., 2019). The study found that visual acuity outcomes were comparable between the two treatment groups at 24 months; tolerating subretinal fluid was not associated with better or worse vision outcomes. Additionally, the subretinal fluid-tolerant arm received an average of just 1.2 fewer injections over 24 months, as compared to the intensive treatment arm, suggesting that a treatment strategy tolerating subretinal fluid has only a modest effect on the number of needed treatments. The FLUID study did affirm the conclusion that subretinal fluid may not have negative consequences for vision as long as patients continue to receive ongoing anti-VEGF treatment. Importantly, these data do not controvert the overarching conclusion that PDA is associated with a requisite high treatment burden and an increased risk of long-term vision loss in affected patients.
2.2.2. Subclinical nonexudative MNV in AMD
Subclinical nonexudative MNV is defined as the presence of an asymptomatic neovascular lesion without active exudation or leakage (Kuehlewein et al., 2015; Palejwala et al., 2015; Roisman et al., 2016; Laiginhas et al., 2020). Historically, the phenomena of inactive “occult” CNV lesions by FA, corresponding to late-staining “plaques” by traditional static indocyanine green angiography (ICGA), has been described (Baumal et al., 1997; Schneider et al., 1997). However, the prevalence, natural history, and clinical significance of nonexudative MNV has not been well characterized until recently (Querques et al., 2013; de Oliveira Dias et al., 2018; Narita et al., 2020), with the emergence of OCT angiography (OCTA) technologies (Borrelli et al., 2018; Spaide et al., 2018). Among patients with unilateral, active exudative NVAMD, the prevalence of subclinical nonexudative MNV in fellow eyes varies among case series but has been reported to range from 6.25% up to 27%. (Palejwala et al., 2015; Roisman et al., 2016; Yanagi et al., 2017; de Oliveira Dias et al., 2018). The presence of subclinical nonexudative MNV, as detected by swept-source OCTA (SS-OCTA), is associated with a substantially higher 24-month risk of active exudative NVAMD disease (up to 13.6 times greater), as compared to AMD eyes without MNV (Yang et al., 2019). Further, while the presence of the nonexudative MNV lesion itself is not typically associated with compromise of visual function or vision loss in the absence of exudation (Laiginhas et al., 2020), the incidence of exudation from the time of first detection of subclinical MNV is 20–21.1% by 12 months and up to 34.5% by 24 months, highlighting the importance of frequent monitoring for exudative disease among these patients (de Oliveira Dias et al., 2018; Heiferman and Fawzi, 2019; Yang et al., 2019). On the other hand, many eyes with nonexudative MNV remain inactive, while a subset of previously treated exudative MNV can become quiescent or “silent” and not require ongoing maintenance treatment (von der Emde et al., 2020). The conversion of inactive nonexudative MNV to active exudative MNV and spontaneous inactivation or quiescence of previously exudative MNV suggest the existence of specific mechanisms that regulate the biology, and particularly the exudative activity, of the neovascular lesion. One such potential pathogenic mechanism for CNV activity may be the activation of choroidal endothelial cells (CEC) by inflammatory mediators such as TNF-α (derived from macrophages or RPE), resulting in loss of CEC tight junctions and increased exudation as well as increased CEC proliferation and migration (Wang et al., 2015). Strategies to downregulate EC activation and promote EC tight junction formation at the CNV complex, such as by increased Rap1 GTPase activity or overexpression (Wang et al., 2015), could represent drug-able pathways or targets to promote sustained normalization or inactivity of MNV. This could in turn substantially improve treatment efficiency and outcomes and reduce treatment burden associated with intravitreal anti-VEGF injections.
Such strategies are particularly intriguing, as several studies have now demonstrated that eyes with subclinical nonexudative MNV have lower rates of progression of adjacent geographic atrophy (GA) foci, as compared to eyes with comparable GA foci without MNV (Heiferman and Fawzi, 2019; Laiginhas et al., 2020). These intriguing data suggest the possibility that the presence of MNV could be protective against progressive atrophic AMD disease in a subset of eyes, perhaps by providing metabolic support to the retina through improved outer retinal circulation (Spaide, 2015). In such cases, strategies to normalize active, exudative MNV to inactive, nonexudative MNV would be especially desirable.
2.3. Paradigms for PDA in NVAMD
Several paradigms have been put forth to understand the basis for PDA and anti-VEGF resistance. While a detailed discussion of each is beyond the scope of this review, we will briefly review them here.
2.3.1. Loss of drug effectiveness
Loss of drug effectiveness is defined as recurrence or worsening of NVAMD disease activity following an initial positive anatomical and vision response in spite of continuation of therapy with a specific anti-VEGF drug. The assumption underlying this phenomenon is that a change of biological responsiveness to anti-VEGF therapy occurred within the NVAMD lesion (i.e., activation of alternative permeation pathways, tachyphylaxis, drug tolerance, etc.).
This phenomenon is uncommon among cases where complete disease quiescence is achieved during initial loading dose. Fauser and colleagues assessed the intra-individual, long-term variability of drug efficacy and treatment interval among patients with suppressed, quiescent disease, and have observed that the drug efficacy and effective treatment interval generally remain constant over time, for both ranibizumab and aflibercept (Muether et al., 2013; Fauser et al., 2014; Fauser and Muether, 2016; Enders et al., 2016). Eghoj and Sorensen analyzed a large retrospective cohort of NVAMD and found only a 2% rate of loss of drug efficacy following primary inactivation of MNV (Eghoj and Sorensen, 2012). Analyses of treat-and-extend studies, such as the TREX-AMD study, have affirmed these findings, particularly with respective to a constant efficacious treatment interval (Wykoff et al. 2015, 2018). Accordingly, many cases of apparent loss of drug effectiveness actually may be cases of subtherapeutic dosing, resulting in worsening of incompletely controlled NVAMD upon premature extension (see subtherapeutic dosing section below).
One potential cause of authentic loss of drug effectiveness is upregulation of other soluble mediators of exudation, following sustained exposure to anti-VEGF drug. For instance, it has been shown that repeated exposure to bevacizumab can be associated with increased levels of VEGF-C and VEGF-D in the aqueous humor of eyes of NVAMD patients. Increased expression of these other mediators could mediate leakage in spite of adequate levels of functional drug targeting VEGF-A, producing an apparent loss of drug effect upon continued exposure to that anti-VEGF drug of interest (Cabral et al., 2018). Based on these data and general rationale, efforts are underway to develop a novel VEGF-C/D “Trap” fusion protein drug (OPT-302, Opthea), to be administered in combination with anti-VEGF(-A) drugs, to reduce PDA and improve vision response (Dugel et al., 2020b).
Another potential cause of loss of drug effectiveness is the development of anti-drug neutralizing antibodies directed against anti-VEGF biologics (especially ranibizumab and bevacizumab, which are humanized mouse antibodies), as part of a systemic immune response that develops with repetitive treatment. In the MARINA Phase 3 ranibizumab study, the rate of anti-ranibizumab neutralizing antibodies in the sera of study participants at baseline was 0.9% in the group receiving ranibizumab 0.3 mg, 0% in the ranibizumab 0.5 mg group, and 0.5% in the sham injection group (Rosenfeld et al., 2006). Following 24 months, rates of anti-ranibizumab antibodies were 4.4% in the 0.3 mg group, 6.3% in the 0.5 mg group, in comparison to 1.1% in the sham injection group, though the titers of neutralizing antibodies was not reported (Rosenfeld et al., 2006). Exploratory subgroup analyses of MARINA did not identify a significant difference in safety and efficacy outcomes between patients with and patients without anti-ranibizumab antibodies. Forooghian and colleagues performed a pilot study in NVAMD patients, collecting sera from patients who were treated with intravitreal bevacizumab and who exhibited loss of drug effectiveness (n = 11), comparing to a control group (n = 12) comprised of both NVAMD patients naïve to bevacizumab and NVAMD patients who had received bevacizumab but did not develop loss of drug effectiveness (Forooghian et al., 2011). The authors found that both groups had relatively low titers of anti-bevacizumab antibodies (5–20 pg/mL), with the loss of drug effectiveness patient group having a slightly higher mean anti-bevacizumab titer (13.6 ± 4.7 pg/mL) than the control patient group (10.9 ± 4.9 pg/mL), with a trend toward statistical significance (Forooghian et al., 2011). The clinical significance of this finding was unclear, and it remains uncertain whether (and to what extent) neutralizing antibodies against anti-VEGF drugs contribute to loss of drug effectiveness in practice.
Many investigators have cited tachyphylaxis or tolerance to a given anti-VEGF drug as potential causes of PDA (Binder, 2012; Eghoj and Sorensen, 2012; Hara et al., 2019), which is largely based on studies showing that switching to a different anti-VEGF drug can be associated with improved short-term treatment response (Fassnacht-Riederle et al., 2014; Ehlken et al., 2014; Ashraf et al., 2018). In pharmacology, tachyphylaxis classically refers to an acute and rapidly diminishing responsiveness to successive doses of a drug; it occurs in cardiac or respiratory systems, where there is downregulation of a receptor targeted by a drug (e.g., beta-blockers) or in central nervous system where neurotransmitters or receptors targeted by a drug are depleted or downregulated. Tachyphylaxis can be overcome by a drug “holiday” (holding the drug for a period of time), or by extending the interval between successive treatments. Neither strategy has been proven to improve the observed efficacy of an anti-VEGF drug and in fact is likely to worsen disease. Therefore, tachyphylaxis is unlikely to be a cause of PDA (Arjamaa and Minn, 2012). Tolerance refers to loss of responsiveness of a biological system to a drug following repeat exposure, due to a change in the interaction between the drug and its target. However, tolerance typically does not occur in an interaction between a neutralizing antibody/fusion protein (anti-VEGF drug) and its target antigen (VEGF). Thus, likewise, tolerance is unlikely to be a cause of PDA.
2.3.2. Subtherapeutic dosing
Another cause of PDA might be subtherapeutic dosing, defined as an insufficient amount of drug available to target and bind VEGF at the new vessel and adjacent retinal tissue, resulting in continued biologic activity. An obvious subgroup in this category would be cases in which an over-aggressive extension of the treatment interval between injections was done before achieving complete quiescence to the initial loading doses, resulting in worsening of fluid or new hemorrhage (Regillo et al., 2008; Barbazetto et al., 2010; Brown et al., 2013a).
Some investigators have postulated that a subset of NVAMD cases demonstrate PDA as a result of progressively increased VEGF(-A) expression at the new vessel that exceeds the amount of bioavailable drug, or as a result of increased expression of VEGF receptors within MNV tissue, which enables continued PDA even at low VEGF tissue levels (Binder, 2012). However, this phenomenon has not been definitively established in NVAMD patients (Muether et al., 2012).
Increasing the dosage of administered drug has been employed as a strategy to address subtherapeutic dosage. The HARBOR study did not find that high-dose ranibizumab 2.0 mg was superior to ranibizumab 0.5 mg for visual acuity gains among newly diagnosed NVAMD patients (Busbee et al., 2013). However, smaller investigator-initiated studies such as the SAVE trial and the LAST study have specifically evaluated high-dose ranibizumab 2.0 mg in NVAMD patients with PDA in spite of ranibizumab 0.5 mg and have demonstrated that higher dosage can produce improved treatment response in some patients with PDA (Fung et al., 2012; Brown et al., 2013b). Similarly, higher dose aflibercept 4.0 mg has been shown to improvement treatment response in some patients with PDA (You et al., 2018). However, the long-term benefit of high-dose treatment is unclear and it is unknown whether higher dosage in this setting is addressing a problem of subtherapeutic dosing, as we have defined, or suboptimal pharmacokinetics (PK), which is defined as a failure to durably maintain target tissue levels of the drug as it is cleared from the posterior segment of the eye (Del Amo et al., 2017). Cases of PDA attributable to suboptimal PK represent a minority of patients. However, this problem could also be addressed by more frequent dosing, or with sustained released technologies for continuous dosing of anti-VEGF drug, such as the ranibizumab port delivery system, which is presently in late-stage clinical development (Campochiaro et al., 2019).
2.3.3. Pharmacogenetics and genetic determinants of anti-VEGF responsiveness
Some investigators have proposed that genetic variants may be associated with response to anti-VEGF therapy. Studies evaluating polymorphism rs1061170 (T1277C, Y402H) in complement factor H (CFH) suggest that patients who are homozygous for the variant risk C allele (CC genotype) have higher rate of PDA as compared to homozygous for T-allele (TT genotype) (Chen et al., 2012). In a separate study, patients homozygous for the CFH Y402H risk allele required an increased number of injections, suggesting the possibility of an association with PDA (Lee et al., 2009). The biological basis for these genetic associations remains highly speculative but could be related to alterations in local ocular or systemic inflammatory phenotype or to variations in pharmacogenomics. Meanwhile, studies of NVAMD patients in CATT did not find a relationship between anti-VEGF treatment response and genotypes known to be associated with AMD ((rs1061170 (CFH), rs10490924 (ARMS2), rs11200638 (HTRA1), and rs2230199 (C3)) (Hagstrom et al., 2013) or with VEGF-A or VEGFR2 gene polymorphisms (Hagstrom et al., 2014).
2.3.4. Heterogeneity of NVAMD pathobiology
While VEGF is clearly a master factor for new vessel formation and for microvascular exudation, it is certainly not the only factor or active disease mechanism in NVAMD. Our preferred paradigm for PDA is that observed resistance to anti-VEGF therapy is caused by heterogeneity of NVAMD pathobiology, wherein anti-VEGF responsive cases reflect a predominance of VEGF-mediated disease mechanisms, and cases with PDA reflect the presence of other pathobiology mechanisms beyond VEGF. While heterogeneity of NVAMD pathobiology represents our preferred paradigm based on our established lines of investigation, the paradigms previously discussed are not necessarily mutually exclusive, and we do not discount the potential contributions of these other paradigms as additional causes of PDA. In the remaining sections, we will review available data from clinical studies and investigations of experimental CNV that support the paradigm of heterogeneity of NVAMD pathobiology.
2.4. Pathobiology of PDA: lessons from the clinic
Over the past decade, numerous clinical trials have assessed the potential of adjunctive treatments to be administered in combination with anti-VEGF drugs to improve clinical outcomes, particularly for patients with PDA, and the vast majority have failed. Without knowledge of the pathobiology of anti-VEGF resistant disease, the rationale for developing disease-relevant combotherapy strategies remains highly speculative, increasing the risk for such development failures.
The clinical pathobiology of NVAMD is frequently assumed to be uniformly caused by formation of pathologic new capillaries under the retina (Green, 1999). Contrary to this assumption, we and others have observed that the vascular morphology (and therefore the pathobiology) of NVAMD is highly variable and heterogeneous (Cousins et al., 2008; Bearelly et al., 2008; Lad et al., 2012; Mettu et al. 2012a, 2016; Serrano et al., 2012; Kokame et al. 2019a, 2019b). These variations can be readily imaged clinically by indocyanine green angiography (ICGA). Traditionally, NVAMD is classified by fluorescein angiography (FA) leakage patterns (i.e., Type 1 MNV (historically defined as occult), Type 2 MNV (historically defined as classic), serous PED with MNV). Water-soluble fluorescein dye tracks with exudation, identifying areas of pathologic leakage. Highly protein-bound ICG remains mostly intravascular, facilitating visualization of vascular morphology and blood flow. Because ICG fluoresces in near infrared, it can be imaged through the retinal and choroidal pigment. Historically, its poor fluorescence efficiency has limited it routine use for NVAMD by retina specialists; however, the use of technological advancements such as scanning laser ophthalmoscopy and rapid frame acquisition greatly enhanced ICGA image resolution and thereby improved its utility in modern clinical practice.
Our group has had a longstanding interest in using high-speed, video ICGA to identify and characterize the morphology of the pathologic new vessel among patients with NVAMD and routinely employ ICGA in the diagnostic evaluation of NVAMD patients at the Duke Center for Macular Diseases (Cousins et al., 2008; Bearelly et al., 2008; Lad et al., 2012; Mettu et al. 2012a, 2016; Serrano et al., 2012). In retrospective cohort studies and the prospective PERSIST study, we have identified at least six distinct morphologic phenotypes (Fig. 2): 1) Arteriolar pattern; 2) Capillary pattern; 3) Mixed Capillary-Arteriolar; 4) Polypoidal choroidal vasculopathy with Branching vascular network (PCV); 5) Retinal angiomatous proliferation (RAP), or Type 3 MNV; and 6) Choroidal leak syndrome (CLS, also referred to as pachychoroid). Arteriolar pattern (Fig. 2A) is characterized by high flow through large-caliber feeder artery, many branching arterioles, and terminal vascular anastomotic loops but minimal capillary components. The branching arterioles are frequently associated with an intermediate reflectivity PED by OCT, consistent with perivascular fibrosis (i.e., increased extracellular matrix deposition around vascular components). In contrast, Capillary pattern (Fig. 2B) is evident as a relatively slow filling, discrete homogenous focus of microvessels (the structure of which is beyond the resolution of ICGA). The Mixed Capillary-Arteriolar pattern (Fig. 2C) shares features of both phenotypes, with capillary morphology arising from discernible arteriole(s).
Fig. 2.
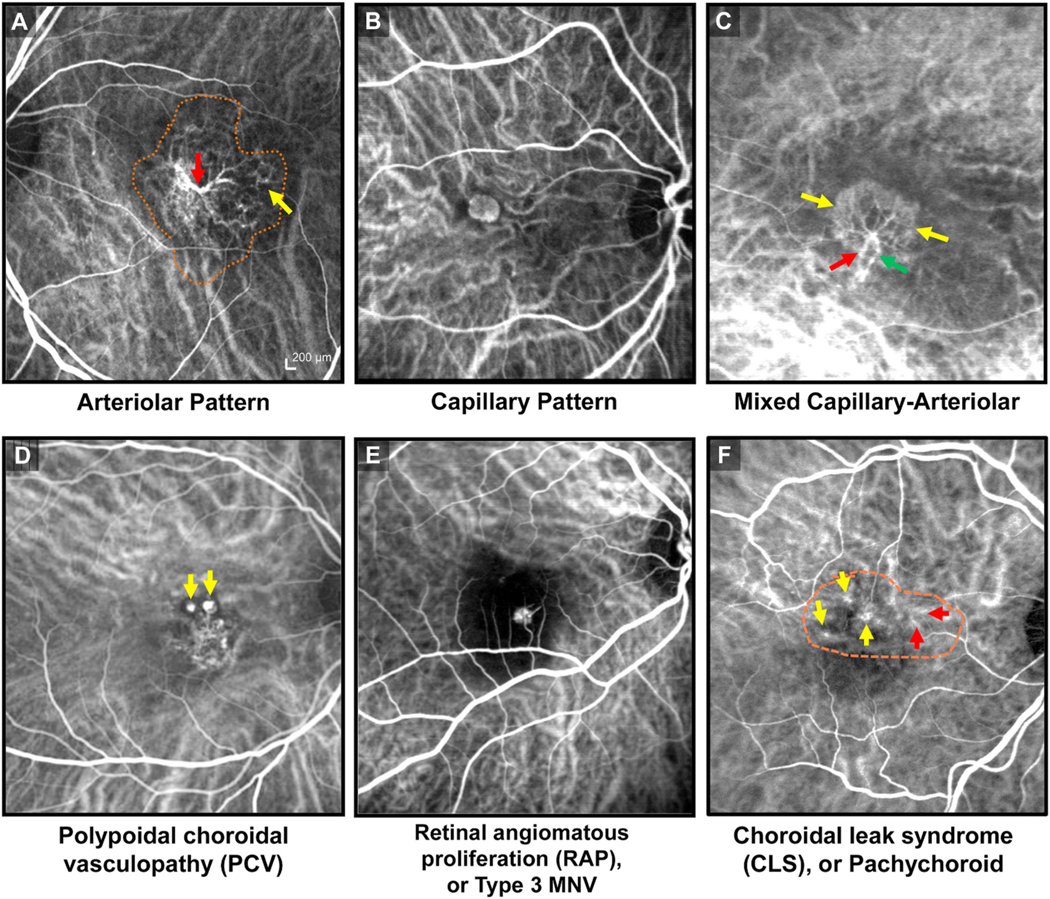
Morphologic subtypes of NVAMD, as visualized by indocyanine green angiography (ICGA). (A) Arteriolar pattern (extent of neovascularization outlined by orange dashes) is characterized by high flow through large-caliber feeder artery (red arrow), which gives rise to many branching arterioles and terminal vascular anastomotic loops (yellow arrow) but minimal capillary components. (B) Capillary pattern is evident as a relatively slow filling, discrete homogenous focus of microvessels. (C) Mixed-Capillary is characterized by presence of feeder artery (red arrow), capillary rim (yellow arrows) and draining venule (green arrow), sharing features from both Arteriolar and Capillary Patterns. (D) Polypoidal choroidal vasculopathy (PCV) subtype of macular neovascularization (MNV) is comprised of aneurysmal, vascular dilatations (yellow arrows), frequently in association with a high-flow, variably organized branching vascular network of arterioles and draining venules. (E) Type 3 MNV, or retinal angiomatous proliferation (RAP), is characterized by intraretinal neovascularization originating from the retinal circulation. (F) Choroidal leak syndrome (CLS), or pachychoroid spectrum of NVAMD, is apparent as choroidal neovascular remodeling (red arrows), irregular and frequently transient hot spots (yellow arrows), and late choroidal hyperpermeability (outlined by orange dashes), in association with variable sub-RPE thickening and subretinal fluid by OCT and coarse pigment mottling of the macula by clinical examination.
PCV (Fig. 2D) is comprised of aneurysmal, vascular dilatations, frequently in association with a high-flow, variably organized network of branching arterioles, sharing some overlap in morphologic features with the Arteriolar Pattern. While PCV is a well-known form of MNV that was initially described by Yannuzzi as a distinct clinical entity (Yannuzzi, 1982; Yannuzzi et al., 1990), it has been increasingly recognized as a subtype of NVAMD. Classically thought to be more common in Asians and in African-Americans, we have observed a high rate of PCV in our patient populations that are predominantly white, with PCV accounting for nearly 20% of newly diagnosed NVAMD patients in our patient series. This is consistent with the findings of other investigators in recent studies of white-predominant populations, with PCV prevalence rates ranging from 21.5% to 31.1% (Hatz and Prunte, 2014; Pereira et al., 2015), suggesting that PCV may be underdiagnosed due to lack of routine clinical use of ICGA (which is the gold standard for PCV diagnosis).
Among newly diagnosed NVAMD patients prior to anti-VEGF treatment, the Capillary and Mixed Capillary-Arteriolar patterns together account for just 20% of NVAMD cases. In contrast, patients with Arteriolar pattern, account for approximately 35% of NVAMD, with along with PCV, together comprise approximately 55% of NVAMD (Lad et al., 2012; Mettu et al. 2012a, 2016; Serrano et al., 2012). Type 3 MNV, or RAP, (Fig. 2E), initially described by Hartnett and colleagues in 1992 (Hartnett et al., 1992) and further characterized by video ICGA by Hartnett in 1996 (Hartnett et al., 1996), accounts for approximately 10% of cases; and CLS, or pachychoroid (Fig. 2F) (Cheung et al., 2019), apparent as choroidal neovascular remodeling, transient hot spots, and late choroidal hyperpermeability by ICGA with subretinal fluid variable sub-RPE thickening by OCT, accounts for 10% of cases. Five percent of cases have no discernible morphologic pattern.
In the retrospective analyses as well as the prospective PERSIST study, we assessed the relationship between neovascular morphologic subtype and treatment response (Lad et al., 2012; Mettu et al. 2012a, 2016; Serrano et al., 2012). We have observed that eyes with capillary and mixed capillary-arteriolar subtype are highly responsive to anti-VEGF therapy and rarely exhibit PDA (<3% of cases) (Figs. 3 and 4). In contrast, eyes with Arteriolar pattern or PCV exhibit manifestations of PDA in approximately 60% of cases and comprise the vast majority of anti-VEGF resistant disease. We have observed that Arteriolar pattern lesions with high flow are more likely to exhibit PDA, suggesting that hemodynamics of high blood flow may mediate aspects of PDA, especially persistent fluid and hemorrhage (Fig. 5). Moreover, among eyes that achieve quiescence, the presence of Arteriolar pattern at baseline diagnosis is associated recurrent leakage on subsequent extension of treatment interval (Mettu et al., 2016). Our findings of a high rate of resistance among PCV patients are consistent with those of other investigators (Hatz and Prunte, 2014; Wong et al., 2016; Kokame et al., 2019a). Based on these findings, we hypothesize that the morphology of the new vessel by ICGA predicts response to anti-VEGF therapy in NVAMD and specifically, that PDA and anti-VEGF resistance in NVAMD occurs in MNV that have high-flow, features of arteriolarization, and in some cases, polyps. OCT angiography (OCT-A) has been shown to delineate neovascular morphology in an analogous fashion and offers a noninvasive alternative to ICGA to further explore this hypothesis (Spaide, 2015; Kuehlewein et al., 2015; Al-Sheikh et al., 2018). Our ongoing work is focused on evaluation of the utility of OCTA for identification of these morphologic subtypes and prediction of response to anti-VEGF therapy, relative to ICGA as a standard.
Fig. 3.

Example of Treatment Response in Capillary Pattern CNV. At baseline, (A) fluorescein angiography (FA) demonstrates a Type 2 MNV pattern and (B) indocyanine green angiography (ICGA) demonstrates Capillary Pattern morphology (red arrows). Post treatment with a single anti-VEGF, (C) FA shows clearance of the Type 2 MNV and (D) ICGA shows regression of the capillary microvascular structure (red arrows).
Fig. 4.
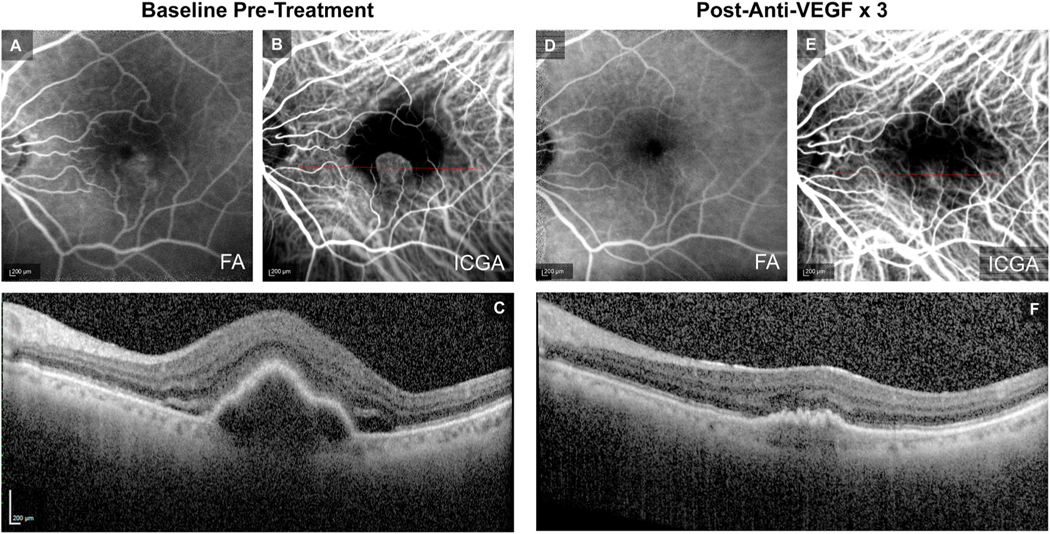
Example of Treatment Response in Mixed Capillary-Arteriolar CNV. At baseline, (A) fluorescein angiography (FA) demonstrates a Type 2 MNV pattern, (B) indocyanine green angiography (ICGA) demonstrates Mixed Capillary-Arteriolar CNV, with a feeder artery (red arrowhead) giving rise to a capillary rim, and (C) optical coherence tomography (OCT) demonstrates a mixed serous and fibrovascular pigment epithelial detachment (PED) and subretinal fluid (SRF). Post-loading dose with three anti-VEGF treatments, (D) FA shows resolution of leakage from the Type 2 MNV, (E) ICGA shows regression of the capillary rim with persistence of the feeder vascular structure (red arrowhead), and (F) OCT demonstrates reduction in PED and clearance of SRF.
Fig. 5.

Example of Treatment Response in Arteriolar Pattern CNV. At baseline, (A) clinical exam and (B) fluorescein angiography (FA) demonstrates evidence of serous pigment epithelial detachment, (C) indocyanine green angiography (ICGA) demonstrates an Arteriolar predominant lesion, with feeder artery (red arrowhead), arteriole (orange arrow), ill-defined marginal rim of vessels (yellow-dotted region, probable capillaries), and draining vein (green arrowhead). Post-loading dose with three anti-VEGF treatments, (D) there is large submacular hemorrhage in the macula by clinical exam, (E) FA demonstrates blockage of fluorescence from the hemorrhage but increased marginal hyperfluorescence indicative of MNV lesion growth, and (F) ICGA demonstrates growth of the CNV lesion, with increased vessel caliber of choroidal feeder artery (red arrowhead), growth of new branching arterioles (orange arrow),extension of arterioles with vascular loops without visible capillaries into the macula (yellow-dotted region), and draining venule (green arrowhead).
Re-contextualizing PDA in light of these findings under the paradigm of heterogeneity of pathobiology, we can theorize about specific potential causes. Persistent fluid exudation in Arteriolar pattern CNV occurs not solely as a result of increased VEGF-mediated permeability, but also as a result of increased exudation from arteriovenous anastomotic terminal loops or poorly formed vascular structures in the setting of high blood flow. Hemorrhage occurs not as a result of endothelial shear stress in fragile, leaky capillaries, but instead due to poorly formed tight junctions at arteriovenous anastomotic terminal loops that are unable to sustain the high rates of flow through Arteriolar pattern CNV. Progressive fibrosis occurs as a result of increased extracellular matrix deposition in association with Arteriolar CNV lesions. We will consider specific pathobiology for these features of Arteriolar pattern CNV in the following sections.
2.5. Pathobiology of capillary CNV: angiogenesis and maturation
Angiogenesis begins when VEGF binds its specific tyrosine kinase receptors VEGFR-2 (primarily) and VEGFR-1 on endothelial cells, which triggers cellular activation via amplification of downstream intracellular signaling pathways (Maharaj and D’Amore, 2007; Penn et al., 2008; Campochiaro, 2015; Apte et al., 2019). In general, the accepted paradigm is that activated endothelial cells are responsible for triggering the subsequent cascade of cellular events that enables new capillary formation, and thus serve to orchestrate most of the biology of capillary angiogenesis (Nieves et al., 2009; Patel-Hett and D’Amore, 2011). For CNV, the primary source of VEGF is thought to be RPE cells (Lopez et al., 1996; Spilsbury et al., 2000; Grossniklaus et al., 2002), which may be upregulated either as a result of focal inflammatory injury or possibly ischemia. Other potential sources of VEGF may be Müller cells, retinal astrocytes, or infiltrating macrophages (Grossniklaus et al., 2002; Kent and Sheridan, 2003; Krause et al., 2014). Upon activation, endothelial cells degrade basement membrane, proliferate, and migrate, remodeling extracellular matrix to facilitate invasion into the surrounding microenvironment in Bruch’s membrane and the sub-RPE space (Grossniklaus and Green, 2004; Costa et al., 2007; Grossniklaus et al., 2010). These endothelial cells then assemble into a network of nascent microvascular capillary tubes.
These nascent capillaries are stabilized by maturation, via recruitment of pericytes, mural cells that ensheath and support the growing network of microvascular endothelial tubes. Maturation is believed to be regulated by the assembling endothelial cells, which recruit pericytes to the nascent capillary network primarily via platelet derived growth factor (PDGF) and transforming growth factor-β (TGF-β), the latter of which is thought to also suppress elongation of capillary tubes (Hirschi et al. 1998, 1999). PDGF-mediated maturation of capillary neovessels prevents their regression even if VEGF is inhibited or removed from the immediate local microenvironment (Benjamin et al., 1998; Hirschi et al., 1999; Armulik et al., 2005; Gaengel et al., 2009; Hellberg et al., 2010), highlighting the importance of dynamic interplay between endothelial cells and pericytes in formation and growth of capillary new vessels. Specifically, heterotypic cell-cell contact between pericytes and endothelial cells promotes maintenance and stability of the capillary neovessel (Hirschi et al., 1997; Darland et al., 2003).
While a full review of capillary angiogenesis biology is beyond the scope of this review, it is notable that other growth factors, such as FGF-2, act as endothelial cell mitogens to promote capillary elongation and growth (Seghezzi et al., 1998; Ramsauer and D’Amore, 2007). Meanwhile, multiple signaling pathways, such as ephrin-B2/Eph pathway, angiopoietin-Tie2 pathway, integrin signaling, Wnt/beta-catenin signaling modulate angiogenesis through regulation of endothelial cell activation and proliferation (Goodwin et al., 2007; Ramsauer and D’Amore, 2007; Silva et al., 2008; Wang et al., 2010; Bryan et al., 2010). In particular, angiopoetin-1 (Ang1) and angiopoetin-2 (Ang2) act in opposition to one another; Ang1 produced by pericytes and mural cells serves to stabilize microvessels in a stable, quiescent state, while, in the presence of VEGF, Ang2 mediates the activation and proliferation of endothelial cells. Other signaling pathways such as Rho GTPase/Rho-associated kinase (ROCK) modulate the extent to which intracellular signaling activity is amplified within endothelial cells following activation by VEGF binding (van Nieuw Amerongen et al., 2003; Bryan et al., 2010).
2.6. Pathobiology of arteriolar CNV: neovascular remodeling (NVR)
In contrast to capillary CNV, the formation of Arteriolar pattern CNV is not well understood. Based on our clinical observations, we propose that the morphology of Arteriolar pattern CNV reflects neovascular remodeling (NVR): the transformation of nascent neovessels into high-flow feeder artery with many branching arterioles and perivascular fibrosis. Importantly, we propose that NVR is a specific and regulatable pathobiology; formation of arteriolarization and fibrosis are neither inevitable, stereotyped endpoints in the growth and development of CNV nor simply a reflection of disease chronicity.
Several key clinical observations support this hypothesis. We have observed that the Arteriolar pattern subtype (as well as PCV subtype) are readily apparent soon after NVAMD conversion (Lad et al., 2012; Mettu et al. 2012a, 2016; Serrano et al., 2012). These lesions do not regress following anti-VEGF treatment and they frequently exhibit progressive growth, which is evident not as capillary formation but instead by the development of new arteriolar vessels and an associated increase in fibrosis. On the other hand, we have not observed that Capillary pattern lesions evolve to become Arteriolar pattern (with rare exception). Patients with Capillary pattern CNV frequently sustain disease quiescence even with extended treatment intervals; when these lesions do reactivate following a prolonged period without treatment, they exhibit exudation and occasionally growth of new capillaries, but they do not “mature” or “evolve” to form Arteriolar pattern (Lad et al., 2012; Mettu et al. 2012a, 2016; Serrano et al., 2012). Further, fellow eye conversions to NVAMD almost always share the same phenotype as the first eye, suggesting the existence of systemic regulation of arteriolarization, rather than only local factors or chronicity. Thus, the specific subtype of neovessel is generally conserved over time, and we propose that the development of the Arteriolar pattern CNV occurs via the distinct pathobiology of NVR.
There are few histopathology studies that have assessed variability in the morphology and structure of CNV in NVAMD. Lutty et al., published a clinicopathologic report that recapitulates the distinct morphologic differences between Capillary and Arteriolar pattern subtypes, readily apparent by UEA-lectin choroidal flatmount: capillary CNV as an ill-defined network of endothelial-lined microvessels and arteriolar CNV as a large complex of feeder artery, intricately interconnected branching arterioles, and terminal vascular loops (Seddon et al., 2016). These remarkable morphologic differences are consistent with the concept that underlying heterogeneity in the mechanisms that drive neovascular formation and growth inform response to anti-VEGF therapy and specifically suggest that the observed resistance to anti-VEGF therapy occurs as a result of additional pathobiologic mechanisms beyond VEGF-mediated angiogenesis.
2.7. Experimental laser-induced CNV: modeling NVAMD pathobiology in mice
Much of what we now know about the biology of angiogenesis for CNV and NVAMD derives from the experimental murine laser-induced CNV model (Grossniklaus et al., 2010). Both long wavelength infrared/red laser (810 nm/650 nm) and green laser (532 nm) can be used to induce CNV development, but they produce different injury responses. With infrared/red laser, the primary site of thermal damage is the inner choroid and RPE, and retinal tissue destruction is minimized (especially with 810 nm). Immediately after laser application, there is displacement of overlying photoreceptor outer segments without extensive fluid movement, the inner retina is intact, and ERG amplitudes are generally preserved (Caicedo et al., 2005a). Application of green (532 nm) laser induces a tissue burn with direct and full thickness retinal injury in addition to RPE/inner choroid injury, resulting in photoreceptor degeneration and Müller cells gliosis that is directly mediated by the laser application. Studies of the laser CNV model using red laser allow a focused evaluation of the biology of new vessel formation while minimizing retinal injury; studies using the green laser assess CNV development as part of a wound healing response to more extensive injury (Strittmatter et al., 2016). Our laboratory has exclusively utilized infrared/red diode laser in laser-induced CNV studies to minimize thermal injury at the neurosensory retina and minimize any confounding biology of the direct laser injury, which is particularly important for the study of retinal pathology overlying CNV lesions (discussed in Section 3).
2.8. Capillary vs. arteriolar CNV in mice: understanding key pathologic differences
Our laboratory has had a longstanding interest in CNV pathobiology with a specific focus on the identification of key determinants of CNV severity. We were the first group to identify age as an independent risk factor for CNV severity (Espinosa-Heidmann et al., 2002). Using the murine laser-induced CNV model in wild-type mice, we observed that old, 16 month old C57BL/6J mice developed more severe CNV as compared to young 2 month old mice, findings that indicated that age-related systemic susceptibility factors, independent of local changes in the retina, are determinants of CNV severity (since these wild-type mice do not have an intrinsic retinal phenotype). Our initial characterization of increased severity in old mice was evident as increased lesion size, increased vascular staining (by FITC-dextran perfusion), and greater cellularity (by propidium iodide staining) (Espinosa-Heidmann et al., 2002). As we have expanded our efforts to include more sophisticated analytical methods to assess lesion activity, cellular composition, and structural morphology, we have definitively established that the observed severity in old mice specifically reflects the biology of NVR (Figs. 6–8). By FA, the severe phenotype in old mice is not only larger in size but also demonstrates more rapid filling and more extensive dye leakage, indicating greater disease activity or exudation as compared to small, well-demarcated lesions with minimal leakage in young mice (Fig. 6). By lectin flatmount, old mice have larger-caliber vessels and terminal vascular loops (Fig. 6F) along with increased perivascular fibrosis (Fig. 7B), as compared to young mice with smaller capillary lesions (Fig. 6C) and minimal fibrosis (Fig. 7A) (Espinosa-Heidmann et al. 2002, 2013).
Fig. 6.

Vascular morphology in experimental CNV lesions by fluorescein angiography and lectin-stained flatmount. Early (~1 min) and late (~4 min) FA photographs were obtained to characterize lesion size and leakage activity of experimental CNV. Lectin-stained vascular flatmounts were obtained to characterize differences in vascular morphology (magnification: × 100; scale bar: 100 μm). Young mice demonstrated small lesions, well-demarcated borders, and mild fluorescein leakage (A, B). Lectin-stained flatmount analysis of one of these lesions (corresponding to red box [B]) demonstrated well-defined, small-diameter capillary networks with minimal discernible large-caliber arterioles (C). FA from old mice demonstrated large, confluent CNV with very active fluorescein leakage (D, E). Lectin-stained flatmount from one of these CNV lesions (corresponding to red box [E]) revealed many large branching arterioles (arrow) and vascular loops at the lesion margins (arrowhead) (F).
Fig. 8.

Vascular morphology and cellular composition in experimental CNV lesions by confocal microscopy of flatmounts stained for CD31 (green) endothelial cells and smooth muscle actin (SMA) (red) vascular smooth muscle cells and myofibroblasts. Scale bar: 100 μm. Young mice demonstrate CNV lesions with (A) CD31+ endothelial cells within a well-demarcated but ill-defined network of microvessels; (B) there is minimal staining with SMA+ within the microvascular structure, reflecting the predominance of pericytes as mural cells and rarity of VSMCs and associated myofibroblasts, features that are all consistent with Capillary morphology. Old mice demonstrate CNV lesions (C) with an extensive network of CD31+ branching large-caliber vascular structures, with terminal loops at the lesion margin interconnecting the branching vessels. (D) Double- staining for SMA + perivascular mural cells reveals the presence of extensive SMA + perivascular cells, including SMA + VSMCs that directly invest and envelop the arteriolar vascular structures as well as SMA + cells within the lesion interstitium, myofibroblasts, which are responsible for deposition of perivascular extracellular matrix deposition (fibrosis).
Fig. 7.

Masson Trichrome demonstrating extracellular matrix deposition in experimental murine laser-induced CNV. As compared to young mice (A), old mice (B) demonstrated thicker CNV lesions with more extensive extracellular matrix deposition, indicative of increased perivascular fibrosis.
Comparative analysis of vascular morphology and differential cellular composition of CNV provides perhaps the most definitive confirmation of lesion biology (Fig. 8). Mild lesions of young mice demonstrate CD31+ endothelial cells within a well-dermarcated but ill-defined network of microvessels; there is minimal staining with smooth muscle actin (SMA+, which labels VSMC and myofibroblasts cells) within the microvascular structure, reflecting the predominance of pericytes as mural cells and rarity of VSMCs and associated myofibroblasts (Fig. 8A–B). These features are all consistent with capillary morphology. In contrast, severe lesions of old mice have an extensive network of branching large-caliber vascular structures, with terminal loops at the lesion margin interconecting the branching vessels (Fig. 8C). Double-staining for SMA + perivascular mural cells reveals that SMA + VSMCs directly invest and envelop the vascular structures, affirming that these are indeed arteriolarized vessels; additional SMA + cells not in direct contact with the vascular structures but within the lesion interstitium represent SMA + myofibroblasts, which are responsible for production of extracellular matrix components (collagen, etc.) comprising the fibrosis that surround and ensheath the arteriolarized vessels (Fig. 8D). This phenotype is strikingly similar to the morphology of the Arteriolar pattern CNV in patients with NVAMD. Importantly, these phenotypic differences in CNV of young and old mice were observed at the same time point (14 days) following CNV induction and are not reflective of a difference in the time course of lesion development (i.e., capillary lesions in young mice do not transform or evolve into arteriolarized lesions at later time points of 4 or 5 weeks post induction). Cross-sectional immunofluorescence analyses offer confirmatory evidence, as there is a significantly increased frequency of (SMA)+ VSMCs and myofibroblasts as compared to CNV of young mice (normalized to total cell count), though there is no difference in the frequency of CD31+ endothelial cells in old vs. young CNV lesions (Espinosa-Heidmann et al. 2002, 2013).
From analysis of the distinguishing pathologic features of the arteriolarized CNV phenotype in old mice, we can infer that NVR requires the formation of vessels with large-caliber lumen as well as the acquisition of vascular smooth muscle cells (VSMCs) as mural support cells and further ensheathment in perivascular fibrosis by myofibroblasts. The morphology of Arteriolar CNV in experimental CNV is clearly distinct from Capillary CNV, paralleling our findings in human NVAMD. Intuitively, the mechanisms that mediate NVR biology should likewise be distinct. However, conceptually and mechanistically, a key question remains: Does NVR occur via the transformation of nascently formed capillary structures into arteriolarized lesions at an early time point, with alterations of the vascular structure and morphology, as a “second step” that follows the conventional paradigm for capillary angiogenesis? Alternatively, do arteriolarized lesions form as the product of a biology that is altogether distinct from traditional capillary angiogenesis and that is established from the outset of lesion induction?
To address this question, we performed a comparative analysis of CNV lesion formation in old vs. young mice over time following laser induction, assessing differences in the dynamic interplay and assembly of endothelial cells and mural support cells at key timepoints in the development of each lesion type (Fig. 9). As expected, at 3 days post- laser induction, nascent capillary lesions in young mice demonstrated an initial migrating wave of CD31+ endothelial cells at the outer margin of the lesion, with a weakly positive focus of SMA + cells at the site of laser injury (Fig. 9A). In contrast, and unexpectedly, nascent lesions in old mice demonstrated a prominent initial “wreath” of SMA + cells encircling well beyond the margins of the site of laser injury, with none to minimal CD31+ endothelial cells present (Fig. 9D). Importantly, laser settings and application were identical for both age groups. Capillary structures were evident by day 7 in young mice (Fig. 9B). However, in the lesions of old mice, by 7 days post-laser induction, the SMA + cells have begun to pattern into a scaffold of tunnel-like structures, and CD31+ endothelial cells have begun to grow at the center of the lesion, with a leading edge of growth outwards into the SMA + scaffold tunnels (Fig. 9E). By 14 days, in CNV of young mice, growth of capillary lesion is complete (Fig. 9C), while in CNV of old mice, CD31+ endothelial cells have completed growth out to the full margin of the lesion, forming branching arteiroles and anastomotic loops at the rim of the arteriolarized complex (Fig. 9F).
Fig. 9.

Time course for dynamic changes in cellular composition and morphology in developing experimental CNV lesions, by confocal microscopy of flatmounts stained for CD31 (green) endothelial cells and smooth muscle actin (SMA) (red) vascular smooth muscle cells and myofibroblasts. Scale bar: 100 μm. At 3 days post-laser induction, (A) nascent lesions in young mice demonstrate an initial migrating wave of CD31+ endothelial cells at the outer margin of the lesion, with a weakly positive focus of SMA + cells at the site of laser injury. By day 7, (B) formation of CD31+ microvascular structures are evident in CNV lesions of young mice, with patterning of SMA + cells within the interstitium of the capillary lesion. By day 14, (C) formation of capillary CNV lesion is complete, with formation of a complete microvascular network. At 3 days post-laser induction in old mice (D), nascent lesions demonstrate a prominent initial “wreath” of SMA + cells encircling well beyond the margins of the site of laser injury, with none to minimal CD31+ endothelial cells present. By day 7, (E) the SMA + cells have begun to pattern into a scaffold of tunnel-like structures (dotted white line), and CD31+ endothelial cells have begun to grow at the center of the lesion, with a leading edge of growth (arrowheads) outwards into the SMA + scaffold tunnels. By 14 days, (F) CD31+ endothelial cells have completed growth out to the full margin of the lesion, forming branching arterioles and anastomotic loops at the rim of the arteriolarized CNV lesion..
These observations suggest a potential alternative paradigm for the biology of NVR. In contrast to angiogenesis where endothelial cells are the primary cells orchestrating formation of microvascular tubes, in NVR, SMA + VSMCs and myofibroblasts control the pathobiology. These cells are the first wave of to infiltrate the incipient lesion and assemble into a scaffold of mesenchymal support cells that establishes the template for endothelial cells to migrate and form large-caliber vascular tubes, as opposed to microvascular tubes. Thus, it is possible that there is a reversal of roles from the conventional angiogenesis paradigm: VSMC and myofibroblasts “lead the way” for endothelial cells, patterning vessel growth. Subsequent dynamic interplay between these mesenchymal cells and endothelial cells (Ferrara, 2010), via paracrine signals and/or heterotypic cell-cell contact, may then enable the formation of large-caliber arterioles, terminal arteriovenous anastomotic loops, and draining venules (see cartoon in Fig. 20). Importantly, the key observation of early recruitment and activation of mesenchymal cells at the incipient lesion suggests the distinct possibility that the arteriolarized phenotype is determined at the outset of lesion development. This is consistent with our observations in NVAMD patients that the Arteriolar pattern CNV is apparent soon after disease conversion and is not a function of the natural history of disease in the absence of treatment and is not simply reflective of disease chronicity. Collectively, these findings support the concept that NVR is a distinct and regulatable pathobiology in NVAMD.
Fig. 20.

Integrated hypothesis for neovascular remodeling. (A) Circulating monocytes infiltrate the site of incipient choroidal neovascularization (CNV) at Bruch’s membrane/sub-RPE space, where they transform into macrophages. (B) Activated macrophages secrete fibrogenic factors that recruit and activate bone-marrow derived mesenchymal precursor cells from the circulation via the choroid. (C) MPCs differentiate into vascular smooth muscle cells and myofibroblasts, which establish the template for the phenotype of neovessel growth early in CNV development by forming a perivascular mesenchymal scaffold, into which endothelial cells grow to form arterioles, venules, and terminal vascular loops. (D) Growth is complete as an Arteriolar CNV.
2.9. Macrophages and NVR
2.9.1. Conceptual framework for mononuclear phagocyte biology in health
In normal health, two subtypes of innate immune mononuclear phagocyte cells surveil the tissue niche to provide vital homeostatic functions: microglia (in the retina) and tissue-resident macrophages (in the choroid). Full characterization of the ontogeny of retinal microglia is beyond the scope of this review but has been thoroughly reviewed elsewhere (Guillonneau et al., 2017; Saban, 2018; McMenamin et al., 2019). In brief, over the past decade, the embryonic origin of central nervous system (CNS) microglia has been firmly established: CNS microglia are derived from precursor cells that seed the CNS during embryonic development (i.e., “yolk-sac” or fetal liver-derived) and can self-renew and maintain their own population during adulthood (Ginhoux et al., 2010). Retinal microglia, like CNS microglia, are not bone marrow derived and are self-renewing (Panagis et al., 2005; Xu et al., 2007; Wohl et al., 2010; O’Koren et al., 2016). While these and other similarities to CNS microglia point toward an embryonic origin for retina microglia as well (Li et al., 2019), embryonic origin for retina microglia has not yet been definitively established (Saban, 2018; McMenamin et al., 2019). As with some other resident macrophage populations, retinal microglia display tissue-specific properties though specific distinguishing cellular markers are lacking (Naito et al., 1996; Faust et al., 1997; Wozniak, 1998).
On the other hand, with age, the choroid has increasing proportion of tissue-resident macrophages that are monocyte-derived, presumably due to the ease with which circulating monocytes can traffic through the interstitium of the choroid (McMenamin et al., 2019). Whether functional differences exist between embryonically derived and monocyte-derived tissue-resident macrophages in the setting of normal health is unknown, though this is area of active investigation in the field of macrophage ontogeny using lineage-tracing tools (Naito et al., 1996; Ginhoux and Guilliams, 2016; Wolf et al., 2019).
2.9.2. Conceptual framework for mononuclear phagocyte biology in disease
In the setting of retinal disease, including AMD, blood-derived macrophages are recruited to the retina and choroid (Grossniklaus et al., 2005; Partsch et al., 2006; Tatar et al., 2009; Lad et al., 2015). Circulating bone marrow-derived monocytes are recruited from the bloodstream, where they infiltrate the retina and choroid and transform into macrophages, in order to mediate effector functions in response to injury (Reinoso et al., 2004; Wang et al., 2019). Blood-derived macrophages typically have high turnover rate, and their continued presence in the tissue is dependent on sustained recruitment to the diseased tissue.
The resident microglia population can also expand and dynamically and alter their distribution within the retina, in the setting of disease (Saban, 2018; McMenamin et al., 2019); potential roles in retinal disease have previously reviewed (Karlstetter et al., 2015; Guillonneau et al., 2017). Since histologic markers and cellular morphology do not reliably distinguish between resident microglia and monocyte-derived macrophages (e.g., both cell populations express Iba-1 and F4/80), it has not been possible to differentiate the two populations of mononuclear phagocytes within the retina until the recent introduction of cell reporter systems, such as the mouse Cx3-chemokine receptor 1 (Cx3cr1) CreER (Cx3cr1CreER) reporter system (Goldmann et al., 2013; Parkhurst et al., 2013). This system enables lineage tracing experiments: following tamoxifen pulse-labeling, microglia retain reporter expression indefinitely, while circulating monocytes retain expression for approximately 2–3 weeks (the time necessary for monocyte turnover from the bone marrow). Thus, following a washout period (approximately 4 weeks), monocytes and microglia can be reliably distinguished by differential expression of the reporter in the previously tamoxifen-exposed mouse.
O’Koren et al. utilized a Cx3cr1eYFP−CreER; R26tdT reporter mouse to ascertain differential distribution of monocytes and microglia in a mouse model of retinal degeneration following light damage (O’Koren et al., 2016). In the Cx3cr1eYFP− CreER; R26tdT reporter mouse, both microglia and monocyte-derived macrophages express YFP but tamoxifen pulse labeling induces a td-Tomato (TdT) reporter that selectively labels microglia following washout period. The authors found that, following retinal light exposure, tdT + microglia migrate to the subretinal space and adhere to the apical surface of the RPE, while monocyte-derived macrophages are found primarily in the plexiform layers and do not extend to the subretinal space. While functional differences between the microglia and monocyte-derived macrophages has not been definitively established, the authors hypothesize that microglia migration to the subretinal space occurs as a protective response in the setting of photoreceptor injury, while the recruitment of monocyte-derived macrophages into the retina exacerbates disease pathology (O’Koren et al., 2016; Yu et al., 2020).
We embrace the perspective that retinal microglia also contribute to AMD (Guillonneau et al., 2017; Saban, 2018; Reyes et al., 2019), though there is clearly much to learn about their potential role(s) and whether they are protective or pathogenic. This reporter system can be applied to distinguish microglia and monocyte-derived macrophages within the retina in mouse models relevant to AMD, including the laser CNV model (and models of subRPE deposit formation). However, since lineage-tracing studies are still novel in retinal biology, to our knowledge, such investigation has not yet been performed in the experimental CNV model. Work is underway in our laboratory to utilize this reporter system to analyze differential contributions of retinal microglia vs. monocyte-derived macrophages, particularly with respect to changes in photoreceptor and synaptic biology in the setting of CNV formation.
In this review, we will concentrate on the roles of blood-derived macrophages in NVAMD disease, since there is a preponderance of human and experimental mouse data characterizing their contributions to CNV pathobiology, including NVR, capillary angiogenesis, CNV induction, and dysfunction of the neurosensory retina overlying CNV.
2.9.3. Biology of blood-derived macrophages
Blood-derived macrophages frequently retain many properties of the circulating monocytes from which they originate (Wolf et al., 2019), and as such, the activation state (as defined by its gene expression) of the circulating monocyte is a key determinant of the specific effector functions that the macrophage manifests upon activation in the tissue (Cousins et al., 2004). However, specific exposures within the microenvironments in which monocytes reside or traffic can alter their activation state. Macrophages utilize a pattern recognition response to identify broad groups of offensive stimuli, including microbe-associated molecules (e.g., pathogen-associated molecular patterns, or PAMPs), toxins, or cellular debris resulting from injury (e.g., damage-associated molecular patterns, or DAMPs) (Gordon, 1999; Moilanen et al., 1999; Hamrick et al., 2000). Broadly, macrophages serve three primary functions: 1) as phagocytic cells that scavenge, engulf, and clear cellular debris and pathogens; 2) as inflammatory effector cells that produce and secrete cytokines, growth factors, oxidants, mediators, and matrix-modifying enzymes; and 3) as antigen presenting cells for T lymphocytes to initiate adaptive immunity mechanisms (Gordon, 1999).
Classically, in innate immunity, macrophages respond to acute injury or infection (Leifer and Medvedev, 2016), but they also play important roles in chronic degenerative diseases like AMD. Furthermore, unlike in primary autoimmune disorders, macrophages directly mediate an innate immune response at the RPE and retina without previous immunization. In the setting of AMD, cellular injury stimuli at the RPE, choroid, and outer retina, which can include drusen and subRPE deposits, oxidized proteins or lipid (van Leeuwen et al., 2018), focal vascular endothelial cell injury at the choriocapillaris, and subretinal deposit formation (Mettu et al., 2012b), can recruit and activate macrophages from the systemic circulation via upregulation of cell adhesion molecules, production of macrophage chemotactic factors, and release of pro-inflammatory mediators (Yang et al., 2016; Wang et al., 2019).
2.9.4. Macrophage-mediated NVR in experimental CNV
In human NVAMD, histopathology of CNV frequently reveals the presence of macrophages, and macrophage frequency is increased in thicker, more fibrotic lesions (Grossniklaus et al., 2005; Partsch et al., 2006; Tatar et al., 2009). In the murine laser-induced CNV model, we have similarly observed that macrophages infilrate CNV lesions and that there is significantly increased macrophage infiltration in arteriolar CNV of old mice as compared to capillary CNV of young mice (Fig. 10). We performed macrophage depletion studies in old mice to understand the role of macrophages in arteriolar CNV development (Espinosa-Heidmann et al., 2003a). We used systemic administration of clodronate liposomes for these studies, since systemically clodronate liposomes efficiently deplete monocytes from the circulation but do not affect local microglia since liposomes do not penetrate the blood-retinal barrier (Van Rooijen, 1989; Huitinga et al., 1990). In old mice, depletion of circulating monocytes with clodronate liposomes (administered prior to laser and throughout lesion development) abrogates the arteriolar CNV phenotype, as compared to control-treated old mice (Fig. 11). Interestingly, systemic monocyte depletion did not abolish CNV formation but instead converted large arteriolar CNV to smaller capillary CNV lesions, similar in appearance to those found in young mice. The observation that capillary CNV formation was maintained in spite of monocyte depletion suggests that blood-derived macrophages are dispensable for angiogenesis in CNV formation in old mice, though we do not discount the likelihood that capillary angiogenesis was at least partially reduced (Sakurai et al., 2003). These findings indicate that NVR specifically requires blood-derived macrophages and that, in old mice, the biology of CNV formation and growth defaults to capillary angiogenesis in the setting of macrophage depletion.
Fig. 10.
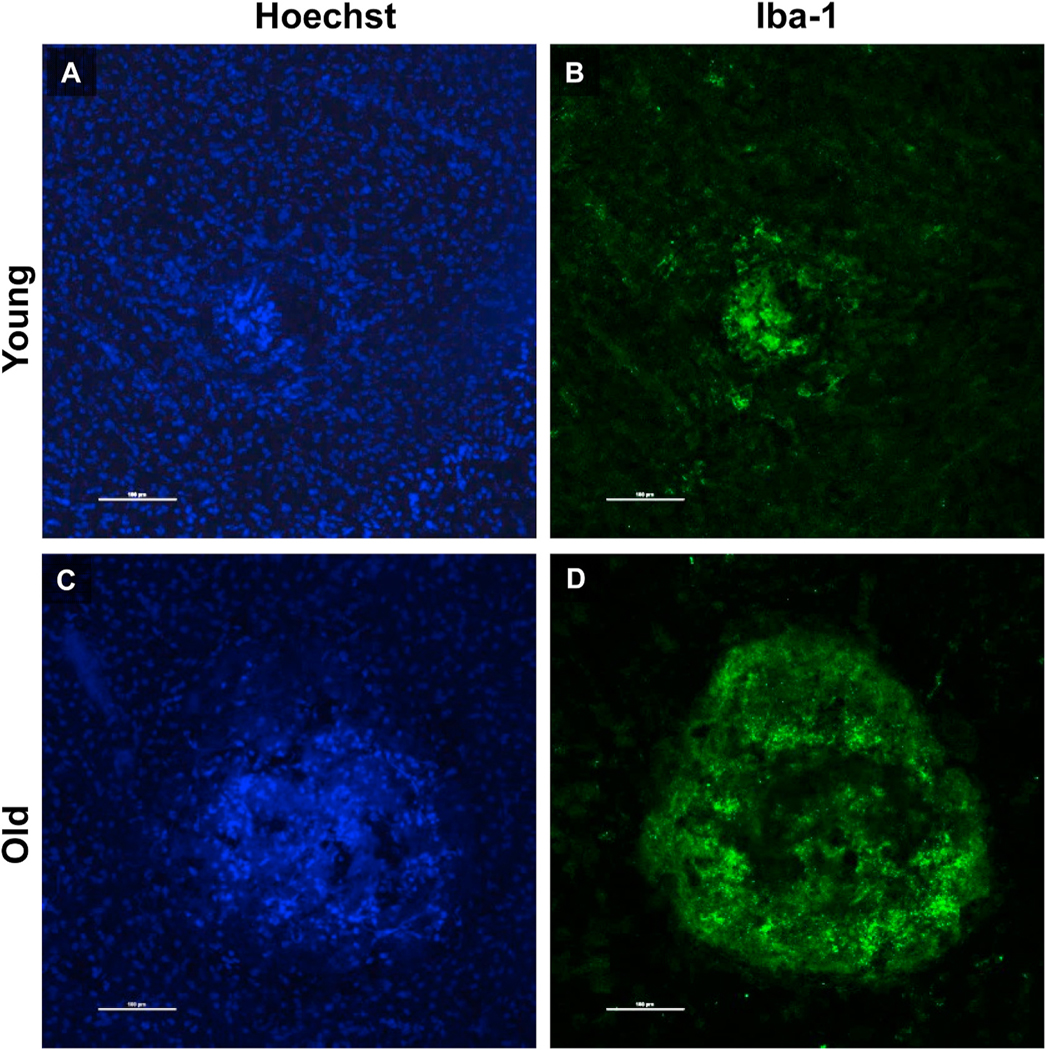
Macrophage CNV infiltration by confocal microscopy of flatmounts stained for Iba-1 (green) macrophages with Hoechst nuclear staining. Scale bar: 100 μm. As compared to CNV lesions of young mice (A) and (B), old mice, (C) and (D) demonstrate more extensive macrophage infiltration within CNV lesions.
Fig. 11.
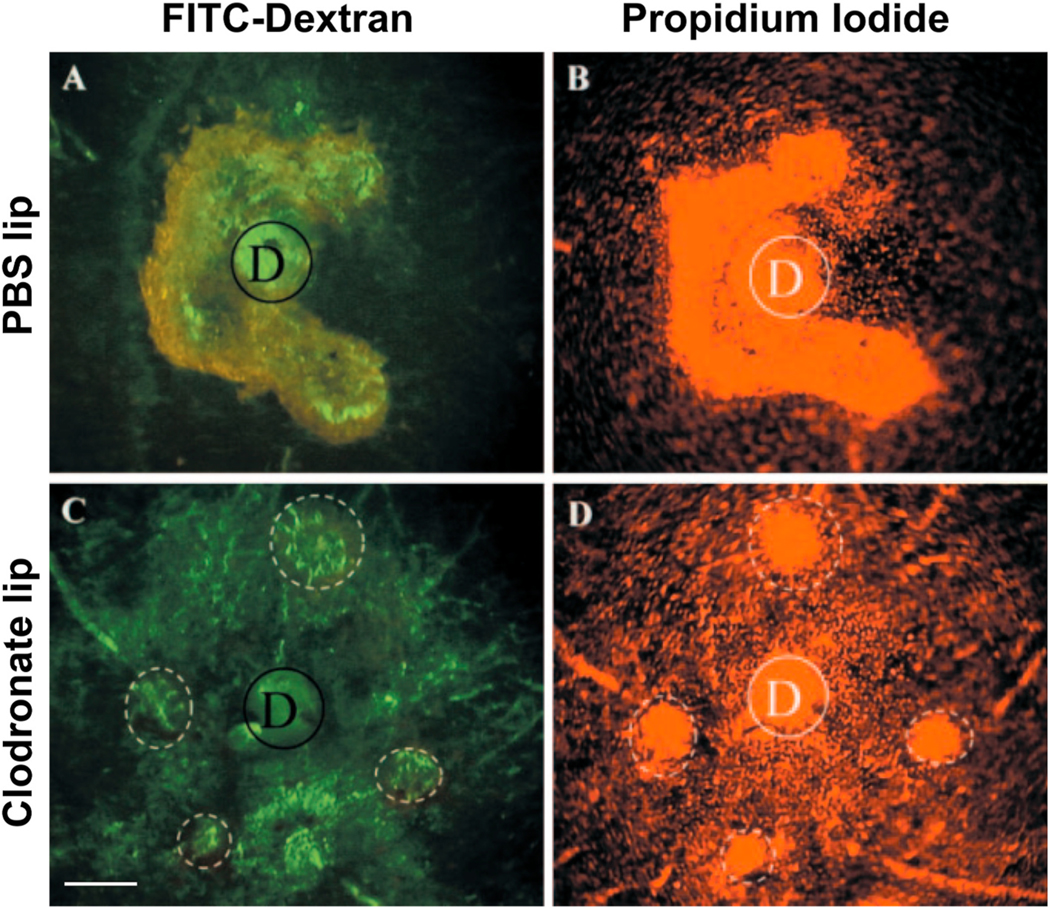
Effects of systemic monocyte depletion on experimental CNV in old mice, by flatmounts (FITC-dextran for vascularity, and propidium iodide for cellularity) of experimental CNV lesions in aged mice treated with either PBS liposomes (PBS lip) (controls) or clodronate liposomes (Clodronate lip) at 2 weeks post induction, D = optic disc. Scale bar: 100 μm. (C) and (D) Depletion of circulating monocytes by systemic administration of clodronate liposomes prevents the development of large arteriolar CNV lesions; clodronate-treated mice instead develop smaller capillary CNV lesions. (A) and (B) Control-PBS lip treated mice demonstrate large CNV lesions characteristic of arteriolar CNV in old mice (which, in this example, are confluent due to extensive lesion growth).
To better understand whether macrophages mediate the early steps of NVR, we have performed additional studies with systemic monocyte depletion by clodronate liposome administration just on the day prior and the day following laser induction in aged mice (Fig. 12). We observed that systemic monocyte depletion in this setting completely abrogates the initial recruitment of mesenchymal cells and the formation of the SMA + cell “wreath”-like scaffold that forms by day 3 of CNV development (Fig. 12D-E-F), indicating that specific and dynamic interactions between macrophages and mesenchymal precursors (that give rise to SMA + cells) establish the template for arteriolar CNV early in lesion development. We hypothesize that macrophages mediate NVR via secretion of specific factors and cytokines that recruit and activate mesenchymal cells, which differentiate into SMA + VSMCs and myofibroblasts and set the template for arteriolar CNV formation. Indeed, potential factors promoting fibrovascular growth, including TGF-β, FGF- 1, FGF-2, IGF-1, and osteopontin, have been found localized to infiltrating macrophages at the site of incipient CNV formation by other investigators in histopathology of laser-induced CNV (Li et al., 2017).
Fig. 12.

Effects of systemic monocyte depletion on experimental CNV development at day 3 following CNV induction in old mice. Scale bar: 100 μm. Control-PBS lip treated mice (A–C) demonstrate the (C) expected formation of a prominent initial “wreath” of SMA + cells, in association (B) with early lesional Iba-1 macrophage infiltration. Depletion of circulating monocytes by systemic administration of clodronate liposomes (D–F) completely abrogates the formation of the wreath of SMA + cells (F) at day 3, in association with a complete suppression of lesional macrophage infiltration (E).
2.9.5. Functional macrophage subsets and NVR
Conceptually, macrophages exist in different levels or states of metabolic and functional activity. Each state represents different “programs” of gene activation and mediator synthesis that ultimately determines specific effector biology of the macrophage cell. There are multiple paradigms that have been put forth to understand differential biology of monocyte and macrophages, the majority of which are focused on differences in inflammatory and effector functions. In the conventional paradigm, infiltrating “quiescent” M0 monocytes can become differentiated into classically activated (“M1”) or alternatively activated (“M2”) macrophages within inflamed tissues (Cao et al., 2011; Sica and Mantovani, 2012; Biswas et al., 2012). Typical stimuli that promote activation of M1 macrophages include bacterial toxins (such as lipopolysaccharide (LPS)), antibody-coated pathogens, complement-coated debris, or certain cytokines (Jiang et al., 1992; Schlegel et al., 1999; Schumann and Latz, 2000). While M1 macrophages are proinflammatory, M2 macrophages mediate tissue repair and resolution of inflammation (Hammerstrom, 1979; Rutherford et al., 1993; Takahashi et al., 1996; Blackwell and Searle, 1999; Apte, 2010). M2 macrophages contribute to physiologic processes in response to injury, including wound repair, neovascularization, extracellular matrix turnover, and fibrosis (Yang et al., 2016; Wang et al., 2019). However, these processes can contribute to chronic tissue injury in the absence of widespread tissue destruction. For example, M2 macrophages play important roles in the pathogenesis of atherosclerosis, glomerulosclerosis, osteoarthritis, keloid formation, pulmonary fibrosis, and other noninflammatory disorders, indicating that the “repair” process is not always beneficial to tissues that have complex morphologies and precise structure-function requirements (Mahdavian Delavary et al., 2011; Chung et al., 2018; Jinnouchi et al., 2019; Griffin and Scanzello, 2019; Warheit-Niemi et al., 2019). The M1-M2 dichotomy in this conventional paradigm derives primarily from in vitro studies and presupposes that the fate and effector function of recruited monocytes are determined as bulk populations of cells by differentiation factors within an inflamed tissue microenvironment, and that macrophages can be “switched” from M1 to M2, and vice versa, either by manipulating factors in the tissue microenvironment or by altering intracellular signaling or transcriptional activity within macrophages that regulate the cell’s M1 or M2 identity. Studies that apply the M1-M2 dichotomy to CNV pathobiology generally embrace the concept that M2 macrophages mediate CNV formation and growth.
Investigators from the monocyte ontogeny field have proposed an alternative paradigm: Functionally distinct monocyte subsets can be identified in the blood according to specific cell surface markers (Geissmann et al., 2003; Woollard and Geissmann, 2010), which include Ly6C in mice and CD14 and CD16 in humans. In mice, “classical” Ly6Chi monocytes (~80% of normal blood monocytes) have high expression of CCR2, low expression of CX3CR1, and are inflammatory, expressing high levels of TNF-α, IL-1β, NOS2, and proteases (Nahrendorf et al., 2007). This subset tends to predominate in the normal physiologic unperturbed “steady state.” “Nonclassical” Ly6Clo monocytes (~10%) have low expression of CCR2, high expression of CX3CR1, and in the setting of infection, injury, or illness, serve a reparative function, expressing pro-fibrogenic and vascular factors such as TGF-β, osteopontin, IGF-1, FGF, CTGF, VEGF and others (Nahrendorf et al., 2007; Donnelly et al., 2011; Hanna et al., 2012).
This alternative paradigm presupposes that monocytes are pre- programmed to one or the other subset within bone marrow, spleen, or lymphoid tissue. Thus, circulating monocytes in the blood are committed to specific effector functions and are triggered to mediate these functions upon tissue entry. While Ly6Chi monocytes enter tissue early in response to injury or infection, Ly6Clo monocyte tissue entry is delayed (i.e., during a second phase) (Nahrendorf et al., 2007; Nahrendorf and Swirski, 2013), creating a biphasic response of early inflammatory response followed by later reparative response. In humans, these subsets include classical CD14++CD16− , intermediate CD14++CD16+, and nonclassical CD14+CD16+ monocytes (Ingersoll et al., 2010; Wong et al., 2011). However, considerably less is understood about the biology of these subsets in humans, with some investigations suggesting that nonclassical and intermediate subsets may serve pro-inflammatory roles, and the classical subset serving more reparative roles (Ingersoll et al., 2010; Wong et al., 2011). There are variable reports on the roles of Ly6Chi and Ly6Clo monocytes in experimental CNV (Tan et al. 2015, 2016; Liyanage et al., 2016), though based on this paradigm, it is plausible that both subsets participate in CNV formation and growth in a coordinated fashion.
To add to the complexity, some studies suggest a third blended paradigm. Dichotomous blood monocyte subsets are present, but tissue Ly6Clo macrophages are presumed to be derived from Ly6Chi infiltrating monocytes, which undergo a “phenotype switch” into a Ly6Cint-Ly6Clo subset (Arnold et al., 2007; Ramachandran et al., 2012). Importantly, these paradigms are not mutually exclusive and may apply variably to different tissues and different disease states.
We are broadly interested in the different effector biologies of macrophages, scavenging, inflammatory, and reparative, and the contributions of each to NVAMD pathobiology, and especially to NVR. We embrace the concept that blood monocytes can be preprogrammed into functionally distinct subsets, which give rise to distinct inflammatory and reparative macrophage subsets in the tissue. We hypothesize that the effect of these monocytes on CNV pathobiology is driven by the biphasic recruitment of the two subsets, the intrinsic biology of each subset, and the relative numbers of each subset in the blood at the time of recruitment to the incipient CNV lesion. For example, inflammatory Ly6Chi monocytes may be recruited to the choroid and outer retina in response to focal injury where they mediate CNV induction. Meanwhile, higher frequency of circulating reparative Ly6Clo monocytes, a phenomenon that occurs in aging and also in association with certain environmental risk factors such as systemic pathogen exposure and cigarette smoke (Zawada et al., 2012; Hanna et al., 2012; Chen et al., 2019; Rozing et al., 2020), can infiltrate the incipient CNV in a coordinated fashion following inflammatory Ly6Chi monocyte recruitment. These reparative macrophages may then produce increased levels of pro-fibrogenic effector molecules according to their activation state, mediating the biology of NVR.
In support of this conceptual hypothesis and specifically highlighting the importance of reparative macrophages to NVR, Lutty and colleagues identified a high frequency of activated HLA-DR + macrophages in association with arteriolarized CNV in human postmortem NVAMD specimens, with cells extensively localized throughout the interstitium of the lesion and especially at the marginal rim in association with anastomotic loops (McLeod et al., 2016). Separately, our group has observed infiltration of CD163+ reparative macrophages in postmortem CNV specimens from NVAMD patients (Lad et al., 2015). Furthermore, comparative analysis of arteriolar vs. capillary CNV specimens reveals numerous CD163+ macrophages in association with arteriolar CNV with perivascular fibrosis but only rare CD163+ cells in capillary CNV lesions (Fig. 13). Profibrotic factors produced by reparative macrophages, such as transforming growth factor-β (TGF-β), insulin-like growth factor 1 (IGF-1), fibroblast growth factor (FGF), osteopontin, and osteonectin have been found localized to macrophages in fibrotic CNV in histopathology of postmortem specimens (Reddy et al., 1995; Kent and Sheridan, 2003).
Fig. 13.

Reparative macrophage infiltration in postmortem CNV specimens from patients with NVAMD. Dashed white lines delimit CNV, yellow stars indicate RPE (which is autofluorescent in (B)). Scale bar: 100 μm. Capillary CNV (A) have minimal SMA+ (green) staining and minimal CD163+ (red) reparative macrophage infiltration, and CNV lesion remains entirely in sub-RPE space within Bruch’s membrane. In contrast, arteriolar CNV is substantially thicker, extends into subretinal space, with presence of large-caliber SMA+ (green) vessel (arrows on either side, seen in cross-section), and extensive CD163+ (red) macrophage infiltration within the CNV lesion.
2.9.6. Macrophage activation in other models of NVR in experimental CNV
Mechanisms of macrophage activation are clearly multifactorial, involving not only aging but also genetics, systemic health factors, and environmental cofactors including infection (LaVerda et al., 1999; Vliegen et al., 2004; Boyle, 2005; Wen et al., 2018). To demonstrate that the biology of NVR is generalizable beyond the aging model, we have investigated whether other potential factors that have been previously associated with NVAMD can promote macrophage-mediated NVR in experimental CNV in association with monocyte/macrophage activation (Suñ;er et al., 2004; Cousins et al., 2012; Mettu et al., 2014).
2.9.6.1. Latent CMV infection of macrophages.
Cytomegalovirus (CMV) is a common virus that infects people of all ages. While primary infection causes little to no symptoms in most people, it is frequently followed by establishment of persistent or latent infection, including in myeloid lineage cells and monocytes that give rise to activated macrophage cell populations in tissues (Hahn et al., 1998; Sweet, 1999). We assessed CMV IgG titers in a case-control study of dry AMD patients, NVAMD patients, and age-matched control subjects without AMD and found that highest titers of CMV IgG were significantly associated with NVAMD, as compared to dry AMD and controls, suggesting increased risk of NVAMD in patients with latent CMV infection (Miller et al., 2004). To understand the role of latent CMV infection in CNV pathobiology, we assessed the effects of latent murine CMV (MCMV) in 7–8 month old mice in the laser-induced CNV model and found that mice with latent MCMV infection exhibited larger CNV lesions with arteriolarization and fibrosis, as compared to control mice receiving UV-inactivated MCMV exposed control mice that had small capillary CNV (Cousins et al., 2012) (Fig. 14). In MCMV-infected mice, lesion size and extent of NVR increased with duration of latent MCMV infection, with maximal effect observed at 12 weeks duration of MCMV infection prior to laser induction. Chronic latent MCMV was not detected in the choroid and RPE of infected mice, indicating that the effects of CMV in formation of CNV are not due to local latent infection within ocular tissues. However, MCMV was detectable in splenic macrophages and bone marrow progenitor cells, and gene expression analysis of splenic macrophages isolated from mice with chronic MCMV infection (12 weeks) demonstrated upregulation of pro-inflammatory, pro-fibrogenic, and pro-angiogenic factors. Moreover, CMV infection has been shown to induce increased numbers of CD163+ reparative monocytes/macrophages that express primarily pro-fibrogenic factors and retain expression of only few pro-inflammatory effector cytokines (Nikitina et al., 2018). We also found that treatment of MCMV-infected mice with the anti-viral ganciclovir suppressed observed gene upregulation in splenic macrophages (Cousins et al., 2012), indicating that macrophage activation was directly mediated by latent MCMV infection.
Fig. 14.
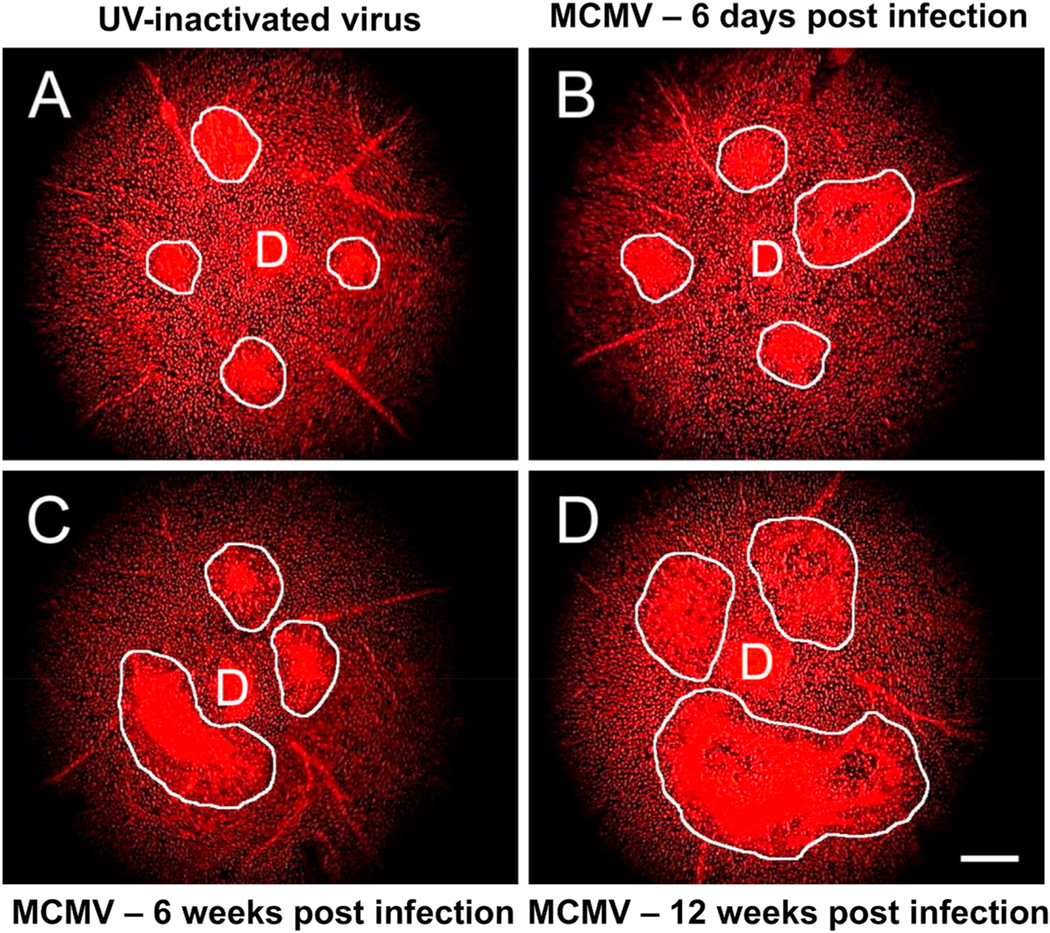
Mice with latent MCMV infection develop more severe arteriolar CNV. Groups of adult 7–8 month old C57BL/6 mice were inoculated intraperitoneally with either MCMV or UV-inactivated virus. At 6 days, 6 weeks, or 12 weeks after inoculation, all mice were subjected to laser treatment to induce CNV, and, four weeks later, flatmount preparations of the posterior pole of mouse eyes were stained with propidium iodide. Flatmount preparations of representative individual mouse eyes showing areas of CNV (white outlines). (D = Optic Disc) (Magnification = 50 ×) (Scale bar = 100 μm)) (A) Mouse inoculated with UV-inactivated MCMV (control) for 12 weeks prior to laser treatment. (B) Mouse infected with MCMV for 6 days prior to laser treatment. (C) Mouse infected with MCMV for 6 weeks prior to laser treatment. (D) Mouse infected with MCMV for 12 weeks prior to laser treatment.
In this model, as latent MCMV-infected myeloid precursor and monocyte cells mature, it is likely specific macrophage effector genes become transactivated by CMV immediate-early gene products that are expressed during latency; these viral immediate-early gene products effectively “hijack” macrophage gene expression and prime them toward reparative effector function (Stenberg, 1996; Hahn et al., 1998; Cinatl et al., 1999; Slobedman et al., 2002). These activated infected monocytes then produce higher levels of pro-fibrogenic factors upon recruitment to the site of RPE/Bruch’s membrane injury, mediating the biology of NVR. Ongoing work in our laboratory is centered on identifying the specific immediate-early gene products that prime precursor cells and monocytes for NVR biology and on characterizing the macrophage-derived factors that specifically mediate NVR in the setting of latent MCMV infection.
2.9.6.2. Pathogen burden and “PAMP” stimulation of macrophages.
Infectious pathogens may modulate AMD disease pathobiology through additional means. One possibility is that the pathogen may directly infiltrate and infect the locus of disease at the RPE, Bruch’s membrane, and choriocapillaris. This hypothesis has been put forth by investigators who detected evidence of Chlamydia pneumoniae in CNV tissue from patients with NVAMD, both by immunohistochemistry (IHC) and polymerase chain reaction (PCR), in contrast to no detectable pathogen in non-AMD tissue specimens (Kalayoglu et al., 2005). While this study raised the intriguing possibility that direct infection may contribute to AMD pathobiology, there is limited evidence for other pathogens detected within CNV and limited additional evidence specifically for C. pneumoniae direct infection in NVAMD (Robman et al., 2007; Guymer and Robman, 2007).
An alternative and distinct concept is total pathogen burden, wherein systemic immune alterations observed in association with AMD occur as a result of cumulative infections with multiple pathogens over the course of life (Zhu et al., 2000). In this hypothesis, immune alterations are not due to one single pathogen or infection; multiple pathogens carry more and cumulative risk (Zhu et al., 2000). Accordingly, we have observed that patients with elevated serum antibody titers against multiple pathogens, including C. pneumoniae and Helicobacter pylori, had increased risk of NVAMD (Miller et al., 2004). The concept of total pathogen burden can be understood through priming effects on the innate immune system. Chronic exposure to microbial components can prime, or partially activate, monocytes in spleen, lymph node, bone marrow, or other sites of exposure or surveillance, altering the expression of certain cytokines and mediators and committing exposed cells to specific effector functions (Obonyo et al., 2007; Ge et al., 2013). These functions then become fully manifest upon recruitment to the disease locus (i.e., RPE and retina) and transformation into fully activated macrophages. This phenomenon has been recognized in diseases such as atherosclerosis, colonic inflammation, and kidney fibrosis (Yang et al., 2008; Campbell et al., 2011; Falck-Hansen et al., 2013). Of more direct relevance, the presence of periodontal disease, which is a known primer of circulating monocytes, has been independently associated with AMD, even after controlling for other more established risk factors (Wagley et al., 2015).
To understand whether the concept of total pathogen burden was potentially relevant to biology of NVR, we studied the effects of systemic pathogen-associated molecular patterns (PAMP) exposure in the murine laser-induced CNV model (Mettu et al., 2014). PAMPs are microbe-associated molecules that are recognized by monocytes/macrophages via cell-surface pattern recognition receptors (PRRs, including the family of toll-like receptors (TLRs)). PAMP binding to PRRs on circulating monocytes primes increased expression of cytokines and soluble mediators (Obonyo et al., 2007; Ge et al., 2013). We exposed 7–9 month old mice to low levels of the PAMP lipopolysaccharide (LPS) via intraperitoneal injection, at levels well below those associated with toxicity, at day − 1 prior to laser induction and again at day +1 (Fig. 15). We subsequently observed that exposure to LPS promoted development of larger CNV lesions with features of arteriolar CNV, especially increased leakage and greater number of SMA + large-caliber arterioles, consistent with the biology of NVR (Fig. 15A-B-C). Similar findings were observed for systemic exposure to low levels of zymosan and TSST-1. Development of NVR in the setting of low-grade systemic LPS exposure was associated with increased frequency of circulating nonclassical Ly6Clo monocytes in the blood at day 3 post-laser, the time point at which NVR biology is established at the incipient CNV lesion (Fig. 16). These data support the hypothesis that total pathogen burden may promote NVR via priming of circulating monocytes. Another non-mutually exclusive mechanism that may contribute to the observed findings is that PAMPs may modulate disease via direct interaction with PRRs expressed by the RPE (e.g. TLR2, which has been shown to be expressed at the RPE apical surface (Feng et al., 2017)).
Fig. 15.

Mice exposed to low level of lipopolysaccharide (LPS, 10 μg) by intraperitoneal injection prior to laser induction develop more severe arteriolar CNV. LPS-stimulated mice (7–9 month old) demonstrate (A) prominent leakage by fluorescein angiogram (FA), (B) large-caliber arterioles (arrows) with vascular loops (arrowheads) by TRITC-lectin flatmount, and (C) increased density of SMA + arterioles by cross-sectional immunofluorescence. In contrast, PBS control-exposed mice have (D) small lesions with mild leakage by FA, (E) capillary morphology by flatmount; and (F) few SMA + vessels. Larger size was confirmed by (G) quantitative analysis of flatmounts (**p < 0.05).
Fig. 16.

Development of Arteriolar CNV in LPS-stimulated mice is associated with increased frequency of nonclassical monocytes. The composition of blood monocyte populations was assessed at day 3 following laser induction, the time period when neovascular remodeling biology is believed to occur. Whereas PBS-exposed mice with capillary lesions had an average ratio of 11% nonclassical Ly6Clo to 89% classical Ly6Chi blood monocytes, LPS-exposed mice had a ratio of 36% nonclassical Ly6Clo to 64% classical Ly6Chi blood monocytes (p < 0.01). Further, LPS exposure produced a major change in absolute number of Ly6Clo blood monocytes: LPS-exposed mice - 4978 cells/μL of blood versus PBS-exposed mice - 727 cells/μL (p < 0.01).
2.9.6.3. Cigarette smoking and nicotine exposure.
Cigarette smoking is the environmental and systemic health factor with the greatest risk for onset and severity of all forms of AMD (Smith et al., 1996), and has been associated with a two-to four-fold increased incidence of NVAMD (Klein et al., 1993; Christen et al., 1996; Seddon et al., 1996; Smith et al., 1996; Tamakoshi et al., 1997; Delcourt et al., 1998). Since nicotine has been shown to contribute to many aspects of toxicity from cigarette smoke (Villablanca, 1998; Jain, 2001; Heeschen et al., 2001) and since both smoking and nicotine have been shown to alter monocyte activation state and promote pro-fibrogenic effector functions in macrophages (Yuan et al., 2014; Yang and Chen, 2018), we have also assessed the effects of nicotine exposure and also cigarette smoke exposure in the laser-induced CNV model (Suñ;er et al., 2004; Suñ;er et al., 2005). Each of nicotine and cigarette smoke exposures promoted larger lesions consistent with NVR biology as compared to non-exposed controls. Cigarette smoking exposure was associated with increased infiltration of activated macrophages to the incipient CNV lesion while nicotine exposure potentiated PDGF-mediated proliferation of choroidal VSMC in vitro and reversed VEGF-induced suppression of MMP-2 activity, suggesting that multiple mechanisms may promote NVR biology in these models. Further study is needed to clarify the effects of cigarette smoke on recruitment and activation of classical and nonclassical monocyte subsets. Other investigators have demonstrated that nicotine exposure produces findings similar to cigarette exposure in experimental murine CNV (Hou et al., 2008; Davis et al., 2012).
Collectively, findings from these investigations affirm the key role of blood-derived macrophages in the development of arteriolar CNV and also demonstrate that certain risk factors for NVAMD can serve as activational stimuli of macrophage effector biology to promote NVR.
2.9.7. Regulation of macrophage effector biology and recruitment in the setting of NVR
Several other studies have assessed specific mechanisms that may contribute to the regulation of macrophage effector function in the setting of NVR. IL-10 is a cytokine that mediates the recruitment and activation of CD163+ reparative macrophages and that upregulates angiogenic and pro-fibrogenic factor expression (Fiorentino et al., 1991; Sun et al., 2011). Apte and colleagues have shown that IL-10 expression is increased in old mice and that targeted inhibition of the IL-10 receptor prevents the severe CNV phenotype (Nakamura et al., 2015). Additionally, markers of reparative macrophages (CD163, Arg1) were downregulated while markers of inflammatory macrophages (TNF-α, IL-6) were elevated, suggesting either a shift in the recruitment of monocyte/macrophage subsets or a shift in macrophage effector biology. They also demonstrated that downstream activation of the transcription factor STAT3 mediates the effects of IL-10 on macrophage effector function in the setting of NVR in old mice, and that STAT3 activation is detectable within CD163+ macrophages in postmortem CNV specimens from NVAMD patients, indicating that IL-10/STAT3 signaling in macrophages may be a regulatory mechanism for macrophage effector function in NVR. This is of particular interest since patients with NVAMD have been shown to have increased expression of STAT3 in intermediate CD14++CD16+ monocytes, suggesting a potential contribution of IL-10/STAT3 signaling to disease phenotype in NVAMD (Chen et al., 2016).
Apte and colleagues also identified impaired cholesterol efflux in macrophages as another potential mechanism that may upregulate reparative effector functions in macrophages, as macrophage-specific deletion of ABCA1, a major cholesterol efflux regulatory protein, resulted in more severe CNV in the laser model, and systemic treatment with LXR agonists, which restore cholesterol efflux, reduced CNV severity in old wild-type mice (Sene et al., 2013). Other mechanisms that may regulate NVR may include modulation of macrophage Rho-associated kinase (ROCK2) (Zandi et al., 2015), and activation of macrophage RIP1 kinase (Ueta et al., 2019), as both pathways have been shown to activate reparative macrophage effector biology in the setting of CNV formation and growth.
In terms of macrophage recruitment and infiltration, both MCP-1 and IL-8 (Kent and Sheridan, 2003) as well as IL-6 may mediate the recruitment of monocytes from the circulation to the incipient CNV (Izumi-Nagai et al., 2007). There is also evidence to support a role for the leukotriene B4 (LTB4)-leukotriene B4 receptor 1 (BLT1) signaling axis, as blockade of LTB4 reduces macrophage infiltration (Sasaki et al., 2018). In the laser CNV model, soluble Fas ligand (sFasL) released from injured RPE is a key mediator that promotes monocyte recruitment and subsequent macrophage activation at the incipient CNV lesion, and this effect is particularly pronounced in old mice (Zhao et al., 2013). Inhibition of complement factor C3 reduces CNV macrophage infiltration, suggesting that complement components may serve as stimuli of inflammatory or reparative macrophage infiltration and activation (Tan et al., 2015).
Activation of the erythropoietin (EPO)/erythropoietin receptor (EPOR) signaling axis in the murine laser CNV model has also been shown to promote increased CNV severity (independent of changes in blood hematocrit) (Bretz et al., 2018). Beyond its well established role in the production of red blood cells (RBCs), EPO has been shown to modulate the biology of various cell types that express EPOR, including macrophages, and has also been shown to promote increased numbers of bone marrow-derived cell types in the systemic circulation (Lifshitz et al., 2010 et al.). Hartnett and colleagues utilized distinct humanized knock-in mouse models, human wild-type EPOR (hWtEPOR) knock-in mice with hypoactive EPOR signaling, and human mutant EPOR (hMtEPOR) knock-in mice with overactive EPOR signaling, and demonstrated that overactive EPOR signaling was sufficient to increase CNV severity in hMtEPOR mice, with increased EPOR signaling associated with 1) increased choroidal macrophage density in hMtEPOR mice, as compared to hWtEPOR mice; 2) increased CNV lesional macrophage infiltration in EPOR-overactive hMtEPOR mice vs. EPOR hypoactive hWtEPOR mice; and 3) increased expression of macrophage-derived effector cytokines (e.g. IL-6, CCL2, etc.) in RPE/choroid of EPOR-overactive hMtEPOR mice (Bretz et al., 2018). Thus, increasing levels of serum EPO and increased EPO/EPOR signaling at monocytes and macrophages could promoted macrophage-mediated CNV severity through increased macrophage recruitment and activation.
Collectively, these data suggest that there may be multiple pathways and mechanisms, rather than a single master factor, that regulate macrophage effector functions and that regulate macrophage infiltration and monocyte recruitment from the systemic circulation in NVR.
2.9.8. Macrophages, NVR, and PDA in NVAMD
To our knowledge, there have not been translational studies that specifically explore the relationship among macrophage biology, clinical morphologic subtypes of NVAMD, and anti-VEGF treatment response. However, available clinical correlative studies offer some clues. Several clinical studies demonstrate that increased intraocular levels of macrophage-derived cytokines are associated with more active (i.e., exudative or leaking) CNV, as compared to eyes with inactive CNV, suggesting a link between macrophage biology and ongoing CNV disease activity; clinicopathologic studies similarly suggest a connection between reparative macrophage infiltration and disease activity (Chalam et al., 2014; Rezar-Dreindl et al., 2016; McLeod et al., 2016). Additional investigations have observed that the proportion of circulating CD11b + monocytes directly correlate with the frequency of anti-VEGF injections necessary for disease control (Subhi et al., 2019). However, local ocular corticosteroids do not improve treatment response or mitigate anti-VEGF resistance and do no prevent perivascular fibrosis and CNV-associated scar formation (Ambati et al., 2013). This is likely due to the fact that corticosteroids have limited effects on the effector function of CD163+ reparative macrophages (Schaer et al., 2002). These observations highlight the need for novel anti-inflammatory drugs that specifically target reparative macrophage biology, to address the problem of anti-VEGF resistant NVAMD.
2.10. Contributions of macrophages to other aspects of CNV pathobiology
In addition to NVR, macrophages have been shown to contribute to other aspects of CNV pathobiology, including CNV induction, or conversion from dry AMD to NVAMD, as well as capillary angiogenesis.
2.10.1. CNV induction: conversion to NVAMD
CNV induction is defined as the early cellular and molecular events that trigger the development of the incipient new vessel, which clinically represents conversion to, or onset of NVAMD. The specific mechanisms that trigger CNV induction in NVAMD remain unknown. Several investigators have advanced a choroidal ischemia or hypoperfusion hypothesis, based on the concept that loss of choriocapillaris and normal choroidal blood flow causes local ischemia that then triggers pathologic neovascularization as a compensatory response (Grunwald et al., 1998; Coleman et al., 2013). However, alterations in choroidal blood flow and choriocapillaris dropout are frequently present in dry AMD in the absence of CNV, and frequently, CNV develop in AMD eyes without clinical or pathologic evidence of hypoxia or ischemia (Mullins et al., 2011; Seddon et al., 2016). Thus, the precise role of choroidal ischemia or hypoperfusion in the induction of CNV remains uncertain.
An alternative stimulus for CNV induction is macrophage-mediated inflammation. Numerous histopathologic studies, including Sarks et al., have demonstrated the presence of macrophages in association with the leading edge of CNV vascular structures, adjacent to or within a thinned and irregular Bruch’s membrane (Sarks et al., 1997). These observations suggest that macrophages might promote the initial development of neovessels via release of angiogenic factors, inflammatory cytokines, or matrix-degrading enzymes, to mediate the initial response to the locus of injured or diseased tissue. Macrophages have also been observed in close association with endothelial progenitor cells (EPCs) in surgically excised CNV specimens (Sheridan et al., 2006), suggesting that macrophages may directly participate in the early recruitment of cellular components to the incipient CNV. In the experimental model of murine laser-induced CNV, pretreatment with intravitreal corticosteroid inhibits incipient CNV development and completely suppresses new formation, with prevention of inflammatory monocyte/macrophage infiltration, suggesting that preventing inflammatory cell infiltration can suppress CNV induction (Ishibashi et al., 1985; Ciulla et al. 2001, 2003).
Data from clinical studies of NVAMD patients also support the concept that macrophages play a key role in CNV induction and conversion to NVAMD. In a case-control study, we isolated circulating monocytes from the peripheral blood of patients with dry AMD, patients with NVAMD, and age-matched control subjects, to assess whether monocyte expression of the inflammatory cytokine TNF-α was associated with disease state (Cousins et al., 2004). We observed a wide variability in monocyte TNF-α expression among different patients, indicating a heterogeneity in monocyte activation state in the general population. Additionally, we observed that patients with monocytes expressing high levels of TNF-α demonstrated the highest prevalence of NVAMD, suggesting that pro-inflammatory monocyte activation state, as reflected by increased expression of TNF-α, is associated with increased risk of NVAMD as compared to patients with dry AMD. In a subsequent case-control study to assess potential association of NVAMD with other monocyte markers, we reproduced this finding, affirming monocyte TNF-α as a key marker associated with NVAMD, and we also found that reduced monocyte expression of complement receptor 1 (CR1) was independently and significantly associated an increased risk of NVAMD. CR1 is found on monocytes and macrophages and serves as a receptor for C3b that when bound, endocytoses and sequesters C3b, effectively limiting activation of complement and downstream inflammatory cascades (Carroll, 2004); potential roles for complement activation in AMD have been extensively reviewed (Toomey et al., 2018). Based on this rationale, monocyte CR1 may serve a primary protective function against NVAMD development by direct negative regulation of complement activation at the locus of disease in dry AMD, and decreased CR1 expression on inflammatory macrophages could thus be permissive for NVAMD progression. Thus, taken together, we would infer that the inflammatory monocytes/macrophages mediate CNV induction. Indeed, using the experimental model of murine laser-induced CNV, several investigators have demonstrated a key role for CCR2+ inflammatory monocytes (which express TNF-α, IL-1β, NOS2, VEGF, and proteases such as MMP-9) in the early development of CNV, as genetic deletion of CCR2 and reduction of this inflammatory monocyte subset substantially reduces CNV induction (Tsutsumi et al., 2003; Lavalette et al., 2011; Liu et al., 2013; Krause et al., 2014). Evaluation of monocyte activation state as a predictor of conversion to NVAMD in a prospective cohort study of dry AMD patients as well as investigations of monocyte subsets in patients with NVAMD is needed to further support this hypothesis.
Other investigators have demonstrated that monocytes from NVAMD patients have altered transcriptomes as compared to controls (Grunin et al., 2016). In addition, inflammatory cytokine receptors CCR1 and CCR2 are co-upregulated on intermediate CD16+ monocytes from NVAMD patients (Grunin et al., 2012) and CD200 upregulated on circulating CD11b + monocytes in NVAMD patients as compared to controls (Singh et al., 2013). Interestingly, in our second case-control study, we found that monocyte VEGF expression was not associated with NVAMD. To further add to this perspective, the PRO-CON clinical trial found that intravitreal anti-VEGF treatment with aflibercept was not efficacious as prophylactic treatment against progression to NVAMD, as compared to sham injection, in high-risk fellow eyes with dry AMD, when assessed either at 1 year or 2 years of treatment (Heier, 2019; Puliafito and Wykoff, 2019). Taken together, these data collectively support the concept and hypothesis that the specific biologic triggers of NVAMD progression and incipient CNV induction include macrophage-mediated inflammation, and further supports exploring the role of other key inflammatory and immune-mediated mechanisms as potential causes of conversion to NVAMD disease. Based on our findings, we speculate that pro-inflammatory monocyte/macrophages may directly trigger NVAMD onset, promoting incipient CNV induction via release of inflammatory meditator such as TNF-α and perhaps other cytokines such as IL-1β or IL-6, enabling subsequent new vessel formation. These data also highlight the potential utility of targeted anti-inflammatory drugs as potential therapies to prevent progression to NVAMD and provide a sound rationale for case-control studies assessing rates of NVAMD in patient populations receiving anti-inflammatory biologic drugs for systemic (e.g. rheumatologic) diseases. In this regard and of relevance to our findings, epidemiologic studies suggest a decreased risk of NVAMD among rheumatoid arthritis patients receiving anti-inflammatories, including targeted anti-TNF-α therapies (McGeer and Sibley, 2005).
2.10.2. Macrophage biology and capillary angiogenesis
While our findings indicate that capillary formation can occur in the absence of blood-derived macrophages in old mice, this does not preclude the possibility that macrophages might still contribute to capillary angiogenesis. The vast majority of studies of experimental laser-induced CNV in the literature assess CNV biology in unmanipulated, wild-type young (i.e., 2–4 month old) mice, which, as we have demonstrated, manifest capillary CNV lesions via angiogenesis and maturation. Numerous studies of young mice have found that following initial CNV induction, macrophages are recruited to the site of incipient neovessel formation and contribute to capillary angiogenesis and maturation via release of growth factors (Liu et al., 2013; Krause et al., 2014). Inflammatory macrophages recruited to the incipient CNV following induction can promote angiogenesis either directly via release of VEGF or indirectly via release of inflammatory cytokines, which stimulate the RPE to produce VEGF and macrophage recruitment factors such as MCP-1 and IL-8 (Kent and Sheridan, 2003). In young mice, CD163+ reparative macrophages begin to infiltrate the lesion at day 4 and express high levels of factors such as VEGF, PDGF, FGF-1, Ang-1, Ang-2, and IL-10, as histopathology with immunostaining of CNV in the laser model demonstrate localization of angiogenic factors with reparative macrophages (Kent and Sheridan, 2003; Li et al., 2017).
Beyond a potential role in NVR biology, IL-10 activation of reparative CD163+ macrophages may also contribute to the biology of capillary angiogenesis, as genetic deletion of IL-10 has also been shown to reduce capillary CNV size in the laser-induced CNV model (Apte et al., 2006; Kelly et al., 2007). Multiple studies have also demonstrated that macrophages may also contribute to capillary angiogenesis, as depletion of circulating monocytes by systemic clodronate administration or by systemic monocyte or local macrophage depletion using CD11b + -DTR system in young mice can reduce capillary CNV lesion size (Sakurai et al., 2003; He and Marneros, 2013).
In correlative studies of NVAMD, circulating monocytes of NVAMD patients have been shown to express high levels of VEGF relative to controls (Chen et al., 2016; Lechner et al., 2017), suggesting that blood-derived macrophages may directly contribute to angiogenesis in NVAMD, while other data suggest that monocytes from NVAMD patients produce higher levels of MCP-1 and IL-8, both macrophage chemotactic factors, indicating that recruited macrophages may also indirectly amplify angiogenesis via ongoing recruitment of monocytes to the site of CNV formation (Lechner et al., 2017). Thus, infiltrating macrophages, of both the inflammatory and reparative subsets, may contribute to capillary CNV formation via production and secretion of relevant growth factors and cytokines that promote and amplify angiogenesis and maturation. This has relevance to understanding the basis of not only a minority subset of NVAMD disease with capillary lesions but also for patients with CNV in the setting of pathologic myopia or secondary to ocular histoplasmosis, who are younger in age and typically manifest Capillary pattern CNV (Wong et al., 2015; Wood et al., 2018).
2.11. Bone-marrow derived mesenchymal precursor cells confer susceptibility to NVR
One of the key distinctions between arteriolar and capillary CNV phenotypes is the type of mural support cell. While pericytes are the predominant mural cell in capillary, development of arteriolar CNV requires the recruitment and activation of VSMCs and collagen-producing myofibroblasts via macrophage-derived pro-fibrogenic factors, as we have demonstrated in previously described studies. Our group has also had a longstanding interest in the origination of various cells types within CNV, including cells that give rise to mural support cells: pericytes in capillary angiogenesis and VSMCs and myofibroblasts in NVR (Espinosa-Heidmann et al. 2003b, 2005; Csaky et al., 2004; Reinoso et al., 2004). In the conventional paradigm for new vessel formation, VSMCs and myofibroblasts arise from one of three local sources within the eye: 1) activated pericytes and VSMCs resident at existing choroidal arteries or arterioles (Luo et al., 2018); 2) activated choroidal fibroblasts; or 3) RPE cells that undergo epithelial to mesenchymal transition (EMT) (Little et al., 2018). Alternatively, we have found that a major (fourth) source of mural support cells in the laser CNV mouse model is the bone marrow (Espinosa-Heidmann et al. 2003b, 2005, 2013).
It is well established that bone marrow contains pluripotent stem cells, which can differentiate into precursor cells for blood cells (leukocytes, red blood cells, platelets). However, bone marrow also contains precursors that for cell types that can form blood vessels, including endothelial precursor cells (EPCs), which become endothelial cells, and mesenchymal precursor cells (MPCs), which can become blood vessel mural support cells: pericytes or VSMCs and myofibroblasts (Asahara et al., 1999; Sata et al., 2002). These cells can exit the bone marrow, enter the circulation, and are recruited to sites of new vessel formation where they differentiate into mature cells, in situ—a phenomenon called postnatal vasculogenesis.
MPCs have been shown to arise from bone marrow hematopoietic precursor cells that are lineage-negative (Lin-) (i.e., negative for lineage markers of mature cells) and that may express one or more “stem” cell surface markers, such as c-kit (CD117), Sca-1, CD34 (Sata et al., 2002; Czarna et al., 2017; Bali et al., 2018). Studies evaluating MPCs have identified the potential of multiple Lin-bone marrow cell populations to give rise to mural support cells, with comparisons across studies made additionally complex by differences in experimental methodologies and models. Thus, consensus markers for bone marrow-derived MPCs in postnatal vasculogenesis have not been definitively established. Importantly, however, MPCs are distinct from mesenchymal stem cells, which are multipotent stromal cells that can differentiate into osteocytes, myocytes, and adipocytes, but not hematopoietic cells (Pittenger et al., 1999).
Our group was the first to demonstrate that bone marrow-derived precursor cells are a source of endothelial and mural support cells in experimental CNV. In analysis of laser-induced CNV in GFP bone marrow chimeric mice (i.e., transplant of GFP-labeled bone marrow to wild-type recipient mice following lethal irradiation), we have observed that CNV contain abundant bone marrow-derived (GFP-labeled) cells that give rise to both endothelial cells and to pericytes and smooth muscle cells in both old and young mice (Espinosa-Heidmann et al., 2003b). We affirmed the contribution of bone marrow-derived EPCs in a separate model of murine CNV, subretinal injection of adenoviral vector expressing VEGF165 (Ad.VEGF165). Lethally irradiated female recipient mice of bone marrow—isolated from male transgenic mice expressing LacZ driven by the endothelial cell specific Tie-2 promoter—had CNV in which 27% of lectin-positive endothelial cells were found to be bone marrow-derived (Csaky et al., 2004).
To characterize the effects of aging on the relative contributions of EPCs and MPCs to CNV phenotype, we undertook a series of studies in the experimental laser CNV, performing transplantation of bone marrow (BMT) from old (16 month old) GFP + donor mice to wild-type young (2 month old) recipient mice and the reciprocal transplantation of young GFP + donor bone marrow to old wild-type recipients (Figs. 17–19) (Espinosa-Heidmann et al., 2013). We observed that BMT from old GFP + donor mice to young recipient mice transferred the Arteriolar phenotype in the laser CNV model (Fig. 17F), whereas control young mice recipients of young GFP + donor bone marrow retained the Capillary phenotype (Fig. 17B) (Espinosa-Heidmann et al., 2013). CNV lesions of young mice recipients of old GFP + donor bone marrow contained approximately 2.5-fold more marrow MPC-derived SMA + VSMCs and myofibroblasts (Fig. 18B and C, lower graph), as compared to CNV lesions of control young mice recipients of young GFP + donor bone marrow (Fig. 18A and C, lower graph), in association with greater arteriolarization and increased deposition of collagen within CNV. The proportion of marrow EPC-derived CD31+ endothelial cells in young mice recipients of old GFP + donor bone marrow was not significantly different as compared to control young mice recipients of young GFP + donor bone marrow (Fig. 18C, upper graph). We also observed that reciprocal transplant of young GFP + donor bone marrow into old wild-type recipients transferred the Capillary phenotype into old recipient mice (Fig. 17C), with few MPC-derived VSMCs and myofibroblasts in old recipients, whereas control old mice recipients of old GFP + donor bone marrow retained the Arteriolar phenotype (Fig. 17E). We found that old bone marrow contains much greater frequency of the SM22+ MPC subtype that gives rise to vascular smooth muscle cells and myofibroblasts than does young marrow, and that MPCs label CD34+ (Espinosa-Heidmann et al., 2013). Additionally, we observed that adoptive transfer of old donor Lin-, CD34+ precursor cells into young recipient mice at the time of laser induction (without irradiation or preceding bone marrow transplant) was sufficient to transfer the arteriolar CNV phenotype, as compared to control adoptive transfer of young donor Lin-, CD34+ precursor cells into young recipient mice, which did not alter the Capillary phenotype (Fig. 19) (Espinosa-Heidmann et al., 2013).
Fig. 17.

Bone marrow transplant confers susceptibility to neovascular remodeling in experimental CNV. Small capillary lesions developed in young unmanipulated mice (A) and also when young BM was transplanted into either young ([B], Y-to-Y) or old mice ([C], Y-to-O). In contrast, old unmanipulated mice (D) and the old-to-old group (E) developed large neovascular lesions that grew to confluence due to extensive growth between adjacent lesions. Additionally, young recipients receiving old BM ([F], O-to-Y) developed intermediate-size lesions, frequently with confluent growth. (G) Quantitative analysis of the surface area of CNV lesions in each group. Significant differences were observed (asterisks) when old BM was transplanted in both young and old recipients in comparison with transplantation of young BM (t-test: P < 0.001). D, optic disc. White line encircles CNV lesions. Magnification: × 50; scale bar: 200 μm.
Fig. 19.

Analysis of CNV lesions in young animals receiving adoptive transfer of CD34+-GFP-labeled cells. CD34+ precursor cells obtained from young or old donors were adoptively transferred to young recipient mice at the time of laser-induced CNV (without prior irradiation or BMT). (A) Mice receiving adoptive transfer of CD34+ cells isolated from a young donor display typical small CNVs. (B) In contrast, animals receiving adoptive transfer of CD34+ cells from old donors developed arteriolar CNVs, larger in size. D, optic disc; white lines encircle CNV lesions. Magnification: × 50; scale bar: 200 μm. (C) Quantitative analysis of surface area showed a significant size increase (asterisk) in animals engrafted with CD34+ cells from old donors as compared with young donors. (D) Immunohistochemistry of mouse eye cross sections with CNV lesion showed recruitment and engraftment of adoptively transferred GFP + cells (arrows) within 3 days after CNV induction. GFP, green; DAPI, blue. Magnification: × 400; scale bar: 20 μm. (E) Quantitative analysis of cellular density after adoptive transfer of young or old CD34+ cells versus young or old macrophages (F4/80). Mice receiving old CD34+ cells developed CNV lesions that are approximately double in size (asterisk) when compared to those in the other groups. Adoptive transfer of young or old F4/80+ splenic macrophages failed to induce any increase in severity, similarly to adoptive transfer of CD34+ cells from old marrow transferred 7 days after CNV induction.
Fig. 18.

Immunofluorescence detection of resident or recruited BM-derived SMA-expressing perivascular mesenchymal cells in CNV lesions after bone marrow transplant (BMT). Young mice received young (A) or old (B) GFP BM, followed by laser-induced CNV. Although both groups demonstrated resident SMA-expressing cells (red, arrowheads) and GFP-labeled BM-derived cells, many more double-positive cells (yellow, arrows), representing BM-derived SMA/GFP-expressing cells, were observed in cross sections from mice receiving old marrow. Quantification of the frequency of total, resident, and BM-derived CD31 endothelial cells ([C], top) showed no difference in resident or BM-derived CD31-expressing endothelial cells between mice receiving young or old marrow. In contrast, significant differences (asterisks) were observed in the frequency of both resident and BM-derived SMA-expressing cells in CNV of mice receiving marrow from young versus old donors ([C], bottom). In particular, mice receiving old marrow had a 2.5-fold increase in marrow-derived SMA-expressing perivascular mesenchymal cells, contributing to nearly half of all SMA-expressing cells in the CNV. SMA, red; GFP, green; colocalization of GFP and SMA, yellow; DAPI, blue. Magnification: × 400; scale bars: 20 μm.
Collectively, these studies of bone marrow-derived precursors indicate that: (1) aging increases the relative frequency of the MPC subtype in bone marrow that gives rise to VSMC and myofibroblasts; (2) MPCs circulate and are recruited to incipient CNV; and (3) within CNV, the MPC subtype predominant in old mice, provisionally identified as Lin-, CD34+, SM22+, differentiates into VSMCs and myofibroblasts, contributing to arteriolarization and fibrosis, while the MPC subtype predominant in young mice gives rise to pericytes, which participate in capillary angiogenesis and maturation. (4) taken together with macrophage biology in laser CNV (Fig. 11) (Espinosa-Heidmann et al., 2003a) and the investigation of early macrophage recruitment and activation of VSMCs and myofibroblasts (Fig. 12), CNV-infiltrating macrophages are most likely the primary cells mediating recruitment and activation of MPCs into CNV. The clinical implications of these findings are that circulating MPCs may play a role in the regulation of NVR. We have developed a method to isolate MPCs from blood of patients and grow them in culture, characterizing functional markers by gene expression analysis and assessing response to pro-fibrogenic factors to affirm their identify as MPCs. Preliminary work has identified circulating MPCs as Lin-, NG2+, SMemb+ and efforts are underway to identify additional putative markers and analyze gene expression of MPCs. In ongoing work in our laboratory, we are testing the hypothesis that there are two distinct subtypes of MPCs, assessing whether NVAMD patients with arteriolar phenotype and PDA, as compared to NVAMD eyes with capillary phenotype and anti-VEGF responsive disease, demonstrate differences in MPCs isolated from peripheral blood.
2.12. Integrated hypothesis for the role of blood-derived macrophages and MPCs in NVR
Importantly, these data do not exclude the impact of age of bone marrow-derived monocytes in NVR. In fact, we hypothesize that both macrophages and MPCs are necessary for the development of NVR (Fig. 20). Collectively, these data support the conceptual hypothesis that NVR occurs as a result of cross-talk between macrophages and MPCs, wherein infiltrating blood-derived macrophages (Fig. 20A) recruit bone-marrow derived MPCs from the circulation (Fig. 20B) and activate them to become vascular smooth muscle cells (VSMC) and myofibroblasts, establishing the template for arteriolar phenotype (Fig. 20C–D), early in the process of CNV formation and development (Espinosa-Heidmann et al., 2013). For example, one such effector signaling mechanism linking macrophages and MPCs may be increased EPO/EPOR signaling (discussed in detail in Section 2.9.5) (Bretz et al., 2018), which 1) has been shown to promote macrophage recruitment to the choroid and is associated with increased inflammatory effector cytokine production in murine laser CNV; and 2) has also been found to be associated with increased number of circulating CD34+ precursor cells in NVAMD patients with active CNV and PDA, as compared to NVAMD patients with stable, inactive CNV (Yodoi et al., 2007). It is quite plausible that other signaling mechanisms may similarly link regulate the cross-talk between macrophages and MPCs in the setting of NVR. The broader implication of this hypothesis is that two distinct bone marrow-derived cell types regulate pathobiology of new vessel formation in the eye. If true, this would indicate that at the onset of NVAMD, the eye is an opportunistic target of a systemic biology.
To better understand whether NVR may represent a systemic biology, we have developed a model for in vivo neovascularization using a Matrigel chamber implant. In this model, a hollow cylindrical silicone tube affixed to a coverslip serves as a reservoir for growth-factor depleted Matrigel, an extracellular matrix substrate. The Matrigel chamber is then implanted into the subcutaneous perineum, with the Matrigel substrate in direct contact with mechanically injured peritoneal tissue. The skin is secured over the implant and vessel ingrowth into Matrigel substrate is allowed to occur in a defined structure and orientation. Two weeks later, the skin is opened to expose the Matrigel implant, and analysis performed by in vivo ICGA and subsequent postmortem histology. We performed the Matrigel chamber assay in old mice as compared to young mice, and our findings were consistent with our observations in the laser CNV model. Old mice demonstrated arteriolarized vessels, with significantly higher frequency of large-caliber SMA + -vessels and increased vascular invasiveness into the implant, in association with increased macrophage infiltration (Fig. 21). These intriguing findings suggest that NVR may be generalizable as a systemic biology to other vascular beds outside of the eye, and work is ongoing to more fully characterize the cell types and specific mediators in this model of neovascularization.
Fig. 21.

Matrigel chamber assay for neovascularization. Hollow silicone chamber affixed to a coverslip is filled with an extracellular matrix substrate, growth-factor depleted Matrigel. The Matrigel chamber was then implanted into the subcutaneous perineum, with the Matrigel substrate in direct contact with mechanically injured peritoneal tissue. The skin was then secured over the implant. At 14 days, the Matrigel implant was exposed and the protective coverslip was removed, to allow imaging of vessel growth by in vivo angiography. Implants were then harvested for histology and immunofluorescence (SMA labeling VSMC and myofibroblasts; MCAM labeling endothelial cells; F4/80 labeling macrophages; NG2 labeling pericytes). Scale bar: 100 μm Old mice demonstrated (D) high-flow, arteriolarized vessels (red arrows), with (E) significantly higher frequency of large-caliber SMA + -vessels, (F) increased macrophage infiltration, with and increased vascular invasiveness into the implant, as compared to young mice (A–C), stars in C indicating leading edge of vessel growth. **p < 0.05.
There remains much to be explored and understood about the regulatory mechanisms for NVR. However, modulation of the cross-talk between reparative macrophages and MPCs may represent a novel therapeutic strategy to target the biology of NVR and address the clinical unmet need of PDA and anti-VEGF resistant NVAMD. While several alternative treatment approaches beyond anti-VEGF monotherapy are presently employed in clinical practice (discussed in next Section 2.12 of this review), none of these currently available approaches specifically target macrophages or MPCs. Future approaches to target macrophages and MPCs, and the biology of NVR are discussed in Section 2.13 of this review.
2.13. Current treatment approaches for PDA in NVAMD
2.13.1. Combotherapy with verteporfin photodynamic therapy for persistent fluid and hemorrhage
Presently, beyond anti-VEGF drugs, there are no other intravitreal drug therapies that have been FDA-approved for the treatment of NVAMD, and there are no treatments that have been specifically approved for the treatment of PDA in spite of anti-VEGF therapy. Many investigators, including our own group, have employed ICGA-directed verteporfin photodynamic therapy (PDT) for the treatment of PDA in NVAMD.
Verteporfin PDT was initially approved by the FDA in 2000 as the first pharmacologic therapy for NVAMD, and specifically for the Type 2 MNV (i.e., predominantly classic CNV) FA leakage subtype, on the basis of the Treatment of Age-related Macular Degeneration With Photodynamic Therapy (TAP) study, which demonstrated that PDT reduced the frequency of 15-letter (i.e., 3-line) vision loss at 12 months, as compared to placebo, in eyes with Type 2 MNV (TAP Study Group 1999). In this treatment, verteporfin is intravenously administered, and as it circulates through the choroidal vasculature, it is activated by directing a nonthermal red laser of 693 nm wavelength at the MNV lesion, with treatment customized by spot size, fluence energy level, and treatment duration. Once photoactivated, verteporfin triggers production of reactive oxygen species that cause damage at the endothelium of the vasculature targeted by the laser. This locally induced phototoxicity is believed to cause platelet aggregation and thrombosis and ultimately vaso-occlusion of the MNV, which remains inactive until there is reperfusion. This treatment superseded thermal laser for Type 2 MNV (i. e. predominantly classic CNV) shortly after its introduction. However, once anti-VEGF therapies changed the treatment paradigm for NVAMD, PDT was abandoned as a first-line therapy.
Multiple studies have evaluated verteporfin PDT as a combotherapy for NVAMD, including DENALI (U.S. and Canada) MONTBLANC (Europe), and EVEREST (Asia, specifically for the PCV subtype), which were all multicenter, double-masked, randomized controlled trials (RCT) that assessed the efficacy of verteporfin PDT + ranibizumab, vs. ranibizumab monotherapy (Larsen et al., 2012; Koh et al., 2012; Kaiser et al., 2012). None of the studies demonstrated that combotherapy was superior to ranibizumab monotherapy, but non-inferiority margins were met in MONTBLANC and EVEREST. In EVEREST, combotherapy was more effective at achieving polyp regression than ranibizumab monotherapy. Subsequently, EVEREST II (multicenter RCT, also in Asia), once more compared verteporfin PDT + ranibizumab, vs. ranibizumab monotherapy, albeit with a different trial design (Koh et al., 2017). All participants received 3 consecutive monthly ranibizumab injections, followed by a rigorous PRN treatment regimen for continued ranibizumab therapy. Participants also received either PDT or sham on day 1 (depending on study group assignment), followed by a PRN regimen for additional PDT or sham, based on the presence of active polypoidal lesions. At 12 months, the combination regimen was not only noninferior to ranibizumab monotherapy for improvement in best-corrected visual acuity (BCVA), but actually superior in analyses of vision improvement (8.3 vs 5.1 ETDRS letters, respectively; mean difference, 3.2 letters) and complete polyp regression (69.3% vs 34.7%, respectively, P < 0.001). Furthermore, adding PDT minimized the ranibizumab injection burden (median of 4.0 vs 7.0, respectively) in the combotherapy group. Thus, data from EVEREST II support the combination of PDT and anti-VEGF therapy as primary treatment for patients with NVAMD of the PCV subgroup, establishing proof of principle that a combotherapy strategy can be efficacious in a well-defined and readily identifiable subpopulation of NVAMD patients.
Traditionally, application of PDT was guided by FA, wherein the spot size was adjusted to the greatest linear diameter that encompasses the entire area of “classic” leakage by FA (TAP Study Group 1999). Although this approach achieves vaso-occlusion in a high percentage of cases, it also carries at least a 1%–2% risk of sudden vision loss secondary to occlusion of normal choroidal vasculature (Newman, 2016). To avoid both inadvertent vaso-occlusion of normal choroid and to limit any local inflammatory response, we use indocyanine green angiography (ICGA)-guided PDT to visualize the discrete feeder vessel to the CNV lesion (especially in Arteriolar or PCV subtypes) and to decrease the spot size application; this approach has been shown to have lower rate of vision loss and adverse effect as compared to traditional FA-guided PDT applied to the entire leakage area (Otani et al., 2007; Koh et al., 2012).
We have utilized ICGA-directed PDT as an adjunctive therapy to manage PDA in spite of monthly anti-VEGF therapy, in patients with Arteriolar pattern CNV (targeting the feeder vessel), PCV (targeting the polyps and feeder vessel originating the branching vascular network), and choroidal leak syndrome (CLS, or pachychoroid) (targeting the area of leakage on FA) (see example, Fig. 22). From our studies of ICGA morphologic subtypes, including the PERSIST study, we have observed that these three subgroups collectively exhibit PDA in approximately 55–60% of cases. In the RESIST study, an open-label, prospective, investigator-initiated study (ClinicalTrials.gov NCT02452840), we assessed the efficacy of adjunctive ICGA-guided verteporfin PDT for the treatment of PDA in spite of anti-VEGF therapy (Mettu et al., 2017). Patients were followed for 12 months after enrollment, with PDT performed and aflibercept administered at the time of enrollment; aflibercept was administered on a treat-and-extend basis (monthly if PDA, extension permitted if quiescent), while PDT could be repeated on an as needed basis at 3-month intervals for persistent or recurrent disease activity. We found that the addition of verteporfin PDT improved anti-VEGF treatment response, such that, by 12 months, 90.9% of patients with prior PDA were able to achieve disease quiescence. Furthermore, 68.2% of these patients who previously had PDA in spite of every 4 week treatment were subsequently able to extend treatment interval to every 6 weeks or every 8 weeks by the 12-month timepoint, while maintaining disease quiescence. As PDT was added following loading dose or points later following initial diagnosis, and the study population reflected a highly selected population with “harder to treat” disease, the addition of combotherapy produced modest visual acuity gains from baseline (+2.5 letters). We have also observed similar results for resolution of PDA in applying ICGA-directed PDT for patients with NVAMD and serous PED (Jindal et al., 2017). In both studies, in many cases of Arteriolar and PCV subtypes, full vaso-occlusion was not essential to achieve resolution of persistent fluid and hemorrhage; in such cases, we observed that reduced blood flow through the CNV lesion was sufficient to achieve a therapeutic effect with subsequent resolution of PDA. Thus, the efficacy of adjunctive PDT for the treatment of PDA by reducing CNV blood flow establishes proof of principle that persistent leakage and hemorrhage in cases of Arteriolar and PCV subtypes is caused by hemodynamics of high blood flow within neovascular lesions and further suggests that verteporfin + PDT can be an effective combotherapy for the treatment of NVAMD patients with PDA.
Fig. 22.

Case example of patient with NVAMD and persistent disease activity undergoing verteporfin photodynamic therapy. 79 year old white female had (A) persistent cystic intraretinal fluid and subretinal fluid by OCT in spite of eight monthly anti-VEGF treatments; (B) indocyanine green angiography (ICGA) demonstrates an Arteriolar CNV with branching arterioles radiating from center feeder artery (outlined with red dashes). Following PDT targeted to the central feeder artery, there is (C) resolution of intraretinal fluid and subretinal fluid by OCT as well as (D) vas-occlusion of the Arteriolar CNV.
2.13.2. Retinal surgery for the treatment of persistent hemorrhage
As discussed, subretinal hemorrhage in the macula, also known as submacular hemorrhage, can be associated with severe vision loss (i.e., loss of three or more lines of visual acuity), especially in the absence of treatment (Avery et al., 1996). Small to medium-sized areas of submacular hemorrhage (e.g., 1–2 disc areas or less) can be treated with continued anti-VEGF injections (Altaweel et al., 2015). However, eyes with large submacular hemorrhages frequently sustain poor outcomes with anti-VEGF treatment in clinical practice. Such patients were excluded from pivotal clinical trials for anti-VEGF drugs. For these patients, an alternative vitreoretinal surgical approach can be considered to manage large submacular hemorrhage. The Submacular Surgery Trial, performed prior to the advent of anti-VEGF drugs, was a randomized clinical trial that compared direct evacuation of the hemorrhage and CNV lesion through one or more retinotomies following pars plana vitrectomy, vs. observation (Bressler et al., 2004). In this setting, surgery did not offer vision benefit and carried a high risk of complications, including retinal detachment and macular fibrosis. For this reason, direct evacuation as a surgical strategy has generally fallen out of favor. An alternative approach is pars plan vitrectomy, with subretinal injection of tissue plasminogen activator (tPA) to attempt to break down clotted blood, and pneumatic displacement of hemorrhage out of the central macula (Haupert et al., 2001). Variations of this approach utilizing fluid-air or fluid-gas exchange with postoperative face down positioning have been described, along with the addition of anti-VEGF administration immediately following surgery. Several series utilizing this approach have demonstrated anatomic success with benefits to visual acuity (with variability in successful vision improvement likely due to size and chronicity of hemorrhage, degree of foveal involvement, and baseline underlying AMD disease state prior to hemorrhage) (Olivier et al., 2004; Lincoff et al., 2008; Treumer et al., 2012). Office-based procedure with intravitreal injection of tPA and pneumatic displacement of hemorrhage have also been described (Ohji et al., 1998; Hesse et al., 1999; Hassan et al., 1999; Chen et al., 2007). In most cases, continued treatment with anti-VEGF therapy following surgery remains a critical component of effective management of disease activity and optimization of vision outcome (Stifter et al., 2007). In general, while anti-VEGF therapy can be used for management of small to medium-sized hemorrhages, surgical approaches may be considered for patients with submacular hemorrhage covering a majority of the macular area and encompassing the central fovea.
2.14. Development of novel combotherapies for NVAMD
Over the past decade, there have been many failed or terminated clinical development initiatives for NVAMD, including novel drugs that have targeted specific aspects of vascular biology: platelet derived growth factor (PDGF) (pegpleranib; Ophthotech), PDGF receptor (PDGFR) (rinucumab; Regeneron), and angiopoietin 2 (Ang2) (nesvacumab; Regeneron), to name a few examples (Rosenfeld and Feuer, 2018; Puliafito and Wykoff, 2019). These failures underscore two critical needs: 1) to more clearly understand the causes of vision loss beyond VEGF-mediated angiogenesis and exudation and to assess whether these causes have a potentially drugable target for restoration of vision; and 2) to identify subpopulations of patients who are likely to benefit from novel drugs directed against such targets. Studies of NVR pathobiology inform the first need. Morphologic phenotyping of MNV by clinical angiography (either ICGA or in the near future, next generation swept-source OCT angiography) and perhaps blood-based biomarkers, such as those investigating monocytes (and potentially MPCs), could in theory be useful companion diagnostics to inform the second need, to rationally select NVAMD patients who might benefit from a novel combotherapy drug candidate.
It is our perspective that additional therapeutic targets identified from studies of capillary angiogenesis in experimental CNV are high risk for failure in drug development because 1) anti-VEGF drugs are already highly effective for patients with capillary CNV lesions in terms of disease control and optimal vision outcomes, and additional drugs targeting capillary angiogenesis biology are unlikely to produce a substantive marginal benefit for improved vision; and 2) such targets may not necessarily inform PDA in patients with arteriolar CNV. As a case in point, efforts to develop drugs targeting PDGF (pegpleranib, Ophthotech) and PDGFR (rincumab, Regeneron) and the relevant biology of capillary maturation failed to produce vision benefit as combotherapy over anti-VEGF monotherapy alone (Jaffe et al., 2017; Dunn et al., 2017; Heier et al., 2020).
2.14.1. Therapies to improve leakage control
Clinically, patients with PDA and arteriolar CNV frequently manifest persistent leakage from the marginal CNV rim, which is comprised of terminal anastomotic arteriovenous loops. Thus, it is possible that persistent leakage in this setting is due to a failure of endothelial tight junction formation within these arteriovenous loops. Within the angiopoietin-Tie2 signaling pathway, Ang2 has been shown to promote disassembly of endothelial cell tight junctions, and experimentally, blockade of Ang2 permits formation of tight junctions, stabilization of vessels, and reduction in exudation (Akwii et al., 2019). Based on this biologic rationale, several efforts to develop Ang2-targeted biologic drugs have been undertaken or are underway. The ONYX Phase 2 clinical trial of combotherapy nesvacumab (Regeneron, targeting Ang2) + aflibercept failed to show a superior vision benefit over aflibercept alone (Hussain et al., 2019). Meanwhile, the LUCERNE and TENAYA Phase 3 clinical trials of faricimab (bi-specific antibody targeting VEGF and Ang2, Roche), as compared to aflibercept, have fully enrolled as of September 2019, and, as of the time of this review, primary study completion is expected in August 2021. A slightly different approach to modulating Tie2 signaling is inhibition of vascular endothelial-protein tyrosine phosphatase (VE-PTP), negative regulator of Tie2 signaling; blockade of VE-PTP could theoretically promote neovascular quiescence through permissive Tie2 activation (Shen et al., 2014).
All of the currently available drugs target VEGF-A (aflibercept also targets VEGF-B and PlGF). As noted, one potential hypothesis for loss of drug effectiveness is upregulation of other factors that promote fluid leakage, such as VEGF-C and VEGF-D (Cabral et al., 2018). OPT-302 (Opthea) is a VEGF-C/D “Trap” molecule, a recombinant fusion protein that binds VEGF-C and VEGF-D and blocks their binding to VEGFR-2 and VEGFR-3 receptors. Phase 2b clinical trial of combotherapy OPT-302 + ranibizumab vs. ranibizumab monotherapy provided intriguing findings, as the study achieved its primary endpoint, demonstrating superiority in visual acuity, with mean + 14.22 letters for combotherapy OPT-302 2.0 mg + ranibizumab, vs. 10.84 letters for ranibizumab alone (p = 0.01) (Al-Khersan et al., 2019). There was a trend toward reduced persistent disease activity in the combotherapy group, though small patient numbers in the post hoc analysis preclude definitive conclusions about whether blockade of VEGF-C and VEGF-D mediates effects via improved control of exudation. Phase 3 testing in underway.
Drugs that directly target endothelial cells to promote tight junction formation, such as agonists of liver X receptor (LXR), a family of nuclear receptors (de Wit et al., 2017), might be also be effective therapies to achieve quiescent disease. Additionally, since macrophage-derived effector molecules may mediate persistent endothelial cell dysfunction and deficient tight junction formation, macrophage-directed therapies may be effective for this subpopulation of patients (see the next Section 2.13.2 for additional discussion of potential macrophage targets).
2.14.2. Limiting progressive fibrosis
Given the role of macrophages and MPCs in the biology of arteriolarization and fibrosis, targeting the cross-talk between these two cell types offers several potential strategies to limit progressive fibrosis, including downregulation of macrophage effector function, targeted inhibition of pro-fibrogenic factors, blockade of MPC recruitment, and/or suppression of activation and differentiation of myofibroblasts. While we have been primarily interested in strategies to limit macrophage effector function in the setting of NVR, strategies directed against MPCs or other mesenchymal cells that give rise to myofibroblasts and vascular smooth muscle cells could be an alternative therapeutic strategy to limit fibrosis.
Macrophages use calcium as a major second messenger system for cellular activation. The intermediate kinase calcium/calmodulin-dependent protein kinase kinase 2 (CaMKK2) is expressed in macrophages and has been shown to regulates macrophage activation in response to lipopolysaccharide (LPS), amplifying expression of effector mediators (Racioppi et al., 2012; Racioppi and Means, 2012). CaMKK2 knockout impairs the ability of macrophages to adhere and extend membrane processes, reducing macrophage accumulation and diminishing cytokine release in response to certain toxins. CaMKK2 activation in macrophages is not specific to LPS but can occur with exposure to any stimuli that increases intracellular calcium and upregulate calmodulin activity (Zhang et al., 2008; Kelly et al., 2010). CaMKK2 activation in tumor-associated macrophages appears to facilitate escape from immunosurveillance and may also contribute to tumor angiogenesis (Racioppi et al., 2019). Importantly, CaMKK2 functions as an amplification circuit for signaling in immune cells, and genetic deletion or pharmacologic inhibition of CaMKK2 in mouse models diminishes inflammation but does not modify calcium homeostasis or cause immunosuppression (Racioppi et al., 2012; Racioppi and Means, 2012). In assessing a potential role for CaMKK2 in NVR, we have observed that CaMKK2 − /− mice exposed to low-grade LPS are protected from the NVR phenotype in experimental CNV (Fig. 23). As compared to wild-type (WT) mice exposed to low levels of LPS, which demonstrate arteriolar CNV, CaMKK2 − /− mice demonstrate smaller capillary lesions with significant reduction in CNV size as assessed by flatmount (Fig. 23). This intriguing finding suggests that CaMKK2 activation is a regulatory node in macrophage-mediated NVR and that targeted inhibition of CaMKK2 may be a viable strategy to limit the development of progressive fibrosis. Work is ongoing in our laboratory to more fully characterize the role of macrophage CaMKK2 in NVR and to determine whether it may be a potentially drugable target in NVAMD.
Fig. 23.

Experimental CNV in CaMKK2 −/− mice exposed to low level of lipopolysaccharide (LPS, 10 μg) by intraperitoneal injection prior to laser induction followed by preparation of TRITC-lectin flatmounts at 14 days post-induction. Scale bar: 100 μm, D = optic disc. (A) Wild-type control mice demonstrate CNV large-caliber vessels with vascular loops, whiles (B) CaMKK2 −/− demonstrates smaller lesions that appear to have capillary morphology. Quantitative analysis of flatmount surface area confirms reduced lesion size for CaMKK2 −/− mice (**p < 0.05).
Several investigators have highlighted other potential mechanisms in macrophages that could be relevant to NVR biology. Modulation of macrophage signal transducer and activator of transcription 3 (STAT3) signaling (Izumi-Nagai et al., 2007), inhibition of macrophage Rho-associated kinase (ROCK2) (Zandi et al., 2015), and inhibition of receptor interacting serine/threonine-protein kinase 1 (RIP1) kinase (Ueta et al., 2019) may downregulate reparative macrophage effector biology in the setting of CNV formation and growth, which may be sufficient to prevent fibrosis. Activation of LXR in macrophages may downregulate reparative macrophage effector function by reverse cholesterol efflux and improved cholesterol homeostasis (Sene et al., 2013). Other potential therapeutic strategies include targeting of cytokines that upregulate reparative macrophage effector function, such as IL-10, and targeting macrophage-derived soluble mediators that promote fibrosis via MPC activation, including transforming growth factor-β (TGF-β), insulin-like growth factor 1 (IGF-1), and fibroblast growth factor (FGF) (Wynn and Barron, 2010). Lastly, targeting the production of matricellular proteins produced by MPCs and fibroblasts, especially connective tissue growth factor (CTGF) and osteonectin, may reduce subsequent development of fibrosis (Sawyer and Kyriakides, 2016).
3. Suboptimal vision recovery (SVR) in NVAMD
3.1. Definition and clinicopathologic manifestations
A significant proportion of NVAMD patients have suboptimal vision outcomes in spite of aggressive treatment and suppression of exudative disease. In clinical trials of anti-VEGF drugs, less than 40% of patients achieve significant improvement in vision (i.e., three or more lines of visual acuity) (Brown et al., 2006; Rosenfeld et al., 2006; Martin et al., 2011; Heier et al., 2012; Dugel et al., 2020a). Further, in both clinical trials as well as long-term outcomes studies in real-world clinical practice, approximately 55–60% of patients have visual acuity worse than 20/40 in spite of adequate dosing (Brown et al., 2006; Rosenfeld et al., 2006; Martin et al., 2011; Peden et al., 2015; Heier et al., 2012; Dugel et al., 2020a). Why do not more of these patients achieve significant vision improvement with anti-VEGF treatment? For some patients, vision loss is irreversible as a result of loss of photoreceptors and/or RPE atrophy, occurring either as part of the NVAMD disease process or due to geographic atrophy in the setting of coincident dry AMD disease, which is reviewed elsewhere (Rosenfeld et al., 2011; Fleckenstein et al., 2018; Rozing et al., 2020). However, many patients have vision loss that is potentially reversible, with preserved photoreceptors, intact RPE, and exudative disease that is quiescent without retinal swelling (Phipps et al., 2003). We have defined this phenomenon and clinical problem as suboptimal vision recovery (SVR).
The exact prevalence of SVR is unknown, largely because the clinical problem has not been well defined or recognized among clinicians and investigators, and remarkably little research has addressed the specific mechanisms for dysfunction of the neurosensory retina in NVAMD. While it is often assumed that photoreceptor death accounts for vision loss, this is not universally true. Frequently, photoreceptors, and cones in particular, seem to be spared in NVAMD patients with major vision deficits (Curcio et al., 1996; Medeiros and Curcio, 2001; Curcio, 2001), suggesting that functional changes may occur in photoreceptors and synaptic connections in the absence of cell death. The general concept of synaptic dysfunction despite neuronal survival has been recognized in neurodegenerative diseases such as multiple sclerosis, traumatic brain injury, and Alzheimer’s disease, and has invited interest in neuroprotective and neurorestorative therapeutic strategies for these diseases (Wyss-Coray and Mucke, 2002; Raff et al., 2002; Coleman and Perry, 2002). While photoreceptor synaptic terminals have demonstrated plasticity in retinal diseases such as retinal detachment and retinal degenerations (Sanyal et al., 1992; Peng et al., 2000; Fisher et al., 2001), the concept of reversible synaptic dysfunction, especially as a cause of SVR, has received comparably little interest in NVAMD.
In pathologic specimens from NVAMD patients with CNV, labeling of the neurosensory retina overlying CNV for synaptic vesicle markers, such as synaptophysin, reveals synaptic disorganization at the outer plexiform layer (Fig. 24B), which is frequently accompanied by an abundance of infiltrating macrophages within the neurosensory retina overlying CNV (Fig. 24B). We have observed that the vast majority of these retina-infiltrating cells stain positive for CD163, which labels reparative macrophages but not microglia (Lad et al., 2015). Further, while CD68+ inflammatory macrophages are often present within CNV lesions, we have found they are rarely present within the neurosensory retina overlying CNV (Lad et al., 2015). These data suggest a potential link between synaptic disorganization and infiltrating, blood-derived reparative macrophages in NVAMD.
Fig. 24.

Histology of neurosensory retina overlying CNV in postmortem specimen from patient with NVAMD. Scale bar: 100 μm. (A) Normal control demonstrates normal pattern of synaptophysin (red) staining at the outer plexiform layer (OPL) and inner plexiform layer (IPL), and presence of occasional Iba-1+ (green) macrophages in the choroid but only minimal Iba-1+ cells in the retina (which likely label either tissue-resident perivascular macrophages or microglia). (B) NVAMD lesion demonstrates loss of normal synaptophysin staining at the OPL but generally preserved staining at the IPL, in association with extensive infiltration of Iba-1 macrophages not only in the CNV lesion but also in the overlying neurosensory retina.
3.2. SVR: lessons from experimental CNV in mice
3.2.1. Physiologic visual loss in experimental CNV is caused by photoreceptor synaptic dysfunction
To better understand the relationship between CNV and onset and development of synaptic dysfunction in the overlying neurosensory retina and to investigate the role of blood-derived macrophages in mediating synaptic dysfunction and physiologic vision loss, we have performed a series of studies in the murine experimental laser-induced CNV model (Caicedo et al. 2005a, 2005b). To ensure that the observed effects in the retina were related to authentic biology of new vessel formation and not to artifact of direct thermal damage, we utilized a low-energy 810 nm infrared laser that induces CNV formation while minimizing injury and structural damage to the overlying retina (see Section 2.6 of this review). To control for potential effects of laser injury, for a separate set of control animals without CNV, we performed laser photocoagulation with a 532 nm diode green laser, which is known to induce direct retinal injury; treatment parameters for the 532 nm laser in controls were chosen to create only focal thermal retinal injury and avoid inner choroid cavitation bubble and CNV induction.
We observed that functional ERG loss occurred early at 1 week following CNV induction, with reduction in b-wave amplitudes but generally, preservation of a-wave amplitudes, in the absence of major histopathologic changes in the retina (Fig. 25) (Caicedo et al., 2005a). The preservation of a-wave amplitudes suggested that photoreceptor function remained intact, with sufficiently preserved RPE support for the photoreceptor visual cycle in spite of CNV formation and growth. ERG b-wave amplitudes were reduced in a time-dependent fashion following laser induction, and there was a direct correlation between reduction in b-wave amplitudes and growth and lateral extension of the CNV lesion. We observed preserved ERG recording immediately after laser application and CNV induction, indicating that the observed functional changes by ERG were due specifically to CNV formation and growth and not due to the artifact of thermal laser injury.
Fig. 25.

Choroidal neovascularization-induced changes in retinal morphology and physiology. A,B: Vertical sections of the eye with choroidal neovascularization (CNV) at 1 week (A) and 2 weeks (B) after laser injury to the choroid (CH). The outer plexiform layer (OPL) was thinner at 2 weeks after CNV (arrow in B). C: Electroretinogram (ERG) recordings showed that the amplitudes of the b-wave were reduced 1 week after CNV. In this particular eye, the amplitude of the a-wave did not change. D: The b-wave amplitudes of the ERG were significantly reduced after CNV (repeated measures ANOVA, F = 6.076, P < 0.01). The amplitudes of the ERG recorded immediately after CNV did not differ from those of the control group (con). Filled circles represent mean ± SEM; open circles are data from individual mice. E: The outer plexiform layer did not show major histological changes 1 week after CNV. At 2 weeks after CNV, the outer nuclear layer had significantly fewer cell rows than that of the adjacent control retina (paired Student’s test, P < 0.01). Further cell loss was present at 4 weeks after CNV. Values are presented as a fraction of control values obtained from the adjacent retina. Asterisks indicate statistical significance. INL, inner nuclear layer; IPL, inner plexiform layer; SC, sclera. Scale bar = 100 μm in A (applies to A,B).
To test whether photoreceptors overlying CNV retain light adaptation function, we immunostained retinal sections for transducin in dark-adapted sections and assessed light-dependent translocation of transducin from outer segments to inner segments of photoreceptors on light stimulation (Caicedo et al., 2005a). We found that the retinal region overlying CNV had the same immunostaining pattern as adjacent retina away from CNV, indicating that transducin translocation capacity of photoreceptors overlying CNV was preserved albeit reduced, and this capacity remained preserved even at 4 weeks after CNV induction (control 532 nm laser application to the retina without CNV induction caused loss of normal transducin translocation capacity in the setting of outer retinal injury). In contrast, studies of activity-dependent FM1–43 uptake, a measure of photoreceptor synaptic function, revealed loss of photoreceptor synaptic function in the outer plexiform layer of the retina overlying CNV as soon as 1 week after CNV induction, versus control retina sections without CNV with intact photoreceptor synaptic function (of note, the inner plexiform layer synaptic function overlying CNV remained intact). These observations were consistent with ERG findings and preceded the histologic evidence of synaptic disruption overlying CNV, described below.
We also observed histologic evidence of disrupted photoreceptor synapses in the outer plexiform layer of the retina overlying CNV, with preserved synaptic marker staining at the inner plexiform layer (Fig. 26) (Caicedo et al., 2005a). Synaptic disruption at the outer plexiform layer in retina overlying CNV was evident as disorganized, irregular staining of the photoreceptor presynaptic protein SV2 (Fig. 26B) and the vesicular glutamate transporter 1 (vGluT1) (Fig. 26D), which were no longer restricted to the outer plexiform layer and was instead mislocalized to the outer nuclear layer, as compared to control and adjacent retina sections without CNV, where staining of these synaptic markers was tightly organized within the outer plexiform layer (Fig. 26A and C). This pattern of synaptic marker disruption overlying CNV was very consistent and was markedly apparent by week 2 following CNV induction (Fig. 26E), with a time-dependent progression in the lateral spread of synaptic marker disruption at the outer plexiform layer paralleling the lateral spread of CNV growth (Fig. 26E–F). Importantly, synaptic changes by immunostaining were apparent while the overall cellular morphology of the retina remained intact, with general preservation of photoreceptor nuclear layer at early timepoints. Evidence of photoreceptor cell death was apparent at 3 weeks and later timepoints. However, this was less pronounced, as compared to the observed synaptic changes, and occurred well after observed synaptic dysfunction and disorganization and onset of physiologic vision loss. Taken together, these findings suggest that early dysfunction of the neurosensory retina overlying CNV results primarily from loss of photoreceptor synaptic function rather than from photoreceptor cell death and raise the intriguing possibility that intervention to reverse synaptic dysfunction could be a viable therapeutic strategy to improve vision in NVAMD.
Fig. 26.

Immunostaining patterns for photoreceptor synaptic markers in the outer plexiform (OPL) layer changed abruptly after CNV. A,B: The precise arrangement of SV2 immunostaining in the OPL (A, adjacent retina) was completely disrupted in the retina over CNV (B, 4 weeks after CNV). By contrast, SV2 staining in the inner plexiform layer was not affected. C,D: vGluT1 immunostaining (red) was similarly disrupted over CNV (D, 4 weeks after CNV). Counterstaining with DAPI (blue) shows that the overall morphology of the retina was not affected. Scale bars 20 μm in A (applies to A,B), and C (applies to C,D). E: Quantification SV2 immunoreactivity in OPL at different time points following CNV induction. The intensity of SV2 immunoreactivity in the OPL was expressed as a fraction of that of the OPL in control, unaffected retinal regions. SV2 immunostaining was significantly decreased at 2 and 4 weeks after CNV induction (one-way ANOVA, P 0.001). F: Quantification of the lateral spread of CNV growth (red circles), of SV2 immunostaining reduction in retinas with CNV (black circles), and of SV2 immunostaining reduction in photocoagulated retinas of controls without CNV (empty circles).
3.2.2. Müller cell activation occurs in retina overlying CNV in association with synaptic disruption
Müller cells span the entire depth of the retina and provide important support and homeostatic functions within the retina, including extending lateral processes into the inner and outer plexiform layers to support photoreceptor synaptic function and recycle glutamate, via the glutamate transporter GLAST. In the setting of cellular injury, Müller cells become “activated,” with upregulation of specific response to injury signaling pathways and cellular processes. We sought to identify evidence of Müller cell dysfunction and activation during CNV formation and growth, assessing specific cellular response to injury markers (Caicedo et al., 2005a). In addition to observing a marked redistribution of GLAST away from the outer plexiform layer by 2 weeks, we also found increased immunostaining for fos-related antigens and c-fos and upregulation of phosphorylated extracellular signal-regulated kinase (pERK) staining (Fig. 27), all as early as 3 days after CNV and peaking at 1 week. Notably, these signs of Müller cell activation generally preceded histologic signs of synaptic disruption (Fig. 27A–F). Furthermore, while the staining for these Müller cell activation markers overlying central aspects of CNV gradually declined over time, retinal regions overlying the leading edge of the CNV lesions continued to display strong immunoreactivities for all three Müller cell activation markers even at later time points (Fig. 27G–H), indicating a strong correlation between Müller cell activation and CNV activity and growth. The early onset of Müller cell activation and the colocalization of Müller cell activation with the leading edge of CNV lesion growth suggest that Müller cell activation may be a key mediator of the observed synaptic dysfunction overlying experimental CNV.
Fig. 27.

A–C: Timecourse of immunostaining for Müller cell activation (Fos-related antigen) and outer plexiform vGluT1 immunostaining; Müller cell activation preceded the development of vGluT1 synaptic disruption. Immunostaining for fos-related antigens (FRA) strongly increased after CNV, reaching peak intensities at 1 week after CNV (B). At 4 weeks after CNV (C), immunostaining levels were lower than at 1 week after CNV but were still higher than in adjacent retinal regions (A). D–F: Immunostaining for vGluT1 began to show slight irregularity at week 1 but did not show extensive disruption over CNV until week 4. Scale bar = 50 μm in C (for A-C), 20 μm in E (for D-F). (G–H) Müller cell activation was most prominent over the leading of CNV growth. Increased pERK immunostaining was still present at 4 weeks after CNV in Müller cells located over the leading edges of CNV. Müller cells were labeled from the inner limiting membrane at the virtual surface to the outer limiting membrane at the level of photoreceptor inner segments. Strongest immunostaining intensities could be seen in Müller cell processes in the outer plexiform layer (arrow). Scale bar in G (for G and H) = 50 μm.
3.2.3. Blood-derived macrophages infiltrate retina overlying CNV
We also observed a substantial infiltration of F4/80+ mononuclear phagocytes in the retina overlying CNV (Caicedo et al., 2005a). This was apparent as early as 3 days following CNV induction and increased over time, with progressive infiltration within the retina spreading laterally along with the leading edge of CNV growth (regions away from the CNV lesion had no or minimal macrophage infiltration). In contrast, in control eyes with photocoagulated retina without CNV, there was a slight increase in F4/80+ cells at 1 week at the site of laser application, but these did not spread laterally and were no longer present at later time points, indicating that macrophage infiltration within the neurosensory retina was specifically associated with CNV growth and not with retinal injury.
Since F4/80 labels both macrophages and microglia, we analyzed GFP bone marrow chimeras (i.e., transplantation of GFP + donor bone marrow to lethally irradiated wild-type recipient mice) in the laser-induced CNV model (Caicedo et al., 2005b) and found that the majority of F4/80+ cells in the retina overlying CNV also label positive for GFP (approximately 55% by 7 days and 70% by 14 days), affirming the presence of infiltrating blood-derived macrophages, and confirming that the frequency of GFP + cells in the retina overlying CNV increased with progressive increase in lateral CNV length (Fig. 28A–E).
Fig. 28.

Bone marrow-derived cells [green fluorescent protein (GFP)-labeled] invaded the retina over choroidal neovascularization (CNV). (A) At 4 weeks after inducing CNV, many GFP-labeled cells were present in the retina over CNV but not in adjacent retinal regions. Stippled line = CNV borders; the center of the CNV lesion does not appear on this micrograph. (B) The number of GFP-labeled cells (bars) increased with time after CNV and correlated with CNV size (circles). # of GFP + cells = number of GFP-labeled cells per retinal sections containing CNV (n = 5 mice). 3d, 1w, 2w, 4w = 3 days, 1 week, 2 weeks, and 4 weeks after laser application, respectively. ONL, outer nuclear layer; INL, inner nuclear layer; IPL, inner plexiform layer; CH, choroid. Scale bars for A = 50 μm. (C–E) GFP-labeled cells (green in C) were immunoreactive for the mononuclear phagocyte marker F4/80 (red in D). In these representative images (4 weeks after CNV), all GFP-labeled bone marrow-derived cells were F4/80 immunoreactive (arrowheads, yellow in E) and had a macrophage phenotype. A resident microglial cell (arrow; not GFP-labeled) could also be seen. Scale bars = 50 μm (in C also applies to D and E).
Retinal blood vessels overlying CNV demonstrated increased immunostaining for endothelial cell adhesion molecules VCAM1, ICAM1, and PECAM (Fig. 29A–J). In the GFP + bone marrow chimera studies, GFP + F4/80+ macrophages were observed in close association to these immunoreactive vessels (Fig. 29C–D, G–J), indicating that infiltrating macrophages are being actively recruited from the bloodstream via the retinal circulation overlying CNV (Caicedo et al., 2005b). Most F4/80+ macrophages were observed to co-localize with activated pERK immunoreactive Müller cells, in particular with Müller cells processes in the outer plexiform layer (Fig. 30A–C). To assess whether infiltrating macrophages may be promoting Müller cell activation, we performed monocyte depletion studies by systemic clodronate administration (after CNV induction and growth) (Fig. 31), which diminished the density of macrophage infiltration in the overlying retina and reduced the density of activated pERK + Müller cells in the retina overlying CNV (Fig. 31B).
Fig. 29.

Immunostaining intensities for vascular cell adhesion molecule 1 (VCAM 1), intercellular cell adhesion molecule 1 (ICAM 1) and platelet-endothelial cell adhesion molecule (PECAM) increased in retinal blood vessels over CNV. (A) At 3 days after inducing CNV, VCAM 1 immunostaining was strong in large blood vessels at the vitreal surface and small blood vessels in other retinal layers over CNV but was barely detectable in adjacent retinal regions. GFP-labeled cells (arrows) were seen close to immunoreactive blood vessels. The CNV lesion also contained GFP-labeled cells as well as VCAM 1 immunoreactivity. (B) At 4 weeks after inducing CNV, blood vessels in the inner retina and outer plexiform retina (arrowheads) were still VCAM 1 immunoreactive. The increased VCAM 1 immunoreactivity extended laterally. VCAM 1 was also upregulated in the outer limiting membrane over CNV. (C) Round and amoeboid GFP-labeled cells were seen closely associated with VCAM 1 immunoreactive blood vessels in the innermost regions of the retina 3 days after CNV. (D) At 4 weeks after CNV, blood vessels were still VCAM 1 immunoreactive. GFP-labeled cells had stellate morphology. (E) Quantification of VCAM 1 immunostaining in retinal blood vessels after CNV. There was a sharp increase in the intensity of VCAM 1 immunoreactivity at 3 days after CNV (3d). Immunoreactivity intensities remained high thereafter (n = 5 mice per time point). C = values from control (adjacent) retina. Control values were similar at all time points and were pooled. a.u. = arbitrary units. (F) The lateral spread of increased VCAM 1 immunoreactivity (empty circles; n = 5 mice per time point) increased with time and paralleled CNV growth (filled circles; n = 5 mice per time point). Scale bars = 100 μm (in B, also applies to A), 20 μm (in D, also applies to C). (G and H) At 1 week after CNV (G), blood vessels at the vitreal surface were ICAM 1 immunoreactive. ICAM 1 immunoreactive blood vessels could also be seen in the outer plexiform layer (H; 4 weeks after CNV). GFP-labeled cells were closely associated with these blood vessels. (I and J) GFP-labeled cells colocalized with strongly PECAM immunoreactive blood vessels in the outer plexiform layer (I) and at the vitreal surface (J) in the retina over CNV. Scale bars = 20 μm (in G also applies to H, in J to I).
Fig. 30.

Bone marrow-derived macrophages were closely associated with activated Müller cells over CNV (4 weeks after laser application). GFP-labeled cells colocalized with c-fos (red in A) and pERK (red in B, C) immunoreactive Müller cells. Macrophage processes were seen intermingling with Müller cell processes in the outer plexiform layer (C). Scale bars = 20 μm (in B also applies to C).
Fig. 31.

Depletion of circulating macrophages diminished macrophage infiltration and Müller cell activation in the retina over CNV. (A) The retina overlying CNV (2 weeks after laser application) of a PBS-treated (control) mouse showed extensive macrophage recruitment (red F4/80 immunofluorescence) and Müller cell activation (green pERK immunofluorescence). (B) Clodronate treatment strongly reduced macrophage recruitment and Müller cell activation. (C) Quantification of macrophage densities in PBS-treated (PBS) and clodronate-treated mice (CLO) in retinal regions over (filled bars) and adjacent to CNV (empty bars). Macrophage densities significantly increased in PBS treated but not in clodronate-treated mice (CLO; n = 8 mice per group; paired Student’s t-test; asterisk denotes significance at p < 0.01). Stippled line = macrophage density of control retinal regions. (D) The density of pERK immunoreactive Müller cells over CNV was reduced in clodronate-treated mice (n = 8 mice per group; Student’s t-test; p < 0.05). Scale bar = 50 μm.
Collectively, these findings suggest that physiologic retinal vision loss in experimental CNV, and possibly NVAMD, may be mediated by the recruitment of blood-derived macrophages from the retinal vasculature into the neurosensory retina overlying CNV, which promote photoreceptor synaptic dysfunction (at least in part) via Müller cell activation. Importantly, these findings do not exclude the possibility that microglia also play a role in this setting. Work is underway using the (Cx3cr1CreER) reporter system (O’Koren et a. 2016)) in the laser CNV model to better characterize differential responses of microglia and blood-derived macrophages and their potential relative contributions to photoreceptor synaptic dysfunction in this setting.
3.3. Potential mechanisms for Müller cell activation, synaptic dysfunction, and SVR in NVAMD
As noted, SVR has not been a well-recognized entity or well-defined clinical problem in NVAMD, so it is not surprising that specific cellular and molecular mechanisms have not been explored. Our findings in the murine laser-induced CNV model clearly demonstrate key roles for blood-derived infiltrating macrophages in initiating, and Müller cells in mediating the pathogenesis of synaptic dysfunction in SVR. While these findings do not exclude the possibility of other triggers that may promote synaptic dysfunction, they do provide a vital starting point to begin to elaborate specific disease mechanisms. We will consider hypotheses for potential mechanisms, including several that our laboratory group is currently investigating.
The co-localization of macrophages with Müller cells and the finding that Müller cell activation generally preceded widespread histologic evidence of synaptic disorganization suggests that the Müller cell is the initial cellular target in SVR. One possibility is that macrophage-derived cytokines (e.g., IGF-1, TGF-β, FGF, TNF-α, IL-1β) bind to receptors on the Müller cell surface, activating downstream immediate early response signaling pathways such as ERK1/2 and p38 mitogen-activated protein kinase (MAPK), which translocate to the nucleus to initiate gene transcription and activate cellular response to injury pathways (Sabio and Davis, 2014). By this mechanism, photoreceptors cells may be indirect targets of these infiltrating macrophages, since they do not express receptors for most cytokines. Müller cell activation may also potentiate the effector function of macrophages, producing a positive feedback loop for macrophage-driven Müller cell activation; ligation of CD40 on Müller cells has been shown to upregulate expression of TNF-α and IL-1β via P2X7 activation in macrophages (Portillo et al., 2017). A second possibility is that infiltrating macrophages release reactive oxygen species (ROS), such as hypochlorous acid, via myeloperoxidase expression (Marikovsky et al., 2003), which can directly injure Müller cells, photoreceptors, and neurons. A third possibility is synaptic dysfunction that is directly mediated by macrophages, wherein macrophages themselves release excessive glutamate resulting in neuronal excitotoxicity (Yawata et al., 2008). Importantly, these potential mechanisms of macrophage-mediated disease are not mutually exclusive. For instance, loss of Müller cell glutamate reuptake due to injury could act in concert with macrophage glutamate release to mediate neuronal excitotoxicity. Additionally, other macrophage-independent or macrophage-indirect mechanisms might also contribute to Müller cell activation and dysfunction. For example, in our studies of experimental CNV, we found that ERK1/2 activation in Müller cells was diminished but not abolished following macrophage depletion, suggesting that soluble mediators directly released from CNV, injured RPE, or activated microglia could also contribute to Müller cell activation (Caicedo et al., 2005b). Exudative disease, particular subretinal and/or intraretinal hemorrhage is likely another cause of Müller cell activation, and it is also possible that soluble mediators produced by other injured neuronal cells or Müller cells could function in a paracrine fashion to propagate Müller cell activation (Bringmann et al., 2009).
3.4. Cellular dysfunction in the setting of Müller cell activation and potential mechanistic hypotheses for SVR
Aberrant interaction between macrophages and Müller cells, along with these other triggers of activation, or cellular response to injury, can promote dysregulation or dysfunction of a number of cellular processes that are vital to Müller cell support of retinal neuronal cells: 1) cytoskeletal regulation and maintenance of cellular contacts; 2) glutamate and neurotransmitter uptake and metabolism; 3) transmembrane ion gradient regulation; 4) regulation of water transport; 5) production of neurotrophic factors (e.g., BDNF), which are necessary for normal health and function of neuronal cells including photoreceptors; 6) oxidant metabolism; and 7) mitochondrial function. As of yet, we have not definitively established which processes and mechanisms are dysregulated or dysfunctional in the laser CNV model or in other mouse models relevant to AMD but are presently working on exploring several lines of investigation. In the following sections, we will explore current knowledge from the literature and our ongoing work in experimental retinal vascular disease to discuss how each of these cellular processes are potentially relevant to synaptic dysfunction and SVR and will consider potential molecular mechanisms relevant for each (see Fig. 32 for illustrations of potential cellular mechanisms).
Fig. 32.

Müller cells provide a number of critical support functions required for normal photoreceptor and neuronal function including glutamate reuptake and metabolism, potassium and water homeostasis, neurovascular coupling, support of cone visual cycle and release of neurotrophic factors. (A) Macrophages extravasate from the retinal vasculature and subsequently contact and injure Müller cells leading to dysfunction of one or more Müller cell functions. Potential injury mechanisms include cytokine release, glutamate release, reactive oxygen species production or others as yet uncharacterized. (B) These injury stimuli may lead to loss of normal cytoskeleton, cell adhesion, glutamate cycle, ion and water regulation and mitochondrial function in Müller cells which are required for synaptic support of photoreceptors and neurons.
3.4.1. Actin cytoskeletal dysregulation and loss of cellular contacts
The Müller cell has a precisely organized cellular architecture that enables cellular subdomain specific functions mediated at specific contacts with cells distributed along the full depth of the retina. These include Müller cell footplates at the internal limiting membrane, contacts with retinal capillaries at the neurovascular bundle, lateral processes arborizing within the outer and inner plexiform layers, and cellular contacts with cone photoreceptors (in support of the cone visual cycle) (Reichenbach et al., 2007; Wang and Kefalov, 2011; Derouiche et al., 2012). Maintenance of these cellular subdomains is required for retinal water transport, homeostasis of the extracellular milieu, neurovascular coupling, and neuronal synaptic support. This precise architecture and organization of Müller cell membrane processes is enabled by tightly regulated cytoskeletal elements including both actin and intermediate filaments and by mediators of both cell-cell contacts and cell-extracellular matrix adhesion (Derouiche et al., 2012). One well characterized regulator of cytoskeleton turnover is heat shock protein 25/27 (Hsp25/27) (Schneider et al., 1998; Haslbeck, 2002; Concannon et al., 2003). This molecule belongs to the family of small heat shock proteins (which includes α-β crystallins) and serves a dual role as cytoskeleton stabilizer and chaperone protein. Phosphorylation of Hsp25/27 is mediated by p38 MAPK signaling cascade, wherein p38 MAPK phosphorylates MAPKAP2 kinase (MK2), which in turn phosphorylates Hsp25/27. Phosphorylated Hsp25/27 acts as a F-actin cap-binding protein, binding to the active edge of a growing microfilament, causing a deficit in cytoskeleton reassembly and repair. Thus, p38 MAPK - Hsp25/27 activation may mediate increased actin cytoskeleton turnover and subsequent loss of Müller cell cytoskeletal architecture, and loss of the tightly regulated morphology. Additionally, this injury response can mediate loss of cellular contacts, through downregulation of those cell adhesive interactions (with other cells and the extracellular matrix) that are dependent on the actin cytoskeleton. Loss of normal cytoskeletal architecture and adhesive interactions could lead to Müller cell dysmorphology, particularly in intricate structures such as the lateral processes and contacts with retinal capillaries. Together, these alterations could lead to retraction of lateral processes and disruption of synaptic support as well as dysfunctional water transport and neurovascular coupling. Indeed, we have observed Müller cell morphological changes in experimental CNV (Caicedo et al., 2005a), and we have extensively characterized alterations in cellular subdomains in a different model of retinal vascular disease, experimental laser-induced retinal vein occlusion (RVO), where we observed loss of perisynaptic contacts and retraction of lateral processes as well as cellular swelling and dysmorphology (Allingham et al., 2018).
3.4.2. Glutamate and neurotransmitter uptake and metabolism
A major support function of Müller cells is re-uptake and recycling of glutamate from synaptic terminals. Interruption of normal glutamate re-uptake can compromise efficiency of synaptic function and, if prolonged, can lead to neuronal toxicity, with persistent over-activation leading to excessive ion influx, osmotic swelling, and free radical degeneration (Izumi et al. 1999, 2002). This can perpetuate a vicious cycle of Müller cell activation, as overactivity of glutamate receptors on Müller cells also activates p38 MAPK, ERK1/2, and other injury response pathways (Ji et al., 2012; Gao et al., 2017). Retraction of lateral processes and/or downregulation of proteins required for Müller cell glutamate uptake and recycling may exacerbate photoreceptor-bipolar synaptic dysfunction, eventually leading to degeneration. Cellular activation also promotes dysfunctional glutamate re-uptake by downregulation of glutamate transporters and subsequent downregulation of glutamine synthetase (Chen and Weber, 2002), an enzyme essential for glutamate recycling. All of these changes have the potential to impair synaptic function due to excess glutamate in the extracellular space and due to disrupted recycling of glutamate back to presynaptic terminals (Bringmann et al., 2009; Bringmann and Wiedemann, 2012). While it has been previously demonstrated that pharmacologic blockade of Müller cell glutamine synthetase results in glutamate depletion in bipolar and retinal ganglion cells (Pow and Robinson, 1994), the fact that photoreceptor presynaptic terminals are capable of glutamate reuptake may render them relatively less sensitive to loss of Müller cell support for glutamate metabolism. However, Müller cells are required for efficient termination of synaptic transmission within the photoreceptor-bipolar cell synapse (Matsui et al., 1999). Additional studies will be required to further clarify the role for disrupted glutamate metabolism on specific retinal cell types in neovascular AMD.
3.4.3. Regulation of retinal ion gradients and water transport
Müller cell Kir4.1 channels maintain extracellular potassium concentrations, contribute to regulation of extracellular glutamate via maintenance of potassium gradients, and contribute to regulation of water transport and Müller cell volume (Reichenbach et al., 2007; Bringmann et al., 2013). Activation of p38 MAPK in Müller cells downregulates Kir 4.1 potassium channels, which can lead to accumulation of extracellular potassium, enhanced undershoot recovery of neuronal stimulation, and reduction in glutamate transport into the Müller cell (Kofuji et al., 2000; Gao et al., 2017). AQP4 is found at the terminal endfeet of Müller cells, especially at the internal limiting membrane and at the junction with retinal capillaries at the neurovascular unit, to facilitate transport and removal of excess water into the vitreous or the retinal circulation, respectively. In these locations, Kir4.1 and AQP4 are co-localized, as water transport is linked to localized potassium flux out of the cell (Newman, 1984; Nagelhus et al., 1999). AQP4 is also located in perisynaptic membranes within the inner and outer plexiform layers where they function independent of Kir4.1 (Nagelhus et al., 1998). In the setting of Müller cell activation, effective colocalization of Kir4.1 and AQP4 at endfeet is lost due to downregulation of Kir4.1 expression and more diffuse distribution of AQP4 within Müller cells, which prevents normal potassium and water transport and can lead to Müller cell swelling and loss of normal synaptic support. We and others have observed these changes in experimental retinal vein occlusion (RVO) (Rehak et al., 2009; Allingham et al., 2018), which, while pathologically distinct, manifests macular edema and cystic intraretinal fluid similar to that seen in NVAMD. Furthermore, glutamate receptor over-activation and subsequent excessive ion influx and free radical generation has been shown to exacerbate the dysregulation of potassium and water transport (Schwartz, 1993; Ji et al., 2012).
3.4.4. Production of neurotrophic factors
As part of their support functions, Müller cells produce neurotrophic factors, such as BDNF and CNTF (Harada et al. 2000, 2002), which are vital for maintenance of neuronal function. Specifically, such neurotrophic factors may serve important roles for maintaining synaptic function, through regulation of neurotransmitter receptor activity (e.g. NMDA receptor, AMPA receptor) on post-synaptic bipolar cells and through regulation of receptor expression and localization and neurotransmitter metabolism. There is also evidence to support a role for synaptogenesis and synaptic remodeling following injury (Koh et al., 2018). Müller cell activation downregulates production of neurotrophic factors, which can have deleterious effects on synaptic function (Harada et al., 2002).
3.4.5. Support of retinal visual cycle for cone photoreceptors
Recent work has characterized a cone-specific visual cycle, wherein Müller cells in contact with cone photoreceptors convert all trans-retinal back to 11-cis-retinol for cone cells, where it is oxidized to the 11-cis-retinal chromophore necessary for pigment regeneration (Collery et al., 2008; Wang and Kefalov, 2011; Xue et al., 2015). This cone visual cycle enables both rapid dark adaptation as well as continuous function under bright and rapidly-changing light conditions. As this is an area of more recent focus among retinal biologists, much remains unknown about this visual cycle in both health and disease. We speculate that disruption of the contacts between Müller cells and cone photoreceptors, increase in production of reactive oxidant species, or compromise in bioenergetics necessary for maintenance of efficient visual cycle support, all of which may occur in the setting of Müller cell activation, could contribute to diminished efficiency of the cone-specific visual cycle, and leading to SVR. While ERG in our studies did not show significantly compromised a-wave (Caicedo et al., 2005a), this result may not inform potential dysfunction of Müller cell visual cycle in NVAMD, since much is not understood about cone function in rod-dominant species such as mice.
3.4.6. Oxidant metabolism and mitochondrial dysfunction
Müller cells produce glutathione, which can reduce and neutralize reactive oxygen species in the extracellular space, limiting oxidant injury to other retinal cells. Glutathione synthetase is downregulated in the setting of Müller cell activation, reducing antioxidant capacity of the Müller cell (Bringmann et al., 2006). In fact, alterations in potassium current and glutamate recycling promotes generation of reactive oxidant species, as noted above. This can result in induction of mitochondrial dysfunction, which can manifest as electron leak from the electron transport chain and generation of additional oxidant species, as compromise of niche bioenergetics needs by reduction in ATP synthesis, and dysregulation of calcium homeostasis. Müller cell active transport of ions and glutamate are dependent on a high transmembrane potential mediated by Na/K ATPase, and maintenance of complex cytoskeletal structures such as those seen in Müller cells is highly energy intensive (Nicholls and Attwell, 1990; Newman, 1993). Thus, mitochondrial dysfunction can promote a persistent state of cellular injury, wherein oxidants contribute to actin cytoskeletal dysregulation, as well as dysregulated glutamate recycling and ion gradient/water homeostasis (Toft-Kehler et al., 2017). Within Müller cells, mitochondria are localized to lateral processes; there is significant debate about the relative importance of mitochondria-derived ATP (versus ATP derived from glycolysis) in synaptic support and the role of mitochondrial dysfunction is uncertain (Vos et al., 2010; Macaskill et al., 2009). It is possible that these mitochondria may provide niche ATP sources to metabolic enzymes required for reuptake and recycling of glutamate and other neurotransmitters (Derouiche et al., 2015). Another possibility is that these mitochondria in lateral processes enhance calcium buffering and optimize normal ion flux (Zenisek and Matthews, 2000; Wan et al., 2010; Macaskill et al., 2009). Mitochondria are also localized within the photoreceptor synapse and the presynaptic and postsynaptic terminals of other retinal neurons, though there is also considerable debate about the role of mitochondria in the bioenergetics necessary for packaging and transport of synaptic vesicles and neurotransmitter reuptake and recycling at the presynaptic terminals (Vos et al., 2010; Macaskill et al., 2009). In this setting as well, mitochondria also maintain calcium ion homeostasis that is essential for synaptic function (Zenisek and Matthews, 2000; Wan et al., 2010; Macaskill et al., 2009). Thus, mitochondrial dysfunction at the lateral processes and at photoreceptor synaptic terminals may contribute to aberrant photoreceptor-bipolar synaptic transmission and consequently physiologic vision loss.
3.4.7. Integrating mechanisms for Müller cell activation and SVR
Müller cell activation by infiltrating macrophages can trigger a host of potential molecular and subcellular response to injury pathways that directly compromise normal Müller cell function and indirectly produce photoreceptor-bipolar synaptic dysfunction, leading to onset of SVR. We have highlighted several that are plausible and likely to contribute to SVR based on available evidence and current understanding of Müller cell biology, though this list is certainly not exhaustive and there are likely to be other mechanisms that also contribute. Importantly, each of these is not a standalone mechanism; as we have elucidated, they are likely to be interdependent and simultaneously activated, since they are part of a broad cellular response to injury and since one mechanism can potentiate one or more other mechanisms. As mentioned, we also do not discount the possibility that other non-macrophage stimuli also mediate these responses to injury mechanisms (Bringmann et al., 2006; Bringmann and Wiedemann, 2012), though we embrace the hypothesis that blood-derived macrophages are the primary mediators of SVR in the setting of NVAMD. Future work from our laboratory and other investigators will need to elucidate and affirm the specific and relative contributions of these mechanisms to synaptic dysfunction and physiologic vision loss in experimental models of NVAMD and other retinal diseases.
3.5. Treatment strategies for SVR in NVAMD
There are no available treatments to improve vision independent of leakage control for patients with SVR, and given our limited understanding of SVR pathobiology, to our knowledge, there are presently no significant development efforts of novel drugs for reversal of photoreceptor synaptic dysfunction in NVAMD. Given the demonstrated role of retina-infiltrating macrophages in experimental CNV, we hypothesize that macrophage-directed therapies may also have a role for SVR. Steroids have not been effective for improvement of vision in NVAMD patients, suggesting that this type of anti-inflammatory will not be effective in the setting of SVR, and this is consistent with the observation that steroids have minimal effect on the function of reparative macrophages (Schaer et al., 2002). In addition to evaluating role of macrophage CaMKK2 in NVR, we are also investigating macrophage CaMKK2 as a potential target to reduce physiologic vision loss in experimental CNV. Theoretically, targeting mechanisms that downregulate vascular endothelial cell adhesion molecules could reduce macrophage infiltration into the retina. However, since multiple vascular adhesion molecules are upregulated in the setting of disease, it is likely that there is redundancy of function of these molecules such that targeting just one of them is insufficient to prevent macrophage infiltration into the retina from the circulation.
Small molecule inhibitors of immediate early response pathways, such as ERK1/2, p38 MAPK, and c-fos, could theoretically downregulate Müller cell activation and cellular response to injury pathways (Lambert et al., 2020), limiting the roles of actin cytoskeleton dysregulation, aberrant ion gradient and water transport, mitochondrial dysfunction, and others, in promoting synaptic dysfunction.
Finally, emerging clinical data points toward a potential role for mitoreparative drugs for the treatment of SVR. Our group conducted a single-site, Phase 1, open-label clinical trial to assess the safety and efficacy of subcutaneous elamipretide (Stealth BioTherapeutics), a mitochondria-targeted peptide that reverses mitochondrial dysfunction (Wood et al., 2020), in patients with either intermediate dry AMD (high-risk drusen) or dry AMD with noncentral GA (Allingham et al., 2019; Cousins et al., 2019). While one eye with dry AMD was designated as the study eye, the fellow, non-study eye was not restricted in terms of disease state, and the study had a subset of patients (n = 13) with NVAMD in the fellow eye with stable acuity and quiescent disease, maintained on stable anti-VEGF regimen. Among these patients, mean visual acuity gain from baseline at 24 weeks was approximately +6.3 letters in the fellow eye with NVAMD, in spite of the diverse and heterogeneous states of NVAMD disease (Mettu et al., 2019). The percentage of fellow eyes with NVAMD that gained 6 or more letters in best-corrected visual acuity (BCVA) was 54%, while the percentage of fellows eyes that gained 10 or more letters in BCVA was 31%. (Mettu et al., 2019). The observed effects were attributable to reversing SVR since exudative disease activity was quiescent while on study drug. From these findings, we can infer that mitochondrial dysfunction may contribute to SVR, and these data provide proof-of-concept that mitoreparative drugs may offer promise to treat SVR. This is particularly intriguing since it represents a novel mechanism of action that is independent of new vessel pathobiology. Since vision treatment response is not reflected by morphologic changes by OCT, development of such a therapy for SVR would require a unique companion diagnostic, perhaps a novel form of physiologic testing, such as direct focal ERG testing of the neurosensory retina localized to the site of MNV to capture and quantify discrete regions of photoreceptor synaptic dysfunction. In terms of knowledge-building studies, additional investigation is necessary to better understand the cells (Müller cell, photoreceptor synapse, bipolar cell, etc.) where mitochondrial dysfunction may be operative within the retina as well as the specific mechanisms whereby mitochondrial repair drugs might improve vision benefit.
4. Future directions and conclusions
In this review, we have explored paradigms to understand disease mechanisms for incomplete response to anti-VEGF therapy and the specific clinical problems of PDA and SVR in NVAMD. We have demonstrated that a major cause of PDA in NVAMD patients is NVR, the formation of Arteriolar pattern with perivascular fibrosis; and that, from studies of experimental CNV, blood-derived reparative macrophages mediate the biology of NVR via recruitment and activation of vascular smoooth muscle cells (VSMCs) and myofibroblasts as the predominant perivascular mural cells early in CNV development (Espinosa-Heidmann et al., 2003a). We have also observed that bone marrow-derived mesenchymal precursor cells (MPCs) confers susceptibility to NVR in experimental CNV (Espinosa-Heidmann et al., 2013). Using studies of GFP bone marrow chimeric mice (i.e., transplant of GFP-labeled bone marrow to wild-type recipient mice following lethal irradiation), we observed that 1) bone marrow-derived MPCs are a major source of mural support cells in experimental CNV; 2) aging increases the relative frequency of the MPC subtype (CD34+ SM22+) in bone marrow that gives rise to VSMC and myofibroblasts; 3) transplantation of bone marrow from old donor mice to young recipients transfers the CNV phenotype of arteriolarization and fibrosis with high frequency of MPC-derived VSMC and myofibroblasts in young recipients; and 4) transplantation of bone marrow from young donor mice to old recipients transfers the capillary CNV phenotype with few MPC-derived VSMC and myofibroblasts in old recipients. Taken together, these findings support the conceptual hypothesis that cross-talk between blood-derived reparative macrophages and MPCs, both recruited from the systemic circulation, drives the development of NVR in NVAMD.
While this work has established initial pathobiologic bases, there remain many questions to answer about the specific biologic mechanisms. For instance, for the development of arteriolar CNV, which subset (s) of monocytes mediate NVR? Are there specific macrophage-derived cytokines that are indispensable for NVR? What are the cellular mechanisms that regulate macrophage effector biology, and are those regulatory mechanisms potentially drugable to modulate macrophage effector function in the setting of disease? What is the precise identity of MPCs that confer susceptibility to NVR and can we identify and quantify them in the blood of NVAMD patients to identify at-risk populations? Are there different sets of MPCs that give rise to pericytes or to VSMCs/myofibroblasts? Can we gain key learnings from the various models of NVR that we have developed, especially the latent CMV infection model and the PAMP stimulation model, to better understand how monocytes and macrophage are preprogrammed for specific effector functions? Our laboratory group is presently focused on elaborating the answers to many of these questions, as we believe that important insights into the biology of NVR may shed light on potential targets for therapy and can also help us affirm whether NVR is in fact a distinct and unique paradigm for new vessel formation, which would have implications not only for NVAMD and ocular diseases but other disorders of new vessel formation (e.g., cancer) elsewhere in the body.
For SVR, we have observed consistent lines of evidence in both postmortem NVAMD specimens and experimental CNV linking progressive synaptic dysfunction to the extent of macrophage infiltration and Müller cell activation, and in turn, to lateral growth of CNV. What are the mechanism(s) whereby CNV growth facilitates recruitment of macrophges into the neurosensory retina from the retinal circulation? Can we identify specific subset(s) of monocytes that give rise to SVR- mediating infiltrating macrophages? What soluble mediators are expressed by infiltrating macrophages to activate Müller cells and trigger photoreceptor synaptic dysfunction? How does the gene expression profile of macrophages in this setting differ from resident microglia, and does this delineate biologically distinct roles for these ontogenically distinct cell types? Are there specific regulatory mechanisms for the effector biology of infiltrating macrophages in the neurosensory retina, and are these mechanisms drugable? Of the various mechanisms that we have laid out for Müller cell activation, which ones contribute significantly to synaptic dysfunction and are the inciting signaling pathways, especially ERK1/2 and p38 MAPK, potentially drugable targets? What are the mechanisms by which Kir4.1 and AQP4 are mislocalized and can these be reversed or prevented? And in which cells and specifically how does mitochondrial dysfunction mediate synaptic dysfunction and physiologic vision loss in experimental CNV, and can mitoreparative drugs be viable therapies to address the problem of SVR and improve vision outcomes?
Nearly two decades into the anti-VEGF era, our community has collectively made significant strides in understanding the pathobiology of NVAMD and as a result, the future of novel treatments for NVAMD appears very promising. As we look beyond anti-VEGF to the next generation of medicines, continued investigations of mechanisms for NVR and for Müller cell activation and photoreceptor synaptic dysfunction, with a specific focus on understanding how blood-derived macrophages mediate vision loss in each, are essential, so that we can discover novel drugable targets, broaden the landscape and paradigms for NVAMD treatment, and extend the promise of improved vision outcomes to patients with incomplete response to anti-VEGF therapy in the years ahead.
Acknowledgements
The authors acknowledge Ananya Mettu for assistance with graphic design.
5. Funding sources
This was work was supported by National Institutes of Health (NIH) R01 EY018880 (SWC), NIH R01 EY013318 (SWC), NIH R03 EY015292 (SWC), NIH K08 EY025325 (PSM), NIH K08 EY026627 (MJA), NIH Core Grant P30 EY005722 (to Duke Eye Center), Unrestricted Grant from Research to Prevent Blindness Grant (to Duke Eye Center), Grant funding from Stealth BioTherapeutics (PSM, MJA, SWC), and Grant funding from Bausch + Lomb (PSM, SWC). The funding sources/sponsors had no role in the design and conduct of studies, data analysis, writing, or decision to publish the work presented herein.
References
- Akwii RG, Sajib MS, Zahra FT, Mikelis CM, 2019. Role of Angiopoietin-2 in Vascular Physiology and Pathophysiology’, Cells, p. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Khersan H, Hussain RM, Ciulla TA, Dugel PU, 2019. ’Innovative therapies for neovascular age-related macular degeneration. Expet Opin. Pharmacother. 20, 1879–1891. [DOI] [PubMed] [Google Scholar]
- Al-Sheikh M, Iafe NA, Phasukkijwatana N, Sadda SR, Sarraf D, 2018. Biomarkers of neovascular activity in age-related macular degeneration using optical coherence tomography angiography’. Retina 38, 220–230. [DOI] [PubMed] [Google Scholar]
- Allingham MJ, Tserentsoodol N, Saloupis P, Mettu PS, Cousins SW, 2018. ’Aldosterone exposure causes increased retinal edema and severe retinopathy following laser-induced retinal vein occlusion in mice. Invest. Ophthalmol. Vis. Sci. 59, 3355–3365. [DOI] [PubMed] [Google Scholar]
- Allingham MJ, Mettu PS, Cousins SW, 2019. ’Elamipretide, a mitochondrial- targeted drug, for the treatment of vision loss in dry AMD with high risk drusen: results of the phase 1 ReCLAIM study. Invest. Ophthalmol. Vis. Sci. 60, 361–61. [Google Scholar]
- Group comparison of age-related macular degeneration treatments trials research, and catt research group comparison of age-related macular degeneration treatments trials. Altaweel MM, Daniel E, Martin DF, Mittra RA, Grunwald JE, Lai MM, Melamud A, Morse LS, Huang J, Ferris FL 3rd, Fine SL, Maguire MG, 2015. Outcomes of eyes with lesions composed of >50% blood in the comparison of age-related macular degeneration treatments trials (CATT) Ophthalmology 122, 391–398 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambati J, Atkinson JP, Gelfand BD, 2013. ’Immunology of age-related macular degeneration. Nat. Rev. Immunol. 13, 438–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apte RS, 2010. ’Regulation of angiogenesis by macrophages. Adv. Exp. Med. Biol. 664, 15–19. [DOI] [PubMed] [Google Scholar]
- Apte RS, Richter J, Herndon J, Ferguson TA, 2006. ’Macrophages inhibit neovascularization in a murine model of age-related macular degeneration. PLoS Med. 3 e310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apte RS, Chen DS, Ferrara N, 2019. ’VEGF in signaling and disease: beyond discovery and development. Cell 176, 1248–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arjamaa O, Minn H, 2012. ’Resistance, not tachyphylaxis or tolerance. Br. J. Ophthalmol. 96, 1153–1154. [DOI] [PubMed] [Google Scholar]
- Armulik A, Abramsson A, Betsholtz C, 2005. Endothelial/pericyte interactions. Circ. Res. 97, 512–523. [DOI] [PubMed] [Google Scholar]
- Arnold L, Henry A, Poron F, Baba-Amer Y, van Rooijen N, Plonquet A, Gherardi RK, Chazaud B, 2007. ’Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J. Exp. Med. 204, 1057–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold JJ, Markey CM, Kurstjens NP, Guymer RH, 2016. ’The role of sub-retinal fluid in determining treatment outcomes in patients with neovascular age-related macular degeneration–a phase IV randomised clinical trial with ranibizumab: the FLUID study. BMC Ophthalmol. 16, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asahara T, Masuda H, Takahashi T, Kalka C, Pastore C, Silver M, Kearne M, Magner M, Jeffrey MI, 1999. Bone marrow origin of endothelial progenitor cells responsible for postnatal vasculogenesis in physiological and pathological neovascularization. Circ. Res. 85, 221–228. [DOI] [PubMed] [Google Scholar]
- Ashraf M, Banaee T, Silva FQ, Singh RP, 2018. ’Switching anti-vascular endothelial growth factors in refractory neovascular age-related macular degeneration. Ophthalmic Surg Lasers Imag. Retina 49, 166–170. [DOI] [PubMed] [Google Scholar]
- Avery RL, Fekrat S, Hawkins BS, Bressler NM, 1996. ’Natural history of subfoveal subretinal hemorrhage in age-related macular degeneration. Retina 16, 183–189. [DOI] [PubMed] [Google Scholar]
- Bali Parul, Sridhar Bammidi, Banik Avijit, Nehru Bimla, Anand Akshay, 2018. ’CD34 and CD117 stemness of lineage-negative cells reverses memory loss induced by amyloid beta in mouse model. Front. Behav. Neurosci 12, 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbazetto I, Saroj N, Shapiro H, Wong P, Freund KB, 2010. Dosing regimen and the frequency of macular hemorrhages in neovascular age-related macular degeneration treated with ranibizumab. Retina 30, 1376–1385. [DOI] [PubMed] [Google Scholar]
- Baumal CR, Reichel E, Duker JS, Wong J, 1997. Indocyanine green hyperfluorescence associated with serous retinal pigment epithelial detachment in age-related macular degeneration. Ophthalmology 104, 761–769. [DOI] [PubMed] [Google Scholar]
- Bearelly S, Espinosa-Heidmann DG, Cousins SW, 2008. ’The role of dynamic indocyanine green angiography in the diagnosis and treatment of retinal angiomatous proliferation. Br. J. Ophthalmol 92, 191–196. [DOI] [PubMed] [Google Scholar]
- Benjamin LE, Hemo I, Keshet E, 1998. ’A plasticity window for blood vessel remodelling is defined by pericyte coverage of the preformed endothelial network and is regulated by PDGF-B and VEGF. Development 125, 1591. [DOI] [PubMed] [Google Scholar]
- Berg K, Pedersen TR, Sandvik L, Bragadottir R, 2015. ’Comparison of ranibizumab and bevacizumab for neovascular age-related macular degeneration according to LUCAS treat-and-extend protocol. Ophthalmology 122, 146–152. [DOI] [PubMed] [Google Scholar]
- Berg K, Hadzalic E, Gjertsen I, Forsaa V, Berger LH, Kinge B, Henschien H, Fossen K, Markovic S, Pedersen TR, Sandvik L, Bragadottir R, 2016. ’Ranibizumab or bevacizumab for neovascular age-related macular degeneration according to the lucentis compared to avastin study treat-and-extend protocol: two- year results. Ophthalmology 123, 51–59. [DOI] [PubMed] [Google Scholar]
- Binder S, 2012. ’Loss of reactivity in intravitreal anti-VEGF therapy: tachyphylaxis or tolerance? Br. J. Ophthalmol 96, 1–2. [DOI] [PubMed] [Google Scholar]
- Biswas SK, Chittezhath M, Shalova IN, Lim JY, 2012. ’Macrophage polarization and plasticity in health and disease. Immunol. Res 53, 11–24. [DOI] [PubMed] [Google Scholar]
- Blackwell JM, Searle S, 1999. ’Genetic regulation of macrophage activation: understanding the function of Nramp1 (=Ity/Lsh/Bcg). Immunol. Lett 65, 73–80. [DOI] [PubMed] [Google Scholar]
- Borrelli E, Sarraf D, Freund KB, Sadda SR, 2018. ’OCT angiography and evaluation of the choroid and choroidal vascular disorders. Prog. Retin. Eye Res 67, 30–55. [DOI] [PubMed] [Google Scholar]
- Boyle JJ, 2005. ’Macrophage activation in atherosclerosis: pathogenesis and pharmacology of plaque rupture’. Curr. Vasc. Pharmacol 3, 63–68. [DOI] [PubMed] [Google Scholar]
- Bressler NM, Bressler SB, Childs AL, Haller JA, Hawkins BS, Lewis H, MacCumber MW, Marsh MJ, Redford M, Sternberg P Jr., Thomas MA, Williams GA, Group Submacular Surgery Trials Research, 2004. ’Surgery for hemorrhagic choroidal neovascular lesions of age-related macular degeneration: ophthalmic findings: SST report no. 13. Ophthalmology 111, 1993–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretz Colin A., Divoky Vladimir, Prchal Josef, Kunz Eric, Simmons Aaron B., Wang Haibo, Hartnett Elizabeth, Mary, 2018. Erythropoietin signaling increases choroidal macrophages and cytokine expression, and exacerbates choroidal neovascularization. Sci. Rep 8, 2161–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bringmann A, Wiedemann P, 2012. ’Müller glial cells in retinal disease. Ophthalmologica 227, 1–19. [DOI] [PubMed] [Google Scholar]
- Bringmann A, Pannicke T, Grosche J, Francke M, Wiedemann P, Skatchkov SN, Osborne NN, Reichenbach A, 2006. ’Müller cells in the healthy and diseased retina. Prog. Retin. Eye Res 25, 397–424. [DOI] [PubMed] [Google Scholar]
- Bringmann A, Iandiev I, Pannicke T, Wurm A, Hollborn M, Wiedemann P, Osborne NN, Reichenbach A, 2009. ’Cellular signaling and factors involved in Müller cell gliosis: neuroprotective and detrimental effects. Prog. Retin. Eye Res 28, 423–451. [DOI] [PubMed] [Google Scholar]
- Bringmann A, Grosche A, Pannicke T, Reichenbach A, 2013. ’GABA and glutamate uptake and metabolism in retinal glial (müller) cells’. Front. Endocrinol 4, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DM, Kaiser PK, Michels M, Soubrane G, Heier JS, Kim RY, Sy JP, Schneider S, 2006. ’Ranibizumab versus verteporfin for neovascular age-related macular degeneration. N. Engl. J. Med 355, 1432–1444. [DOI] [PubMed] [Google Scholar]
- Brown DM, Tuomi L, Shapiro H, Group Pier Study, 2013a. Anatomical measures as predictors of visual outcomes in ranibizumab-treated eyes with neovascular age- related macular degeneration. Retina 33, 23–34. [DOI] [PubMed] [Google Scholar]
- Brown DM, Chen E, Mariani A, Major JC Jr., Save Study Group, 2013b. ’Super- dose anti-VEGF (SAVE) trial: 2.0 mg intravitreal ranibizumab for recalcitrant neovascular macular degeneration-primary end point. Ophthalmology 120, 349–354. [DOI] [PubMed] [Google Scholar]
- Bryan BA, Dennstedt E, Mitchell DC, Walshe TE, Noma K, Loureiro R, Saint- Geniez M, Campaigniac JP, Liao JK, D’Amore PA, 2010. ’RhoA/ROCK signaling is essential for multiple aspects of VEGF-mediated angiogenesis. Faseb. J 24, 3186–3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busbee BG, Ho AC, Brown DM, Heier JS, Suñer IJ, Li Z, Rubio RG, Lai P, 2013. ’Twelve-month efficacy and safety of 0.5 mg or 2.0 mg ranibizumab in patients with subfoveal neovascular age-related macular degeneration. Ophthalmology 120, 1046–1056. [DOI] [PubMed] [Google Scholar]
- Cabral T, Lima LH, Mello LGM, Polido J, Correa EP, Oshima A, Duong J, Serracarbassa P, Regatieri CV, Mahajan VB, Belfort R Jr., 2018. Bevacizumab injection in patients with neovascular age-related macular degeneration increases angiogenic biomarkers. Ophthalmol Retina 2, 31–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caicedo A, Espinosa-Heidmann DG, Hamasaki D, Pina Y, Cousins SW, 2005a. ’Photoreceptor synapses degenerate early in experimental choroidal neovascularization. J. Comp. Neurol 483, 263–277. [DOI] [PubMed] [Google Scholar]
- Caicedo A, Espinosa-Heidmann DG, Pina Y, Hernandez EP, Cousins SW, 2005b. ’Blood-derived macrophages infiltrate the retina and activate Muller glial cells under experimental choroidal neovascularization. Exp. Eye Res 81, 38–47. [DOI] [PubMed] [Google Scholar]
- Campbell MT, Hile KL, Zhang H, Asanuma H, Vanderbrink BA, Rink RR, Meldrum KK, 2011. ’Toll-Like receptor 4: a novel signaling pathway during renal fibrogenesis. J. Surg. Res 168, e61–e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campochiaro PA, 2015. ’Molecular pathogenesis of retinal and choroidal vascular diseases. Prog. Retin. Eye Res 49, 67–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campochiaro PA, Marcus DM, Awh CC, Regillo C, Adamis AP, Bantseev V, Chiang Y, Ehrlich JS, Erickson S, Hanley WD, Horvath J, Maass KF, Singh N, Tang F, Barteselli G, 2019. The port delivery system with ranibizumab for neovascular age-related macular degeneration: results from the randomized phase 2 ladder clinical trial. Ophthalmology 126, 1141–1154. [DOI] [PubMed] [Google Scholar]
- Cao X, Shen D, Patel MM, Tuo J, Johnson TM, Olsen TW, Chan CC, 2011. ’Macrophage polarization in the maculae of age-related macular degeneration: a pilot study. Pathol. Int 61, 528–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll MC, 2004. The complement system in regulation of adaptive immunity. Nat. Immunol 5, 981–986. [DOI] [PubMed] [Google Scholar]
- Casswell AG, Kohen D, Bird AC, 1985. ’Retinal pigment epithelial detachments in the elderly: classification and outcome’. Br. J. Ophthalmol 69, 397–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarthy U, Harding SP, Rogers CA, Downes SM, Lotery AJ, Wordsworth S, Reeves BC, Ivan Study Investigators, 2012. ’Ranibizumab versus bevacizumab to treat neovascular age-related macular degeneration: one-year findings from the IVAN randomized trial. Ophthalmology 119, 1399–1411. [DOI] [PubMed] [Google Scholar]
- Chalam KV, Grover S, Sambhav K, Balaiya S, Murthy RK, 2014. Aqueous interleukin-6 levels are superior to vascular endothelial growth factor in predicting therapeutic response to bevacizumab in age-related macular degeneration. J Ophthalmol 2014, 502174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Weber AJ, 2002. ’Expression of glial fibrillary acidic protein and glutamine synthetase by Müller cells after optic nerve damage and intravitreal application of brain-derived neurotrophic factor. Glia 38, 115–125. [DOI] [PubMed] [Google Scholar]
- Chen CY, Hooper C, Chiu D, Chamberlain M, Karia N, Heriot WJ, 2007. ’Management of submacular hemorrhage with intravitreal injection of tissue plasminogen activator and expansile gas. Retina 27, 321–328. [DOI] [PubMed] [Google Scholar]
- Chen H, Yu K-D, Xu G-Z, 2012. ’Association between variant Y402H in age-related macular degeneration (AMD) susceptibility gene CFH and treatment response of AMD: a meta-analysis. PloS One 7 e42464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Lechner J, Zhao J, Toth L, Hogg R, Silvestri G, Kissenpfennig A, Chakravarthy U, Xu H, 2016. ’STAT3 activation in circulating monocytes contributes to neovascular age-related macular degeneration. Curr. Mol. Med 16, 412–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Luo C, Zhao J, Devarajan G, Xu H, 2019. ’Immune regulation in the aging retina. Prog. Retin. Eye Res 69, 159–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong KX, Teo KYC, Cheung CMG, 2020. ‘Influence of pigment epithelial detachment on visual acuity in neovascular age-related macular degeneration’. Surv. Ophthalmol In press 10.1016/j.survophthal.2020.05.003. [DOI] [PubMed] [Google Scholar]
- Cheung CMG, Lee WK, Koizumi H, Dansingani K, Lai TYY, Freund KB, 2019. ’Pachychoroid disease. Eye 33, 14–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang A, Chang LK, Yu F, Sarraf D, 2008. ’Predictors of anti-VEGF-associated retinal pigment epithelial tear using FA and OCT analysis. Retina 28, 1265–1269. [DOI] [PubMed] [Google Scholar]
- Christen WG, Glynn RJ, Manson JE, Ajani UA, Buring JE, 1996. ’A prospective study of cigarette smoking and risk of age-related macular degeneration in men. Jama 276, 1147–1151. [PubMed] [Google Scholar]
- Chuang EL, Bird AC, 1988. ’Repair after tears of the retinal pigment epithelium. Eye 2 (Pt 1), 106–113. [DOI] [PubMed] [Google Scholar]
- Chung S, Overstreet JM, Li Y, Wang Y, Niu A, Wang S, Fan X, Sasaki K, Jin GN, Khodo SN, Gewin L, Zhang MZ, Harris RC, 2018. ’TGF-beta promotes fibrosis after severe acute kidney injury by enhancing renal macrophage infiltration. JCI Insight 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cinatl J Jr., Vogel JU, Kotchetkov R, Scholz M, Doerr HW, 1999. ’Proinflammatory potential of cytomegalovirus infection. specific inhibition of cytomegalovirus immediate-early expression in combination with antioxidants as a novel treatment strategy? Intervirology 42, 419–424. [DOI] [PubMed] [Google Scholar]
- Ciulla TA, Criswell MH, Danis RP, Hill TE, 2001. ’Intravitreal triamcinolone acetonide inhibits choroidal neovascularization in a laser-treated rat model. Arch. Ophthalmol 119, 399–404. [DOI] [PubMed] [Google Scholar]
- Ciulla TA, Criswell MH, Danis RP, Fronheiser M, Yuan P, Cox TA, Csaky KG, Robinson MR, 2003. ’Choroidal neovascular membrane inhibition in a laser treated rat model with intraocular sustained release triamcinolone acetonide microimplants. Br. J. Ophthalmol 87, 1032–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemens CR, Alten F, Termühlen J, Mihailovic N, Rosenberger F, Heiduschka P, Eter N, 2020. ’Prospective PED-study of intravitreal aflibercept for refractory vascularized pigment epithelium detachment due to age-related macular degeneration: morphologic characteristics of non-responders in optical coherence tomography. Graefes Arch. Clin. Exp. Ophthalmol 258, 1411–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman MP, Perry VH, 2002. ’Axon pathology in neurological disease: a neglected therapeutic target. Trends Neurosci. 25, 532–537. [DOI] [PubMed] [Google Scholar]
- Coleman DJ, Silverman RH, Rondeau MJ, Lloyd HO, Khanifar AA, Chan RV, 2013. ’Age-related macular degeneration: choroidal ischaemia? Br. J. Ophthalmol 97, 1020–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collery R, McLoughlin S, Vendrell V, Finnegan J, Crabb JW, Saari JC, Kennedy BN, 2008. ’Duplication and divergence of zebrafish CRALBP genes uncovers novel role for RPE- and Muller-CRALBP in cone vision. Invest. Ophthalmol. Vis. Sci 49, 3812–3820. [DOI] [PubMed] [Google Scholar]
- Concannon CG, Gorman AM, Samali A, 2003. On the role of Hsp27 in regulating apoptosis. Apoptosis : Int. J. Programmed Cell Death 8, 61–70. [DOI] [PubMed] [Google Scholar]
- Congdon N, O’Colmain B, Klaver CC, Klein R, Munoz B, Friedman DS, Kempen J, Taylor HR, Mitchell P, Group Eye Diseases Prevalence Research, 2004. ’Causes and prevalence of visual impairment among adults in the United States’. Arch. Ophthalmol 122, 477–485. [DOI] [PubMed] [Google Scholar]
- Costa C, Incio J, Soares R, 2007. ’Angiogenesis and chronic inflammation: cause or consequence? Angiogenesis 10, 149–166. [DOI] [PubMed] [Google Scholar]
- Cousins SW, Espinosa-Heidmann DG, Csaky KG, 2004. ’Monocyte activation in patients with age-related macular degeneration: a biomarker of risk for choroidal neovascularization? Arch. Ophthalmol 122, 1013–1018. [DOI] [PubMed] [Google Scholar]
- Cousins SW, Bearelly S, Reinoso MA, Chi SL, Espinosa-Heidmann DG, 2008. ’Dynamic indocyanine green angiography-guided focal thermal laser treatment of fibrotic choroidal neovascularization. Graefes Arch. Clin. Exp. Ophthalmol 246, 1677–1683. [DOI] [PubMed] [Google Scholar]
- Cousins SW, Espinosa-Heidmann DG, Miller DM, Pereira-Simon S, Hernandez EP, Chien H, Meier-Jewett C, Dix RD, 2012. ’Macrophage activation associated with chronic murine cytomegalovirus infection results in more severe experimental choroidal neovascularization. PLoS Pathog. 8 e1002671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cousins SW, Allingham MJ, Mettu PS, 2019. ’Elamipretide, a mitochondria- targeted drug, for the treatment of vision loss in dry AMD with noncentral geographic atrophy: results of the phase 1 ReCLAIM study. Invest. Ophthalmol. Vis. Sci 60, 974–74. [Google Scholar]
- Csaky KG, Baffi JZ, Byrnes GA, Wolfe JD, Hilmer SC, Flippin J, Cousins SW, 2004. ’Recruitment of marrow-derived endothelial cells to experimental choroidal neovascularization by local expression of vascular endothelial growth factor’. Exp. Eye Res 78, 1107–1116. [DOI] [PubMed] [Google Scholar]
- Curcio CA, 2001. ’Photoreceptor topography in ageing and age-related maculopathy. Eye 15, 376–383. [DOI] [PubMed] [Google Scholar]
- Curcio CA, Medeiros NE, Millican CL, 1996. ’Photoreceptor loss in age-related macular degeneration. Invest. Ophthalmol. Vis. Sci 37, 1236–1249. [PubMed] [Google Scholar]
- Czarna Anna, Sanada Fumihiro, Matsuda Alex, Kim Junghyun, Signore Sergio, Pereira João D., Sorrentino Andrea, Ramaswamy Kannappan, Cannatà Antonio, Hosoda Toru, Rota Marcello, Crea Filippo, Anversa Piero, Leri Annarosa, 2017. Single-cell analysis of the fate of c-kit-positive bone marrow cells. NPJ Regenerative Med. 2, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel E, Toth CA, Grunwald JE, Jaffe GJ, Martin DF, Fine SL, Huang J, Ying GS, Hagstrom SA, Winter K, Maguire MG, Group Comparison of age- related macular degeneration treatments trials research, 2014. Risk of scar in the comparison of age-related macular degeneration treatments trials. Ophthalmology 121, 656–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darland DC, Massingham LJ, Smith SR, Piek E, Saint-Geniez M, D’Amore PA, 2003. ’Pericyte production of cell-associated VEGF is differentiation-dependent and is associated with endothelial survival. Dev. Biol 264, 275–288. [DOI] [PubMed] [Google Scholar]
- Davis SJ, Lyzogubov VV, Tytarenko RG, Safar AN, Bora NS, Bora PS, 2012. The effect of nicotine on anti-vascular endothelial growth factor therapy in a mouse model of neovascular age-related macular degeneration. Retina 32, 1171–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Oliveira Dias JR, Zhang Q, Garcia JMB, Zheng F, Motulsky EH, Roisman L, Miller A, Chen CL, Kubach S, de Sisternes L, Durbin MK, Feuer W, Wang RK, Gregori G, Rosenfeld PJ, 2018. ’Natural history of subclinical neovascularization in nonexudative age-related macular degeneration using swept- source OCT angiography. Ophthalmology 125, 255–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit NM, Vanmol J, Kamermans A, Hendriks J, de Vries HE, 2017. ’Inflammation at the blood-brain barrier: the role of liver X receptors. Neurobiol. Dis 107, 57–65. [DOI] [PubMed] [Google Scholar]
- Del Amo EM, Rimpelä AK, Heikkinen E, Kari OK, Ramsay E, Lajunen T, Schmitt M, Pelkonen L, Bhattacharya M, Richardson D, Subrizi A, Turunen T, Reinisalo M, Itkonen J, Toropainen E, Casteleijn M, Kidron H, Antopolsky M, Vellonen KS, Ruponen M, Urtti A, 2017. ’Pharmacokinetic aspects of retinal drug delivery. Prog. Retin. Eye Res 57, 134–185. [DOI] [PubMed] [Google Scholar]
- Delcourt C, Diaz JL, Ponton-Sanchez A, Papoz L, 1998. ’Smoking and age-related macular degeneration. The POLA Study. Pathologies Oculaires Liées à l’Age. Arch. Ophthalmol 116, 1031–1035. [DOI] [PubMed] [Google Scholar]
- Derouiche A, Pannicke T, Haseleu J, Blaess S, Grosche J, Reichenbach A, 2012. ’Beyond polarity: functional membrane domains in astrocytes and Müller cells. Neurochem. Res 37, 2513–2523. [DOI] [PubMed] [Google Scholar]
- Derouiche A, Haseleu J, Korf HW, 2015. Fine astrocyte processes contain very small mitochondria: glial oxidative capability may fuel transmitter metabolism. Neurochem. Res 40, 2402–2413. [DOI] [PubMed] [Google Scholar]
- Donnelly DJ, Longbrake EE, Shawler TM, Kigerl KA, Lai W, Tovar CA, Ransohoff RM, Popovich PG, 2011. ’Deficient CX3CR1 signaling promotes recovery after mouse spinal cord injury by limiting the recruitment and activation of Ly6Clo/iNOS+ macrophages. J. Neurosci 31, 9910–9922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugel PU, Koh A, Ogura Y, Jaffe GJ, Schmidt-Erfurth U, Brown DM, Gomes AV, Warburton J, Weichselberger A, Holz FG, Hawk, Harrier Study Investigators, 2020a. ’HAWK and HARRIER: phase 3, multicenter, randomized, double-masked trials of brolucizumab for neovascular age-related macular degeneration. Ophthalmology 127, 72–84. [DOI] [PubMed] [Google Scholar]
- Dugel PU, Boyer DS, Antoszyk AN, Steinle NC, Varenhorst MP, Pearlman JA, Gillies MC, Finger RP, Baldwin ME, Leitch IM, 2020b. ’Phase 1 study of OPT- 302 inhibition of vascular endothelial growth factors C and D for neovascular age- related macular degeneration. Ophthalmol Retina 4, 250–263. [DOI] [PubMed] [Google Scholar]
- Dunn EN, Hariprasad SM, Sheth VS, 2017. ’An overview of the fovista and rinucumab trials and the fate of anti-PDGF medications. Ophthalmic Surg Lasers Imag. Retina 48, 100–104. [DOI] [PubMed] [Google Scholar]
- Eghoj MS, Sorensen TL, 2012. ’Tachyphylaxis during treatment of exudative age- related macular degeneration with ranibizumab. Br. J. Ophthalmol 96, 21–23. [DOI] [PubMed] [Google Scholar]
- Ehlken C, Jungmann S, Böhringer D, Agostini HT, Junker B, Pielen A, 2014. ’Switch of anti-VEGF agents is an option for nonresponders in the treatment of AMD. Eye 28, 538–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enders P, Scholz P, Muether PS, Fauser S, 2016. ’Variability of disease activity in patients treated with ranibizumab for neovascular age-related macular degeneration. Eye 30, 1072–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ersoz MG, Karacorlu M, Arf S, Sayman Muslubas I, Hocaoglu M, 2017. Retinal pigment epithelium tears: classification, pathogenesis, predictors, and management. Surv. Ophthalmol 62, 493–505. [DOI] [PubMed] [Google Scholar]
- Espinosa-Heidmann DG, Suñer I, Hernandez EP, Frazier WD, Csaky KG, Cousins SW, 2002. ’Age as an independent risk factor for severity of experimental choroidal neovascularization’. Invest. Ophthalmol. Vis. Sci 43, 1567–1573. [PubMed] [Google Scholar]
- Espinosa-Heidmann DG, Suñer IJ, Hernandez EP, Monroy D, Csaky KG, Cousins SW, 2003a. ’Macrophage depletion diminishes lesion size and severity in experimental choroidal neovascularization. Invest. Ophthalmol. Vis. Sci 44, 3586–3592. [DOI] [PubMed] [Google Scholar]
- Espinosa-Heidmann DG, Caicedo A, Hernandez EP, Csaky KG, Cousins SW, 2003b. ’Bone marrow-derived progenitor cells contribute to experimental choroidal neovascularization. Invest. Ophthalmol. Vis. Sci 44, 4914–4919. [DOI] [PubMed] [Google Scholar]
- Espinosa-Heidmann DG, Reinoso MA, Pina Y, Csaky KG, Caicedo A, Cousins SW, 2005. ’Quantitative enumeration of vascular smooth muscle cells and endothelial cells derived from bone marrow precursors in experimental choroidal neovascularization. Exp. Eye Res 80, 369–378. [DOI] [PubMed] [Google Scholar]
- Espinosa-Heidmann DG, Malek G, Mettu PS, Caicedo A, Saloupis P, Gach S, Dunnon AK, Hu P, Spiga MG, Cousins SW, 2013. ’Bone marrow transplantation transfers age-related susceptibility to neovascular remodeling in murine laser-induced choroidal neovascularization. Invest. Ophthalmol. Vis. Sci 54, 7439–7449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans RN, Reeves BC, Phillips D, Muldrew KA, Rogers C, Harding SP, Chakravarthy U, 2020. Long-term visual outcomes after release from protocol in patients who participated in the inhibition of VEGF in age-related choroidal neovascularisation (IVAN) trial. Ophthalmology 127, 1191–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falck-Hansen M, Kassiteridi C, Monaco C, 2013. ’Toll-Like receptors in atherosclerosis’. Int. J. Mol. Sci 14, 14008–14023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fassnacht-Riederle H, Becker M, Graf N, Michels S, 2014. ’Effect of aflibercept in insufficient responders to prior anti-VEGF therapy in neovascular AMD. Graefes Arch. Clin. Exp. Ophthalmol 252, 1705–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauser S, Muether PS, 2016. ’Clinical correlation to differences in ranibizumab and aflibercept vascular endothelial growth factor suppression times. Br. J. Ophthalmol 100, 1494–1498. [DOI] [PubMed] [Google Scholar]
- Fauser S, Schwabecker V, Muether PS, 2014. ’Suppression of intraocular vascular endothelial growth factor during aflibercept treatment of age-related macular degeneration. Am. J. Ophthalmol 158, 532–536. [DOI] [PubMed] [Google Scholar]
- Faust N, Huber MC, Sippel AE, Bonifer C, 1997. ’Different macrophage populations develop from embryonic/fetal and adult hematopoietic tissues. Exp. Hematol 25, 432–444. [PubMed] [Google Scholar]
- Feng L, Ju M, Lee KYV, Mackey A, Evangelista M, Iwata D, Adamson P, Lashkari K, Foxton R, Shima D, Ng YS, 2017. ’A proinflammatory function of toll-like receptor 2 in the retinal pigment epithelium as a novel target for reducing choroidal neovascularization in age-related macular degeneration. Am. J. Pathol 187, 2208–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara N, 2010. ’Pathways mediating VEGF-independent tumor angiogenesis. Cytokine Growth Factor Rev. 21, 21–26. [DOI] [PubMed] [Google Scholar]
- Fiorentino DF, Zlotnik A, Mosmann TR, Howard M, Garra A, 1991. ’IL-10 inhibits cytokine production by activated macrophages. J. Immunol 147, 3815. [PubMed] [Google Scholar]
- Fisher SK, Stone J, Rex TS, Linberg KA, Lewis GP, 2001. ’Experimental retinal detachment: a paradigm for understanding the effects of induced photoreceptor degeneration. Prog. Brain Res. 131, 679–698. [DOI] [PubMed] [Google Scholar]
- Fleckenstein M, Mitchell P, Freund KB, Sadda S, Holz FG, Brittain C, Henry EC, Ferrara D, 2018. The progression of geographic atrophy secondary to age- related macular degeneration. Ophthalmology 125, 369–390. [DOI] [PubMed] [Google Scholar]
- Forooghian Farzin, Emily Y, Chew Catherine B, Meyerle Cukras, Catherine Wong, Wai T, 2011. ’Investigation of the role of neutralizing antibodies against bevacizumab as mediators of tachyphylaxis. Acta Ophthalmol. 89, e206–e207. [DOI] [PubMed] [Google Scholar]
- Fung AT, Kumar N, Vance SK, Slakter JS, Klancnik JM, Spaide RS, Freund KB, 2012. ’Pilot study to evaLuate the role of high-dose rAnibizumab 2.0 mg in the management of neovascular age-related macular degeneration in patients with perSistent/recurrenT macular fluid <30 days following treatment with intravitreal anti-VEGF therapy (the LAST Study). Eye 26, 1181–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaengel K, Genové G, Armulik A, Betsholtz C, 2009. Endothelial-mural cell signaling in vascular development and angiogenesis. Arterioscler. Thromb. Vasc. Biol 29, 630–638. [DOI] [PubMed] [Google Scholar]
- Gao F, Li F, Miao Y, Xu LJ, Zhao Y, Li Q, Zhang SH, Wu J, Sun XH, Wang Z, 2017. ’Involvement of the MEK-ERK/p38-CREB/c-fos signaling pathway in Kir channel inhibition-induced rat retinal Müller cell gliosis. Sci. Rep 7, 1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Y, Mansell A, Ussher JE, Brooks AES, Manning K, Wang CJH, Taylor JA, 2013. ’Rotavirus NSP4 triggers secretion of proinflammatory cytokines from macrophages via toll-like receptor 2. J. Virol 87, 11160–11167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissmann F, Jung S, Littman DR, 2003. ’Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 19, 71–82. [DOI] [PubMed] [Google Scholar]
- Ginhoux F, Guilliams M, 2016. ’Tissue-Resident macrophage ontogeny and homeostasis. Immunity 44, 439–449. [DOI] [PubMed] [Google Scholar]
- Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF, Conway SJ, Ng LG, Stanley ER, Samokhvalov IM, Merad M, 2010. ’Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330, 841–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glatt H, Machemer R, 1982. ’Experimental subretinal hemorrhage in rabbits’. Am. J. Ophthalmol 94, 762–773. [DOI] [PubMed] [Google Scholar]
- Goldmann Tobias, Peter Wieghofer, Müller Philippe F., Wolf Yochai, Varol Diana, Simon Yona, Brendecke Stefanie M., Kierdorf Katrin, Staszewski Ori, Datta Moumita, Tom Luedde, Heikenwalder Mathias, Jung Steffen, Prinz Marco, 2013. ’A new type of microglia gene targeting shows TAK1 to be pivotal in CNS autoimmune inflammation. Nat. Neurosci 16, 1618–1626. [DOI] [PubMed] [Google Scholar]
- Goodwin AM, Kitajewski J, D’Amore PA, 2007. ’Wnt1 and Wnt5a affect endothelial proliferation and capillary length; Wnt2 does not. Growth Factors 25, 25–32. [DOI] [PubMed] [Google Scholar]
- Gordon S, 1999. Macrophages and the Immune Response. Lippencott-Raven Publishers, Philadelphia: ). [Google Scholar]
- Green WR, 1999. ’Histopathology of age-related macular degeneration. Mol. Vis 5, 27. [PubMed] [Google Scholar]
- Griffin TM, Scanzello CR, 2019. ’Innate inflammation and synovial macrophages in osteoarthritis pathophysiology. Clin. Exp. Rheumatol 37 (Suppl. 120), 57–63. [PMC free article] [PubMed] [Google Scholar]
- Grossniklaus HE, Green WR, 2004. ’Choroidal neovascularization’. Am. J. Ophthalmol 137, 496–503. [DOI] [PubMed] [Google Scholar]
- Grossniklaus HE, Ling JX, Wallace TM, Dithmar S, Lawson DH, Cohen C, Elner VM, Elner SG, Sternberg P Jr., 2002. ’Macrophage and retinal pigment epithelium expression of angiogenic cytokines in choroidal neovascularization. Mol. Vis 8, 119–126. [PubMed] [Google Scholar]
- Grossniklaus HE, Miskala PH, Green WR, Bressler SB, Hawkins BS, Toth C, Wilson DJ, Bressler NM, 2005. ’Histopathologic and ultrastructural features of surgically excised subfoveal choroidal neovascular lesions: submacular surgery trials report no. 7. Arch. Ophthalmol 123, 914–921. [DOI] [PubMed] [Google Scholar]
- Grossniklaus HE, Kang SJ, Berglin L, 2010. ’Animal models of choroidal and retinal neovascularization. Prog. Retin. Eye Res 29, 500–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunin M, Burstyn-Cohen T, Hagbi-Levi S, Peled A, Chowers I, 2012. ’Chemokine receptor expression in peripheral blood monocytes from patients with neovascular age-related macular degeneration. Invest. Ophthalmol. Vis. Sci 53, 5292–5300. [DOI] [PubMed] [Google Scholar]
- Grunin M, Hagbi-Levi S, Rinsky B, Smith Y, Chowers I, 2016. ’Transcriptome analysis on monocytes from patients with neovascular age-related macular degeneration. Sci. Rep 6, 29046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunwald JE, Hariprasad SM, DuPont J, Maguire MG, Fine SL, Brucker AJ, Maguire AM, Ho AC, 1998. ’Foveolar choroidal blood flow in age-related macular degeneration. Invest. Ophthalmol. Vis. Sci 39, 385–390. [PubMed] [Google Scholar]
- Guber J, Praveen A, Saeed MU, 2013. ’Higher incidence of retinal pigment epithelium tears after ranibizumab in neovascular age-related macular degeneration with increasing pigment epithelium detachment height. Br. J. Ophthalmol 97, 1486–1487. [DOI] [PubMed] [Google Scholar]
- Guillonneau X, Eandi CM, Paques M, Sahel JA, Sapieha P, Sennlaub F, 2017. ’On phagocytes and macular degeneration. Prog. Retin. Eye Res 61, 98–128. [DOI] [PubMed] [Google Scholar]
- Guymer R, Robman L, 2007. ’Chlamydia pneumoniae and age-related macular degeneration: a role in pathogenesis or merely a chance association? Clin. Exp. Ophthalmol 35, 89–93. [DOI] [PubMed] [Google Scholar]
- Guymer RH, Markey CM, McAllister IL, Gillies MC, Hunyor AP, Arnold JJ, Investigators, Fluid, 2019. ’Tolerating subretinal fluid in neovascular age-related macular degeneration treated with ranibizumab using a treat-and-extend regimen: FLUID study 24-month results. Ophthalmology 126, 723–734. [DOI] [PubMed] [Google Scholar]
- Hagstrom SA, Ying GS, Pauer GJT, Sturgill-Short GM, Huang J, Callanan DG, Kim IK, Klein ML, Maguire MG, Martin DF, 2013. ’Pharmacogenetics for genes associated with age-related macular degeneration in the Comparison of AMD Treatments Trials (CATT). Ophthalmology 120, 593–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagstrom SA, Ying GS, Pauer GJ, Sturgill-Short GM, Huang J, Maguire MG, Martin DF, 2014. ’VEGFA and VEGFR2 gene polymorphisms and response to anti- vascular endothelial growth factor therapy: comparison of age-related macular degeneration treatments trials (CATT). JAMA Ophthalmol 132, 521–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn G, Jores R, Mocarski ES, 1998. ’Cytomegalovirus remains latent in a common precursor of dendritic and myeloid cells. Proc. Natl. Acad. Sci. U. S. A 95, 3937–3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammerstrom J, 1979. ’Human macrophage differentiation in vivo and in vitro. A comparison of human peritoneal macrophages and monocytes. Acta Pathol Microbiol Scand [C] 87C, 113–120. [PubMed] [Google Scholar]
- Hamrick TS, Havell EA, Horton JR, Orndorff PE, 2000. ’Host and bacterial factors involved in the innate ability of mouse macrophages to eliminate internalized unopsonized Escherichia coli. Infect. Immun 68, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna RN, Shaked I, Hubbeling HG, Punt JA, Wu R, Herrley E, Zaugg C, Pei H, Geissmann F, Ley K, Hedrick CC, 2012. ’NR4A1 (Nur77) deletion polarizes macrophages toward an inflammatory phenotype and increases atherosclerosis. Circ. Res 110, 416–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara C, Wakabayashi T, Fukushima Y, Sayanagi K, Kawasaki R, Sato S, Sakaguchi H, Nishida K, 2019. ’Tachyphylaxis during treatment of exudative age- related macular degeneration with aflibercept. Graefes Arch. Clin. Exp. Ophthalmol 257, 2559–2569. [DOI] [PubMed] [Google Scholar]
- Harada T, Harada C, Nakayama N, Okuyama S, Yoshida K, Kohsaka S, Matsuda H, Wada K, 2000. ’Modification of glial-neuronal cell interactions prevents photoreceptor apoptosis during light-induced retinal degeneration. Neuron 26, 533–541. [DOI] [PubMed] [Google Scholar]
- Harada T, Harada C, Kohsaka S, Wada E, Yoshida K, Ohno S, Mamada H, Tanaka K, Parada LF, Wada K, 2002. ’Microglia-Müller glia cell interactions control neurotrophic factor production during light-induced retinal degeneration. J. Neurosci 22, 9228–9236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartnett ME, Weiter JJ, Garsd A, Jalkh AE, 1992. ’Classification of retinal pigment epithelial detachments associated with drusen. Graefes Arch. Clin. Exp. Ophthalmol 230, 11–19. [DOI] [PubMed] [Google Scholar]
- Hartnett ME, Weiter JJ, Staurenghi G, Elsner AE, 1996. Deep retinal vascular anomalous complexes in advanced age-related macular degeneration. Ophthalmology 103, 2042–2053. [DOI] [PubMed] [Google Scholar]
- Haslbeck M, 2002. ’sHsps and their role in the chaperone network. Cell. Mol. Life Sci. : CM 59, 1649–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan AS, Johnson MW, Schneiderman TE, Regillo CD, Tornambe PE, Poliner LS, Blodi BA, Elner SG, 1999. ’Management of submacular hemorrhage with intravitreous tissue plasminogen activator injection and pneumatic displacement. Ophthalmology 106, 1900–1906. ; discussion 06–7. [DOI] [PubMed] [Google Scholar]
- Hatz K, Prunte C, 2014. ’Polypoidal choroidal vasculopathy in Caucasian patients with presumed neovascular age-related macular degeneration and poor ranibizumab response. Br. J. Ophthalmol 98, 188–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haupert CL, McCuen BW 2nd, Jaffe GJ, Steuer ER, Cox TA, Toth CA, Fekrat S, Postel EA, 2001. ’Pars plana vitrectomy, subretinal injection of tissue plasminogen activator, and fluid-gas exchange for displacement of thick submacular hemorrhage in age-related macular degeneration. Am. J. Ophthalmol 131, 208–215. [DOI] [PubMed] [Google Scholar]
- He L, Marneros AG, 2013. ’Macrophages are essential for the early wound healing response and the formation of a fibrovascular scar. Am. J. Pathol 182, 2407–2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Hahn P, Iacovelli J, Wong R, King C, Bhisitkul R, Massaro-Giordano M, Dunaief JL, 2007. ’Iron homeostasis and toxicity in retinal degeneration. Prog. Retin. Eye Res 26, 649–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heeschen C, Jang JJ, Weis M, Pathak A, Kaji S, Hu RS, Tsao PS, Johnson FL, Cooke JP, 2001. ’Nicotine stimulates angiogenesis and promotes tumor growth and atherosclerosis. Nat. Med 7, 833–839. [DOI] [PubMed] [Google Scholar]
- Heier JS, 2019. Intravitreal Aflibercept versus Sham as Prophylaxis against Conversion to Neovascular AMD, PRO-CON Study: Year 1 results. In: Angiongenesis, Exudation, and Degeneration 2019 Annual Meeting. Miami, FL. [Google Scholar]
- Heier JS, Brown DM, Chong V, Korobelnik JF, Kaiser PK, Nguyen QD, Kirchhof B, Ho A, Ogura Y, Yancopoulos GD, Stahl N, Vitti R, Berliner AJ, Soo Y, Anderesi M, Groetzbach G, Sommerauer B, Sandbrink R, Simader C, Schmidt-Erfurth U, 2012. Intravitreal aflibercept (VEGF trap-eye) in wet age- related macular degeneration. Ophthalmology 119, 2537–2548. [DOI] [PubMed] [Google Scholar]
- Heier JS, Wykoff CC, Waheed NK, Kitchens JW, Patel SS, Vitti R, Perlee L, Chu KW, Leal S, Asmus F, Son V, Schmelter T, Brown DM, 2020. ’Intravitreal combined aflibercept + anti-platelet-derived growth factor receptor β for neovascular age-related macular degeneration: results of the phase 2 CAPELLA trial. Ophthalmology 127, 211–220. [DOI] [PubMed] [Google Scholar]
- Heiferman MJ, Fawzi AA, 2019. ’Progression of subclinical choroidal neovascularization in age-related macular degeneration. PloS One 14 e0217805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellberg C, Ostman A, Heldin CH, 2010. ’PDGF and vessel maturation. Recent Results Canc. Res 180, 103–114. [DOI] [PubMed] [Google Scholar]
- Hesse L, Schmidt J, Kroll P, 1999. ’Management of acute submacular hemorrhage using recombinant tissue plasminogen activator and gas. Graefes Arch. Clin. Exp. Ophthalmol 237, 273–277. [DOI] [PubMed] [Google Scholar]
- Hirschi KK, Rohovsky SA, D’Amore PA, 1997. ’Cell-cell interactions in vessel assembly: a model for the fundamentals of vascular remodelling. Transpl. Immunol 5, 177–178. [DOI] [PubMed] [Google Scholar]
- Hirschi KK, Rohovsky SA, D’Amore PA, 1998. ’PDGF, TGF-beta, and heterotypic cell-cell interactions mediate endothelial cell-induced recruitment of 10T1/2 cells and their differentiation to a smooth muscle fate. J. Cell Biol. 141, 805–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschi KK, Rohovsky SA, Beck LH, Smith SR, D’Amore PA, 1999. ’Endothelial cells modulate the proliferation of mural cell precursors via platelet-derived growth factor-BB and heterotypic cell contact. Circ. Res 84, 298–305. [DOI] [PubMed] [Google Scholar]
- Hou HY, Wang YS, Xu JF, Wang BR, 2008. ’Nicotine promotes contribution of bone marrow-derived cells to experimental choroidal neovascularization in mice. Exp. Eye Res 86, 983–990. [DOI] [PubMed] [Google Scholar]
- Huitinga I, van Rooijen N, de Groot CJ, Uitdehaag BM, Dijkstra CD, 1990. ’Suppression of experimental allergic encephalomyelitis in Lewis rats after elimination of macrophages. J. Exp. Med 172, 1025–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain RM, Neiweem AE, Kansara V, Harris A, Ciulla TA, 2019. ’Tie-2/ Angiopoietin pathway modulation as a therapeutic strategy for retinal disease. Expet Opin. Invest. Drugs 28, 861–869. [DOI] [PubMed] [Google Scholar]
- Ingersoll MA, Spanbroek R, Lottaz C, Gautier EL, Frankenberger M, Hoffmann R, Lang R, Haniffa M, Collin M, Tacke F, Habenicht AJ, Ziegler-Heitbrock L, Randolph GJ, 2010. ’Comparison of gene expression profiles between human and mouse monocyte subsets. Blood 115, e10–e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishibashi T, Miki K, Sorgente N, Patterson R, Ryan SJ, 1985. ’Effects of intravitreal administration of steroids on experimental subretinal neovascularization in the subhuman primate. Arch. Ophthalmol 103, 708–711. [DOI] [PubMed] [Google Scholar]
- Izumi Y, Kirby CO, Benz AM, Olney JW, Zorumski CF, 1999. ’Müller cell swelling, glutamate uptake, and excitotoxic neurodegeneration in the isolated rat retina. Glia 25, 379–389. [PubMed] [Google Scholar]
- Izumi Y, Shimamoto K, Benz AM, Hammerman SB, Olney JW, Zorumski CF, 2002. ’Glutamate transporters and retinal excitotoxicity. Glia 39, 58–68. [DOI] [PubMed] [Google Scholar]
- Izumi-Nagai K, Nagai N, Ozawa Y, Mihara M, Ohsugi Y, Kurihara T, Koto T, Satofuka S, Inoue M, Tsubota K, Okano H, Oike Y, Ishida S, 2007. ’Interleukin-6 receptor-mediated activation of signal transducer and activator of transcription-3 (STAT3) promotes choroidal neovascularization. Am. J. Pathol 170, 2149–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe GJ, Kaiser PK, Thompson D, Gibson A, Saroj N, Vitti R, Berliner AJ, Heier JS, 2016. Differential response to anti-VEGF regimens in age-related macular degeneration patients with early persistent retinal fluid. Ophthalmology 123, 1856–1864. [DOI] [PubMed] [Google Scholar]
- Jaffe GJ, Ciulla TA, Ciardella AP, Devin F, Dugel PU, Eandi CM, Masonson H, Monés J, Pearlman JA, Quaranta-El Maftouhi M, Ricci F, Westby K, Patel SC, 2017. Dual antagonism of PDGF and VEGF in neovascular age-related macular degeneration: a phase IIb, multicenter, randomized controlled trial. Ophthalmology 124, 224–234. [DOI] [PubMed] [Google Scholar]
- Jain RK, 2001. ’Clearing the smoke on nicotine and angiogenesis. Nat. Med 7, 775–777. [DOI] [PubMed] [Google Scholar]
- Ji M, Miao Y, Dong LD, Chen J, Mo XF, Jiang SX, Sun XH, Yang XL, Wang Z, 2012. ’Group I mGluR-mediated inhibition of Kir channels contributes to retinal Müller cell gliosis in a rat chronic ocular hypertension model. J. Neurosci 32, 12744–12755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Beller DI, Frendl G, Graves DT, 1992. ’Monocyte chemoattractant protein- 1 regulates adhesion molecule expression and cytokine production in human monocytes. J. Immunol 148, 2423–2428. [PubMed] [Google Scholar]
- Jindal S, Cousins SW, Mettu PS, 2017. ’Indocyanine green angiography-directed verteporfin photodynamic therapy for treatment of serous pigment epithelium detachments in neovascular age-related macular degeneration. Invest. Ophthalmol. Vis. Sci 58, 1941–41.28384715 [Google Scholar]
- Jinnouchi H, Guo L, Sakamoto A, Torii S, Sato Y, Cornelissen A, Kuntz S, Paek KH, Fernandez R, Fuller D, Gadhoke N, Surve D, Romero M, Kolodgie FD, Virmani R, Finn AV, 2019. Diversity of macrophage phenotypes and responses in atherosclerosis. Cell. Mol. Life Sci. 77, 1919–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser PK, Boyer DS, Cruess AF, Slakter JS, Pilz S, Weisberger A, 2012. ’Verteporfin plus ranibizumab for choroidal neovascularization in age-related macular degeneration: twelve-month results of the DENALI study. Ophthalmology 119, 1001–1010. [DOI] [PubMed] [Google Scholar]
- Kalayoglu MV, Bula D, Arroyo J, Gragoudas ES, D’Amico D, Miller JW, 2005. ’Identification of Chlamydia pneumoniae within human choroidal neovascular membranes secondary to age-related macular degeneration. Graefes Arch. Clin. Exp. Ophthalmol 243, 1080–1090. [DOI] [PubMed] [Google Scholar]
- Karlstetter M, Scholz R, Rutar M, Wong WT, Provis JM, Langmann T, 2015. ’Retinal microglia: just bystander or target for therapy? Prog. Retin. Eye Res 45, 30–57. [DOI] [PubMed] [Google Scholar]
- Kelly J, Ali Khan A, Yin J, Ferguson TA, Apte RS, 2007. ’Senescence regulates macrophage activation and angiogenic fate at sites of tissue injury in mice. J. Clin. Invest 117, 3421–3426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly EK, Wang L, Ivashkiv LB, 2010. ’Calcium-activated pathways and oxidative burst mediate zymosan-induced signaling and IL-10 production in human macrophages. J. Immunol 184, 5545–5552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent D, Sheridan C, 2003. ’Choroidal neovascularization: a wound healing perspective. Mol. Vis 9, 747–755. [PubMed] [Google Scholar]
- Khanani AM, Eichenbaum D, Schlottmann PG, Tuomi L, Sarraf D, 2018. Optimal management of pigment epithelial detachments in eyes with neovascular age-related macular degeneration. Retina 38, 2103–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein R, Klein BE, Linton KL, DeMets DL, 1993. ’The Beaver Dam Eye Study: the relation of age-related maculopathy to smoking. Am. J. Epidemiol 137, 190–200. [DOI] [PubMed] [Google Scholar]
- Kofuji P, Ceelen P, Zahs KR, Surbeck LW, Lester HA, Newman EA, 2000. ’Genetic inactivation of an inwardly rectifying potassium channel (Kir4.1 subunit) in mice: phenotypic impact in retina. J. Neurosci 20, 5733–5740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh A, Lee WK, Chen LJ, Chen SJ, Hashad Y, Kim H, Lai TY, Pilz S, Ruamviboonsuk P, Tokaji E, Weisberger A, Lim TH, 2012. ’EVEREST study: efficacy and safety of verteporfin photodynamic therapy in combination with ranibizumab or alone versus ranibizumab monotherapy in patients with symptomatic macular polypoidal choroidal vasculopathy. Retina 32, 1453–1464. [DOI] [PubMed] [Google Scholar]
- Koh A, Lai TYY, Takahashi K, Wong TY, Chen LJ, Ruamviboonsuk P, Tan CS, Feller C, Margaron P, Lim TH, Lee WK, 2017. ’Efficacy and safety of ranibizumab with or without verteporfin photodynamic therapy for polypoidal choroidal vasculopathy: a randomized clinical trial. JAMA Ophthalmol 135, 1206–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh S, Chen WJ, Dejneka NS, Harris IR, Lu B, Girman S, Saylor J, Wang S, Eroglu C, 2018. ’Subretinal human umbilical tissue-derived cell transplantation preserves retinal synaptic connectivity and attenuates müller glial reactivity. J. Neurosci 38, 2923–2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokame GT, deCarlo TE, Kaneko KN, Omizo JN, Lian R, 2019a. Anti-vascular endothelial growth factor resistance in exudative macular degeneration and polypoidal choroidal vasculopathy. Ophthalmol Retina 3, 744–752. [DOI] [PubMed] [Google Scholar]
- Kokame GT, Liu K, Kokame KA, Kaneko KN, Omizo JN, 2019b. Clinical characteristics of polypoidal choroidal vasculopathy and anti-vascular endothelial growth factor treatment response in caucasians. Ophthalmologica 1–9. [DOI] [PubMed] [Google Scholar]
- Krause TA, Alex AF, Engel DR, Kurts C, Eter N, 2014. ’VEGF-production by CCR2-dependent macrophages contributes to laser-induced choroidal neovascularization. PloS One 9 e94313–e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuehlewein L, Bansal M, Lenis TL, Iafe NA, Sadda SR, Bonini Filho MA, De Carlo TE, Waheed NK, Duker JS, Sarraf D, 2015. ’Optical coherence tomography angiography of type 1 neovascularization in age-related macular degeneration. Am. J. Ophthalmol 160, 739–748.e2. [DOI] [PubMed] [Google Scholar]
- Lad EM, Grunwald L, Mettu PS, Serrano NP, Crowell S, Cousins SW, 2012. ’Lesion morphology on indocyanine green angiography in age-related macular degeneration with classic choroidal neovascular membrane: implications for response to anti-VEGF treatment. Invest. Ophthalmol. Vis. Sci 53, 5161.22743327 [Google Scholar]
- Lad EM, Hammill BG, Qualls LG, Wang F, Cousins SW, Curtis LH, 2014. ’Anti- VEGF treatment patterns for neovascular age-related macular degeneration among Medicare beneficiaries’. Am. J. Ophthalmol 158, 537–543.e2. [DOI] [PubMed] [Google Scholar]
- Lad EM, Cousins SW, Van Arnam JS, Proia AD, 2015. ’Abundance of infiltrating CD163+ cells in the retina of postmortem eyes with dry and neovascular age-related macular degeneration. Graefes Arch. Clin. Exp. Ophthalmol 253, 1941–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laiginhas R, Yang J, Rosenfeld PJ, Falcão M, 2020.’Nonexudative macular neovascularization - a systematic review of prevalence, natural history, and recent insights from OCT angiography. Ophthalmol Retina 4, 651–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert WS, Pasini S, Collyer JW, Formichella CR, Ghose P, Carlson BJ, Calkins DJ, 2020. ’Of mice and monkeys: neuroprotective efficacy of the p38 inhibitor BIRB 796 depends on model duration in experimental glaucoma. Sci. Rep 10, 8535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen M, Schmidt-Erfurth U, Lanzetta P, Wolf S, Simader C, Tokaji E, Pilz S, Weisberger A, 2012. ’Verteporfin plus ranibizumab for choroidal neovascularization in age-related macular degeneration: twelve-month MONT BLANC study results. Ophthalmology 119, 992–1000. [DOI] [PubMed] [Google Scholar]
- Lavalette S, Raoul W, Houssier M, Camelo S, Levy O, Calippe B, Jonet L, Behar- Cohen F, Chemtob S, Guillonneau X, Combadiere C, Sennlaub F, 2011. ’Interleukin-1beta inhibition prevents choroidal neovascularization and does not exacerbate photoreceptor degeneration. Am. J. Pathol 178, 2416–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaVerda D, Kalayoglu MV, Byrne GI, 1999. ’Chlamydial heat shock proteins and disease pathology: new paradigms for old problems? Infect. Dis. Obstet. Gynecol 7, 64–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechner J, Chen M, Hogg RE, Toth L, Silvestri G, Chakravarthy U, Xu H, 2017. ’Peripheral blood mononuclear cells from neovascular age-related macular degeneration patients produce higher levels of chemokines CCL2 (MCP-1) and CXCL8 (IL-8). J. Neuroinflammation 14, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AY, Raya AK, Kymes SM, Shiels A, Brantley MA Jr., 2009. ’Pharmacogenetics of complement factor H (Y402H) and treatment of exudative age- related macular degeneration with ranibizumab. Br. J. Ophthalmol 93, 610–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leifer CA, Medvedev AE, 2016. ’Molecular mechanisms of regulation of Toll-like receptor signaling. J. Leukoc. Biol 100, 927–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Heiduschka P, Alex AF, Niekamper D, Eter N, 2017. ’Behaviour of CD11b- positive cells in an animal model of laser-induced choroidal neovascularisation. Ophthalmologica 237, 29–41. [DOI] [PubMed] [Google Scholar]
- Li F, Jiang D, Samuel MA, 2019. ’Microglia in the developing retina. Neural Dev. 14, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lifshitz L, Tabak G, Gassmann M, Mittelman M, Neumann D, 2010. ’Macrophages as novel target cells for erythropoietin. Haematologica 95, 1823–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lincoff H, Kreissig I, Stopa M, Uram D, 2008. ’A 40 degrees gaze down position for pneumatic displacement of submacular hemorrhage: clinical application and results. Retina 28, 56–59. [DOI] [PubMed] [Google Scholar]
- Little K, Ma JH, Yang N, Chen M, Xu H, 2018. ’Myofibroblasts in macular fibrosis secondary to neovascular age-related macular degeneration - the potential sources and molecular cues for their recruitment and activation. EBioMedicine 38, 283–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Copland DA, Horie S, Wu WK, Chen M, Xu Y, Paul Morgan B, Mack M, Xu H, Nicholson LB, Dick AD, 2013. ’Myeloid cells expressing VEGF and arginase-1 following uptake of damaged retinal pigment epithelium suggests potential mechanism that drives the onset of choroidal angiogenesis in mice. PloS One 8, e72935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liyanage SE, Gardner PJ, Ribeiro J, Cristante E, Sampson RD, Luhmann UF, Ali RR, Bainbridge JW, 2016. ’Flow cytometric analysis of inflammatory and resident myeloid populations in mouse ocular inflammatory models. Exp. Eye Res 151, 160–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lommatzsch A, Heimes B, Gutfleisch M, Spital G, Zeimer M, Pauleikhoff D, 2009. ’Serous pigment epithelial detachment in age-related macular degeneration: comparison of different treatments. Eye 23, 2163–2168. [DOI] [PubMed] [Google Scholar]
- Lopez PF, Sippy BD, Lambert HM, Thach AB, Hinton DR, 1996. ’Transdifferentiated retinal pigment epithelial cells are immunoreactive for vascular endothelial growth factor in surgically excised age-related macular degeneration- related choroidal neovascular membranes. Invest. Ophthalmol. Vis. Sci 37, 855–868. [PubMed] [Google Scholar]
- Luo Xueting, Yang Shiqi, Liang Jian, Zhai Yuanqi, Shen Mengxi, Sun Junran, Feng Yiji, Lu Xinmin, Zhu Hong, Wang Fenghua, Sun Xiaodong, 2018. ’Choroidal pericytes promote subretinal fibrosis after experimental photocoagulation. Dis. Models Mechanisms 11 dmm032060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macaskill AF, Rinholm JE, Twelvetrees AE, Arancibia-Carcamo IL, Muir J, Fransson A, Aspenstrom P, Attwell D, Kittler JT, 2009. ’Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron 61, 541–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguire MG, Martin DF, Ying GS, Jaffe GJ, Daniel E, Grunwald JE, Toth CA, Ferris FL 3rd, Fine SL, 2016. ’Five-Year outcomes with anti-vascular endothelial growth factor treatment of neovascular age-related macular degeneration: the comparison of age-related macular degeneration treatments trials. Ophthalmology 123, 1751–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maharaj AS, D’Amore PA, 2007. ’Roles for VEGF in the adult. Microvasc. Res 74, 100–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahdavian Delavary B, van der Veer WM, van Egmond M, Niessen FB, Beelen RH, 2011. ’Macrophages in skin injury and repair. Immunobiology 216, 753–762. [DOI] [PubMed] [Google Scholar]
- Marikovsky M, Ziv V, Nevo N, Harris-Cerruti C, Mahler O, 2003. ’Cu/Zn superoxide dismutase plays important role in immune response. J. Immunol 170, 2993–3001. [DOI] [PubMed] [Google Scholar]
- Martin DF, Maguire MG, Ying GS, Grunwald JE, Fine SL, Jaffe GJ, Catt Research Group, 2011. ’Ranibizumab and bevacizumab for neovascular age-related macular degeneration. N. Engl. J. Med 364, 1897–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin DF, Maguire MG, Fine SL, Ying GS, Jaffe GJ, Grunwald JE, Toth C, Redford M, Ferris FL 3rd, 2012. ’Ranibizumab and bevacizumab for treatment of neovascular age-related macular degeneration: two-year results. Ophthalmology 119, 1388–1398. [DOI] [PubMed] [Google Scholar]
- Matsui Ko, Hosoi Nobutake, Tachibana Masao, 1999. Active role of glutamate uptake in the synaptic transmission from retinal nonspiking neurons. J. Neurosci 19, 6755–6766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeer PL, Sibley J, 2005. ’Sparing of age-related macular degeneration in rheumatoid arthritis. Neurobiol. Aging 26, 1199–1203. [DOI] [PubMed] [Google Scholar]
- McLeod DS, Bhutto I, Edwards MM, Silver RE, Seddon JM, Lutty GA, 2016. ’Distribution and quantification of choroidal macrophages in human eyes with age- related macular degeneration. Invest. Ophthalmol. Vis. Sci 57, 5843–5855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMenamin PG, Saban DR, Dando SJ, 2019. ’Immune cells in the retina and choroid: two different tissue environments that require different defenses and surveillance. Prog. Retin. Eye Res 70, 85–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medeiros NE, Curcio CA, 2001. ’Preservation of ganglion cell layer neurons in age- related macular degeneration. Invest. Ophthalmol. Vis. Sci 42, 795–803. [PubMed] [Google Scholar]
- Mehta H, Tufail A, Daien V, Lee AY, Nguyen V, Ozturk M, Barthelmes D, Gillies MC, 2018. ’Real-world outcomes in patients with neovascular age-related macular degeneration treated with intravitreal vascular endothelial growth factor inhibitors’. Prog. Retin. Eye Res 65, 127–146. [DOI] [PubMed] [Google Scholar]
- Mettu PS, Crowell S, Shaw J, Grunwald L, Lad EM, Serrano N, Cousins SW, 2012a. ’Neovascular morphology on ICG angiography predicts response to anti- VEGF therapy in eyes with serous pigment epithelial detachments and age-related macular degeneration. Invest. Ophthalmol. Vis. Sci 53, 2654. [Google Scholar]
- Mettu PS, Wielgus AR, Ong SS, Cousins SW, 2012b. Retinal pigment epithelium response to oxidant injury in the pathogenesis of early age-related macular degeneration. Mol. Aspect. Med 33, 376–398. [DOI] [PubMed] [Google Scholar]
- Mettu PS, Saloupis P, Cousins SW, 2014. ’PAMP stimulation of macrophages promotes neovascular remodeling in experimental choroidal neovascularization. Invest. Ophthalmol. Vis. Sci 55, 1198–98. [Google Scholar]
- Mettu PS, Allingham MJ, Nicholas PC, Cousins SW, 2016. ’Neovascular morphology by ICG angiography and response to loading-dose anti-VEGF therapy in patients with neovascular AMD. Invest. Ophthalmol. Vis. Sci 57, 1346. [Google Scholar]
- Mettu PS, Allingham MJ, Nicholas PC, Cousins SW, 2017. ’Adjunctive indocyanine green angiography-directed verteporfin photodynamic therapy for the treatment of persistent disease activity in neovascular AMD. Invest. Ophthalmol. Vis. Sci 58, 1931–31. [Google Scholar]
- Mettu PS, Allingham MJ, Cousins SW, 2019. ’Effects of the mitochondria-targeted drug elamipretide on leakage-independent vision loss in fellow eyes with neovascular AMD in the ReCLAIM study. Invest. Ophthalmol. Vis. Sci 60, 358–58.30682208 [Google Scholar]
- Miller DM, Espinosa-Heidmann DG, Legra J, Dubovy SR, Suñer IJ, Sedmak DD, Dix RD, Cousins SW, 2004. ’The association of prior cytomegalovirus infection with neovascular age-related macular degeneration. Am. J. Ophthalmol 138, 323–328. [DOI] [PubMed] [Google Scholar]
- Moilanen W, Whittle B, Moncada S, 1999. ’Nitric Oxide as a Factor in Inflammation. In: Gallin JI, Synderman R (Eds.), Inflammation: Basic Principles and Clinical Correlates, third ed. Lippincott Williams and Wilkins, Philadelphia. [Google Scholar]
- Muether PS, Hermann MM, Viebahn U, Kirchhof B, Fauser S, 2012. ’Vascular endothelial growth factor in patients with exudative age-related macular degeneration treated with ranibizumab. Ophthalmology 119, 2082–2086. [DOI] [PubMed] [Google Scholar]
- Muether PS, Hermann MM, Droge K, Kirchhof B, Fauser S, 2013. ’Long-term stability of vascular endothelial growth factor suppression time under ranibizumab treatment in age-related macular degeneration. Am. J. Ophthalmol 156, 989–993 e2. [DOI] [PubMed] [Google Scholar]
- Mullins RF, Johnson MN, Faidley EA, Skeie JM, Huang J, 2011. ’Choriocapillaris vascular dropout related to density of drusen in human eyes with early age-related macular degeneration. Invest. Ophthalmol. Vis. Sci 52, 1606–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagelhus EA, Veruki ML, Torp R, Haug FM, Laake JH, Nielsen S, Agre P, Ottersen OP, 1998. ’Aquaporin-4 water channel protein in the rat retina and optic nerve: polarized expression in Müller cells and fibrous astrocytes. J. Neurosci 18, 2506–2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagelhus EA, Horio Y, Inanobe A, Fujita A, Haug FM, Nielsen S, Kurachi Y, Ottersen OP, 1999. ’Immunogold evidence suggests that coupling of K+ siphoning and water transport in rat retinal Müller cells is mediated by a coenrichment of Kir4.1 and AQP4 in specific membrane domains. Glia 26, 47–54. [DOI] [PubMed] [Google Scholar]
- Nahrendorf M, Swirski FK, 2013. ’Monocyte and macrophage heterogeneity in the heart. Circ. Res 112, 1624–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, Libby P, Weissleder R, Pittet MJ, 2007. ’The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J. Exp. Med 204, 3037–3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naito M, Umeda S, Yamamoto T, Moriyama H, Umezu H, Hasegawa G, Usuda H, Shultz LD, Takahashi K, 1996. ’Development, differentiation, and phenotypic heterogeneity of murine tissue macrophages. J. Leukoc. Biol 59, 133–138. [DOI] [PubMed] [Google Scholar]
- Nakamura R, Sene A, Santeford A, Gdoura A, Kubota S, Zapata N, Apte RS, 2015. ’IL10-driven STAT3 signalling in senescent macrophages promotes pathological eye angiogenesis. Nat. Commun 6, 7847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita C, Wu Z, Rosenfeld PJ, Yang J, Lyu C, Caruso E, McGuinness M, Guymer RH, 2020. ’Structural OCT signs suggestive of subclinical nonexudative macular neovascularization in eyes with large drusen. Ophthalmology 127, 637–647. [DOI] [PubMed] [Google Scholar]
- National Eye Institute, 2020. Age-related macular degeneration (AMD) data and statistics. ’. National Eye Instiute. https://www.nei.nih.gov/learn-about-eye-health /resources-for-health-educators/eye-health-data-and-statistics/age-related-macular- degeneration-amd-data-and-statistics.
- Newman EA, 1984. ’Regional specialization of retinal glial cell membrane. Nature 309, 155–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA, 1993. ’Inward-rectifying potassium channels in retinal glial (Müller) cells’. J. Neurosci 13, 3333–3345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman DK, 2016. Photodynamic therapy: current role in the treatment of chorioretinal conditions. Eye 30, 202–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls D, Attwell D, 1990. ’The release and uptake of excitatory amino acids. Trends Pharmacol. Sci 11, 462–468. [DOI] [PubMed] [Google Scholar]
- Nieves BJ, D’Amore PA, Bryan BA, 2009. The function of vascular endothelial growth factor. Biofactors 35, 332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikitina E, Larionova I, Choinzonov E, Kzhyshkowska J, 2018. ’Monocytes and macrophages as viral targets and reservoirs. Int. J. Mol. Sci 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obonyo M, Sabet M, Cole SP, Ebmeyer J, Uematsu S, Akira S, Guiney DG, 2007. ’Deficiencies of myeloid differentiation factor 88, toll-like receptor 2 (TLR2), or TLR4 produce specific defects in macrophage cytokine secretion induced by Helicobacter pylori. Infect. Immun 75, 2408–2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohji M, Saito Y, Hayashi A, Lewis JM, Tano Y, 1998. ’Pneumatic displacement of subretinal hemorrhage without tissue plasminogen activator. Arch. Ophthalmol 116, 1326–1332. [DOI] [PubMed] [Google Scholar]
- Olivier S, Chow DR, Packo KH, MacCumber MW, Awh CC, 2004. ’Subretinal recombinant tissue plasminogen activator injection and pneumatic displacement of thick submacular hemorrhage in Age-Related macular degeneration. Ophthalmology 111, 1201–1208. [DOI] [PubMed] [Google Scholar]
- Otani A, Sasahara M, Yodoi Y, Aikawa H, Tamura H, Tsujikawa A, Yoshimura N, 2007. ’Indocyanine green angiography: guided photodynamic therapy for polypoidal choroidal vasculopathy. Am. J. Ophthalmol 144, 7–14. [DOI] [PubMed] [Google Scholar]
- O’Koren EG, Mathew R, Saban DR, 2016. ’Fate mapping reveals that microglia and recruited monocyte-derived macrophages are definitively distinguishable by phenotype in the retina. Sci. Rep 6, 20636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palejwala NV, Jia Y, Gao SS, Liu L, Flaxel CJ, Hwang TS, Lauer AK, Wilson DJ, Huang D, Bailey ST, 2015. Detection of nonexudative choroidal neovascularization in age-related macular degeneration with optical coherence tomography angiography’. Retina 35, 2204–2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panagis L, Thanos S, Fischer D, Dermon CR, 2005. ’Unilateral optic nerve crush induces bilateral retinal glial cell proliferation. Eur. J. Neurosci 21, 2305–2309. [DOI] [PubMed] [Google Scholar]
- Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR 3rd, Lafaille JJ, Hempstead BL, Littman DR, Gan WB, 2013. ’Microglia promote learning- dependent synapse formation through brain-derived neurotrophic factor. Cell 155, 1596–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partsch MC, Tatar O, Adam A, Volker M, Shinoda K, Lüke M, Bart¨ z–Schmidt K, Grisanti S, Tuebingen Bevacizumab Study Group, 2006. ’Immunohistopathologic evaluation of choroidal neovascular membranes following intravitreal bevacizumab (Avastin®) therapy. Invest. Ophthalmol. Vis. Sci 47, 864–64.16505018 [Google Scholar]
- Patel-Hett S, D’Amore PA, 2011. ’Signal transduction in vasculogenesis and developmental angiogenesis. Int. J. Dev. Biol 55, 353–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauleikhoff D, Löffert D, Spital G, Radermacher M, Dohrmann J, Lommatzsch A, Bird AC, 2002. Pigment epithelial detachment in the elderly. Clinical differentiation, natural course and pathogenetic implications. Graefes Arch. Clin. Exp. Ophthalmol 240, 533–538. [DOI] [PubMed] [Google Scholar]
- Peden MC, Suñer IJ, Hammer ME, Grizzard WS, 2015. ’Long-term outcomes in eyes receiving fixed-interval dosing of anti-vascular endothelial growth factor agents for wet age-related macular degeneration. Ophthalmology 122, 803–808. [DOI] [PubMed] [Google Scholar]
- Peng YW, Hao Y, Petters RM, Wong F, 2000. Ectopic synaptogenesis in the mammalian retina caused by rod photoreceptor-specific mutations. Nat. Neurosci 3, 1121–1127. [DOI] [PubMed] [Google Scholar]
- Penn JS, Madan A, Caldwell RB, Bartoli M, Caldwell RW, Hartnett ME, 2008. ’Vascular endothelial growth factor in eye disease. Prog. Retin. Eye Res 27, 331–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennington KL, DeAngelis MM, 2016. ’Epidemiology of age-related macular degeneration (AMD): associations with cardiovascular disease phenotypes and lipid factors. Eye Vis (Lond) 3, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira FB, Veloso CE, Kokame GT, Nehemy MB, 2015. ’Characteristics of neovascular age-related macular degeneration in Brazilian patients. Ophthalmologica 234, 233–242. [DOI] [PubMed] [Google Scholar]
- Phipps JA, Guymer RH, Vingrys AJ, 2003. ’Loss of cone function in age-related maculopathy. Invest. Ophthalmol. Vis. Sci 44, 2277–2283. [DOI] [PubMed] [Google Scholar]
- Pittenger Mark F., Mackay Alastair M., Beck Stephen C., Jaiswal Rama K., Douglas Robin, Mosca Joseph, D., Moorman Mark A., Simonetti Donald W., Craig Stewart, Marshak Daniel R., 1999. Multilineage potential of adult human mesenchymal stem cells. Science 284, 143–147. [DOI] [PubMed] [Google Scholar]
- Portillo JC, Lopez Corcino Y, Miao Y, Tang J, Sheibani N, Kern TS, Dubyak GR, Subauste CS, 2017. ’CD40 in retinal müller cells induces P2X7-dependent cytokine expression in macrophages/microglia in diabetic mice and development of early experimental diabetic retinopathy. Diabetes 66, 483–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pow DV, Robinson SR, 1994. ’Glutamate in some retinal neurons is derived solely from glia. Neuroscience 60, 355–366. [DOI] [PubMed] [Google Scholar]
- Puliafito CA, Wykoff CC, 2019. ’Looking ahead in retinal disease management: highlights of the 2019 angiogenesis, exudation and degeneration symposium. Int. J. Retina Vitreous 5, 22–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Querques G, Srour M, Massamba N, Georges A, Ben Moussa N, Rafaeli O, Souied EH, 2013. ’Functional characterization and multimodal imaging of treatment-naive "quiescent" choroidal neovascularization. Invest. Ophthalmol. Vis. Sci 54, 6886–6892. [DOI] [PubMed] [Google Scholar]
- Racioppi L, Means AR, 2012. ’Calcium/calmodulin-dependent protein kinase kinase 2: roles in signaling and pathophysiology. J. Biol. Chem 287, 31658–31665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racioppi L, Noeldner PK, Lin F, Arvai S, Means AR, 2012. ’Calcium/calmodulin- dependent protein kinase kinase 2 regulates macrophage-mediated inflammatory responses. J. Biol. Chem 287, 11579–11591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racioppi L, Nelson ER, Huang W, Mukherjee D, Lawrence SA, Lento W, Masci AM, Jiao Y, Park S, York B, Liu Y, Baek AE, Drewry DH, Zuercher WJ, Bertani FR, Businaro L, Geradts J, Hall A, Means AR, Chao N, Chang CY, McDonnell DP, 2019. ’CaMKK2 in myeloid cells is a key regulator of the immune-suppressive microenvironment in breast cancer. Nat. Commun 10, 2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raff MC, Whitmore AV, Finn JT, 2002. ’Axonal self-destruction and neurodegeneration. Science 296, 868–871. [DOI] [PubMed] [Google Scholar]
- Ramachandran P, Pellicoro A, Vernon MA, Boulter L, Aucott RL, Ali A, Hartland SN, Snowdon VK, Cappon A, Gordon-Walker TT, Williams MJ, Dunbar DR, Manning JR, van Rooijen N, Fallowfield JA, Forbes SJ, Iredale JP, 2012. ’Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc. Natl. Acad. Sci. U. S. A 109, E3186–E3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsauer M, D’Amore PA, 2007. ’Contextual role for angiopoietins and TGFbeta1 in blood vessel stabilization. J. Cell Sci. 120, 1810–1817. [DOI] [PubMed] [Google Scholar]
- Reddy VM, Zamora RL, Kaplan HJ, 1995. ’Distribution of growth factors in subfoveal neovascular membranes in age-related macular degeneration and presumed ocular histoplasmosis syndrome. Am. J. Ophthalmol 120, 291–301. [DOI] [PubMed] [Google Scholar]
- Regillo CD, Brown DM, Abraham P, Yue H, Ianchulev T, Schneider S, Shams N, 2008. ’Randomized, double-masked, sham-controlled trial of ranibizumab for neovascular age-related macular degeneration: PIER Study year 1. Am. J. Ophthalmol 145, 239–248. [DOI] [PubMed] [Google Scholar]
- Rehak M, Hollborn M, Iandiev I, Pannicke T, Karl A, Wurm A, Kohen L, Reichenbach A, Wiedemann P, Bringmann A, 2009. ’Retinal gene expression and Müller cell responses after branch retinal vein occlusion in the rat. Invest. Ophthalmol. Vis. Sci 50, 2359–2367. [DOI] [PubMed] [Google Scholar]
- Reichenbach A, Wurm A, Pannicke T, Iandiev I, Wiedemann P, Bringmann A, 2007. ’Müller cells as players in retinal degeneration and edema. Graefes Arch. Clin. Exp. Ophthalmol 245, 627–636. [DOI] [PubMed] [Google Scholar]
- Reinoso MA, Caicedo A, Espinosa–Heidmann DG, Csaky KH, Cousins SW, 2004. ’Blood–derived macrophages and bone marrow derived progenitor cells contribute to intrachoroidal vascular changes in experimental choroidal neovascularization (CNV)’. Invest. Ophthalmol. Vis. Sci 45, 3386–86. [Google Scholar]
- Reyes NJ, Mathew R, Saban DR, 2019. ’Fate mapping in vivo to distinguish bona fide microglia versus recruited monocyte-derived macrophages in retinal disease. Methods Mol. Biol 1834, 153–164. [DOI] [PubMed] [Google Scholar]
- Rezar-Dreindl S, Sacu S, Eibenberger K, Pollreisz A, Buhl W, Georgopoulos M, Krall C, Weigert G, Schmidt-Erfurth U, 2016. ’The intraocular cytokine profile and therapeutic response in persistent neovascular age-related macular degeneration. Invest. Ophthalmol. Vis. Sci 57, 4144–4150. [DOI] [PubMed] [Google Scholar]
- Robman L, Mahdi OS, Wang JJ, Burlutsky G, Mitchell P, Byrne G, Guymer R, Taylor H, 2007. ’Exposure to Chlamydia pneumoniae infection and age-related macular degeneration: the Blue Mountains Eye Study. Invest. Ophthalmol. Vis. Sci 48, 4007–4011. [DOI] [PubMed] [Google Scholar]
- Rofagha S, Bhisitkul RB, Boyer DS, Sadda SR, Zhang K, Seven-Up Study Group, 2013. ’Seven-year outcomes in ranibizumab-treated patients in ANCHOR, MARINA, and HORIZON: a multicenter cohort study (SEVEN-UP). Ophthalmology 120, 2292–2299. [DOI] [PubMed] [Google Scholar]
- Roisman L, Zhang Q, Wang RK, Gregori G, Zhang A, Chen CL, Durbin MK, An L, Stetson PF, Robbins G, Miller A, Zheng F, Rosenfeld PJ, 2016. ’Optical coherence tomography angiography of asymptomatic neovascularization in intermediate age-related macular degeneration. Ophthalmology 123, 1309–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld PJ, Feuer WJ, 2018. ’Lessons from recent phase III trial failures: don’t design phase III trials based on retrospective subgroup Analyses from phase II trials. Ophthalmology 125, 1488–1491. [DOI] [PubMed] [Google Scholar]
- Rosenfeld PJ, Brown DM, Heier JS, Boyer DS, Kaiser PK, Chung CY, Kim RY, 2006. ’Ranibizumab for neovascular age-related macular degeneration. N. Engl. J. Med 355, 1419–1431. [DOI] [PubMed] [Google Scholar]
- Marina, and Anchor Study Groups Rosenfeld, P.J., Shapiro H, Tuomi L, Webster M, Elledge J, Blodi B, 2011. ’Characteristics of patients losing vision after 2 years of monthly dosing in the phase III ranibizumab clinical trials. Ophthalmology 118, 523–530. [DOI] [PubMed] [Google Scholar]
- Rozing MP, Durhuus JA, Krogh Nielsen M, Subhi Y, Kirkwood TB, Westendorp RG, Sørensen TL, 2020. Age-related macular degeneration: a two- level model hypothesis. Prog. Retin. Eye Res 76, 100825. [DOI] [PubMed] [Google Scholar]
- Rutherford MS, Witsell A, Schook LB, 1993. ’Mechanisms generating functionally heterogeneous macrophages: chaos revisited. J. Leukoc. Biol 53, 602–618. [DOI] [PubMed] [Google Scholar]
- Saban DR, 2018. New concepts in macrophage ontogeny in the adult neural retina. Cell. Immunol 330, 79–85. [DOI] [PubMed] [Google Scholar]
- Sabio G, Davis RJ, 2014. ’TNF and MAP kinase signalling pathways. Semin. Immunol 26, 237–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai E, Anand A, Ambati BK, van Rooijen N, Ambati J, 2003. ’Macrophage depletion inhibits experimental choroidal neovascularization. Invest. Ophthalmol. Vis. Sci 44, 3578–3585. [DOI] [PubMed] [Google Scholar]
- Sanyal S, Hawkins RK, Jansen HG, Zeilmaker GH, 1992. ’Compensatory synaptic growth in the rod terminals as a sequel to partial photoreceptor cell loss in the retina of chimaeric mice. Development 114, 797–803. [DOI] [PubMed] [Google Scholar]
- Sarks JP, Sarks SH, Killingsworth MC, 1997. ’Morphology of early choroidal neovascularisation in age-related macular degeneration: correlation with activity. Eye 11 (Pt 4), 515–522. [DOI] [PubMed] [Google Scholar]
- Sarraf D, London NJ, Khurana RN, Dugel PU, Gune S, Hill L, Tuomi L, 2016. ’Ranibizumab treatment for pigment epithelial detachment secondary to neovascular age-related macular degeneration: post hoc analysis of the HARBOR study. Ophthalmology 123, 2213–2224. [DOI] [PubMed] [Google Scholar]
- Sasaki F, Koga T, Ohba M, Saeki K, Okuno T, Ishikawa K, Nakama T, Nakao S, Yoshida S, Ishibashi T, Ahmadieh H, Kanavi MR, Hafezi-Moghadam A, Penninger JM, Sonoda KH, Yokomizo T, 2018. ’Leukotriene B4 promotes neovascularization and macrophage recruitment in murine wet-type AMD models. JCI Insight 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sata M, Saiura A, Kunisato A, Tojo A, Okada S, Tokuhisa T, Hirai H, Makuuchi M, Hirata Y, Nagai R, 2002. ’Hematopoietic stem cells differentiate into vascular cells that participate in the pathogenesis of atherosclerosis. Nat. Med 8, 403–409. [DOI] [PubMed] [Google Scholar]
- Sawyer AJ, Kyriakides TR, 2016. ’Matricellular proteins in drug delivery: therapeutic targets, active agents, and therapeutic localization. Adv. Drug Deliv. Rev 97, 56–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaer DJ, Boretti FS, Schoedon G, Schaffner A, 2002. ’Induction of the CD163- dependent haemoglobin uptake by macrophages as a novel anti-inflammatory action of glucocorticoids. Br. J. Haematol 119, 239–243. [DOI] [PubMed] [Google Scholar]
- Schlegel RA, Krahling S, Callahan MK, Williamson P, 1999. ’CD14 is a component of multiple recognition systems used by macrophages to phagocytose apoptotic lymphocytes. Cell Death Differ. 6, 583–592. [DOI] [PubMed] [Google Scholar]
- Schmidt-Erfurth U, Waldstein SM, Deak GG, Kundi M, Simader C, 2015. Pigment epithelial detachment followed by retinal cystoid degeneration leads to vision loss in treatment of neovascular age-related macular degeneration. Ophthalmology 122, 822–832. [DOI] [PubMed] [Google Scholar]
- Schneider U, Gelisken F, Inhoffen W, Kreissig I, 1997. ’Indocyanine green angiographic findings in fellow eyes of patients with unilateral occult neovascular age-related macular degeneration. Int. Ophthalmol 21, 79–85. [DOI] [PubMed] [Google Scholar]
- Schneider GB, Hamano H, Cooper LF, 1998. ’In vivo evaluation of hsp27 as an inhibitor of actin polymerization: hsp27 limits actin stress fiber and focal adhesion formation after heat shock. J. Cell. Physiol 177, 575–584. [DOI] [PubMed] [Google Scholar]
- Schumann RR, Latz E, 2000. ’Lipopolysaccharide-binding protein. Chem. Immunol 74, 42–60. [DOI] [PubMed] [Google Scholar]
- Schwartz EA, 1993. ’L-glutamate conditionally modulates the K+ current of Müller glial cells. Neuron 10, 1141–1149. [DOI] [PubMed] [Google Scholar]
- Seddon JM, Willett WC, Speizer FE, Hankinson SE, 1996. ’A prospective study of cigarette smoking and age-related macular degeneration in women. Jama 276, 1141–1146. [PubMed] [Google Scholar]
- Seddon JM, McLeod DS, Bhutto IA, Villalonga MB, Silver RE, Wenick AS, Edwards MM, Lutty GA, 2016. ’Histopathological insights into choroidal vascular loss in clinically documented cases of age-related macular degeneration. JAMA Ophthalmol 134, 1272–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seghezzi G, Patel S, Ren CJ, Gualandris A, Pintucci G, Robbins ES, Shapiro RL, Galloway AC, Rifkin DB, Mignatti P, 1998. ’Fibroblast growth factor-2 (FGF-2) induces vascular endothelial growth factor (VEGF) expression in the endothelial cells of forming capillaries: an autocrine mechanism contributing to angiogenesis. J. Cell Biol. 141, 1659–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sene A, Khan AA, Cox D, Nakamura RE, Santeford A, Kim BM, Sidhu R, Onken MD, Harbour JW, Hagbi-Levi S, Chowers I, Edwards PA, Baldan A, Parks JS, Ory DS, Apte RS, 2013. ’Impaired cholesterol efflux in senescent macrophages promotes age-related macular degeneration. Cell Metabol. 17, 549–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano NP, Shaw J, Mettu PS, Lad EM, Crowell S, Cousins SW, 2012. ’High- speed indocyanine green angiography in age related macular degeneration with fibrovascular pigment epithelial detachments. Invest. Ophthalmol. Vis. Sci 53, 1151. [Google Scholar]
- Sharma S, Toth CA, Daniel E, Grunwald JE, Maguire MG, Ying GS, Huang J, Martin DF, Jaffe GJ, Group Comparison of age-related macular degeneration treatments trials research, 2016. ’macular morphology and visual acuity in the second year of the comparison of age-related macular degeneration treatments trials. Ophthalmology 123, 865–875.26783095 [Google Scholar]
- Shen J, Frye M, Lee BL, Reinardy JL, McClung JM, Ding K, Kojima M, Xia H, Seidel C, Lima e Silva R, Dong A, Hackett SF, Wang J, Howard BW, Vestweber D, Kontos CD, Peters KG, Campochiaro PA, 2014. ’Targeting VE- PTP activates TIE2 and stabilizes the ocular vasculature. J. Clin. Invest 124, 4564–4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheridan CM, Rice D, Hiscott PS, Wong D, Kent DL, 2006. ’The presence of AC133-positive cells suggests a possible role of endothelial progenitor cells in the formation of choroidal neovascularization. Invest. Ophthalmol. Vis. Sci 47, 1642–1645. [DOI] [PubMed] [Google Scholar]
- Sica A, Mantovani A, 2012. ’Macrophage plasticity and polarization: in vivo veritas. J. Clin. Invest 122, 787–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva R, D’Amico G, Kairbaan MH-D, Louise ER, 2008. Integrins’, Arteriosclerosis, Thrombosis, and Vascular Biology, vol. 28, pp. 1703–1713. [DOI] [PubMed] [Google Scholar]
- Singer MA, Awh CC, Sadda S, Freeman WR, Antoszyk AN, Wong P, Tuomi L, 2012. ’HORIZON: an open-label extension trial of ranibizumab for choroidal neovascularization secondary to age-related macular degeneration. Ophthalmology 119, 1175–1183. [DOI] [PubMed] [Google Scholar]
- Singh A, Falk MK, Hviid TV, Sorensen TL, 2013. ’Increased expression of CD200 on circulating CD11b+ monocytes in patients with neovascular age-related macular degeneration. Ophthalmology 120, 1029–1037. [DOI] [PubMed] [Google Scholar]
- Slobedman B, Mocarski ES, Arvin AM, Mellins ED, Abendroth A, 2002. ’Latent cytomegalovirus down-regulates major histocompatibility complex class II expression on myeloid progenitors. Blood 100, 2867–2873. [DOI] [PubMed] [Google Scholar]
- Smith W, Mitchell P, Leeder SR, 1996. ’Smoking and age-related maculopathy. The blue mountains eye study. Arch. Ophthalmol 114, 1518–1523. [DOI] [PubMed] [Google Scholar]
- Smith BT, Kraus CL, Apte RS, 2009. Retinal pigment epithelial tears in ranibizumab-treated eyes. Retina 29, 335–339. [DOI] [PubMed] [Google Scholar]
- Spaide RF, 2015. ’Optical coherence tomography angiography signs of vascular abnormalization with antiangiogenic therapy for choroidal neovascularization. Am. J. Ophthalmol 160, 6–16. [DOI] [PubMed] [Google Scholar]
- Spaide RF, Fujimoto JG, Waheed NK, Sadda SR, Staurenghi G, 2018. ’Optical coherence tomography angiography. Prog. Retin. Eye Res 64, 1–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaide RF, Jaffe GJ, Sarraf D, Freund KB, Sadda SR, Staurenghi G, Waheed NK, Chakravarthy U, Rosenfeld PJ, Holz FG, Souied EH, Cohen SY, Querques G, Ohno-Matsui K, Boyer D, Gaudric A, Blodi B, Baumal CR, Li X, Coscas GJ, Brucker A, Singerman L, Luthert P, Schmitz-Valckenberg S, Schmidt-Erfurth U, Grossniklaus HE, Wilson DJ, Guymer R, Yannuzzi LA, Chew EY, Csaky K, Monés JM, Pauleikhoff D, Tadayoni R, Fujimoto J, 2020. ’Consensus nomenclature for reporting neovascular age-related macular degeneration data: consensus on neovascular age-related macular degeneration nomenclature study group. Ophthalmology 127, 616–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spilsbury K, Garrett KL, Shen WY, Constable IJ, Rakoczy PE, 2000. ’Overexpression of vascular endothelial growth factor (VEGF) in the retinal pigment epithelium leads to the development of choroidal neovascularization. Am. J. Pathol 157, 135–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenberg RM, 1996. The human cytomegalovirus major immediate-early gene. Intervirology 39, 343–349. [DOI] [PubMed] [Google Scholar]
- Stifter E, Michels S, Prager F, Georgopoulos M, Polak K, Hirn C, Schmidt- Erfurth U, 2007. ’Intravitreal bevacizumab therapy for neovascular age-related macular degeneration with large submacular hemorrhage. Am. J. Ophthalmol 144, 886–892. [DOI] [PubMed] [Google Scholar]
- Strittmatter K, Pomeroy H, Marneros AG, 2016. ’Targeting platelet-derived growth factor receptor β(+) scaffold formation inhibits choroidal neovascularization. Am. J. Pathol 186, 1890–1899. [DOI] [PubMed] [Google Scholar]
- Subhi Y, Nielsen MK, Molbech CR, Falk MK, Singh A, Hviid TVF, Nissen MH, Sorensen TL, 2019. ’Association of CD11b+ monocytes and anti-vascular endothelial growth factor injections in treatment of neovascular age-related macular degeneration and polypoidal choroidal vasculopathy. JAMA Ophthalmol 137, 515–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Louie MC, Vannella KM, Wilke CA, LeVine AM, Moore BB, Shanley TP, 2011. ’New concepts of IL-10-induced lung fibrosis: fibrocyte recruitment and M2 activation in a CCL2/CCR2 axis. Am. J. Physiol. Lung Cell Mol. Physiol 300, L341–L353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suñer IJ, Espinosa-Heidmann DG, Marin-Castano ME, Hernandez EP, Pereira- Simon S, Cousins SW, 2004. ’Nicotine increases size and severity of experimental choroidal neovascularization. Invest. Ophthalmol. Vis. Sci 45, 311–317. [DOI] [PubMed] [Google Scholar]
- Suñer IJ, Espinosa–Heidmann DG, Pereira–Simon S, Pina Y, Cousins SW, 2005. Cigarette smoke increases severity of experimental choroidal neovascularization (CNV): role of inflammation. Invest. Ophthalmol. Vis. Sci 46, 3507–7.16186327 [Google Scholar]
- Sweet C, 1999. The pathogenicity of cytomegalovirus. FEMS (Fed. Eur. Microbiol. Soc.) Microbiol. Rev 23, 457–482. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Naito M, Takeya M, 1996. ’Development and heterogeneity of macrophages and their related cells through their differentiation pathways. Pathol. Int 46, 473–485. [DOI] [PubMed] [Google Scholar]
- Tamakoshi A, Yuzawa M, Matsui M, Uyama M, Fujiwara NK, Ohno Y, 1997. ’Smoking and neovascular form of age related macular degeneration in late middle aged males: findings from a case-control study in Japan. Research Committee on Chorioretinal Degenerations. Br. J. Ophthalmol 81, 901–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan X, Fujiu K, Manabe I, Nishida J, Yamagishi R, Nagai R, Yanagi Y, 2015. ’Choroidal neovascularization is inhibited via an intraocular decrease of inflammatory cells in mice lacking complement component C3. Sci. Rep 5, 15702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan X, Fujiu K, Manabe I, Nishida J, Yamagishi R, Terashima Y, Matsushima K, Kaburaki T, Nagai R, Yanagi Y, 2016. ’Choroidal neovascularization is inhibited in splenic-denervated or splenectomized mice with a concomitant decrease in intraocular macrophage. PloS One 11 e0160985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatar O, Shinoda K, Kaiserling E, Claes C, Eckardt C, Eckert T, Pertile G, Boeyden V, Scharioth GB, Yoeruek E, Szurman P, Bartz-Schmidt KU, Grisanti S, 2009. ’Implications of bevacizumab on vascular endothelial growth factor and endostatin in human choroidal neovascularisation. Br. J. Ophthalmol 93, 159–165. [DOI] [PubMed] [Google Scholar]
- Treatment of age-related macular degeneration with photodynamic therapy) study group TAP Study Group, 1999. Photodynamic therapy of subfoveal choroidal neovascularization in age-related macular degeneration with verteporfin: one-year results of 2 randomized clinical trials—TAP report 1’. Arch. Ophthalmol 117, 1329–1345. [PubMed] [Google Scholar]
- Toft-Kehler AK, Skytt DM, Svare A, Lefevere E, Van Hove I, Moons L, Waagepetersen HS, Kolko M, 2017. Mitochondrial function in Müller cells - does it matter? Mitochondrion 36, 43–51. [DOI] [PubMed] [Google Scholar]
- Toomey CB, Johnson LV, Bowes Rickman C, 2018. ’Complement factor H in AMD: bridging genetic associations and pathobiology’. Prog. Retin. Eye Res 62, 38–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth CA, Morse LS, Hjelmeland LM, Landers MB 3rd, 1991. ’Fibrin directs early retinal damage after experimental subretinal hemorrhage. Arch. Ophthalmol 109, 723–729. [DOI] [PubMed] [Google Scholar]
- Treumer F, Roider J, Hillenkamp J, 2012. ’Long-term outcome of subretinal coapplication of rtPA and bevacizumab followed by repeated intravitreal anti-VEGF injections for neovascular AMD with submacular haemorrhage. Br. J. Ophthalmol 96, 708–713. [DOI] [PubMed] [Google Scholar]
- Tsutsumi C, Sonoda KH, Egashira K, Qiao H, Hisatomi T, Nakao S, Ishibashi M, Charo IF, Sakamoto T, Murata T, Ishibashi T, 2003. ’The critical role of ocular- infiltrating macrophages in the development of choroidal neovascularization. J. Leukoc. Biol 74, 25–32. [DOI] [PubMed] [Google Scholar]
- Ueta T, Ishihara K, Notomi S, Lee JJ, Maidana DE, Efstathiou NE, Murakami Y, Hasegawa E, Azuma K, Toyono T, Paschalis EI, Aihara M, Miller JW, Vavvas DG, 2019. ’RIP1 kinase mediates angiogenesis by modulating macrophages in experimental neovascularization. Proc. Natl. Acad. Sci. U. S. A 116, 23705–23713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Leeuwen EM, Emri E, Merle BMJ, Colijn JM, Kersten E, Cougnard-Gregoire A, Dammeier S, Meester-Smoor M, Pool FM, de Jong EK, Delcourt C, Rodrigez-Bocanegra E, Biarnés M, Luthert PJ, Ueffing M, Klaver CCW, Nogoceke E, den Hollander AI, Lengyel I, 2018. ’A new perspective on lipid research in age-related macular degeneration. Prog. Retin. Eye Res 67, 56–86. [DOI] [PubMed] [Google Scholar]
- van Nieuw Amerongen GP, Koolwijk P, Versteilen A, van Hinsbergh VW, 2003. ’Involvement of RhoA/Rho kinase signaling in VEGF-induced endothelial cell migration and angiogenesis in vitro. Arterioscler. Thromb. Vasc. Biol 23, 211–217. [DOI] [PubMed] [Google Scholar]
- Van Rooijen N, 1989. ’The liposome-mediated macrophage ’suicide’ technique. J. Immunol. Methods 124, 1–6. [DOI] [PubMed] [Google Scholar]
- Villablanca AC, 1998. ’Nicotine stimulates DNA synthesis and proliferation in vascular endothelial cells in vitro. J. Appl. Physiol 84, 2089–2098, 1985. [DOI] [PubMed] [Google Scholar]
- Vliegen I, Duijvestijn A, Stassen F, Bruggeman C, 2004. ’Murine cytomegalovirus infection directs macrophage differentiation into a pro-inflammatory immune phenotype: implications for atherogenesis. Microb. Infect 6, 1056–1062. [DOI] [PubMed] [Google Scholar]
- von der Emde L, Thiele S, Pfau M, Nadal J, Meyer J, Möller PT, Schmid M, Fleckenstein M, Holz FG, Schmitz-Valckenberg S, 2020. ’Assessment of exudative activity of choroidal neovascularization in age-related macular degeneration by OCT angiography. Ophthalmologica 243, 120–128. [DOI] [PubMed] [Google Scholar]
- Vos M, Lauwers E, Verstreken P, 2010. Synaptic mitochondria in synaptic transmission and organization of vesicle pools in health and disease. Front. Synaptic Neurosci. 2, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagley S, Marra KV, Salhi RA, Gautam S, Campo R, Veale P, Veale J, Arroyo JG, 2015. Periodontal dsease and age-related macular degeneratoin: results from the national health and nutrition examination survey III. Retina 35, 982–988. [DOI] [PubMed] [Google Scholar]
- Wan QF, Zhou ZY, Thakur P, Vila A, Sherry DM, Janz R, Heidelberger R, 2010. ’SV2 acts via presynaptic calcium to regulate neurotransmitter release. Neuron 66, 884–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JS, Kefalov VJ, 2011. ’The cone-specific visual cycle. Prog. Retin. Eye Res 30, 115–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Nakayama M, Pitulescu ME, Schmidt TS, Bochenek ML, Sakakibara A, Adams S, Davy A, Deutsch U, Lüthi U, Barberis A, Benjamin LE, Makinen T, Nobes CD, Adams RH, 2010. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature 465, 483–486. [DOI] [PubMed] [Google Scholar]
- Wang H, Fotheringham L, Wittchen ES, Hartnett ME, 2015. ’Rap1 GTPase inhibits tumor necrosis factor-α-induced choroidal endothelial migration via NADPH oxidase- and NF-κB-Dependent activation of Rac1. Am. J. Pathol 185, 3316–3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Koenig AL, Lavine KJ, Apte RS, 2019. ’Macrophage plasticity and function in the eye and heart. Trends Immunol. 40, 825–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warheit-Niemi HI, Hult EM, Moore BB, 2019. ’A pathologic two-way street: how innate immunity impacts lung fibrosis and fibrosis impacts lung immunity. Clin Transl Immunol. 8 e1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen X, Hu X, Miao L, Ge X, Deng Y, Bible PW, Wei L, 2018. ’Epigenetics, microbiota, and intraocular inflammation: new paradigms of immune regulation in the eye’. Prog. Retin. Eye Res 64, 84–95. [DOI] [PubMed] [Google Scholar]
- Wohl Stefanie G., Schmeer Christian W., Witte Otto W., Isenmann Stefan, 2010. ’Proliferative response of microglia and macrophages in the adult mouse eye after optic nerve lesion. Invest. Ophthalmol. Visual Sci. 51, 2686–2696. [DOI] [PubMed] [Google Scholar]
- Wolf AA, Yanez A, Barman PK, Goodridge HS, 2019. The ontogeny of monocyte subsets. Front. Immunol 10, 1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong KL, Tai JJ, Wong WC, Han H, Sem X, Yeap WH, Kourilsky P, Wong SC, 2011. Gene expression profiling reveals the defining features of the classical, intermediate, and nonclassical human monocyte subsets. Blood 118, e16–31. [DOI] [PubMed] [Google Scholar]
- Wong Tien Y., Ohno-Matsui Kyoko, Leveziel Nicolas, Holz Frank G., Lai Timothy Y., Yu Gon, Hyeong Lanzetta, Paolo Chen, Youxin Tufail, Adnan, 2015. ’Myopic choroidal neovascularisation: current concepts and update on clinical management. Br. J. Ophthalmol 99, 289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong CW, Yanagi Y, Lee WK, Ogura Y, Yeo I, Wong TY, Cheung CMG, 2016. ’Age-related macular degeneration and polypoidal choroidal vasculopathy in Asians. Prog. Retin. Eye Res 53, 107–139. [DOI] [PubMed] [Google Scholar]
- Wood EH, Whitted RJ, Stone TW, Isernhagen RD, Wood WJ, Holcomb DM, Kitchens JW, 2018. ’PRN ranibizumab in the treatment of choroidal neovascularization secondary to ocular histoplasmosis. Ophthalmic Surg Lasers Imag. Retina 49, 20–26. [DOI] [PubMed] [Google Scholar]
- Wood EH, Korot E, Storey PP, Muscat S, Williams GA, Drenser KA, 2020. ’The retina revolution: signaling pathway therapies, genetic therapies, mitochondrial therapies, artificial intelligence. Curr. Opin. Ophthalmol 31, 207–214. [DOI] [PubMed] [Google Scholar]
- Woollard KJ, Geissmann F, 2010. Monocytes in atherosclerosis: subsets and functions. Nat. Rev. Cardiol 7, 77–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak W, 1998. ’Origin and the functional role of microglia. Folia Morphol (Warsz) 57, 277–285. [PubMed] [Google Scholar]
- Wykoff CC, Croft DE, Brown DM, Wang R, Payne JF, Clark L, Abdelfattah NS, Sadda SR, Trex-Amd Study Group, 2015. ’Prospective trial of treat-and-extend versus monthly dosing for neovascular age-related macular degeneration: TREX- AMD 1-year results. Ophthalmology 122, 2514–2522. [DOI] [PubMed] [Google Scholar]
- Wykoff CC, Ou WC, Croft DE, Payne JF, Brown DM, Clark WL, Abdelfattah NS, Sadda SR, Trex-Amd Study Group, 2018. ’Neovascular age-related macular degeneration management in the third year: final results from the TREX-AMD randomised trial. Br. J. Ophthalmol 102, 460–464. [DOI] [PubMed] [Google Scholar]
- Wynn TA, Barron L, 2010. ’Macrophages: master regulators of inflammation and fibrosis. Semin. Liver Dis. 30, 245–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss-Coray T, Mucke L, 2002. ’Inflammation in neurodegenerative disease–a double- edged sword. Neuron 35, 419–432. [DOI] [PubMed] [Google Scholar]
- Xu Heping, Chen Mei, Mayer Eric J., Forrester John V., Dick Andrew D., 2007. ’Turnover of resident retinal microglia in the normal adult mouse. Glia 55, 1189–1198. [DOI] [PubMed] [Google Scholar]
- Xue Y, Shen SQ, Jui J, Rupp AC, Byrne LC, Hattar S, Flannery JG, Corbo JC, Kefalov VJ, 2015. ’CRALBP supports the mammalian retinal visual cycle and cone vision. J. Clin. Invest 125, 727–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagi Y, Mohla A, Lee WK, Lee SY, Mathur R, Chan CM, Yeo I, Wong TY, Cheung CMG, 2017. ’Prevalence and risk factors for nonexudative neovascularization in fellow eyes of patients with unilateral age-related macular degeneration and polypoidal choroidal vasculopathy. Invest. Ophthalmol. Vis. Sci 58, 3488–3495. [DOI] [PubMed] [Google Scholar]
- Yang DC, Chen CH, 2018. ’Cigarette smoking-mediated macrophage reprogramming: mechanistic insights and therapeutic implications. J. Nat. Sci 4. [PMC free article] [PubMed] [Google Scholar]
- Yang X-F, Yin Y, Wang H, 2008. ’Vascular inflammation and atherogenesis are activated via receptors for PAMPs and suppressed by regulatory T cells’. Drug Discov. Today Ther. Strat 5, 125–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Liu F, Tang M, Yuan M, Hu A, Zhan Z, Li Z, Li J, Ding X, Lu L, 2016. ’Macrophage polarization in experimental and clinical choroidal neovascularization. Sci. Rep 6, 30933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Zhang Q, Motulsky EH, Thulliez M, Shi Y, Lyu C, de Sisternes L, Durbin MK, Feuer W, Wang RK, Gregori G, Rosenfeld PJ, 2019. ’Two-Year risk of exudation in eyes with nonexudative age-related macular degeneration and subclinical neovascularization detected with swept source optical coherence tomography angiography. Am. J. Ophthalmol 208, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yannuzzi LA, 1982. Idiopathic polypoidal choroidal vasculopathy. In: 1982 Macula Society Annual Meeting. Miami, FL. [Google Scholar]
- Yannuzzi LA, Sorenson J, Spaide RF, Lipson B, 1990. Idiopathic polypoidal choroidal vasculopathy (IPCV). Retina 10, 1–8. [PubMed] [Google Scholar]
- Yawata I, Takeuchi H, Doi Y, Liang J, Mizuno T, Suzumura A, 2008. ’Macrophage-induced neurotoxicity is mediated by glutamate and attenuated by glutaminase inhibitors and gap junction inhibitors. Life Sci. 82, 1111–1116. [DOI] [PubMed] [Google Scholar]
- Ying GS, Kim BJ, Maguire MG, Huang J, Daniel E, Jaffe GJ, Grunwald JE, Blinder KJ, Flaxel CJ, Rahhal F, Regillo C, Martin DF, Catt Research Group, 2014. ’Sustained visual acuity loss in the comparison of age-related macular degeneration treatments trials. JAMA Ophthalmol 132, 915–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying GS, Maguire MG, Pan W, Grunwald JE, Daniel E, Jaffe GJ, Toth CA, Hagstrom SA, Martin DF, Catt Research Group, 2018. ’Baseline predictors for five-year visual acuity outcomes in the comparison of AMD treatment trials. Ophthalmol Retina 2, 525–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yodoi Yuko, Sasahara Manabu, Kameda Takanori, Yoshimura Nagahisa, Otani Atsushi, 2007. ’Circulating hematopoietic stem cells in patients with neovascular age-related macular degeneration. Invest. Ophthalmol. Visual Sci. 48, 5464–5472. [DOI] [PubMed] [Google Scholar]
- You QS, Gaber R, Meshi A, Ramkumar HL, Alam M, Muftuoglu IK, Freeman WR, 2018. High-dose high-frequency aflibercept for recalcitrant neovascular age- related macular degeneration. Retina 38, 1156–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C, Roubeix C, Sennlaub F, Saban DR, 2020. ’Microglia versus monocytes: distinct roles in degenerative diseases of the retina. Trends Neurosci. 43, 433–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan F, Fu X, Shi H, Chen G, Dong P, Zhang W, 2014. Induction of murine macrophage M2 polarization by cigarette smoke extract via the JAK2/STAT3 pathway. PloS One 9 e107063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zandi S, Nakao S, Chun KH, Fiorina P, Sun D, Arita R, Zhao M, Kim E, Schueller O, Campbell S, Taher M, Melhorn MI, Schering A, Gatti F, Tezza S, Xie F, Vergani A, Yoshida S, Ishikawa K, Yamaguchi M, Sasaki F, Schmidt-Ullrich R, Hata Y, Enaida H, Yuzawa M, Yokomizo T, Kim YB, Sweetnam P, Ishibashi T, Hafezi-Moghadam A, 2015. ’ROCK-isoform-specific polarization of macrophages associated with age-related macular degeneration. Cell Rep. 10, 1173–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zawada AM, Rogacev KS, Schirmer SH, Sester M, Bohm M, Fliser D, Heine GH, 2012. ’Monocyte heterogeneity in human cardiovascular disease. Immunobiology 217, 1273–1284. [DOI] [PubMed] [Google Scholar]
- Zenisek D, Matthews G, 2000. The role of mitochondria in presynaptic calcium handling at a ribbon synapse. Neuron 25, 229–237. [DOI] [PubMed] [Google Scholar]
- Zhang X, Wheeler D, Tang Y, Guo L, Shapiro RA, Ribar TJ, Means AR, Billiar TR, Angus DC, Rosengart MR, 2008. ’Calcium/calmodulin-dependent protein kinase (CaMK) IV mediates nucleocytoplasmic shuttling and release of HMGB1 during lipopolysaccharide stimulation of macrophages. J. Immunol 181, 5015–5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Roychoudhury J, Doggett TA, Apte RS, Ferguson TA, 2013. ’Age- dependent changes in FasL (CD95L) modulate macrophage function in a model of age-related macular degeneration. Invest. Ophthalmol. Vis. Sci 54, 5321–5331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Quyyumi AA, Norman JE, Csako G, Waclawiw MA, Shearer GM, Epstein SE, 2000. ’Effects of total pathogen burden on coronary artery disease risk and C-reactive protein levels’. Am. J. Cardiol 85, 140–146. [DOI] [PubMed] [Google Scholar]