

Abstract

Improving the photocatalytic efficiency of a fully noble-metal-free system for CO2 reduction remains a fundamental challenge, which can be accomplished by facilitating electron delivery as a consequence of exploiting intermolecular interactions. Herein, we have designed two Cu(I) photosensitizers with different pyridyl pendants at the phenanthroline moiety to enable dynamic coordinative interactions between the sensitizers and a cobalt macrocyclic catalyst. Compared to the parent Cu(I) photosensitizer, one of the pyridine-tethered derivatives boosts the apparent quantum yield up to 76 ± 6% at 425 nm for selective (near 99%) CO2-to-CO conversion. This value is nearly twice that of the parent system with no pyridyl pendants (40 ± 5%) and substantially surpasses the record (57%) of the noble-metal-free systems reported so far. This system also realizes a maximum turnover number of 11 800 ± 1400. In contrast, another Cu(I) photosensitizer, in which the pyridine substituents are directly linked to the phenanthroline moiety, is inactive. The above behavior and photocatalytic mechanism are systematically elucidated by transient fluorescence, transient absorption, transient X-ray absorption spectroscopies, and quantum chemical calculations. This work highlights the advantage of constructing coordinative interactions to fine-tune the electron transfer processes within noble-metal-free systems for CO2 photoreduction.
Keywords: noble-metal-free system, CO2 photoreduction, transient, X-ray, absorption spectroscopy, electron transfer, coordinative, interaction
Introduction
Sunlight-driven reduction of CO2 continues to attract attention as a sustainable, carbon-neutral route to produce renewable fuels.1−4 Due to the sluggish multielectron reaction kinetics and competition with H2 evolution,5,6 considerable efforts have been made to develop efficient and selective systems for photocatalytic CO2 reduction. Up to now, various researchers have studied transition metal complexes as molecular photosensitizers (PSs) and catalysts, since they are highly optimizable via synthetic methods and amenable to the detailed mechanistic investigations.7,8 In the last several decades, noble-metal-based PSs, like Ru, Ir, and Re PSs, have been extensively adopted to photochemically drive CO2 reduction.9−11 However, earth-abundant molecular systems, enabling large-scale applications, have also attracted considerable attention in recent years.12−15 Among them, Cu(I)-based PSs have received great attention owing to their versatile redox characteristics as well as the highly variable ligand scaffolds, permitting the fine-tuning of their redox and excited-state properties.16−24 For example, Ishitani and co-workers have designed a diphosphine-tethered phenanthroline ligand to stabilize a dimeric Cu(I) PS which exhibits a notable stability during the photocatalytic CO2 reduction along with Fe25,26 or Mn27 catalysts. The tetradentate nature of the ligand effectively prevents the dissociation of diphosphine donors bound to the Cu(I) centers, leading to a maximum quantum yield (Φ) of 57% in the coproduction of CO and HCOOH. Beller and co-workers in situ deployed Cu(I) PSs by mixing Cu(I) precursors and the individual ligands to drive the photochemical CO2 reduction catalyzed by either Fe cyclopentadienone12 or Mn diimine complexes28 as catalysts. Furthermore, a water-soluble Cu(I) PS, having a tetrasulfonated form of [CuI(xantphos)(bcp)]+ (CuBCP in Figure 1; xantphos = 9,9-dimethyl-4,5-bis(diphenylphosphino)xanthene, bcp = bathocuproine), was also developed for efficient and selective CO2-to-CO conversion with several water-soluble Co porphyrins as catalysts, demonstrating the good selectivity (near 90%) and high turnover numbers (TONs, up to 4000) even operated in fully aqueous media where H2 evolution is often facilitated.29−31
Figure 1.

Cu(I) PSs. (a) Chemical structures, (b) UV–vis absorption, (c) emission, and (d) excited-state decay traces of the Cu(I) PSs. Deaerated 50 μM CH3CN solutions were used for the above spectra. Trace color: CuBCP, blue; CuPBCP, red; CuPPBCP, gold.
The above reports succeeded in revealing some promising performances of Cu(I) PSs in light-driven CO2 reduction. However, there still remains a possibility of improving the photocatalytic performance, since the noble-metal-containing systems could demonstrate TON > 107 and Φ > 80%.32,33 One can therefore expect further improvement by the rational optimization of the fundamental properties of both catalysts and PSs as well as by more delicately controlling the electron transfer (ET) efficiency among the PS, catalyst, and reductive equivalents. Up to now, various strategies have been explored to improve the ET efficiency in CO2 photoreduction. These include the covalent bonding,34−36 H-bonding,37 coordinate bonding,38 and π–π stacking24,39−41 interactions between the PS and catalyst. However, most of them relied on the use of noble-metal-based PSs and/or catalysts. In other words, studies attempting to improve the ET efficiency among the noble-metal-free PS and catalyst have seldom been reported to date.24
In this context, we have fabricated noble-metal-free PSs having pyridyl tethers to permit their dynamic interactions with catalysts via coordinate bonding (Figure 1), where the “dynamic interactions” are intended to express that the bond formation and dissociation are dynamically taking place during the photocatalysis. Among the available interaction modes between PS and the catalyst, coordinate bonds can be highly advantageous because of the following reasons. The metal–ligand bonds can avoid the irreversible loss of association between the PS and catalyst due to their ability to retrieve the linkage depending upon the metal oxidation state, which cannot be expected for the covalent linkages made of C–C, C–O, C–N, etc. Moreover, the metal–ligand bond dissociation may play a role in suppressing undesirable back electron transfer (BET). For example, the low-spin d7 Co(II) ion possesses either an axially elongated square pyramidal or octahedral geometry and thereby serves as a dynamically exchanging coordination site in giving a linkage between the PS and catalyst.38 The fast exchange is expected due to the fundamental lability of the Co(II)-ligand bonds as well as the relatively long axial bond distances caused by the so-called Jahn–Teller effect. Furthermore, upon driving the ET from the reductively quenched PS to the covalently associated catalyst, the Co(II) center may turn into the d8 Co(I) ion which favors a square-planar geometry,42 resulting in the dissociation of the catalyst from the PS to suppress the undesirable BET.
To this end, we demonstrate that the pyridyl tethers introduced to CuBCP indeed bring about substantial catalytic enhancement in photocatalytic CO2 reduction, presenting our proof of concept on the dynamic coordinative interaction into precious-metal-free systems. Cobalt phthalocyanine tetracarboxylic acid (CoTCPc) is employed as a promising molecular catalyst, where its electron-withdrawing43 carboxyl groups may enable a high catalytic performance. The carboxyl groups are located at two random β-positions due to the different regioisomers arising from the synthesis. To evaluate the effect of pyridyl tethers on CuBCP as the prototype, we designed and synthesized two new types of heteroleptic Cu(I) PSs depicted in Figure 1: [CuI(xantphos)(pbcp)]PF6 (CuPBCP; pbcp = 2,9-dimethyl-4,7-di(pyridin-4-yl)-1,10-phenanthroline) and [CuI(xantphos)(ppbcp)]PF6 (CuPPBCP; ppbcp = 2,9-dimethyl-4,7-bis(4-(pyridin-4-yl)phenyl)-1,10-phenanthroline). The former possesses two pyridyl groups directly bonded to the 2,9-dimethyl-1,10-phenanthroline backbone instead of the phenyl groups in parent bcp. The latter has two pyridyl groups bonded to the 4-positions of the phenyl groups of bcp in the prototype. Our study reveals that the CuPPBCP-driven CO2 reduction catalyzed by CoTCPc (i.e., the CuPPBCP/CoTCPc system) achieves an excellent quantum yield (Φ = 76 ± 6% at 425 nm) with a near quantitative selectivity in CO formation. This efficiency is almost twice higher than that of the prototypical CuBCP/CoTCPc system (Φ = 40 ± 5% at 425 nm). Moreover, the CuPPBCP/CoTCPc system could achieve a maximum TON of 11800 ± 1400. These performances also considerably surpass the previous molecular system with dynamic interactions24,37−41 (Φ < 50%, TON < 1500). On the other hand, the CuPBCP/CoTCPc system is found to exhibit rather poor photocatalytic activity, which is even lower than that of the prototype system. However, this result in turn demonstrates the importance of delicate control in achieving highly efficient photocatalytic systems with desirable ET efficiency. Notably, transient X-ray absorption spectroscopic techniques have been utilized to elucidate the photophysics of photosensitizers and study the electron transfer kinetics.
Results
Syntheses and Photophysics of Cu(I) PSs
Three heteroleptic Cu(I) complexes of the type [Cu(P∧P)(N∧N)]+ were prepared as PSs (Figure 1a and experimental details in the Supporting Information (SI)), with three kinds of N∧N ligands, bcp, pbcp, and ppbcp. Two pyridyl pendants were employed to replace the two phenyl groups at the 4,7-position of the bcp prototype in CuPBCP, and two 4-phenylpyridyl groups were employed in the case of CuPPBCP, respectively, with the aim to generate coordinative interactions with the macrocyclic Co(II) catalyst. The purities and structures of these ligands and complexes were clarified by elemental analysis and 1H NMR spectroscopy (Table S1). The above compounds were further characterized by 1H–1H NMR correlation spectroscopy (COSY; Figures S1–S4), 13C and 19P NMR (for Cu complexes, Figures S5–S8), mass spectroscopy (Figures S9–S12), and single-crystal X-ray diffraction (Figure S13 for CuPBCP only; Tables S2 and S6) methods, respectively.
We then adopted various methods to determine the redox and photophysical properties of the three Cu(I) PSs, as summarized in Table 1. Cyclic voltammetry was first applied to investigate their redox properties, where reversible one-electron reduction waves and irreversible oxidation waves were all observed for the three Cu(I) complexes in degassed CH3CN (Figure S14). Notably, the electron-withdrawing effects from the pyridyl pendants lead to more positive reduction potentials (Ered; −1.88/–1.97 vs −2.04 V vs Fc+/0) of CuPBCP/CuPPBCP than the parent CuBCP, respectively, consistent with the less reducing forces of the reduced-state Cu(I) PSs. The excited-state reduction potentials (Ered,ex)46 of the Cu(I) PSs (Table 1 and Figure S14) were then estimated using the ground-state redox parameters together with the emission energies (E0–0, eV). It can be seen that the Ered,ex values of pyridine-tethered Cu(I) PSs (1.00 and 0.81 V vs Fc+/0) are both more positive than that of the prototype CuBCP (0.75 V), indicative of the stronger ability to oxidize sacrificial reductant.
Table 1. Photophysical and Electrochemical Data: Summary of the Photophysical and Redox Properties of the Cu(I) PSsa.
| Cu PS | Ered (V) | λem (nm) | λab (nm) | ε405 (M–1 cm–1) | Φembc | τb (ns) | kr (103 s–1)d | knr (106 s–1)e | isosbestic point (nm)f | E0–0 (eV)f | Ered,ex (V) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| CuBCP | –2.04 | 580 | 285, 388 | 1760 | 0.23% ± 0.02% | 289 ± 5 | 8.0 | 3.5 | 445 | 2.79 | 0.75 |
| CuPBCP | –1.88 | 590 | 281, 395 | 3070 | 0.03% ± 0.01% | 33.0 ± 3.1; τ1 = 3.5, 33%; τ2 = 48.4, 67% | 9.1 | 30.3 | 430 | 2.88 | 1.00 |
| CuPPBCP | –1.97 | 590 | 289, 389 | 4000 | 0.16% ± 0.02% | 206 ± 4 | 7.8 | 4.8 | 446 | 2.78 | 0.81 |
Measured in in deaerated CH3CN solution at 298 K. Experimental error within 5%. Potentials are versus Fc+/0.
Excited at 405 nm. The error bars represent the standard deviations of three independent measurements.
Determined via the relative method with RuBPY (= Ru(bpy)3(PF6)2; bpy = 2,2′-bipyridine) as the standard, 0.095% at 298 K.44
kr = Φem/τ.45
knr= (1 – Φem)/τ.45
Isosbestic point was obtained from the intersection of normalized absorption and emission spectra and E0–0 values = 1240/isosbestic point (eV).45
Next, the photophysical properties of the PSs in deaerated CH3CN were investigated using UV–vis absorption and steady-state fluorescent spectroscopies. Comparison of their UV–vis absorption spectra clarifies the similarity in their absorption features (see Figure 1b), although the molar absorptivity at ca. 400 nm decreases in the order CuPPBCP > CuPBCP > CuBCP. By contrast, their steady-state emission spectra (Figure 1c) are structureless, in which the emission maxima (λem) for the two pyridyl tethered PSs are by ca. 10 nm red-shifted relative to that of the parent CuBCP, reflecting the electron-withdrawing effect of the pyridyl groups. The emission quantum yields (Φem) reveal a descending order as follows; CuBCP (0.23%) > CuPPBCP (0.16%) > CuPBCP (0.03%). The relatively low Φem values are ascribed to the exciplex quenching effect posed by the CH3CN coordination to the excited Cu dye.47 We further determined the excited-state lifetimes by monitoring the fluorescence decays using time-correlated single photon counting (TCSPC; Figure 2d). The observed lifetimes display a trend similar to that seen in Φem by affording the values of CuBCP, CuPPBCP and CuPBCP as 289, 206, and 33.0 ns, respectively (Table 1). The biexponential decay traces of excited CuPBCP suggest the presence of additional decay pathways. Since the lifetimes of [Cu(P∧P)(N∧N)]+ PSs are generally around 10–7 s in coordinative CH3CN,27,28 the shorter-lived (at 10–8 s) excited state of CuPBCP with its biexponential decay traces suggest the presence of additional decay pathways. We further calculated their radiative (kr) and nonradiative (knr) decay rate constants45 of these Cu(I) chromophores, showing their similar kr values while a much faster knr was estimated for CuPBCP than the other two, also indicative of more decay pathways. The LUMO-HUMO gaps were also determined for their ground- and excited-state structures (Table S3). The excited-state structures of CuBCP, CuPBCP and CuPPBCP display LUMO–HOMO energy gaps of −0.954, −0.845, and −0.881 eV, respectively, with increasing energy gaps of CuPBCP < CuPPBCP < CuBCP. The observed trends of energy gaps are in agreement with the less long-lived excited state48 observed for CuPBCP (33.0 ns) compared with those of CuPPBCP (206 ns) and CuBCP (289 ns), respectively.
Figure 2.
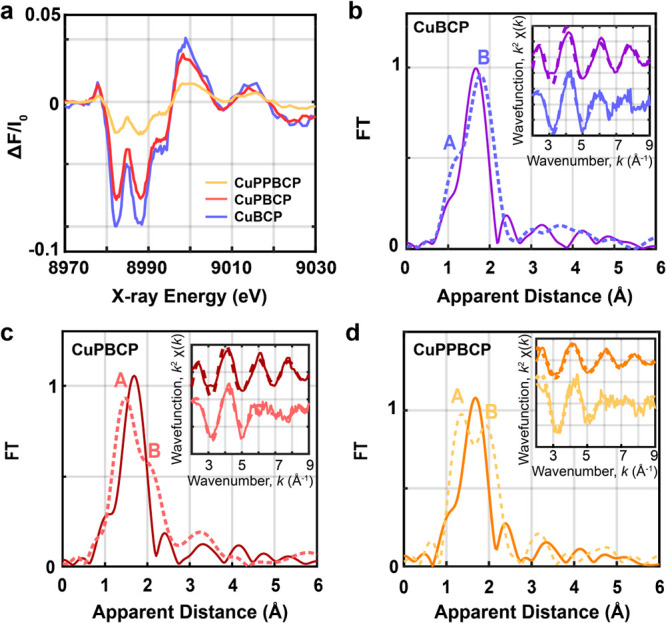
(a) Tr-XAS spectra (laser on/off) corresponding to the excited states of 0.8 mM CuBCP (blue), CuPBCP (red), and CuPPBCP (gold) via 3MLCT transitions at a delay of 100 ps between laser and X-ray pulses. Experimental Fourier transforms of k2-weighted Cu EXAFS of the laser-off (solid lines) and reconstructed (dotted lines) excited states of (b) CuBCP assuming 35% excited state, (c) CuPBCP assuming 27% excited state, and (d) CuPPBCP assuming 9.5% excited state. Insets in (b–d) show the back Fourier transformed experimental (solid lines) and fitted (dashed lines) k2[χ(k)] of the laser off and reconstructed spectra for k values of 2–9 Å–1.
The above observations suggest that the covalent attachment of pyridyl moieties to the N∧N ligand of the Cu(I) PS results in a decreased luminescent quantum yield and excited-state lifetime, which are especially substantial for CuPBCP. The direct attachment of the pyridyl groups in CuPBCP should serve as the sites to accelerate the nonradiative decay process. One possible explanation is that the directly attached pyridines induce the hydrogen bonding interactions with solvent molecules, as documented for the luminescence switch-on behavior of [Ru(bpy)2(dppz)]2+ (dppz = dipyridophenazine) upon insertion to DNA base pairs.49 Alternatively, they might also pose influence on the geometric flattening of the 3MLCT state species, presumably by interacting solvent molecules or counterions to form a low-energy excited state known as an “exciplex”, which subsequently decays to the ground state and thus shortens the excited-state lifetime.50−52 Indeed, the losses in emission lifetime and quantum yield in CuPPBCP are rather small, attributable to the decreased electronic interactions between the phen and pyridyl groups by the insertion of an extra phenylene group at each tether.
To further verify the origins of the modest luminescent properties of CuPBCP, the electronic and conformational properties of the ground and excited states of the Cu(I) PSs were studied through steady-state and time-resolved X-ray absorption near-edge (XANES) and extended X-ray absorption fine structure (EXAFS) spectroscopies (Figure S15). The ground-state XANES spectra of CuBCP, CuPBCP and CuPPBCP in degassed CH3CN (Figure S15a) display identical features in the rising edge and main edge regions from 8980 to 9020 eV, indicative of their high similarity in the coordination environments of the Cu(I) centers among the three PSs. Moreover, all the Cu(I) PSs display a main peak at 8984 eV in their XANES spectra, reflecting the nature of the 4pz orbital in the pseudotetrahedral coordination environments50,51,53 of the Cu(I) complexes (Figure S15a). Each EXAFS spectrum of all PSs further shows a prominent peak (Peak I) corresponding to the average Cu–N and Cu–P bond distances (Figure S15b). Analysis of these EXAFS spectra clearly reveals two similar Cu–N distances at 2.08 ± 0.01 Å (CuBCP), 2.09 ± 0.01 Å (CuPBCP), 2.05 ± 0.02 Å (CuPPBCP), and two comparable Cu–P distances at 2.30 ± 0.02 Å (CuBCP/CuPBCP) and 2.28 ± 0.02 Å (CuPPBCP) (Table S4 and S5, Figure S15). The structural parameters experimentally determined by EXAFS are in good agreement with structural parameters from their single-crystal structural analysis. These parameters also agree well with the DFT optimized coordinates (Tables S5–S8), confirming the suitability of the level of our DFT calculations adopted in this study.
Following the above steady-state XANES and EXAFS measurements, picosecond time-resolved X-ray absorption (tr-XAS) spectroscopy was employed to capture the snapshots of excited-state conformations of PSs by using a 400 nm laser as the pump source. The transient signal monitored at 100 ps features two intense bleach transitions at 8982 and 8988 eV together with a broad positive peak at ∼9000 eV (Figure 2), corresponding to the ground-state bleaching of the Cu(I) PSs and formation of the formally oxidized Cu(II) and 3MLCT excited state. As expected, the MLCT transition results in the promotion of one of the (3d)10 electrons into the low-lying π*(phen) orbital, thus leading to a d-level occupational change from 3d10 to 3d9. A pre-edge feature corresponding to the 1s to 3d quadrupole transition is consequently observed at low photon energies at 8979 eV (Figure 2) which gains intensity due to 4p mixing54 into the Cu 3d shell.
The decay of the 3MLCT excited state was additionally monitored by fixing the X-ray photon energy at 9000 eV and varying the time delay between the laser and X-ray pulses (Figures S16 and S17). The 3MLCT states of CuBCP, CuPBCP and CuPPBCP are formed promptly within the ∼100 ps pulse duration of the X-ray and decay within 287 ± 63, 37 ± 9 and 191 ± 40 ns, respectively, in agreement with the previous time-resolved fluorescent experiments (Table 1 and Figure 1d). The excited state fractions of CuBCP, CuPBCP and CuPPBCP were determined by comparing their laser-off and laser-on XANES spectra (Figure S18) with those previously reported for the heteroleptic Cu(I) complexes possessing similar coordination environments.55 A relative chemical shift in energy of 2 eV is typically observed between Cu(I) and Cu(II) reference complexes, such that the proportions of the excited state of 35%, 27% and 9.5% are estimated in the laser-on spectra of CuBCP, CuPBCP and CuPPBCP, respectively. These spectral features were then used to plot the actual or reconstructed XANES (Figure S18) and EXAFS (Figures 2 and S19) spectra of the excited states.
The reconstructed 3MLCT states of all Cu(I) PSs exhibit a pre-edge transition at 8979 eV and two 1s to 4p rising edge transitions at 8984 and 8992 eV (Figure S19), in good agreement for 4-fold coordinated Cu(II) ions.55 This analysis thus confirms the correct percentages of excited state fractions used in the estimation of the laser on spectra of the Cu-based photosensitizers described here. The derived excited states of the Cu(I) PSs reveal 2 prominent peaks (peaks A and B) corresponding to the distinct contributions of the shortened Cu–N and elongated Cu–P bonds (Figure 2, Tables S4 and S5), as previously observed in Cu(I)-based heteroleptic PSs.55 Analysis of the excited-state EXAFS of the Cu(I) PSs reveals two shortened Cu–N distances at 2.01 ± 0.02 Å (CuBCP), 1.99 ± 0.02 Å (CuPBCP), 1.92 ± 0.02 Å (CuPPBCP) and two elongated Cu–P distances at 2.39 ± 0.01 Å (CuBCP), 2.43 ± 0.01 Å (CuPBCP), and 2.38 ± 0.02 Å (CuPPBCP). These changes are consistent with the formation of a flattened geometry within the 3MLCT state induced by interactions with solvents, the aforementioned “exciplex” effect.50−52 From this perspective, we further noticed that the DFT optimizations on the excited-state vs the ground-state conformation of CuPBCP display a larger change in the torsion of the pendant pyridine/benzene rings of −0.7° and −0.2°, in comparison to CuBCP which displays decreased torsions of −0.1° and −0.2° and CuPPBCP with torsions close to 0° (Table S8). We note that the geometric definitions of the torsion angle changes of the pendant pyridyl or benzyl groups are graphically shown in Table S8. The DFT calculations further show that the excited-state CuPBCP displays a more flattened configuration with torsional angle (the dihedral angle between the planes of the diimine and diphosphine ligands) of 78.5° vs the excited-state structures of CuBCP and CuPPBCP which show torsional angles of 79.6° and 78.6°, respectively (Table S4). More substantially, analysis of the EXAFS within the first coordination sphere of CuPBCP also displays more elongated Cu–P distances of 2.43 ± 0.02 Å compared to CuBCP and CuPPBCP (2.38 ± 0.02–2.39 ± 0.01 Å; Figure 2b–d, Tables S4 and S5). Consequently, the larger torsions in the pyridine rings of CuPBCP with its more flattened state geometry are presumed to lead to the decreased steric hindrance and enhanced solvent accessibility in its excited-state conformation, namely, a stronger exciplex effect, markedly decreasing the excited-state lifetime of CuPBCP.
Photocatalytic CO2 Reduction
The photochemical CO2 reduction driven by the three PSs was examined in a CH3CN/TEA solution containing each PS, CoTCPc as the catalyst, BIH (1,3-dimethyl-2-phenyl-2,3-dihydro-1H-benzo[d]imidazole) as the sacrificial electron donor and phenol (5.0 vol %) as the proton source38,43 to accelerate the catalytic CO2 reduction. Here TEA functions as a proton acceptor to deprotonate the oxidized BIH.39 A commonly observed drawback for the heteroleptic [Cu(P∧P)(N∧N)]+ complexes has been the dissociation of the diphosphine ligand during the photocatalysis due to the formation of Cu(II) species.56,57 Therefore, a general protocol for photocatalysis in the presence of the free diphosphine xantphos ligand (2.0 eq. of PS; 1.0 mM in our experiments) was deployed to promote the repair of the PS during the photocatalysis12 which indeed enhanced the stability of photocatalytic performance (Figure S20 and Table S9). With 50 μM CoTCPc, CuBCP and CuPPBCP both enabled the selective formation of CO with only a trace amount of H2 and no products in the solution phase, resulting in CO selectivity over 97%. All components were confirmed to be necessary to evolve CO with a similarly high efficiency (Table S9). The reaction with 13CO2 atmosphere exhibited the formation of 13CO (m/z = 29; Figure S21) as a major product, evidencing that the source of CO is the gaseous CO2 rather than those that might be given by the decomposition of organic moieties in the mixture. Impressively, the CuPPBCP/CoTCPc system displayed a substantially high Φ of 76 ± 6% at 425 nm (see the SI for determination details). This Φ is comparable to the state-of-the-art molecular systems for photocatalytic CO2 reduction and even better than some noble-metal-based pioneers (Table S10), like fac-[Re(4,4′-(MeO)2bpy)(CO)3{P(OEt)3}]/fac-[Re(bpy)(CO)3(CH3CN)]+ (ΦCO = 59% at 365 nm),58 and a Ru–Re dyad (ΦCO = 45% at 480 nm),59 as well as the previous works on the dynamic interactions between noble-metal PSs and respective catalysts (ΦCO = 14.3% ∼ 46.6%).24,37−41 It is only slightly lower than the record-high system composed of fac-[Re(bpy)(CO)3(CH3CN)]+ catalyst and a trinuclear Re PS (ΦCO = 82% at 436 nm).33 Most importantly, this Φ value is highest among the noble-metal-free systems (Table S11), including Cu purpurin/FeTDHPP (ΦCO = 6% at 450 nm; FeTDHPP = chloroiron(III) 5,10,15,20-tetrakis(2′,6′-dihydroxyphenyl)-porphyrin),19 and [Cu2(P2bph)2]2+/fac-Mn(4,4′-(OMe)2bpy)(CO)3Br (ΦHCOOH+CO = 57% at 436 nm; P2bph = 4,7-diphenyl-2,9-bis(diphenylphosphinotetramethylene)-1,10-phenanthroline).27
Several comparisons were made to highlight the excellent performance of our CuPPBCP/CoTCPc system. Foremost, a lower Φ of 40 ± 5% at 425 nm was achieved with the parent CuBCP under identical conditions, reflecting that the enhanced catalytic performance relies on the dynamic interactions based on the peripheral pyridyl coordination to the catalytically active center. In addition, the CO production rate for the CuPPBCP/CoTCPc gradually decreased after 3 h of reaction, presumably due to the decomposition of Cu(I) PS56,57 or the consumption of BIH. Nevertheless, the CuBCP/CoTCPc system showed even an earlier cease in CO formation after ca. 2 h. Therefore, the dynamic coordinative interactions in the CuPPBCP/CoTCPc system may also be responsible for its increased Φ and longevity achieved in the photocatalysis. Decreasing the catalyst concentrations down to 5.0 and 0.5 μM (Figures S22 and S23; Table 2) further increased the TON up to 11800 ± 1400 based on the catalyst amount, which is superior to most of the noble-metal-free molecular systems (Table S11), albeit with a moderate TON based on the PS amount (Table 2). These excellent performances were also compared with a benchmark noble-metal-based PS, IrBPY (= [Ir(bpy)(ppy)2]PF6, ppy = 2-phenylpyridine; Figure S24), where the initial Φ at 425 nm for our CuPPBCP (76 ± 6% vs 30 ± 4%) is ca. 2.5 times higher than that of IrBPY (Figure 3 and Table 2). This clearly highlights the outstanding photocatalytic performance of the CuPPBCP/CoTCPc system. In sharp contrast, the CuPBCP/CoTCPc system is nearly inactive, showing a 1/140 of TON of CuPPBCP/CoTCPc (5.0 vs 700). The inferior performance of CuPBCP in photochemically driving the catalysis is attributable to its substantially shorter-lived character in the excited state compared with the remainders, as described later in detail.
Table 2. Photoreduction of CO2: Results of Photocatalytic CO2 Reduction Experimentsa.
| entry | PS | [CoTCPc] (μM) | n(CO) (μmol) | n(H2) (μmol) | TON(CO)b | TON’(CO)c | CO% | Φ5 min |
|---|---|---|---|---|---|---|---|---|
| 1 | CuBCP | 50.0 | 85.5 ± 5.3 | 1.33 ± 0.39 | 428 ± 26 | 42.8 ± 2.6 | 98.5 | 40 ± 5% |
| 2 | CuPBCP | 50.0 | 1.10 ± 0.15 | 0.38 ± 0.13 | 5.50 ± 0.75 | 0.55 ± 0.08 | 74.3 | 0.5 ± 0.2% |
| 3 | CuPPBCP | 50.0 | 140 ± 7 | 1.95 ± 0.31 | 700 ± 33 | 70.0 ± 3.3 | 98.6 | 76 ± 6% |
| 4 | IrBPY | 50.0 | 78.4 ± 5.7 | 1.13 ± 0.20 | 392 ± 28 | 39.2 ± 2.8 | 98.6 | 30 ± 4% |
| 5 | CuPPBCP | 5.00 | 38.1 ± 5.0 | 2.48 ± 0.44 | 1910 ± 250 | 19.1 ± 2.5 | 93.9 | 45 ± 4% |
| 6 | CuPPBCP | 0.500 | 23.6 ± 2.8 | 1.70 ± 0.24 | 11 800 ± 1400 | 11.8 ± 1.4 | 93.2 | 9.7 ± 2.2% |
Standard condition: PS (0.5 mM), xantphos ligand (1.0 mM), phenol (5.0 v%), and BIH (50 mM) in 4 mL CH3CN/TEA (v:v = 5:1) within 3 h of 425 nm irradiation (20 mW cm–2) under 1 atm CO2. The errors represent the standard deviations of three independent measurements.
Calculated for the TON values based on the catalyst amount (0.4 μmol).
Calculated for the TON values based on the PS amount (4.0 μmol).
Figure 3.

Photoreduction of CO2. Time profiles of photocatalytic CO (star) and H2 (circle) formation using 0.5 mM PS and 50 μM CoTCPc (red) under 425 nm of irradiation (20 mW cm–2). Trace colors: CuBCP, blue; CuPBCP, red; CuPPBCP, gold; IrBPY, green. The error bars represent the standard deviations of three independent measurements.
Dynamic Coordinative Interaction
As a type of covalent bonds, the labile coordinate bond not only endows the dynamic stability in the coordinatively interacted system, but also additionally facilitates the ET processes by utilizing the inner-sphere ET (ISET) via the coordinative interaction.60 To examine the coordinative interaction between the catalyst and sensitizer in solution, we conducted the 1H NMR titration experiments, where d7-DMF was adopted as the most suitable solvent instead of CD3CN to prepare highly concentrated CoTCPc solutions.38 The proton peak assignments for CuPBCP and CuPPBCP in d7-DMF were thus made prior to the titration experiments (Table S2, Figures S25 and S26). Then, the addition of CoTCPc into the d7-DMF solution of CuPBCP induced notable shifts together with the broadening/relaxation in the proton signals responsible for the pbcp ligand, indicating the interaction between CoTCPc and CuPBCP via the pbcp ligand, most probably by the coordinative interaction discussed above (Figure S27). The binding constant38,61 can be calculated using the variations in the chemical shifts, giving a 1:1 binding constant (K11) of 943 ± 13 M–1 in non-cooperative 1:2 model (see detailed analysis in Experimental Section and Table S12). The K11 value is approaching to the order of 103 M–1, corresponding to the value determined for the coordinative interaction between a pyridine-tethered Ir(III) PS and Co(II) phthalocyanine catalysts.38 The results clearly evidence the presence of a coordinative interaction between CuPBCP and CoTCPc. The observed peak patterns also reveal that the ligand exchange rate is much faster than the NMR time scale, reflecting the dynamic exchange processes occurring in solution. The parallel 1H NMR titration of the CuPPBCP/CoTCPc system revealed a K11 of 3628 ± 236 M–1 in non-cooperative 2:1 model (Figure S28 and Table S13). The higher binding constant of CuPPBCP suggests more facile binding to CoTCPc with its more distant pyridine units. In contrast, the titration of CoTCPc into a solution of CuBCP yielded a negligible shift in all proton signals (Figure S29), ascertaining the requirement of pyridyl tethers to promote the associations between the catalyst and PS.
We further examined whether the components, TEA and phenol, added in the photocatalysis solution interfere with the interaction between CuPBCP and CoTCPc. In Figure S30, in contrast to the CH3CN solution containing 50 μM CoTCPc, we first observed negligible spectral differences in the presence of either phenol or sodium phenolate by UV–vis absorption spectroscopy, excluding their interferences via interactions with CoTCPc. In contrast, the Q-band of CoTCPc at 673 nm became narrower and more intense in the presence of 17 vol % TEA (CH3CN:TEA (v:v) = 5:1), which can be attributed to either the axial coordination of TEA38,62 or the deprotonation of CoTCPc. However, the latter is less possible, since the complete deprotonation of CoTCPc by NaOH decreased the intensity of the Q-band at 673 nm instead. Further addition of phenol induced no changes, suggesting the TEA-interaction is still dominant. The 1H NMR titration was therefore carried out in the presence of excess TEA. During the 1H NMR titration in the CuPBCP/CoTCPc/TEA system as the example, the shifts were still visible (Figure S31), and the K11 was calculated as 901 ± 44 M–1 in non-cooperative 1:2 model (Table S14). This value is quite consistent with that determined in the TEA-free system (see above, 943 ± 13 M–1), clearly indicating that the binding constant with TEA is negligibly smaller than that by the pyridyl tethers in CuPBCP. Therefore, the above results demonstrate that substantial coordinative interaction has been achieved between each pyridine-appended Cu(I) PS and CoTCPc, which can be a key reason for the efficiency improvement from CuBCP to CuPPBCP.
ET Kinetics
To clarify the photocatalytic mechanism, the photoinduced ET processes (eqs 1–5) were investigated by observing the excited states of Cu(I) PSs. Due to the overlapped absorbance between CoTCPc and Cu(I) PSs, the quenching of the excited state of the Cu(I) PSs was examined by the time-resolved luminescence spectroscopy (Figures S32–S34 and Table 3). As expected, all Cu(I) PSs can be effectively quenched by BIH, which suggests a reductive quenching pathway (eqs 1–3). The dynamic28 reductive quenching constants for BIH (kq(BIH)), as determined by Stern–Volmer (eq 4) plots,38 follows a decreasing order to be CuPBCP, CuPPBCP, and CuBCP (20.7, 10.3, and 9.35 × 109 M–1 s–1; Table 3), in good agreement with the tendency of their Ered,ex values (1.00, 0.81, and 0.77 V; Table 1). On the other hand, negligible quenching was observed by the addition of CoTCPc, indicating the lack of oxidative quenching by the catalyst (eqs 1, 5 and 6). We also calculated the quenching fractions (ηq)63 to calculate the formation efficiency of the reduced-state PSs (eq 7; Table 3), in which all the three systems are all approaching to 100%, showing insignificant negative effects from the poor emission ability of CuPBCP.
| 1 |
| 2 |
| 3 |
| 4 |
| 5 |
| 6 |
| 7 |
| 8 |
Table 3. ET Kinetic Data: Related Data and Calculated Reaction Constants from Time-Resolved Spectroscopies in Degassed CH3CN Solutionsa.
| fluorescence
quenching |
TAS |
Tr-XAS |
||||||
|---|---|---|---|---|---|---|---|---|
| PS | τ0 (ns) | kq(BIH) (× 109 M–1 s–1) | ηq | τ0′ (ns) | τ1 (μs) | kET (× 108 M–1 s–1) | τ0″ (ns) | τ2 (μs) |
| CuBCP | 289 ± 5 | 9.35 ± 0.55 | 99.3 ± 0.2% | 302 ± 13 | 75.8 ± 5.2 | 3.72 ± 0.51 | 287 ± 63 | 27.6 ± 2.6 |
| CuPBCP | 33.0 ± 3.1 | 20.7 ± 1.1 | 97.2 ± 1.1% | 47.0 ± 6.2 | 234 ± 26 | 4.77 ± 0.92 | 37.0 ± 9.0 | 6.01 ± 2.83 |
| CuPPBCP | 206 ± 4 | 10.3 ± 0.6 | 99.1 ± 0.4% | 214 ± 10 | 280 ± 36 | 5.47 ± 0.85 | 191 ± 40 | 4.40 ± 1.86 |
Errors are provided as the standard deviation from three parallel experimental data. τ0: excited-state lifetime measured by time-resolved luminescence spectroscopy. kq(BIH).: reductive quenching rate constant with BIH from fluorescence quenching. ηq: quenching fractions with BIH ([BIH] = 0.05 M) from fluorescence quenching. τ0′: excited-state lifetime measured by TAS. τ1: reduced-state lifetime measured by TAS. kET.: reaction rate constant between reduced PS and CoTCPc determined by TAS. τ0″: excited-state lifetime measured by tr-XAS. τ2: formation time scales for the Co(I) species measured by tr-XAS.
We then moved to transient absorption spectroscopy (TAS; Figure 4 and Table 3 with experimental details in the SI) for analyzing the different states of the Cu(I) PSs and the kinetics of the second ET step. For CuBCP, the excitation at 420 nm leads to significant positive absorption peaks at 330 and 530 nm, where the latter is assignable to the absorption by the 3MLCT state of the PS (Figure 4a). The transient species has a lifetime (τ0′) of 301.6 ns, which is close to the result of the time-resolved emission lifetime (τ0 = 289.0 ns). The addition of an excess of BIH affords a long-lived species (τ1 = 75.8 μs; Figure 4b) with a growth of major absorption at 350 nm, indicative of the formation of a reductively quenched product, CuBCP- according to eq 2. The subsequent process is regarded as the ET from CuBCP- to CoTCPc (eq 3), which is evidenced by the decreased longevity of CuBCP– with an increase in the concentration of CoTCPc (Figure 4c). By using eq 8, the second-order rate constant for this step can be calculated as kET = 3.72 × 108 M–1 s–1 (Figure 4d).38
Figure 4.

Nanosecond TAS. Nanosecond TAS results of (a–d) CuBCP, (e–h) CuPBCP, and (i–l) CuPPBCP. TAS evolution of Cu(I) PSs (a,e,i) without or (b,f,j) with excess BIH. (c,g,k) Kinetic traces of Cu(I) PSs with BIH and CoTCPc. (d,h,i) Plot of (τ1/τ – 1) versus [CoTCPc] with linear fitting and error bars from the deviations of decay traces. The data were collected by following the spectra at 360 nm in Ar-saturated CH3CN upon excitation at 425 nm.
Under the same conditions, the TAS behavior of CuPBCP or CuPPBCP upon excitation (Figure 4e and i) together with the response to the addition of BIH (Figure 4f and j) is similar to CuBCP. It can be noticed that the lifetimes of CuPBCP– and CuPPBCP– are much longer than those of CuBCP– (Table 3). Importantly, the reaction of CuPBCP– with CoTCPc at increasing concentrations gives rise to a higher kET = 4.77 × 108 M–1 s–1 than that observed by CuBCP–. Moreover, the increase in kET for CuPPBCP– (5.47 × 108 M–1 s–1) is even higher. The significantly faster ET rates by these pyridyl tethered systems rather contradict with the trend expected from the driving forces of ET, since the reduction potentials of CuPBCP and CuPPBCP (Ered = −1.88 and –1.97 V) are anodically shifted with regard to that of CuBCP (−2.04 V; Table 1). This contradiction in turn supports the major role of the coordinative interaction between the pyridyl substituents and CoTCPc in substantially accelerating the ET by overcoming the disfavored driving force of the reaction.
Tr-XAS was again employed to directly monitor the electronic configurations and ET kinetics of the photogenerated Co(I) state64 under photocatalytic conditions in a multimolecular assembly consisting of the Cu(I) PSs, CoTCPc catalyst and BIH (Figures 5, S35, and S36). Figure 5a shows the Co tr-XAS spectra of all three multimolecular assemblies at an average time delay of 12.3 μs between the laser and X-ray pulses. A prominent peak with a shoulder feature at 7714/7718 eV (I,II) together with a broad dip (III) at 7724 eV is obtained which corresponds to the formation of the reduced Co(I) species65 and the ground-state bleaching of the Co(II) compound, respectively. These energy transitions in turn show that the K-edge of the Co center shifts to lower energy, indicating the reduction of Co(II) and confirming the formation of Co(I) through ET from the Cu-based PSs. The kinetics for the Co(I) formation were further investigated by fixing the photon energy to 7714 eV to monitor the formation of Co(I), with varying time delays between the laser and X-ray pulses (Figures 5 and S36). The formation time scales (τ2) for the Co(I) species with CuPBCP and CuPPBCP are 6.01 ± 2.83 and 4.40 ± 1.86 μs, respectively. In sharp contrast, the Co(I) intermediate is formed with CuBCP at a significantly slower pace within 27.6 ± 2.6 μs, strongly confirming the facilitated electron transfer from the pyridine-decorated Cu(I) PSs to CoTCPc benefited from dynamic coordinative interactions. It is important here to remark that the time scales of formation of Co(I) in the tr-XAS experiments vary differently from those decay kinetics of the reduced-state Cu PSs from TAS. This is due to the higher concentrations of CoTCPc and Cu(I) PSs employed in the tr-XAS measurements for a sufficient transient signal response together with the diffusion-governed nature of these interactions.66
Figure 5.

Tr-XAS for ET kinetics. (a) Experimental differential spectrum (laser on-laser off) corresponding to the Co(I) transient signal at a time delay of ∼12.3 μs between laser and X-ray pulses (red) in the Cu/Co multimolecular assemblies. (b) Pump–probe time delay scans recorded at 7714 eV reflecting the formation of the Co(I) photoinduced species in the multimolecular Co/Cu assemblies consisting of 0.75 mM Cu complex, 1.5 mM CoTCPc with 5 mM BIH in 47% CH3CN/47% DMF/6% H2O, from left to right for CuBCP (blue), CuPBCP (red), and CuPPBCP (gold). Kinetic fits for the Co(I) formation time scales (τ2) are shown.
Overall, the above multiple spectroscopic results clearly demonstrate that the catalytic systems of the Cu(I) PSs follow the reductive quenching pathway and that the ET processes from the two pyridine-tethered Cu(I) PSs to CoTCPc can be facilitated via dynamic coordinative interaction. Via the dynamic coordinative interaction with binding constants at 103 M–1 order of magnitude, the ground-state PS-catalyst complex should not be the main species for catalysis.38 Thus, the reaction between the PS and catalyst should proceed mainly in a bimolecular manner rather than a unimolecular way. Beside the conventional outer-sphere electron transfer (OSET) by the random collision between the PS and catalyst, the intermolecular electron transfer can be additionally facilitated in a manner of ISET via the coordinative interaction as either a stable adduct or a transition state.60 Such facilitations have been demonstrated by the TAS and tr-XAS measurements. Importantly, among the three reaction steps (eqs 1–3) related to the Cu(I) PSs, the rapid reaction rates of 2 and 3 involving CuPBCP manifest that its inactivity in photocatalytic CO2 reduction should mainly originate from its short-lived excited state character (eq 1).
Proposed Mechanism
With verifications on the overall ET kinetics, further mechanistic studies were operated on CoTCPc to elucidate the overall mechanism. First, the redox properties of CoTCPc can be revealed by its CVs in the presence of TEA (Figure S37). Under N2, three reduction events appeared in the CV of CoTCPc. The first reversible one should be attributed to the CoII/I reduction (Ered,1 = −0.88 V), while the two subsequent reduction waves may be induced by the reduction on the phthalocyanine ring.67,68 The introduction of CO2 induces a current enhancement at the second reduction wave (Ered,2 = −1.40 V), indicating that the two-electron reduced species can initiate the catalytic reduction of CO2 Further, an additional catalytic wave appeared at the CoII/I reduction wave in the CV upon the addition of phenol under CO2, showing the decreased overpotential required for effective catalysis by enabling the proton-dependent transfer.69,70 The quite positive catalytic potential for CO2 reduction should contribute to the high catalytic activity of CoTCPc in photocatalysis.
DFT calculation at M0671 level further shed light on the catalytic mechanism of CoTCPc proposed in Figure 6. The calculated reduction potentials for the two successive reduction events of CoTCPc are in good agreement with the measured ones (dashed box in Figure 6), displaying that the calculation methods are suitable for this system and that the initial species in the presence of TEA is less likely to be the deprotonated form of CoTCPc. In the presence of highly reducing CuPPBCP- (Ered = −1.97 V), the Co(II) catalyst can be doubly reduced according to the above CV results discussed above. With BIH as a two-electron reductant, the involvement of highly reducing BI· radical (Ered = −2.06 V) from the TEA-deprotonated BIH+ can also serve as another driving force.59 After double reduction, a low-spin Co(I)-ligand radical species was calculated as the intermediate for subsequent CO2 binding. The formation of this species is favored with a negligible energy change of 0.05 kcal mol–1, followed by protonation to yield a low-spin Co-COOH species43 with a slight uphill energy of 8.24 kcal mol–1. The coordinated CO2 is partially activated due to its slightly bent configuration (Figure S38). Followed by the cleavage of the C–OH bond, a low-spin Co-CO intermediate can be generated with a favorable energy change of −19.72 kcal mol–1. The final release of CO with the regeneration of the catalyst is thermodynamically favorable with a decreasing energy of −2.12 kcal mol–1. Negligible distortion of the phthalocyanine ring can be noticed for all calculated intermediates. With the above insights, an overall catalytic cycle for CuPPBCP/CoTCPc can be proposed (Figure 6). The ET kinetic data are also presented.
Figure 6.

Proposed photocatalytic mechanism. Suggested photocatalytic mechanism for the reduction of CO2 using the CuPPBCP/CoTCPc system was a representative example.
Discussion
In summary, the construction of dynamic coordinative interactions affords a high-performance system for photocatalytic reduction of CO2 to CO by using earth-abundant elements only. An outstanding apparent quantum yield of 76 ± 6% at 425 nm has been achieved with a near-unity selectivity and a maximum turnover number of 11 800 ± 1400. The remarkable performances are attributed the accelerated intramolecular electron transfer pathway based on the dynamic coordinative interactions between the photosensitizer and the catalyst. In particular, the sharply different activity of the two pyridine-tethered photosensitizers reveals the importance of delicate design of linkers to preserve long emission lifetime. We also highlight the promising and versatile use of time-resolved X-ray absorption spectroscopy, which not only verifies the structure–activity relationship among the Cu(I) photosensitizers, but also unveils the electron transfer kinetics required to understand the photocatalysis. We believe that our work herein opens avenues toward the development of highly cost-efficient molecular systems for photocatalytic CO2 reduction.
Experimental Section
Materials
CuBCP72 was prepared following the previously reported methods. RuBPY (97%, Sigma), IrBPY (98%, BLD China), sodium phenolate (98%, Sigma), and other chemicals were commercially available and used without further purification.
Instruments
NMR data were obtained on Bruker advance III instruments (400/500 MHz). Electrochemical measurements were carried out using an electrochemical workstation (CHI 660D). Gas chromatographic analysis was conducted on an Agilent 7820A gas chromatography. The isotopic labeling experiment was conducted under 13CO2 atmosphere and the gas in the headspace was analyzed by a quantitative mass spectrometer attached Agilent 7890A gas chromatography. The liquid phase of the reaction system was analyzed by an ion chromatograph (Metrohm) to detect the presence of formate. UV–vis spectra were collected on a Shimadzu UV-3600 spectrophotometer. The emission experiments were conducted on a modular fluorescent life and steady-state fluorescence spectrometer (FLSP1000, Edinburgh Instruments Ltd.). The TAS was carried out on a laser flash photolysis instrument (LP980, Edinburgh Instruments Ltd.). All experiments were operated at room temperature (24–25 °C) unless otherwise stated.
Synthesis of pbcp and ppbcp Ligands
The pyridine-tethered ligands was prepared via Suzuki coupling according to the reported procedure for the pbcp ligand73 with modifications. One mmol of 4,7-dibromo-2,9-dimethyl-1,10-phenanthroline, 2.5 mmol of (4-(pyridin-4-yl)phenyl)boronic acid (for ppbcp) or 4-phenylboronic acid (for pbcp), 15 mol % P(PPh3)4, and 3 mmol of K2CO3 were dissolved in 50 mL of dioxane/H2O (v:v = 5:1) mixed solvent and degassed with argon. After heating at 100 °C for 1 day, the reaction mixture was cooled to room temperature and subjected to extraction with DCM, followed by washing with brine twice and then drying over MgSO4. The column chromatography with DCM/CH3OH (2%) afforded a pale yellow powder with moderate yields (ppbcp: 32%; pbcp: 35%).
For pbcp: Q-TOF ESI-MS(+): [pbcp+H]+ (measured: 363.1603; simulated: 363.1604). 1H NMR (CD2Cl2, 400 MHz, 25 °C): δ (ppm) = 8.82–8.77 (m, 4H), 7.73 (s, 2H), 7.51 (s, 2H), 7.51–7.48 (m, 4H), 2.99 (s, 6H). Elemental analysis: Calculated (C24H18N4), C, 79.54; H, 5.01; N, 15.46; Measured, C, 79.44; H, 5.08; N, 15.48.
For ppbcp: Q-TOF ESI-MS(+): [ppbcp+H]+ (measured: 515.2230; simulated: 515.2230); 1H NMR (CD2Cl2, 400 MHz, 25 °C): δ [ppm] = 8.77–8.69 (m, 4H), 7.92–7.85 (m, 4H), 7.85 (s, 2H), 7.76–7.69 (m, 4H), 7.68–7.62 (m, 4H), 7.56 (s, 2H), 2.99 (s, 6H). Elemental analysis: Calculated (C36H26N4), C, 84.02; H, 5.09; N, 10.89; Measured, C, 83.98; H, 5.13; N, 10.89.
Synthesis of CuPBCP and CuPPBCP
The synthesis follows a similar procedure for preparing heteroleptic Cu(I) PSs.72 In a 50 mL Schlenk tube, [Cu(CH3CN)4]PF6 (186 mg, 0.5 mmol) and xantphos (289 mg, 0.5 mmol) are dissolved in 10 mL of dry DCM at room temperature under N2 and darkness. The resulting solution is stirred at 40 °C for 2 h. The reaction mixture is then added with the pbcp ligand (180 mg, 0.5 mmol) or ppbcp ligand (257 mg, 0.5 mmol) under N2 flow. The resulting mixture is then heated at 45 °C for 2 h. The reaction mixture was then allowed to cool to room temperature, and the solvent was evaporated under reduced pressure. Then a minimal volume of dry DCM (ca. 5 mL) was added to dissolve most the orange residue, which was filtrated via a 0.45 μm PTFE membrane, followed by the addition of excess xantphos (ca. 70 mg) into the filtrate to prevent the formation of [Cu(N∧N)2]+. Finally, excess dry hexane (50 mL) was added to precipitate CuPBCP/CuPPBCP as a yellow crystalline powder (76%/65% yield). It should be noted that the presence of trace water will make the product oily, leading to the high difficulties in cultivating single crystals.
For CuPBCP: Q-TOF ESI-MS(+): [Cu(xantphos)(pbcp)]+ (measured: 1003.2767; simulated: 1003.2750). 1H NMR (CD3CN, 400 MHz, 25 °C): δ (ppm) = 8.82 (dd, 4H, J = 4.4, 1.6), 7.80 (dd, 2H, J = 7.9, 1.3), 7.69 (s, 2H), 7.58 (s, 2H), 7.51 (dd, 4H, J = 4.4, 1.6), 7.35–7.27 (m, 6H), 7.20–7.06 (m, 18H), 2.37 (s, 6H), 1.76 (s, 6H). 13C NMR (126 MHz, CD3CN) δ 158.51, 154.85, 150.27, 146.99, 144.34, 143.27, 143.26, 133.93, 133.02, 132.96, 132.90, 131.56, 131.43, 131.30, 130.25, 130.05, 128.66, 128.63, 128.59, 128.03, 125.63, 125.42, 124.99, 124.16, 123.17, 121.46, 35.96, 27.93, 26.90. 31P NMR (202 MHz, CD3CN) δ −9.77. Elemental analysis: Calculated (CuC63H50N4OF6P3), C, 65.82; H, 4.38; N, 4.87; Measured, C, 66.01; H, 4.42; N, 4.96.
For CuPPBCP: Q-TOF ESI-MS(+): [Cu(xantphos)(ppbcp)]+ (measured: 1155.3384; simulated: 1155.3376). 1H NMR (CD3CN, 400 MHz, 25 °C) δ (ppm) = 8.74–8.68 (m, 4H), 8.01–7.93 (m, 4H), 7.83–7.77 (m, 4H), 7.75–7.66 (m, 8H), 7.60 (s, 2H), 7.34–7.27 (m, 6H), 7.23–7.03 (m, 18H), 2.36 (s, 6H), 1.76 (s, 6H). 13C NMR (126 MHz, CD3CN) δ: 158.21, 154.90, 150.47, 148.95, 146.85, 143.50, 138.64, 137.48, 133.94, 133.05, 132.99, 132.93, 131.67, 131.55, 131.42, 130.45, 130.25, 130.02, 128.66, 128.62, 128.58, 127.97, 127.47, 125.67, 125.45, 125.41, 123.26, 121.52, 35.97, 27.91, 26.91. 31P NMR (202 MHz, CD3CN) δ −9.91. Elemental analysis: Calculated (CuC75H58N4OF6P3), C, 69.20; H, 4.49; N, 4.30; Measured, C, 69.64; H, 4.44; N, 4.41.
Acknowledgments
We are grateful for the TA spectroscopy and analysis assisted by Prof. Zhi-Ming Zhang, Xiao-Liang Ma and Dr. Ping Wang from Tianjin University of Technology, NMR analysis by Mr. Martin Rentschler from Technische Universität Braunschweig, as well as the fluorescence spectroscopies supported by Ms. Yu-Xin Chen and the single-crystal X-ray diffraction by Dr. Long Jiang both from the Instrumental Analysis and Research Center in Sun Yat-sen University. Gangfeng Ouyang thanks the National Natural Science Foundation of China (21737006, 22076222, and 22036003). Jia-Wei Wang acknowledges Prof. Antoni Llobet (ICIQ, present supervisor of Dr. Jia-Wei Wang and Zhi-Mei Luo) for providing scholar resources during manuscript preparation, and the financial supports from Guangdong Basic and Applied Basic Research Foundation (2020A1515110017 and 2021A1515012033). Dooshaye Moonshiram acknowledges supports the Spanish Ministerio de Ciencia, Innovacion y Universidades grant (PID2019-111086RA-I00), oriented project to the ecological transition and the digital transition (TED2021-132757B–I00), PIE grant from CSIC-ICMM (20226AT001) and the Ramon y Cajal Fellowship (RYC2020-029863-I). This research also used resources of the Advanced Photon Source (Beamline 11 ID-D); a U.S. DOE Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacsau.3c00218.
Author Contributions
Conceptualization: J.-W.W. Methodology: J.-W.W, Xian Z., Z.-M.L., and D.M. Investigation: J.-W.W., L.V., Z.-M.L., Z.L., Y.H., K.Y., J.Y., W.H., Xiaoyi Z., and D.M. Visualization: J.-W.W., Xian Z., M.K., and D.M. Supervision: J.-W.W., D.M., and G.O. Writing—original draft: J.-W.W. and D.M. Writing—review and editing: J.-W.W., Xian Z., K.S., M.K., and G.O. CRediT: Jia-Wei Wang conceptualization, data curation, formal analysis, funding acquisition, investigation, methodology, project administration, resources, validation, visualization, writing-original draft, writing-review & editing; Lucia Velasco data curation, investigation, software, visualization, writing-review & editing; Michael Karnahl formal analysis, methodology, project administration, writing-review & editing; Wenhui Hu data curation, formal analysis.
Gangfeng Ouyang: National Natural Science Foundation of China 21737006, 22076222 and 22036003. Jia-Wei Wang: Guangdong Basic and Applied Basic Research Foundation 2020A1515110017 and 2021A1515012033. Dooshaye Moonshiram: Spanish Ministerio de Ciencia, Innovacion y Universidades grant PID2019–111086RA-I00, TED2021-132757B-I00 German Research Foundation grant AB 1234/1–1; PIE grant from CSIC-ICMM 20226AT001; Ramon y Cajal Fellowship (RYC2020-029863-I)
The authors declare no competing financial interest.
Supplementary Material
References
- Dalle K. E.; Warnan J.; Leung J. J.; Reuillard B.; Karmel I. S.; Reisner E. Electro- and Solar-Driven Fuel Synthesis with First Row Transition Metal Complexes. Chem. Rev. 2019, 119, 2752–2875. 10.1021/acs.chemrev.8b00392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrei V.; Wang Q.; Uekert T.; Bhattacharjee S.; Reisner E. Solar Panel Technologies for Light-to-Chemical Conversion. Acc. Chem. Res. 2022, 55, 3376–3386. 10.1021/acs.accounts.2c00477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng X.; Li R.; Yang J.; Xu S.; Zhang C.; You K.; Ma B.; Guan H.; Ding Y. Hexanuclear Ring Cobalt Complex for Photochemical CO2 to CO Conversion. Chin. J. Catal. 2022, 43, 2414–2424. 10.1016/S1872-2067(22)64144-5. [DOI] [Google Scholar]
- Sun W.; Zhu J.; Zhang M.; Meng X.; Chen M.; Feng Y.; Chen X.; Ding Y. Recent Advances and Perspectives in Cobalt-Based Heterogeneous Catalysts for Photocatalytic Water Splitting, CO2 Reduction, and N2 Fixation. Chin. J. Catal. 2022, 43, 2273–2300. 10.1016/S1872-2067(21)63939-6. [DOI] [Google Scholar]
- Boutin E.; Merakeb L.; Ma B.; Boudy B.; Wang M.; Bonin J.; Anxolabehere-Mallart E.; Robert M. Molecular Catalysis of CO2 Reduction: Recent Advances and Perspectives in Electrochemical and Light-Driven Processes with Selected Fe, Ni and Co Aza Macrocyclic and Polypyridine Complexes. Chem. Soc. Rev. 2020, 49, 5772–5809. 10.1039/D0CS00218F. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Wang Y.; Gu M.; Wang M.; Zhang Z.; Pan W.; Jiang Z.; Zheng H.; Lucero M.; Wang H.; Sterbinsky G. E.; Ma Q.; Wang Y.-G.; Feng Z.; Li J.; Dai H.; Liang Y. Molecular Engineering of Dispersed Nickel Phthalocyanines on Carbon Nanotubes for Selective CO2 Reduction. Nat. Energy 2020, 5, 684–692. 10.1038/s41560-020-0667-9. [DOI] [Google Scholar]
- Wang J.-W.; Liu W.-J.; Zhong D.-C.; Lu T.-B. Nickel Complexes as Molecular Catalysts for Water Splitting and CO2 Reduction. Coord. Chem. Rev. 2019, 378, 237–261. 10.1016/j.ccr.2017.12.009. [DOI] [Google Scholar]
- Huang Y.; Dai H.; Moonshiram D.; Li Z.; Luo Z.-M.; Zhang J.-H.; Yang W.; Shen Y.; Wang J.-W.; Ouyang G. Impaired Conjugation Boosts CO2 Electroreduction by Ni(II) Macrocyclic Catalysts Immobilized on Carbon Nanotubes. J. Mater. Chem. A 2023, 11, 2969–2978. 10.1039/D2TA08781B. [DOI] [Google Scholar]
- Yamazaki Y.; Takeda H.; Ishitani O. Photocatalytic Reduction of CO2 Using Metal Complexes. J. Photochem. Photobiol., C 2015, 25, 106–137. 10.1016/j.jphotochemrev.2015.09.001. [DOI] [Google Scholar]
- Wang J.-W.; Qiao L.-Z.; Nie H.-D.; Huang H.-H.; Li Y.; Yao S.; Liu M.; Zhang Z.-M.; Kang Z.-H.; Lu T.-B. Facile Electron Delivery from Graphene Template to Ultrathin Metal-Organic Layers for Boosting CO2 Photoreduction. Nat. Commun. 2021, 12, 813. 10.1038/s41467-021-21084-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P.; Dong R.; Guo S.; Zhao J.; Zhang Z.-M.; Lu T.-B. Improving Photosensitization for Photochemical CO2-to-CO Conversion. Natl. Sci. Rev. 2020, 7, 1459–1467. 10.1093/nsr/nwaa112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas-Hernández A.; Steinlechner C.; Junge H.; Beller M. Earth-Abundant Photocatalytic Systems for the Visible-Light-Driven Reduction of CO2 to CO. Green Chem. 2017, 19, 2356–2360. 10.1039/C6GC03527B. [DOI] [Google Scholar]
- Wenger O. S. Photoactive Complexes with Earth-Abundant Metals. J. Am. Chem. Soc. 2018, 140, 13522–13533. 10.1021/jacs.8b08822. [DOI] [PubMed] [Google Scholar]
- Wegeberg C.; Wenger O. S. Luminescent First-Row Transition Metal Complexes. JACS Au 2021, 1, 1860–1876. 10.1021/jacsau.1c00353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. W.; Ma F.; Jin T.; He P.; Luo Z. M.; Kupfer S.; Karnahl M.; Zhao F.; Xu Z.; Jin T.; Lian T.; Huang Y. L.; Jiang L.; Fu L. Z.; Ouyang G.; Yi X. Y. Homoleptic Al(III) Photosensitizers for Durable CO2 Photoreduction. J. Am. Chem. Soc. 2023, 145, 676–688. 10.1021/jacs.2c11740. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Schulz M.; Wächtler M.; Karnahl M.; Dietzek B. Heteroleptic Diimine–Diphosphine Cu(I) Complexes as an Alternative Towards Noble-Metal Based Photosensitizers: Design Strategies, Photophysical Properties and Perspective Applications. Coord. Chem. Rev. 2018, 356, 127–146. 10.1016/j.ccr.2017.10.016. [DOI] [Google Scholar]
- Hossain A.; Bhattacharyya A.; Reiser O. Copper’s Rapid Ascent in Visible-Light Photoredox Catalysis. Science 2019, 364, eaav9713 10.1126/science.aav9713. [DOI] [PubMed] [Google Scholar]
- Forero Cortés P. A.; Marx M.; Trose M.; Beller M. Heteroleptic Copper Complexes with Nitrogen and Phosphorus Ligands in Photocatalysis: Overview and Perspectives. Chem. Catal. 2021, 1, 298–338. 10.1016/j.checat.2021.05.005. [DOI] [Google Scholar]
- Yuan H.; Cheng B.; Lei J.; Jiang L.; Han Z. Promoting Photocatalytic CO2 Reduction with a Molecular Copper Purpurin Chromophore. Nat. Commun. 2021, 12, 1835. 10.1038/s41467-021-21923-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaudelot J.; Oger S.; Perusko S.; Phan T. A.; Teunens T.; Moucheron C.; Evano G. Photoactive Copper Complexes: Properties and Applications. Chem. Rev. 2022, 122, 16365–16609. 10.1021/acs.chemrev.2c00033. [DOI] [PubMed] [Google Scholar]
- Zott M. D.; Canestraight V. M.; Peters J. C. Mechanism of a Luminescent Dicopper System That Facilitates Electrophotochemical Coupling of Benzyl Chlorides Via a Strongly Reducing Excited State. ACS Catal. 2022, 12, 10781–10786. 10.1021/acscatal.2c03215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan T. A.; Armaroli N.; Saavedra Moncada A.; Bandini E.; Delavaux-Nicot B.; Nierengarten J. F.; Armspach D. Stable Luminescent [Cu(NN)(PP)](+) Complexes Incorporating a Beta-Cyclodextrin-Based Diphosphane Ligand with Metal-Confining Properties. Angew. Chem., Int. Ed. 2023, 62, e202214638 10.1002/anie.202214638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K. K.; Qin C. C.; Ding M. J.; Guo S.; Lu T. B.; Zhang Z. M. Broadband and Strong Visible-Light-Absorbing Cuprous Sensitizers for Boosting Photosynthesis. Proc. Natl. Acad. Sci. U. S. A. 2022, 119, e2213479119 10.1073/pnas.2213479119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. W.; Li Z.; Luo Z. M.; Huang Y.; Ma F.; Kupfer S.; Ouyang G. Boosting CO2 Photoreduction by π-π-Induced Preassembly between a Cu(I) Sensitizer and a Pyrene-Appended Co(II) Catalyst. Proc. Natl. Acad. Sci. U. S. A. 2023, 120, e2221219120 10.1073/pnas.2221219120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda H.; Ohashi K.; Sekine A.; Ishitani O. Photocatalytic CO2 Reduction Using Cu(I) Photosensitizers with a Fe(II) Catalyst. J. Am. Chem. Soc. 2016, 138, 4354–4357. 10.1021/jacs.6b01970. [DOI] [PubMed] [Google Scholar]
- Takeda H.; Monma Y.; Ishitani O. Highly Functional Dinuclear CuI-Complex Photosensitizers for Photocatalytic CO2 Reduction. ACS Catal. 2021, 11, 11973–11984. 10.1021/acscatal.1c03336. [DOI] [Google Scholar]
- Takeda H.; Kamiyama H.; Okamoto K.; Irimajiri M.; Mizutani T.; Koike K.; Sekine A.; Ishitani O. Highly Efficient and Robust Photocatalytic Systems for CO2 Reduction Consisting of a Cu(I) Photosensitizer and Mn(I) Catalysts. J. Am. Chem. Soc. 2018, 140, 17241–17254. 10.1021/jacs.8b10619. [DOI] [PubMed] [Google Scholar]
- Steinlechner C.; Roesel A. F.; Oberem E.; Päpcke A.; Rockstroh N.; Gloaguen F.; Lochbrunner S.; Ludwig R.; Spannenberg A.; Junge H.; Francke R.; Beller M. Selective Earth-Abundant System for CO2 Reduction: Comparing Photo- and Electrocatalytic Processes. ACS Catal. 2019, 9, 2091–2100. 10.1021/acscatal.8b03548. [DOI] [Google Scholar]
- Zhang X.; Cibian M.; Call A.; Yamauchi K.; Sakai K. Photochemical CO2 Reduction Driven by Water-Soluble Copper(I) Photosensitizer with the Catalysis Accelerated by Multi-Electron Chargeable Cobalt Porphyrin. ACS Catal. 2019, 9, 11263–11273. 10.1021/acscatal.9b04023. [DOI] [Google Scholar]
- Zhang X.; Yamauchi K.; Sakai K. Earth-Abundant Photocatalytic CO2 Reduction by Multielectron Chargeable Cobalt Porphyrin Catalysts: High CO/H2 Selectivity in Water Based on Phase Mismatch in Frontier MO Association. ACS Catal. 2021, 11, 10436–10449. 10.1021/acscatal.1c02475. [DOI] [Google Scholar]
- Sueyoshi F.; Zhang X.; Yamauchi K.; Sakai K. Controlling the Photofunctionality of a Polyanionic Heteroleptic Copper(I) Photosensitizer for CO2 Reduction Using Its Ion-Pair Formation with Polycationic Ammonium in Aqueous Media. Angew. Chem., Int. Ed. 2023, 62, e202217807 10.1002/anie.202217807. [DOI] [PubMed] [Google Scholar]
- Hu Y.; Zhan F.; Wang Q.; Sun Y.; Yu C.; Zhao X.; Wang H.; Long R.; Zhang G.; Gao C.; Zhang W.; Jiang J.; Tao Y.; Xiong Y. Tracking Mechanistic Pathway of Photocatalytic CO2 Reaction at Ni Sites Using Operando, Time-Resolved Spectroscopy. J. Am. Chem. Soc. 2020, 142, 5618–5626. 10.1021/jacs.9b12443. [DOI] [PubMed] [Google Scholar]
- Morimoto T.; Nishiura C.; Tanaka M.; Rohacova J.; Nakagawa Y.; Funada Y.; Koike K.; Yamamoto Y.; Shishido S.; Kojima T.; Saeki T.; Ozeki T.; Ishitani O. Ring-Shaped Re(I) Multinuclear Complexes with Unique Photofunctional Properties. J. Am. Chem. Soc. 2013, 135, 13266–13269. 10.1021/ja406144h. [DOI] [PubMed] [Google Scholar]
- Nakada A.; Koike K.; Maeda K.; Ishitani O. Highly Efficient Visible-Light-Driven CO2 Reduction to CO Using a Ru(II)–Re(I) Supramolecular Photocatalyst in an Aqueous Solution. Green Chem. 2016, 18, 139–143. 10.1039/C5GC01720C. [DOI] [Google Scholar]
- Kuramochi Y.; Fujisawa Y.; Satake A. Photocatalytic CO2 Reduction Mediated by Electron Transfer Via the Excited Triplet State of Zn(II) Porphyrin. J. Am. Chem. Soc. 2020, 142, 705–709. 10.1021/jacs.9b12712. [DOI] [PubMed] [Google Scholar]
- Windle C. D.; George M. W.; Perutz R. N.; Summers P. A.; Sun X. Z.; Whitwood A. C. Comparison of Rhenium-Porphyrin Dyads for CO2 Photoreduction: Photocatalytic Studies and Charge Separation Dynamics Studied by Time-Resolved IR Spectroscopy. Chem. Sci. 2015, 6, 6847–6864. 10.1039/C5SC02099A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung P. L.; Kapper S. C.; Zeng T.; Thompson M. E.; Kubiak C. P. Improving Photocatalysis for the Reduction of CO2 through Non-Covalent Supramolecular Assembly. J. Am. Chem. Soc. 2019, 141, 14961–14965. 10.1021/jacs.9b07067. [DOI] [PubMed] [Google Scholar]
- Wang J. W.; Jiang L.; Huang H. H.; Han Z.; Ouyang G. Rapid Electron Transfer Via Dynamic Coordinative Interaction Boosts Quantum Efficiency for Photocatalytic CO2 Reduction. Nat. Commun. 2021, 12, 4276. 10.1038/s41467-021-24647-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. W.; Huang H. H.; Wang P.; Yang G.; Kupfer S.; Huang Y.; Li Z.; Ke Z.; Ouyang G. Co-Facial π-π Interaction Expedites Sensitizer-to-Catalyst Electron Transfer for High-Performance CO2 Photoreduction. JACS Au 2022, 2, 1359–1374. 10.1021/jacsau.2c00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu L.-Q.; Chen K.-H.; Yang Z.-W.; He L.-N. A Rhenium Catalyst with Bifunctional Pyrene Groups Boosts Natural Light-Driven CO2 Reduction. Green Chem. 2020, 22, 8614–8622. 10.1039/D0GC03111A. [DOI] [Google Scholar]
- Nasrallah H.; Lyu P.; Maurin G.; El-Roz M. Highly Efficient CO2 Reduction under Visible-Light on Non-Covalent Ru···Re Assembled Photocatalyst: Evidence on the Electron Transfer Mechanism. J. Catal. 2021, 404, 46–55. 10.1016/j.jcat.2021.09.007. [DOI] [Google Scholar]
- Wang J.-W.; Yamauchi K.; Huang H.-H.; Sun J.-K.; Luo Z.-M.; Zhong D.-C.; Lu T.-B.; Sakai K. A Molecular Cobalt Hydrogen Evolution Catalyst Showing High Activity and Outstanding Tolerance to CO and O2. Angew. Chem., Int. Ed. 2019, 58, 10923–10927. 10.1002/anie.201904578. [DOI] [PubMed] [Google Scholar]
- Li Z.; Wang J.-W.; Huang Y.; Ouyang G. Enhancing CO2 Photoreduction Via the Perfluorination of Co(II) Phthalocyanine Catalysts in a Noble-Metal-Free System. Chin. J. Catal. 2023, 49, 160–167. 10.1016/S1872-2067(23)64433-X. [DOI] [Google Scholar]
- Ishida H.; Bünzli J.-C.; Beeby A. Guidelines for Measurement of Luminescence Spectra and Quantum Yields of Inorganic and Organometallic Compounds in Solution and Solid State (IUPAC Technical Report). Pure Appl. Chem. 2016, 88, 701–711. 10.1515/pac-2014-0706. [DOI] [Google Scholar]
- Kjaer K. S.; Kaul N.; Prakash O.; Chabera P.; Rosemann N. W.; Honarfar A.; Gordivska O.; Fredin L. A.; Bergquist K. E.; Haggstrom L.; Ericsson T.; Lindh L.; Yartsev A.; Styring S.; Huang P.; Uhlig J.; Bendix J.; Strand D.; Sundstrom V.; Persson P.; Lomoth R.; Warnmark K. Luminescence and Reactivity of a Charge-Transfer Excited Iron Complex with Nanosecond Lifetime. Science 2019, 363, 249–253. 10.1126/science.aau7160. [DOI] [PubMed] [Google Scholar]
- Larsen C. B.; Wenger O. S. Photoredox Catalysis with Metal Complexes Made from Earth-Abundant Elements. Chem. - Eur. J. 2018, 24, 2039–2058. 10.1002/chem.201703602. [DOI] [PubMed] [Google Scholar]
- Chen L. X.; Shaw G. B.; Novozhilova I.; Liu T.; Jennings G.; Attenkofer K.; Meyer G. J.; Coppens P. Mlct State Structure and Dynamics of a Copper(I) Diimine Complex Characterized by Pump-Probe X-Ray and Laser Spectroscopies and DFT Calculations. J. Am. Chem. Soc. 2003, 125, 7022–7034. 10.1021/ja0294663. [DOI] [PubMed] [Google Scholar]
- Griffith J. S.; Orgel L. E. Ligand-Field Theory. Quarterly Reviews, Chemical Society 1957, 11, 381–393. 10.1039/qr9571100381. [DOI] [Google Scholar]
- Boynton A. N.; Marcelis L.; Barton J. K. [Ru(Me4phen)2dppz](2+), a Light Switch for DNA Mismatches. J. Am. Chem. Soc. 2016, 138, 5020–5023. 10.1021/jacs.6b02022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler L.; Hadt R. G.; Hayes D.; Chen L. X.; Mulfort K. L. Synthesis, Structure, and Excited State Kinetics of Heteroleptic Cu(I) Complexes with a New Sterically Demanding Phenanthroline Ligand. Dalton. Trans. 2017, 46, 13088–13100. 10.1039/C7DT02476B. [DOI] [PubMed] [Google Scholar]
- Kohler L.; Hayes D.; Hong J.; Carter T. J.; Shelby M. L.; Fransted K. A.; Chen L. X.; Mulfort K. L. Synthesis, Structure, Ultrafast Kinetics, and Light-Induced Dynamics of Cuhetphen Chromophores. Dalton. Trans. 2016, 45, 9871–9883. 10.1039/C6DT00324A. [DOI] [PubMed] [Google Scholar]
- Penfold T. J.; Karlsson S.; Capano G.; Lima F. A.; Rittmann J.; Reinhard M.; Rittmann-Frank M. H.; Braem O.; Baranoff E.; Abela R.; Tavernelli I.; Rothlisberger U.; Milne C. J.; Chergui M. Solvent-Induced Luminescence Quenching: Static and Time-Resolved X-Ray Absorption Spectroscopy of a Copper(I) Phenanthroline Complex. J. Phys. Chem. A 2013, 117, 4591–4601. 10.1021/jp403751m. [DOI] [PubMed] [Google Scholar]
- Chen L. X.; Jennings G.; Liu T.; Gosztola D. J.; Hessler J. P.; Scaltrito D. V.; Meyer G. J. Rapid Excited-State Structural Reorganization Captured by Pulsed X-Rays. J. Am. Chem. Soc. 2002, 124, 10861–10867. 10.1021/ja017214g. [DOI] [PubMed] [Google Scholar]
- Westre T. E.; Kennepohl P.; DeWitt J. G.; Hedman B.; Hodgson K. O.; Solomon E. I. A Multiplet Analysis of Fe K-Edge 1s → 3d Pre-Edge Features of Iron Complexes. J. Am. Chem. Soc. 1997, 119, 6297–6314. 10.1021/ja964352a. [DOI] [Google Scholar]
- Rentschler M.; Iglesias S.; Schmid M.-A.; Liu C.; Tschierlei S.; Frey W.; Zhang X.; Karnahl M.; Moonshiram D. The Coordination Behaviour of CuI Photosensitizers Bearing Multidentate Ligands Investigated by X-Ray Absorption Spectroscopy. Chem. - Eur. J. 2020, 26, 9527–9536. 10.1002/chem.201905601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moonshiram D.; Garrido-Barros P.; Gimbert-Surinach C.; Picon A.; Liu C.; Zhang X.; Karnahl M.; Llobet A. Elucidating the Nature of the Excited State of a Heteroleptic Copper Photosensitizer by Using Time-Resolved X-Ray Absorption Spectroscopy. Chem. - Eur. J. 2018, 24, 6464–6472. 10.1002/chem.201800330. [DOI] [PubMed] [Google Scholar]
- Fischer S.; Hollmann D.; Tschierlei S.; Karnahl M.; Rockstroh N.; Barsch E.; Schwarzbach P.; Luo S.-P.; Junge H.; Beller M.; Lochbrunner S.; Ludwig R.; Brückner A. Death and Rebirth: Photocatalytic Hydrogen Production by a Self-Organizing Copper–Iron System. ACS Catal. 2014, 4, 1845–1849. 10.1021/cs500387e. [DOI] [Google Scholar]
- Takeda H.; Koike K.; Inoue H.; Ishitani O. Development of an Efficient Photocatalytic System for CO2 Reduction Using Rhenium(I) Complexes Based on Mechanistic Studies. J. Am. Chem. Soc. 2008, 130, 2023–2031. 10.1021/ja077752e. [DOI] [PubMed] [Google Scholar]
- Tamaki Y.; Koike K.; Morimoto T.; Ishitani O. Substantial Improvement in the Efficiency and Durability of a Photocatalyst for Carbon Dioxide Reduction Using a Benzoimidazole Derivative as an Electron Donor. J. Catal. 2013, 304, 22–28. 10.1016/j.jcat.2013.04.002. [DOI] [Google Scholar]
- Taube H.; Myers H.; Rich R. L. Observations on the Mechanism of Electron Transfer in Solution. J. Am. Chem. Soc. 1953, 75, 4118–4119. 10.1021/ja01112a546. [DOI] [Google Scholar]
- Thordarson P. Determining Association Constants from Titration Experiments in Supramolecular Chemistry. Chem. Soc. Rev. 2011, 40, 1305–1323. 10.1039/C0CS00062K. [DOI] [PubMed] [Google Scholar]
- Zhang P.; Wang M.; Li C.; Li X.; Dong J.; Sun L. Photochemical H2 with Noble-Metal-Free Molecular Devices Comprising a Porphyrin Photosensitizer and a Cobaloxime Catalyst. Chem. Commun. 2009, 46, 8806–8808. 10.1039/c0cc03154b. [DOI] [PubMed] [Google Scholar]
- Ozawa K.; Tamaki Y.; Kamogawa K.; Koike K.; Ishitani O. Factors Determining Formation Efficiencies of One-Electron-Reduced Species of Redox Photosensitizers. J. Chem. Phys. 2020, 153, 154302 10.1063/5.0023593. [DOI] [PubMed] [Google Scholar]
- Lentz C.; Schott O.; Auvray T.; Hanan G. S.; Elias B. Design and Photophysical Studies of Iridium(II)–Cobalt(III) Dyads and Their Application for Dihydrogen Photo-Evolution. Dalton. Trans. 2019, 48, 15567–15576. 10.1039/C9DT01989H. [DOI] [PubMed] [Google Scholar]
- Moonshiram D.; Gimbert-Surinach C.; Guda A.; Picon A.; Lehmann C. S.; Zhang X.; Doumy G.; March A. M.; Benet-Buchholz J.; Soldatov A.; Llobet A.; Southworth S. H. Tracking the Structural and Electronic Configurations of a Cobalt Proton Reduction Catalyst in Water. J. Am. Chem. Soc. 2016, 138, 10586–10596. 10.1021/jacs.6b05680. [DOI] [PubMed] [Google Scholar]
- Gotico P.; Moonshiram D.; Liu C.; Zhang X.; Guillot R.; Quaranta A.; Halime Z.; Leibl W.; Aukauloo A. Spectroscopic Characterisation of a Bio-Inspired Ni-Based Proton Reduction Catalyst Bearing a Pentadentate N2S3 Ligand with Improved Photocatalytic Activity. Chem. - Eur. J. 2020, 26, 2859–2868. 10.1002/chem.201904934. [DOI] [PubMed] [Google Scholar]
- Yue Z.; Ou C.; Ding N.; Tao L.; Zhao J.; Chen J. Advances in Metal Phthalocyanine Based Carbon Composites for Electrocatalytic CO2 Reduction. ChemCatChem. 2020, 12, 6103–6130. 10.1002/cctc.202001126. [DOI] [Google Scholar]
- Roy S.; Miller M.; Warnan J.; Leung J. J.; Sahm C. D.; Reisner E. Electrocatalytic and Solar-Driven Reduction of Aqueous CO2 with Molecular Cobalt Phthalocyanine–Metal Oxide Hybrid Materials. ACS Catal. 2021, 11, 1868–1876. 10.1021/acscatal.0c04744. [DOI] [Google Scholar]
- Costentin C.; Drouet S.; Robert M.; Saveant J. M. A Local Proton Source Enhances CO2 Electroreduction to CO by a Molecular Fe Catalyst. Science 2012, 338, 90–94. 10.1126/science.1224581. [DOI] [PubMed] [Google Scholar]
- Wang J. W.; Huang H. H.; Sun J. K.; Ouyang T.; Zhong D. C.; Lu T. B. Electrocatalytic and Photocatalytic Reduction of CO2 to CO by Cobalt(II) Tripodal Complexes: Low Overpotentials, High Efficiency and Selectivity. ChemSusChem 2018, 11, 1025–1031. 10.1002/cssc.201702280. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Truhlar D. G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals. Theor. Chem. Acc. 2008, 120, 215–241. 10.1007/s00214-007-0310-x. [DOI] [Google Scholar]
- Luo S. P.; Mejia E.; Friedrich A.; Pazidis A.; Junge H.; Surkus A. E.; Jackstell R.; Denurra S.; Gladiali S.; Lochbrunner S.; Beller M. Photocatalytic Water Reduction with Copper-Based Photosensitizers: A Noble-Metal-Free System. Angew. Chem., Int. Ed. 2013, 52, 419–423. 10.1002/anie.201205915. [DOI] [PubMed] [Google Scholar]
- Li C.; Schwab M.; Zhao Y.; Chen L.; Bruder I.; Münster I.; Erk P.; Müllen K. A Phenanthroline Derivative as Exciton Blocking Material for Organic Solar Cells. Dyes Pigm. 2013, 97, 258–261. 10.1016/j.dyepig.2012.12.003. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.