

Key Points
-
•
Molecular profiling of MZL may assist in the rational use of BTK inhibitor therapy.
-
•
BTK and PLCG2 mutations confer acquired resistance of MZL to BTK inhibition.
Visual Abstract
Abstract
Using tissue whole exome sequencing (WES) and circulating tumor cell–free DNA (ctDNA), this Australasian Leukaemia & Lymphoma Group translational study sought to characterize primary and acquired molecular determinants of response and resistance of marginal zone lymphoma (MZL) to zanubrutinib for patients treated in the MAGNOLIA clinical trial. WES was performed on baseline tumor samples obtained from 18 patients. For 7 patients, ctDNA sequence was interrogated using a bespoke hybrid-capture next-generation sequencing assay for 48 targeted genes. Somatic mutations were correlated with objective response data and survival analysis using Fisher exact test and Kaplan-Meier (log-rank) method, respectively. Baseline WES identified mutations in 33 of 48 (69%) prioritized genes. NF-κB, NOTCH, or B-cell receptor (BCR) pathway genes were implicated in samples from 16 of 18 patients (89%). KMT2D mutations (n = 11) were most common, followed by FAT1 (n = 9), NOTCH1, NOTCH2, TNFAIP3 (n = 5), and MYD88 (n = 4) mutations. MYD88 or TNFAIP3 mutations correlated with improved progression-free survival (PFS). KMT2D mutations trended to worse PFS. Acquired resistance mutations PLCG2 (R665W/R742P) and BTK (C481Y/C481F) were detected in 2 patients whose disease progressed. A BTK E41K noncatalytic activating mutation was identified before treatment in 1 patient who was zanubrutinib-refractory. MYD88, TNFAIP3, and KMT2D mutations correlate with PFS in patients with relapsed/refractory MZL treated with zanubrutinib. Detection of acquired BTK and PLCG2 mutations in ctDNA while on therapy is feasible and may herald clinical disease progression. This trial was registered at https://anzctr.org.au/ as #ACTRN12619000024145.
Introduction
Marginal zone lymphoma (MZL) is a heterogeneous, indolent, but incurable B-cell non-Hodgkin lymphoma (NHL) characterized by cellular dependency on B-cell receptor (BCR) signaling, leading to the activation of NF-κB and related pathways.1 According to the WHO 2022 classification of hematological malignancies, 4 recognized subtypes of MZL in adults exist: extranodal MZL of mucosa-associated lymphoid tissue (MALT), splenic (SMZL), nodal (NMZL), and primary cutaneous MZL.2 The genomic landscape of MZL is less well-defined than that of other B-cell NHLs, but genes involving pathways regulating the marginal zone development, such as the NOTCH pathway (NOTCH1, NOTCH2, SPEN, CREBBP, and DTX1), NF-κB signaling (MYD88, TNFAIP3 (A20), BIRC3, TRAF3, and CXCR4), or BCR signaling (CARD11, CXCR4, and KLHL6) are often affected.3, 4, 5, 6 Notably, primary activating mutations of BTK, such as the E41K mutation, which are rarely reported in diffuse large B-cell lymphoma have not been observed in indolent lymphomas such as MZL.7,8 Although responses to frontline chemoimmunotherapy for the treatment of MZL are often favorable, the disease is characterized by frequent relapses, and there is no established standard of care for subsequent lines of treatment.9
A previous single-arm, open-label, phase 2 clinical trial of 63 patients with relapsed/refractory MZL (rrMZL) treated with the BTK inhibitor (BTKi) ibrutinib demonstrated a 58% objective response rate (ORR) with 10% complete response (CR), a median duration of response (DOR) of 27.6 months (95% confidence interval [CI]: 12.1 months to not estimable [NE]), a median progression-free survival (PFS) of 15.7 months (95% CI: 12.2-30.4 months), and a median overall survival not reached (95% CI: NE-NE).10 Responses were observed across all MZL subtypes, with biomarker studies identifying patients bearing lymphoma with mutated TNFAIP3 (A20) and MYD88 as more likely to respond. In contrast, lymphomas with mutated KMT2D (MLL) and CARD11 were less likely to respond to ibrutinib. This study did not, however, assess for the emergence of acquired mutations affecting BTK or its enzymatic substrate, PLCG2.11, 12, 13, 14, 15 Mutations in either of these lead to acquired BTKi resistance in chronic lymphocytic leukemia, but data in MZL are limited to a single case study of a patient who was ibrutinib-treated and developed large cell transformation and molecular profiling, identifying acquired BTK (C481S) and PLCG2 (R665W) mutations.11,16
Zanubrutinib, a second-generation BTKi, occupies the BTK-binding site in a concentration-dependent manner similar to that of ibrutinib but with more than 3 times the potency.17 It is also more selective, with fewer off-target effects and, hence, fewer adverse reactions.17,18 MAGNOLIA, a phase 2 study of zanubrutinib in rrMZL, demonstrated excellent tolerability and efficacy.19 At a median follow-up of 15.7 months, the ORR was 68.2% (95% CI: 55.56-79.11) with a CR of 25.8%; the median DOR and PFS were not reached (95% CI: NE-NE), with a 12-month DOR and PFS of 93% and 83%, respectively (95% CI: 79.8-97.7; 95% CI: 70.55-89.93). Only 4 patients (6%) discontinued treatment because of adverse events, none of which were considered treatment-related. The ACE-LY-003 study has also demonstrated favorable clinical activity of acalabrutinib for the treatment of relapsed MZL, with an ORR of 53%, including 13% CR, and a median PFS of 27.4 months.20
The present cooperative trial group correlative study, sponsored by the Australasian Leukaemia and Lymphoma Group, sought to determine whether a baseline molecular profile using whole exome sequencing (WES) could predict primary resistance to zanubrutinib and whether the emergence of resistance mutations in circulating tumor DNA (ctDNA), a component of cell-free DNA (cfDNA), heralds clinical progression in patients treated on the MAGNOLIA clinical trial.
Methods
This study was conducted in accordance with the provisions of the Declaration of Helsinki and approved by the local governing institutional review board. Eighteen patients of the Australasian Leukaemia and Lymphoma Group LS21 correlative study were part of the MAGNOLIA study and provided informed consent before procurement of clinical materials.19
DNA from primary tumor, buccal swabs, and Streck Cell-Free DNA BCT tubes (La Vista, NE) was isolated using commercial isolation kits (Qiagen, Venlo, The Netherlands). Additional information on processing and evaluation of quality metrics are provided in the supplemental Methods. All NGS libraries were constructed using Agilent XTHS reagents and protocols incorporating unique molecular barcoding (Agilent Technologies, CA). For primary tumor, WES was performed using Agilent WES Ver7 reagents; however, bioinformatics analysis was restricted to 48 candidate genes, as listed in supplemental Table 1. This set of genes was selected based on current literature, focusing on those previously reported in MZL studies: those affecting NF-κB, NOTCH, BCR pathways, tumor suppressors/oncogenes, and genes involved in MZL development and related transcription factors/chromatin remodeling as well as those that are commonly occurring.3,4,6 For the sequencing of cfDNA, an NGS bespoke bait capture set for the same 48 genes was used (443 kbp capture area, including a copy number variation [CNV] backbone; SureDesign, Agilent Technologies, CA). Sequencing was performed on a NovaSeq 6000 instrument (Illumina, SP flow cell; 2 × 150 bp chemistry).
Data processing of FASTQ files was performed via an in-house bioinformatic pipeline incorporating unique molecular identifiers (UMI) deduplexing, VarDict for variant calling, and CNVkit for CNV analysis. Variant calls in patient samples (identified using VarDict) were manually curated by inspecting Binary Aligment Map (BAM) files in the Integrative Genomics Viewer.21 Only nonsynonymous mutations were included in the final data set, as per predetermined curation criteria (supplemental Methods). Pathogenicity was assessed using OpenCRAVAT,22 which annotates variants with an impact on protein structure (eg, stop-gain, frame-shift deletions/insertions, and complex substitutions) and integrates online database information, such as ClinVar (version 2022.06.14) and COSMIC (version 94.0.0).23
For the 17 patients (94%) with tumor samples available for WES and treated with zanubrutinib, mutational analysis was correlated with investigator-assessed ORR and PFS using Fisher exact test and the Kaplan-Meier (log-rank) method, respectively (GraphPad Prism version 9.3.1). Mutational plots were visualized using GenVisR, and the represented protein structural model of BTK is described in supplemental Methods. Complete NGS data (primary tumor and ctDNA) is available in the NCBI/SRA repository.
Results
NF-κB, NOTCH, and BCR mutations commonly occur in rrMZL
WES was performed on 19 tumor samples from 18 patients before zanubrutinib therapy. Seventeen patients were administered with zanubrutinib during the study, and 1 patient failed screening but was still eligible for molecular characterization of tumor. Germ line comparison was available for 4 of the patients. The median patient age was 71 years (range: 37-86 years), with a male predominance (67%). All patients received at least 1 prior line of chemoimmunotherapy (median: 1.5; range: 1-4). Patient and tumor sample characteristics are summarized in supplemental Table 2.
Ninety mutations were identified, with multiple mutations of the same gene detected in 7 of 19 tumor samples (37%). Thirty-three (69%) of the candidate genes interrogated were affected (Figure 1; supplemental Table 3). A median of 5 mutations were detected per tumor sample (range: 0-12), with missense mutations predominating (76%; Figure 1). Paired lymph node and gastric tissue were analyzed in 1 patient (MZ17): NOTCH1, NOTCH2, and KMT2D mutations were identified in both samples, but TNFAIP3 and FAS mutations were identified only in the lymph node. One sample with limited tumor tissue available for analysis failed to yield any variants.
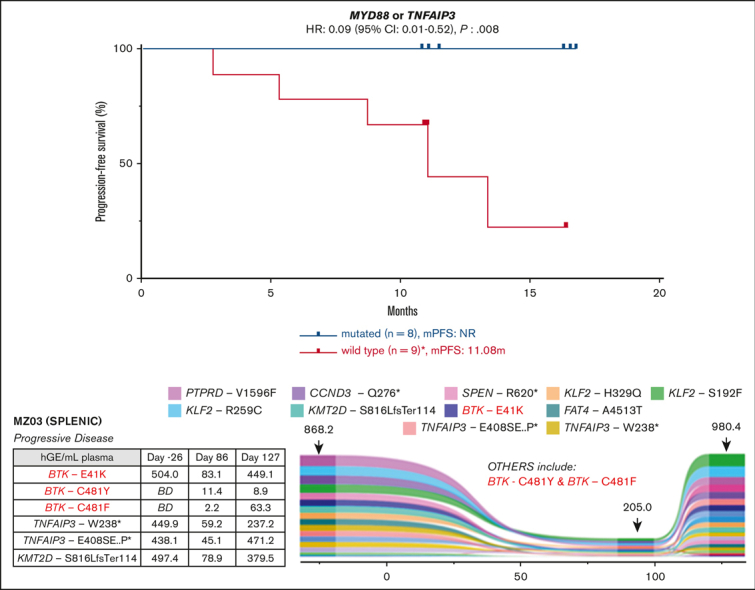
Figure 1.

Comutation plot for variants detected in primary tumor at study entry. (Left) Clustered according to best clinical response: CR Complete response, PR Partial response, SD Stable disease, PD Progressive disease. Unique sample identifier and MZL subtype also represented. Mutation burden is expressed relative to sample with highest number of mutations detected. Only one mutation per gene per tumor sample is represented. (Right) Ribbon diagram of BTK structure showing location of the non-catalytic E41K mutation distant from zanubrutinib binding site.
NF-κB, NOTCH, and BCR pathway gene mutations were detected in 14 (78%), 10 (56%), and 6 (33%) patient samples, respectively. Sixteen of the 18 patients (89%) had 1 of these pathways affected. The remaining genes predominantly affected were related to MZL development, chromatin remodeling, transcription regulation, and cell cycle regulation.
Baseline-screened ctDNA samples were also analyzed for 2 patients (MZ03 and MZ07), for whom tumor WES samples were not available (Figure 1). Before zanubrutinib therapy, TNFAIP3 and KMT2D mutations were detected in both patients, whereas MZ03 ctDNA also harbored a BTK E41K mutation (Figure 1).
NF-κB pathway gene mutations may predict response to zanubrutinib in MZL
Seventeen patients (94% of the cohort) with tumor samples available for WES were treated with zanubrutinib. One patient died from COVID-19 infection while on therapy. The median follow-up was 11.1 months (range, 2.76-16.8 months). There was no correlation between MZL subtype or number of previous lines of therapy and survival (median PFS not reached [NR]; P = 0.876; median PFS NR vs NR vs 10.85; P = 0.439). There was also no correlation between patient tumor mutational profile and response rates; however, most of the cohort achieved a response (ORR: 88% and CR: 24%; supplemental Table 4). The mutational profile did associate with PFS: 7 patients with a tumor sample containing at least 1 KMT2D mutation (total mutations: 11) had a shortened PFS despite 6 of 7 achieving an objective response (median PFS 13.4 months vs NR; P = 0.05; HR: 6.15; 95% CI: 1.00-37.78; Figure 2A). Two of these patients (MZ11 and MZ17) had tumors with concomitant TNFAIP3 mutations and did not undergo disease progression during the follow-up period.
Figure 2.

Molecular predictors of response to zanubrutinib. Kaplan-Meier survival curves showing progression-free survival according to presence or absence of somatic mutations of (A) MYD88 or TNFAIP3 (B) KMT2D, (C) NOTCH1 or NOTCH2. Median follow-up: 11.1 months (2.76-16.80). ∗includes one COVID-19-related death.
MYD88 mutations (including 1 non-L265P) were observed in 4 samples (MZ01, MZ02, MZ14, and MZ33), and TNFAIP3 mutations were observed in 4 samples (MZ11, MZ16, MZ17, and MZ34). MYD88 and TNFAIP3 mutations were mutually exclusive. All 8 of these patients with tumors harboring mutations of MYD88 or TNFAIP3 achieved an objective response and had prolonged PFS (median PFS, NR vs 11.1 months; P = 0.008; HR: 0.09; 95% CI: 0.01-0.52; Figure 2B).
NOTCH1 and NOTCH2 mutations were each observed in 4 samples and co-occurred in MZ17. NOTCH mutations were not associated with the outcome (median PFS, NR vs NR; P = 0.49; HR: 1.89; 95% CI: 0.31-11.34; Figure 2C). CARD11 mutations were not identified in our cohort.
Baseline mutations of PLCG2 were detected in 2 samples (MZ02 and MZ21); however, these mutations (H244R and L704V) are not known to confer BTKi resistance. MZ02 also harbored a MYD88 L265P mutation; the patient achieved a partial response (PR), and their disease did not progress during the census period (PFS: 11.51 months). MZ21 harbored 2 KMT2D mutations and 1 TP53 mutation (affecting the DNA-binding domain in the latter); this patient never achieved an objective response and had disease progression after 2.5 months of zanubrutinib therapy. A TP53 R290C mutation was also detected in MZ15; this patient achieved a CR and did not have disease progression (the follow-up period was 10.85 months). The mutation was in a non-DNA–binding domain with a variant allele frequency (VAF) of 46%.
FAT genes (FAT1, FAT3, or FAT4) were the most frequently affected genes in our cohort, with 13 mutations detected in 6 tumor samples, including 9 in FAT1. However, they did not correlate with clinical outcomes (median PFS NR vs NR; P = 0.54; HR: 0.56; 95% CI: 0.08-3.66). The frequency of other mutations detected (3 or fewer) was too low for meaningful clinical correlation. CNV analysis was performed but was uninformative in terms of association with response to zanubrutinib (supplemental Figure 1).
ctDNA can be used to track mutations and detect acquisition of new mutations conferring BTKi resistance
Longitudinal monitoring to detect the acquisition of resistance mutations was performed in 7 patients at multiple time points using cfDNA isolated from plasma with comparative germ line DNA; 4 of 7 also had baseline formalin-fixed, paraffin-embedded tumor samples available; the other 3 of 7 did not have baseline tumor samples available for WES analysis. The mean and median cfDNA yield were 10.6 ng/mL and 6.9 ng/mL, respectively (range: 2.5-105 ng/mL), with levels increasing at the time of progression. ctDNA analysis was also performed on multiple samples from the 7 patients: 3 responders (MZ01, MZ02, and MZ04), 3 progressors (MZ03, MZ05, and MZ07), and 1 patient (MZ06) with multiple mutations (n = 5) in WES who had failed screening and did not receive zanubrutinib.
Changes in ctDNA burden during therapy
In the responder cohort, MZ01 and MZ02 had the MYD88 L265P mutation identified in tumor sample WES; both patients achieved PR and demonstrated decreasing mutation burden in ctDNA during therapy (Figure 3A-B). Baseline ctDNA from both patients also demonstrated mutations at screening that were not detected in WES: the KMT2D Q2416H mutation (with decreasing VAF) and the TP53 R248W (stable VAF) in MZ01 and MZ02, respectively. No acquired mutations associated with resistance to BTKi were observed in this cohort.
Figure 3.

Detection of ctDNA mutations and evolution during zanubrutinib therapy for 5 patients. Represented is the relative abundance for each detected mutation (plotted as hGE/mL plasma, calculated from AF and plasma cfDNA concentration) and variation over time (x-axis: days). Numbers indicated above the plot at the sample time-points are the calculated total ctDNA (hGE/mL plasma). Associated tables present values for all BTK, PLCG2, TNFAIP3, KMT2D, MYD88, NOTCH2 and TP53 mutations. MZ04 not shown as only one time point (D254) was available for analysis. BD = below detection.
Emergence of new mutations in ctDNA conferring resistance to BTKi during therapy
Baseline tumor WES was not available for the 3 patients who experienced disease progression on zanubrutinib and had ctDNA available (the progressor cohort). Samples from patients MZ03 (Figure 3C) and MZ07 (Figure 3E) demonstrated the acquisition of mutations associated with BTKi resistance. Patient MZ07 achieved a PR but subsequently had disease progression on day 253, with PLCG2 R665W and L742P mutations observed in the ctDNA at progression but not at baseline (Figure 3E). MZ03 ctDNA demonstrated a BTK E41K mutation before zanubrutinib therapy; this patient’s disease progressed early (day 86) and had new detectable BTK C481F and C481Y mutations, in addition to the persistence of the BTK E41K mutation. This patient continued therapy for another 41 days, with a repeat sample showing the VAF of these respective mutations changing over time (Figure 3C). The BTK E41K mutation was detected at all 3 time points, including screening. MZ05 did not have baseline tumor WES or ctDNA samples available (Figure 3D). Samples for ctDNA were available after the commencement of zanubrutinib and did not demonstrate the acquisition of BTK or PLCG2 mutations, but notably, a mutation affecting BIRC3 and 3 TP53 mutations (R280G, I255F, and R213∗) were detected at the earliest time point sample available (day 84) as well as progression (day 334) samples.
Differences between ctDNA and baseline tissue WES
Three patients (MZ01, MZ02, and MZ06) had tumor WES and screening ctDNA available for comparison. Of the 13 mutations detected in the WES from these patients, 8 (62%) mutations were detected in the ctDNA. In contrast, 2 mutations detected in the screening ctDNA were not present in the WES. Five mutations, including KMT2D and TNFAIP3, identified in the tumor sample of the screen-failure patient (MZ06), were detectable in the ctDNA. Four additional mutations (CACNA1H, EP300, and 2 TBL1XR1) were detected in the ctDNA but not present in the WES. A PLCG2 H244R mutation detected in the WES of MZ02 was not detected in the ctDNA. The complete list of genes detected via WES and ctDNA can be found in supplemental Tables 3 and 5, respectively.
Discussion
Chronic active BCR–mediated signaling has been identified as a critical step in MZL pathogenesis, providing a rationale for BTKi as a therapeutic tool.1 Furthermore, the role of genes affecting BCR and related pathways, particularly NF-κB and NOTCH, has been established in several studies of B-cell malignancies, including MZL.3,4,6,24 In this cohort of patients with rrMZL, all but 2 patients (89%) had at least 1 of these pathways affected by a mutation determined by baseline WES. Our findings are consistent with those of the available literature: Noy et al described a response to ibrutinib among patients with MZL harboring the MYD88 or TNFAIP3 mutation and a worse outcomes in those with KMT2D mutations.10 This indicates an expected overlap between the response and resistance determinants of ibrutinib and zanubrutinib, consistent with a common therapeutic target. NOTCH mutations, which are associated with improved responses to BTKi in other lymphoproliferative disorders such as mantle cell lymphoma and chronic lymphocytic leukemia, did not appear to be associated with response in our MZL cohort nor that of Noy et al, although this observation is limited by the small sample size.10,25,26
The biological mechanisms underpinning BTKi responsiveness have not been fully elucidated but, at least in the case of the MYD88 mutation, the finding is not surprising. Somatic activating mutations of MYD88 promote toll-like receptor activation via BTK interaction and NF-κB signaling.27, 28, 29 BTK inhibition has proven efficacious in patients with Waldenström macroglobulinemia, >90% of whom harbor the MYD88 L265P mutation.2,30 BTKis can also be effective in patients with Waldenström macroglobulinemia harboring wild-type MYD88, likely because of the presence of other mutations in NF-κB pathway genes.31 Loss of the ubiquitin-editing enzyme TNFAIP3 results in increased NF-κB signaling, which can be counteracted by a BTKi.32,33 Our findings support those recently reported in which only one mutation affecting NF-κB pathway signaling (MYD88 or TNFAIP3) is sufficient to affect BTKi response, because these mutations were mutually exclusive in both our WES-assessed cohort and other studies.3,4
The biological consequences of KMT2D mutations in MZL development are less well established. KMT2D encodes a histone methyltransferase, which can function as a tumor suppressor; it is affected by NF-κB signaling, and a deficiency of KMT2D perturbs germinal center B-cell development and promotes lymphomagenesis.34,35 Interestingly, 2 of the patients harboring KMT2D-mutated MZL whose disease did not progress during zanubrutinib therapy also harbored TNFAIP3 mutations.
We also interrogated genes known to be commonly mutated in MZL.3,4,6 Of these, mutation of FAT genes was most frequently observed. FAT genes encode atypical cadherins, which can exhibit known tumor suppressor activity in solid organ malignancies.36 Although commonly mutated in MZL, their role in the disease’s development remains unclear, and their presence was not associated with progression outcomes in our cohort treated with zanubrutinib.
Using a bespoke hybrid-capture bait technology with unique molecular indexes, we were able to detect and track the MYD88 L265P mutation in 2 of our patients with a sensitivity of 0.1%. This is relevant given the predictive value of the MYD88 mutation for response to BTKis. The decreasing, but persistent, VAF is consistent with the PRs achieved by both patients, which were ongoing at the time of final sampling.
Two patients with disease progression had detectable PLCG2 (R665W, RL42P) and BTK C481Y/F mutations at time of progression, confirming that patients with MZL appear susceptible to the same acquired resistance mutations seen in CLL.11 The latter case was informative for 2 reasons: firstly, the baseline ctDNA sample (before zanubrutinib commencement) harbored the BTK E41K mutation previously reported in diffuse large B-cell lymphoma but not in the genomic landscapes of indolent NHL, including MZL.7,8 This mutation in a noncatalytic site in the pleckstrin-homology domain is remote from the catalytic site where zanubrutinib binding occurs and has previously been validated as an activating mutation using preclinical modeling.8,37 To our knowledge, this novel finding in our cohort represents the first report of an activating BTK mutation before BTKi in a patient with indolent NHL.8 Secondly, there was evidence of clonal selection at a nucleotide level within the BTK catalytic site, consistent with the complex resistance patterns that occur at the single-cell level.38 Such mutations would not have been detected via digital droplet polymerase chain reaction, which is one of the recommended technologies for ctDNA detection.39 The third patient in the progressor cohort did not have mutations of BTK or PLCG2 detected; however, mutation of BIRC3 may potentially account for the disease progression, because this has been demonstrated to confer resistance to BTKi in other B-cell NHL via a noncanonical NF-κB pathway activation.40,41 Patients who develop the BTK C481S mutation may respond to noncovalent BTKi (pirtobrutinib, ARQ-351, fenebrutinib, and vecabrutinib), though these other agents do not overcome other select BTK or downstream PLCG2 mutations.8,42
Our study is limited by small numbers and the germ line comparator being available for only 22% of the patients. We also cannot exclude that some of the mutations are germ-line variants, especially those with VAFs approximating 50% (eg, MZ15 TP53 R290C), and may not be pathogenic in certain oncogenic contexts. Furthermore, we predominantly limited our analysis to the coding regions of the 48 genes implicated in signaling pathways related to BTKi and MZL development. Although our candidate gene list was comprehensive for these pathways, we cannot exclude that there may be other mutations, including those affecting noncoding regions that may be associated with response to BTKi.
Furthermore, not all the mutations detected in WES were detected in ctDNA. Concordance between tumor-based and ctDNA-based genotyping is reported at >70%, but it is related to tumor shedding and resultant cfDNA concentrations.39 In general, low-grade lymphomas have lower cfDNA concentrations than aggressive lymphomas.39,43 The cfDNA concentration in our cohort (mean: 10.6 ng/mL; median: 6.9 ng/mL) compares favorably to that of ctDNA studies in other low-grade lymphomas of ∼1.15 to 6.5 ng/mL but is still significantly lower than aggressive lymphomas (∼650 ng/mL).43,44 Even with an assay sensitivity of 0.1%, not all mutations detected in the tumor will be present in the ctDNA without sufficient tumor shedding.
In contrast, several mutations present in ctDNA were not identified in WES. This is commonly observed and likely represents spatial tumor heterogeneity, as demonstrated in the patient where we sampled 2 different tumor samples.39 We cannot exclude that other patients may have demonstrated intratumoral heterogeneity, but obtaining multiple tissue samples from patients was not feasible in this study. There is also the possibility that some of the mutations do not originate in the lymphoma itself but represent either clonal hematopoiesis of indeterminate potential or shedding from other undetected malignancies (ie, TP53 R248W in MZ02).
In summary, the correlative studies described herein have demonstrated that mutations in MYD88 and TNFAIP3 associate with improved PFS, and mutations in KMT2D may associate with reduced PFS for patients with rrMZL treated with zanubrutinib. The novel finding of the noncatalytic BTK E41K mutation in our cohort also describes a potential primary resistance mechanism to BTKi treatment. The hypothesized ability to detect mutations potentially predicting response via noninvasive sampling (as exemplified by the detection of the MYD88 L265P mutation in ctDNA) was demonstrated. Furthermore, our hypothesis of acquired resistance to BTKi mediated through acquired BTK and PLCG2 mutations was supported and may herald clinical progression. These studies have been informative for the use of BTKis for rrMZL in terms of predicting the primary response or resistance and demonstrating acquired resistance mechanisms. Larger cohort studies should be performed to provide the power to validate our observations with a view to optimizing the selection of patients with rrMZL that may derive clinical benefit from therapeutic BTK inhibition.
Conflict-of-interest disclosure: J.S. has received research funding from Amgen, Astex, and Bristol Myers Squibb/Celgene; served on advisory board or received consultancy fees from Astellas, Bristol Myers Squibb, Novartis, Mundipharma and Otsuka; and speakers bureau from Mundipharma. E.A.H. has served on advisory boards for Roche, Gilead, Antengene, LINK, Novartis, BeiGene, and Merck Sharp & Dohme and speaker bureau for AstraZeneca, Roche, and Merck, Sharp & Dohme; and received research funding from Roche, Bristol Myers Squibb, Merck KgA, AstraZeneca, Janssen, BeiGene, AbbVie, and Takeda. E.A.H. also reports personal support (Bristol Myers Squibb). J.T. has received research funding from BeiGene, Janssen, Pharmacyclics, Roche, Takeda, and Cellectar. V.R., M.C., and J.L. are employees of and own stock in BeiGene. S.S.O. has received research funding, served as a member of advisory boards, and received honoraria from BeiGene. G.P.G. has received research funding from BeiGene, AbbVie, Janssen, and Merck; advisory board/consultancy fees from Roche, Janssen, Gilead, Bristol Myers Squibb, Merck, Novartis, and Clinigen LINK; and serves on the speakers bureau of Roche. The remaining authors declare no competing financial interests.
Acknowledgments
The Australasian Leukaemia & Lymphoma Group (ALLG) acknowledges the support of our participating member hospitals and patients who consented to participate. ALLG acknowledges the National Blood Cancer Registry for its role in data and the Blood Cancer Therapeutics Laboratory at Monash Health for its role in the laboratory work. Four ALLG-associated sites contributed 4 tumor and buccal samples and cfDNA from 7 patients. BeiGene Ltd provided 14 tumor samples from international patients that had provided consent for tissue use for biomarker studies.
BeiGene Ltd provided financial support for the studies and reviewed the final manuscript.
J.S. is supported by an Australian NHMRC EL2 Fellowship.
Authorship
Contribution: M.T., M.W., J.T., S.S.O., and G.P.G. designed the study; M.T. and M.W. performed experimental studies and data analysis; and all authors contributed to data collection and writing of the manuscript.
Footnotes
Presented in poster form at the European Hematology Association Congress, 12 May 2022.
Complete NGS data (primary tumor and ctDNA) is available in the NCBI/SRA repository (Accession number: PRJNA953925).
The full-text version of this article contains a data supplement.
Supplementary Material
References
- 1.Young RM, Staudt LM. Targeting pathological B cell receptor signalling in lymphoid malignancies. Nat Rev Drug Discov. 2013;12(3):229–243. doi: 10.1038/nrd3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alaggio R, Amador C, Anagnostopoulos I, et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia. 2022;36(7):1720–1748. doi: 10.1038/s41375-022-01620-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rossi D, Trifonov V, Fangazio M, et al. The coding genome of splenic marginal zone lymphoma: activation of NOTCH2 and other pathways regulating marginal zone development. J Exp Med. 2012;209(9):1537–1551. doi: 10.1084/jem.20120904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Spina V, Khiabanian H, Messina M, et al. The genetics of nodal marginal zone lymphoma. Blood. 2016;128(10):1362–1373. doi: 10.1182/blood-2016-02-696757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Braggio E, Dogan A, Keats JJ, et al. Genomic analysis of marginal zone and lymphoplasmacytic lymphomas identified common and disease-specific abnormalities. Mod Pathol. 2012;25(5):651–660. doi: 10.1038/modpathol.2011.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jaramillo Oquendo C, Parker H, Oscier D, Ennis S, Gibson J, Strefford JC. Systematic Review of Somatic Mutations in Splenic Marginal Zone Lymphoma. Sci Rep. 2019;9:1–9. doi: 10.1038/s41598-019-46906-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reddy A, Zhang J, Davis NS, et al. Genetic and functional drivers of Diffuse Large B cell Lymphoma. Cell. 2017;171(2):481–494.e15. doi: 10.1016/j.cell.2017.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang S, Mondal S, Zhao C, et al. Noncovalent inhibitors reveal BTK gatekeeper and auto-inhibitory residues that control its transforming activity. JCI Insight. 2019;4(12):e127566. doi: 10.1172/jci.insight.127566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Broccoli A, Zinzani PL. How do we sequence therapy for marginal zone lymphomas? Hematology Am Soc Hematol Educ Program. 2020;2020(1):295–305. doi: 10.1182/hematology.2020000157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Noy A, de Vos S, Coleman M, et al. Durable ibrutinib responses in relapsed/refractory marginal zone lymphoma: long-term follow-up and biomarker analysis. Blood Adv. 2020;4(22):5773–5784. doi: 10.1182/bloodadvances.2020003121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Woyach JA, Furman RR, Liu TM, et al. Resistance mechanisms for the Bruton's tyrosine kinase inhibitor ibrutinib. N Engl J Med. 2014;370(24):2286–2294. doi: 10.1056/NEJMoa1400029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou Q, Lee GS, Brady J, et al. A hypermorphic missense mutation in PLCG2, encoding phospholipase Cgamma2, causes a dominantly inherited autoinflammatory disease with immunodeficiency. Am J Hum Genet. 2012;91(4):713–720. doi: 10.1016/j.ajhg.2012.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Handunnetti SM, Tang CPS, Nguyen T, et al. BTK Leu528Trp - a potential secondary resistance mechanism specific for patients with chronic lymphocytic leukemia treated with the next generation BTK inhibitor zanubrutinib. Blood. 2019;134(Suppl 1) 170-170. [Google Scholar]
- 14.Zhang SQ, Smith SM, Zhang SY, Lynn Wang Y. Mechanisms of ibrutinib resistance in chronic lymphocytic leukaemia and non-Hodgkin lymphoma. Br J Haematol. 2015;170(4):445–456. doi: 10.1111/bjh.13427. [DOI] [PubMed] [Google Scholar]
- 15.Sedlarikova L, Petrackova A, Papajik T, Turcsanyi P, Kriegova E. Resistance-associated mutations in chronic lymphocytic leukemia patients treated with novel agents. Front Oncol. 2020;10(894):1–10. doi: 10.3389/fonc.2020.00894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Epperla N, Shana’ah AY, Jones D, et al. Resistance mechanism for ibrutinib in marginal zone lymphoma. Blood Adv. 2019;3(4):500–502. doi: 10.1182/bloodadvances.2018029058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu X-J, Xu L, Pang X-J, et al. Progress in the development of small molecular inhibitors of the Bruton’s tyrosine kinase (BTK) as a promising cancer therapy. Bioorg Med Chem. 2021;47:116358. doi: 10.1016/j.bmc.2021.116358. [DOI] [PubMed] [Google Scholar]
- 18.Beigene Ltd . Zanubrutinib (BGB-3111) Investigator’s Brochure. Beigene Ltd; 2022. [Google Scholar]
- 19.Opat S, Tedeschi A, Linton K, et al. The MAGNOLIA trial: zanubrutinib, a next-generation bruton tyrosine kinase inhibitor, demonstrates safety and efficacy in relapsed/refractory marginal zone lymphoma. Clin Cancer Res. 2021;27(23):6323–6332. doi: 10.1158/1078-0432.CCR-21-1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Strati P, Coleman M, Champion R, et al. A phase 2, multicentre, open-label trial (ACE-LY-003) of acalabrutinib in patients with relapsed or refractory marginal zone lymphoma. Br J Haematol. 2022;199(1):76–85. doi: 10.1111/bjh.18368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Robinson JT, Thorvaldsdóttir H, Wenger AM, Zehir A, Mesirov JP. Variant review with the integrative genomics viewer. Cancer Res. 2017;77(21):e31–e34. doi: 10.1158/0008-5472.CAN-17-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pagel KA, Kim R, Moad K, et al. Integrated informatics analysis of cancer-related variants. JCO Clin Cancer Inform. 2020;4:310–317. doi: 10.1200/CCI.19.00132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Skidmore ZL, Wagner AH, Lesurf R, et al. GenVisR: genomic visualizations in R. Bioinformatics. 2016;32(19):3012–3014. doi: 10.1093/bioinformatics/btw325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Braggio E, Dogan A, Keats JJ, et al. Genomic analysis of marginal zone and lymphoplasmacytic lymphomas identified common and disease-specific abnormalities. Modern Pathology. 2012;25(5):651–660. doi: 10.1038/modpathol.2011.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Del Poeta G, Laurenti L, Chiarenza A, et al. Clinical relevance of NOTCH1 mutations in ibrutinib-treated chronic lymphocytic leukemia (CLL) Blood. 2018;132(Suppl 1) 4396-4396. [Google Scholar]
- 26.Agarwal R, Chan Y-C, Tam CS, et al. Dynamic molecular monitoring reveals that SWI–SNF mutations mediate resistance to ibrutinib plus venetoclax in mantle cell lymphoma. Nat Med. 2019;25(1):119–129. doi: 10.1038/s41591-018-0243-z. [DOI] [PubMed] [Google Scholar]
- 27.Leleu X, Eeckhoute J, Jia X, et al. Targeting NF-κB in Waldenstrom macroglobulinemia. Blood. 2008;111(10):5068–5077. doi: 10.1182/blood-2007-09-115170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Watters TM, Kenny EF, O'Neill LAJ. Structure, function and regulation of the Toll/IL-1 receptor adaptor proteins. Immunol Cell Biol. 2007;85(6):411–419. doi: 10.1038/sj.icb.7100095. [DOI] [PubMed] [Google Scholar]
- 29.Ntanasis-Stathopoulos I, Gavriatopoulou M, Fotiou D, Dimopoulos MA. Current and novel BTK inhibitors in Waldenström’s macroglobulinemia. Ther Adv Hematol. 2021;12:1–14. doi: 10.1177/2040620721989586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tam CS, Opat S, D'Sa S, et al. A randomized phase 3 trial of zanubrutinib vs ibrutinib in symptomatic Waldenström macroglobulinemia: the ASPEN study. Blood. 2020;136(18):2038–2050. doi: 10.1182/blood.2020006844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dimopoulos M, Sanz RG, Lee HP, et al. Zanubrutinib for the treatment of MYD88 wild-type Waldenström macroglobulinemia: a substudy of the phase 3 ASPEN trial. Blood Adv. 2020;4(23):6009–6018. doi: 10.1182/bloodadvances.2020003010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vereecke L, Beyaert R, van Loo G. The ubiquitin-editing enzyme A20 (TNFAIP3) is a central regulator of immunopathology. Trends Immunol. 2009;30(8):383–391. doi: 10.1016/j.it.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 33.Wenzl K, Manske MK, Sarangi V, et al. Loss of TNFAIP3 enhances MYD88(L265P)-driven signaling in non-Hodgkin lymphoma. Blood Cancer J. 2018;8(10) doi: 10.1038/s41408-018-0130-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang J, Dominguez-Sola D, Hussein S, et al. Disruption of KMT2D perturbs germinal center B cell development and promotes lymphomagenesis. Nat Med. 2015;21(10):1190–1198. doi: 10.1038/nm.3940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ortega-Molina A, Boss IW, Canela A, et al. The histone lysine methyltransferase KMT2D sustains a gene expression program that represses B cell lymphoma development. Nat Med. 2015;21(10):1199–1208. doi: 10.1038/nm.3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Katoh M. Function and cancer genomics of FAT family genes (review) Int J Oncol. 2012;41(6):1913–1918. doi: 10.3892/ijo.2012.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li T, Tsukada S, Satterthwaite A, et al. Activation of Bruton's tyrosine kinase (BTK) by a point mutation in its pleckstrin homology (PH) domain. Immunity. 1995;2(5):451–460. doi: 10.1016/1074-7613(95)90026-8. [DOI] [PubMed] [Google Scholar]
- 38.Thompson ER, Nguyen T, Kankanige Y, et al. Single-cell sequencing demonstrates complex resistance landscape in CLL and MCL treated with BTK and BCL2 inhibitors. Blood Adv. 2022;6(2):503–508. doi: 10.1182/bloodadvances.2021006211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lauer EM, Mutter J, Scherer F. Circulating tumor DNA in B-cell lymphoma: technical advances, clinical applications, and perspectives for translational research. Leukemia. 2022;36(9):2151–2164. doi: 10.1038/s41375-022-01618-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rahal R, Frick M, Romero R, et al. Pharmacological and genomic profiling identifies NF-kappaB-targeted treatment strategies for mantle cell lymphoma. Nat Med. 2014;20(1):87–92. doi: 10.1038/nm.3435. [DOI] [PubMed] [Google Scholar]
- 41.Diop F, Moia R, Favini C, et al. Biological and clinical implications of BIRC3 mutations in chronic lymphocytic leukemia. Haematologica. 2020;105(2):448–456. doi: 10.3324/haematol.2019.219550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang E, Mi X, Thompson MC, et al. Mechanisms of resistance to noncovalent Bruton’s tyrosine kinase inhibitors. N Engl J Med. 2022;386(8):735–743. doi: 10.1056/NEJMoa2114110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schroers-Martin JG, Kurtz DM, Soo J, et al. Determinants of circulating tumor dna levels across lymphoma histologic subtypes. Blood. 2017;130 4018-4018. [Google Scholar]
- 44.Andrade-Campos MM, Salar A, Sanchez-Gonzalez B, et al. Assessment of cell-free dna (cfdna) in 221 patients with lymphoproliferative malignancies at diagnosis and during follow-up. Blood. 2019;134(Suppl 1) 492-492. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.