

Abstract
Brain swelling causes morbidity and mortality in various brain injuries and diseases but lacks effective treatments. Brain swelling is linked to the influx of water into perivascular astrocytes through channels called aquaporins. Water accumulation in astrocytes increases their volume, which contributes to brain swelling. Using a mouse model of severe ischemic stroke, we identified a potentially targetable mechanism that promoted the cell surface localization of aquaporin 4 (AQP4) in perivascular astrocytic endfeet, which completely ensheathe the brain’s capillaries. Cerebral ischemia increased the abundance of the heteromeric cation channel SUR1-TRPM4 and of the Na+/Ca2+ exchanger NCX1 in the endfeet of perivascular astrocytes. The influx of Na+ through SUR1-TRPM4 induced Ca2+ transport into cells through NCX1 operating in reverse mode, thus raising the intra-endfoot concentration of Ca2+. This increase in Ca2+ stimulated calmodulin-dependent translocation of AQP4 to the plasma membrane and water influx, which led to cellular edema and brain swelling. Pharmacological inhibition or astrocyte-specific deletion of SUR1-TRPM4 or NCX1 reduced brain swelling and improved neurological function in mice to a similar extent as an AQP4 inhibitor and was independent of infarct size. Thus, channels in astrocyte endfeet could be targeted to reduce post-ischemic brain swelling in stroke patients.
Introduction
Brain swelling is a major cause of morbidity and mortality in brain tumors, cerebral malaria, traumatic brain injury, and hemorrhagic and ischemic stroke. In large hemispheric stroke, brain swelling places patients at high risk for neurological deterioration and is largely responsible for the high mortality rate of 50–80% (1). Approved treatments are limited to unproven osmotherapies or surgical removal of a large part of the cranium and are administered only after brain swelling has already compromised neurological function. Development of preventative treatments based on the cellular and molecular mechanisms of brain swelling may improve both clinical prognosis and patient quality of life.
After ischemia, brain swelling is caused by an inflow into brain tissues of cerebrospinal fluid (2) and fluid of vascular origin – protein-free “ionic edema” and an ultrafiltrate of plasma, “vasogenic edema” – that arises from dysfunction of the blood-brain barrier (BBB) (3, 4). The BBB is comprised of an inner endothelial layer, an intermediate basement membrane containing pericytes, and an outer layer of astrocyte endfeet. The endothelial layer, whose cells are joined by impermeable tight junctions, acts as the physical barrier of the BBB (5). Loss of integrity of the endothelial barrier leads to the inflow of edema that contributes to brain swelling (6, 7). Perivascular astrocyte endfeet are highly specialized (8, 9), diffusion-limited (10) cellular processes that fully and dynamically ensheathe the vertebrate cerebral vasculature (11, 12). Astrocyte endfeet are recognized as critical mediators of differentiation and maintenance of the BBB (13–15).
In the brain, the water channel aquaporin 4 (AQP4) is densely abundant in perivascular and subpial astrocyte endfeet (16, 17). AQP4 plays an important role in post-ischemic edema and edema clearance (18, 19). Water transport by AQP4 is governed by the transmembrane osmotic gradient, and by the rate limiting number of AQP4 channels present in the cell membrane (20). Our understanding of AQP4 function in disease has been confounded by contradictory reports of increased or decreased expression (21). Moreover, it’s unclear whether its cell-membrane localization, which is required for its function, is dynamically regulated in disease. Here, we investigated AQP4 regulation and the role of astrocyte endfeet in post-ischemic BBB dysfunction in mice and we identified an ion transport mechanism that underlies brain swelling.
Results
SUR1-TRPM4 is newly expressed in astrocyte endfeet post-ischemia
In brain tissue from mice subjected to middle cerebral artery occlusion followed by reperfusion (MCAO/R), we detected SUR1 expression in ipsilateral but not contralateral microvessels (Fig. 1A). The earliest increase occurred in perivascular astrocyte endfeet after 6 hours reperfusion, where microvascular SUR1 colocalized with AQP4, outside of the CD31+ endothelial layer (Fig. 1B–D). Later, after 24 hours reperfusion, SUR1 abundance was evident in the endothelium, pericytes, and astrocyte somata (fig. S1A–C).
Fig. 1. SUR1-TRPM4 and NCX1 are expressed in post-ischemic astrocyte endfeet.
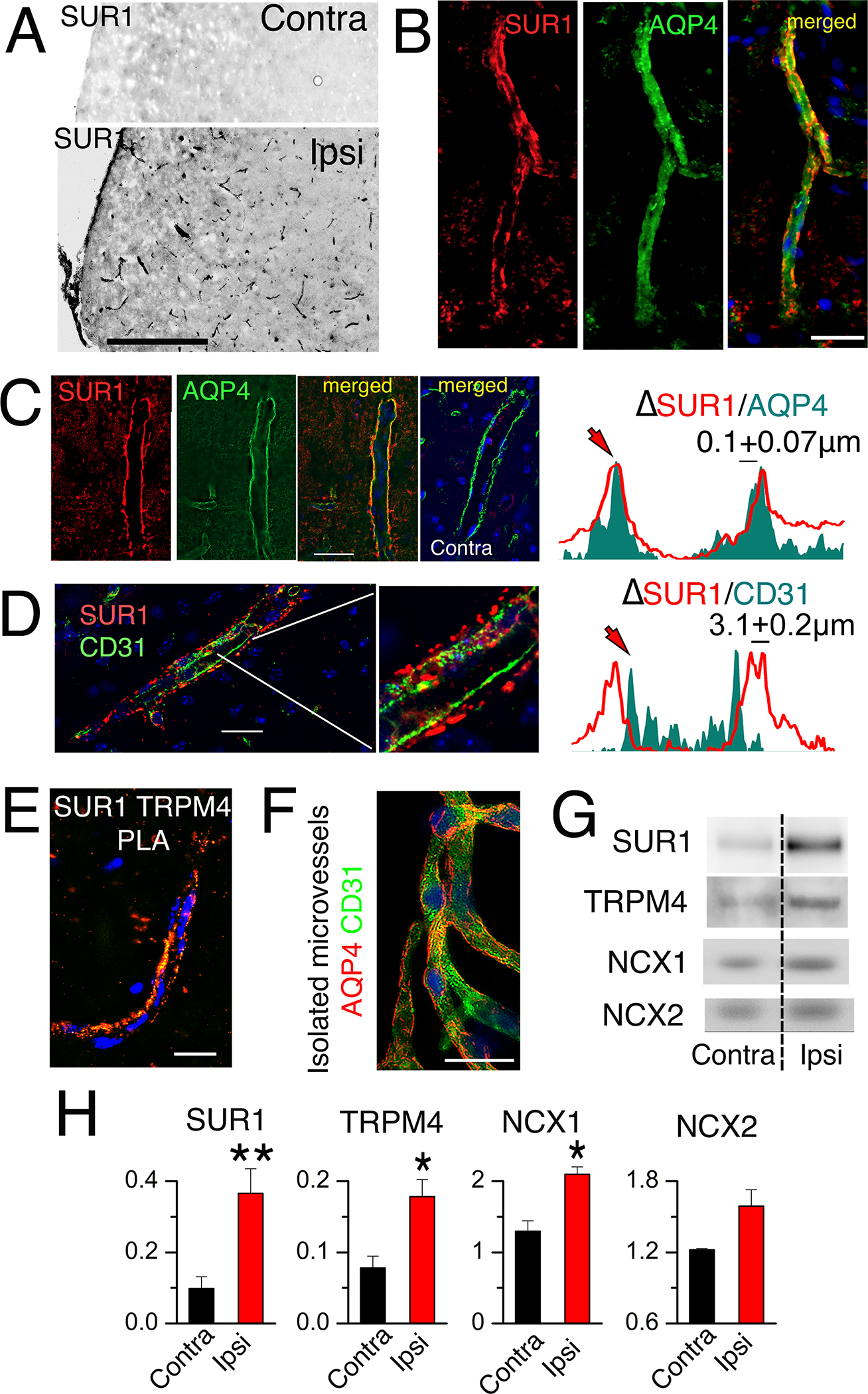
(A) Immunohistochemical analysis of expression of SUR1 (black) in ipsilateral (Ipsi) and contralateral (Contra) microvessels in brain tissue sections from mice after MCAO for 2 hours followed by reperfusion for 24 hours. Scale bar, 200 μm. (B to D) As described in (A) but with 6-hour reperfusion, immunohistochemistry for expression of SUR1 (red) in AQP4+ astrocyte endfeet (green; merged image) in microvessels within ipsilateral (B,C) and contralateral tissues (C), and for SUR1 (red) and CD31 (endothelium marker; green) in ipsilateral tissues (D). Nuclei are stained blue with DAPI (4′,6-diamidino-2-phenylindole). Right, fluorescent intensity profiles for the respective pairs. Profiles are representative and data are mean ± S.E. from N = 30 (C) and 24 (D) measurements in ipsilateral tissues from 3 mice. Scale bars, 25 μm. (E) PLA imaging for SUR1-TRPM4 heteromers lining a microvessel in ipsilateral tissues isolated from mice as described in (B–D). Image is representative of 5 mice. Scale bar, 25 μm. (F) Immunolabelling for CD31 (green) and AQP4 (red) in isolated microvessels from ipsilateral tissues from mice as described in (B–D). Image is representative of 4 mice. Scale bar, 25 μm. (G and H) In mice as described in (B–D), microvessels isolated from ipsilateral (Ipsi) and contralateral (Contra) tissues were studied by immunoblot for SUR1, TRPM4, NCX1, and NCX2 (G). Data (H) are mean ± S.E. from 3 or 4 mice per group, normalized to β-actin. *P<0.05 and **P<0.01 by t-test.
In humans and rats following cerebral ischemia, SUR1 forms heteromeric channels with TRPM4 (22). Proximity ligation assays (PLA) of the mouse brain tissues after MCAO/R showed that SUR1 formed heteromers with TRPM4 that localized to perivascular astrocyte endfeet beginning at 6 hours after reperfusion (Fig. 1E, fig. S2A–D). Immunoblotting of isolated microvessels with astrocyte endfeet still attached (Fig. 1F) (9) corroborated the statistically significant increases in SUR1 and TRPM4 abundance in the ipsilateral cortex (Fig. 1G,H; fig. S2E,F).
SUR1-TRPM4 activation induces Ca2+ influx in astrocyte endfeet
Calcium signaling plays numerous critical roles in astrocyte function. To determine whether newly expressed SUR1-TRPM4 channels are involved, we performed Ca2+ imaging analysis on brain slices from mice expressing the Ca2+ reporter, GCaMP6f, under control of the astrocyte-specific promoter, gfaABC1D (Fig. 2A). In middle cerebral artery territory tissues from sham-injured mice, bath application of the potent SUR1 activator NN414 (tifenazoxide) (23) caused minimal increases in endfoot Ca2+ (Fig. 2, B and C), which may suggest low SUR1 expression. Conversely, in tissues from MCAO/R mice, NN414 induced robust perivascular Ca2+ signals (Fig. 2B,C), consistent with functional activity of newly expressed SUR1-TRPM4 channels in astrocyte endfeet.
Fig. 2. SUR1-TRPM4 and NCX1 function together to increase Ca2+ in post-ischemic astrocyte endfeet.
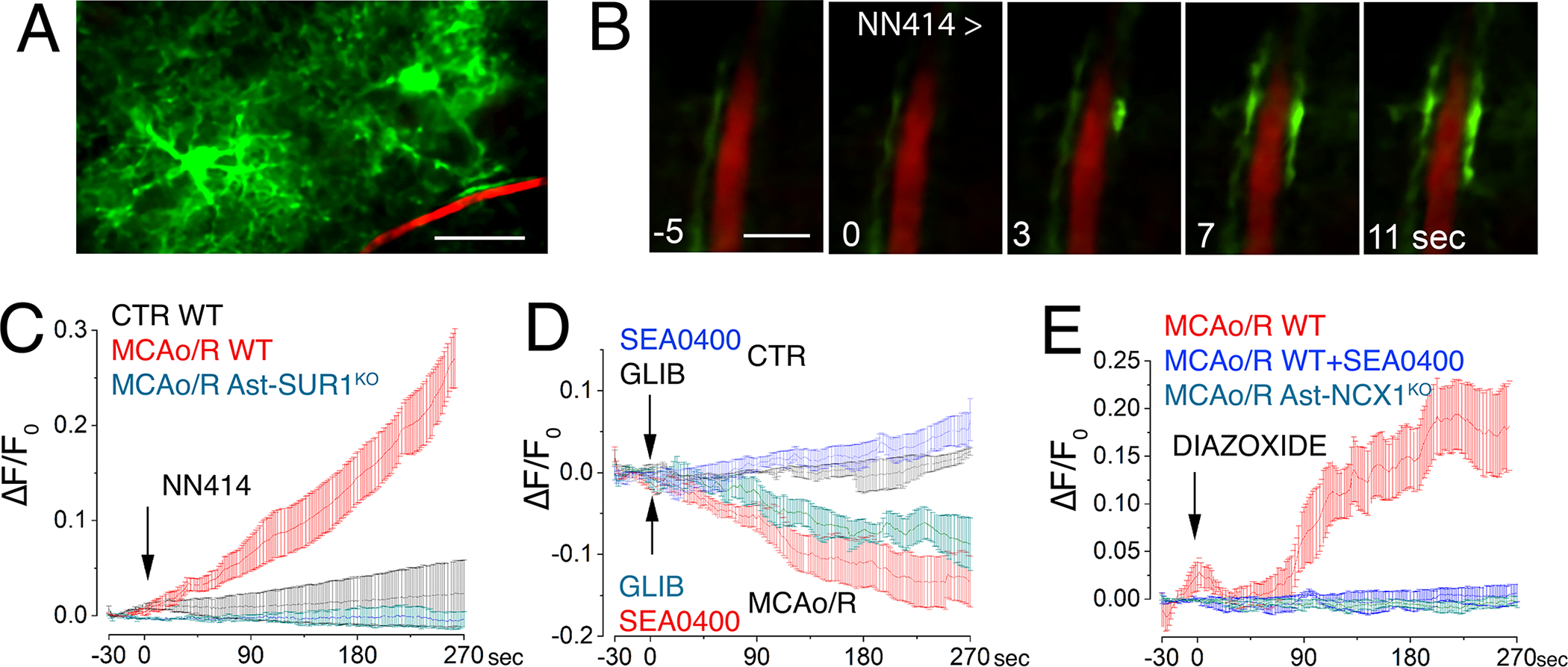
(A) Image of astrocytes expressing GCaMP6f (green) near a microvessel with fluorescent dextran (red) in brain tissue slices from mice that expressed the Ca2+ reporter GCaMP6f under control of the astrocyte-specific promoter gfaABC1D and had undergone MCAO for 2 hours then reperfusion for 24 hours. Scale bar: 25 μm. (B) Temporal sequence of Ca2+ imaging (green) in perivascular astrocyte endfeet in slices from mice described in (A) following SUR1 activation by NN414. Inset numbers are time in seconds with respect to NN414 application. Scale bar: 5 μm. Images are representative of 5 slices from 3 mice. (C) Temporal changes in intracellular Ca2+ (ΔF/Fo) in perivascular astrocyte endfeet following SUR1 activation with NN414 in slices from mice described in (A and B) post-MCAO/R, either WT mice (red) or Ast-SUR1KO (cyan), and in slices from control non-ischemic WT mice (CTR; black). Data are mean ± S.E. from 5 regions of interest (ROIs) in 5 slices from 3 mice per group; baseline trends before adding test agents were subtracted. (D) Assay and analysis as described in (C), following inhibition of SUR1 (glibenclamide; GLIB) or NCX1 (SEA0400) in control non-ischemic or post-MCAO/R WT mice. (E) Assay and analysis as described in (C) in post-MCAO/R WT with or without SEA0400 or post-MCAO/R Ast-NCX1KO mice following SUR1 activation with diazoxide.
In post-MCAO/R brain slices from mice with astrocyte-specific knockout of Abcc8/SUR1 (Ast-SUR1KO; fig. S3A–D), NN414 had no effect on endfoot Ca2+ (Fig. 2C), confirming both the specificity of NN414 for SUR1 and the role of SUR1 in astrocytes in the Ca2+ responses.
SUR1-mediated Ca2+ influx in astrocyte endfeet requires NCX1
The finding that SUR1 activation by NN414 induced Ca2+ influx post-MCAO/R was unexpected, because SUR1-TRPM4 is a non-selective monovalent cation channel that does not transport Ca2+ (24). The effects of NN414 were independent of the Ca2+ transporter, TRPV4 (fig. S4), identified previously as a mechanism of Ca2+ influx in cultured astrocytes (25, 26). Given that the activation of SUR1-TRPM4 results in Na+ influx (27), this led us to speculate about the possible involvement of Na+-Ca2+ exchanger (NCX) as a Na+-dependent mechanism mediating Ca2+ influx. Astrocytes express all three isoforms: NCX1, NCX2, and NCX3 (28).
NCX transports Ca2+ and Na+ across the cell membrane in both directions, depending upon the cation electrochemical gradients. Normally, NCX promotes the extrusion of Ca2+, but in cells such as astrocytes, whose membrane potential is near the reversal potential of NCX, modest increases in Na+ concentration can switch NCX into Ca2+ entry mode (CaEnt-NCX) (29). We hypothesized that in the foregoing experiment, Na+ influx due to SUR1-TRPM4 activated CaEnt-NCX, thereby resulting in the observed increase in Ca2+.
The benzyloxyphenyl derivatives SN-6 and SEA0400 inhibit NCX1 and, with less potency, NCX2 (30, 31). In brain slices from sham-injured mice, SEA0400 caused a small gradual increase in Ca2+ (Fig. 2D), consistent with SEA0400 blocking normal Ca2+ extrusion by NCX. However, in brain slices from post-MCAO/R mice, SEA0400 progressively reduced endfoot Ca2+ concentration (Fig. 2D), consistent with inhibition of a tonically activated Ca2+ influx transporter—CaEnt-NCX—post-MCAO/R.
A similarly progressive reduction in endfoot Ca2+ was observed with the SUR1 inhibitor, glibenclamide (Fig. 2D), suggesting that tonic activation of CaEnt-NCX was due to tonic activation of SUR1-TRPM4 post-MCAO/R. There was no effect of glibenclamide in non-ischemic sham controls (Fig. 2D).
In post-MCAO/R brain slices from WT mice, SEA0400 blocked the rise in endfoot Ca2+ normally induced by SUR1 activation, as shown here with diazoxide (Fig. 2E). Astrocyte-specific deletion of Slc8a, the gene encoding NCX1 (Ast-NCX1KO; fig. S5A,B) (Fig. 2E), also blocked the rise in endfoot Ca2+ normally induced by SUR1 activation, consistent with a requirement for NCX1 in astrocytes.
Immunohistochemistry confirmed that perivascular NCX1 was localized within astrocyte endfeet (fig. S6A–C). Immunoblotting of isolated microvessels with astrocyte endfeet still attached from post-MCAO/R brains (9) corroborated statistically significant increases in NCX1 but not NCX2 (Fig. 1G,H; fig. S6D).
Ca2+ influx induces cell-membrane localization of AQP4
We hypothesized that Ca2+ influx mediated by NCX1 activates calmodulin (CaM) to drive cell-membrane localization of AQP4 in post-ischemic perivascular astrocyte endfeet (26). Following ischemia, AQP4 becomes progressively “dysregulated”, meaning it moves away from astrocyte endfeet to occupy other regions of the astrocyte cell membrane (32). Nevertheless, our results indicated that AQP4 remains abundant up to 24 hours in perivascular regions (fig. S7A–C).
In primary cortical rat astrocytes and in transfected HEK293 cells, trafficking of AQP4 to the cell membrane requires binding of CaM to AQP4 and serine phosphorylation of AQP4 (33). We corroborated both requirements in vivo using PLA. Following MCAO/R, with ischemia verified by MAP2 immunolabeling (fig. S8A), the MAP2-negative region of the ipsilateral hemisphere exhibited abundant CaM/AQP4 PLA punctae (Fig. 3A), as did the anterior cerebral artery (ACA)-MCA watershed region (fig. S8B,D), consistent with binding of CaM to AQP4. The MAP2-negative region of the ipsilateral hemisphere (Fig. 3B) as well as the ACA-MCA watershed region (fig. S8C,E) showed abundant phosphoserine/AQP4 PLA punctae, consistent with serine phosphorylation of AQP4. Because PLA would detect phosphorylation on serine residues in AQP4 as well as on any closely (less than 40 nm) associated protein, we verified the increase in AQP4 phosphorylation by blotting for phosphoserine in immunoprecipitates of AQP4 (fig. S8F) (34). In both the MAP2-negative region (Fig. 3A,B) as well as in the ACA-MCA watershed region (fig. S8D,E), glibenclamide greatly reduced the number of PLA punctae for both AQP4/CaM and AQP4/phosphoserine compared to vehicle.
Fig. 3. Cell membrane localization of aquaporin-4 increases after ischemia and requires SUR1-TRPM4 and NCX1.
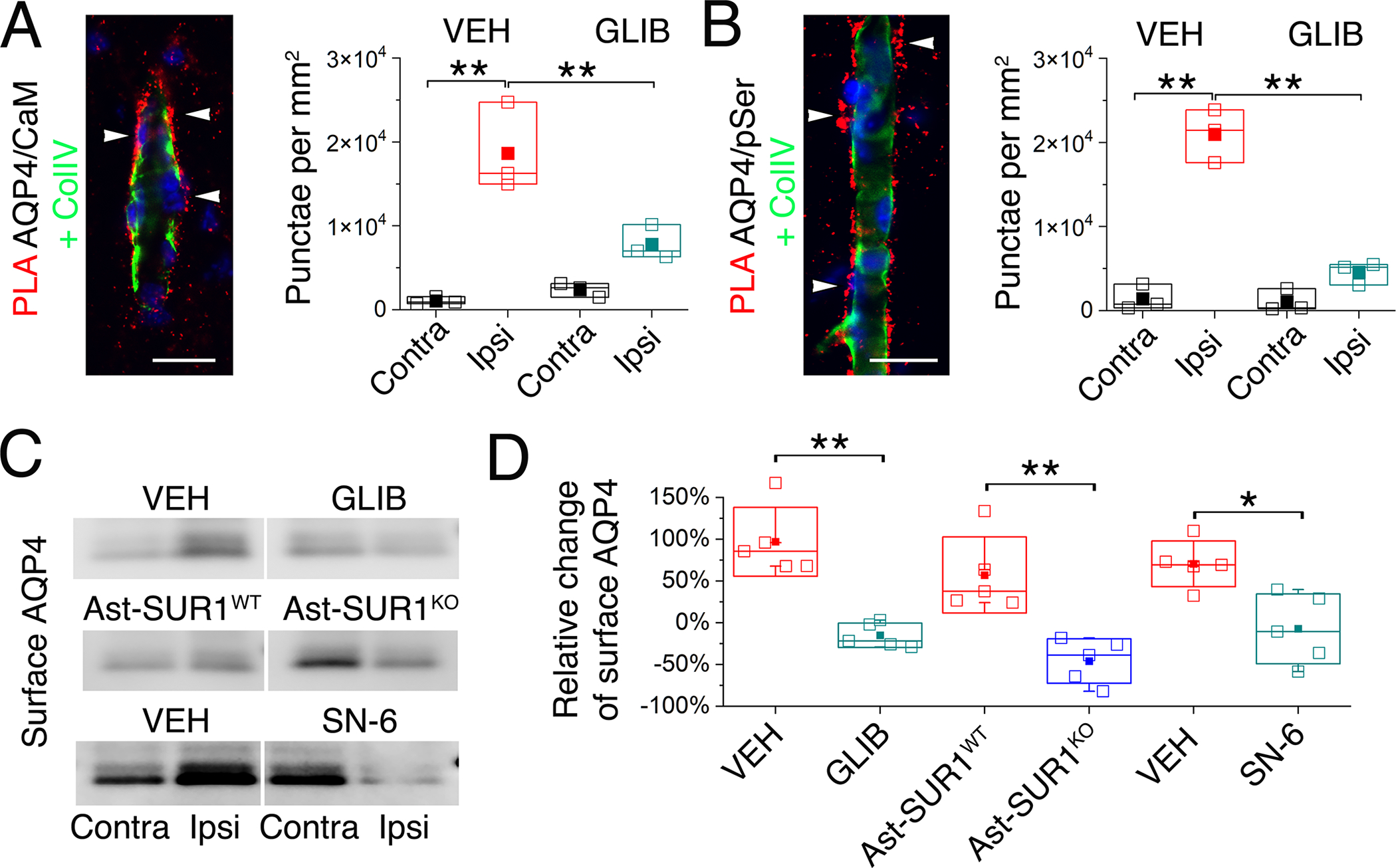
(A and B) Proximity ligation assay (PLA) (red) showing co-association of AQP4 with calmodulin (CaM) (A), and serine phosphorylation (pSer) of AQP4 (B), both located outside of the microvascular collagen IV layer (ColIV; green) following MCAO/R (2 hours MCAO with 6 hours reperfusion). The number of PLA punctae (arrowheads) per mm2, mean ± S.E. from 3 mice per group, is plotted for ipsilateral (Ipsi) and contralateral (Contra) hemispheres from mice administered vehicle (VEH) or glibenclamide (GLIB) at reperfusion. Nuclei stained blue with DAPI **P<0.01 for contra vs. ipsi; ##P<0.01 for VEH vs. GLIB, both by ANOVA. Scale bars: 25 μm. (C and D) In mice as described in (A,B), and additionally WT mice administered SN-6, and in a mouse with astrocyte-specific deletion of Abcc8/SUR1 (Ast-SUR1KO) and a littermate control (Ast-SUR1WT), immunoblot was performed for ipsilateral (Ipsi) and contralateral (Contra) cell-membrane AQP4 (C), with densitometric data for cell-membrane AQP4 (ipsilateral relative to contralateral) quantified (mean ± S.E.) for the 6 conditions (D); *, P<0.05; **, P<0.01 by ANOVA; 5 mice/group.
We next sought to determine whether binding of CaM to AQP4 and serine phosphorylation of AQP4 in vivo after MCAO/R would promote the trafficking of AQP4 to the cell membrane, as reported in primary cortical rat astrocytes and in transfected HEK293 cells (33). We thus studied the cell-membrane localization of AQP4 in coronal brain slices. WT mice with MCAO/R (2/6 hours) intraperitoneally administered vehicle at reperfusion exhibited a marked increase in cell-membrane localized AQP4 in the ipsilateral MCA territory relative to the contralateral (Fig. 3C–D; fig. S9A). By contrast, no changes were observed in total AQP4 (fig. S9, A and D) or in cell-membrane localization of the Na+/K+ ATPase subunits, ATP1A1 and ATP1A2 (fig. S9, A and E), the former expressed by both astrocytes and neurons, and the latter expressed predominantly by astrocytes (35). In WT mice with MCAO/R, glibenclamide blocked the increase in cell-membrane AQP4 (Fig. 3C–D, and fig. S9A). Similarly, the increase in cell-membrane-localized AQP4 was blocked in Ast-SUR1KO mice compared with littermate controls (Fig. 3C–D, and fig. S9B), and in WT mice administered SN-6 compared to those administered vehicle (Fig. 3C–D, and fig. S9C).
Together, the results demonstrated that SUR1-TRPM4 and NCX1 together promoted an increase in the intracellular Ca2+ concentration and the cell-membrane localization of AQP4 within post-ischemic perivascular astrocyte endfeet, as depicted in our proposed model (Fig. 4).
Fig. 4. Proposed model of the functional axis comprised of SUR1-TRPM4, NCX1 and AQP4 in pre-ischemic vs. post-ischemic astrocyte endfeet.
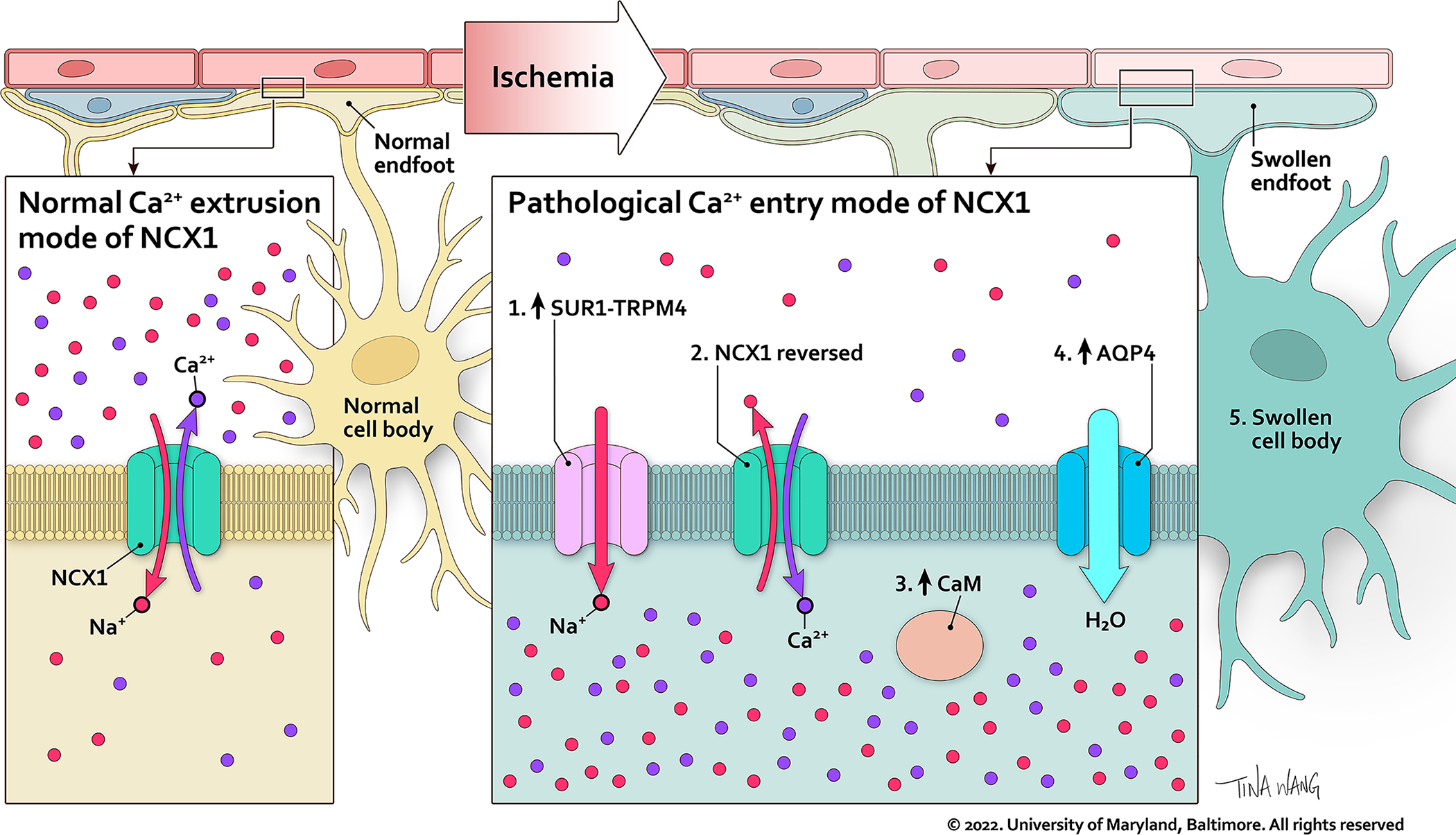
In the pre-ischemic astrocyte endfoot (left), NCX1 operates in calcium extrusion mode, and SUR1-TRPM4 is absent. In the post-ischemic astrocyte endfoot (right), newly increased SUR1-TRPM4 mediates Na+ influx (#1) which drives Ca2+ influx through the calcium entry mode of NCX1 (#2). The increase in Ca2+ activates CaM (#3), which stimulates trafficking of AQP4 to the cell membrane (#4), where AQP4 facilitates water influx that contributes to swelling.
SUR1-TRPM4 and NCX1 determine brain swelling
Brain edema and brain swelling historically have been found to be proportional to infarct size (36, 37). A reduction in swelling obtained with a reduction in infarct size is exemplified by studies with glibenclamide in rat (38, 39) and mouse (fig. S10A–D) models with salvageable penumbra. However, proper study of mechanisms of swelling requires that brain swelling be evaluated in a manner independent of a reduction in infarct size, for example using models with minimal salvageable penumbra. Using our mouse model of severe prolonged ischemia (2 hours MCAO) followed by 24 hours reperfusion, we analyzed outcome data exclusively from animals with large infarcts, >40 mm3 in both the control and the treatment groups. Controls included vehicle treatment of WT mice and floxed WT littermates of transgenic mice. In the multiple series of paired experiments, mean infarct volumes were ~75 mm3, with no differences in infarct volumes, by design, between controls and pharmacological or genetic treatments (fig. S11A–D).
Both constitutive and conditional Ast-SUR1KO mice had reduced hemispheric swelling (Fig. 5A–C; fig. S11A). Mice administered glibenclamide to target SUR1 (40) had hemispheric swelling significantly less than vehicle controls (Fig. 5C; fig. S11A), similar to Ast-SUR1KO mice.
Fig. 5. SUR1-TRPM4 plays a critical role in brain swelling independent of infarct size.
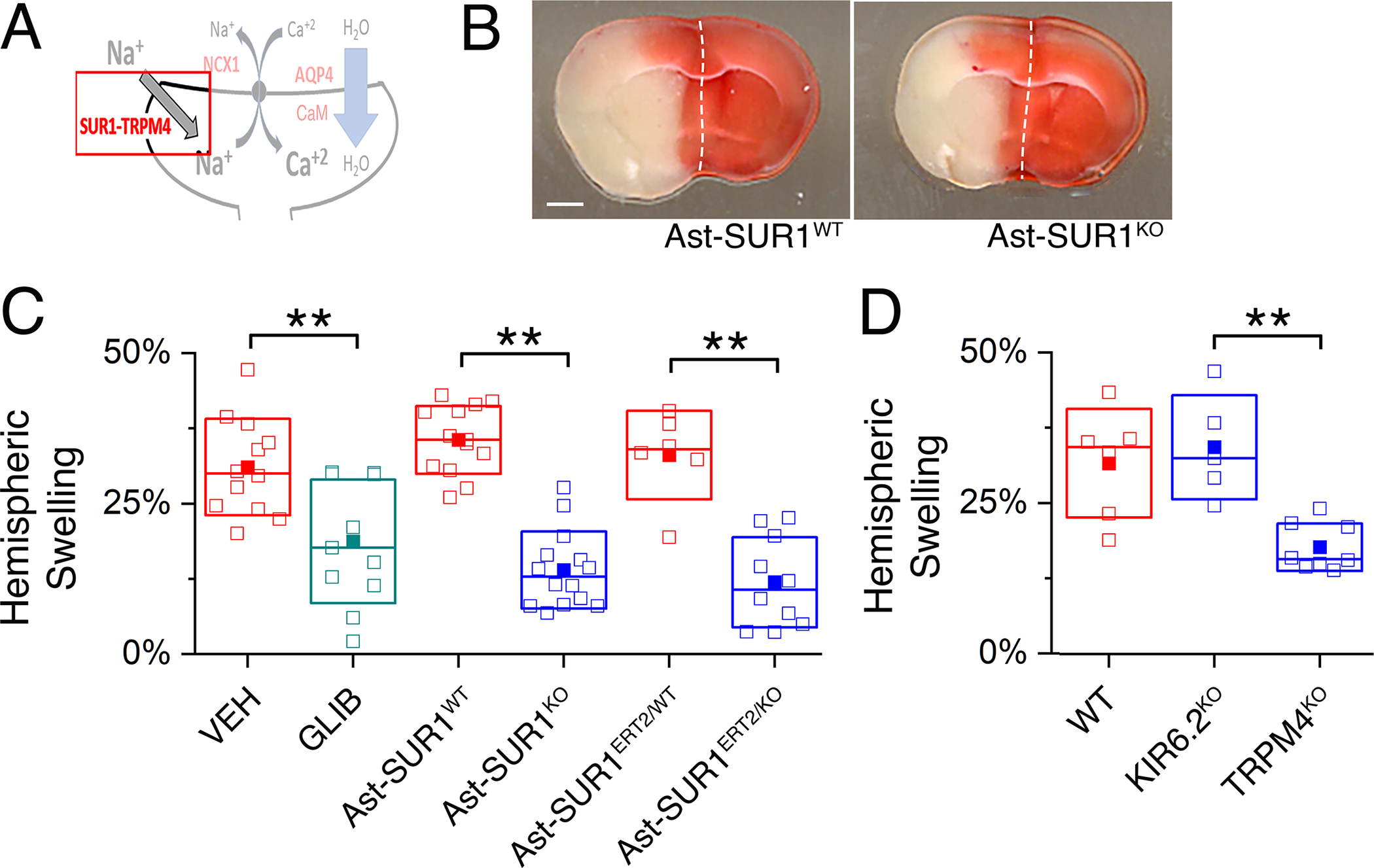
(A) Proposed model of the functional axis comprised of SUR1-TRPM4, NCX1, and AQP4, with emphasis on SUR1-TRPM4. (B) Images of TTC-stained coronal brain sections following MCAO/R in a mouse with deletion of Abcc8/SUR1 in astrocytes (Ast-SUR1KO) and a littermate control (Ast-SUR1WT). Scale bar: 1 mm. Ischemia was induced by MCAO for 2 hours followed by reperfusion for 24 hours. Images are representative of 14 mice per group. (C and D) In mice as described in (B) with infarct volumes >40 mm3, ipsilateral hemispheric swelling following MCAO/R was quantified in slices from (C) WT mice administered vehicle (VEH) or glibenclamide (GLIB) at reperfusion and mice with constitutive or conditional astrocyte-specific deletion of Abcc8/SUR1 (Ast-SUR1KO, Ast-SUR1ERT2/KO) and littermate controls (Ast-SUR1WT and Ast-SUR1ERT2/WT), and (D) in slices from mice with global deletion of the gene encoding KIR6.2 or TRPM4 (KIR6.2KO, TRPM4KO) and littermate controls of the latter (TRPM4WT [WT]). KIR6.2WT littermates were not studied. Data are mean ± S.E. from N = 5 to 14 mice per group. **P<0.01 by ANOVA.
SUR1 assembles with two different pore-forming subunits, KIR6.2 and TRPM4, to form KATP and SUR1-TRPM4 channels, respectively. Mice with global deletion of the gene encoding KIR6.2 (KIR6.2KO) were not protected from swelling (Fig. 5D; fig. S11B), as reported with KIR6.1KO (41). By contrast, global deletion of the gene encoding TRPM4 (TRPM4KO) yielded reduced hemispheric swelling (Fig. 5D; fig. S11B), similar to that seen in Ast-SUR1KO mice.
Mice administered SN-6 or SEA0400, which block NCX1 and, with less potency, NCX2, had significantly less swelling than vehicle controls, as did Ast-NCX1KO mice (Fig. 6A–C; fig. S11C). Mice administered YM-244769, which blocks NCX3, had hemispheric swelling equivalent to vehicle controls (Fig. 6C; fig. S11C), ruling out involvement of NCX3.
Fig. 6. NCX1 and AQP4 play critical roles in brain swelling independent of infarct size.
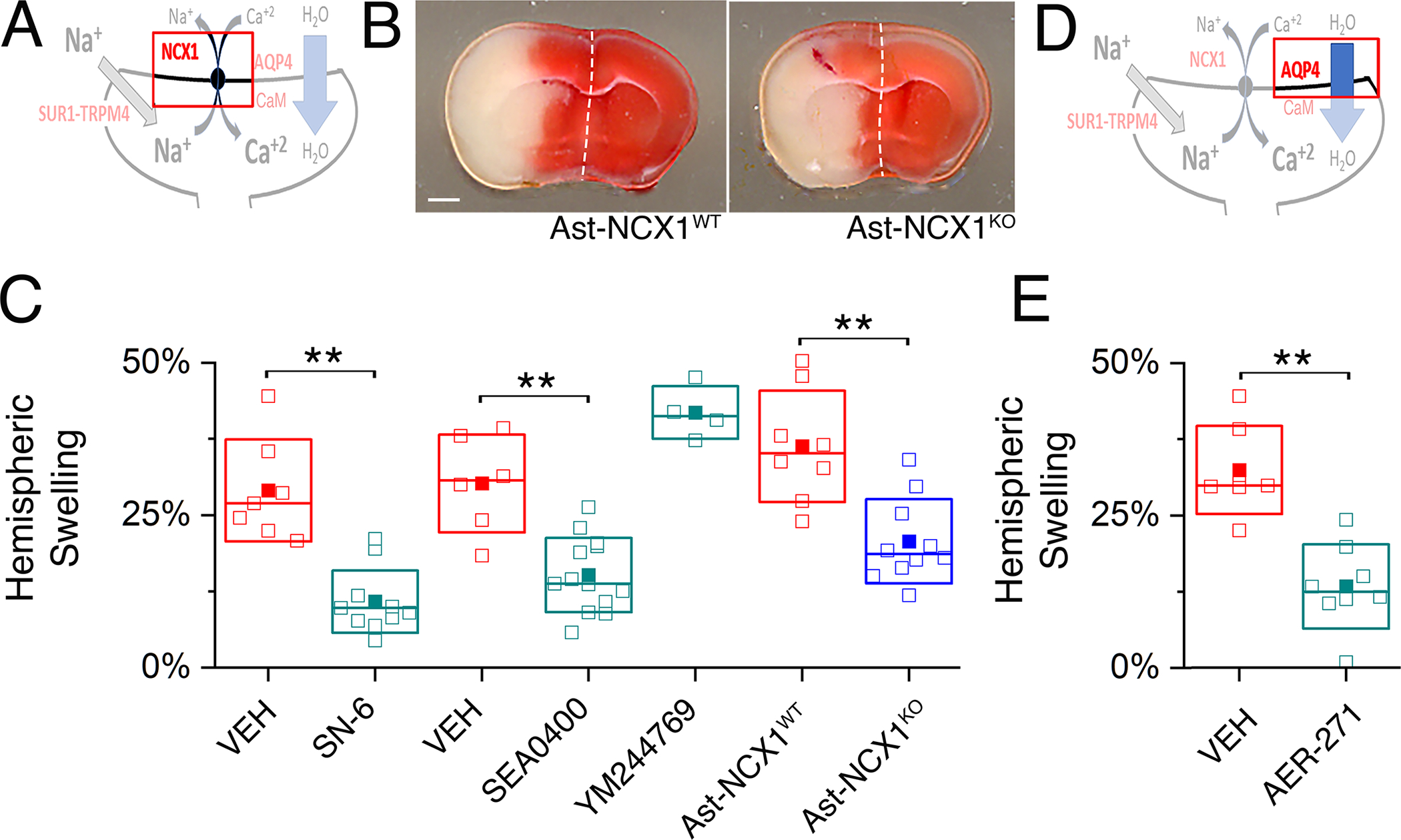
(A) Proposed model of the functional axis comprised of SUR1-TRPM4, NCX1 and AQP4, with emphasis on NCX1. (B) Images of TTC-stained coronal brain sections following MCAO/R in a mouse with deletion of Slc8a1/NCX1 in astrocytes (Ast-NCX1KO) and a littermate control (Ast-NCX1WT). Scale bar: 1 mm. Ischemia was induced by MCAO for 2 hours followed by reperfusion for 24 hours. Images are representative of 8 mice per group. (C) In mice as described in (B) with infarct volumes >40 mm3, ipsilateral hemispheric swelling following MCAO/R was quantified in slices from WT mice administered vehicle (VEH) or SN-6 or SEA0400 or YM244769 at reperfusion, and mice with conditional astrocyte-specific deletion of Slc8a1/NCX1 (Ast-NCX1KO) and littermate controls (Ast-NCX1WT). Data are mean ± S.E. from 4 to 13 mice per group. **P<0.01 by ANOVA. (D) Proposed model of the functional axis comprised of SUR1-TRPM4, NCX1 and AQP4, with emphasis on AQP4. (E) In mice as described in (B) with infarct volumes >40 mm3, ipsilateral hemispheric swelling following MCAO/R was quantified in slices from WT mice administered vehicle (VEH) or AER-271 at reperfusion. Data are mean ± S.E. from N = 7 or 8 mice per group. **P<0.01 by ANOVA.
For comparison, we evaluated the effect of AER-271, which was shown to inhibit brain swelling in a rat MCAO model (42). AER-271 is under development as a small molecule inhibitor of AQP4 (42), although it also inhibits IκB kinase (43). Mice administered AER-271 had significantly less hemispheric swelling than vehicle controls (Fig. 6D,E; fig. S11D). AER-271 reduced brain swelling to the same extent as inhibition of SUR1-TRPM4 or NCX.
Ischemia-induced hemispheric swelling is a manifestation of BBB dysfunction, which we independently evaluated by measuring the extravasation of Evans Blue (EB). Mice administered glibenclamide, SN-6, or AER-271 had significantly less EB extravasation at 24 hours compared to vehicle-treated animals (fig. S12), paralleling observations on hemispheric swelling.
Neurological function mirrored hemispheric swelling, and verified the benefit of reduced swelling, despite equivalent infarct volumes. Mice administered glibenclamide, SN-6, SEA0400 or AER-271, as well as Ast-SUR1KO, Ast-NCX1KO and TRPM4KO mice had better Garcia neuroscores than controls (fig. S11).
Together, the results demonstrated that SUR1-TRPM4 and NCX1 together promoted not only an increase in the intracellular Ca2+ concentration and the cell-membrane localization of AQP4 within post-ischemic perivascular astrocyte endfeet, but also that these effects were closely linked to brain swelling, independent of infarct size.
Discussion
In astrocytes in vivo, cell-membrane anchoring of AQP4 at endfeet requires α-syntrophin and other members of dystrophin associated protein complex whose fate post-ischemia is poorly characterized (44, 45). The pivotal studies of Kitchen et al. (25, 26, 33) demonstrated that, subsequent to various stimuli, trafficking of AQP4 to the cell membrane in cultured cells is dynamically modulated by protein kinase A phosphorylation and by Ca2+/calmodulin binding (26). However, in cultured cells, AQP4 is not expressed in a polarized manner, nor is it anchored to specific cell compartments, raising questions regarding the applicability of Kitchen et al.’s findings to in vivo conditions. Here, using brain tissues following MCAo/R, we validated the concept of dynamic cell-surface localization of AQP4 in ischemia in vivo, we characterized the upstream cation transport mechanism responsible for activating calmodulin required for AQP4 cell-membrane localization, and we showed that targeting this mechanism effectively reduced both cell-membrane localization of AQP4 and brain swelling.
Our data showed that SUR1-TRPM4 and NCX1 functioned together to generate Ca2+ influx in perivascular astrocyte endfeet and promote AQP4 cell-membrane localization, as reflected in our proposed model. The drugs used to block SUR1-TRPM4 or NCX1 have potential off-target effects (46–48) but in all cases the effects of pharmacological inhibition were replicated by cell-specific gene deletion. The influx of Ca2+ induced by activating SUR1 in astrocyte endfeet was blocked by both pharmacological inhibition and astrocyte-specific deletion of SUR1 and NCX1 and was observed reliably in post-ischemic brain slices but not in uninjured tissues, consistent with ischemia-induced increases in SUR1-TRPM4 and NCX1. Similarly, pharmacological inhibition of both SUR1 and NCX1, as well as astrocyte-specific deletion, inhibited the increase in cell-membrane localization of AQP4 observed with ischemia. TRPV4 was identified previously as a mechanism of Ca2+ influx to promote AQP4 cell-membrane localization in cultured cells (25, 26) but studies of its role in vivo have been contradictory (49, 50). In post-ischemic brain tissues, we found that Ca2+ influx in perivascular astrocyte endfeet induced by SUR1 activation was inhibited not by blocking TRPV4 but by blocking NCX1.
Ca2+ influx mediated by NCX is well known in astrocytes, with CaEnt-NCX being activated by Na+ influx from a variety of cation transporters (29, 51). Our data indicated that SUR1 activation was a critical source of Na+ responsible for activating CaEnt-NCX1 in post-ischemic astrocyte endfeet. The SUR1-TRPM4 channel was first discovered in post-ischemic astrocytes, where its unique role in cell swelling was first described (24, 27). In astrocytes, SUR1-TRPM4 assembles with AQP4 to form a macromolecular complex that facilitates astrocyte swelling (52). SUR1-TRPM4 has been linked to post-ischemic brain edema formation in both preclinical (38, 53, 54) and early phase clinical (55–57) studies. SUR1-TRPM4 is the target of glibenclamide, a drug approved for diabetes mellitus that is currently under study in a Phase 3 trial to reduce brain swelling in large hemispheric infarctions (NCT04950972). However, the channel’s role specifically in astrocyte endfeet in the post-ischemic brain was previously unknown, and indeed the findings reported here were surprising, given the previous emphasis on the role of SUR1-TRPM4 in endothelium (58). The work presented here identifies SUR1-TRPM4 at perivascular astrocyte endfeet as a target of glibenclamide, and AQP4 cell-membrane localization as a key mechanism of its action in reducing swelling.
The models used to study brain swelling can impact advances in understanding of underlying mechanisms. In addition to a non-salvageable infarct core, an ischemic insult produces an ischemic penumbra that may or may not be salvageable depending upon the promptness and adequacy of reperfusion. Most models of acute ischemic stroke incorporate a salvageable penumbra, and treatments found to reduce swelling or edema generally do so in proportion to the reduction in infarct size conferred by the treatment (36, 37). To disentangle swelling from stroke size, we developed models with an unsalvageable penumbra, thus eliminating a reduction in infarct size as a confounder in the reduction in swelling and facilitating the study of swelling independent of infarct size.
In multiple mouse and rat models of stroke as well as in humans with stroke, targeting SUR1 with glibenclamide or antisense oligodeoxynucleotide or gene deletion invariably reduced post-ischemic brain swelling, as shown here and elsewhere (22, 38, 39, 53–55, 59, 60). In the reports from our laboratory, we purposely varied the severity of the ischemic insult or the time-to-treatment, to influence the volume of salvageable penumbra. In standard models with salvageable penumbra, treatment reduced both swelling and infarct size (38, 53). In models designed to have minimal salvageable penumbra, the reduction in swelling had no effect on infarct size, showing that swelling can be treated independent of infarct size (22, 54, 60). In human stroke, the concept of the salvageable penumbra has grown in clinical importance since the introduction of mechanical thrombectomy. However, clinical management of stroke requires molecularly informed treatments to reduce brain swelling, especially with large infarctions (61), regardless of the presence or volume of a salvageable penumbra.
Brain swelling is widely recognized as a major cause of morbidity and mortality in numerous brain conditions. In the clinic, strategies to prevent brain swelling are not yet available, and strategies for its treatment are reactionary and non-specific. Targeting AQP4 directly, which has been examined in a Phase 1 safety-only clinical trial of AER-271 (NCT03804476), may protect against water influx during the onset of edema but also may prolong the edematous state by interfering with brain water efflux during the resolution phase (62). A promising alternative is to target the mechanism that drives post-ischemic cell-membrane localization of AQP4, such as CaM (26) or the ion transporters in astrocyte endfeet that mediate the increase in intracellular Ca2+ responsible for activating CaM. In this regard, both NCX1 and SUR1-TRPM4 may be appropriate, druggable targets. Notably, blocking NCX would not reduce the potentially harmful Na+ overload due to SUR1-TRPM4, whereas blocking SUR1-TRPM4 upstream of NCX would reduce both Na+ and Ca2+ overload.
Materials and Methods
Experimental Design
A key element of our experimental design was to perform all experiments using brain tissues from mice following ischemia / reperfusion. We avoided experiments on cultured cells to preclude results that might not be applicable to in vivo neurobiology. Experiments on calcium imaging, protein-protein interaction, and measurements of cell membrane AQP4 were carried out using brain slices from mice following MCAO/R, directly reflecting pathological in vivo neurobiology. In addition, we studied a murine model of severe stroke, choosing to investigate brain swelling in mice with large infarcts that were not reduced by treatment, even though with lesser ischemic insults the same treatments reduced infarct volume (fig. S10) (38, 53). These unique aspects of our study allowed us to make claims regarding brain swelling without the usual confounder of a reduction in infarct volume, which by itself can reduce swelling.
A power analysis was used to determine the minimum number of animals required for the experiments on treatments of MCAO/R mice. Experiments were conducted using random allocation to treatment group with treatments and outcome measures performed by investigators blinded to group identity. For key experiments on brain swelling, mice were excluded if infarct volumes did not exceed a pre-specified volume of 40 mm3. All experiments were replicated, with the number of replicates indicated in the text.
Ethics statement
Animal experiments were performed under a protocol approved by the Institutional Animal Care and Use Committee (IACUC #0819004) of the University of Maryland, Baltimore, and in accordance with the relevant guidelines and regulations as stipulated in the National Research Council Publication, “Guide for the Care and Use of Laboratory animals”. Data are reported in accordance with the ARRIVE guidelines (63).
Subjects
Male mice were used for experiments when they were ~3 months old (~25 gm). Wild-type (WT) C57BL/6 mice were obtained from Envigo (Indianapolis, IN, USA). Abcc8fl/fl mice (loxP sites inserted to flank exon 2 of the Abcc8 gene) on a C57BL6 background were generated, characterized, and gifted by Dr. Joseph Bryan (Pacific Northwest Research Institute, Seattle, WA, USA) (64). Slc8a1fl/fl mice (loxP sites inserted to flank exon 11 of the Slc8a1 gene) on a C57BL6 background were generated and characterized by Henderson et al. (65), and breeding pairs were provided by Dr. Mordecai P. Blaustein (University of Maryland, Baltimore MD, USA). Mice with global deletion of Trpm4 (Trpm4–/–) on a C57BL6 background were generated, characterized and gifted by Prof. Dr. Marc Freichel and Prof. Dr. Veit Flockerzi (Universität des Saarlandes, Homburg, Germany) (66). Mice with global deletion of Kcnj11 (Kcnj11–/–) on a C57BL6 background were generated and characterized by Prof. Susumu Seino (Kobe University, Japan) (67). Breeding pairs were provided by Dr. Sonia M. Najjar (Heritage College of Osteopathic Medicine, Athens, OH, USA). Gfap-cre mice (B6.Cg-Tg(Gfap-cre)73.12Mvs/J; stock #012886) and GFAP-cre/ERT2 mice (B6.Cg-Tg(GFAP-cre/ERT2)505Fmv/J; stock #012849), in which Cre recombinase is under the control of the murine Gfap or human GFAP promoter, either constitutive or conditional, were obtained from The Jackson Laboratory (Bar Harbor, ME, USA).
In mice with cre recombinase regulated by ERT2, astrocyte-specific deletion was induced using tamoxifen (68). Genotyping of mice used for breeding and experiments was performed by Transnetyx (Cordova, TN, USA). Mice were given free access to food and water, except during experiments. Validation of Ast-SUR1KO and Ast-NCX1KO mice is presented in fig. S3 and fig. S5.
Middle cerebral artery occlusion/reperfusion (MCAo/R)
MCAo was induced under general anesthesia [induction, 3.0% isoflurane; maintenance, 1.5–2.0% isoflurane with a mixture of O2 and N2O (20 and 80% respectively)] as described (69–72) using a silicon filament occluder (602356PK5Re Doccol Corp, Redlands CA, USA) for 2-hour or 45-minute occlusion followed by common carotid artery ligation. Relative cerebral blood flow (rCBF), measured by laser Doppler flowmetry (Moor Instruments, DRT4), was reduced 65% or more: (mean ± SD) 77.4 ± 5.8 % (<65% was an exclusion criterion). Mice with 2-hour occlusion were euthanized at 6 hours after reperfusion for measurements of cell-membrane AQP4 and for some immunohistochemistry experiments, or at 24 hours after reperfusion for other experiments. Measurements of infarct size and brain swelling, or Evans Blue extravasation were carried out at 24 hours after reperfusion in mice with 2-hour or 45-minute occlusion.
Sample size calculation
For experiments on post-MCAo/R brain swelling, we based our sample size calculation on a previous study in which antisense oligodeoxynucleotide targeting Abcc8 was shown to reduce swelling but not infarct size in a rat permanent MCAO model (22). Values derived from that study suggested an effect size (Cohen’s d) of 2.67, where d=(M1–M2)/SDpooled, M1 and M2 are the means, and SDpooled=[(SD1+SD2)/2]½ . Using the following assumptions: two-tailed hypothesis, α, 0.05; desired power, 90%; d, 2.67, the sample size calculation indicated a minimum sample size of 5 subjects per group. Generally, groups of >5 were prepared to allow for anticipated exclusions.
Drug treatments
Individual drug treatments were studied contemporaneously with vehicle controls, with the specific treatment for each animal determined randomly by a scientist not performing the MCAO/R procedure. SN-6 (#4544; Tocris Bioscience, Minneapolis, MN, USA) was prepared as a stock solution in dimethyl sulfoxide (DMSO) (10 mg/mL), diluted to a final concentration of 1.25 mg/mL in phosphate buffered saline (PBS), and 200 μL (~10 mg/kg) was injected intraperitoneal (IP) (73, 74). SAE0400 (#6164; Tocris) was prepared as a stock solution in DMSO (5 mg/mL), diluted to a final concentration of 1.25 mg/mL in normal saline (NS), and 200 μL (~10 mg/kg) was injected intravenously (IV) (73, 74). YM-244769 (#2184; Tocris) was prepared as a stock solution in DMSO (5 mg/mL), diluted to a final concentration of 0.714 mg/mL in NS, and 350 μL (~10 mg/kg) was injected IV (48, 75). IP injections of SN-6 or vehicle were performed once, at the time of reperfusion, using a 27-gauge needle with the depth of the injection limited to 3 mm by a sleeve of PE20 tubing placed over the needle. IV injections of SEA0400, YM-244769 or vehicle were performed once, at the time of reperfusion, following surgical exposure of the external jugular vein. Controls received the same volume of vehicle using the same route.
Glibenclamide (#G2539; MilliporeSigma, Rockville, MD, USA) was prepared as a stock solution in DMSO (2.5 mg/mL). The stock solution was diluted to 50 μg/mL in PBS, and 200 μL (10 μg/mouse; ~0.4 mg/kg) was injected IP as a loading dose at reperfusion. The stock solution, diluted to 100 μg/mL in PBS with NaOH (4 μL of 10 N NaOH per mL), was loaded into mini-osmotic pumps (Alzet pump 1003D; 100 μL volume; 1 μL/hour) to deliver 100 ng/hour subcutaneously beginning at reperfusion. Controls received the same volume of vehicle by IP injection and mini-osmotic pump.
AER-271 (#SML2737; MilliporeSigma) was administered by continuous infusion IP (42). A slurry of AER-271 (5 mg) in 125 μL of 10% DMSO in corn oil was prepared. The slurry was loaded into a 25 mm-long piece of tubing (Alzet jugular catheter; #0007710; Durect Corp., Cupertino, CA, USA), which was fixed to the outlet of a mini-osmotic pump (Alzet 1007D; Durect Corp.) preloaded and primed with PBS, which expelled the slurry from the PE tubing at a rate of 1 μL/hour. At the time of reperfusion, the body of the pump was implanted subcutaneously in the flank, and the tip of the PE tubing was inserted into the peritoneal cavity for continuous infusion IP. This system delivered 1.6 mg/kg/hr of AER-271 IP. Controls received the same volume of vehicle by mini-osmotic pump.
Neurological function
For MCAO/R protocols of 2/6 hours and 2/24 hours, circling behavior was evaluated at the time of reperfusion (2 hours) and mice were excluded if they failed to show circling. For experiments on brain swelling (MCAO/R 2/24 hours), neurological function was evaluated at 24 hours after reperfusion by investigators blinded to treatment group using the modified Garcia scoring system (76), after which the mice were euthanized.
Infarct volume, hemispheric swelling, and BBB integrity
After euthanasia, brains were harvested, and 3 to 4 consecutive 2-mm coronal sections were prepared using a chilled brain slicer matrix (Ted Pella, Inc., Redding, CA, USA). Brain slices were immersed in 2,3,5-triphenyltetrazolium chloride (TTC) at 22 °C ×10 minutes. Stained sections were imaged at 2400 dpi using a flatbed scanner. Images were processed using the National Institutes of Health ImageJ software 1.52a with an open-source, semi-automated “plug-in”, which featured automatic-thresholding that yields reliable, unbiased measurements of TTC– and hemisphere areas (77). Infarct volume (mm3) was calculated by multiplying the software-determined TTC– infarct area (mm2) by the slice thickness (2 mm) and summing infarct volumes across slices. To correct for tissue swelling, the total infarct volume was divided by the swelling factor, calculated as ipsilateral hemisphere area / contralateral hemisphere area. Hemispheric swelling was calculated as (ipsilateral hemisphere volume / contralateral hemisphere volume) – 1, expressed as percent.
BBB integrity was assessed by quantifying Evans Blue (EB) extravasation. At 3 hours before euthanasia, mice were administered EB (200 μL of 2% EB in normal saline IP) (78, 79). Tissues were processed as above, except that after the TTC-stained sections were imaged, brain tissues from each hemisphere were collected separately and homogenized, and EB was quantified spectrophotometrically (78–80). Data are reported as optical density at 620 nm.
Inclusion/exclusion criteria
Pre-established exclusion criteria following MCAO were: (1) LDF reduction <65%; (2) absence of circling behavior after emerging from anesthesia; (3) death prior to completing the prespecified requirement for 24 hours of reperfusion (incidence, 4.3%); (4) subarachnoid hemorrhage identified at necropsy (incidence, 2.4%). For brain swelling experiments with MCAO/R (2/24 hours), an additional pre-established exclusion criterion was infarct volumes <40 mm3. This last exclusion resulted in uniformly large infarcts across groups, regardless of treatment, and allowed the study of brain swelling independent of infarct volume.
Calcium imaging of perivascular astrocyte endfeet
All calcium imaging experiments on brain slices were carried out 2–3 weeks after cortical injections of an AAV5 virus encoding a Ca2+ reporter under control of an astrocyte-specific promoter (AAV5 gfaABC1D-cyto-GCaMP6f, #52925-AAV5; Addgene, Watertown, MA, USA) (81). A viral suspension with a final titer of 7×1012 vg/mL was prepared. Animals were anesthetized (ketamine: 80 mg/kg; xylazine: 10 mg/kg) and mounted in a stereotaxic apparatus (David Kopf Instruments, Tujunga, CA, USA). A scalp incision was made, and 2 small burr holes were drilled in the skull at the following x, y coordinates relative to bregma, targeting the ACA-MCA watershed territory: −1.2, +2.5 mm; −0.2, +2.5. A Hamilton syringe with a 27-g cone tipped needle was lowered to z = −1.5 mm. The AAV5 suspension was delivered at the rate of 0.15 μL/minute for a total of 1.5 μL at each of the 2 locations.
After euthanasia, mice were transcardially perfused with ice-cold normal saline (NS) followed in some cases by transcardial injection of 4 mg (10 mg/mL in PBS) TRITC-dextran (D1868, ThermoFisher Scientific, Waltham, MA, USA). The cerebrum was quickly removed, and 300 μm coronal slices were prepared (VT1200S, Leica Biosystems, Deer Park, IL, USA) in ice-cold slicing artificial cerebrospinal fluid (aCSF) containing (mM): 222.1 sucrose, 27 NaHCO3, 1.4 NaH2PO4, 2.5 KCl, 0.5 ascorbic acid, 1 CaCl2, 7 MgSO4, and bubbled with carbogen (95%/5% O2/CO2). Slices were recovered in aCSF (33–35 °C) for 30 minutes.
Calcium imaging was performed using a spinning disc confocal microscope (Nikon CSU-W1, Melville, NY, USA) system. Slices were perfused with experimental aCSF containing (mM): 126 NaCl, 26 NaHCO3, 1.24 NaH2PO4, 2.5 KCl, 10 D-glucose, 2.4 CaCl2, 1.3 MgCl2. Thirty seconds of baseline data were collected with perfusate that contained vehicle, whereupon the perfusate was changed to one containing a test agent, and images were obtained for an additional 4.5 minutes. Test agents included diazoxide (100 μM), NN414 (1 μM), glibenclamide (10 μM), SEA0400 (1 μM), HC-067047 (1 μM). For analysis, regions-of-interest (ROI) were drawn over perivascular astrocyte endfeet, and mean fluorescence was expressed as F/F0. Photobleaching curves were estimated from the baseline data and subtracted from the timeseries.
Isolation of microvessels with attached astrocyte endfeet
Cerebral microvessels were isolated from mouse brain using the method described by Boulay et al. (82, 83). These microvessels remain coated with β-dystroglycan+ and AQP4+ astrocyte endfeet that had been “amputated” from their parent astrocyte somata, as we showed (9).
Brain slice cell-membrane biotinylation and immunoblotting (84, 85)
Following MCAO/R, mice were sacrificed using a lethal dose of sodium pentobarbial IP and cervical dislocation. The cerebrum was dissected, then quickly transferred to ice-cold slicing aCSF (see above). Using a vibratome (VT1200S, Leica Biosystems), 400 μm-thick coronal slices from +1.0 mm to −2.0 mm relative to bregma were prepared in ice-cold slicing aCSF bubbled with carbogen (95% O2 / 5% CO2). A single 400 μm slice at the epicenter was prepared and incubated in 2% TTC at room temperature to confirm an ischemic injury. The subfornical organ, which contains AQP4-expressing ependymal cells (16), was removed. Hemislices contra- or ipsilateral to the MCAO/R injury were washed then placed in EZ-link™ sulfo-NHS-SS-biotin (1 mg/mL, ThermoFisher Scientific) in ice-cold aCSF for 30 minutes with gentle rocking. The biotinylation reaction was quenched by washing the slices 3× over 30 minutes using ice-cold Tris-buffered (50 mM, Gibco, ThermoFisher Scientific) aCSF. Slices were lysed by mechanical homogenization, then repeatedly vortexed in ice-cold Tris-lysis buffer (10 mM Tris, ½X DPBS, 1% Triton X-100) supplemented with the cOmplete™ protease inhibitor cocktail (MilliporeSigma) for 30 minutes. Soluble lysates were cleared by centrifugation for 10 minutes, and protein amounts were determined using the Bradford assay (Bio-Rad, Hercules, CA, USA). Then, 4 mg of soluble lysates was incubated in streptavidin-Dynabeads™ (Invitrogen, ThermoFisher Scientific) with end-over-end rotation overnight at 4 °C. Magnetically immobilized streptavidin beads were washed 3×. Biotinylated proteins were eluted using 2X NuPAGE LDS sample buffer (Invitrogen) and warmed for 30 minutes at 37 °C.
Pulled down biotinylated proteins (surface) and non-pulled down lysate (total) were resolved using gel electrophoresis then transferred to 0.45-μm PVDF membranes (LC2005, Invitrogen). Membranes were incubated with primary antibodies against AQP4 (AB3594), Na+/K+ ATPase subunits, (ATP1A1, ATP1A2) or HSC70 (table S1) overnight at 4 °C in 5% nonfat milk/1X TBST blocking buffer. Following 3× washes in 1X TBST, membranes were incubated with species-appropriate HRP-conjugated secondary antibodies (Cell Signaling Technology, Danvers, MA, USA) for 1 hour at room temperature. After thorough washes in 1X TBST, membranes were incubated in chemiluminescent substrate (SuperSignal West Pico PLUS Chemiluminescent Substrate, ThermoFisher Scientific) and imaged with a chemiluminescent imaging system (ImageQuant LAS-4000, Fujifilm, Lake Forest, IL, USA). Detected signals were analyzed by densitometry using the ImageJ software. Total AQP4 was normalized to HSC70. The change in ipsilateral total AQP4 is presented as [(TI/HSC70I)/(TC/HSC70C)]*100%, where T=total AQP4, I=ipsilateral and C=contralateral. The change in ipsilateral surface AQP4 is presented as [(SI/TI)/(SC/TC)]*100%, where S=surface AQP4, T=total AQP4, I=ipsilateral and C=contralateral.
In initial experiments, we studied cell-surface localization of AQP4 after 2-hour MCAO / 24-hour reperfusion. This experiment yielded abundant biotinylated HSC70, which we took as evidence of post-ischemic cellular necrosis that would invalidate accurate determination of surface protein expression due to contamination by intracellular protein. Repeating the experiment with 2-hour MCAO / 6-hour reperfusion yielded no detectable biotinylated HSC70. All experiments reported here on cell-surface localization were performed with MCAO/R (2/6 hours).
Serine phosphorylation of AQP4
Brain hemispheres rapidly dissected from post-ischemic mice were Dounce homogenized in ice-cold lysis buffer (1/2X DPBS, 1% Triton X-100) supplemented with the cOmplete™ protease inhibitor and PhosStop™ phosphatase inhibitor (Millipore) cocktails. Following centrifugation of lysate and Bradford estimation of precleared proteins, lysates were immunoprecipitated with the AQP4 antibody (sc-32739; table S1) and Dynabeads Protein G (ThermoFisher) overnight in 4 °C. Aliquots of AQP4-immunoprecipitated fraction were either treated or not treated with alkaline phosphatase (100 U/mL; Millipore) at 37 °C for 30 minutes prior to elution in the 2X NuPAGE LDS sample buffer. Following gel electrophoresis and transfer to 0.2 μm PVDF membranes, membranes were probed for phosphoserine (table S1) in 5% v/v BSA in 1X TBST. Following chemiluminescent detection, membranes were stripped and re-probed for immunoprecipitated AQP4, using AQP4 antibody (AB3594; table S1), as described above. Specific AQP4 phosphoserine signals were identified by comparing the abolishment of signals at the appropriate molecular mass in the phosphatase-treated fractions.
Immunohistochemistry
Under deep anesthesia, mice were euthanized, underwent transcardial perfusion with normal saline (NS) (20 mL) followed by 10% neutral buffered formalin (20 mL) or 4% paraformaldehyde (20 mL). Brains were harvested and post-fixed. Tissues were cryoprotected with 30% sucrose, frozen in OCT and cryosectioned (10 μm). Immunohistochemistry was performed as we described (86, 87) in a non-blinded manner. In some cases, sections were first processed for antigen retrieval in Epitope Retrieval Solution (cat# IW-1100; IHCWORLD, Woodstock, MD, USA) using an Epitope Retrieval Steamer (cat# IW-1102; IHCWORLD) for 10–15 minutes, followed by cooling for 30 minutes, then washing in distilled H2O. For all immunolabelings, sections were incubated at 4 °C overnight with primary antibodies (table S1). After several rinses in PBS, sections were incubated with species-appropriate fluorescent secondary antibodies (Alexa Fluor 488 and 555, Molecular Probes, ThermoFisher Scientific) for 1 hour at room temperature. Controls for immunohistochemistry included the omission of primary antibodies. Unbiased assessments of specific labeling were obtained using NIS-Elements AR software (Nikon Instruments, Melville, NY, USA) from sections (one section per mouse) immunolabeled as a single batch. All images for a given signal were captured using uniform parameters of magnification, area, exposure and gain.
Proximity ligation assay (PLA)
PLA, the recognition of two epitopes detected only if they are less than 40 nm apart (88), was used to visualize in situ protein-protein interactions and AQP4 serine phosphorylation and confirmed by immunoblot. Following antigen retrieval, tissues were processed as above with primary antibodies directed against the paired targets: SUR1/TRPM4, AQP4/CaM, and AQP4/phosphoserine (table S1). PLAs were performed according to the manufacturer’s protocol, as we described (87), using the Duolink® In Situ Orange Starter Kit (MilliporeSigma), including in situ PLA probe anti-goat plus (DUO92003), in situ PLA probe anti-rabbit minus (DUO92005), in situ wash buffer, fluorescent (DUO82049) and fluorescent detection reagent (orange) (DUO92007). Controls included the use of sham tissues and the omission of primary antibodies with injured tissues.
For analysis, separate ROIs of 240 × 160 μm were placed on multiple random ipsilateral MAP2− areas, multiple ipsilateral ACA-MCA watershed areas (identified at the edge of MAP2 labelings), and multiple random contralateral MAP2+ cortical areas. We computed a histogram of pixel intensity for a particular ROI, and pixels were classified as having specific labeling based on signal intensity greater than 3× that of background. We quantified the number of punctae per mm2 using the “object count” module in the NIS-Elements AR software. The average of 3 ROIs for each mouse was computed.
Statistical analyses
Nominal data are presented as mean ± SE unless otherwise noted. The Kolmogorov-Smirnov test was used to assess normality of the data. PLA (4 conditions), cell-membrane AQP4 (6 conditions), infarct volumes (18 conditions), hemispheric swelling (18 conditions), and EB extravasation (5 conditions) were analyzed using ANOVA with post-hoc Bonferroni corrections. Otherwise, nominal data were analyzed using a t-test. Pairs of Garcia neuroscores were analyzed using the Mann-Whitney test. Statistical tests were performed using Origin Pro (V8; OriginLab, North Hampton, MA, USA) or GraphPad Prism (8.3.0; GraphPad Software, San Diego, CA, USA). Significance threshold was P<0.05.
Supplementary Material
Acknowledgements:
We thank Prof. Susumu Seino, Kobe University, Japan, and Dr. Sonia M. Najjar, Heritage College of Osteopathic Medicine, Athens, OH, USA for providing mice with global deletion of Kcnj11. Prof. Em. Kenneth Philipson, UCLA, Los Angeles, CA, USA, originally provided the NCX1-floxed (Slc8a1fl/fl) mice to M.P.B. We thank Prof. Dr. Marc Freichel and Prof. Dr. Veit Flockerzi, Universität des Saarlandes, Homburg, Germany for providing mice with global deletion of Trpm4. We acknowledge with gratitude the expert assistance of the Confocal Microscopy Core, Center for Innovative Biomedical Resources, University of Maryland, Baltimore (NIH grant, S10 OD026698).
Funding:
J.M.S. is supported by grants from the Department of Veterans Affairs (I01RX003060, I01BX004652), the National Heart, Lung and Blood Institute (R01HL082517) and the National Institute of Neurological Disorders and Stroke (NINDS) (R01NS102589; R01NS105633; R01NS127986); V.G. is supported by a grant from NINDS (R01NS107262); K.T.K. is supported by grants from NINDS (R01NS117609; R01NS111029); R.M.J. is supported by a grant from NINDS (R01NS115815).
Footnotes
Competing Interests: J.M.S. holds a US patent (7,285,574), “A novel non-selective cation channel in neural cells and methods for treating brain swelling.” J.M.S. is a member of the Board of Directors and holds shares in Remedy Pharmaceuticals and is a paid consultant for Biogen. No support, direct or indirect, was provided to J.M.S. or for this project by Remedy Pharmaceuticals or by Biogen. All other authors declare that they have no competing interests.
Data and materials availability:
All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
References and Notes
- 1.Arch AE, Sheth KN, Malignant cerebral edema after large anterior circulation infarction: a review. Curr. Treat. Options. Cardiovasc. Med 16, 275 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Mestre H, Du T, Sweeney AM, Liu G, Samson AJ, Peng W, Mortensen KN, Staeger FF, Bork PAR, Bashford L, Toro ER, Tithof J, Kelley DH, Thomas JH, Hjorth PG, Martens EA, Mehta RI, Solis O, Blinder P, Kleinfeld D, Hirase H, Mori Y, Nedergaard M, Cerebrospinal fluid influx drives acute ischemic tissue swelling. Science 367, eaax7171 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Simard JM, Kent TA, Chen M, Tarasov KV, Gerzanich V, Brain oedema in focal ischaemia: molecular pathophysiology and theoretical implications. Lancet Neurol 6, 258–268 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stokum JA, Gerzanich V, Simard JM, Molecular pathophysiology of cerebral edema. J Cereb Blood Flow Metab 36, 513–538 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang CX, Shuaib A, Critical role of microvasculature basal lamina in ischemic brain injury. Prog Neurobiol 83, 140–148 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Underly RG, Levy M, Hartmann DA, Grant RI, Watson AN, Shih AY, Pericytes as Inducers of Rapid, Matrix Metalloproteinase-9-Dependent Capillary Damage during Ischemia. J Neurosci 37, 129–140 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cai W, Liu H, Zhao J, Chen LY, Chen J, Lu Z, Hu X, Pericytes in Brain Injury and Repair After Ischemic Stroke. Transl Stroke Res 8, 107–121 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Derouiche A, Pannicke T, Haseleu J, Blaess S, Grosche J, Reichenbach A, Beyond polarity: functional membrane domains in astrocytes and Muller cells. Neurochem Res 37, 2513–2523 (2012). [DOI] [PubMed] [Google Scholar]
- 9.Stokum JA, Shim B, Huang W, Kane M, Smith JA, Gerzanich V, Simard JM, A large portion of the astrocyte proteome is dedicated to perivascular endfeet, including critical components of the electron transport chain. J Cereb Blood Flow Metab, 2546–2560 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nuriya M, Yasui M, Endfeet serve as diffusion-limited subcellular compartments in astrocytes. J Neurosci 33, 3692–3698 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mathiisen TM, Lehre KP, Danbolt NC, Ottersen OP, The perivascular astroglial sheath provides a complete covering of the brain microvessels: an electron microscopic 3D reconstruction. Glia 58, 1094–1103 (2010). [DOI] [PubMed] [Google Scholar]
- 12.Mills WA 3rd, Woo AM, Jiang S, Martin J, Surendran D, Bergstresser M, Kimbrough IF, Eyo UB, Sofroniew MV, Sontheimer H, Astrocyte plasticity in mice ensures continued endfoot coverage of cerebral blood vessels following injury and declines with age. Nat Commun 13, 1794 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liebner S, Plate KH, Differentiation of the brain vasculature: the answer came blowing by the Wnt. J Angiogenes Res 2, 1 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cho C, Smallwood PM, Nathans J, Reck and Gpr124 Are Essential Receptor Cofactors for Wnt7a/Wnt7b-Specific Signaling in Mammalian CNS Angiogenesis and Blood-Brain Barrier Regulation. Neuron 95, 1056–1073 e1055 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heithoff BP, George KK, Phares AN, Zuidhoek IA, Munoz-Ballester C, Robel S, Astrocytes are necessary for blood-brain barrier maintenance in the adult mouse brain. Glia 69, 436–472 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nielsen S, Nagelhus EA, Amiry-Moghaddam M, Bourque C, Agre P, Ottersen OP, Specialized membrane domains for water transport in glial cells: high-resolution immunogold cytochemistry of aquaporin-4 in rat brain. J Neurosci 17, 171–180 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gleiser C, Wagner A, Fallier-Becker P, Wolburg H, Hirt B, Mack AF, Aquaporin-4 in astroglial cells in the CNS and supporting cells of sensory organs-a comparative perspective. Int J Mol Sci 17, 1411 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vella J, Zammit C, Di Giovanni G, Muscat R, Valentino M, The central role of aquaporins in the pathophysiology of ischemic stroke. Front Cell Neurosci 9, 108 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang X, Andjelkovic AV, Zhu L, Yang T, Bennett MVL, Chen J, Keep RF, Shi Y, Blood-brain barrier dysfunction and recovery after ischemic stroke. Prog Neurobiol 163–164, 144–171 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nagelhus EA, Ottersen OP, Physiological roles of aquaporin-4 in brain. Physiol Rev 93, 1543–1562 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Szczygielski J, Kopanska M, Wysocka A, Oertel J, Cerebral Microcirculation, Perivascular Unit, and Glymphatic System: Role of Aquaporin-4 as the Gatekeeper for Water Homeostasis. Front Neurol 12, 767470 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Woo SK, Tsymbalyuk N, Tsymbalyuk O, Ivanova S, Gerzanich V, Simard JM, SUR1-TRPM4 channels, not KATP, mediate brain swelling following cerebral ischemia. Neurosci Lett 718, 134729 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dabrowski M, Larsen T, Ashcroft FM, Bondo Hansen J, Wahl P, Potent and selective activation of the pancreatic beta-cell type K(ATP) channel by two novel diazoxide analogues. Diabetologia 46, 1375–1382 (2003). [DOI] [PubMed] [Google Scholar]
- 24.Chen M, Simard JM, Cell swelling and a nonselective cation channel regulated by internal Ca2+ and ATP in native reactive astrocytes from adult rat brain. J Neurosci 21, 6512–6521 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Salman MM, Kitchen P, Woodroofe MN, Brown JE, Bill RM, Conner AC, Conner MT, Hypothermia increases aquaporin 4 (AQP4) plasma membrane abundance in human primary cortical astrocytes via a calcium/transient receptor potential vanilloid 4 (TRPV4)- and calmodulin-mediated mechanism. Eur J Neurosci 46, 2542–2547 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kitchen P, Salman MM, Halsey AM, Clarke-Bland C, MacDonald JA, Ishida H, Vogel HJ, Almutiri S, Logan A, Kreida S, Al-Jubair T, Winkel Missel J, Gourdon P, Tornroth-Horsefield S, Conner MT, Ahmed Z, Conner AC, Bill RM, Targeting aquaporin-4 subcellular localization to treat central nervous system edema. Cell 181, 784–799 e719 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen M, Dong Y, Simard JM, Functional coupling between sulfonylurea receptor type 1 and a nonselective cation channel in reactive astrocytes from adult rat brain. J Neurosci 23, 8568–8577 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Minelli A, Castaldo P, Gobbi P, Salucci S, Magi S, Amoroso S, Cellular and subcellular localization of Na+-Ca2+ exchanger protein isoforms, NCX1, NCX2, and NCX3 in cerebral cortex and hippocampus of adult rat. Cell Calcium 41, 221–234 (2007). [DOI] [PubMed] [Google Scholar]
- 29.Rose CR, Ziemens D, Verkhratsky A, On the special role of NCX in astrocytes: Translating Na+-transients into intracellular Ca2+ signals. Cell Calcium 86, 102154 (2020). [DOI] [PubMed] [Google Scholar]
- 30.Iwamoto T, Forefront of Na+/Ca2+ exchanger studies: molecular pharmacology of Na+/Ca2+ exchange inhibitors. J Pharmacol Sci 96, 27–32 (2004). [DOI] [PubMed] [Google Scholar]
- 31.Ozdemir S, Bito V, Holemans P, Vinet L, Mercadier JJ, Varro A, Sipido KR, Pharmacological inhibition of Na+/Ca2+ exchange results in increased cellular Ca2+ load attributable to the predominance of forward mode block. Circ Res 102, 1398–1405 (2008). [DOI] [PubMed] [Google Scholar]
- 32.Frydenlund DS, Bhardwaj A, Otsuka T, Mylonakou MN, Yasumura T, Davidson KG, Zeynalov E, Skare O, Laake P, Haug FM, Rash JE, Agre P, Ottersen OP, Amiry-Moghaddam M, Temporary loss of perivascular aquaporin-4 in neocortex after transient middle cerebral artery occlusion in mice. Proc Natl Acad Sci U S A 103, 13532–13536 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kitchen P, Day RE, Taylor LH, Salman MM, Bill RM, Conner MT, Conner AC, Identification and molecular mechanisms of the rapid tonicity-induced relocalization of the aquaporin 4 channel. J Biol Chem 290, 16873–16881 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li Y, Schmidt-Edelkraut U, Poetz F, Oliva I, Mandl C, Holzl-Wenig G, Schonig K, Bartsch D, Ciccolini F, gamma-Aminobutyric A receptor (GABA(A)R) regulates aquaporin 4 expression in the subependymal zone: relevance to neural precursors and water exchange. J Biol Chem 290, 4343–4355 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mann CN, Devi SS, Kersting CT, Bleem AV, Karch CM, Holtzman DM, Gallardo G, Astrocytic alpha2-Na(+)/K(+) ATPase inhibition suppresses astrocyte reactivity and reduces neurodegeneration in a tauopathy mouse model. Sci Transl Med 14, eabm4107 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kondo T, Reaume AG, Huang TT, Carlson E, Murakami K, Chen SF, Hoffman EK, Scott RW, Epstein CJ, Chan PH, Reduction of CuZn-superoxide dismutase activity exacerbates neuronal cell injury and edema formation after transient focal cerebral ischemia. J Neurosci 17, 4180–4189 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park CK, Jun SS, Cho SH, Kang JK, Assessment of the relationship between ischemic damage and brain swelling in frozen brain slices. Acta Neurochir Suppl 70, 17–19 (1997). [DOI] [PubMed] [Google Scholar]
- 38.Simard JM, Yurovsky V, Tsymbalyuk N, Melnichenko L, Ivanova S, Gerzanich V, Protective effect of delayed treatment with low-dose glibenclamide in three models of ischemic stroke. Stroke 40, 604–609 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wali B, Ishrat T, Atif F, Hua F, Stein DG, Sayeed I, Glibenclamide Administration Attenuates Infarct Volume, Hemispheric Swelling, and Functional Impairments following Permanent Focal Cerebral Ischemia in Rats. Stroke Res Treat 2012, 460909 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Woo SK, Kwon MS, Ivanov A, Gerzanich V, Simard JM, The sulfonylurea receptor 1 (Sur1)-transient receptor potential melastatin 4 (Trpm4) channel. J Biol Chem 288, 3655–3667 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhong CJ, Chen MM, Lu M, Ding JH, Du RH, Hu G, Astrocyte-specific deletion of Kir6.1/K-ATP channel aggravates cerebral ischemia/reperfusion injury through endoplasmic reticulum stress in mice. Exp Neurol 311, 225–233 (2019). [DOI] [PubMed] [Google Scholar]
- 42.Farr GW, Hall CH, Farr SM, Wade R, Detzel JM, Adams AG, Buch JM, Beahm DL, Flask CA, Xu K, LaManna JC, McGuirk PR, Boron WF, Pelletier MF, Functionalized phenylbenzamides inhibit aquaporin-4 reducing cerebral edema and improving outcome in two models of CNS injury. Neuroscience 404, 484–498 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Onai Y, Suzuki J, Kakuta T, Maejima Y, Haraguchi G, Fukasawa H, Muto S, Itai A, Isobe M, Inhibition of IkappaB phosphorylation in cardiomyocytes attenuates myocardial ischemia/reperfusion injury. Cardiovasc Res 63, 51–59 (2004). [DOI] [PubMed] [Google Scholar]
- 44.Inoue M, Wakayama Y, Liu JW, Murahashi M, Shibuya S, Oniki H, Ultrastructural localization of aquaporin 4 and alpha1-syntrophin in the vascular feet of brain astrocytes. Tohoku J Exp Med 197, 87–93 (2002). [DOI] [PubMed] [Google Scholar]
- 45.Hoddevik EH, Khan FH, Rahmani S, Ottersen OP, Boldt HB, Amiry-Moghaddam M, Factors determining the density of AQP4 water channel molecules at the brain-blood interface. Brain Struct Funct 222, 1753–1766 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Perregaux DG, McNiff P, Laliberte R, Hawryluk N, Peurano H, Stam E, Eggler J, Griffiths R, Dombroski MA, Gabel CA, Identification and characterization of a novel class of interleukin-1 post-translational processing inhibitors. J Pharmacol Exp Ther 299, 187–197 (2001). [PubMed] [Google Scholar]
- 47.Lamkanfi M, Mueller JL, Vitari AC, Misaghi S, Fedorova A, Deshayes K, Lee WP, Hoffman HM, Dixit VM, Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J Cell Biol 187, 61–70 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yamashita K, Watanabe Y, Kita S, Iwamoto T, Kimura J, Inhibitory effect of YM-244769, a novel Na(+)/Ca(2+) exchanger inhibitor on Na(+)/Ca(2+) exchange current in guinea pig cardiac ventricular myocytes. Naunyn Schmiedebergs Arch Pharmacol 389, 1205–1214 (2016). [DOI] [PubMed] [Google Scholar]
- 49.Pivonkova H, Hermanova Z, Kirdajova D, Awadova T, Malinsky J, Valihrach L, Zucha D, Kubista M, Galisova A, Jirak D, Anderova M, The Contribution of TRPV4 Channels to Astrocyte Volume Regulation and Brain Edema Formation. Neuroscience 394, 127–143 (2018). [DOI] [PubMed] [Google Scholar]
- 50.Tanaka K, Matsumoto S, Yamada T, Yamasaki R, Suzuki M, Kido MA, Kira JI, Reduced Post-ischemic Brain Injury in Transient Receptor Potential Vanilloid 4 Knockout Mice. Front Neurosci 14, 453 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Song S, Luo L, Sun B, Sun D, Roles of glial ion transporters in brain diseases. Glia 68, 472–494 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stokum JA, Kwon MS, Woo SK, Tsymbalyuk O, Vennekens R, Gerzanich V, Simard JM, SUR1-TRPM4 and AQP4 form a heteromultimeric complex that amplifies ion/water osmotic coupling and drives astrocyte swelling. Glia 66, 108–125 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Simard JM, Chen M, Tarasov KV, Bhatta S, Ivanova S, Melnitchenko L, Tsymbalyuk N, West GA, Gerzanich V, Newly expressed SUR1-regulated NC(Ca-ATP) channel mediates cerebral edema after ischemic stroke. Nat Med 12, 433–440 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Simard JM, Tsymbalyuk N, Tsymbalyuk O, Ivanova S, Yurovsky V, Gerzanich V, Glibenclamide is superior to decompressive craniectomy in a rat model of malignant stroke. Stroke 41, 531–537 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sheth KN, Elm JJ, Molyneaux BJ, Hinson H, Beslow LA, Sze GK, Ostwaldt AC, Del Zoppo GJ, Simard JM, Jacobson S, Kimberly WT, Safety and efficacy of intravenous glyburide on brain swelling after large hemispheric infarction (GAMES-RP): a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Neurol 15, 1160–1169 (2016). [DOI] [PubMed] [Google Scholar]
- 56.Kimberly WT, Bevers MB, von Kummer R, Demchuk AM, Romero JM, Elm JJ, Hinson HE, Molyneaux BJ, Simard JM, Sheth KN, Effect of IV glyburide on adjudicated edema endpoints in the GAMES-RP Trial. Neurology 91, e2163–e2169 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vorasayan P, Bevers MB, Beslow LA, Sze G, Molyneaux BJ, Hinson HE, Simard JM, von Kummer R, Sheth KN, Kimberly WT, Intravenous Glibenclamide Reduces Lesional Water Uptake in Large Hemispheric Infarction. Stroke 50, 3021–3027 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gerzanich V, Kwon MS, Woo SK, Ivanov A, Simard JM, SUR1-TRPM4 channel activation and phasic secretion of MMP-9 induced by tPA in brain endothelial cells. PLoS One 13, e0195526 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Igarashi T, Sastre C, Wolcott Z, Kimberly WT, Continuous Glibenclamide Prevents Hemorrhagic Transformation in a Rodent Model of Severe Ischemia-Reperfusion. J Stroke Cerebrovasc Dis 30, 105595 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Simard JM, Woo SK, Tsymbalyuk N, Voloshyn O, Yurovsky V, Ivanova S, Lee R, Gerzanich V, Glibenclamide-10-h Treatment Window in a Clinically Relevant Model of Stroke. Transl Stroke Res 3, 286–295 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ng FC, Yassi N, Sharma G, Brown SB, Goyal M, Majoie C, Jovin TG, Hill MD, Muir KW, Saver JL, Guillemin F, Demchuk AM, Menon BK, San Roman L, Liebeskind DS, White P, Dippel DWJ, Davalos A, Bracard S, Mitchell PJ, Wald MJ, Davis SM, Sheth KN, Kimberly WT, Campbell BCV, Collaborators H, Cerebral Edema in Patients With Large Hemispheric Infarct Undergoing Reperfusion Treatment: A HERMES Meta-Analysis. Stroke 52, 3450–3458 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Amiry-Moghaddam M, Otsuka T, Hurn PD, Traystman RJ, Haug FM, Froehner SC, Adams ME, Neely JD, Agre P, Ottersen OP, Bhardwaj A, An alpha-syntrophin-dependent pool of AQP4 in astroglial end-feet confers bidirectional water flow between blood and brain. Proc Natl Acad Sci U S A 100, 2106–2111 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Percie du Sert N, Hurst V, Ahluwalia A, Alam S, Avey MT, Baker M, Browne WJ, Clark A, Cuthill IC, Dirnagl U, Emerson M, Garner P, Holgate ST, Howells DW, Karp NA, Lazic SE, Lidster K, MacCallum CJ, Macleod M, Pearl EJ, Petersen OH, Rawle F, Reynolds P, Rooney K, Sena ES, Silberberg SD, Steckler T, Wurbel H, The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. J Cereb Blood Flow Metab 40, 1769–1777 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nakamura Y, Bryan J, Targeting SUR1/Abcc8-type neuroendocrine KATP Channels in Pancreatic Islet Cells. PLoS One 9, e91525 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Henderson SA, Goldhaber JI, So JM, Han T, Motter C, Ngo A, Chantawansri C, Ritter MR, Friedlander M, Nicoll DA, Frank JS, Jordan MC, Roos KP, Ross RS, Philipson KD, Functional adult myocardium in the absence of Na+-Ca2+ exchange: cardiac-specific knockout of NCX1. Circ Res 95, 604–611 (2004). [DOI] [PubMed] [Google Scholar]
- 66.Vennekens R, Olausson J, Meissner M, Bloch W, Mathar I, Philipp SE, Schmitz F, Weissgerber P, Nilius B, Flockerzi V, Freichel M, Increased IgE-dependent mast cell activation and anaphylactic responses in mice lacking the calcium-activated nonselective cation channel TRPM4. Nat Immunol 8, 312–320 (2007). [DOI] [PubMed] [Google Scholar]
- 67.Miki T, Nagashima K, Tashiro F, Kotake K, Yoshitomi H, Tamamoto A, Gonoi T, Iwanaga T, Miyazaki J, Seino S, Defective insulin secretion and enhanced insulin action in KATP channel-deficient mice. Proc Natl Acad Sci U S A 95, 10402–10406 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, Lein ES, Zeng H, A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13, 133–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Engel O, Kolodziej S, Dirnagl U, Prinz V, Modeling stroke in mice - middle cerebral artery occlusion with the filament model. J Vis Exp, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ansari S, Azari H, McConnell DJ, Afzal A, Mocco J, Intraluminal middle cerebral artery occlusion (MCAO) model for ischemic stroke with laser doppler flowmetry guidance in mice. J Vis Exp, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bertrand L, Dygert L, Toborek M, Induction of Ischemic Stroke and Ischemia-reperfusion in Mice Using the Middle Artery Occlusion Technique and Visualization of Infarct Area. J Vis Exp, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Llovera G, Simats A, Liesz A, Modeling Stroke in Mice: Transient Middle Cerebral Artery Occlusion via the External Carotid Artery. J Vis Exp, (2021). [DOI] [PubMed] [Google Scholar]
- 73.Matsuda T, Arakawa N, Takuma K, Kishida Y, Kawasaki Y, Sakaue M, Takahashi K, Takahashi T, Suzuki T, Ota T, Hamano-Takahashi A, Onishi M, Tanaka Y, Kameo K, Baba A, SEA0400, a novel and selective inhibitor of the Na+-Ca2+ exchanger, attenuates reperfusion injury in the in vitro and in vivo cerebral ischemic models. J Pharmacol Exp Ther 298, 249–256 (2001). [PubMed] [Google Scholar]
- 74.Morimoto N, Kita S, Shimazawa M, Namimatsu H, Tsuruma K, Hayakawa K, Mishima K, Egashira N, Iyoda T, Horie I, Gotoh Y, Iwasaki K, Fujiwara M, Matsuda T, Baba A, Komuro I, Horie K, Takeda J, Iwamoto T, Hara H, Preferential involvement of Na(+)/Ca(2)(+) exchanger type-1 in the brain damage caused by transient focal cerebral ischemia in mice. Biochem Biophys Res Commun 429, 186–190 (2012). [DOI] [PubMed] [Google Scholar]
- 75.Iwamoto T, Watanabe Y, Kita S, Blaustein MP, Na+/Ca2+ exchange inhibitors: a new class of calcium regulators. Cardiovasc Hematol Disord Drug Targets 7, 188–198 (2007). [DOI] [PubMed] [Google Scholar]
- 76.Shimamura N, Matchett G, Tsubokawa T, Ohkuma H, Zhang J, Comparison of silicon-coated nylon suture to plain nylon suture in the rat middle cerebral artery occlusion model. J Neurosci Methods 156, 161–165 (2006). [DOI] [PubMed] [Google Scholar]
- 77.Friedlander F, Bohmann F, Brunkhorst M, Chae JH, Devraj K, Kohler Y, Kraft P, Kuhn H, Lucaciu A, Luger S, Pfeilschifter W, Sadler R, Liesz A, Scholtyschik K, Stolz L, Vutukuri R, Brunkhorst R, Reliability of infarct volumetry: Its relevance and the improvement by a software-assisted approach. J Cereb Blood Flow Metab 37, 3015–3026 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kuts R, Frank D, Gruenbaum BF, Grinshpun J, Melamed I, Knyazer B, Tarabrin O, Zvenigorodsky V, Shelef I, Zlotnik A, Boyko M, A Novel Method for Assessing Cerebral Edema, Infarcted Zone and Blood-Brain Barrier Breakdown in a Single Post-stroke Rodent Brain. Front Neurosci 13, 1105 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Frank D, Gruenbaum BF, Grinshpun J, Melamed I, Severynovska O, Kuts R, Semyonov M, Brotfain E, Zlotnik A, Boyko M, Measuring Post-Stroke Cerebral Edema, Infarct Zone and Blood-Brain Barrier Breakdown in a Single Set of Rodent Brain Samples. J Vis Exp, (2020). [DOI] [PubMed] [Google Scholar]
- 80.Manaenko A, Chen H, Kammer J, Zhang JH, Tang J, Comparison Evans Blue injection routes: Intravenous versus intraperitoneal, for measurement of blood-brain barrier in a mice hemorrhage model. J Neurosci Methods 195, 206–210 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Del Franco AP, Chiang PP, Newman EA, Dilation of cortical capillaries is not related to astrocyte calcium signaling. Glia, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Boulay AC, Saubamea B, Adam N, Chasseigneaux S, Mazare N, Gilbert A, Bahin M, Bastianelli L, Blugeon C, Perrin S, Pouch J, Ducos B, Le Crom S, Genovesio A, Chretien F, Decleves X, Laplanche JL, Cohen-Salmon M, Translation in astrocyte distal processes sets molecular heterogeneity at the gliovascular interface. Cell Discov 3, 17005 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Boulay AC, Saubamea B, Decleves X, Cohen-Salmon M, Purification of Mouse Brain Vessels. J Vis Exp, e53208 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gabriel LR, Wu S, Melikian HE, Brain slice biotinylation: an ex vivo approach to measure region-specific plasma membrane protein trafficking in adult neurons. J Vis Exp, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wheeler DS, Underhill SM, Stolz DB, Murdoch GH, Thiels E, Romero G, Amara SG, Amphetamine activates Rho GTPase signaling to mediate dopamine transporter internalization and acute behavioral effects of amphetamine. Proc Natl Acad Sci U S A 112, E7138–7147 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gerzanich V, Stokum JA, Ivanova S, Woo SK, Tsymbalyuk O, Sharma A, Akkentli F, Imran Z, Aarabi B, Sahuquillo J, Simard JM, Sulfonylurea Receptor 1, Transient Receptor Potential Cation Channel Subfamily M Member 4, and KIR6.2:Role in Hemorrhagic Progression of Contusion. J Neurotrauma 36, 1060–1079 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tsymbalyuk O, Gerzanich V, Mumtaz A, Andhavarapu S, Ivanova S, Makar TK, Sansur CA, Keller A, Nakamura Y, Bryan J, Simard JM, SUR1, newly expressed in astrocytes, mediates neuropathic pain in a mouse model of peripheral nerve injury. Mol Pain 17, 17448069211006603 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Soderberg O, Gullberg M, Jarvius M, Ridderstrale K, Leuchowius KJ, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson LG, Landegren U, Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods 3, 995–1000 (2006). [DOI] [PubMed] [Google Scholar]
- 89.Bentivoglio M, Kristensson K, Tryps and trips: cell trafficking across the 100-year-old blood-brain barrier. Trends Neurosci 37, 325–333 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Makar TK, Gerzanich V, Nimmagadda VK, Jain R, Lam K, Mubariz F, Trisler D, Ivanova S, Woo SK, Kwon MS, Bryan J, Bever CT, Simard JM, Silencing of Abcc8 or inhibition of newly upregulated Sur1-Trpm4 reduce inflammation and disease progression in experimental autoimmune encephalomyelitis. J Neuroinflammation 12, 210 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gerzanich V, Makar TK, Guda PR, Kwon MS, Stokum JA, Woo SK, Ivanova S, Ivanov A, Mehta RI, Morris AB, Bryan J, Bever CT, Simard JM, Salutary effects of glibenclamide during the chronic phase of murine experimental autoimmune encephalomyelitis. J Neuroinflammation 14, 177 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Everaerts W, Zhen X, Ghosh D, Vriens J, Gevaert T, Gilbert JP, Hayward NJ, McNamara CR, Xue F, Moran MM, Strassmaier T, Uykal E, Owsianik G, Vennekens R, De Ridder D, Nilius B, Fanger CM, Voets T, Inhibition of the cation channel TRPV4 improves bladder function in mice and rats with cyclophosphamide-induced cystitis. Proc Natl Acad Sci U S A 107, 19084–19089 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Popp A, Jaenisch N, Witte OW, Frahm C, Identification of ischemic regions in a rat model of stroke. PLoS One 4, e4764 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yang B, Zador Z, Verkman AS, Glial cell aquaporin-4 overexpression in transgenic mice accelerates cytotoxic brain swelling. J Biol Chem 283, 15280–15286 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Neely JD, Christensen BM, Nielsen S, Agre P, Heterotetrameric composition of aquaporin-4 water channels. Biochemistry 38, 11156–11163 (1999). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.