

Abstract
Background
Severe coronavirus disease 2019 (COVID‐19) can cause thrombotic events that lead to severe complications or death. Antiplatelet agents, such as acetylsalicylic acid, have been shown to effectively reduce thrombotic events in other diseases: they could influence the course of COVID‐19 in general.
Objectives
To assess the efficacy and safety of antiplatelets given with standard care compared to no treatment or standard care (with/without placebo) for adults with COVID‐19.
Search methods
We searched the Cochrane COVID‐19 Study Register (which comprises MEDLINE (PubMed), Embase, ClinicalTrials.gov, WHO ICTRP, medRxiv, CENTRAL), Web of Science, WHO COVID‐19 Global literature on coronavirus disease and the Epistemonikos COVID‐19 L*OVE Platform to identify completed and ongoing studies without language restrictions to December 2022.
Selection criteria
We followed standard Cochrane methodology. We included randomised controlled trials (RCTs) evaluating antiplatelet agents for the treatment of COVID‐19 in adults with COVID‐19, irrespective of disease severity, gender or ethnicity.
Data collection and analysis
We followed standard Cochrane methodology.
To assess bias in included studies, we used the Cochrane risk of bias tool (RoB 2) for RCTs. We rated the certainty of evidence using the GRADE approach for the outcomes.
Main results
Antiplatelets plus standard care versus standard care (with/without placebo)
Adults with a confirmed diagnosis of moderate to severe COVID‐19
We included four studies (17,541 participants) that recruited hospitalised people with a confirmed diagnosis of moderate to severe COVID‐19. A total of 8964 participants were analysed in the antiplatelet arm (either with cyclooxygenase inhibitors or P2Y12 inhibitors) and 8577 participants in the control arm. Most people were older than 50 years and had comorbidities such as hypertension, lung disease or diabetes. The studies were conducted in high‐ to lower middle‐income countries prior to wide‐scale vaccination programmes.
Antiplatelets compared to standard care:
– probably result in little to no difference in 28‐day mortality (risk ratio (RR) 0.95, 95% confidence interval (CI) 0.85 to 1.05; 3 studies, 17,249 participants; moderate‐certainty evidence). In absolute terms, this means that for every 177 deaths per 1000 people not receiving antiplatelets, there were 168 deaths per 1000 people who did receive the intervention (95% CI 151 to 186 per 1000 people);
– probably result in little to no difference in worsening (new need for invasive mechanical ventilation or death up to day 28) (RR 0.95, 95% CI 0.90 to 1.01; 2 studies, 15,266 participants; moderate‐certainty evidence);
– probably result in little to no difference in improvement (participants discharged alive up to day 28) (RR 1.00, 95% CI 0.96 to 1.04; 2 studies, 15,454 participants; moderate‐certainty evidence);
– probably result in a slight reduction of thrombotic events at longest follow‐up (RR 0.90, 95% CI 0.80 to 1.02; 4 studies, 17,518 participants; moderate‐certainty evidence);
– may result in a slight increase in serious adverse events at longest follow‐up (Peto odds ratio (OR) 1.57, 95% CI 0.48 to 5.14; 1 study, 1815 participants; low‐certainty evidence), but non‐serious adverse events during study treatment were not reported;
– probably increase the occurrence of major bleeding events at longest follow‐up (Peto OR 1.68, 95% CI 1.29 to 2.19; 4 studies, 17,527 participants; moderate‐certainty evidence).
Adults with a confirmed diagnosis of asymptomatic SARS‐CoV‐2 infection or mild COVID‐19
We included two RCTs allocating participants, of whom 4209 had confirmed mild COVID‐19 and were not hospitalised. A total of 2109 participants were analysed in the antiplatelet arm (treated with acetylsalicylic acid) and 2100 participants in the control arm. No study included people with asymptomatic SARS‐CoV‐2 infection.
Antiplatelets compared to standard care:
– may result in little to no difference in all‐cause mortality at day 45 (Peto OR 1.00, 95% CI 0.45 to 2.22; 2 studies, 4209 participants; low‐certainty evidence);
– may slightly decrease the incidence of new thrombotic events up to day 45 (Peto OR 0.37, 95% CI 0.09 to 1.46; 2 studies, 4209 participants; low‐certainty evidence);
– may make little or no difference to the incidence of serious adverse events up to day 45 (Peto OR 1.00, 95% CI 0.60 to 1.64; 1 study, 3881 participants; low‐certainty evidence), but non‐serious adverse events were not reported.
The evidence is very uncertain about the effect of antiplatelets on the following outcomes (compared to standard care plus placebo):
– admission to hospital or death up to day 45 (Peto OR 0.79, 95% CI 0.57 to 1.10; 2 studies, 4209 participants; very low‐certainty evidence);
– major bleeding events up to longest follow‐up (no event occurred in 328 participants; very low‐certainty evidence).
Quality of life and adverse events during study treatment were not reported.
Authors' conclusions
In people with confirmed or suspected COVID‐19 and moderate to severe disease, we found moderate‐certainty evidence that antiplatelets probably result in little to no difference in 28‐day mortality, clinical worsening or improvement, but probably result in a slight reduction in thrombotic events. They probably increase the occurrence of major bleeding events. Low‐certainty evidence suggests that antiplatelets may result in a slight increase in serious adverse events.
In people with confirmed COVID‐19 and mild symptoms, we found low‐certainty evidence that antiplatelets may result in little to no difference in 45‐day mortality and serious adverse events, and may slightly reduce thrombotic events. The effects on the combined outcome admission to hospital or death up to day 45 and major bleeding events are very uncertain. Quality of life was not reported.
Included studies were conducted in high‐ to lower middle‐income settings using antiplatelets prior to vaccination roll‐outs.
We identified a lack of evidence concerning quality of life assessments, adverse events and people with asymptomatic infection. The 14 ongoing and three completed, unpublished RCTs that we identified in trial registries address similar settings and research questions as in the current body of evidence. We expect to incorporate the findings of these studies in future versions of this review.
Keywords: Adult, Humans, Aspirin, Asymptomatic Infections, COVID-19, Platelet Aggregation Inhibitors, SARS-CoV-2
Plain language summary
Are antiplatelets an effective treatment for people with COVID‐19?
Key messages
Antiplatelets are a group of different medicines that can prevent potentially fatal clot formation in the blood vessels ('thrombotic events'). For hospitalised patients with COVID‐19, they probably slightly reduce thrombotic events but probably do not affect deaths, clinical worsening or improvement in COVID‐19 compared with placebo or standard care.
However, antiplatelets may result in a slight increase in serious unwanted effects ('adverse events') and probably increase major bleeding.
Similarly, in non‐hospitalised people, antiplatelets slightly decrease thrombotic events. They may result in little to no difference in deaths or serious unwanted effects, however the evidence on admission to hospital or death and major bleeding events is very uncertain.
There are 14 further studies that have not completed yet, and the results of three other completed studies are not yet available.
What are antiplatelets?
Antiplatelets are a group of drugs which, in different ways, stop blood from changing to a gel‐like substance called a clot, which we mean by the term 'thrombotic events'. They are taken mainly by mouth, usually by people who are at high risk of developing a clot (people who have already experienced or who tend to build up a clot).
A clot could lead to a stroke, coronary heart disease, poor blood circulation in the legs, thromboses in the legs or clot obstruction ('embolism') in the lung circulation that could lead to shortness of breath, heart failure and death.
How might antiplatelets treat COVID‐19?
People with COVID‐19 might be at risk from blood clots. Antiplatelets prevent clots from forming in the body, and this could in turn prevent complications that lead to death and clinical deterioration.
What did we want to find out?
We wanted to know whether antiplatelets in addition to usual care are effective for adults with COVID‐19, when compared to usual care with or without a placebo (a treatment that looks and tastes the same as the study drug but with no active ingredient), and whether they cause unwanted effects. We were particularly interested in:
– number of deaths from any cause up to 28 days after treatment, or longer if reported; – whether people got better or worse after treatment (including unwanted effects of the disease itself, like thrombotic events); – unwanted effects of the treatment (especially major bleeding).
What did we do?
We searched for studies that reported on people with COVID‐19 who received antiplatelets together with usual care, or usual care alone (with/without placebo). We summarised the results of the studies and rated our confidence in the evidence, based on common criteria about the reliability of the evidence.
What did we find?
We identified four studies, including 17,541 people with moderate to severe COVID‐19 (hospitalised). Of these, one compared acetylsalicylic acid (aspirin) with usual care, two studies compared 'P2Y12 inhibitors' (e.g. clopidogrel, prasugrel, ticagrelor) with usual care, and a fourth one compared either acetylsalicylic acid or P2Y12 inhibitors with usual care. The studies included people with a confirmed or suspected diagnosis of SARS‐CoV‐2 infection. Two studies compared acetylsalicylic acid to placebo in 4209 non‐hospitalised people with confirmed mild COVID‐19.
We also found 14 ongoing studies, two studies without published results and another that had been withdrawn after a preprint version. We found no studies that included people with COVID‐19 infection but no symptoms.
Main results
Antiplatelets:
– probably make little or no difference to deaths by 28 or 180 days, worsening up to day 28 (new invasive mechanical ventilation or death) or improvement (discharged alive) up to day 28, and they probably slightly decrease thrombotic events;
– may result in a slight increase in serious unwanted effects and probably increase major bleeding events.
In people not in hospital, with mild disease, antiplatelets may result in little to no difference in death within 45 days, or in the incidence of serious adverse events, and they may slightly decrease the incidence of thrombotic events. The evidence for these participants is very uncertain about the effects on worsening (hospitalisation or death within 45 days) and on major bleeding events. Studies did not report on quality of life and general unwanted effects.
What are the limitations of the evidence?
The studies were conducted in populations from high‐ to middle‐income countries, many of them prior to the roll‐out of COVID‐19 vaccination programmes and before Omicron became the most prevalent variant. We have moderate confidence in the evidence for mortality, worsening/improvement up to day 28, and major bleeding events or thrombotic events in hospitalised people. We have low confidence in the evidence for the effects on serious unwanted effects, because they occurred rarely. For non‐hospitalised people, our confidence in the evidence is low for the effects on death, thrombotic events and serious adverse events, and very low for the effects on worsening and major bleeding events (those events were rare and the time period between symptom onset and treatment was long). Information about other unwanted effects and quality of life was not available.
How up‐to‐date is this evidence?
Our evidence is up‐to‐date to 22 December 2022.
Summary of findings
Summary of findings 1. Antiplatelets in hospitalised adults with moderate to severe COVID‐19.
| Antiplatelets (plus standard care) compared to standard care (with/without placebo) in hospitalised adults with COVID‐19 (moderate to severe disease) | ||||||
|
Patient or population: hospitalised adults with COVID‐19 (moderate to severe disease) Setting: inpatients Intervention: antiplatelets (plus standard care) Comparison: standard care (with/without placebo) | ||||||
| Outcomes | Estimated absolute effectsa (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with standard care (with/without placebo) | Risk with antiplatelets (plus standard care) | |||||
| 28‐day all‐cause mortality | 177 per 1000 | 168 per 1000 (151 to 186) | RR 0.95 (0.85 to 1.05 |
17,249b,c (3 RCTs) |
⊕⊕⊕⊖ Moderated |
Antiplatelets probably result in little to no difference in mortality up to day 28. |
| Worsening up to day 28: participants with new need for invasive mechanical ventilation or death | 230 per 1000 | 218 per 1000 (207 to 232) | RR 0.95 (0.90 to 1.01) |
15,266 (2 RCTs) |
⊕⊕⊕⊖ Moderated |
Antiplatelets probably result in little to no difference in worsening up to day 28. |
| Improvement up to day 28: participants discharged alive | 743 per 1000 | 743 per 1000 (714 to 773) | RR 1.00 (0.96 to 1.04) |
15,454 (2 RCTs) |
⊕⊕⊕⊖ Moderated |
Antiplatelets probably result in little to no difference in improvement up to day 28. |
| Thrombotic events at longest follow‐up* | 57 per 1000 | 52 per 1000 (46 to 58) |
RR 0.90 (0.80 to 1.02) |
17,518 (4 RCTs) |
⊕⊕⊕⊖ Moderated |
Antiplatelets probably result in a slight reduction in thrombotic events. |
| Serious adverse events at longest follow‐up** | 5 per 1000 | 7 per 1000 (2 to 24) | Peto OR 1.57 (0.48 to 5.14) |
1815 (1 RCT) |
⊕⊕⊖⊖ Lowe |
Antiplatelets may result in a slight increase in serious adverse events. |
| Adverse events during study treatment | Not reported | — | — | — | — | We did not identify any study reporting this outcome. |
| Major bleeding events at longest follow‐up*** | 10 per 1000 | 16 per 1000 (12 to 21) | Peto OR 1.68 (1.29 to 2.19) |
17,527 (4 RCTs) |
⊕⊕⊕⊖ Moderated |
Antiplatelets probably increase the occurrence of major bleeding events. |
| CI: confidence interval; COVID‐19: coronavirus disease 2019; OR: odds ratio, RCT: randomised controlled trial, RR: risk ratio | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aThe risk in the control group was assumed from the pooled effects of the control group from included studies; the intervention risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). bAdditionally, there was a study with 292 participants reporting all‐cause mortality at longest follow‐up (RR 1.11, 95% CI 0.75 to 1.64) (Bohula 2022 (COVID‐PACT)). We did not include this study in the meta‐analysis due to an unclear duration of follow‐up and high risk of bias. cREMAP‐CAP 2022 reported 180‐day mortality in a second publication. Treatment with antiplatelets may result in little to no difference in 180‐day mortality (RR 0.97, 95% CI 0.82 to 1.15; 1,312 participants; low‐certainty evidence). dDowngraded one level for serious risk of bias: in Horby 2021 (RECOVERY), an open‐label study, 10% in the intervention group did not receive acetylsalicylic acid; in the open‐label study Bohula 2022 (COVID‐PACT), where 30% in the intervention group discontinued the intervention, the co‐intervention (therapeutic or prophylactic anticoagulation) showed a high level of discontinuation or cross‐over. eDowngraded two levels for very serious imprecision: only one out of three studies contributed data, with very low event rates and a very wide CI.
*The time frames for 'the longest follow‐up' were 28 days (Berger 2022; Bohula 2022 (COVID‐PACT); Horby 2021 (RECOVERY)) or unclear (REMAP‐CAP 2022).
**The time frames for 'the longest follow‐up' were reported for up to 28 days in three studies (Berger 2022; Horby 2021 (RECOVERY); REMAP‐CAP 2022).
***The time frames for 'the longest follow‐up' were reported for up to 28 days in three studies (Berger 2022; Horby 2021 (RECOVERY)). The longest follow‐up in REMAP‐CAP 2022 for major bleeding was 14 days.
Summary of findings 2. Antiplatelets in non‐hospitalised adults with COVID‐19.
| Antiplatelets in non‐hospitalised adults with asymptomatic infection or mild COVID‐19 | ||||||
|
Patient or population: non‐hospitalised adults with mild COVID‐19 Setting: outpatients Intervention: antiplatelets (plus standard care) Comparison: standard care plus placebo | ||||||
| Outcomes | Estimated absolute effectsa (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo (plus standard care) | Risk with antiplatelets (plus standard care) | |||||
| All‐cause mortality at day 45 | 6 per 1000 | 6 per 1000 (3 to 13) | Peto OR 1.00 (0.45 to 2.22) | 4209 (2 RCTs) | ⊕⊕⊖⊖ Lowb | Antiplatelets may result in little to no difference in all‐cause mortality. |
| Admission to hospital or death up to day 45 | 39 per 1000 | 31 per 1000 (23 to 43) | Peto OR 0.79 (0.57 to 1.10) | 4209 (2 RCTs) | ⊕⊖⊖⊖ Very lowb,c | The evidence is very uncertain about the effect of antiplatelets on admission to hospital or death up to day 45. |
| Quality of life up to longest follow‐up* | Not reported | — | — | — | — | We did not identify any study reporting this outcome. |
| Thrombotic events up to day 45 | 3 per 1000 | 1 per 1000 (0 to 4) | Peto OR 0.37 (0.09 to 1.46) | 4209 (2 RCTs) | ⊕⊕⊖⊖ Lowb | Antiplatelets may slightly decrease the incidence of new thrombotic events. |
| Adverse events during study treatment | Not reported | — | — | — | — | We did not identify any study reporting this outcome. |
| Serious adverse events up to day 45 | 16 per 1000 | 16 per 1000 (10 to 26) |
Peto OR 1.00 (0.60 to 1.64) | 3881 (1 RCT) | ⊕⊕⊖⊖ Lowb |
Antiplatelets may make little or no difference to the incidence of serious adverse events. |
| Major bleeding events up to longest follow‐up** | There was no major bleeding or clinically relevant non‐major bleeding event among all participants of the study (same number of participants in each group). | Not estimable | 328 (1 RCT) | ⊕⊖⊖⊖ Very lowd | The evidence is very uncertain about the effect of antiplatelets on the incidence of new major bleeding events. | |
| CI: confidence interval; COVID‐19: coronavirus disease 2019; OR: odds ratio; RCT: randomised controlled trial | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aThe risk in the control group was assumed from the pooled effects of the control groups from included studies; the intervention risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). bDowngraded two levels due to very serious imprecision: low number of events and wide CI. cDowngraded one level due to serious indirectness (one study reports death only due to cardiovascular events, the other study includes major thrombosis in this outcome definition). dDowngraded three levels due to extremely serious imprecision; low sample size/number of events; only one RCT reporting this outcome.
*The time frame for 'at longest follow‐up' in REMAP‐CAP 2022 was at day 180. **The time frame for 'at longest follow‐up' in Connors 2021 (ACTIV‐4B) and Eikelboom 2022 (ACTCOVID) was up to 45 days. Connors 2021 (ACTIV‐4B) reported data after an additional 30‐day safety follow‐up.
Background
This work is part of a series of Cochrane reviews investigating treatments and therapies for coronavirus disease 2019 (COVID‐19). Reviews of this series share information in the background section and methodology based on the first published reviews about monoclonal antibodies (Kreuzberger 2021) and convalescent plasma (Iannizzi 2023), and are part of the German research project 'CEOsys' (COVID‐19 evidence‐ecosystem; CEOsys).
Description of the condition
COVID‐19 is a rapidly spreading infectious disease caused by severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2; WHO2021a). On 22 March 2020, the World Health Organization (WHO) declared the COVID‐19 outbreak a pandemic. Despite intensive international efforts to contain its spread, as of April 2022, COVID‐19 has resulted in more than 753 million confirmed cases and over 6.8 million deaths worldwide (WHO 2023), and estimated global all‐age excess mortality is much higher (COVID‐19 Excess Mortality Collaborators). Several vaccines against COVID‐19 have been distributed across countries and more than an additional hundred vaccine candidates are in development (WHO 2022c). As of February 2023, 13 billion vaccinations could be registered worldwide (WHO 2023).
Specific risk factors for severe disease, hospitalisation and mortality have been identified: individuals aged 65 years or older, smokers and those with certain underlying medical conditions, such as cancer, chronic kidney disease, chronic obstructive pulmonary disease (COPD), heart conditions, immunocompromised state, obesity, sickle cell disease or diabetes mellitus are more likely to have severe courses of the disease (Huang 2020; Liang 2020; WHO2021a; Williamson 2020). COVID‐19 case fatality varies widely between countries and reporting periods (from 0.1% to more than 19%; Johns Hopkins 2021). However, these numbers may be misleading due to varying testing frequency, lag in reporting dates, incomplete capturing of all cases and variations in case definitions since the beginning of the pandemic.
The median incubation period is estimated to be between five and six days. 97.5% of symptomatic cases develop symptoms within 11.5 days of exposure (Lauer 2020). Sore throat, cough, fever, headache, fatigue and myalgia or arthralgia are the most commonly reported symptoms (Struyf 2020). Other symptoms include dyspnoea, chills, nausea or vomiting, diarrhoea, loss of taste and smell and nasal congestion (WHO2021a). The majority of infected people have mild symptoms (approximately 80%, Wu 2020) or remain completely asymptomatic (Buitrago‐Garcia 2020). A smaller proportion (approximately 14%) are affected by severe or critical disease, with intensive care unit (ICU) admittance due to respiratory failure, septic shock or multiple organ dysfunction (Wu 2020). Moreover, there is an elevated risk of thrombotic complications in hospitalised COVID‐19 patients (Cui 2020) and COVID‐19 patients in the ICU (Klok 2020). A meta‐analysis suggests that about 2 in 10 COVID‐19 patients (mostly those treated in hospital) develop pulmonary embolism (Liao 2020). Numbers for deep vein thrombosis are similarly high (Middeldorp 2020). Furthermore, the high cumulative incidence of thrombotic complications in critically ill patients with COVID‐19 pneumonia is confirmed (Al Duhailib 2022; Bompard 2020; Katsoularis 2022; Klok 2020).
Nowadays, several drugs for the treatment of COVID‐19 are available, mostly for patients with severe or critical disease (e.g. corticosteroids, remdesivir, tocilizumab, baricitinib) (Ghosn 2021; Grundeis 2023; Kramer 2022; Wagner 2022), which are recommended in clinical guidelines (WHO living guideline 2023). For patients with mild disease and risk factors for severe disease, nirmatrelvir/ritonavir is available (Reis 2022). In addition, treatment consists of supportive care with oxygen supply in moderately severe cases, and non‐invasive ventilation, high‐flow nasal cannula therapy or even invasive mechanical ventilation, as well as prone positioning, and sometimes extracorporeal membrane oxygenation, in very severe cases (WHO 2021e).
Description of the intervention
In the early pandemic, retrospective assessments showed a potential benefit of pre‐diagnosis acetylsalicylic acid treatment on 30‐day mortality (Osborne 2021). As thrombotic macroangiopathy (e.g. deep vein thrombosis, ischaemic stroke, acute myocardial infarction or lung artery emboly) and microangiopathy (microangiopathic pulmonary dysfunction) can substantially influence the course of COVID‐19 (Menter 2020), preventing those events could reduce the burden on our healthcare systems, i.e. hospitalisation, clinical deterioration or death.
Antiplatelets are a group of drugs, administered via oral, rectal or intravenous routes, which inhibit platelet aggregation through different mechanisms of action (Eikelboom 2012). Based on the mechanism of action, antiplatelets can be classified as follows (Hashemzadeh 2008; Krötz 2008).
Platelet aggregation inhibitors: cyclooxygenase inhibitors as acetylsalicylic acid (brand name: Aspirin® and others) and adenosine diphosphate (ADP) receptor/P2Y12 inhibitors (clopidogrel, ticlopidine, prasugrel, cangrelor and elinogrel)
Glycoprotein IIb/IIIa platelet inhibitors (abciximab, eptifibatide and tirofiban)
Protease‐activated receptor‐1 antagonists (vorapaxar)
Others: nucleoside transport inhibitors interfering with cyclic adenosine monophosphate (dipyridamole and cilostazol)
Antiplatelets have been used as prophylactic or therapeutic interventions for a number of conditions, such as: primary and secondary cardiovascular prevention of acute ischaemic stroke (Sandercock 2014); coronary heart disease (acute coronary syndromes or stented coronaries), initially even with dual platelet aggregation inhibition (Khan 2020); and intermittent claudication (Wong 2011), amongst others (Minhas 2022).
The most common adverse events related to antiplatelet medications are bleeding events (upper gastrointestinal bleeding, ecchymosis, haematuria, epistaxis) acetylsalicylic acid‐induced asthma, nasal polyps, thrombocytopenia (Kalyanasundaram 2011), and renal dysfunction (Awtry 2000).
How the intervention might work
Acetylsalicylic acid is the most widely used antiplatelet agent and acts by irreversibly inhibiting the activity of the cyclooxygenase enzyme (COX) during the synthesis of prostaglandin H2, a precursor of thromboxane A2 and prostaglandin I2 (prostacyclin). Thromboxane A2 induces platelet aggregation and vasoconstriction (COX‐1 mediates its production), and prostaglandin I2 inhibits platelet aggregation and induces vasodilatation (mediated by COX‐2). Acetylsalicylic acid at low doses (75 mg to 150 mg) completely or partially blocks COX‐1, thus inhibiting the production of thromboxane A2, while high doses inhibit COX‐2 (Warner 2011). This could be the clue to reducing thromboembolic outcomes associated with COVID‐19 (in microscopic small as well as in huge vessels) and might at the same time reduce the hyperinflammatory reaction to viral infection. It has been suggested that the SARS‐CoV‐2 virus uses platelets to modulate the immune responses, including the circulation of platelet‐neutrophil aggregates, contributing to the hypercoagulability state observed in severe COVID‐19 patients (Najm 2021). Acetylsalicylic acid, with the brand name Aspirin®, is available as both oral tablets or rectal suppository, or as an intravenously applicable medication if the patient cannot take the drug orally.
ADP receptor/P2Y12 inhibitors are another group of antiplatelet drugs that includes thienopyridines (clopidogrel, ticagrelor, prasugrel and cangrelor) and cyclo‐pentyltriazolo‐pyrimidines (ticagrelor, cangrelor and elinogrel). They block phosphoinositide hydrolysis, thromboxane A2 formation, protein phosphorylation and inhibit the increase in cytosolic Ca++, which is usually the result of ADP receptor activation and a major mechanism of platelet activation (Kunapuli 2003). ADP receptor/P2Y12 inhibitors bind the ADP‐P2Y12 receptor on the surface of platelets and inhibit activation of the glycoprotein IIb/IIIa receptor complex. This receptor complex plays an important role in platelet aggregation and thrombus formation (Al‐Abdouh 2022). Other studies have shown that in addition to their antithrombotic properties, P2Y12 inhibitors, particularly ticagrelor, can reduce inflammation; other beneficial effects have been reported in experimental models and in clinical inflammatory diseases, including sepsis and acute lung injury (Berger 2022).
The antithrombotic properties especially, but also the anti‐inflammatory properties, of antiplatelets could be a valuable approach in the treatment of COVID‐19.
Why it is important to do this review
The burden of thrombotic events in individuals with COVID‐19 is high, which warrants effective treatments and prevention. There is still a lack of information regarding treatment of COVID‐19. Antiplatelets are low‐cost interventions that might be especially useful in low‐resource settings. Therefore, the evidence from this review is needed in order to effectively decrease the burden of COVID‐19.
Objectives
To assess the efficacy and safety of antiplatelets given with standard care compared to no treatment or standard care (with/without placebo) for adults with COVID‐19.
Methods
Criteria for considering studies for this review
Types of studies
The main description of methods is based on the standard template of the Cochrane Haematology review group and is in line with a series of Cochrane Reviews investigating treatments and therapies for COVID‐19, mostly written under the auspices of CEOsys. Specific adaptions related to the research question were made if necessary; for this review we added thrombotic and bleeding events to our core outcome set, but we did not use those outcomes to determine whether to include a trial or not. The protocol for this review was registered with the international prospective register of systematic reviews (PROSPERO) (Piechotta 2021).
To assess the effectiveness and safety of antiplatelets for the treatment of COVID‐19, we included randomised controlled trials (RCTs), as this study design, if performed appropriately, provides the best evidence for experimental therapies in highly controlled therapeutic settings. We used the methods recommended in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2021a). We had planned to also accept cross‐over RCT designs, but we would have only considered results from the first period before cross‐over because COVID‐19 is not a chronic condition and its exact course and long‐term effects are yet to be defined.
We excluded cluster‐RCTs, controlled non‐randomised studies of interventions and observational studies. We also excluded animal studies, pharmacokinetic studies and in vitro studies.
We included the following formats, if sufficient information was available on study design, characteristics of participants, interventions and outcomes.
Full‐text publications
Preprint versions
Abstract publications
Results published in trials registries
We included preprints and conference abstracts to have a complete overview of the ongoing research activity, especially for tracking newly emerging studies about antiplatelets in COVID‐19. We did not apply any limitation with respect to the length of follow‐up.
Types of participants
We included adults with a confirmed diagnosis of COVID‐19 (as described in the studies) and we did not exclude any studies based on gender, ethnicity, disease severity or setting.
We excluded studies that evaluate antiplatelets for the treatment of other coronavirus diseases such as SARS (severe acute respiratory syndrome) or MERS (Middle East respiratory syndrome), or other viral diseases, such as influenza. If studies enroled populations with or exposed to mixed viral diseases, we had planned to only include these if trial authors provided subgroup data for SARS‐CoV‐2 infection. We excluded studies investigating antiplatelets for long‐COVID treatment.
Types of interventions
We considered the following comparisons.
Antiplatelets with standard care versus standard care (with/without placebo).
Antiplatelets with standard care versus active treatment with standard care.
Different formulations, doses, schedules of antiplatelets.
Antiplatelets, in any dose or duration of treatment, alone or in combination with other interventions, provided that it has been possible to assess the effects of the antiplatelet agent alone (similar co‐interventions in the compared groups). We allowed any co‐interventions, including supportive care, anticoagulants, anti‐viral drugs and corticosteroids, for example. Studies comparing different formulations, doses, schedules of antiplatelets were also included. Co‐interventions were allowed, but must have been comparable between intervention groups.
We excluded studies with the following comparison.
Antiplatelets with standard care in combination with active treatment A versus another active treatment (treatment B, e.g. monoclonal antibodies (mAbs)).
Types of outcome measures
We included studies evaluating antiplatelets for the treatment of COVID‐19 independently of reported outcomes.
We evaluated core outcomes based on the Core Outcome Measures in Effectiveness Trials (COMET) Initiative for COVID‐19 patients (COMET 2021; Marshall 2020), and additional outcomes that have been prioritised by consumer representatives and the German guideline panel for therapy of hospitalised people with COVID‐19.
We defined outcome sets for two populations. Those are: (i) hospitalised individuals with a confirmed diagnosis of COVID‐19 and moderate to severe disease, and (ii) ambulatory managed individuals with a confirmed diagnosis of SARS‐CoV‐2 infection and asymptomatic or mild disease, according to the WHO Clinical Progression Scale (Marshall 2020).
Individuals with a confirmed diagnosis of COVID‐19 and moderate to severe disease
Effectiveness of antiplatelets
Prioritised outcomes (included in the summary of findings tables)
All‐cause mortality up to day 28, day 60, time‐to‐event, and up to the longest follow‐up.
-
Clinical status at day 28, day 60, and up to the longest follow‐up, including:
-
worsening of clinical status
participants with clinical deterioration (new need for invasive mechanical ventilation) or death up to day 28;
-
improvement of clinical status
participants discharged alive up to day 28.
-
Any thrombotic event up to longest follow‐up (e.g. pulmonary embolism, deep vein thrombosis, venous thromboembolism, myocardial infarction, extracorporeal membrane oxygenation (ECMO) or dialyse filter clotting).
Safety of antiplatelets
Serious adverse events at longest follow‐up available during the study period, defined as number of participants with any serious event.
Adverse events (any grade) during study treatment during the study period, defined as number of participants with any event.
-
Major bleeding up to longest follow‐up defined by fulfilment of at least one of the following three criteria:
fatal haemorrhagic events; and/or
bleeding in a critical area or organ (e.g. intracranial, intraspinal, intraocular, retroperitoneal, intraarticular, pericardial, intramuscular with compartment syndrome); and/or
a decrease in haemoglobin concentration of ≥ 2 g/dL, or requirement of a transfusion of two or more units of blood.
Additional outcomes (not included in the summary of findings tables)
Time‐to‐event mortality data: mortality up to longest follow‐up.
Quality of life, including fatigue and neurological status, assessed with standardised scales (e.g. WHO‐Quality of life‐100 (WHOQOL‐100)) at up to seven days, up to 28 days and longest follow‐up available.
Duration of hospitalisation, or time to discharge from hospital.
Admission to the intensive care unit (ICU).
Ventilator‐free days; ventilator‐free defined as WHO Clinical Progression Scale ≤ 6 for the subgroup of participants requiring invasive mechanical ventilation at baseline, i.e. WHO ≥ 7).
Need for new dialysis (up to 28 days).
Individuals with a confirmed diagnosis of asymptomatic SARS‐CoV‐2 infection or mild disease
Effectiveness of antiplatelets
Prioritised outcomes (included in the summary of findings tables)
All‐cause mortality at day 28, day 60, time‐to‐event and up to the longest follow‐up.
Admission to hospital or death within 45 days, day 60, time‐to‐event and up to the longest follow‐up.
-
Symptom resolution:
All initial symptoms resolved (asymptomatic) at day 14, day 28 and up to the longest follow‐up.
Duration to symptom resolution.
Quality of life, including fatigue and neurological status, assessed with standardised scales (e.g. WHOQOL‐100) at up to 7 days, up to 28 days and the longest follow‐up available.
Any thrombotic event (e.g. pulmonary embolism, deep vein thrombosis, venous thromboembolism, myocardial infarction, ECMO or dialyse filter clotting) up to the longest follow‐up
Safety of antiplatelets
Serious adverse events at longest follow‐up available during the study period, defined as the number of participants with any serious event.
Adverse events (any grade) during study treatment during the study period, defined as number of participants with any event.
-
Major bleeding defined by fulfilment of at least one of the following three criteria:
fatal haemorrhagic events; and/or
bleeding in a critical area or organ (e.g. intracranial, intraspinal, intraocular, retroperitoneal, intraarticular, pericardial, intramuscular with compartment syndrome); and/or
a decrease in haemoglobin concentration of ≥ 2 g/dL, or requirement of a transfusion of two or more units of blood.
Additional outcomes (not included in the summary of findings tables)
Time to symptom onset.
Length of hospital stay and time to discharge, for the subgroup of participants hospitalised during the course of the disease.
Admission to the intensive care unit (ICU).
Timing of outcome measurement
In the case of time‐to‐event analysis, e.g. for all‐cause mortality (survival) or time to discharge from hospital, we included the outcome measure based on the longest follow‐up time. We also collected information on outcomes from all other time points reported in the publications (for example, REMAP‐CAP 2022 also reported 90‐day mortality).
We included adverse events occurring during active treatment and included long‐term adverse events as well. If sufficient data were available, we grouped the measurement time points of eligible outcomes, for example, adverse events and serious adverse events, into those measured directly after treatment (up to seven days after treatment), medium‐term outcomes (up to 15 days after treatment) and longer‐term outcomes (more than 30 days after treatment).
Search methods for identification of studies
Electronic searches
Our Information Specialist (IM) conducted systematic searches, which were reviewed by another Information Specialist, in the following sources from inception. Following a living systematic review approach, six searches were conducted between 28 April 2021 and 22 December 2022 (date of the last search for all databases), with no restrictions on the language of publication.
-
Cochrane COVID‐19 Study Register (CCSR) (www.covid-19.cochrane.org), comprising:
MEDLINE (PubMed), weekly updates
Embase.com, weekly updates
ClinicalTrials.gov (www.clinicaltrials.gov), daily updates
World Health Organization International Clinical Trials Registry Platform (ICTRP) (trialsearch.who.int/), weekly updates
medRxiv (www.medrxiv.org), weekly updates
Cochrane Central Register of Controlled Trials (CENTRAL), monthly updates
-
Web of Science Core Collection:
Science Citation Index Expanded (from 1945)
Emerging Sources Citation Index (from 2015)
WHO COVID‐19 Global literature on coronavirus disease (search.bvsalud.org/global-literature-on-novel-coronavirus-2019-ncov/)
Epistemonikos COVID‐19 L*OVE Platform (app.iloveevidence.com/loves)
For detailed search strategies, see Appendix 1.
Searching other resources
We identified other potentially eligible studies or ancillary publications by searching the reference lists of included studies, relevant systematic reviews and meta‐analyses. In addition, we contacted the investigators of included studies to obtain additional information on the retrieved studies. In addition, we searched current treatment guidelines for further literature.
We searched for grey literature, which we defined as searching trials registers such as ClinicalTrials.gov and WHO ICTRP contained in the CCSR, as well as searching preprint servers and grey literature indexes contained in the CCSR and the WHO COVID‐10 Global Literature database.
Once we established our set of included studies, we searched for preprints via Europe PMC, to check if any preprints for ongoing studies were published since our database search. We also compared our identified studies with results from projects that aim to track COVID‐19 intervention research, i.e.www.covid-trials.organd covid-nma.com/dataviz.
Data collection and analysis
In multi‐arm studies, we extracted and analysed only the antiplatelet domain groups to avoid arbitrary omission of relevant groups and double‐counting of participants. As we found no relevant differences in the outcomes between acetylsalicylic acid and P2Y12 inhibitors, and no other types of antiplatelets contributed to our outcomes, we decided to merge the antiplatelet groups.
Selection of studies
Two out of four review authors (AF, NS, MA, SM) independently screened the results of the search strategies for eligibility for this review by reading the abstracts using EndNote software (EndNote 2013). We coded the abstracts as either 'include' or 'exclude'. In the case of disagreement or if it was unclear whether we should retrieve the abstract or not, we obtained the full‐text publication for further discussion. Two out of three review authors (AF, NS, MA) assessed the full‐text articles of selected studies. If the two review authors were unable to reach a consensus, they consulted a third review author (VP) to reach a final decision.
We documented the study selection process in a flow chart, as recommended in the PRISMA statement (Moher 2009), and showed the total numbers of retrieved references and the numbers of included and excluded studies. We listed all studies that we excluded after full‐text assessment and the reasons for their exclusion in the Characteristics of excluded studies section.
Data extraction and management
We conducted data extraction according to the guidelines proposed by Cochrane (Li 2021). Two out of three review authors (ALF, MA, SM) extracted data independently and in duplicate, using a customised data extraction form developed in Microsoft Excel (Microsoft 2018). The data selection form was piloted (in other projects under the auspices of CEOsys) to ensure consistency in the process of data extraction. We solved disagreements by discussion. If no agreement was obtained, a third review author was involved to solve the disagreement. We found only English‐language trials, so none of them had to be translated.
Two out of three review authors (ALF, MA, SM) independently assessed eligible studies obtained in the process of study selection (as described above) for methodological quality and risk of bias. If the review authors were unable to reach a consensus, a third review author was consulted.
We extracted the following information if reported.
General information: author, title, source, publication date, country, language, duplicate publications
Study characteristics: trial design, setting and dates, source of participants, inclusion/exclusion criteria, comparability of groups, treatment cross‐overs, compliance with assigned treatment, length of follow‐up
Participant characteristics: age, gender, ethnicity, number of participants recruited/allocated/evaluated, additional diagnoses, severity of disease, previous treatments, concurrent treatments and standard care, complementary medicine (e.g. quercetin, elderberry, zinc)
Interventions: dose, frequency, timing, duration and route of administration, setting (e.g. hospitalised, non‐hospitalised), duration of follow‐up
Control intervention: placebo, no treatment; dose, frequency, timing, duration and route of administration, setting, duration of follow‐up.
Outcomes: as specified under Types of outcome measures
Risk of bias assessment: study design, confounding, definition of risk estimates, selection bias, attrition bias, detection bias, reporting bias
Assessment of risk of bias in included studies
We used the Risk of Bias 2 (RoB 2) tool to analyse the risk of bias of study results (Sterne 2019). Of interest for this review is the effect of the assignment to the intervention (the intention‐to‐treat (ITT) effect), thus we performed all assessments with RoB 2 on this effect. The outcomes that we assessed are those specified for inclusion in the summary of findings table.
Two out of five review authors (ALM, ALF, MA, RR, SM) independently assessed the risk of bias for each outcome. In case of discrepancies amongst their judgements and inability to reach consensus, we consulted a third review author to reach a final decision. We assessed the following types of bias, as outlined in Chapter 8 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2021b).
Bias arising from the randomisation process
Bias due to deviations from the intended interventions
Bias due to missing outcome data
Bias in measurement of the outcome
Bias in selection of the reported result
To address these types of bias we used the signalling questions recommended in RoB 2 and made a judgement using the following options.
'Yes': if there is firm evidence that the question is fulfilled in the study (i.e. the study is at low or high risk of bias for the given the direction of the question).
'Probably yes': a judgement has been made that the question is fulfilled in the study (i.e. the study is at low or high risk of bias given the direction of the question).
'No': if there is firm evidence that the question is unfulfilled in the study (i.e. the study is at low or high risk of bias for the given the direction of the question).
'Probably no': a judgement has been made that the question is unfulfilled in the study (i.e. the study is at low or high risk of bias given the direction of the question).
'No information': if the study report does not provide sufficient information to allow any judgement.
We used the algorithms proposed by RoB 2 to assign each domain one of the following levels of bias.
Low risk of bias
Some concerns
High risk of bias
Subsequently, we derived an overall risk of bias rating for each prespecified outcome in each study in accordance with the following suggestions.
'Low risk of bias': we judge the trial to be at low risk of bias for all domains for this result.
'Some concerns': we judge the trial to raise some concerns in at least one domain for this result, but not to be at high risk of bias for any domain.
'High risk of bias': we judge the trial to be at high risk of bias in at least one domain for the result, or we judge the trial to have some concerns for multiple domains in a way that substantially lowers confidence in the results.
We used the RoB 2 Excel tool to implement RoB 2 (available from riskofbias.info) and stored and presented our detailed RoB 2 assessments as supplementary online material: https://zenodo.org/record/7637454.
We made the risk of bias assessments between November 2021 and January 2023 (November 2021: Connors 2021 (ACTIV‐4B), Horby 2021 (RECOVERY); January 2022: Berger 2022; March 2022: REMAP‐CAP 2022; January 2023: Bohula 2022 (COVID‐PACT), Eikelboom 2022 (ACTCOVID), REMAP‐CAP 2022).
Measures of treatment effect
For dichotomous outcomes, we recorded the number of events and total number of participants in both treatment and control groups. We reported the pooled risk ratio (RR) with a 95% confidence interval (CI) (Higgins 2021c). If the number of observed events was small (less than 5% of sample per group), and if studies had a balanced number of participants in all treatment groups, we reported the Peto odds ratio (OR) with 95% CI (Higgins 2021c).
We extracted hazard ratios (HRs) for time‐to‐event outcomes (e.g. time to death). If HRs were not available, we made every effort to estimate the HR as accurately as possible from available data using the methods proposed by Parmar and Tierney (Parmar 1998; Tierney 2007). If sufficient studies provided HRs, we used HRs rather than RRs or mean differences (MDs) in a meta‐analysis, as they provide more information. As only REMAP‐CAP 2022 reported the 90‐day‐mortality data, we decided to present mortality data as a dichotomous outcome for day 28, to use a common mortality outcome parameter and to report time‐to‐event data narratively.
For continuous outcomes, we recorded the mean, standard deviation and total number of participants in both treatment and control groups. Where continuous outcomes used the same scale, we would have performed analyses using the MD with 95% CIs. For continuous outcomes measured with different scales, we had planned to perform analyses using the standardised mean difference (SMD). To interpret SMDs, we would have re‐expressed SMDs in the original units of a particular scale with the most clinical relevance and impact (e.g. clinical symptoms with the WHO Clinical Progression Scale (Marshall 2020)).
Unit of analysis issues
The aim of this review is to summarise trials that analyse data at the level of the individual participant. We collated multiple reports of one study so that the study, and not the report, is the unit of analysis. As COVID‐19 is an acute condition, in case of cross‐over trials, we would have included only data from the first period to avoid double counting of participants or events. However, we did not identify cross‐over trials.
We analysed the time points as reported in the section Types of outcome measures.
Studies with multiple treatment groups
As recommended in Chapter 6 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2021d), for studies with multiple treatment groups of the same intervention (i.e. different agent, dose, route of administration), we evaluated arms separately and compared with the common comparator. For pair‐wise meta‐analysis, we split the ‘shared’ group into two or more groups with smaller sample sizes, and included two or more (reasonably independent) comparisons. For this purpose, for dichotomous outcomes, both the number of events and the total number of participants was divided, and for continuous outcomes, the total number of participants would have been divided with unchanged means and SDs.
Dealing with missing data
Chapter 10 of the Cochrane Handbook for Systematic Reviews of Interventions suggests a number of potential sources for missing data, which we took into account: at study level, at outcome level and at summary data level (Higgins 2021c). At all levels, it is important to differentiate between data 'missing at random', which may often be unbiased, and 'not missing at random', which may bias study and thus review results.
If data were missing, we requested these data from the principal investigators (i.e. we contacted Bohula 2022 (COVID‐PACT); Horby 2021 (RECOVERY); NCT04363840; NCT04483960; NCT04808895; REMAP‐CAP 2022). We received responses from Dr Peto regarding the Horby 2021 (RECOVERY) trial, from Dr Baird‐zars regarding the Bohula 2022 (COVID‐PACT) trial and from Dr Lau regarding the NCT04363840 trial. If, after this, data were still missing, we had to make explicit assumptions of any methods the included studies used.
Assessment of heterogeneity
We assessed heterogeneity of treatment effects between trials by visual inspection of our forest plots and using a Chi² test with a significance level at P < 0.1. We used the I² statistic and visual examination to assess possible heterogeneity (I² statistic 30% to 75% taken to signify moderate heterogeneity, I² statistic > 75% taken to signify considerable heterogeneity; Higgins 2021c). If heterogeneity was suspected to be high, we had planned to explore potential causes through sensitivity and subgroup analyses. If we could not find a reason for heterogeneity, we would not have performed a meta‐analysis, but planned to comment on results from all studies and present these in tables.
Assessment of reporting biases
As mentioned above, we searched trials registries to identify completed trials that have not been published elsewhere, to minimise or determine publication bias. We intended to explore potential publication bias by generating a funnel plot and statistically testing this by conducting a linear regression test for meta‐analyses involving at least 10 trials (Sterne 2019), but we found fewer than 10 trials and therefore could not perform this test. We would have considered P < 0.1 as significant for this test.
Data synthesis
If the clinical and methodological characteristics of individual studies were sufficiently homogeneous, we pooled the data in meta‐analysis. We performed analyses according to the recommendations of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2021c). We analysed trials including different severities of disease separately, grouping them into mild (non‐hospitalised), and moderate to severely ill (hospitalised), as these are different populations in different settings, resulting in differing outcomes (see Types of outcome measures). We treated placebo and no treatment as the same intervention, as well as standard care at different institutions and time points.
We used the Review Manager Web (RevMan Web) software for analyses (RevMan Web 2022). One out of three review authors (VP, ALF, SM) entered the data into the software, and a second review author (AM, SM, ALF) checked the data for accuracy. We used the random‐effects model for the analyses as we anticipated that true effects are related, but are not the same for included studies. We used a fixed‐effect model for the Peto OR. If we could not perform a meta‐analysis we commented on the results, with the results from all studies presented in tables or narratively. If meta‐analysis was possible, we assessed the effects of potential biases in sensitivity analyses (see Sensitivity analysis). By using the Mantel‐Haenszel method for binary outcomes, we were able to calculate an estimate of the amount of variation between the studies we analysed.
We had planned to use the inverse variance method for continuous outcomes, or outcomes where HRs were available. We had planned to explore heterogeneity above 80% with subgroup analyses. If we had not found a cause for the heterogeneity, we had planned not to perform a meta‐analysis, but to comment on the results narratively and present them in tables if studies were not homogenous enough.
Subgroup analysis and investigation of heterogeneity
We performed subgroup analyses of 28‐day mortality using the following characteristics.
Different antiplatelets, as they have different mechanisms of action and pathways, and could therefore have different effects on the outcome.
Additional anticoagulation versus no additional anticoagulation.
Additionally, we had planned to perform subgroup analyses of the following characteristics, but had insufficient data.
Age of participants (divided into applicable age groups, e.g. children, 18 to 65 years, 65 years and older), suggesting that older participants may benefit more from treatment with antiplatelets, as they are at higher risk for thrombotic events.
Pre‐existing conditions (diabetes, respiratory disease, hypertension, immunosuppression), suggesting that participants with some diseases that increase thrombotic risk may benefit more.
Timing of first dose administration with illness onset, suggesting that an earlier treatment may prevent deterioration.
We used the tests for interaction to test for differences between subgroup results and considered P < 0.01 as statistically significant for all subgroup analyses.
Sensitivity analysis
We performed sensitivity analysis of the following characteristics for mortality at 28 days.
Fixed‐effect versus random‐effects model
Risk of bias assessment components (excluding studies with a high risk of bias)
Additionally, we had planned to perform sensitivity analyses of the following characteristics, but did not find sufficient data to perform them.
Comparison of preprints of COVID‐19 interventions versus peer‐reviewed articles
Comparison of studies with premature termination versus completed studies
Summary of findings and assessment of the certainty of the evidence
Summary of findings
We used the MAGICapp software to create summary of findings tables (MAGICapp). For time‐to‐event outcomes, we had planned to calculate absolute effects at specific time points, as recommended in the GRADE guidance 27 (Skoetz 2020), but we have not reported HR in our summary of findings tables.
According to Chapter 14 of the updated Cochrane Handbook for Systematic Reviews of Interventions, the “most critical and/or important health outcomes, both desirable and undesirable, limited to seven or fewer outcomes” should be included in the summary of findings table(s) (Higgins 2021e). We included outcomes prioritised according to the Core Outcome Set for intervention studies (COMET 2021) and patient relevance, as follows.
Hospitalised individuals with a confirmed diagnosis of COVID‐19 and moderate to severe disease
28‐day all‐cause mortality
-
Worsening of clinical status up to day 28
Participants with clinical deterioration (new need for invasive mechanical ventilation) or death
-
Improvement of clinical status up to day 28
Participants discharged alive
Thrombotic events (at longest follow‐up)
Serious adverse events (at longest follow‐up)
Adverse events (at longest follow‐up)
Major bleeding events (at longest follow‐up)
Antiplatelets in non‐hospitalised individuals with asymptomatic infection or mild COVID‐19
All‐cause mortality at day 45
-
Worsening of clinical status up to day 45
Admission to hospital or death
Quality of life up to longest follow‐up
Thrombotic events up to day 45
Adverse events up to day 45
Serious adverse events up to day 45
Major bleeding events up to longest follow‐up
Assessment of the certainty of the evidence
We used the Grading of Recommendations Assessment, Development and Evaluation (GRADE) approach to assess the certainty of the evidence for the outcomes listed in the previous section.
The GRADE approach uses five domains (risk of bias, consistency of effect, imprecision, indirectness and publication bias) to assess the certainty in the body of evidence for each prioritised outcome.
We downgraded our certainty of evidence as follows.
Serious (‐1) or very serious (‐ 2) risk of bias
Serious (‐1) or very serious (‐ 2) inconsistency
Serious (‐1) or very serious (‐ 2) uncertainty about directness
Serious (‐1) or very serious (‐ 2) or extremely (‐3) imprecise or sparse data
Serious (‐1) or very serious (‐ 2) probability of reporting bias
The GRADE system uses the following criteria for assigning grade of evidence.
High: we are very confident that the true effect lies close to that of the estimate of the effect.
Moderate: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of effect, but there is a possibility that it is substantially different.
Low: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect.
Very low: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect.
We followed the current GRADE guidance for these assessments in its entirety as recommended in the Cochrane Handbook for Systematic Reviews of Interventions, Chapter 14 (Higgins 2021e).
We used the overall risk of bias judgement, derived from the RoB 2 Excel tool, to inform our decision on downgrading for risk of bias. We phrased the findings and certainty of the evidence as suggested in the informative statement guidance (Santesso 2020).
Results
Description of studies
Results of the search
We searched all databases and screened the resulting records up to 22 December 2022.
Our searches retrieved 806 records for the specific database searches. After removing 52 duplicates, we screened 754 records based on their titles and abstracts. We excluded 719 records that did not meet the prespecified inclusion criteria. After screening, we identified 35 full texts that could meet our inclusion criteria. Three (five records) of these 35 studies are awaiting classification, so we could not include them in our analyses. We had to exclude two studies (two records) because they did not meet the inclusion criteria. Fourteen studies (15 records) were still ongoing, so we also could not include them. In the end, we were able to include six RCTs (13 records) that met our inclusion criteria. Details of our electronic searches can be found in the corresponding PRISMA flow diagram (Figure 1).
1.
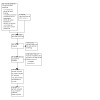
Study flow diagram
Six RCTs are included in this review (Berger 2022; Bohula 2022 (COVID‐PACT); Connors 2021 (ACTIV‐4B); Eikelboom 2022 (ACTCOVID); Horby 2021 (RECOVERY); REMAP‐CAP 2022), of which four RCTs included participants with moderate to severe COVID‐19 (Berger 2022; Bohula 2022 (COVID‐PACT); Horby 2021 (RECOVERY); REMAP‐CAP 2022), and two included participants with mild disease (Connors 2021 (ACTIV‐4B); Eikelboom 2022 (ACTCOVID)).
14 RCTs are currently ongoing (NCT05073718; NCT04808895; Bohula 2022 (COVID‐PACT); CTRI/2020/08/027503; CTRI/2021/03/032059; CTRI/2021/06/034254; IRCT20180205038626N7; NCT04768179; NCT04363840; NCT04937088; NCT04445623; NCT04703608; NCT04365309; Sharma 2021).
Three RCTs are awaiting classification (NCT04391179; NCT04659109; RESIST 2021).
Two RCTs were excluded (Eikelboom 2022 (ACTCOVID19 hospitalised); NCT04483960), because Eikelboom 2022 (ACTCOVID19 hospitalised) used a combined intervention of a platelet inhibitor plus rivaroxaban and NCT04483960 did not use antiplatelets.
Details on the interventions planned in the ongoing trials can be found in Table 3 (Characteristics of ongoing studies).
1. Characteristics of ongoing studies.
| Study ID | Title | Intervention | Comparator | Other study interventions | Planned number of participants | Setting | Planned completion date | Link |
| ChiCTR2000030055 | Multicentre study for the treatment of Dipyridamole with novel coronavirus pneumonia | Dipyridamole | Standard care | None | 460 | Hospital | NR | covid-19.cochrane.org/studies/crs-13247467 |
| IRCT20180205038626N7 | The Investigation of the Effectiveness of ASA Usage on the Incidence of Cardiovascular Events in Patients with Corona Virus (COVID‐19) A Clinical Trial Study | Acetylsalicylic acid | Placebo | None | 36 | Hospital | NR | en.irct.ir/trial/52374 |
| NCT04703608 | Prevention and Treatment for COVID ‐19 (Severe Acute Respiratory Syndrome Coronavirus 2 SARS‐CoV‐2) Associated Severe Pneumonia in the Gambia | Acetylsalicylic acid | Placebo | Ivermectin | 1200 | Hospital and private household | March 2022 | clinicaltrials.gov/ct2/show/record/NCT04703608?cond=NCT%3A+04703608&draw=2&rank=1 |
| NCT04808895 | Acetylsalicylic Acid in the Prevention of Severe SARS‐CoV2 Pneumonia in Hospitalised Patients With COVID‐19 | Acetylsalicylic acid | Placebo | None | 204 | NR | July 2021 | clinicaltrials.gov/ct2/show/record/NCT04808895?cond=NCT%3A+04808895&draw=2&rank=1 |
| CTRI/2020/08/027503 | Low Dose Aspirin in Moderate to Severe SARS‐ CoV‐2 Infected Patients: A Pilot Randomized Controlled Trial | Acetylsalicylic acid | Standard care | None | 60 | Hospital | NR | www.ctri.nic.in/Clinicaltrials/pdf_generate.php?trialid=44980&EncHid=&modid=&compid=%27,%2744980det%27 |
| NCT04768179 | Safety and Efficacy of Low Dose Aspirin / Ivermectin Combination Therapy for Treatment of Covid‐19 Patients | Acetylsalicylic acid | Standard care | Ivermectin | 490 | Hospital | June 2021 | clinicaltrials.gov/ct2/show/NCT04768179?cond=NCT04768179&draw=1&rank=1 |
| NCT04363840 | The LEAD COVID‐19 Trial: Low‐risk, Early Aspirin and Vitamin D to Reduce COVID‐19 Hospitalizations | Acetylsalicylic acid | Observation | Vitamin D | 1080 | Hospital | December 2020 | clinicaltrials.gov/ct2/show/NCT04363840?cond=04363840&draw=2&rank=1 |
| NCT04445623 | Prasugrel in Severe COVID‐19 Pneumonia | Prasugrel hydrochloride | Placebo | None | 128 | Hospital | January 2021 | clinicaltrials.gov/ct2/show/NCT04445623?cond=NCT%3A+04445623&draw=1&rank=1 |
| NCT04365309 | Protective Effect of Aspirin on COVID‐19 Patients | Acetylsalicylic acid | Standard care | None | 128 | Hospital | June 2020 | clinicaltrials.gov/ct2/show/NCT04365309?cond=NCT%3A+04365309&draw=2&rank=1 |
| NCT05073718 | Acetylsalicylic Acid in COVID‐19 (ASA‐SARS) | Acetylsalicylic acid | Placebo | Prophylactic doses of LMWH | 398 | NR | October 2023 | https://clinicaltrials.gov/ct2/show/NCT05073718 |
| CTRI/2021/03/032059 | Investigator Initiated Study to Evaluate the Effect of Marketed Colchicine 0.5mg Given Along Marketed Asprin 75mg with SOC and Marketed Aspirin 75mg with SOC on COVID 19 patients | Acetylsalicylic acid | Colchicine | None | 63 | Outpatient | NR | www.ctri.nic.in/Clinicaltrials/pdf_generate.php?trialid=53115&EncHid=&modid=&compid=%27,%2753115det%27 |
| NCT04937088 | Outpatient Liquid Aspirin (OLA) (OLA COVID) | Acetylsalicylic acid | Placebo | None | 200 | Outpatient | June 2022 | clinicaltrials.gov/ct2/show/NCT04937088 |
| CTRI/2021/06/034254 | Clinical trial of APMV2020 in Covid 19 Subjects | Acetylsalicylic acid | Multivitamin and multimineral tablet | Promethazine hydrochloride | 60 | Outpatient | NR | www.cochranelibrary.com/central/doi/10.1002/central/CN-02327762/full |
| Sharma 2021 | A Randomized Open‐Label Trial to Evaluate the Efficacy and Safety of Triple Therapy with Aspirin, Atorvastatin, and Nicorandil in Hospitalised Patients with SARS Cov‐2 Infection | Acetylsalicylic acid | Standard care | Atorvastatin, nicorandil | 396 | Hospital | NR | trialsjournal.biomedcentral.com/articles/10.1186/s13063-021-05361-y |
NR: not reported
Included studies
Design and sample size
We included six randomised controlled trials (RCTs) according to our inclusion criteria, involving 21,750 participants (17,541 moderate to severe COVID‐19; 4209 asymptomatic SARS‐CoV‐2 infection or mild COVID‐19:) (Berger 2022; Bohula 2022 (COVID‐PACT); Connors 2021 (ACTIV‐4B); Eikelboom 2022 (ACTCOVID); Horby 2021 (RECOVERY); REMAP‐CAP 2022). All studies were published in journals.
Two of the studies are still recruiting to other drugs (not antiplatelets) as they are adaptive platform trials (Horby 2021 (RECOVERY); REMAP‐CAP 2022), and one study has results but was terminated because the event rates were lower than anticipated (Connors 2021 (ACTIV‐4B)). The estimated study completion dates ranged from December 2022 to December 2023. One study stopped randomisation early due to a decrease in ICU patients with COVID‐19 (Bohula 2022 (COVID‐PACT)).
Berger 2022 is a Bayesian, adaptive randomised clinical trial, with completion planned in November 2023 and an estimated number of 3000 participants. Connors 2021 (ACTIV‐4B) is a multicentre adaptive, randomised, double‐blind, placebo‐controlled platform trial, which was terminated with an actual enrolment of 657 participants, of whom 328 contributed data to our analyses as the remaining participants were randomised to either prophylactic or therapeutic doses of apixaban (an antithrombotic drug not inhibiting platelet aggregation). The completion date of this study was 5 August 2021. Horby 2021 (RECOVERY) is an open‐label, randomised clinical adaptive multicentric trial, currently recruiting as of July 2023 with completion planned in November 2023 with 50,000 participants. Similarly, REMAP‐CAP 2022 is a Bayesian, adaptive, open‐label randomised platform trial evaluating multiple interventions in multiple domains. The estimated completion date is December 2023 with 10,000 participants. Bohula 2022 (COVID‐PACT) is a multicentre, 2 x 2 factorial, open‐label, randomised trial whose results were published in August 2022. This trial randomised 390 participants. Eikelboom 2022 (ACTCOVID) is an open‐label, parallel‐group, factorial, randomised controlled trial with 3917 participants. The estimated completion date of this study is June 2023.
Setting and participants
All studies were multicentre trials. The Berger 2022 study was conducted in different centres in the USA, Brazil, Italy and Spain. Connors 2021 (ACTIV‐4B) took place at different sites in the USA. Horby 2021 (RECOVERY) was conducted at different global sites in the United Kingdom, Indonesia and Nepal. REMAP‐CAP 2022 acquired data in eight countries, including Canada, France, Germany, India, Italy, Nepal, the Netherlands or the United Kingdom. The Eikelboom 2022 (ACTCOVID) trial was conducted at 48 clinical sites in different countries, including Brazil, Canada, Colombia, Ecuador, Egypt, India, Nepal, Pakistan, Philippines, Russian Federation, South Africa and the United Arab Emirates. Bohula 2022 (COVID‐PACT) was also a multicentre trial that took place in 34 centres in the United States.
Except for two studies (Connors 2021 (ACTIV‐4B); Eikelboom 2022 (ACTCOVID)), all studies examined participants with moderate to severe COVID‐19 who were hospitalised.
In all studies, a SARS‐CoV‐2 infection was necessary for inclusion. Connors 2021 (ACTIV‐4B) included symptomatic but clinically stable participants with mild COVID‐19 who were diagnosed with a SARS‐Cov‐2 infection in the last 14 days. Horby 2021 (RECOVERY) included hospitalised participants with suspected or test‐confirmed SARS‐CoV‐2 infection; 97% of the participants in both groups had a positive test, 1% per strata tested negative. Berger 2022 explored hospitalised participants with a laboratory‐confirmed SARS‐CoV‐2 infection without the need for intensive care (non‐critically ill cohort). REMAP‐CAP 2022 included participants who were admitted to hospital with acute illness due to a suspected or proven COVID‐19 infection, and 97% of included participants had a positive PCR result. Bohula 2022 (COVID‐PACT) included participants with an acute SARS‐CoV‐2 infection admitted to an ICU. Eikelboom 2022 (ACTCOVID) included participants with a symptomatic and laboratory‐confirmed diagnosis of COVID‐19 within seven days of diagnosis.
The median age of the participants in Berger 2022 was 52.7 years, and 41.5% were female. Additional reported diagnoses included cardiovascular diseases for 43.7% in the intervention group and 55.8% in the standard care group. Other additional diseases were chronic conditions such as diabetes, chronic kidney diseases, liver diseases or respiratory diseases (asthma or COPD). Previous treatments were largely balanced in both groups. Additional treatments were glucocorticoids (65.5% versus 62.5%), remdesivir (56.0% versus 47.6%), acetylsalicylic acid (15.0% versus 13.4%), anticoagulant therapy (10.6% versus 14.5%) and interleukin‐6 (IL‐6) inhibitors (2.7% versus 3.9%). Concerning respiratory support at randomisation, there were participants without oxygen application (12.2% versus 10.9%), on low flow oxygen (77.8% versus 78.4%) or on high‐flow nasal cannula (0.8% versus 0.4%).
In Connors 2021 (ACTIV‐4B), the median age was 54 years and 59.1% of the participants were female. Additional reported diagnoses were hypertension for 33.5% in the acetylsalicylic acid group and 54% in the placebo group, and diabetes (17.7% versus 14.6%). There were no reported previous treatments at baseline.
In Horby 2021 (RECOVERY), the median age in the intervention group was 59.2 years (62% female) and in the control group 59.3 years (61% female). There were several additional diagnoses reported: 22% of both groups reported diabetes, 11% (intervention group) and 10% (standard care group) had heart diseases, 19% in each group reported chronic lung diseases and 43% of both groups indicated any of the above. 94% of both groups received corticosteroids as a concomitant therapy. The trial included participants with no respiratory support or simple oxygen (67% in both strata), but also on non‐invasive ventilation (28% for both) and invasive mechanical ventilation (5% for both).
In REMAP‐CAP 2022, the median age in the intervention groups (acetylsalicylic acid and P2Y12 inhibitor groups) was 57.0 years, and in the control group it was 57 years. In the intervention groups, 35.2% (acetylsalicylic acid) and 30.5% (P2Y12 inhibitor) were female, and in the control group 34.6% were female. The participants were divided into a critically ill group and a non‐critically ill group. Additional diagnoses were diabetes (21.9%), respiratory diseases (18.8%), kidney diseases (3.5%), severe cardiovascular diseases (4.0%) and any immunosuppressive condition (4.0%). Previous treatments within 48 hours of recruitment that were reported were steroids (97.0%), remdesivir (21.9%), tocilizumab (38.7%), sarilumab (9.3%), low‐molecular‐weight heparin or unfractionated heparin in different doses or direct oral anticoagulants. This study reported two groups of participants separately: the severely ill population, with around 36% of participants invasively mechanically ventilated (IMV), and the remaining participants, mostly on noninvasive ventilation (NIV) or high‐flow nasal cannula (HFNC). In the moderately ill population most participants needed no respiratory support or only oxygen (87%) and only a small number of participants needed NIV, HFNC or IMV at baseline.
In Bohula 2022 (COVID‐PACT), the median age in the intervention group (IG) was 59 years (38 % female) and 62 years in the control group (CG) (43% female). There were several additional diagnosis reported: hypertension (56% IG versus 62% CG), diabetes (38% IG versus 26% CG), atherosclerotic cardiovascular disease (15% IG versus 13% CG), active cancer (5.2% IG versus 3.7% CG), chronic kidney disease (11% IG versus 11% CG) and pulmonary disease (22% IG versus 19% CG). All participants in both groups needed some form of oxygen therapy: 1.6% versus 0.5% needed oxygen by mask or nasal canula, 79% versus 88% needed NIV or HFNC, and 19% versus 12% needed invasive ventilation.
In Eikelboom 2022 (ACTCOVID), the median age in the intervention group was 45.2 years (38.6% female) and in the control group it was 44.8 years (40.3% female). They reported five additional diagnoses: diabetes (12.7% versus 14.1%), hypertension (22.6% versus 21.5%), dyslipidaemia (8.9% versus 8.1%), cardiovascular disease (5.1% versus 4.5%) and chronic lung disease (7.1% versus 8.3%). There were no reported previous treatments at baseline.
Interventions and comparators
In Berger 2022, the intervention group received a therapeutic dose of anticoagulants and P2Y12 inhibitor (heparin was standard care with an added P2Y12 inhibitor). 63.2% of the included participants received ticagrelor and 36.8% received clopidogrel for 14 days or until hospital discharge, whichever occurred first (Berger 2022). The control group of this study received a therapeutic dose of anticoagulation for 14 days or until hospital discharge (whichever occurred first).
Two RCTs compared acetylsalicylic acid to standard care with placebo (Connors 2021 (ACTIV‐4B)) or without placebo (Horby 2021 (RECOVERY)).
Connors 2021 (ACTIV‐4B) investigated the effect of acetylsalicylic acid compared with a matching placebo once a day. The application form was oral. The day's dosage was 81 mg for 45 days. In this trial, two additional intervention groups were treated with either prophylactic or therapeutic doses of anticoagulants (apixaban twice daily 2.5 or 5 mg), but those groups did not meet our inclusion criteria and data were therefore not extracted. We found no information about additional anticoagulant medication in the groups included in our meta‐analysis.
In Horby 2021 (RECOVERY), 150 mg acetylsalicylic acid once daily until discharge was compared to standard care, which was defined as usual hospital care. Only 6587 participants (90%) in the intervention group received at least one dose of acetylsalicylic acid and 210 participants (3%) in the control group received at least one dose of acetylsalicylic acid (Horby 2021 (RECOVERY)), resulting in unplanned treatment switching.
In REMAP‐CAP 2022, the control group received standard care, possibly with other interventions of the trial. The intervention group was divided into two arms (acetylsalicylic acid or a P2Y12 inhibitor). Participants in the acetylsalicylic acid arm received 75 mg to 100 mg once daily. The participants in the P2Y12 inhibitor arm received clopidogrel 75 mg once daily without a loading dose, prasugrel (60 mg loading dose followed by 10 mg daily (if aged < 75 years and weight ≥ 60 kg) or 5 mg daily (if aged ≥ 75 years or weight < 60 kg)), ticagrelor 60 mg twice daily without a loading dose (REMAP‐CAP 2022).
In Bohula 2022 (COVID‐PACT), the control group received standard care plus prophylactic or therapeutic anticoagulation (enoxaparin 40 mg subcutaneously once daily, unfractionated heparin 5000 IU subcutaneously three times daily, unfractionated heparin intravenously continuous targeting the activated partial thromboplastin time (aPTT) at a level that is 1.5 to 2.5 times longer than the control value, or enoxaparin 1 mg/kg subcutaneously every 12 hours). The intervention group received antiplatelet therapy with clopidogrel plus standard care and prophylactic anticoagulation or therapeutic anticoagulation: the intervention group with prophylactic anticoagulation received clopidogrel 300 mg orally once, followed by clopidogrel 75 mg orally once daily plus enoxaparin 40 mg subcutaneously once daily or unfractionated heparin 5000 IU subcutaneously three times daily. Participants with antiplatelet therapy plus full‐dose anticoagulation received clopidogrel 300 mg orally once, followed by clopidogrel 75 mg orally once daily plus unfractionated heparin intravenously continuous targeting the activated partial thromboplastin time (aPTT) at a level that is 1.5 to 2.5 times longer than the control value, or enoxaparin 1 mg/kg subcutaneously every 12 hours.
In Eikelboom 2022 (ACTCOVID), the control group received standard care or standard care plus additionally (in a first randomisation) colchicine 0.6 mg twice daily for three days and then 0.6 mg once daily for 25 days. The intervention group received oral acetylsalicylic acid 100 mg once daily as a tablet for 28 days, or oral acetylsalicylic acid 100 mg once daily as a tablet for 28 days plus (in a first randomisation) colchicine 0.6 mg twice daily for 3 days and then 0.6 mg once daily for 25 days.
Outcomes
Three trials reported 28‐day mortality (Berger 2022; Horby 2021 (RECOVERY); REMAP‐CAP 2022); REMAP‐CAP 2022 also reported 90‐day mortality and 180‐day mortality. Bohula 2022 (COVID‐PACT) reported all‐cause mortality without concrete description of the time frame of follow‐up, but probably up to day 21.
Two studies reported all‐cause mortality up to longest follow‐up (45 days) for non‐hospitalised participants with mild COVID‐19 (Connors 2021 (ACTIV‐4B); Eikelboom 2022 (ACTCOVID)).
The worsening of clinical status up to day 28, defined as new need for invasive mechanical ventilation or death, was reported for the hospitalised participants in Horby 2021 (RECOVERY) and REMAP‐CAP 2022.
The improvement of clinical status up to day 28, defined as participants discharged alive from hospital, was reported in Horby 2021 (RECOVERY) and at day 21 in the Berger 2022 trial.
Thrombotic events up to the longest follow‐up were reported in four trials for participants with moderate to severe COVID‐19 (Berger 2022; Bohula 2022 (COVID‐PACT); Horby 2021 (RECOVERY); REMAP‐CAP 2022) and by two studies for participants with mild COVID‐19 who were not hospitalised (Connors 2021 (ACTIV‐4B); Eikelboom 2022 (ACTCOVID)). The time frames for the longest follow‐up in participants with moderate to severe COVID‐19 were 28 days (Berger 2022; Bohula 2022 (COVID‐PACT); Horby 2021 (RECOVERY)) or unclear (REMAP‐CAP 2022). In participants with mild COVID‐19, Connors 2021 (ACTIV‐4B) reported this outcome at 45 days (and added 30‐day safety follow‐up to the 45 days) and Eikelboom 2022 (ACTCOVID) reported thrombotic events that had occurred up to day 45.
The safety outcomes major bleeding and serious adverse events during study treatment were reported for up to 28 days for the hospitalised participants in three studies (Berger 2022; Horby 2021 (RECOVERY); REMAP‐CAP 2022), although the longest follow‐up of REMAP‐CAP 2022 for major bleeding was 14 days. These outcomes were reported in the outpatient setting by Connors 2021 (ACTIV‐4B) and Eikelboom 2022 (ACTCOVID) for up to 45 days (again, Connors 2021 (ACTIV‐4B) reported data after the additional 30‐day safety follow‐up). Admission to hospital or death was reported for the outpatient setting in Connors 2021 (ACTIV‐4B) and Eikelboom 2022 (ACTCOVID) up to day 45.
The need for new dialysis was reported in Horby 2021 (RECOVERY) at day 28 for non‐hospitalised participants.
REMAP‐CAP 2022 reported quality of life of participants at day 180, including use of the EQ‐5D‐5L (EuroQol‐5 dimensions ‐5 levels questionnaire), which is a preference‐based health‐related quality of life (HRQoL) instrument comprising five dimensions: mobility, self‐care, usual activities, pain/discomfort and anxiety/depression. The EQ‐5D‐5L utility score is calculated from the individual response to each item and ranges from −0.593 (score of 0 is equivalent to death) to 1.00 (full health), with higher values indicating better health states. In addition, respondents were asked to indicate their present health state on a visual analogue scale (EQ VAS) ranging from the worst imaginable health state (0) to the best imaginable health state (100).
Ongoing studies
We identified 14 ongoing RCTs (ChiCTR2000030055; CTRI/2020/08/027503; CTRI/2021/03/032059; CTRI/2021/06/034254; IRCT20180205038626N7; NCT04363840; NCT04365309; NCT04445623; NCT04703608; NCT04768179; NCT04808895; NCT04937088; NCT05073718; Sharma 2021). The majority of studies are investigating acetylsalicylic acid. One study is investigating other antiplatelets, namely prasugrel hydrochloride (NCT04445623).
The 14 ongoing RCTs plan to recruit between 36 and 1200 participants. According to the trial registries, two studies were due to complete in 2020, three studies aimed to complete in 2021, two studies were to have finished in 2022, and one is due to complete during 2023. Six other studies did not give information on a planned completion date.
Four of the ongoing trials are still recruiting (Bohula 2022 (COVID‐PACT); IRCT20180205038626N7; NCT04703608; NCT04483960). Six have not recruited yet (CTRI/2020/08/027503; CTRI/2021/03/032059; NCT04445623; NCT04768179; NCT04808895; NCT05073718), and one study is enrolling by invitation (NCT04365309).
Please refer to Table 3 for further details on the planned completion dates and planned number of participants per study.
Studies awaiting assessment
We classified three studies as awaiting assessment. One was published, but later withdrawn from the preprint server it was originally published on (RESIST 2021). Two studies are listed as having completed according to the trial registries, but no results have been published yet (NCT04391179; NCT04659109). If eligible, they will be considered in the next review update.
Excluded studies
We excluded two studies at full‐text screening as these studies did not evaluate antiplatelets (NCT04483960; Eikelboom 2022 (ACTCOVID19 hospitalised)).
Risk of bias in included studies
Using the risk of bias 2 (RoB 2) tool recommended in Chapter 8 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2021a), we assessed the methodological quality and risk of bias for the outcomes of the results of six RCTs (Berger 2022; Bohula 2022 (COVID‐PACT); Connors 2021 (ACTIV‐4B); Eikelboom 2022 (ACTCOVID); Horby 2021 (RECOVERY); REMAP‐CAP 2022). The RoB 2 table is accessible at zenodo.org/record/7637454.
Overall judgement in studies including individuals with a confirmed diagnosis of COVID‐19 and moderate to severe disease
For Horby 2021 (RECOVERY), we rated the risk of bias for the outcome '28‐day mortality' for hospitalised participants to be high due to deviations from the intended intervention. In the intervention group of this study, 10% of participants never obtained the treatment and 31% did not receive it consistently. In the corresponding control group, 210 (3%) participants received treatment with acetylsalicylic acid. Reasons for deviation from the intended intervention were not given. This was also the reason to rate high risk for 'improvement up to day 28: discharged alive', and 'need for new dialysis up to day 28'. Concerning the outcomes 'major bleeding up to longest follow‐up' and 'thrombotic events up to longest follow‐up', we additionally had some concerns regarding bias in the measurement of those events, since the study was unblinded and the overall bias for those endpoints was rated high risk due to deviations from intended interventions.
Assessing Bohula 2022 (COVID‐PACT), we rated the risk of bias to be high due to imbalanced high rates of cross‐overs (deviations from intended interventions) for the outcomes 'major bleeding up to longest follow‐up', 'thrombotic events up to longest follow‐up' and 'all‐cause mortality up to the longest follow‐up'. Additionally, the planned duration of treatment was not specified nor given in the study/registry/publication, so we were not able to judge whether there were deviations from intended interventions. We also identified unbalanced co‐interventions and unclear time frames of the treatments and follow‐up.
In Berger 2022, we rated the risk of bias to be low for the outcomes 'major bleeding up to longest follow‐up', 'any thrombotic event up to longest follow‐up', '28‐day mortality 'and 'improvement up to day 28: discharged alive' for hospitalised participants, since those outcomes were assessed according to the protocol and the allocation was randomised. The trial was an open‐label design.
For REMAP‐CAP 2022, we rated the risk of bias for the outcome 28‐day mortality for hospitalised participants to be 'some concerns' due to deviations from the intended intervention and a bias in the measurement of the outcome. The deviations from intended interventions were also the reason why we rated the risk of bias to be 'some concerns' for the outcomes 'any thrombotic event', 'major bleeding up to longest follow‐up' and 'serious adverse events up to longest follow‐up' in this study. Regarding the outcome 'worsening up to day 28: participants with new need for invasive mechanical ventilation or death', we rated the risk of bias to be high in this study. The reasons for this assessment were two‐fold: firstly due to missing outcome data, as we found that outcome reported for only 60.5% (1104 out of 1824) of the randomised participants. Secondly, some concerns arose due to deviations from intended interventions (missing information whether there were cross‐overs or whether the assigned medication was really taken). The study undertook a follow‐up after 180 days for the outcome '180‐day mortality', and we assessed there to be some concerns about bias because there was no information about whether there were cross‐overs or whether the assigned medication was really taken. Additionally, the design was unblinded and there were missing outcome data (only some of the study centres reported this outcome).
Overall judgements in studies including individuals with a confirmed diagnosis of COVID‐19 and asymptomatic or mild disease
Concerning Connors 2021 (ACTIV‐4B), we rated the risk of bias to be low for the outcome 'all‐cause mortality up to day 45' since this outcome was assessed according to the protocol, the allocation was randomised and the trial was double‐blind. These reasons were also applied for the endpoints 'admission to hospital or death up to day 45', 'major bleeding up to longest follow‐up' and 'thrombotic events up to longest follow‐up', where we detected no risk of bias using the RoB 2 tool.
In Eikelboom 2022 (ACTCOVID), we rated the risk of bias for the outcomes 'thrombotic events up to longest follow‐up', 'admission to hospital or death up to day 45', 'all cause mortality up to day 45' and 'serious adverse events up to longest follow‐up' as some concerns because 7.9% of the participants discontinued the intervention. It is not clear whether this happened due to the trial context or how this could affect the outcome.
Effects of interventions
Individuals with a confirmed diagnosis of COVID‐19 and moderate to severe disease
We present our certainty in the evidence for prioritised outcomes for antiplatelets (plus standard care) compared to standard care (with/without placebo) in hospitalised individuals with COVID‐19 (moderate to severe disease) in Table 1.
Prioritised outcomes
All‐cause mortality up to day 28, day 60, time‐to‐event and up to longest follow‐up
We identified three studies reporting this outcome for 17,249 hospitalised participants (Berger 2022; Horby 2021 (RECOVERY); REMAP‐CAP 2022): 168 of 1000 participants treated with antiplatelets died within 28 days. Antiplatelets probably result in little to no difference in 28‐day mortality (RR 0.95, 95% CI 0.85 to 1.05; risk difference (RD) 9 fewer per 1000, 95% CI from 26 fewer to 9 more; I2 = 26%; moderate‐certainty evidence; Analysis 1.1). Our main reason to downgrade was serious risk of bias due to deviations from intended interventions.
1.1. Analysis.

Comparison 1: Individuals with a confirmed diagnosis of COVID‐19 and moderate to severe disease, Outcome 1: All‐cause mortality up to day 28
REMAP‐CAP 2022 reported 180‐day mortality at longest follow‐up in an extra publication (REMAP‐CAP 2023). Therapy with antiplatelets led to an adjusted hazard ratio of 0.85 (credibility interval 0.71 to 1.03) compared with standard care, reported from the fully adjusted Bayesian model (adjusted for other treatments from other domains, site, time, sex and age). Antiplatelets may result in little to no difference in 180‐day mortality (RR 0.97, 95% CI 0.82 to 1.15; 1,312 participants; RD 10 fewer per 1000, 95% CI from 58 fewer to 49 more; I2 = not applicable; low‐certainty evidence; Analysis 1.8). Upon personal request to the authors of the study, it was impossible to obtain the frequentist confidence intervals of the adjusted hazard ratio. As there was only one study reporting this outcome and due to risk of bias, we rated the evidence as low certainty for this outcome.
1.8. Analysis.

Comparison 1: Individuals with a confirmed diagnosis of COVID‐19 and moderate to severe disease, Outcome 8: 180‐day mortality
Clinical status at day 28 and up to longest follow‐up
Worsening of clinical status up to day 28: participants with clinical deterioration (new need for invasive mechanical ventilation) or death
We identified two studies with 15,266 participants reporting this outcome (Horby 2021 (RECOVERY); REMAP‐CAP 2022). Considering the reported event rates across participants, we estimated that 218 of 1000 participants experience this outcome up to 28 days when treated with antiplatelets or standard care alone. Antiplatelets probably result in little to no difference in worsening up to day 28 (RR 0.95, 95% CI 0.90 to 1.01; RD 12 fewer per 1000, 95% CI 29 fewer to 30 more; I2 = 0%; moderate‐certainty evidence; Analysis 1.2). Our main reason to downgrade was serious risk of bias due to deviations from intended interventions. This outcome was not reported for other time points.
1.2. Analysis.

Comparison 1: Individuals with a confirmed diagnosis of COVID‐19 and moderate to severe disease, Outcome 2: Worsening up to day 28: participants with new need for invasive mechanical ventilation or death
Improvement of clinical status up to day 28: participants discharged alive
We identified two studies with 15,454 participants reporting this outcome (Horby 2021 (RECOVERY); REMAP‐CAP 2022). Considering the reported event rates across participants, we estimated that 743 of 1000 participants experience this outcome at up to 28 days when treated with antiplatelets. Antiplatelets probably result in little to no difference in improvement (discharged alive) up to day 28 (RR 1.00, 95% CI 0.96 to 1.04; RD 0 participants per 1000, 95% CI 29 fewer to 30 more; I2 = 71%; moderate‐certainty evidence; Analysis 1.3). Our main reason to downgrade was serious risk of bias due to deviations from intended interventions. This outcome was not reported for other time points.
1.3. Analysis.

Comparison 1: Individuals with a confirmed diagnosis of COVID‐19 and moderate to severe disease, Outcome 3: Improvement up to day 28: discharged alive
Thrombotic events up to longest follow‐up
All included studies reported thrombotic events up to longest follow‐up for 17,518 participants (Berger 2022; Horby 2021 (RECOVERY); REMAP‐CAP 2022). Fifty‐two per 1000 participants assigned to be treated with antiplatelets experienced a thrombotic event (such as pulmonary embolism, myocardial infarction, ischaemic cerebrovascular event, systemic arterial thromboembolism and deep venous thrombosis). Antiplatelets probably result in a slight reduction of thrombotic events (RR 0.90, 95% CI 0.80 to 1.02; RD 5 fewer per 1000, 95% CI 11 fewer to 1 more; I2 = 0%; moderate‐certainty evidence; Analysis 1.4). Our main reason to downgrade was serious risk of bias due to deviations from intended interventions.
1.4. Analysis.

Comparison 1: Individuals with a confirmed diagnosis of COVID‐19 and moderate to severe disease, Outcome 4: Thrombotic events
Safety of antiplatelets up to longest follow‐up
Serious adverse events up to longest follow‐up
One study reported this outcome for 1815 participants (REMAP‐CAP 2022). Considering the reported event rates across participants, we estimated that 7 of 1000 participants experience a serious adverse event when treated with antiplatelets. Antiplatelets may result in a slight increase in serious adverse events (Peto OR 1.57, 95% CI 0.48 to 5.14; RD 2 more per 1000, 95% CI 3 fewer to 19 more; I2 not applicable; low‐certainty evidence; Analysis 1.5). Our main reason to downgrade was very serious imprecision: only one out of three studies contributed data, with very low event rates and a very wide confidence interval.
1.5. Analysis.

Comparison 1: Individuals with a confirmed diagnosis of COVID‐19 and moderate to severe disease, Outcome 5: Serious adverse events
Adverse events during study treatment
None of the included studies reported non‐serious adverse events according to our definition (i.e. adverse events that occurred during the study period, defined as number of participants with any event). Horby 2021 (RECOVERY) only reported suspected unexpected serious adverse reactions (SUSAR), but other serious or non‐serious adverse events were not recorded (only information about use of renal dialysis, documented new major cardiac arrhythmia, major bleeding, thrombotic events or non‐coronavirus infections).
Major bleeding up to longest follow‐up
All included studies reported major bleeding, for a total of 17,527 participants (Berger 2022; Bohula 2022 (COVID‐PACT); Horby 2021 (RECOVERY); REMAP‐CAP 2022). In the safety population, 16 of 1000 participants treated with antiplatelets had a major bleeding event. Antiplatelets probably increase the occurrence of major bleeding events (Peto OR 1.68, 95% CI 1.29 to 2.19; RD 6 more per 1000, 95 % CI 2 more to 11 more; I2 = 11%; moderate‐certainty evidence; Analysis 1.6). Our main reason to downgrade was serious risk of bias due to deviations from intended interventions.
1.6. Analysis.

Comparison 1: Individuals with a confirmed diagnosis of COVID‐19 and moderate to severe disease, Outcome 6: Major bleeding events
Additional outcomes
Quality of life
This outcome was reported for day 180 for a very small subgroup of participants (N = 84 acetylsalicylic acid, N = 83 clopidogrel, N = 96 placebo) in one study (REMAP‐CAP 2022). Results are therefore very uncertain.
Using the EQ‐5D‐5L utility score, antiplatelet treatment lead to a mean (SD) of 0.69 (0.27) (N = 163) versus 0.63 (0.30) (N = 95) in the control group (scale from 0 to 1, higher values represent better outcomes). Single domains of the questionnaire were presented separately for acetylsalicylic acid, P2Y12 inhibitors (clopidogrel) and control.
The study also reported the EQ VAS for day 180. Treatment with acetylsalicylic acid resulted in a mean (SD) score of 65.7 (22.3). The score for treatment with P2Y12 inhibitors was 71.3 (20.1), and the score for standard care was 66.1 (21.0) (scale from 0 to 100, higher values represent better outcomes).
Duration of hospitalisation, or time to discharge from hospital
Horby 2021 (RECOVERY) reported the median time to hospital discharge up to day 28, with the median being eight days in the acetylsalicylic acid group and nine days in the standard care group (each IQR 5 to > 28 days).
Need for new dialysis (up to 28 days)
We identified one study with participants randomised to either acetylsalicylic acid or standard care reporting the new need for dialysis up to day 28 in 14,771 participants (Horby 2021 (RECOVERY)). Considering the reported event rates across participants, we estimated that 37 of 1000 participants experience this outcome at up to 28 days when treated with platelet inhibitors. Treatment with antiplatelets resulted in no difference in the need for dialysis at up to day 28 (Peto OR 0.99, 95% CI 0.84 to 1.18; RD 1 fewer per 1000, 95% CI 6 fewer to 6 more; Analysis 1.7).
1.7. Analysis.

Comparison 1: Individuals with a confirmed diagnosis of COVID‐19 and moderate to severe disease, Outcome 7: Need for new dialysis up to day 28
Other planned outcomes were not reported: admission to the ICU, ventilator‐free days (ventilator‐free defined as WHO Clinical Progression Scale ≤ 6 for the subgroup of participants requiring invasive mechanical ventilation at baseline, i.e WHO Clinical Progression Scale ≥ 7).
Subgroup analyses
We performed a subgroup analysis for the prioritised outcome 28‐day mortality (Analysis 2.1), stratified by the type of antiplatelet agent that was used (cyclooxygenase inhibitor, i.e. acetylsalicylic acid; or P2Y12 inhibitors, i.e. either clopidogrel, ticagrelor or prasugrel). Of our included studies conducted amongst participants with moderate to severe COVID‐19, Horby 2021 (RECOVERY) and REMAP‐CAP 2022 used a cyclooxygenase inhibitor. Berger 2022 and the multi‐armed trial REMAP‐CAP 2022 compared P2Y12 inhibitors to standard care. Overall, there was no evidence for subgroup differences for the outcome all‐cause mortality up to day 28 (P = 0.82).
2.1. Analysis.

Comparison 2: Subgroup analysis: Effects on mortality up to day 28, Outcome 1: Subgroup analysis (type of antiplatelet agent): cyclooxygenase inhibitors or ADP receptor/P2Y12 inhibitors in participants with moderate to severe COVID‐19
We performed a subgroup analysis related to standard care or standard care with therapeutic anticoagulation. Berger 2022 treated participants with therapeutic dose heparin. Horby 2021 (RECOVERY) medicated participants in the reported subgroup with lower/higher dose low molecular weight heparins as thromboprophylaxis. We found data for the critically ill participants in REMAP‐CAP 2022 who were either treated with low/intermediate doses of heparin or who got subtherapeutic/therapeutic doses of heparin. The study did not report those subgroups for the moderately ill participants. We did not identify subgroup differences for the outcome all‐cause mortality at up to day 28 (P = 0.12) (Analysis 2.2).
2.2. Analysis.

Comparison 2: Subgroup analysis: Effects on mortality up to day 28, Outcome 2: Subgroup analysis: concomitant therapeutic dose anticoagulation or prophylactic anticoagulation
Sensitivity analyses
Please find our sensitivity analyses in Table 4. We performed the following analyses.
2. Sensitivity analysis for the primary outcome all‐cause mortality up to day 28 for people with moderate to severe COVID‐19.
| Outcome | Main analysis using random‐effects model | Main analysis using fixed‐effect model | Risk of bias (excluding studiesa at high risk of bias) |
| All‐cause mortality up to day 28 | RR 0.95, 95% CI 0.85 to 1.05; 3 RCTs, 17,249 participants | RR 0.96, 95% CI 0.90 to 1.02; 3 RCTs, 17,249 participants | RR 1.02, 95% CI 0.63 to 1.62; 2 RCTs, 2357 participants |
CI: confidence interval; RCT: randomised controlled trial; RR: risk ratio
aExcluded study with high risk of bias: Horby 2021 (RECOVERY)
Comparison of the fixed‐effect versus random‐effects statistical model. Performing a sensitivity analysis of the fixed‐effect model did not make any relevant change to the effect.
Comparison of studies with a low risk of bias or some concerns versus studies with a high risk of bias. When excluding Horby 2021 (RECOVERY) due to a high risk of bias, the direction of the effect changed, but without relevant impact on the overall little to no difference on all‐cause mortality up to day 28 (RR 1.02, 95% CI 0.63 to 1.62, 2357 participants).
Comparison of preprints versus peer‐reviewed articles. For the outcome 28‐day mortality, we found only a preprint that was withdrawn later (RESIST 2021); therefore, we did not conduct this sensitivity analysis.
Comparison of premature termination of studies with completed studies. As we did not identify any prematurely ended studies, we did not perform this sensitivity analysis.
Individuals with a confirmed diagnosis of SARS‐CoV‐2 infection and asymptomatic or mild disease
We present our certainty in the evidence for prioritised outcomes for antiplatelets in non‐hospitalised individuals with COVID‐19 (asymptomatic and mild disease) in Table 2.
We found no study including participants with asymptomatic SARS‐CoV‐2 infection. Two included studies considered people with mild disease (Connors 2021 (ACTIV‐4B); Eikelboom 2022 (ACTCOVID)).
Prioritised outcomes
All‐cause mortality at day 28, day 60, time‐to‐event and up to longest follow‐up
We identified two studies with 4209 participants randomised to either acetylsalicylic acid or placebo reporting this outcome (Connors 2021 (ACTIV‐4B); Eikelboom 2022 (ACTCOVID)). Considering the reported event rates across participants, we estimated that 6 of 1000 participants die at up to 45 days when treated with placebo or standard care alone. Antiplatelets may result in little to no difference in all‐cause mortality up to day 45 (Peto OR 1.00, 95% CI 0.45 to 2.22; RD 0 fewer per 1000, 95% CI 3 fewer to 7 more; 2 studies; 4209 participants; low‐certainty evidence; Analysis 3.1). The main reason to downgrade was very serious imprecision (low number of events and wide confidence interval).
3.1. Analysis.

Comparison 3: Individuals with a confirmed diagnosis of SARS‐CoV‐2 infection and asymptomatic to mild disease, Outcome 1: All‐cause mortality up to longest follow‐up (45 days)
Admission to hospital or death at longest follow‐up
We identified two studies with 4209 participants randomised to either acetylsalicylic acid or placebo reporting this outcome (Connors 2021 (ACTIV‐4B); Eikelboom 2022 (ACTCOVID)). Considering the reported event rates across participants, we estimated that 31 of 1000 participants had to be hospitalised or died at up to 45 days when treated with placebo or standard care alone. The evidence is very uncertain about the effects of antiplatelets on admission to hospital (for cardiovascular reasons) or death up to day 45 (Peto OR 0.79, 95% CI 0.57 to 1.10; RD 8 fewer per 1000, 95% CI 16 fewer to 4 more; 2 studies, 4209 participants; very low‐certainty evidence; Analysis 3.2). The main reasons to downgrade were very serious imprecision (low number of events and wide confidence interval) and serious indirectness (one study only reported death due to a cardiovascular event, the other study included major thrombosis in this outcome's definition).
3.2. Analysis.

Comparison 3: Individuals with a confirmed diagnosis of SARS‐CoV‐2 infection and asymptomatic to mild disease, Outcome 2: Admission to hospital or death up to day 45
Symptom resolution
We found no studies that reported symptom resolution outcomes (all initial symptoms resolved (asymptomatic) at day 14, day 28 and up to longest follow‐up or duration to symptom resolution).
Quality of life
This outcome was not reported.
Any thrombotic event up to longest follow‐up
We identified two studies with 4209 participants randomised to either acetylsalicylic acid or placebo reporting this outcome (Connors 2021 (ACTIV‐4B); Eikelboom 2022 (ACTCOVID)). Considering the reported event rates across participants, we estimated that 1 of 1000 participants experience a thrombotic event at up to 45 days when treated with placebo or standard care alone. Antiplatelets may slightly decrease the incidence of new thrombotic events (Peto OR 0.37, 95% CI 0.09 to 1.46; RD 2 fewer per 1000, 95% CI 3 fewer to 1 more; 2 studies, 4209 participants; low‐certainty evidence; Analysis 3.3). The main reason to downgrade was very serious imprecision (low number of events and wide confidence interval).
3.3. Analysis.

Comparison 3: Individuals with a confirmed diagnosis of SARS‐CoV‐2 infection and asymptomatic to mild disease, Outcome 3: Thrombotic events
Safety of antiplatelets up to longest follow‐up
Serious adverse events up to longest follow‐up
We identified one study with 3881 participants randomised to either acetylsalicylic acid or placebo reporting this outcome (Eikelboom 2022 (ACTCOVID)). Considering the reported event rates across participants, we estimated that 16 of 1000 participants experience serious adverse events when treated with antiplatelets. Antiplatelets may make little to no difference on the incidence of serious adverse events (Peto OR 1.00, 95% CI 0.60 to 1.64; RD 0 fewer per 1000, 95% CI 6 fewer to 10 more; 1 study; 3881 participants; low‐certainty evidence; see Analysis 3.4). The main reason to downgrade was very serious imprecision (low number of events and wide confidence interval).
3.4. Analysis.

Comparison 3: Individuals with a confirmed diagnosis of SARS‐CoV‐2 infection and asymptomatic to mild disease, Outcome 4: Serious adverse events
Adverse events during study treatment
None of the included studies reported non‐serious adverse events according to our definition in participants with mild COVID‐19 (i.e. adverse events that occurred during the study period, defined as number of participants with any event).
Major bleeding up to longest follow‐up
We identified one study with 328 participants reporting this outcome (Connors 2021 (ACTIV‐4B)). The evidence is very uncertain about the effects of antiplatelets on the incidence of new major bleeding events, as there were no events in 164 participants treated with acetylsalicylic acid or 164 participants treated with placebo. The evidence is very uncertain about the effects of antiplatelets on major bleeding up to day 45 (RR not applicable; 1 study, 328 participants; very low‐certainty evidence). We downgraded three levels due to extremely serious imprecision: low sample size, low number of events, only one small RCT reporting this outcome.
Additional outcomes
The included studies did not report on additional outcomes, such as time to symptom onset, length of hospital stay, time to discharge (for any subgroup of participants hospitalised during course of disease) and admission to the ICU.
Discussion
Summary of main results
We included six studies in this review, of which four reported the effects of antiplatelets plus standard care compared to standard care alone (with/without placebo) in 17,541 hospitalised participants with moderate to severe COVID‐19 (see Table 1), and two studies reported effects in 4209 non‐hospitalised participants with mild COVID‐19 (see Table 2).
Moderate‐certainty evidence suggests that antiplatelets probably make little to no difference to all‐cause mortality up to 28 days, or to either worsening or improvement of clinical status (up to day 28), but probably result in a slight reduction of thrombotic events up to the longest follow‐up in participants with moderate or severe COVID‐19. The time frames for 'the longest follow‐up' were 28 days (Berger 2022; Bohula 2022 (COVID‐PACT); Horby 2021 (RECOVERY)) or unclear (REMAP‐CAP 2022) in participants with moderate to severe COVID‐19. In participants with mild COVID‐19, Connors 2021 (ACTIV‐4B) reported this outcome at 45 days (and added 30‐day safety follow‐up to the 45 days), and Eikelboom 2022 (ACTCOVID) reported thrombotic events that had occurred up to day 45. There is low‐certainty evidence that antiplatelets may result in little to no difference in 180‐day mortality. They may result in a slight increase in serious adverse events up to the longest follow‐up and probably slightly increase the occurrence of major bleeding events up to the longest follow‐up. Adverse events were not reported.
In participants with mild COVID‐19, we found low‐certainty evidence that antiplatelets may result in little to no difference in 45‐day mortality and serious adverse events, but may slightly decrease the incidence of new thrombotic events. The effects on the combined outcome 'admission to hospital or death up to day 45' and on major bleeding events are very uncertain. Quality of life and non‐serious adverse events were not reported.
Overall completeness and applicability of evidence
Overall completeness
Overall, this review identified a lack of evidence about the effect of antiplatelets for the following outcomes and groups.
Adverse events and serious adverse events related to antiplatelets (only two of six studies reported serious adverse events and none reported adverse events according to standard definition).
People with asymptomatic SARS‐CoV‐2 infection or mild COVID‐19 (only two studies contributed data for people with mild COVID‐19, with very low event rates for all reported outcomes).
Quality of life among people with any severity of the disease.
We identified 14 ongoing RCTs and three studies awaiting classification that compare antiplatelets to placebo, standard care alone or other treatments. We aim to include these in updates of this review.
Applicability of evidence
We identified the following issues that may reduce the applicability of the evidence.
A small portion of included participants had negative/unknown test results (in Horby 2021 (RECOVERY); REMAP‐CAP 2022 only 97% of participants had a positive test result).
The impact of vaccination status on the different outcomes was not accessible, because the outcomes were not separately reported for the subgroup of vaccinated participants; the same was the case for subgroups stratified by sex and age (there were only some age subgroups reported by Horby 2021 (RECOVERY)).
The studies took place in high‐ to moderate‐income countries; no low‐income countries contributed to the evidence we found. The six included studies were conducted in 12 different countries, mainly from high‐income countries (Canada, France, Germany, Italy, the Netherlands, Spain, the UK, the USA) and upper‐middle‐income countries (Brazil), but also from lower‐middle‐income countries (India, Indonesia, Nepal), see World Bank 2022.
The applicability of the Connors 2021 (ACTIV‐4B) study is limited: "median time from diagnosis to randomization was 7 days (IQR, 3‐10 days), and the median time from randomization to initiation of study treatment was 3 days (IQR, 2‐5 days)" (Connors 2021 (ACTIV‐4B)), so probably those participants with a more severe course of the disease would have already been admitted to hospital before randomisation took place (i.e. the sickest people were already in hospital so were not included in the randomisation). Moreover, the study reported only the admission to hospital "due to cardiovascular events" and not due to all causes.
Critical appraisal of selected outcomes (thrombotic event, major bleeding, serious adverse events) considering death as a competing event: as most of the studies reported the outcome itself and a combination of the outcome with death (such as 'thrombotic event or death'), we discussed the option to report those combined endpoints to adjust better for the competing risk of death (i.e. in people who died the event could still have occurred, so the deaths could disguise the true value). However, because the event rates were rather low compared to mortality event rates (which were often a multiple of the outcome's event rates), we judged that the combined outcomes would not be representative of the effect of interest. Finally, it seemed more appropriate to us to report those outcomes as separate events, but caution must be taken when interpreting those effect estimates.
Potentially decreased applicability to Omicron, the variant most prevalent later in the pandemic (studies were conducted between September 2020 and June 2021; the Omicron variant was first described in November 2021 (WHO 2021c)).
Quality of the evidence
Antiplatelets plus standard care versus standard care for the treatment of people with moderate to severe COVID‐19
We have moderate certainty in the evidence for: 28‐day all‐cause mortality, worsening or improvement of clinical status, thrombotic events and major bleeding events; and we have low certainty in the identified evidence for safety outcomes (serious adverse events) and 180‐day mortality. Our main reason for downgrading certainty for effectiveness outcomes was serious study limitations due to deviations from intended interventions, especially in unblinded studies. An additional concern related to safety outcomes was very serious imprecision, as the largest study did not report safety data and the only study contributing data had very low event rates and a very wide confidence interval.
Antiplatelets plus standard care versus standard care (plus placebo) for the treatment of people with asymptomatic SARS‐CoV‐2 infection or mild COVID‐19
We have low certainty in the identified evidence for the outcomes all‐cause mortality (at day 45), thrombotic events (up to day 45) and serious adverse events (up to day 45), due to the low number of events and wide confidence intervals. Therefore, we downgraded two levels due to serious imprecision.
We have very low certainty in the identified evidence for the composite outcome admission to hospital or death (up to day 45) due to serious imprecision and serious indirectness, as very few events (deaths/hospital admissions) occurred and confidence intervals were wide. Concerning the outcome major bleeding events (up to the longest follow‐up), we have very low certainty due to extremely serious imprecision. There was only one study reporting this outcome, with a low number of event rates and small sample size. Therefore, we downgraded three levels for extremely serious imprecision.
Potential biases in the review process
We were committed at all times to following the guidance provided in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2021a), to avoid potential biasses in the review process.
An experienced medical information specialist developed an all‐encompassing search strategy, which was peer‐reviewed by another information specialist. We included all identified published studies, but also kept track of studies that were either still ongoing or labelled as completed in the study registry, to avoid overlooking any upcoming publication. The search included relevant electronic databases as well as clinical trial registries. We contacted principal investigators and requested additional information of our interest. In addition to peer‐reviewed full‐text articles, we also included preprints (one of which was withdrawn and one of which was shortly later published as a peer‐reviewed version). We are confident that we identified all relevant studies to date and will monitor ongoing studies closely, as well as full publication of preprints, after the publication of this review.
We performed sensitivity analyses with a fixed‐effect model and excluding studies at high risk of bias for the main outcome, but did not find any relevant differences in all‐cause mortality up to day 28. One study reported longer follow‐up (REMAP‐CAP 2022, 90‐day and 180‐day mortality) and found a potential small effect (adjusted hazard ratio with credibility intervals) favouring treatment with antiplatelets. For 180‐day mortality, the corresponding risk ratio suggests an insignificant very small effect with a wide confidence interval crossing the zero effect line. Due to imprecision and risk of bias, we downgraded by two levels. We would need more studies reporting comparably long‐term results to ascertain any effect on 90‐day mortality.
We performed all steps of this review in duplicate, and consulted with at least one other review author (mostly in the team) in case of any disagreement. Since the publication of the review protocol, we have considered competing events carefully. We discussed whether we should revise our outcomes to take this issue into account, but decided to report outcomes as 'worsening up to day 28', 'thrombotic events', 'serious adverse events' and 'major bleeding' without the combination with death to avoid impact on the incidences of those events. We described each included study in full detail and made explicit judgements on individual risk of bias.
Agreements and disagreements with other studies or reviews
Despite there being reviews of other COVID‐19 treatments, we did not identify many systematic reviews evaluating antiplatelets. Most reviews did not perform the essential steps for a systematic research synthesis, such as a comprehensive literature search, risk of bias assessment and evidence certainty grading. More importantly, they used only retrospective cohort studies, or pooled randomised controlled trials with retrospective cohort studies. Two systematic reviews reported a large effect favouring antiplatelets in terms of mortality (Wang 2021; Wijaya 2021). However, they included observational studies, which could be confounded. As we performed a rigorous evaluation of all available randomised controlled trials, our results differ substantially from these two systematic reviews.
Authors' conclusions
Implications for practice.
Based on the current evidence, in adults with moderate to severe coronavirus disease 2019 (COVID‐19) the use of antiplatelets plus standard care in comparison to standard care probably results in little to no difference to mortality (up to day 28) or clinical progression (worsening or improvement). They probably slightly decrease thrombotic events but slightly increase the occurrence of major bleeding events. Low‐certainty evidence suggests that antiplatelets may make little or no difference to 180‐day mortality and may result in a slight increase in serious adverse events. We do not know whether antiplatelets have any impact on adverse events, as these results were reported too heterogeneously. For all outcomes, the 95% confidence interval includes both benefit and harm and our certainty in the evidence is moderate to low. We acknowledge that the true effect may slightly differ from the reported effect.
For people with a confirmed diagnosis of mild COVID‐19, antiplatelets may have little to no effect on mortality up to day 45 or on the incidence of serious adverse events when compared to standard care with/without placebo, but they may slightly decrease the incidence of new thrombotic events. The evidence is very uncertain about the effect of antiplatelets compared to standard care with/without placebo regarding the outcomes 'admission to hospital or death up to day 45' and major bleeding. The included trials did not report on quality of life or non‐serious adverse events during study treatment.
Implications for research.
There is a need for more long‐term data (e.g. 180‐day mortality) and data on patient‐relevant outcomes such as adverse events and quality of life; most studies did not report these outcomes, or they were reported in an inconclusive way.
Moderate‐certainty evidence suggests that there is probably little to no difference in 28‐day all‐cause mortality for hospitalised participants, so further research on this population might be low priority in future.
Because the virus has evolved and adapted rapidly in recent years, it is important to keep the science up to date. Therefore, new studies should report subgroup results for vaccinated persons and individuals with different COVID‐19 variants, such as Omicron. Above all, good‐quality research can help to fight the pandemic. Health‐based policy interventions generally require scientifically justifiable studies that research prognostic effectiveness before policy implementation, and for evaluations afterwards. This highlights the urgent need for adequately powered randomised controlled trials (RCTs) with comparable study arms.
As we only identified two RCTs that included non‐hospitalised people with mild disease treated with antiplatelets, efforts should be made to collect good‐quality data regarding these people, and for those who are asymptomatic.
We identified 14 ongoing RCTs. Publications of these ongoing studies may resolve some uncertainties and allow better judgement regarding the effectiveness of antiplatelets for the treatment of COVID‐19. In addition, three studies are already completed but have not yet been published, therefore their results are not included. In accordance with the approach of this review, we will update our search and include eligible trials.
Notes
Reviews of this series partly share information in the background section and methodology based on the first published reviews about monoclonal antibodies (Kreuzberger 2021) and convalescent plasma (Iannizzi 2023).
Risk of bias
Risk of bias for analysis 1.1 All‐cause mortality up to day 28.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Berger 2022 | Low risk of bias | Participants were appropriate randomized via computer generated random numbering in a 1:1 ratio to receive standard treatment and the allocazion sequence was concealed. There are no baseline differences that would suggest a problem with randomization. | Low risk of bias | An appropriate analysis to assess an effect of assignment were conducted on all randomised participants, therefore any deviations from interventions are unlikely to impact the results. | Low risk of bias | Data for this outcome was available for all participants ramodmized. | Low risk of bias | No differences in the measurement or ascertainment of the outcome between intervention groups were noticed or indicated. | Low risk of bias | Data was analysed as specified in a previously published protocol. The outcome was pre‐specified by the review authors. | Low risk of bias | For this outcome, there is a low risk of bias for all the domains. |
| Horby 2021 (RECOVERY) | Low risk of bias | A web‐based randomisation with allocation concealment was used and the baseline characteristics were balanced between groups. | High risk of bias | In the intervention group, 10% never obtained the treatment and 31% did not receive it consistently. There were even 210 (3%) participants in the control group receiving the treatment. | Low risk of bias | Data for this outcome were available for 99% of the included participants. ITT analysis conducted. | Low risk of bias | The measurement of the outcome was appropriate. We suppose, that the assessment of an objective outcome as death could not be influenced by the knowledge of the intervention. | Low risk of bias | The SAP was finalized before unblinded outcome data were available, there were no multiple elegible outcome measurements/data for this outcome. | High risk of bias | Overall, we rate the high risk of bias to be high due to deviations from the intended intervention. |
| REMAP‐CAP 2022 | Low risk of bias | Due to platform study design, allocation concealment for randomisation between Cox‐inhibitor and P2Y12 inhibitor was not given, but as we analyzed both interventions together, the impact seems negligible. Any baseline differences are probably due to chance. | Some concerns | We rate some concerns due to missing information whether there were crossovers or whether the assigned medication was really taken. The rate of withdrewn consent was low among all strata. | Low risk of bias | The outcome data was available for nearly all participants randomised. | Some concerns | The outcome was measured making phone calls and due to the unblinded design, we rate some concerns in this domain. | Low risk of bias | The outcome was assessed and analyzed as planned. | Some concerns | Overall rated some concerns due to missing information about how many participants recieved their assigned intervention and due to the unblinded design. |
Risk of bias for analysis 1.2 Worsening up to day 28: participants with new need for invasive mechanical ventilation or death.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Horby 2021 (RECOVERY) | Low risk of bias | A web‐based randomisation with allocation concealment was used and the baseline characteristics were balanced between groups. | High risk of bias | In the intervention group, 10% never obtained the treatment and 31% did not recieve it consistently. There were even 210 (3%) participants in the control group receiving the intervention. | Low risk of bias | Data for this outcome were available for 99% of the included participants. | Low risk of bias | The measurement of the outcome was appropriate. We suppose, that the assessment of such an objective outcome could not have been influenced by the knowledge of the intervention. | Low risk of bias | SAP finalized before assessment. | High risk of bias | Overall, we rate the risk of bias to be high due to deviations from the intended intervention. |
| REMAP‐CAP 2022 | Low risk of bias | Due to platform study design, allocation concealment for randomisation between Cox‐inhibitor and P2Y12 inhibitor was not given, but as we analyzed both interventions together, the impact seems negligible. Any baseline differences are probably due to chance. | Some concerns | We rate some concerns due to missing information whether there were crossovers or whether the assigned medication was really taken. The rate of withdrewn consent was low among all strata. | High risk of bias | Rated high due to a huge amount of participants randomized without reported outcome: We found that outcome reported for 60.5% (1.104 out of 1.824 ) of the randomized participants. | Low risk of bias | We rate low risk of bias in this domain, although there was a lack of information about how concretely the outcome was assessed. | Low risk of bias | Rated low, as the SAP was finalized before unbliding and the outcome was measured and analyzed as planned. | High risk of bias | Overall rated high due to a huge amount (>20%) of participants randomized had no reported outcome. |
Risk of bias for analysis 1.6 Major bleeding events.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Berger 2022 | Low risk of bias | Participants were appropriate randomized via computer generated random numbering in a 1:1 ratio to receive standard treatment and the allocazion sequence was concealed. There are no baseline differences that would suggest a problem with randomization. | Low risk of bias | Patients were aware; carers were aware ; no placebo. An appropriate analysis to assess an effect of assignment were conducted on all randomised participants, therefore any deviations from interventions are unlikely to impact the results. | Low risk of bias | No indications that missingness depend on the true value of the outcome were observed. | Low risk of bias | No differences in the measurement or ascertainment of the outcome between intervention groups were noticed or indicated. | Low risk of bias | Data was analysed as specified in a previously published protocol. The outcome was pre‐specified by the review authors. | Low risk of bias | For this outcome, there is a low risk of bias for all the domains. |
| Bohula 2022 (COVID‐PACT) | Low risk of bias | Due to blockwise electronic randomsation with 2x2 factorial design and almost no baseline differences, we rated low risk of bias in this domain. The minor differences on Baseline were probably due to small sample sizes. | High risk of bias | Rated high due to imbalanced high rates of crossovers. Additionally, the planned duration of treatment wasn't specified nor in the study registry nor in the publication, so we cannot judge whether there were deviations from intended interventions. | Low risk of bias | The outcome was reported for all randomised participants. | Low risk of bias | he measurement of the outcome was appropriate and it is unlikely that it differed between intervention groups. The outcome assessors were aware of the intervention received, but it is unlikely that knowledge of intervention received could have affected outcome measurement. | Some concerns | Rated some concerns, because no SAP was available and mutliple outcome measure possible due to timeframe of follow up that was not prespecified nor reported (deviations from intended interventions potential selection of the reported result). | High risk of bias | Overall rated high due to many and imbalanced crossovers, unbalanced cointerventions, unclear timeframe of treatment and follow up. |
| Horby 2021 (RECOVERY) | Low risk of bias | A web‐based randomisation with allocation concealment was used and the baseline characteristics were balanced between groups. | High risk of bias | In the intervention group, 10% never obtained the treatment and 31% did not recieve it consistently. There were even 210 (3%) participants in the control group receiving the intervention. | Low risk of bias | Data for this outcome were available for 99% of the included participants. ITT analysis was conducted. | Some concerns | The measurement of the outcome was appropriate. We suppose, that it is unlikely that the assessment of such a well‐defined outcome was influenced by the knowledge of intervention, but there remains a risk due to the unblinded study design. | Low risk of bias | The SAP was finalized before unblinded outcome data were available, there were no multiple elegible outcome measurements/data for this outcome. | High risk of bias | Overall, we rate a high risk of bias due to deviations from the intended intervention and some concerns due to the unblinded study design. |
| REMAP‐CAP 2022 | Low risk of bias | Due to platform study design, allocation concealment for randomisation between Cox‐inhibitor and P2Y12 inhibitor was not given, but as we analyzed both interventions together, the impact seems negligible. Any baseline differences are probably due to chance. | Some concerns | We rate some concerns due to missing information whether there were crossovers or whether the assigned medication was really taken. The rate of withdrewn consent was low among all strata. | Low risk of bias | The outcome data was available for all participants randomised. | Low risk of bias | Rated low due to well‐defined and hard outcome. | Low risk of bias | Rated low as prespecified in trial protocol and SAP. | Some concerns | Overall rated some concerns due to missing information about whether participants recieved the allocated intervention. |
Risk of bias for analysis 1.7 Need for new dialysis up to day 28.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Horby 2021 (RECOVERY) | Low risk of bias | A web‐based randomisation with allocation concealment was used and the baseline characteristics were balanced between groups. | High risk of bias | In the intervention group, 10% never obtained the treatment and 31% did not recieve it consistently. There were even 210 (3%) participants in the control group receiving the intervention. | Low risk of bias | Data for this outcome were available for 99% of the included participants. | Low risk of bias | The measurement of the outcome was appropriate. The decision to start a new renal replacement therapy could have been influenced by the open‐label design, but the influence of the assessment by knowledge of the intervention is almost impossible. | Low risk of bias | The SAP was finalized before unblinded outcome data were available, there were no multiple elegible outcome measurements/data for this outcome. | High risk of bias | Overall, we rate a high risk of bias due to deviations from the intended intervention. |
Risk of bias for analysis 1.9 All‐cause mortality up to longest follow‐up.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Bohula 2022 (COVID‐PACT) | Low risk of bias | Due to blockwise electronic randomsation with 2x2 factorial design and almost no baseline differences, we rated low risk of bias in this domain. The minor differences on Baseline were probably due to small sample sizes. | High risk of bias | Rated high due to imbalanced high rates of crossovers. Additionally, the planned duration of treatment wasn't specified nor in the study registry nor in the publication, so we cannot judge whether there were deviations from intended interventions. | Low risk of bias | The outcome was reported for all randomised participants. | Low risk of bias | The measurement of the outcome was appropriate and it is unlikely that it differed between intervention groups. The outcome assessors were aware of the intervention received, but it is unlikely that knowledge of intervention received could have affected outcome measurement. | Some concerns | Rated some concerns, because no SAP was available and mutliple outcome measure possible due to timeframe of follow up that was not prespecified nor reported (deviations from intended interventions potential selection of the reported result). | High risk of bias | Overall rated high due to many and imbalanced crossovers, unbalanced cointerventions, unclear timeframe of treatment and follow up. |
Risk of bias for analysis 3.4 Serious adverse events.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Eikelboom 2022 (ACTCOVID) | Low risk of bias | Participants were randomized via computer generated random numbering in a 2:2 ratio to receive standard treatment and the allocation sequence was concealed. There are no baseline differences that would suggest a problem with randomization. | Some concerns | 8.4% of participants discontinued intervention, but in an ambulatory unblinded study this seems to be an affordable rate; therefore rated some concerns. | Low risk of bias | Data for this outcome was available for almost all participants randomized. | Low risk of bias | The measurement of the outcome was appropriate and it is unlikely that it differed between intervention groups. The outcome assessors were aware of the intervention received, but it is unlikely that knowledge of intervention received could have affected outcome measurement. | Low risk of bias | The data that produced this result was analysed in accorance with the pre specified analysis plan and the outcome was reported as planned in the protocol. | Some concerns | Overall rated some concerns due to the unblinded design and due to 8.4% intervention discontinuations that could have skewed the effect. |
Acknowledgements
This review was published in collaboration with the Cochrane Central Editorial Service. We particularly thank our Sign‐off Editor (final editorial decision): Toby Lasserson, Deputy Editor‐in‐Chief, Cochrane Evidence Production and Methods Directorate; Managing Editor (selected peer reviewers, collated peer reviewer comments, provided editorial guidance to authors, edited the article): Marwah Anas El‐Wegoud, Cochrane Central Editorial Service; Editorial Assistant (conducted editorial policy checks and supported editorial team): Lisa Wydrzynski, Cochrane Central Editorial Service; and our copy editor (copy editing and production): Andrea Takeda, Cochrane Central Production Service, for their helpful suggestions.
We thank Kathrin Grummich (Information Retrieval Specialist, Cochrane Germany) for peer reviewing the search strategy. We would also like to thank our peer reviewers (provided comments and recommended an editorial decision): Charles Marc SAMAMA, MD, PhD, FCCP, FESAIC Professor of Anaesthesiology Chair, Department of Anaesthesia, Intensive Care and Perioperative Medicine GHU AP‐HP. Centre ‐ Université Paris‐Cité ‐ Cochin Hospital. 27 rue du Faubourg St Jacques 75014 Paris, France (clinical review); Hugo ten Cate, MD, PhD, Maastricht University Medical Center, Maastricht, the Netherlands (clinical review); Zoe McQuilten, Department of Epidemiology and Preventive Medicine, Monash University, Melbourne, Australia (clinical review); Dr. Marcial Fallas (consumer review); Kerry Dwan, Cochrane Methods Support Unit (methods review); Nuala Livingstone, Cochrane Evidence Production and Methods Directorate (methods review); and Robin Featherstone, Cochrane Central Editorial Service (search review). One additional peer reviewer provided clinical peer review but chose not to be publicly acknowledged.
The research was part of a project supported by the German Federal Ministry of Education and Research (NaFoUniMedCovid19, funding number: 01KX2021; part of the project "CEOsys"). The contents of this document reflect only the authors' views and the German Ministry is not responsible for any use that may be made of the information it contains.
We thank Dr Peto and the RECOVERY study team for providing us with information regarding the RECOVERY trial. We thank the COVID‐Pact trial for the email conversation when they were still recruiting that the study was still ongoing. We thank Dr Lau for providing us with information on the LEAD COVID‐19 trial.
This work is part of a series of reviews investigating treatments and therapies for COVID‐19 as part of the project CEOsys. Text passages in the background section (e.g. Description of the condition and Why it is important to do this review) are shared between reviews of this series (Iannizzi 2023; Kreuzberger 2021). We thank the authors of the first published reviews of this series for providing and sharing this information. Moreover, we thank the Cochrane Haematology working group for using the template for the description of methods. Furthermore, we thank our colleagues for their work at the hospitals (Universitätsklinikum Leipzig, University of Cologne) that made it possible for us to take the time for this review.
Appendices
Appendix 1. Search strategies
Cochrane COVID‐19 Study Register
"platelet antiaggregant" OR "platelet antiaggregants" OR "platelet anti‐aggregant" OR "platelet anti‐aggregants" OR "agglutination inhibitor" OR "agglutination inhibitors" OR "aggregation inhibitor" OR "aggregation inhibitors" OR "platelet inhibitor" OR "platelet inhibitors" OR antiplatelet* OR "anti‐platelet" OR "anti‐platelets" OR antiaggreg* OR "platelet anti‐aggregation" OR aspirin* OR "acetylsalicylic acid" OR "acetyl salicylic acid" OR acetysal* OR acylpyrin* OR aloxiprimum* OR colfarit* OR dispril* OR easprin* OR ecotrin* OR endosprin* OR magnecyl* OR micristin* OR polopirin* OR polopiryna* OR solprin* OR solupsan* OR zorprin* OR aggrenox*
Study characteristics:
1) "Intervention assignment": randomised, unclear
2) "Study design": parallel/crossover, unclear
Web of Science
#1 TI=(COVID OR COVID19 OR "SARS‐CoV‐2" OR "SARS‐CoV2" OR SARSCoV2 OR "SARSCoV‐2" OR "SARS coronavirus 2" OR "2019 nCoV" OR "2019nCoV" OR "2019‐novel CoV" OR "nCov 2019" OR "nCov 19" OR "severe acute respiratory syndrome coronavirus 2" OR "novel coronavirus disease" OR "novel corona virus disease" OR "corona virus disease 2019" OR "coronavirus disease 2019" OR "novel coronavirus pneumonia" OR "novel corona virus pneumonia" OR "severe acute respiratory syndrome coronavirus 2) OR AB=(COVID OR COVID19 OR "SARS‐CoV‐2" OR "SARS‐CoV2" OR SARSCoV2 OR "SARSCoV‐2" OR "SARS coronavirus 2" OR "2019 nCoV" OR "2019nCoV" OR "2019‐novel CoV" OR "nCov 2019" OR "nCov 19" OR "severe acute respiratory syndrome coronavirus 2" OR "novel coronavirus disease" OR "novel corona virus disease" OR "corona virus disease 2019" OR "coronavirus disease 2019" OR "novel coronavirus pneumonia" OR "novel corona virus pneumonia" OR "severe acute respiratory syndrome coronavirus 2)
#2 TI=(antiplatelet* OR anti‐platelet* OR antiaggreg* OR anti‐aggreg* OR (platelet* NEAR/5 inhibit* OR antagonis*) OR (thrombocyt* NEAR/5 inhibit* OR antagonis*) ) OR AB=(antiplatelet* OR antiplatelet* OR antiaggreg* OR antiaggreg* OR (platelet* NEAR/5 inhibit* OR antagonis*) OR (thrombocyt* NEAR/5 inhibit* OR antagonis*))
#3 TI=(aspirin* OR "acetylsalicylic acid" OR "acetyl salicylic acid" OR acetysal* OR acylpyrin* OR aloxiprimum* OR colfarit* OR dispril* OR easprin* OR ecotrin* OR endosprin* OR magnecyl* OR micristin* OR polopirin* OR polopiryna* OR solprin* OR solupsan* OR zorprin* OR aggrenox*) OR AB=(aspirin* OR "acetylsalicylic acid" OR "acetyl salicylic acid" OR acetysal* OR acylpyrin* OR aloxiprimum* OR colfarit* OR dispril* OR easprin* OR ecotrin* OR endosprin* OR magnecyl* OR micristin* OR polopirin* OR polopiryna* OR solprin* OR solupsan* OR zorprin* OR aggrenox*)
#4 #2 OR #3
#5 TI=(random* OR placebo OR trial OR groups OR "phase 3" OR "phase3" OR p3 OR "pIII") OR AB=(random* OR placebo OR trial OR groups OR "phase 3" OR "phase3" OR p3 OR "pIII")
#6 #1 AND #4 AND #5
Searched in Science Citation Index Expanded and Emerging Sources Citation Index
World Health Organization COVID‐19 Global literature on coronavirus disease
Advanced search in the search fields: title, abstract, subject
platelet inhibitor* or antiplatelet* or anti‐platelet* or antiaggreg* or anti‐aggreg* or aspirin* or acetylsalicylic acid or "acetyl salicylic acid" or acetysal* or acylpyrin* or aloxiprimum* or colfarit* or dispril* or easprin* or ecotrin* or endosprin* or magnecyl* or micristin* or polopirin* or polopiryna* or solprin* or solupsan* or zorprin* or aggrenox*
AND
random* or placebo or trial or groups or "phase 3" or "phase3" or p3 or "pIII"
Epistemonikos, L*OVE List Coronavirus disease (COVID‐19)
LOVE list / COVID‐19
searched by PICO:
prevention or treatment:
-
pharmacological interventions
-
antithrombotics
Antiplatelets (only primary studies; filter results by RCT)
-
Data and analyses
Comparison 1. Individuals with a confirmed diagnosis of COVID‐19 and moderate to severe disease.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1.1 All‐cause mortality up to day 28 | 3 | 17249 | Risk Ratio (M‐H, Random, 95% CI) | 0.95 [0.85, 1.05] |
| 1.2 Worsening up to day 28: participants with new need for invasive mechanical ventilation or death | 2 | 15266 | Risk Ratio (M‐H, Random, 95% CI) | 0.95 [0.90, 1.01] |
| 1.3 Improvement up to day 28: discharged alive | 2 | 15454 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.96, 1.04] |
| 1.4 Thrombotic events | 4 | 17518 | Risk Ratio (M‐H, Random, 95% CI) | 0.90 [0.80, 1.02] |
| 1.5 Serious adverse events | 1 | 1815 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.57 [0.48, 5.14] |
| 1.6 Major bleeding events | 4 | 17527 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.68 [1.29, 2.19] |
| 1.7 Need for new dialysis up to day 28 | 1 | 14771 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.99 [0.84, 1.18] |
| 1.8 180‐day mortality | 1 | 1312 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.82, 1.15] |
| 1.9 All‐cause mortality up to longest follow‐up | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected |
1.9. Analysis.

Comparison 1: Individuals with a confirmed diagnosis of COVID‐19 and moderate to severe disease, Outcome 9: All‐cause mortality up to longest follow‐up
Comparison 2. Subgroup analysis: Effects on mortality up to day 28.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 2.1 Subgroup analysis (type of antiplatelet agent): cyclooxygenase inhibitors or ADP receptor/P2Y12 inhibitors in participants with moderate to severe COVID‐19 | 3 | 17249 | Risk Ratio (M‐H, Random, 95% CI) | 0.95 [0.89, 1.02] |
| 2.1.1 Cyclooxygenase inhibitor (acetylsalicylic acid) | 2 | 15859 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.90, 1.02] |
| 2.1.2 P2Y12 inhibitor (clopidogrel, ticagrelor, prasugrel) | 2 | 1390 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.61, 1.69] |
| 2.2 Subgroup analysis: concomitant therapeutic dose anticoagulation or prophylactic anticoagulation | 3 | 15828 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.83, 1.13] |
| 2.2.1 Antiplatelet agents plus standard care (including prophylactic anticoagulation) | 2 | 15024 | Risk Ratio (M‐H, Random, 95% CI) | 0.92 [0.77, 1.10] |
| 2.2.2 Antiplatelet agents plus therapeutic anticoagulation | 2 | 804 | Risk Ratio (M‐H, Random, 95% CI) | 1.27 [0.88, 1.84] |
Comparison 3. Individuals with a confirmed diagnosis of SARS‐CoV‐2 infection and asymptomatic to mild disease.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 3.1 All‐cause mortality up to longest follow‐up (45 days) | 2 | 4209 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.00 [0.45, 2.22] |
| 3.2 Admission to hospital or death up to day 45 | 2 | 4209 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.79 [0.57, 1.10] |
| 3.3 Thrombotic events | 2 | 4209 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.37 [0.09, 1.46] |
| 3.4 Serious adverse events | 1 | 3881 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.00 [0.60, 1.64] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Berger 2022.
| Study characteristics | |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes | Composite primary outcome
Components of the primary outcome
Secondary outcomes (at day 28) and individual components
Components of the secondary outcomes
|
| Notes |
|
Bohula 2022 (COVID‐PACT).
| Study characteristics | |
| Methods |
|
| Participants |
|
| Interventions | Intervention: antiplatelet therapy with clopidogrel PLUS standard care, or clopidogrel PLUS therapeutic anticoagulation PLUS standard care
|
| Outcomes | Primary outcome: venous or arterial thrombotic event Hierarchical composite: death due to venous or arterial thrombosis, pulmonary embolism, clinically evident deep vein thrombosis, type 1 MI, ischaemic stroke, systemic embolism or acute limb ischaemia, or clinically silent DVT Secondary outcomes: clinically evident venous or arterial thrombotic events Hierarchical composite: death due to venous or arterial thrombosis, pulmonary embolism, clinically evident deep vein thrombosis, type 1 MI, ischaemic stroke, systemic embolism or acute limb ischaemia Additional outcomes Bleeding, cardiovascular death/non‐cardiovascular and unknown deaths |
| Notes |
|
Connors 2021 (ACTIV‐4B).
| Study characteristics | |
| Methods |
|
| Participants |
|
| Interventions | Study arms
Details of intervention
|
| Outcomes | Primary outcome: composite of symptomatic deep venous thrombosis, pulmonary embolism, arterial thromboembolism, myocardial infarction, ischaemic stroke, hospitalisation for cardiovascular or pulmonary events and all‐cause mortality for up to 45 days after treatment initiation Secondary outcomes: individual components of the primary study end point as well as mortality without antecedent hospitalisation |
| Notes | Sponsor: This study was, in part, funded by National Institutes of Health (NIH) Agreement 1OT2HL156812‐01. Specifically, the ACTIV‐4B trial was supported by Other Transition Authorities from the National Heart, Lung, and Blood Institute (NHLBI). Grantee institutions included the University of Pittsburgh; the University of Illinois Chicago; and the Brigham and Women's Hospital. The trial drugs and matching placebo were donated by the Bristol Myers Squibb–Pfizer Alliance. COI
|
Eikelboom 2022 (ACTCOVID).
| Study characteristics | |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Notes | Sponsor/funding: Canadian Institutes for Health Research, Bayer, Population Health Research Institute, Hamilton Health Sciences Research Institute, and Thistledown Foundation |
Horby 2021 (RECOVERY).
| Study characteristics | |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes | Primary outcome
Secondary outcomes
Additional outcomes
|
| Notes |
|
REMAP‐CAP 2022.
| Study characteristics | |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes | Primary outcomes: organ support‐free days to day 21, survival to hospital discharge Secondary outcomes: 90‐day mortality, progression to intubation, ECMO or death, days free from organ support (respiratory, cardiovascular), length of stay in the hospital, hospital mortality, death or major thrombotic events, major bleeding, serious adverse events, etc. |
| Notes | Sponsor: the Platform for European Preparedness Against (Re‐)Emerging Epidemics (PREPARE) consortium of the European Union, FP7‐HEALTH‐2013‐INNOVATION‐1 (grant 602525), the Rapid European COVID‐19 Emergency Research Response (RECOVER) consortium of the European Union’s Horizon 2020 Research and Innovation Programme (grant 101003589), the Australian National Health and Medical Research Council (grant APP1101719), the Health Research Council of New Zealand (grant 16/631), the Canadian Institute of Health Research Strategy for Patient‐Oriented Research Innovative Clinical Trials Program (grant 158584), the NIHR and the NIHR Imperial Biomedical Research Centre, the Health Research Board of Ireland (grant CTN 2014‐012), the University of Pittsburgh Medical Center (UPMC) Learning While Doing Program, the Translational Breast Cancer Research Consortium, the French Ministry of Health (grant PHRC‐20‐0147), the Minderoo Foundation, and the Wellcome Trust Innovations Project (grant 215522). Dr Shankar‐Hari is funded by an NIHR clinician scientist fellowship (grant CS‐2016‐16‐011) and Dr Gordon is funded by an NIHR research professorship (grant RP‐2015‐06‐18). 19 authors had pharmaceutical grants |
ASA: acetylsalicylic acid; COI: conflicts of interest; CNS: central nervous system; COPD; chronic obstructive pulmonary disease; DVT: deep vein thrombosis; ECMO: extracorporeal membrane oxygenation; eGFR: estimated glomerular filtration rate; ICU: intensive care unit; IQR: interquartile range; IV: intravenous; n.i.: no information; NR: not reported; RCT: randomised controlled trial; SD: standard deviation
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Eikelboom 2022 (ACTCOVID19 hospitalised) | Ineligible intervention |
| NCT04483960 | No platelet inhibitor |
Characteristics of studies awaiting classification [ordered by study ID]
NCT04391179.
| Methods |
|
| Participants |
|
| Interventions | Intervention: dipyridamole 100 mg by mouth 4 times a day for 14 days Control: placebo given by mouth 4 times a day for 14 days Concomitant therapy: NR Treatment cross‐overs: NR Duration of follow‐up: NR Compliance with assigned treatment: NR |
| Outcomes | Primary outcomes
Secondary outcomes
|
| Notes |
|
NCT04659109.
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Notes |
|
RESIST 2021.
| Methods |
|
| Participants |
|
| Interventions |
Concomitant therapy: NR Treatment cross‐overs: NR Duration of follow‐up: all study participants were followed up for 10 days or until hospital discharge, whichever was later Treatment cross‐overs: NR Compliance with assigned treatment: NR |
| Outcomes | Primary outcomes
Secondary outcomes
Additional outcomes: NR |
| Notes |
|
ECMO: extracorporeal membrane oxygenation; ICU: intensive care unit; IV: intravenous; NR: not reported; RCT: randomised controlled trial
Characteristics of ongoing studies [ordered by study ID]
ChiCTR2000030055.
| Study name | Multicentre study for the treatment of Dipyridamole with novel coronavirus pneumonia (COVID‐19) |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | March 2020 |
| Contact information | Qingling Zhang
|
| Notes | Funding: National Key R&D Program of China, National Natural Science Foundation of China, Taikang Insurance Group Co., Ltd and Beijing Taikang Yicai Foundation and philanthropy donation from individuals. |
CTRI/2020/08/027503.
| Study name | Low Dose aspirin in Moderate to Severe SARS‐ CoV‐2 Infected Patients: A Pilot Randomized Controlled Trial |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes | Primary outcome: SpO2/ FiO2 ratio in day 1 to 7 post randomisation in both the groups Secondary outcomes
Additional outcomes: NR |
| Starting date | NR |
| Contact information | Contact: Dr Souvik Maitra, 08146727891, souvikmaitra@live.com |
| Notes |
|
CTRI/2021/03/032059.
| Study name | Investigator initiated study to evaluate the effect of Marketed Colchicine 0.5mg given along marketed Asprin 75mg with SOC and Marketed aspirin 75mg with SOC on COVID 19 patients |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes | Primary outcomes
Secondary outcomes: safety Additional outcomes: NR |
| Starting date | Enrolment planned for 2‐ March 2021 |
| Contact information | Dr Vispute Abhay Shantaram, 9819428656, surgician@gmail.com |
| Notes | Sponsors/funding: SRV Hospital, Dr Mandakini Parihar Marg Opposite LokmanyaTilak Terminus, Tilak Nagar, Chembur, Mumbai, Maharashtra |
CTRI/2021/06/034254.
| Study name | Clinical trial of APMV2020 in Covid 19 subjects |
| Methods |
|
| Participants |
|
| Interventions |
Along with standard care as per MoHFW guidelines for 10 days
|
| Outcomes | Primary outcomes
Secondary outcomes
Additional outcomes: NR |
| Starting date | NR |
| Contact information | Mr Taasin Ahmed Shah,
912225817126, tdcruz@meyer.co.in Ms Tania Dcruz, 912225817126, tdcruz@meyer.co.in |
| Notes | Sponsors/funding: Meyer Organics Pvt. Ltd A‐303, Road No. 32, Wagle Estate, Thane ‐ 400 604. (India) |
IRCT20180205038626N7.
| Study name | The investigation of the effectiveness of ASA usage on the incidence of cardiovascular events in patients with coronavirus (COVID‐19) A clinical trial study |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes | Primary outcomes
Secondary outcomes
Additional outcomes: NR |
| Starting date | NR |
| Contact information | Contact: Zahra Ahmadnia, +98 13 3361 8177, zahmadnia@gums.ac.ir |
| Notes |
|
NCT04363840.
| Study name | The LEAD COVID‐19 Trial: Low‐risk, Early aspirin and Vitamin D to Reduce COVID‐19 Hospitalizations (LEAD COVID‐19) |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes | Primary outcomes: hospitalisation Secondary outcomes: NR Additional outcomes: NR |
| Starting date | 1 May 2020 |
| Contact information | Contact: Frank H Lau, MD 504 412 1240, flau@lsuhsc.edu |
| Notes | Due to missing funding the trial was not started
|
NCT04365309.
| Study name | Protective Effect of Aspirin on COVID‐19 Patients (PEAC) |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes | Primary outcomes
Secondary outcomes: NR Additional outcomes: NR |
| Starting date | 10 February 2020 |
| Contact information | NR |
| Notes |
|
NCT04445623.
| Study name | Prasugrel in Severe COVID‐19 Pneumonia (PARTISAN) |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes | Primary outcomes
Secondary outcomes
Additional Outcomes: NR |
| Starting date | 1 July 2020 |
| Contact information |
|
| Notes |
|
NCT04703608.
| Study name | Prevention and Treatment for COVID ‐19 (Severe Acute Respiratory Syndrome Coronavirus 2 SARS‐CoV‐2) Associated Severe Pneumonia in the Gambia (PaTS‐COVID) |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes | Primary outcomes
Secondary outcomes
Additional outcomes
|
| Starting date | 22 January 2021 |
| Contact information | Contact: Anna Roca, PhD, +220 4495442 ext 2305, aroca@mrc.gm |
| Notes |
|
NCT04768179.
| Study name | Safety & Efficacy of Low Dose aspirin / Ivermectin Combination Therapy for Treatment of Covid‐19 Patients (IVCOM) |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes | Primary outcomes
Secondary outcomes
|
| Starting date | 19 February 2020 |
| Contact information |
|
| Notes |
|
NCT04808895.
| Study name | Acetylsalicylic Acid in the Prevention of Severe SARS‐CoV2 Pneumonia in Hospitalised Patients With COVID‐19 (Asperum) |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes | Primary outcomes
Secondary outcomes
Additional outcomes: NR |
| Starting date | 1 April 2021 |
| Contact information | Contact: Pietro Minuz, Professor, +39 045‐8124414, pietro.minuz@univr.it Contact: Marco Cattaneo, Professor, +390250323095, marco.cattaneo@unimi.it |
| Notes |
|
NCT04937088.
| Study name | Outpatient Liquid Aspirin (OLA) |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes | Primary outcomes
Secondary outcomes: NR Additional outcomes: NR |
| Starting date | 6 December 2021 |
| Contact information | Frank Lau, MD 504‐412‐1240, flau@lsuhsc.edu |
| Notes | Louisiana State University Health Sciences Center in New Orleans, Innovate UK |
NCT05073718.
| Study name | ASA‐SARS / NCT05073718 |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes | Primary outcomes (time frame: up to 37 weeks)
Secondary outcomes (time frame: up to 37 weeks)
Additional outcomes: NR |
| Starting date | Estimated start date: 15 January 2022 |
| Contact information | Laura Garcia, Dr. +34932275400, laura.garcia@isglobal.org |
| Notes | Sponsor/collaborators: Barcelona Institute for Global Health, Hospital Universitario de Torrejon Madrid, Hospital Universitario Infanta Leonor, Fundacio Institut de Recerca de l'Hospital de la Santa Creu i Sant Pau, Hospital del Mar, Hospital Central de Maputo Mozambique |
Sharma 2021.
| Study name | A Randomized Open‐Label Trial To Evaluate The Efficacy And Safety Of Triple Therapy With aspirin, Atorvastatin, And Nicorandil In Hospitalised Patients With SARS Cov‐2 Infection: A Structured Summary Of A Study Protocol For A Randomized Controlled Trial |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes | Primary outcomes
Secondary outcomes
Additional outcomes: NR |
| Starting date | 8 April 2021 |
| Contact information | Ambudhar Sharma, 9418048268, moc.liamg@414rahdubma Charu Sharma, moc.liamg@4891rahdubma Sujeet Raina, moc.liamg@aniarteejus |
| Notes |
COI: conflict of interest; ECMO: extracorporeal membrane oxygenation; NR: not reported; RCT: randomised controlled trial
Differences between protocol and review
We revised improvement and worsening outcomes together with the German COVID‐19 guideline panel to consider the competing event of death. Our definition of clinical status was less dependent on the need of respiratory support ‐ that is the reason why some outcomes became secondary outcomes and the following outcomes were deleted.
Liberation from supplemental oxygen in patients, i.e. WHO ≤ 4 on the Clinical Progression Scale (for the subgroup of participants requiring any supplemental oxygen or ventilator support at baseline, i.e WHO ≥ 5), including duration to liberation.
Weaning or liberation from invasive mechanical ventilation in patients, i.e. WHO ≤ 6 (for the subgroup of participants requiring invasive mechanical ventilation at baseline, i.e. WHO ≥ 7), including duration from liberation.
Need for non‐invasive mechanical ventilation or high‐flow oxygen, i.e. WHO = 6 (for the subgroup of participants not requiring non‐invasive or non‐invasive mechanical ventilation, or high‐flow oxygen at baseline, i.e. WHO ≤ 5);
Need for oxygen by mask or nasal prongs, i.e. WHO = 5 (for the subgroup of participants not requiring any supplemental oxygen or ventilator support at baseline, i.e. WHO ≤ 4).
Quality of life outcomes were moved to secondary outcomes, since we did not want to overload the summary of findings tables and the safety outcomes seemed more important to us.
The subgroup analysis of severity was not possible due to a lack of information in the studies.
Originally, we had planned to perform a subgroup analyses of the following characteristics.
Age of participants (divided into applicable age groups, e.g. children, 18 to 65 years, 65 years and older), suggesting that older participants may benefit more from treatment with antiplatelet agents, as they are at higher risk for thrombotic events.
Pre‐existing conditions (diabetes, respiratory disease, hypertension, immunosuppression), suggesting that participants with some diseases that increase thrombotic risk may benefit more.
Timing of first dose administration with illness onset, suggesting that an earlier treatment may prevent deterioration.
Due to insufficient data, we were not able to perform these subgroup analyses.
We had a longer reflectional process involving clinical experts about whether we should meta‐analyse different antiplatelet types together, as we did in our review. We decided to perform a subgroup analysis stratified by the antiplatelet agent that was used, to rule out a relevant differences between drugs.
Contributions of authors
AF: clinical expertise, conception and writing of the review, study selection, data extraction, risk of bias, GRADE assessments, proofreading of the manuscript.
SM: conception and writing of the review, data extraction, screening, risk of bias, proofreading of the manuscript
LE: conception, clinical and methodological expertise and advice, proofreading of the manuscript.
AM: methodological expertise and advice, risk of bias assessment, writing and proofreading of the manuscript.
MA: conception, methodological expertise and advice, study selection, data extraction, proofreading of the manuscript.
MS: clinical input, proofreading of the manuscript
IM: development of the search strategy
SW: methodological expertise and advice, data extraction, risk of bias assessment, proofreading of the manuscript.
RP: conception, methodological expertise and advice, data assessment, writing and proofreading of the manuscript.
RR: methodological expertise and advice, risk of bias assessment, writing and proofreading of the manuscript.
NS: conception, methodological expertise and advice, study selection, data assessment, conception, writing and proofreading of the manuscript.
Sources of support
Internal sources
-
University Hospital of Cologne, Germany
Evidence‐based Oncology, Department I of Internal Medicine, Faculty of Medicine and University Hospital Cologne, University of Cologne, Cologne, Germany
-
Charité – Universitätsmedizin Berlin, corporate member of Freie Universität Berlin and Humboldt‐Universität zu Berlin, Berlin, Germany
Department of Infectious Diseases and Respiratory Medicine
-
University Hospital Leipzig, Germany
Department of Anesthesiology and Intensive Care Medicine
External sources
-
Federal Ministry of Education and Research of Germany, Germany
This review is part of the CEOsys project funded by the Network of University Medicine (Nationales Forschungsnetzwerk der Universitätsmedizin (NUM)) by the Federal Ministry of Education and Research of Germany (Bundesministerium für Bildung und Forschung (BMBF)), grant number 01KX2021.
Declarations of interest
AF: works as an Intensive Care Medicine Physician and is member of the CEOsys project funded by the Network of University Medicine (Nationales Forschungsnetzwerk der Universitätsmedizin (NUM)) by the Federal Ministry of Education and Research of Germany (Bundesministerium für Bildung und Forschung (BMBF)), grant number 01KX2021, paid to the institution.
SM: none known; she is a staff member of Cochrane Haematology, but was not involved in the editorial process for this review.
RR: none known.
AM: none known.
MS: none known.
LE: she is Co‐ordinating Editor of Cochrane Haematology, but was not involved in the editorial process for this review. She is an investigator on the REMAP‐CAP trial but was not involved in the data extraction or risk of bias assessment of this trial.
SW: none known; she is Content Editor of Cochrane Anaesthesia, but was not involved in the editorial process for this review.
IM: none known; she is an Information Specialist of Cochrane Haematology, but was not involved in the editorial process for this review.
MA: none known.
RP: none known.
NS: none known; she is Co‐ordinating Editor of Cochrane Haematology, but was not involved in the editorial process for this review.
These authors should be considered joint first author.
These authors contributed equally to this work.
New
References
References to studies included in this review
Berger 2022 {published data only}
- Berger JS, Kornblith LZ, Gong MN, Reynolds HR, Cushman M, Hochmann JS, et al. Effect of P2Y12 inhibitors on survival free of organ support among non–critically ill hospitalized patients with COVID-19: a randomized clinical trial. JAMA 2022;327(3):227-36. [DOI: 10.1001/jama.2021.23605] [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT04505774. Accelerating COVID-19 Therapeutic Interventions and Vaccines 4 ACUTE (ACTIV-4A). clinicaltrials.gov/ct2/show/NCT04505774 (first received 10 August 2020).
Bohula 2022 (COVID‐PACT) {published data only}
- Bohula EA, Berg DD, Lopes MS, Connors JM, Babar I, Barnett CF, et al. Anticoagulation and antiplatelet therapy for prevention of venous and arterial thrombotic events in critically ill patients with COVID-19: (COVID-PACT). Circulation 2022;146:1344–56. [DOI: 10.1161/CIRCULATIONAHA.122.061533] [DOI] [PMC free article] [PubMed] [Google Scholar]
Connors 2021 (ACTIV‐4B) {published data only}
- Connors JM, Brooks MM, Sciurba FC, Krishnan JA, Bledsoe JR, Kindzelski A, et al. Effect of antithrombotic therapy on clinical outcomes in outpatients with clinically stable symptomatic COVID-19: The ACTIV-4B randomized clinical trial. JAMA 2021;326(17):1703-12. [DOI: 10.1001/jama.2021.17272] [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT04498273. COVID-19 Positive Outpatient Thrombosis Prevention in Adults Aged 40-80. clinicaltrials.gov/ct2/show/NCT04498273 (first received 4 August 2020).
Eikelboom 2022 (ACTCOVID) {published data only}www.phri.ca/research/act‐covid‐19/
- Eikelboom JW, Jolly SS, Belley-Cote EP, Whitlock RP, Rangarajan S, Xu L, et al. Colchicine and aspirin in community patients with COVID-19 (ACT): an open-label, factorial, randomised, controlled trial. Lancet Respiratory Medicine 2022;10(12):1160–8. [DOI: 10.1016/ S2213-2600(22)00299-5] [DOI] [PMC free article] [PubMed] [Google Scholar]
Horby 2021 (RECOVERY) {published and unpublished data}
- Horby PW, Pessoa-Amorim G, Staplin N, Emberson JR, Spata E, Campbell M. Aspirin in patients admitted to hospital with COVID-19 (RECOVERY): a randomised, controlled, open-label, platform trial. medRxiv 2021;June 8:1-27. [DOI: 10.1101/2021.06.08.21258132] [DOI] [Google Scholar]
- Horby PW, Pessoa-Amorim G, Staplin N, Emberson JR, Spata E, Campbell M. Aspirin in patients admitted to hospital with COVID-19(RECOVERY): a randomised, controlled, open-label, platform trial. The Lancet 17 November 2021;Published Online:1-9. [DOI: 10.1016/ S0140-6736(21)01825-0] [Google Scholar]
- NCT04381936. Randomised Evaluation of COVID-19 Therapy (RECOVERY). clinicaltrials.gov/ct2/show/NCT04381936 (first received 11 May 2020).
- RECOVERY Collaborative Group. Aspirin in patients admitted to hospital with COVID-19 (RECOVERY): a randomised, controlled, open-label, platform trial. Lancet 2022 Jan 8;399:143 to 151. [DOI: 10.1016/S0140-6736(21)01825-0] [DOI] [PMC free article] [PubMed] [Google Scholar]
REMAP‐CAP 2022 {published data only}
- NCT02735707. Randomized, Embedded, Multifactorial Adaptive Platform Trial for Community- Acquired Pneumonia (REMAP-CAP). clinicaltrials.gov/ct2/show/NCT02735707 (first received 13 April 2016).
- REMAP-CAP Writing Committee for the REMAP-CAP Investigators. Effect of antiplatelet therapy on survival and organ support-free days in critically ill patients with COVID-19: a randomized clinical trial. JAMA 2022;13:1247-59. [DOI: 10.1001/jama.2022.2910] [DOI] [PMC free article] [PubMed] [Google Scholar]
- REMAP-CAP Writing Committee for the REMAP-CAP Investigators. Long-term (180-Day) outcomes in critically ill patients with COVID-19 in the REMAP-CAP randomized clinical trial. JAMA January 2023;329(1):39-51. [DOI: 10.1001/jama.2022.23257] [DOI] [PMC free article] [PubMed] [Google Scholar]
References to studies excluded from this review
Eikelboom 2022 (ACTCOVID19 hospitalised) {published data only}pubmed.ncbi.nlm.nih.gov/36228641
- Eikelboom JW, Jolly SS, Belley-Cote EO, Whitlock RP, Rangarajan S, Xu L, et al. Colchicine and the combination of rivaroxaban and aspirin in patients hospitalised with COVID-19 (ACT): an open-label,factorial, randomised, controlled trial. Lancet Respiratory Medicine 10 October 2022;10:1169–77. [DOI: 10.1016/ S2213-2600(22)00298-3] [DOI] [PMC free article] [PubMed] [Google Scholar]
NCT04483960 {unpublished data only}
- NCT04483960. Australasian COVID-19 Trial (ASCOT) ADAptive Platform Trial (ASCOT ADAPT). clinicaltrials.gov/ct2/show/NCT04483960 (first received 23 July 2020).
References to studies awaiting assessment
NCT04391179 {published data only}
- NCT04391179. Dipyridamole to Prevent Coronavirus Exacerbation of Respiratory Status (DICER) in COVID-19. clinicaltrials.gov/ct2/show/study/NCT04391179 (first received May 18th 2020).
NCT04659109 {published data only}
- NCT04659109. Glenzocimab in SARS-Cov-2 Acute Respiratory DistrEss syNdrome Related to COVID-19 (GARDEN). clinicaltrials.gov/ct2/show/NCT04659109 (first received 9 December 2020).
RESIST 2021 {published data only}
- CTRI/2020/07/026791. Statin and aspirin in SARS-CoV-2 infection. ctri.nic.in/Clinicaltrials/pmaindet2.php?trialid=44814 (first received 25 July 2020).
- Ghati N, Deepti S, Bhatnagar S, Mahendran M, Thakur A, Prasad K, et al. A randomised control trial of statin and aspirin as adjuvant therapy in patients with SARS-CoV-2 infection (RESIST Trial). SSRN 2021. [DOI: 10.2139/ssrn.3820512] [DOI] [PMC free article] [PubMed]
- Ghati N, Roy A, Bhatnagar S, Bhati S, Bhushan S, Mahendran M, et al. Atorvastatin and aspirin as adjuvant therapy in patients with SARS-CoV-2 infection: A structured summary of a study protocol for a randomised controlled trial. Trials 2020;21:902. [DOI: 10.1186/s13063-020-04840-y] [DOI] [PMC free article] [PubMed] [Google Scholar]
References to ongoing studies
ChiCTR2000030055 {published data only}ChiCTR2000030055
- ChiCTR2000030055. Multicenter study for the treatment of Dipyridamole with novel coronavirus pneumonia (COVID-19). www.chictr.org.cn/showprojen.aspx?proj=49864 (first received 22 February 2020).
CTRI/2020/08/027503 {published data only}
- CTRI/2020/08/027503. Low Dose Aspirin in Moderate to Severe SARS- CoV-2 Infected Patients: A Pilot Randomized Controlled Trial. ctri.nic.in/Clinicaltrials/pmaindet2.php?trialid=44980 (first received 31 August 2020).
CTRI/2021/03/032059 {published data only}
- CTRI/2021/03/032059. Investigator initiated study to evaluate the effect of Marketed Colchicine 0.5mg given along marketed Asprin 75mg with SOC and Marketed aspirin 75mg with SOC on COVID 19 patients. ctri.nic.in/Clinicaltrials/pdf_generate.php?trialid=53115 (first received 17 March 2021).
CTRI/2021/06/034254 {published data only}
- CTRI/2021/06/034254. Randomized controlled clinical trial to assess the efficacy and safety of APMV2020 in mildsymptomatic subjects of COVID 19. ctri.nic.in/Clinicaltrials/pdf_generate.php?trialid=56647 (first received 15 June 2021).
IRCT20180205038626N7 {published data only}
- IRCT20180205038626N7. The investigation of the effectiveness of ASA usage on the incidence of cardiovascular events in patients with corona virus (COVID-19) A clinical trial study. en.irct.ir/trial/52374 (first received 27 December 2020).
NCT04363840 {published data only}
- NCT04363840. The LEAD COVID-19 Trial: Low-risk, Early Aspirin and Vitamin D to Reduce COVID-19 Hospitalizations (LEAD COVID-19). clinicaltrials.gov/ct2/show/NCT04363840 (first received 27 April 2020).
NCT04365309 {published data only}
- NCT04365309. Protective Effect of Aspirin on COVID-19 Patients (PEAC). clinicaltrials.gov/ct2/show/NCT04365309 (first received 28 April 2020).
NCT04445623 {published data only}
- NCT04445623. Prasugrel in Severe COVID-19 Pneumonia (PARTISAN). clinicaltrials.gov/ct2/show/NCT04445623 (first received 24 June 2020).
NCT04703608 {published data only}
- NCT04703608. Prevention and Treatment for COVID -19 (Severe Acute Respiratory Syndrome Coronavirus 2 SARS-CoV-2) Associated Severe Pneumonia in the Gambia (PaTS-COVID). www.clinicaltrials.gov/ct2/show/NCT04703608 (first received 11 January 2021).
NCT04768179 {published data only}
- NCT04768179. Safety & Efficacy of Low Dose Aspirin / Ivermectin Combination Therapy for Treatment of Covid-19 Patients (IVCOM). clinicaltrials.gov/ct2/show/NCT04768179 (first received 24 February 2021).
NCT04808895 {published data only}
- NCT04808895. Acetylsalicylic Acid in the Prevention of Severe SARS-CoV2 Pneumonia in Hospitalised Patients With COVID-19 (Asperum). clinicaltrials.gov/ct2/show/NCT04808895 (first received 22 March 2021).
NCT04937088 {published data only}
- NCT04937088. Outpatient Liquid Aspirin (OLA) (OLA COVID). clinicaltrials.gov/ct2/show/NCT04937088 (first received 23 June 2021).
NCT05073718 {published data only}
- NCT05073718. Acetylsalicylic Acid in COVID-19 (ASA-SARS). clinicaltrials.gov/ct2/show/NCT05073718 (first received 11 October 2021).
Sharma 2021 {published data only}
- CTRI: 2021/04/032648. Study to access effectiveness of new drug therapy (aspirin,atorvastatin and nicorandil) in covid-19. trialsearch.who.int/Trial2.aspx?TrialID=CTRI/2021/04/032648 (first received 4 August 2021).
- Sharma A, Sharma C, Raina S, Singh B, Dadhwal DS, Dogra V, et al. A randomized open-label trial to evaluate the efficacy and safety of triple therapy with aspirin, atorvastatin, and nicorandil in hospitalised patients with SARS Cov-2 infection: A structured summary of a study protocol for a randomized controlled trial. Trials 2021;22(451):1-3. [DOI: 10.1186/s13063-021-05361-y] [DOI] [PMC free article] [PubMed] [Google Scholar]
Additional references
Al Duhailib 2022
- Al Duhailib Z, Oczkowski S, Polok K, Fronczek J, Szczeklik W, Piticaru J et al. Venous and arterial thrombosis in COVID-19: An updated narrative review. Journal of Infection and Public Health June 2022;15(6):689-702. [DOI: 10.1016/j.jiph.2022.05.003.] [DOI] [PMC free article] [PubMed] [Google Scholar]
Al‐Abdouh 2022
- Al-Abdouh A, Abusnina W, Mhanna M, Radideh Q, Alzu'bi H, Rmilah AA, et al. P2Y12 inhibitors versus aspirin monotherapy for long-term secondary prevention of atherosclerotic cardiovascular disease events: A systematic review and meta-analysis. Current Problems in Cardiology 2022;47(10):101292. [DOI: 10.1016/j.cpcardiol.2022.101292] [DOI] [PubMed] [Google Scholar]
Awtry 2000
- Awtry EH, Loscalzo J. Aspirin. Circulation 2000;101(10):1206-18. [DOI: 10.1161/01.cir.101.10.1206] [DOI] [PubMed] [Google Scholar]
Bompard 2020
- Bompard F, Monnier H, Saab I, Tordjman M, Abdoul H, Fournier L, et al. Pulmonary embolism in patients with COVID-19 pneumonia. European Respiratory Journal 30 July 2020;56(56):1. [DOI: 10.1183/13993003.01365-2020] [DOI] [PMC free article] [PubMed] [Google Scholar]
Buitrago‐Garcia 2020
- Buitrago-Garcia D, Egli-Gany D, Counotte M J, Hossmann S, Imeri H, Ipekci Aziz M, et al. Occurrence and transmission potential of asymptomatic and presymptomatic SARS-CoV-2 infections: a living systematic review and meta-analysis. PLoS Medicine 2020;17(9):e1003346-e1003346. [DOI: 10.1371/journal.pmed.1003346] [DOI] [PMC free article] [PubMed] [Google Scholar]
CEOsys
- CEOsys: Living evidence synthesis as the basis for decisions in the COVID-19 pandemic. covid-evidenz.de/what-is-ceosys/.
COMET 2021
- Core outcome set developers’ response to COVID-19. Available from www.comet-initiative.org/Studies/Details/1538 (accessed 22 March 2021).
COVID‐19 Excess Mortality Collaborators
- COVID-19 Excess Mortality Collaborators. Estimating excess mortality due to the COVID-19 pandemic: a systematic analysis of COVID-19-related mortality, 2020-21. Lancet 10 March 2022;S0140-6736(21)02796-3:1513-1536. [DOI: ] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Cui 2020
- Cui S, Chen S, Li X, Liu S, Wang F. Prevalence of venous thromboembolism in patients with severe novel coronavirus pneumonia. Journal of Thrombosis and Haemostasis 2020;18(18):1421-24. [DOI: 10.1111/jth.14830] [DOI] [PMC free article] [PubMed] [Google Scholar]
Eikelboom 2012
- Eikelboom JW, Hirsh J, Spencer FA, Baglin TP, Weitz JI. Antiplatelet drugs: Antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012;141(2 Suppl):e89S-e119. [DOI] [PMC free article] [PubMed] [Google Scholar]
EndNote 2013 [Computer program]
- EndNote X9. The EndNote Team. Clarivate, 2013.
Ghosn 2021
- Ghosn L, Chaimani A, Evrenoglou T, Davidson M, Graña C, Schmucker C, et al. Interleukin-6 blocking agents for treating COVID-19: a living systematic review. Cochrane Database of Systematic Reviews 18. March 2021, Issue 3. Art. No: CD013881. [DOI: 10.1002/14651858.CD013881] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Grundeis 2023
- Grundeis F, Ansems K, Dahms K, Thieme V, Metzendorf MI, Skoetz N, et al. Remdesivir for the treatment of COVID-19. Cochrane Database of Systematic Reviews 25. January 2023, Issue 1. Art. No: CD014962. [DOI: 10.1002/14651858.CD014962] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hashemzadeh 2008
- Hashemzadeh M, Furukawa M, Goldsberry S, Movahed MR. Chemical structures and mode of action of intravenous glycoprotein IIb/IIIa receptor blockers: A review. Journal of Clinical and Experimental Cardiology 2008;13(4):192-7. [PMC free article] [PubMed] [Google Scholar]
Higgins 2021a
- Higgins JPT, Thomas J, Chandler J, Cumpston M, Li T, Page M J, Welch VA ( editors ). Cochrane Handbook for Systematic Reviews of Interventions version 6.2 (updated February 2021). Available from: training.cochrane.org/handbook 2021.
Higgins 2021b
- Higgins JP, Savović J, Page MJ, Elbers RG, Sterne JA. Chapter 8: Assessing risk of bias in a randomized trial. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.2 (updated February 2021). Available from training.cochrane.org/handbook 2021.
Higgins 2021c
- Deeks JJ, Higgins JP, Altman DG, editor(s). Chapter 10: Analysing data and undertaking meta-analyses. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al, editor(s). Cochrane Handbook for Systematic Reviews of Interventionsversion 6.2 (updated February 2021). Cochrane, 2021. Available from training.cochrane.org/handbook.
Higgins 2021d
- Higgins JP, Tianging L, Deeks J J, editor(s). Chapter 6: Choosing effect measures and computing estimates of effect. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.2 (updated September 2021). Available from training.cochrane.org/handbook 2021.
Higgins 2021e
- Schünemann HJ, Higgins JP, Vist GE, Glasziou P, Akl EA, Skoetz N, et a l. Chapter 14: Completing ‘Summary of findings’ tables and grading the certainty of the evidence. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al (editors). Cochrane Handbook for Systematic Reviews of Interventions version 6.2 (updated February 2021). Cochrane, 2021. Available from www.training.cochrane.org/handbook.
Huang 2020
- Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020;395(10223):497-506. [DOI: 10.1016/S0140-6736(20)30183-5] [DOI] [PMC free article] [PubMed] [Google Scholar]
Iannizzi 2023
- Iannizzi C, Chai KL, Piechotta V, Valk SJ, Kimber C, Monsef I, et al. Convalescent plasma for people with COVID‐19: a living systematic review. Cochrane Database of Systematic Reviews 2023, Issue 1. Art. No: CD013600. [DOI: 10.1002/14651858.CD013600.pub5] [DOI] [PMC free article] [PubMed] [Google Scholar]
Johns Hopkins 2021
- Johns Hopkins University & Medicine Coronavirus Resource Center. Mortality analyses. Available from coronavirus.jhu.edu/data/mortality (accessed at 4 January 2021).
Kalyanasundaram 2011
- Kalyanasundaram A, Lincoff AM. Managing adverse effects and drug-drug interactions of antiplatelet agents. Nature Reviews Cardiology 2011;8(10):592-600. [DOI] [PubMed] [Google Scholar]
Katsoularis 2022
- Katsoularis I, Fonseca-Rodriguez O, Farrington P, Jerndal H, Lundevaller EH, Sund M et al. Risks of deep vein thrombosis, pulmonary embolism, and bleeding after covid-19: nationwide self-controlled cases series and matched cohort study. BMJ 06 April 2022;e069590:377. [DOI: 10.1136/bmj-2021-069590] [DOI] [PMC free article] [PubMed] [Google Scholar]
Khan 2020
- Khan SU, Singh M, Valavoor S, Khan MU, Lone AN, Khan MZ, Khan MS, Mani P, Kapadia SR, Michos ED, Stone GW, Kalra A, Bhatt DL. Dual Antiplatelet Therapy After Percutaneous Coronary Intervention and Drug-Eluting Stents: A Systematic Review and Network Meta-Analysis. Circulation 13 October 2013;142(15):1425-1436. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Klok 2020
- Klok FA, Kruip MJHA, Meer NJM, Arbous MS, Gommers D, Kant KM, et al. Confirmation of the high cumulative incidence of thrombotic complications in critically ill ICU patients with COVID-19: An updated analysis. Thrombosis Research 2020;191:148-50. [DOI: 10.1016/j.thromres.2020.04.041] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kramer 2022
- Kramer A, Prinz C, Fichtner F, Fischer AL, Thieme V, Grundeis F, et al. Janus kinase inhibitors for the treatment of COVID-19. Cochrane Database of Systematic Reviews 13. June 202, Issue 6. Art. No: CD015209. [DOI: 10.1002/14651858.CD015209] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kreuzberger 2021
- Kreuzberger N, Hirsch C, Chai KL, Tomlinson E, Khosravei Z, Popp M et al. SARS‐CoV‐2‐neutralising monoclonal antibodies for treatment of COVID‐19. Cochrane Database of Systematic Reviews 2021, Issue 9. Art. No: CD013825. [DOI: 10.1002/14651858.CD013825.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Krötz 2008
- Krötz F, Sohn HY, Klauss V. Antiplatelet drugs in cardiological practice: established strategies and new developments. Vascular Health and Risk Management 2008;4(3):637-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kunapuli 2003
- Kunapuli SP, Dorsam RT, Kim S, Quinton TM. Platelet purinergic receptors. Elsevier 22 January 2003;3(2):175-180. [DOI: 10.1016/S1471-4892(03)00007-9] [DOI] [PubMed] [Google Scholar]
Lauer 2020
- Lauer SA, Grantz KH, Bi Q, Jones FK, Zheng Q, Meredith HR, et al. The incubation period of coronavirus disease 2019 (COVID-19) from publicly reported confirmed cases: estimation and application. Annals of Internal Medicine 2020;172(9):577–82-577–82. [DOI: 10.7326/M20-0504] [DOI] [PMC free article] [PubMed] [Google Scholar]
Li 2021
- Li T, Higgins JP, Deeks JJ. Chapter 5: Collecting data. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.2 (updated February 2021). Cochrane, 2021. Available from training.cochrane.org/handbook.
Liang 2020
- Liang W, Guan W, Chen R, Wang W, Li J, Xu K, et al. Cancer patients in SARS-CoV-2 infection: a nationwide analysis in China. Lancet Oncology 2020;21(3):335-7. [DOI: 10.1016/S1470-2045(20)30096-6] [DOI] [PMC free article] [PubMed] [Google Scholar]
Liao 2020
- Liao SC, Shao SC, Chen YT, Chen YC, Ming JH. Incidence and mortality of pulmonary embolism in COVID-19: a systematic review and meta-analysis. Critical Care 2020;24:464. [DOI: 10.1186/s13054-020-03175-z] [DOI] [PMC free article] [PubMed] [Google Scholar]
MAGICapp [Computer program]
- MAGICapp. Brønnøysund (NOR): Norwegian MAGIC Evidence Ecosystem Foundation (powered by UserVoice Inc.), 2020.
Marshall 2020
- Marshall JC, Murthy S, Diaz J, Adhikari NK, Angus DC, Arabi YM, et al. A minimal common outcome measure set for COVID-19 clinical research. Lancet Infectious Diseases 2020;20(8):e192-7. [DOI: 10.1016/S1473-3099(20)30483-7] [DOI] [PMC free article] [PubMed] [Google Scholar]
Menter 2020
- Menter T, Haslbauer JD, Nienhold R, Savic S, Hopfer H, Deigendesch A, et al. Postmortem examination of COVID-19 patients reveals diffuse alveolar damage with severe capillary congestion and variegated findings in lungs and other organs suggesting vascular dysfunction. Histopathology 5. July 2020;77(2):198-209. [DOI: 10.1111/his.14134] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Microsoft 2018 [Computer program]
- Mircosoft Excel. Microsoft Corporation. Microsoft Corporation, 2018.
Middeldorp 2020
- Middeldorp S, Coppens M, Haaps TF, Foppen M, Vlaar AP, Müller MC, et al. Incidence of venous thromboembolism in hospitalized patients with COVID‐19. Journal of Thrombosis and Haemostasis 2020;18:1995-2002. [DOI: 10.1111/jth.14888] [DOI] [PMC free article] [PubMed] [Google Scholar]
Minhas 2022
- Minhas JS, Chithiramohan T, Wang X, Barnes SC, Clough RH, Kadicheeni M, et al. Oral antiplatelet therapy for acute ischaemic stroke. Cochrane Database of Systematic Reviews 2022, Issue 1(1). Art. No: CD000029. [DOI: 10.1002/14651858.CD000029.pub4] [DOI] [PMC free article] [PubMed] [Google Scholar]
Moher 2009
- Moher D, Liberati A, Tetzlaff J, Altman DG. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLOS Medicine 2009;6(7):e1000097. [DOI: 10.1371/journal.pmed.1000097] [DOI] [PMC free article] [PubMed] [Google Scholar]
Najm 2021
- Najm A, Alunno A, Mariette X, Terrier B, De Marco G, Emmel J, et al. Pathophysiology of acute respiratory syndrome coronavirus 2 infection: a systematic literature review to inform EULAR points to consider. RMD Open 2021;7(1):e001549. [DOI: 10.1136/rmdopen-2020-001549] [DOI] [PMC free article] [PubMed] [Google Scholar]
Osborne 2021
- Osborne TF, Veigulis ZP, Arreola DM, Mahajan SM, Röösli E, Curtin CM. Association of mortality and aspirin prescription for COVID-19 patients at the Veterans Health Administration. PLoS One 2021;16(2):e0246825. [DOI: 10.1371/journal.pone.0246825] [DOI] [PMC free article] [PubMed] [Google Scholar]
Parmar 1998
- Parmar MK, Torri V, Stewart L. Extracting summary statistics to perform meta-analyses of the published literature for survival endpoints. Statistics in Medicine 1998;17(24):2815-34. [DOI: ] [DOI] [PubMed] [Google Scholar]
Piechotta 2021
- Piechotta V, Andreas M, Fischer AL, Estcourt LJ, Monsef I, Fichtner F, et al. Antiplatelet agents for the treatment of COVID-19 (part of German Ecosystem CEO-Sys). www.crd.york.ac.uk/prospero/display_record.php?ID=CRD42021256351 2021.
Reis 2022
- Reis S, Metzendorf MI, Kuehn R, Popp M, Gagyor I, Kranke P, et al. Nirmatrelvir combined with ritonavir for preventing and treating COVID-19. Cochrane Database of Systematic Reviews 20. September 2022, Issue 9. Art. No: CD015395. [DOI: 10.1002/14651858.CD015395.pub2] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
REMAP‐CAP 2023
- REMAP-CAP Writing Committee for the REMAP-CAP Investigators. Long-term (180-day) outcomes in critically ill patients with COVID-19 in the REMAP-CAP randomized clinical trial. JAMA 2023;329(1):39-51. [DOI: 10.1001/jama.2022.23257] [DOI] [PMC free article] [PubMed] [Google Scholar]
RevMan Web 2022 [Computer program]
- Review Manager Web (RevMan Web). Version Version: 4.10.0. Cochrane, 2022. Available at revman.cochrane.org.
Sandercock 2014
- Sandercock PAG, Counsell C, Tseng MC, Cecconi E. Oral antiplatelet therapy for acute ischaemic stroke. Cochrane Database of Systematic Reviews 2014, Issue 3. Art. No: CD000029. [DOI: 10.1002/14651858.CD000029.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Santesso 2020
- Santesso N, Glenton C, Dahm P, Garner P, Akl A, Alper B, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. Journal of Clinical Epidemiology 2020;119:126-35. [DOI: 10.1016/j.jclinepi.2019.10.014] [DOI] [PubMed] [Google Scholar]
Skoetz 2020
- Skoetz N, Goldkuhle M, Van Dalen EC, Akl EA, Trivella M, Mustafa RA, et al. GRADE guidelines 27: how to calculate absolute effects for time-to-event outcomes in summary of findings tables and Evidence Profiles. Journal of Clinical Epidemiology 2020;118:124-31. [DOI: 10.1016/j.jclinepi.2019.10.015] [DOI] [PubMed] [Google Scholar]
Sterne 2019
- Sterne JAC, Savović J, Page MJ, Elbers RG, Blencowe NS, Boutron I, et al. RoB 2: a revised tool for assessing risk of bias in randomised trials. BMJ 2019;366:l4898. [DOI: 10.1136/bmj.l4898] [DOI] [PubMed] [Google Scholar]
Struyf 2020
- Struyf T, Deeks JJ, Dinnes J, Takwoingi Y, Davenport C, Leeflang MM, et al. Signs and symptoms to determine if a patient presenting in primary care or hospital outpatient settings has COVID-19 disease. Cochrane Database of Systematic Reviews 2020, Issue 7. Art. No: CD013665. [DOI: 10.1002/14651858.CD013665] [DOI] [PMC free article] [PubMed] [Google Scholar]
Tierney 2007
- Tierney JF, Stewart LA, Ghersi D, Burdett S, Sydes MR. Practical methods for incorporating summary time-to-event data into meta-analysis. Trials 2007;8:16. [DOI: 10.1186/1745-6215-8-16] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wagner 2022
- Wagner C, Griesel M, Mikolajewska A, Metzendorf MI, Fischer AL, Stegemann M, et al. Systemic corticosteroids for the treatment of COVID-19: Equity-related analyses and update on evidence. Cochrane Database of Systematic Reviews 2022, Issue 11. Art. No: CD014963. [DOI: 10.1002/14651858.CD014963.pub2] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wang 2021
- Wang Y, Ao G, Nasr B, Qi X. Effect of antiplatelet treatments on patients with COVID-19 infection: A systematic review and meta-analysis. The American Journal of Emergency Medicine 2021;43:27-30. [DOI: 10.1016/j.ajem.2021.01.016] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Warner 2011
- Warner TD, Nylander S, Whatling C. Anti-platelet therapy: cyclo-oxygenase inhibition and the use of aspirin with particular regard to dual anti-platelet therapy. British Journal of Clinical Pharmacology 2011;72(4):619-33. [DOI: 10.1111/j.1365-2125.2011.03943.x] [DOI] [PMC free article] [PubMed] [Google Scholar]
WHO 2021c
- World Health Organization. Update on Omicron. www.who.int/news/item/28-11-2021-update-on-omicron 2021.
WHO 2021e
- World Health Organization. COVID-19 Clinical management: living guidance, 25 January 2021. apps.who.int/iris/handle/10665/338882 (accessed 15 April 2021).
WHO 2022c
- World Health Organization. Draft landscape of COVID-19 candidate vaccines. www.who.int/publications/m/item/draft-landscape-of-covid-19-candidate-vaccines (accessed 25 June 2022).
WHO 2023
- World Health Organization. WHO Coronavirus Disease (COVID-19) Dashboard. covid19.who.int (accessed 11 February 2023).
WHO living guideline 2023
- WHO living guideline. Therapeutics and COVID-19: Living guideline. www.who.int/publications/i/item/WHO-2019-nCoV-therapeutics-2023.1 13.01.2023.
WHO2021a
- World Health Organization. Report of the WHO‐China Joint Mission on coronavirus disease 2019 (COVID‐19). www.who.int/docs/default-source/coronaviruse/who-china-joint-mission-on-covid-19-final-report (accessed 22 March 2021).
Wijaya 2021
- Wijaya I, Andhika R, Huang I, Purwiga A, Budiman KY. The effects of aspirin on the outcome of COVID-19: A systematic review and meta-analysis. Clinical Epidemiology and Global Health 2021;12:100883. [DOI: 10.1016/j.cegh.2021.100883] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Williamson 2020
- Williamson E, Walker AJ, Bhaskaran KJ, Bacon S, Bates C, Morton CE, et al. Factors associated with COVID-19-related death using OpenSAFELY. Nature 2020;584:430-6. [DOI: 10.1038/s41586-020-2521-4] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wong 2011
- Wong PF, Chong LY, Mikhailidis DP, Robless P, Stansby G. Antiplatelet agents for intermittent claudication. Cochrane Database of Systematic Reviews 2011, Issue 11. Art. No: CD001272. [DOI: 10.1002/14651858.CD001272.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
World Bank 2022
- GNI per capita, Atlas method (current US$). https://data.worldbank.org/indicator/NY.GNP.PCAP.CD (assessed xx month year).
Wu 2020
- Wu Z, McGoogan J M. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: summary of a report of 72314 cases from the Chinese Center for Disease Control and Prevention. JAMA 2020;323(13):1239-1242. [DOI: 10.1001/jama.2020.2648] [DOI] [PubMed] [Google Scholar]