

Abstract
A multistate energy decomposition analysis (MS-EDA) method is described to dissect the energy components in molecular complexes in excited states. In MS-EDA, the total binding energy of an excimer or an exciplex is partitioned into a ground-state term, called local interaction energy, and excited-state contributions that include exciton excitation energy, superexchange stabilization, and orbital and configuration-state delocalization. An important feature of MS-EDA is that key intermediate states associated with different energy terms can be variationally optimized, providing quantitative insights into widely used physical concepts such as exciton delocalization and superexchange charge-transfer effects in excited states. By introducing structure-weighted adiabatic excitation energy as the minimum photoexcitation energy needed to produce an excited-state complex, the binding energy of an exciplex and excimer can be defined. On the basis of the nature of intermolecular forces through MS-EDA analysis, it was found that molecular complexes in the excited states can be classified into three main categories, including (1) encounter excited-state complex, (2) charge-transfer exciplex, and (3) intimate excimer or exciplex. The illustrative examples in this Perspective highlight the interplay of local excitation polarization, exciton resonance, and superexchange effects in molecular excited states. It is hoped that MS-EDA can be a useful tool for understanding photochemical and photobiological processes.
Keywords: Multistate energy decomposition analysis, excited-state energy components, exciton resonance, charge transfer, superexchange, MS-EDA, MSDFT
1. Introduction
Energy decomposition analysis (EDA) is widely used and plays an important role in the understanding of intermolecular interactions in molecular systems.1−6 These studies provide insights into the interplay of Pauli exclusion, polarization and charge transfer effects that contribute to intermolecular forces. In turn, the information gained from these investigations can be useful to designing and optimizing empirical potential energy functions for condensed-phase and biomolecular simulations.7,8 A large number of EDA models have been proposed, but they are almost exclusively limited to molecular complexes in the ground state.4,6,9−49 On the other hand, a major current frontier of theoretical chemistry is to study chemical processes taking place in electronically excited states in areas such as photochemistry, photovoltaic devices,50 photosynthesis and photoreception in biology,51−54 catalysis, and even reactions in fuel cells and at the electrodes.55 Electronic coupling among local states in these systems can be used to determine the rate of excited-state energy transfer. Unfortunately, little attention has been paid to energy decomposition analysis of intermolecular interactions in excited states,5,56−58 although several studies of energy components of excited states have been reported.59,60 Moreover, it is not always clear if the intuitive terms for the ground state can be adapted to molecular systems in the excited states. In this Perspective, we present a multistate energy decomposition analysis (MS-EDA) introduced in a recent preliminary report5 to define the energy terms relevant to intermolecular interactions in excited states. This is now possible following the development of multistate density functional theory (MSDFT) to treat the ground state, excited states and diabatic states on an equal footing with the inclusion of electron correlation.61−64
Energy decomposition analysis was first introduced by Morokuma and Kitaura on the basis of Hartree–Fock theory in the 1970s,9,65 who defined intuitive terms such as electrostatic, exchange repulsion, polarization and charge transfer energies, which are intimately related to concepts used in chemical research. The method was an instant success and has become extremely popular in computational chemistry.66 Subsequently, a wide range of models have been proposed, attacking the energy components from different perspectives for different situations. Roughly speaking, these methods may be grouped into three categories: (1) physical approaches, either based on a formal perturbation theory or by intuitive construction and modification of the interaction terms; (2) variational methods, developed by constructing well-defined intermediate molecular wave functions that can be variationally optimized; and (3) perturbation theories using quantum chemical methods or classical electrostatics theory.
The original Kitaura–Morokuma EDA belongs to the first category through a gradual annihilation of the Fock and overlap matrix elements relevant to intermolecular interactions.65 Other models that partition the energy terms through an electron density formulation such as the interacting quantum atom (IQA) method according to zero-flux density surfaces,67 and the natural energy decomposition analysis (NEDA) by making use of natural bond orbitals.13,68 EDA models that do not include variational energy minimization of the intermediate states can lead to an overestimation of the resonance energy and charge transfer effects.69−71
EDA models belonging to the second category were developed with certain elements of variational optimization of key reference intermediate states for energy partition. In Mo’s block-localized wave function (BLW)72 approach and the absolutely localized molecular orbital analysis,6,25 the strictly block-localized orbitals for every intermediate in the EDA are fully variationally optimized, providing a set of well-defined diabatic intermediate states for interpretation of the resonance energies of aromaticity, anomeric effects and forward-and-backward bonding interactions.3,46,73 Other illustrative examples include the early constrained space orbital variations (CSOV) method by successively mixing occupied and virtual orbitals of different molecules,11,74,75 and the reduced variational space self-consistent-field (RVS-SCF) model in which the orbitals of one fragment is optimized in the presence of the frozen orbitals of other fragments.12 Another class of EDA theory features the extended transition state (ETS) scheme,30,76,77 along with natural orbitals for chemical valence (NOCV) theory, in which not only noncovalent intermolecular interactions are decomposed, but also the energy of a chemical bond can be separated into unpaired electron contributions.24,29 The ETS-NOCV method allows the analysis of bonding characteristics such as σ,π,δ bond types.77
In category three,35 perhaps the best known example is the popular symmetry-adapted perturbation theory (SAPT), which can be applied both to wave function theory and density functional theory.36,78 The SAPT method relies on a series of monomer Fock, perturbation and interaction operators. The first order polarization and exchange corrections are assigned to electrostatic and exchange energies, and higher order terms result in induction and dispersion terms. However, separation of charge-transfer energy can be challenging in perturbation approaches.79−81
Energy decomposition analysis for molecular complexes in the excited states is rare. Methods exist to extract local information and charge transfer contribution in excited states by analyzing density difference and transition density matrices.58−60,82−86 In principle, EDA methods for the ground state can be adapted to describe interactions in the excited states. One application was recently reported by Ge et al., who transferred the corresponding energy terms in the ground-state EDA to excited complexes.57,87 In particular, the interaction energy for an exciplex is separated into frozen (froz), polarization (pol) and charge transfer (CT) terms, similar to that in the ground state: ΔEint* = ΔEfroz + ΔEpol* + ΔECT.56 To obtain “frozen” excitations, the orbitals for each isolated monomer and its excitation amplitudes are combined to construct a supramolecular operator. Later, an excitation splitting term was incorporated to describe interactions between degenerate monomer states in a symmetric excimer.56 While this excited-state decomposition scheme revealed interesting information in the lens of a ground-state microscope, the intermediate states cannot be variationally optimized because interfragment interactions were reconstructed from separate monomer terms. Yet, there is an interest in defining intermediate diabatic states which can be used to represent excitations localized on individual monomers to understand the reaction rates of excited-state energy transfer and excitation induced oxidation–reduction reactions.88 Recently, we introduced a multistate approach,5 making use of multistate density functional theory (MSDFT),61 to define variationally optimizable intermediate states to interpret properties associated with excited-state processes,89,90 including local excitation, exciton resonance, superexchange, and orbital-and-configuration delocalization. This Perspective further expands that work in several fronts. In addition to understanding intermolecular interactions in excited-state complexes, the energy terms and the associated diabatic states in this analysis can be used to investigate ultrafast photochemical reactions.50,51,54
In the following, we first present the theory of multistate energy decomposition analysis (MS-EDA), and the fundamental principles and computational algorithms of MSDFT. Then, we illustrate the MS-EDA method with a set of applications to excited-state complexes which can be loosely classified as encounter exciplex, excitation-induced charge-transfer complex, and intimate excimer and exciplex. We validate the computational procedure by comparison with results from time-dependent density functional theory (TDDFT) that works well for most of these exciplexes and their monomers as well as with available experimental data. In some cases, the computational MS-EDA results shed light on experimental findings with new interpretations. It is hoped that the new MS-EDA method shall stimulate further analysis and interpretation of computational results on excited states from delocalized wave function theories and TDDFT.
2. Theory
We aim at developing a method for energy analysis of molecular complexes in an excited state. For the photochemical reaction producing the exciplex (XY)* between compounds X and Y
| 1 |
we define the binding energy of (XY)* in its Kth excited state as
| 2 |
where the superscript “0” denotes the energy and wave function of a molecule in the ground state, and E(XY)*K is the energy of the Kth excited state of the exciplex (XY)* associated with the wave function Ψ(XY)*, and hν̅0K is the energy for chromophore excitation whose definition will be addressed next (not necessarily Y alone as indicated).
Before we discuss the energy components, we first take a closer look at the meaning of “binding energy” ΔEbK of an exciplex. It turns out that it is not as straightforward as that of a bimolecular complex in the ground state. From a viewpoint of the physical process, the external energy supplied (i.e., photoexcitation) to initiate the formation of an excited-state complex from two isolated molecules in their ground state is the vertical transition of one relevant monomer (or more) to the Franck–Condon region. Then, the excited species can quickly relax to its equilibrium geometry of a specific excited state (e.g., K), which in turn binds to another monomer and together relaxes to the equilibrium exciplex (Scheme 1). Alternatively, two molecules form a ground-state complex, the dimer absorbs a photon and gets excited into an exciplex, which then directly relaxes to its final geometry. The precise mechanism is not always known exactly, but the final state is well-defined (eq 1), and its energy can be related to the emission spectrum of the exciplex. However, the energy difference between the final exciplex and the vertical excitation energy is not ΔEb since some of the vertical excitation energy may have dissipated (without discussing its possible mechanisms) into the environment in the relaxation process. Furthermore, it is not always clear if one, two or a fraction of each monomer are excited in the first place, e.g., in the case of a symmetric or nearly symmetric excimer where the excited state is delocalized over both monomers. Therefore, we need to define an effective external energy hν̅0K to produce the unbound monomers to form the corresponding exciplex in the Kth excited state.
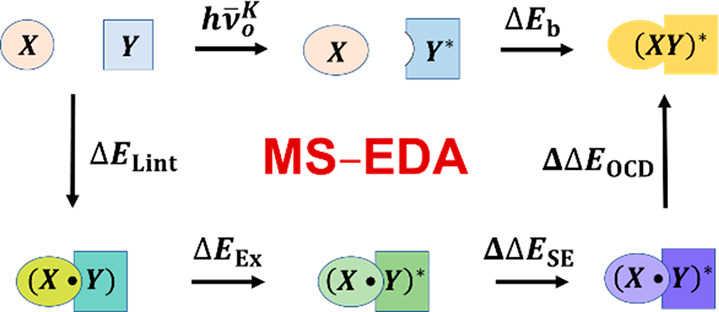
Scheme 1. Thermodynamic Cycle Relating the Energy Components in Multistate Energy Decomposition Analysis of Exciplex Binding Energy ΔEb.
The top row corresponds to the physical process of exciplex formation (eq 2). The remaining illustration highlights the energy terms defined by eq 4. The shapes of objects represent molecular geometries, while different colors depict the polarized wave functions under different conditions. Blocks with boundaries denote fragment-localized wave functions, all of which can be variationally optimized. The lower panels in light-yellow shade specify the minimal active space (MAS) in nonorthogonal state interacion (NOSI) calculations, respectively, for the exciton state, derived from local excited states (Lex) shown in pink, and for the super-exchange (SE) contribution due to charge transfer diabatic states. Adapted with modifications from ref (5). Copyright 2023 American Chemical Society.
Here, we define hν̅0K in eq 1 as the minimum excitation energy associated with the formation of the exciplex in the Kth excited state, which is the weighted sum of the adiabatic excitation energies of the monomers at their geometries in the exciplex complex. It is related to the “vertical” emission energies of the monomers in the exciplex geometries and the geometrical distortion energy in going from the equilibrium structures of the ground-state monomers to the distorted geometries (X[SK] and Y[SKEC]) in the excited complex.
| 3 |
where wX(K) and wY are the structure weights of monomer excited states in the exciton complex defined in section 2.2. Thus, hν̅0K is the effective external energy used to deform the monomer geometries and to excite them to the corresponding states at the fractions found in the exciton complex. Clearly, hν̅0 is state-specific. Then, the energy change in going from the isolated, “fractionally” excited monomers to the final complex is the exciplex binding energy ΔEbK.
2.1. Multistate-Energy Decomposition Analysis (MS-EDA)
In MS-EDA, on the basis of the thermodynamic cycle in Scheme 1, we separate the binding energy of an exciplex in eq 2 into the following terms:
| 4 |
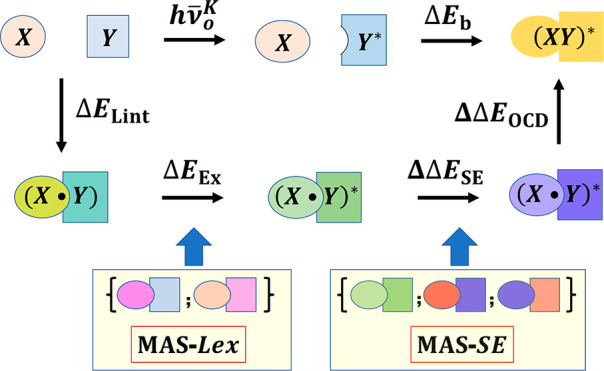
where ΔELint[(X•Y)] denotes the interaction energy between monomers X and Y in the ground state, ΔEExK[(X•Y)*] is the excitation energy of the exciton (Ex) state and hν̅0 is defined in eq 3, ΔΔESEK[(X±•Y∓)*] specifies superexchange stabilization, and ΔΔEOCD[(XY)*] represents the energy change due to orbital and configuration-state delocalization (OCD). The progressive energy terms in the MS-EDA procedure are schematically illustrated in Scheme 2 with a sequence of intermediate states leading to the final exciplex formation. Each state depicted in Scheme 1 has a well-defined wave function (or MSDFT configuration states) that can be variationally optimized.
Scheme 2. Energy Correlation Diagram, Energy Terms, and Associated States in Multistate Energy Decomposition Analysis.
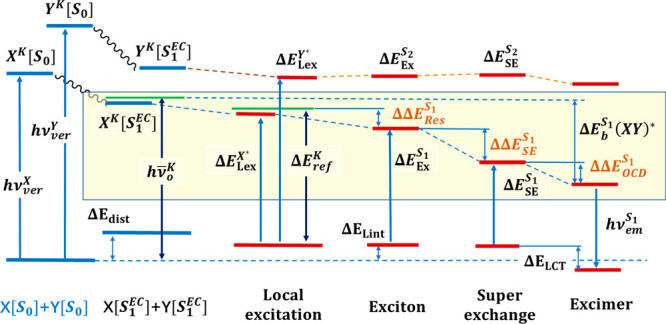
[S0] and [S1EC] indicate geometries of X and Y in the respective equilibrium ground state and that in optimized exciplex complex (EC) in the first excited state. Blue lines are monomer compounds, and maroon lines denote dimer structures. Energies that can be observed spectroscopically are indicated by hν, except for local excitations given in relative energies. Resonance and delocalization energies are given in brown. Energy changes in light yellow shade are associated with the exciplex binding energy.
Although the binding energy of an excited-state complex is state-dependent, we will sometimes omit the superscript K for simplicity, but one needs to keep in mind that the intermediate states and energy terms in MS-EDA correspond to the same Kth state of the exciplex (not necessarily in the same order) and they must be matched in the analysis. Often, the lowest excited state of a given spin is of particular interest, although symmetry allowed and avoided curve crossings can take place and the order of states may differ at different stages of the MS-EDA scheme, in which case, analysis of more than one state is necessary.
The individual energy components in eq 4 can be further decomposed into more specific terms to gain insights into the intricacy of intermolecular interactions in an excited state. Some of these energy terms can be directly measured experimentally or extracted from experimental data. In this section, we define the interaction energy terms. In the next section, we analyze the additional components that constitute some of these terms in the exciplex.
Throughout this Perspective, we use Ψ and Φ to denote a physical adiabatic state or an intermediate diabatic state with further specification indicated by a subscript and the particular excited state specified by a superscript. Ψ and Φ are typically multistate wave functions, which are also used to represent the matrix density. The Greek letter Ξ is used to specify a single Slater determinant wave function for a given electronic configuration, typically monomer-block localized, to represent the corresponding electron density in block-localized excitation (BLE) and target state optimized density functional theory (TSO-DFT) calculations.91,92 Occupied Kohn–Sham molecular spin orbitals, typically block localized, are denoted by χi, χj, ..., and unoccupied (virtual) orbitals are given as χa, χb, .... In the present MS-EDA method, we use the monomer block-localized Kohn–Sham (BLKS) determinant, Ξ(X•Y)KS, as a reference state, which is used to generate the initial guess for singly excited configurations Ξi, all of which are variationally optimized individually using BLE and TSO–DFT for block-localized excitation.91,92 Together, these variationally optimized determinants states form a minimum active space denoted by a single subscript {ΞA; A = 1, ...}.
2.1.1. Ground-State Interaction Energy
The formation of a molecular aggregate in an excited state has energy contributions both from binding interactions in the ground state and from energy terms induced by photoexcitation (Scheme 1), keeping in mind that in many situations an exciplex is only stable in the excited state (thus, the ground-state effect could be repulsive). Since EDA has been thoroughly investigated for molecular interactions in the ground state, we separate ground-state and excited-state energies in the present multistate analysis. Consequently, the energy components leading to the monomer block-localized complex in the ground state are grouped into a single term called local interaction (Lint) energy in MS-EDA.
| 5 |
where (X•Y) denotes a bimolecular complex whose wave function is block-localized, indicated by the symbol “•”, the parentheses specify an antisymmetrized product wave function from the monomers, Ψ(X•Y) = Â{ΩXΩY} with ΩX and ΩY denoting products of BLKS orbitals, and ΞXo and ΞY are Kohn–Sham determinants for molecules X and Y in isolation in their equilibrium geometries (Figure 1). Here, the mutual polarization between the two monomers are naturally included in Ψ(X•Y) as a result of variational optimization of the wave function; Ψ(X•Y) can be a multistate wave function in which case it is replaced by a linear combination of configuration state functions in the active space. Note that an energy deformation term has been included in ΔELint because the molecular geometry of the exciplex is used to determine E(X•Y)[Ψ(X•Y)] in eq 5.
Figure 1.

Reference state (dashed lines in red) for local excitations (lines black) and exciton-resonance stabilization energy (blue arrows). Local and exciton excitation energies relative to the block-localized complex are indicated by red arrows.
ΔELint[(X•Y)] is called local-interaction energy because molecular orbitals are not globally delocalized over the full complex. In addition to the structural deformation energy, it includes an electrostatic term called Heitler–London energy, Ψ(X•Y)o = Â(ΩX•ΩY0), and a polarization energy in the variationally optimized state Ψ(X•Y) = Â{ΩX•ΩY}.3,27 Note that ΔELint[(X•Y)] is not the binding energy of the bimolecular complex (XY) in the ground state because charge-transfer delocalization is not included. Additional details for ground-state analysis can be found in references.1−3,6
2.1.2. Exciton Excitation Energy
In MS-EDA, we first define an exciton state, denoted by (X•Y)* (Scheme 1). Then, the energy difference between the exciton state and the ground state of the block-localized complex is called the exciton-excitation energy ΔEExK.
| 6 |
where E(X•Y)*K and E(X•Y) are the energies of the Kth exciton state and the ground state, respectively, corresponding to the wave functions Ψ(X•Y)* and Ψ(X•Y) (eq 5). According to the Frenkel exciton model,93 the wave function of the exciton state is expressed as follows.
| 7 |
where Ψ(X*•Y)KX and Ψ(X•Y*) are the spin-adapted, configuration state functions of the two locally excited monomers (MAS-Lex in Scheme 1), and a1K and a2K are the state coefficients. The superscripts KX and KY specify, respectively, the states of the locally excited states of monomers X and Y that contribute to the exciton state K (again, the subscripts or even K are typically omitted without causing confusion). In general, both Ψ(X*•Y)KX and Ψ(X•Y*) are multiconfigurational wave functions, and more than one state from each monomer may be included.
Obviously, eq 7 can be extended to any number of monomers in an aggregate of n monomers or to condensed phases:
where Ψ(X1•···•XA*•···•Xn)KA denotes the local excitation to the KAth state of monomer XA* in the presence of the rest of the n – 1 monomers in the ground state. In solution, the local excitation energy of the chromophore includes a solvatochromic shift in comparison with the excitation energy of the isolated monomer. To limit the scope of this Perspective, we shall not further discuss condensed-phase properties including exciton delocalization in general situations.
2.1.3. Charge Transfer States and Superexchange Energy
The exciton state above from the interactions among strictly localized monomer states includes the mutual polarization induced by chromophore excitation, but charge transfer (CT) contributions are excluded by construction (i.e., monomer localization). In the second step of MS-EDA, we introduce CT resonance states by combining the forward and backward CT states:
| 8 |
We note that for a symmetrical excimer the amount of forward and backward CT is the same, leaving a net zero “CT”. Nevertheless, charge-transfer resonance in eq 8 can still stabilize the excimer complex significantly. On the other hand, in asymmetrical exciplexes, the significance of CT resonance strongly depends on the relative energies of the two directions of CT. Then, the CT state in the direction with the lower energy typically plays a dominant role, whereas the effect in the opposite direction of CT can be ignored. To encompass both scenarios, we borrow a term from electron transfer theory,50,94 giving the name superexchange (SE) stabilization to the decrease in energy when superexchange states (eq 8) are admixed to the exciton states (eq 7).
| 9 |
where the wave function for the superexchange intermediate state in MS-EDA is given by
| 10 |
It should be emphasized that the notation |X+•Y–⟩ (and |X–•Y+⟩) defines a spin-adapted state obtained from MSDFT-NOSI for the spin-pairing interactions between the two spin-mixed determinants with single-electron transfer in α or β spin (MAS-SE in Scheme 1). The two resulting eigenstates correspond to a singlet state and a triplet component with Ms = 0 (see below).
2.1.4. Orbital and Configuration-State Delocalization
In the final step of MS-EDA, we relax the strictly localized BLKS orbitals to the fully delocalized molecular (or Kohn–Sham) orbitals over the entire exciplex system. In addition, the number of basis configurations used to define the intermediate states in the exciton and superexchange step is relatively small, which is useful for interpretation of excited-state energies. Thus, the difference between excited-state energies from TDDFT calculations for the delocalized exciplex and MSDFT-NOSI in analysis is also included. We denote the Kth adiabatic excited state of the exciplex as Ψ(XY)*K where the removal of the dot “•” emphasizes spatial delocalization of the orbitals and configuration space. The energy change in this step is called orbital and configuration-state delocalization (OCD) energy.
| 11 |
In principle, SE and OCD energies can be combined as the overall CT effects, similar to that in ground-state EDA, but it is useful to make the distinction and separation here in view of the difference in the number of basis configuration state functions to treat the intermediate states and the adiabatic states of the exciplex.
The ΔΔEKOCD term is defined in terms of excited-state energies (eq 11), but, in fact, it also includes ground-state effects, corresponding to the charge-transfer energy term in ground-state EDA, denoted by ΔELCT (Scheme 2). It can be extracted as the difference between the energy of the (XY) complex in the ground state and the local binding state (X•Y). Thus, the contribution to exciplex binding from the total ground-state effect is ΔELb = ΔELint + ΔELCT. Then, the OCD effect on the excited state is ΔΔEOCD = ΔΔEOCDK – ΔELb. In this paper, we do not further separate these two OCD terms, except in EOCD In some specific cases, we comment on the numerical results of these terms.
On computation of the excited-state energy of an exciplex, multiconfiguration self-consistent-field (MCSCF) or configuration interaction-based approaches such as multireference configuration interaction (MR-CI) in wave function theory can be used to determine the energies and excited-state wave functions Ψ(XY)*K. Alternatively, MSDFT and linear-response TDDFT method can be used to obtain E(XY)* and ρ(XY)*K in density functional theory. Given that MSDFT is a new variational DFT method and the development of approximate matrix correlation functionals hat not yet advanced to a level as that in KS-DFT, we employ TDDFT for estimating the excited state energies of the final exciplexes. Nevertheless, the accuracy of MSDFT in the MS-EDA analysis can be validated by comparison of the computed excitation energies for the monomer states with those obtained using TDDFT, employing a common approximate KS density functional and the same basis set. In this regard, the present MS-EDA method can also be viewed and used as an interpretative tool to dissect and understand the energies from TDDFT calculations.
2.2. Block-Localized Excited States
In the above MS-EDA, the exciton state is viewed as the resonance delocalization among monomer-localized excitations of individual chromophores.93 Of interest is the relative contributions of locally excited states and the overall resonance energy in exciton formation (eq 7). In this regard, the term local excitation refers to the excited state of an individual monomer chromophore in the presence of other compounds in their ground state. This is achieved in TSO-DFT calculations by using the BLMO (in WFT) or BLKS orbitals (in MSDFT) that are strictly localized within each monomer by construction. These BLKS orbitals are clearly different from localized orbitals obtained through a particular symmetry orthogonal transformation.95,96 Local excitation has a physical interpretation that can be directly related to experimental solvatochromic shifts induced by solvation (though this important physical property will not be addressed in this Perspective in view of space). In this section, we further decompose ΔEExK into specific components related to local excitation and resonance delocalization. This analysis also provides meaning to the structure weights used in the definition of hν̅o in eqs 2 and 3.
2.2.1. Local Excitation and Polarization Effects
The excitation energy of a locally excited state of monomer X is given by ΔELexKX(X*•Y):
| 12 |
where νLexKX(X*•Y) is the excitation frequency of X if one considers Y as an environmental bath such as a solvent or simply another molecule in the present analysis (Scheme 2). The introduction of a frequency notation emphasizes the possibility that this quantity may be compared with the experimental absorption spectrum of a chromophore in solution or in a matrix of cluster molecules. Then, the solvatochromic shift of chromophore X in the presence of monomer Y (or in solution) relative to that in isolation (or in another reference solvent denoted by the subscript o), νo(X*), is given by
| 13 |
νoKX(X*) can have the same energy as that of the hν̅0 term in eqs 1 and 2 for a single chromophore in special cases such as a symmetrical excimer.
Local excitation of monomer X changes its electrostatic field which polarizes the charge distribution of the other monomer Y in the ground state (or solvent). In turn, the reaction field of the ground state of Y further affects the energy of the excited monomer. The equilibrium state ΨKX(X*•Y) is a result of their mutual polarization, and an understanding of its magnitude is of interest in MS-EDA. In fact, the local excitation energy of X* in the block-localized complex (X•Y) (eq 12) may be considered as photoexcitation of X in the hypothetic state ([Xo*]•Y) without interacting with Y in the complex i.e., the gas-phase adiabatic excitation energy hν0KX(X*), followed by the mutual polarization of X* with its environment Y to the local excited state,
| 14 |
corresponding to the following two steps (see also Scheme 1):
Consequently, the polarization energy arising from local excitation ΔELpolKX(X*•Y) is identical to the observable spectral shift:
| 15 |
For a symmetric dimeric excimer, the polarization energies for the two local excitations are identical. In this case, the block-localized excited states (X*•X) and (X•X*) are strongly correlated between two degenerate states, each corresponding to a diabatic state, which is also of interest in the study of excited-state energy transfer and exciton delocalization. The emission spectrum of the resonance state of the excimer may be used to determine the magnitude of the resonance energy relative to the variationally optimized diabatic states.97 Note that the energies expressed in eq 14 can indeed be determined variationally by using the block-localized excitation (BLE) method,92 or the targeted state optimization (TSO) approach.98
2.2.2. Exciton Resonance Energy
To determine the resonance energy of an exciton state, a variationally optimizable reference state in the absence of resonance (delocalized) excitation must be defined for comparison. For a perfectly symmetric excimer, it is straightforward simply to use the energy of one of the local excitation energies since both local excitations contribute equally. However, in the case of an asymmetric excimer or an exciplex of different monomers in which the two monomers contribute differently, a clear definition of the reference state is needed. Here, we introduce a structure-weighted reference state for estimating the resonance energy in an exciton state.
| 16 |
where the energies of the locally excited states ΨKX(X*•Y) and ΨKY(X•Y*) are variationally minimized, and wXK and wY are the Chirgwin–Coulson configuration weights99 of the Kth exciton state in eq 7.
The structure weights for locally excited states, Ψ(X*•Y)KX and Ψ(X•Y*), in the resonance exciton state (eq 7) can be determined according to the Chirgwin–Coulson partition:
| 17a |
| 17b |
where S12 is the overlap integral between the two locally excited states, S12 = S21 = ∫dr{Ψ(X*•Y)KX}*Ψ(X•Y*).
Here, an exciton-state specific reference is defined because each locally excited state has different contributions in different exciton states. This is illustrated in Figure 1 in which two locally excited states in an exciplex interact to produce two exciton states, corresponding to the in-phase and out-phase combinations of Ψ(X*•Y)KX and Ψ(X•Y*). It would have been unfair if the energy of any individual state or the average energy of both states were used as the reference to determine exciton-resonance (ER) energy because the former does not consider the contribution from the other state, and the latter does not truly reflect the relative significance of individual states in the exciton state (eq 17a). The reference defined in eq 16 provides an adequate consideration of these factors. In the limit of an identical excimer, the weighted reference state is the same as the energy of one locally excited monomer.
With the same analogy, the structural weights determined at the exciton state are used to define a weighted adiabatic excitation energy for exciplex formation (eqs 2 and 3).
Having defined a reference energy, the exciton resonance energy (Figure 1) due to locally excited state-interaction is easily determined:
| 18 |
The use of eq 18 implies that the sum of the resonance energies for the resonance-stabilized and destabilized exciton states is not exactly the resonance splitting of the delocalized states (Figure 1). We further remark on the use of a weighted local-excitation energy as the reference state. First, the local excitations of all monomers contribute to the resonance-delocalized exciton state. Figure 1 highlights schematically the relative energies of the individual energy terms, including the weighted local-excitation energy (reference state), and the resonance stabilization energy for a given exciton state in the complex. In the case where monomers X and Y are asymmetric, the reference states for the in-phase and out-phase combinations of the locally excited states are different. In the former combination, the lower energy state (state K+ in Figure 1) is stabilized, whereas the higher energy exciton state (state L (K–) in Figure 1) is destabilized. Since the stabilization and destabilization effects are relative contributions from all locally excited states, the state-specific, weighted local-excitation energy takes into account the relative significance to resonance energy and antiresonance energy. For an excimer complex, since both local-excitation energies are the same, a single reference state is naturally used for both resonance and antiresonance effects.
2.3. Multistate Density Functional Theory
Multistate density functional theory (MSDFT) is used to determine the intermediate states in multistate energy-decomposition analysis. Here, we summarize the fundamental theorems of MSDFT,61,62 the definition of a Hamiltonian matrix density functional and the nonorthogonal state interaction (NOSI) method.64,100,101 NOSI distinguishes from nonorthogonal configuration interaction (NOCI) in that the former includes dynamic correlation in each excited basis state in an active space, whereas the latter configuration space generally requires expensive corrections for dynamic correlation to yield quantitative results.63
2.3.1. Fundamental Theorems
Although MSDFT has been used in a range of applications in the past,64,100 it was in 2021 that Lu and Gao proved three fundamental theorems, establishing MSDFT as a rigorous density functional theory of quantum mechanics for any number of N states.61 For a system described by the Hamiltonian Ĥ = Ĥ° + vext(r), where Ĥ° consists of kinetic energies and electronic interaction energies, and vext(r) is the local external potential, the first theorem establishes the correspondence of the Hamiltonian and the N-dimensional multistate matrix density D(r). Theorem 2 is a variational principle, resulting in the energies and densities of all N eigenstates. Theorem 3 defines the representation of an N-dimensional multistate matrix density, enabling the design of different practical algorithms and procedures for computation.
-
(1)Given the subspace
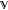 spanned by the lowest N eigenstates of a molecular system described by Ĥ, the Hamiltonian Ĥ° is a universal matrix functional of the N-dimensional
multistate density D(r),
spanned by the lowest N eigenstates of a molecular system described by Ĥ, the Hamiltonian Ĥ° is a universal matrix functional of the N-dimensional
multistate density D(r), 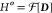 , independent of the external potential vext(r). Then,
the total Hamiltonian matrix density functional (HMDF)
is
, independent of the external potential vext(r). Then,
the total Hamiltonian matrix density functional (HMDF)
is
Here, the multistate density D(r) is a matrix of electron densities {ρi(r) ≡ ρii (r); i = 1, ..., N} and transition densities between any two basis states {ρij(r); i ≠ j, i, j = 1, ..., N}. In other words, Theorem 1 establishes a one-to-one correspondence between D and
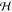 .
.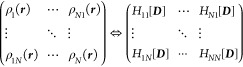
-
(2)For any N-dimensional trial matrix density D'(r), the trace of
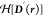 , defined as the multistate energy EMS[D'(r)] = ΣiHii', is greater than
or equal
to the multistate energy EMS[D] of the subspace
, defined as the multistate energy EMS[D'(r)] = ΣiHii', is greater than
or equal
to the multistate energy EMS[D] of the subspace 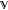 :
:
The equal sign holds true if the trial density is a matrix density of the subspace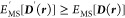
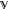 , i.e., D’(r) = D(r). Then, diagonalization of the Hamiltonian
matrix
, i.e., D’(r) = D(r). Then, diagonalization of the Hamiltonian
matrix 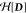 yields exactly all N eigenstate
energies (and densities), including the ground state and N – 1 excited states.
yields exactly all N eigenstate
energies (and densities), including the ground state and N – 1 excited states. -
(3)
The N-dimensional multistate matrix density D(r) can be sufficiently represented by N2 independent Slater determinant wave functions.
It is straightforward to see that the Hohenberg–Kohn
theorem is a special case of Theorem 1 for the ground state, where N = 1. In this case, the HMDF  reduces to a 1 × 1 matrix for one
state, the ground state, becoming a scalar energy density functional
of the ground state density ρ0(r): E0 = E[ρ0(r)]. Then, according to Theorem
3, the density D(r)
≡ ρ0(r) can
be represented by a single Slater determinant, as in the Kohn–Sham
formulation of HK-DFT.
reduces to a 1 × 1 matrix for one
state, the ground state, becoming a scalar energy density functional
of the ground state density ρ0(r): E0 = E[ρ0(r)]. Then, according to Theorem
3, the density D(r)
≡ ρ0(r) can
be represented by a single Slater determinant, as in the Kohn–Sham
formulation of HK-DFT.
A major departure, however, from the HKS-DFT is that state interaction is essential to representing the multistate density and determining the energies of excited states, rather than using a noninteracting KS reference system to represent the electron density of the ground state. Importantly, the implication of Theorem 3 is that for a given number of N states of interest, a finite number of N2 determinant wave functions is sufficient to represent exactly the multistate density D(r). Consequently, it defines an upper bound of basis configurational states, i.e., a minimal active space (MAS), in MSDFT. Obviously, the number of determinants (N2) to exactly represent the multistate density D(r) is significantly smaller than that needed to represent the corresponding wave functions as in a full CI treatment or even in an approximate method employing a complete active space (CAS).
2.3.2. Correlation Matrix Functional
We next introduce the correlation matrix functional, leading to a method for optimizing the multistate matrix density via one-electron orbitals. Given the basis determinants {ΞA} of a MAS, representing exactly the multistate matrix density D(r) for the N states of interest {ΨK; K = 1, ..., N}, the Hamiltonian matrix density functional is written as
| 19 |
where the four terms on the right-hand side of the equation are, respectively, the matrix density functionals for the kinetic, Hartree-exchange, external potential, and correlation energies. The matrix elements for the first three terms in eq 19 are determined from the corresponding Slater determinants,
The correlation matrix functional 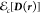 is formally defined as follows according
to Theorem 1,61
is formally defined as follows according
to Theorem 1,61
| 20 |
Note that for the ground state, i.e., N = 1, eq 20 is equivalent to the exchange-correlation functional in KS-DFT,
However, a difference from KS-DFT is that 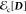 defines the part of correlation energy
not explicitly included in the minimal active space used to represent D(r).63
defines the part of correlation energy
not explicitly included in the minimal active space used to represent D(r).63
2.3.3. Nonorthogonal State Interaction
The adiabatic ground and intermediate states in the present MS-EDA analysis are written as linear combinations of determinant basis states {ΞA} that form a minimal active space (MAS), consisting of the Kohn–Sham reference and singly excited configurations,
| 21 |
where Ξ(X•Y)KS is the reference BLKS determinant for the block-localized complex
(eq 5), Ξia is a singly excited configuration, and {c} are configuration coefficients determined by NOSI by diagonalizing
the Hamiltonian matrix functional  . Although orbitals and {c} can be optimized simultaneously, in NOSI,63 the determinant states Ξ(X•Y) and
in eq 21 are variationally
optimized first using the block-localized excitation (BLE) method.91,92,98 In particular, each constrained
BLKS-determinant wave function is written
. Although orbitals and {c} can be optimized simultaneously, in NOSI,63 the determinant states Ξ(X•Y) and
in eq 21 are variationally
optimized first using the block-localized excitation (BLE) method.91,92,98 In particular, each constrained
BLKS-determinant wave function is written
| 22 |
where χjU (j=1, ⋯, NU) is the jth block-localized spinorbital that is expanded over the basis functions located on atoms of monomer U (U = X, Y), NU is the number of electrons, and χa indicates that the ith occupied spinorbital of monomer X in the BLKS reference Ξ(X•Y)KS is replaced by the ath virtual orbital. In MSDFT, we include all spin-complement configurations in the MAS, all of which are together labeled by a single index {ΞA}.
Subsequently, the matrix elements of the
Hamiltonian matrix functional  in the basis of {ΞA} are determined by
in the basis of {ΞA} are determined by
| 23 |
where the first term can be evaluated by a
standard method for nonorthogonal matrix elements.102−104 For the diagonal elements  of the correlation matrix functional, the
approximate density functional energy developed for KS-DFT is used,
of the correlation matrix functional, the
approximate density functional energy developed for KS-DFT is used,
| 24 |
keeping in mind that ρA(r) is obtained from a block-localized determinant ΞA with non-aufbau occupations for locally excited state.
The off-diagonal element 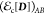 called transition density functional (TDF)100 is new, and does not exist in KS-DFT; currently,
an explicit approximate TDF has not yet been developed. Thus, further
approximations are needed. For spin coupling interactions between
determinants that yield a pair of singlet and triplet (Ms = 0) states, the value of the TDF energy can be obtained
consistently by enforcing the energy degeneracy among the components
of a triplet state,105−108 corresponding to a KS density functional used for the diagonal terms:
called transition density functional (TDF)100 is new, and does not exist in KS-DFT; currently,
an explicit approximate TDF has not yet been developed. Thus, further
approximations are needed. For spin coupling interactions between
determinants that yield a pair of singlet and triplet (Ms = 0) states, the value of the TDF energy can be obtained
consistently by enforcing the energy degeneracy among the components
of a triplet state,105−108 corresponding to a KS density functional used for the diagonal terms:
| 25 |
where ExcKS[ρAB(r)] is the “Kohn–Sham” correlation energy with the electron density ρAB↑↑(r) of the spin-up triplet state ΞAB, and ExcKS[ρA(r)] is the KS correlation energy using ρA↑↓(r) (equivalently with ρB) corresponding to the spin-mixed determinant state. The superscript sp in eq 25 indicates spin-pairing interactions. The subscript AB in ρAB↑↑(r) is used to indicate that the determinant ΞAB refers to the triplet state with respect to the two spin complement determinants ΞA↑↓ and ΞB. Applying the TDF term in eq 25 in NOSI yields a pair of singlet and triplet states (Ms = 0), thereby, the singlet–triplet energy gap without spin contamination. For all other situations, we use the overlap-weighted average correlation energy of the two interacting states to approximate their TDF:64
| 26 |
We note that the use of a standard exchange-correlation functional or a hybrid density functional for the diagonal terms typically yields more accurate results than employing a correlation-only functional. In this case, the corresponding exchange term is reciprocally removed in the first term of eq 23. In other words, standard KS-DFT energies are used for the diagonal terms except that the determinant is block-localized nonaufbau occupation constrained.
3. Computational Details
Monomer and exciplex structures have been optimized using KS-DFT or TDDFT with the Minnesota M06-2X density functional109 and the cc-pVTZ and cc-pVDZ basis sets.110 The Gaussian16 program is used in these calculations.111 For the present multistate energy decomposition analysis, the MSDFT-NOSI method has been implemented into the Qbics program developed in our laboratories112 and a separate program for BLW-ED analysis interfaced with the GAMESS-US program for electronic integrals.113 In NOSI calculations, we used a fine grid consisting of 96 radial shells and 302 angular points. In addition, EOM-CCSD, FIC-NEVPT2, and MRCI were performed with two basis sets for comparison in the Supporting Information. Throughout this Perspective, energies are given in electron volts (eV), widely used in spectroscopy of excited states.
4. Results and Discussion
Based on the nature of stabilizing forces in exciplex formation, we roughly group the excited-state complexes into three categories: (1) encounter excited-state complex, (2) charge-transfer exciplex, and (3) intimate excimer or exciplex. Excited-state complexes in Category 1 originate dominantly from the same type of intermolecular forces as that in the ground state, in which one molecule is in an excited state and the geometry of the other molecule in the complex remains in the ground state with only a minor geometrical deformation induced by (nonphotochemical) intermolecular interactions. The excimer complex between two acetone molecules and the exciplex between acetone and methyl vinyl ether belong to this category, in which one acetone molecule is excited to its n → π* state that is loosely bound to the second molecule. In Category 2, interfragment charge-transfer excitation has a lower energy than or similar to that of valence excitations of either species in a bimolecular complex. The driving force for exciplex formation is electrostatic Coulomb interaction. Examples include complexes involving the strong electron acceptor tetracyanoethylene (TCNE). Category 3 complexes are produced as a result of the resonant excitation of all individual species in the complex. The pentacene excimer complex in the S1 state is a vivid illustration.
We first discuss the performance of nonorthogonal state interaction (NOSI) method of MSDFT by comparison with results from TDDFT calculations employing the same approximate density functional (M06-2X). Then, we present findings from MS-EDA analysis of the three categories of excited-state complexes.
4.1. Excitation Energies and Validation of Method
The performance of the MSDFT-NOSI method along with a small MAS is validated on the computed excitation energies (Figure 2). Since intermediate reference states in the EDA procedure are electronic diabatic states, which are not direct observable experimentally, we focus on comparison of the excitation energies of the monomer compounds. Table 1 lists the total energies of the ground state and the vertical excitation energies of the first two singlet excited states from MSDFT-NOSI calculations for the monomer compounds considered in this work and related molecules. In addition, included in Table 1 are the emission energies using the optimized structures of acetone and pentacene in the first excited states. The NOSI results are compared with TDDFT values along with experimental data. For these simple organic chromophores, TDDFT is known to perform well in benchmark studies,114 providing a good reference for comparison with the NOSI method employing the same approximate density functional and basis set. Table 1 shows that the agreement between results from the two computational methods is excellent with a root-mean-square deviation (RMSD) of 0.11 eV for the S1 states, 0.25 eV for the S2 states, and 0.19 eV for all vertical excitation energies. A further indicator is the mean-signed-average (MSA) and mean-unsigned-error (MUE), which are −0.06 and 0.14 eV, exhibiting no systematic errors for the singlet excited states. We anticipate that this trend will be carried over to the energies of intermediate states in MS-EDA.
Figure 2.
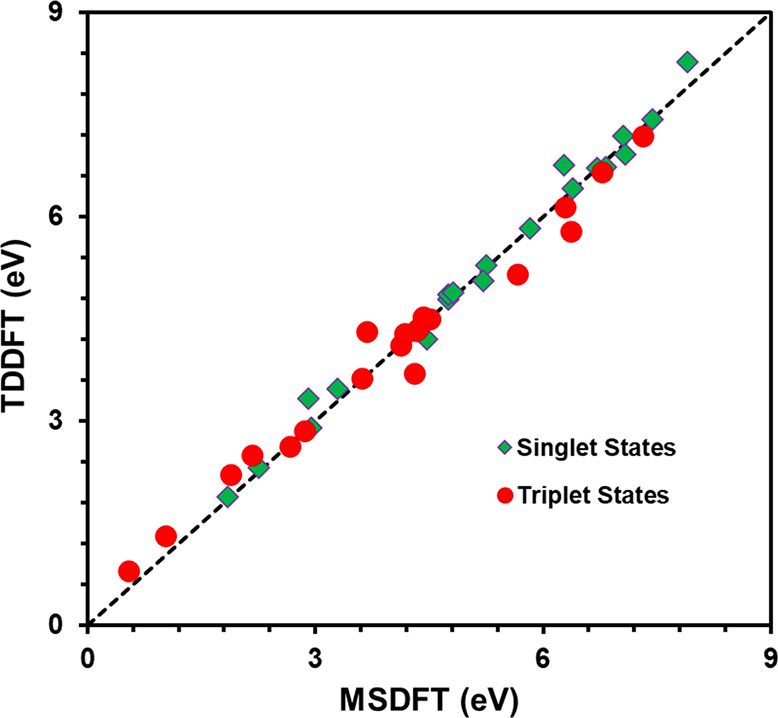
Comparison of vertical excitation energies obtained using multistate density functional theory (MSDFT) and time-dependent density functional theory (TDDFT). The Minnesota M06-2X functional is used.
Table 1. Computed Total Energies (au) for the Ground State and Excitation Energies (eV)a.
|
S0 (au) |
S1 |
S1 |
||||||
|---|---|---|---|---|---|---|---|---|
| moleculeb | MSDFT | ΔEKS | MSDFT | TDDFT | expc | MSDFT | TDDFT | expc |
| naphthalene | –385.76533 | 0.00000 | 4.75 | 4.78 | 4.7 | 4.81 | 4.89 | 3.97 |
| pentacene | –846.54223 | 0.00000 | 2.25 | 2.32 | 2.3 | 3.29 | 3.47 | |
| PCA | –845.54485 | –0.00007 | 5.25 | 5.29 | 5.08 | 5.83 | 5.83 | 6.41 |
| TCNE | –447.41678 | 0.00000 | 4.75 | 4.85 | 4.75, 4.84 | 5.21 | 5.06 | 5.67 |
| t-DME | –307.51100 | –0.00015 | 6.71 | 6.71 | 6.2 | 7.08 | 6.91 | |
| c-DME | –307.50997 | –0.00028 | 6.39 | 6.42 | 6.16 | 6.82 | 6.73 | |
| MVE | –193.03249 | –0.00043 | 7.06 | 7.19 | 6.48 | 7.44 | 7.42 | 9.6 |
| Me2C=O | –193.07551 | –0.00055 | 4.47 | 4.20 | 4.38 | 7.90 | 8.27 | 6.35 |
| Me2C=O (S1) | –193.04394 | –0.00197 | 2.94 | 2.90 | 3.06 | 6.27 | 6.75 | |
| pentacene (S1) | –846.53459 | 0.00000 | 1.85 | 1.89 | 2.91 | 3.33 | ||
The Minnesota M06-2X density functional is used both in NOSI and TDDFT calculations. Relative energy given in electron volts (eV) between results from MSDFT-NOSI and KS-DFT, ΔEKS = ENOSI – EKSDFT. This is a reasonable indicator of possible double counting of electron correlation for single-reference molecules.
PCA: para-chloromethyl anisole, TCNE: tetracyanoethylene, MVE: methyl vinyl ether, DME: 1,2-dimethoxyethene.
Another important finding is revealed in Table 1 by comparing the
total ground-state energies
from NOSI with those by KS-DFT calculations (Figure 2). The former method is a multistate, multiconfigurational
DFT in which state interaction is key in determining
both the ground state and excited state energies, whereas the latter
employs a noninteracting system consisting of a single determinant
wave function purely for representing the ground-state density. One
lingering question in multiconfigurational DFT (MC-DFT) has been double
counting of electron correlation since KS-DFT, in principle, includes
all electron correlation, which is added to an MCSCF energy.100,121−123 MSDFT differs from other MC-DFT models in
that the total electron density from a MCSCF wave function is not
directly used in KS-DFT correlation energy evaluations. Rather, the
MSDFT correlation functional is an implicit functional of the multistate
density and the correlation functional developed for KS-DFT is used
solely to approximate the correlation energy of a determinant state
(diagonal terms) of the Hamiltonian matrix density functional  . Consequently, we find that the difference
in the ground-state energy between MSDFT and KS-DFT is negligibly
small, even though the KS-DFT state is included as one member of the
MAS in all NOSI calculations. Acetone exhibits a small amount of multistate
stabilization of 0.55 meV (0.01 kcal/mol) in the ground-state geometry
and ca. 2 meV (0.05 kcal/mol) using the S1 state geometry, while multistate effects as compared to KS-DFT are
generally less than 0.5 meV for other compounds. The data in Table 1 demonstrate that
there is little double-counting of correlation in these systems when
an approximate density functional developed for KS-DFT is directly
used for the determinant states in MSDFT.
. Consequently, we find that the difference
in the ground-state energy between MSDFT and KS-DFT is negligibly
small, even though the KS-DFT state is included as one member of the
MAS in all NOSI calculations. Acetone exhibits a small amount of multistate
stabilization of 0.55 meV (0.01 kcal/mol) in the ground-state geometry
and ca. 2 meV (0.05 kcal/mol) using the S1 state geometry, while multistate effects as compared to KS-DFT are
generally less than 0.5 meV for other compounds. The data in Table 1 demonstrate that
there is little double-counting of correlation in these systems when
an approximate density functional developed for KS-DFT is directly
used for the determinant states in MSDFT.
The computed triplet state energies are given in Table 2. For the lowest triplet T1 state, KS-DFT is typically sufficient, whereas the T2 states are obtained using TDDFT based on this reference. Table 2 shows that the accord between NOSI and TDDFT (KS-DFT) results is also reasonably good. In particular, the RMSD errors are 0.25 eV for T1 states, 0.35 eV for T2 states, and 0.31 eV overall. Further, the MSE and MUE are, respectively, 0.00 and 0.20 eV for all triplet states. However, the NOSI T1 energies of pentacene (both in the S0 and S1 structures) and para-chloromethyl anisole (PCA) have somewhat greater deviations from KS-DFT values than other cases. We attribute the increased deviation to multiconfiguration characteristics in the T1 triplet states for these two aromatic compounds, which is confirmed by the Chirgwin–Coulson structure weights, having values of 0.92 and 0.94 for the dominant configuration in the two (S0 and S1) pentacene structures, and a value of only 0.65 for the dominant configuration in PCA. Thus, the use of a single determinant in KS-DFT for the T1 state may be questionable for these compounds and could indeed have large errors. In this regard, the results from MSDFT-NOSI may be more reliable. This is reflected in the computed T1 energies, where NOSI yields a lower value than KS-DFT.
Table 2. Computed Total Triplet-State Energies (au) and Excitation Energies (eV) for the First Two Triplet States Relative to the Singlet Ground Statea.
|
T1(X) |
T1 |
T2 |
||||||
|---|---|---|---|---|---|---|---|---|
| MSDFT | ΔEKS | MSDFT | TDDFT | expb | MSDFT | TDDFT | exp | |
| naphthalene | –385.63259 | –0.00050 | 3.61 | 3.63 | 3.0 | 4.18 | 4.28 | 3.9 |
| pentacene | –846.50452 | –0.01088 | 1.03 | 1.32 | 0.86 | 2.17 | 2.50 | >2.0 |
| PCAb | –845.40970 | –0.03489 | 3.68 | 4.32 | 4.42 | 4.53 | ||
| TCNE | –447.31172 | 0.00000 | 2.86 | 2.86 | 5.66 | 5.16 | ||
| t-DME | –307.35069 | –0.00004 | 4.36 | 4.36 | 6.77 | 6.65 | ||
| c-DME | –307.35079 | –0.00026 | 4.33 | 4.33 | 6.29 | 6.14 | ||
| MVE | –192.86691 | 0.00000 | 4.51 | 4.50 | 4.2 | 7.31 | 7.19 | 8.8 |
| Me2C=O | –192.92376 | 0.00000 | 4.13 | 4.11 | 4.18 | 6.36 | 5.78 | 5.88 |
| Me2C=O (S1) | –192.94603 | –0.00001 | 2.66 | 2.63 | 4.30 | 3.70 | ||
| pentacene (S1) | –846.51462 | –0.00923 | 0.54 | 0.80 | 1.88 | 2.22 | ||
4.2. Encounter Excited State Complexes
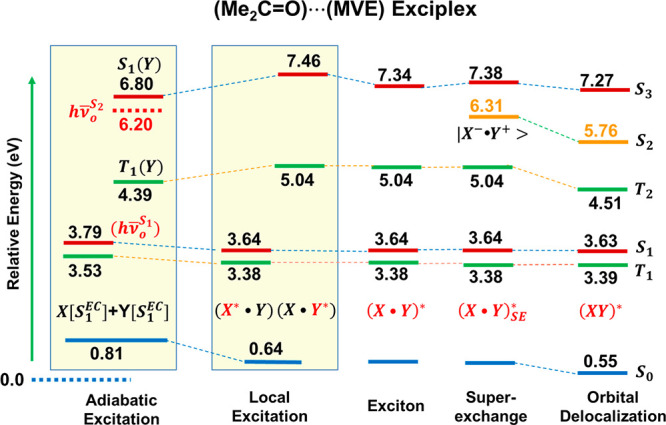
The optimized structures (Figure 3) of the excimer complex of two acetone molecules and the exciplex between acetone and methyl vinyl ether in their first excited state using TDDFT/M06-2X/cc-pVTZ reveal that they are characterized as a single acetone excitation to its n → π* state with a bent geometry interacting with its binding partner in the ground state. This is clearly reflected by the geometrical distortion energies of 0.75 and 0.82 eV for the photochemically excited acetone in the two complexes (Table 3), respectively, corresponding to the change from the ground state in equilibrium to that found in the excited-state complexes. For the other partners in the complexes the energy changes are negligible (the small negative values are probably due to multistate effects). Table 3 lists the adiabatic and intermediate excited-state energies of the monomer and dimer species, all of which are given relative to that at the equilibrium ground state for direct comparison (Scheme 1). Therefore, the adiabatic excitation energies of acetone are about 3.8 eV in both complexes, significantly smaller than the vertical excitation energy of 4.47 eV (Table 1), but the vertical emission (hνad−ΔEdist) energies of 3.05 and 2.97 eV of the excited acetone in the exciplex geometries in the two complexes are similar to that (2.94 eV) of the optimized S1 structure of acetone (Table 1) from MSDFT-NOSI calculations. On the other hand, the “adiabatic” excitation energies of the second monomers in the two exciplexes are, respectively, 4.39 and 6.80 eV, close to the vertical excitation energies of the isolated monomers (4.47 and 7.06 eV). Thus, these two exciplexes are best described as dipolar van der Waals complexes.
Figure 3.

Optimized structures in the first singlet excited states of the acetone excimer and acetone···methyl vinyl ether exciplex.
Table 3. Excitation Energies of Adiabatic, Diabatic, and Intermediate States in the Lowest Singlet and Triplet Excited States Associated with the Acetone Dimeric and Acetone-Methyl Vinyl Ether (MVE) Exciplex Complexesa.
| (X)···(Y) |
(Me2C=O)···(Me2C=O) |
(Me2C=O)···(MVE) |
|||
|---|---|---|---|---|---|
| energy terms | singlet | triplet | singlet | triplet | |
| hνad | X*[EC] | 3.80 | 3.53 | 3.79 | 3.53 |
| Y*[EC] | 4.39 | 4.08 | 6.80 | 4.39 | |
| ΔEdist | X[EC] | 0.75 | 0.75 | 0.82 | 0.82 |
| Y[EC] | –0.02 | –0.02 | –0.01 | –0.01 | |
| ELex | (X*•Y) | 3.58 | 3.31 | 3.64 | 3.38 |
| (X•Y*) | 4.97 | 4.67 | 7.46 | 5.04 | |
| EEx | (X•Y)+* | 3.58 | 3.31 | 3.64 | 3.38 |
| (X•Y)−* | 4.97 | 4.67 | 7.34 | 5.04 | |
| ESE | (X•Y)+SE | 3.52 | 3.26 | 3.64 | 3.38 |
| (X•Y)−SE | 4.85 | 4.55 | 6.31 (CT),b 7.38 | 5.04 | |
| E(XY) | (XY)1* | 3.53 | 3.22 | 3.63 | 3.39 |
| (XY)2* | 4.66 | 4.46 | 5.76 (CT),b 7.27 | 4.51 | |
All energies are given in electron volts (eV), relative to the ground state energies of a monomer, X[S0] and Y[S0], or two separate molecules at their optimized structures, i.e., adiabatic ground-state energies are set equal to zero. Square brackets indicate optimized geometry used in energy calculations with S0 for the ground state and EC as exciplex complex. All calculations for bimolecular complexes adopt the [EC] structure. The M06-2X density functional and the cc-pVTZ basis set are used in NOSI calculations. The energies terms may be compared with the illustration of Scheme 1.
Charge transfer (CT) state.
The triplet state energies are also shown in Table 3, exhibiting similar trends, but they are obtained using the same monomer and excimer geometries optimized for the singlet states. Thus, they are not further discussed nor listed for these complexes or any of the other systems below.
The local excitation energies of the excited acetone and its binding partner in both complexes exhibit different trends (Tables 1 and 2), with a large Stokes shift of 0.84 eV (3.80 eV excitation and 2.96 eV emission) in the acetone excimer and 0.79 eV (3.79 eV excitation and 3.00 eV emission) in the acetone-MVE exciplex for the excited acetone molecule, but with essentially no spectral change for the second monomer in the complex as it largely remains in the ground-state geometry. A relevant quantity is the exciton polarization energy, ΔELpolK, corresponding to the vertical excitation energy difference of the chromophore (acetone) in the same geometry with and without the presence of the second binding partner molecule, which is −0.09 eV (2.96 eV–3.05 eV) for the acetone excimer, and +0.03 eV (3.00 eV–2.97 eV) in the acetone-MVE exciplex. The polarization effects are small for the ground-state monomers, with energy changes of +0.06 and +0.01 eV in the two complexes, respectively. Note that the seemingly positive energy changes are compensated for by the binding energy of the isolated, distorted monomers to form the local complexes (−0.11 and −0.17 eV, respectively). The excited-state energies and relative energies in Table 4 are displayed in Figure 4 for the acetone-MVE exciplex, corresponding to the energy terms defined in Scheme 2, whereas results for the acetone excimer can be found in ref (5). Fluorescence quenching by alkyl vinyl ethers has been reported by Schore and Turro who attributed the observed quenching rates to the formation of acetone···ether exciplexes with charge transfer playing a role.129 A correlation to the observed spectral shift of tetracyanoethylene···alkyl vinyl ethers discussed next was found.
Table 4. Relative Energies (eV) in Multistate Energy Decomposition Analysis of the Lowest Singlet and Triplet Excited States in the Acetone Excimer and Acetone-Methyl Vinyl Ether (MVE) Exciplexa.
| (X)···(Y) | (Me2C=O)···(Me2C=O) |
(Me2C=O)···(MVE) |
||||||
|---|---|---|---|---|---|---|---|---|
| energy terms | S1 | S2 | T1 | T2 | S1 | S2 | T1 | T2 |
| hν̅oK | 3.80 | 4.39 | 3.53 | 4.08 | 3.79 | 6.20 | 3.53 | 4.39 |
| ΔErefK | 2.96 | 4.35 | 2.69 | 4.05 | 3.00 | 6.70 | 2.74 | 4.40 |
| ΔELint | 0.62 | 0.62 | 0.62 | 0.62 | 0.64 | 0.64 | 0.64 | 0.64 |
| ΔEExK | 2.96 | 4.35 | 2.69 | 4.05 | 3.00 | 6.70 | 2.74 | 4.40 |
| ΔΔEResK | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| ΔΔESEK | –0.06 | –0.12 | –0.05 | –0.12 | 0.00 | –1.03,b 0.04 | 0.00 | 0.00 |
| ΔΔEOCDK | 0.01 | –0.19 | –0.04 | –0.09 | –0.01 | –0.55,b −0.11 | 0.01 | –0.53 |
| ΔΔELCTS0 | –0.07 | –0.07 | –0.07 | –0.07 | –0.09 | –0.09 | –0.09 | –0.09 |
| ΔEb | –0.27 | 0.27 | –0.41 | 0.38 | –0.16 | –0.44,b 1.07 | –0.14 | 0.12 |
Figure 4.
Ground and excited-state energies (eV) for the first singlet exciplex of acetone and methyl vinyl ether in multistate energy decomposition analysis. The energies of adiabatic ground state of isolated monomers in equilibrium geometries have been set equal to zero. The effective adiabatic excitation energies of the separate monomers in the exciplex geometries are given in red. Singlet (maroon) and triplet (green) states are labeled on the far right for exciplex complex. Energy levels are not drawn in scale.
The remainder of the energy terms for the two loosely associated excimer complexes are largely unremarkable except the second excited state of the acetone-MVE exciplex (recall that geometry is optimized for the S1 state), where a charge transfer state emerges to be lower than the covalent excitation of MVE (state in brown in the superexchange intermediate in Figure 4). Thus, relative to the S2 exciton state, corresponding to the first local excited state of MVE in the dimer complex, the superexchange stabilization is −1.03 eV, which is further lowered by −0.55 eV by orbital and configuration-state delocalization (OCD term). Therefore, the net binding energy of the S2 exciplex state (S1 state geometry) is −0.44 eV, relative to MVE reference valence excitation (hν̅oS2). For the S1 exciplexes of acetone dimer and acetone-MVE, the net binding energies (eq 4) are −0.27 and −0.33 eV, with a net ground-state contribution of 0.55 eV in both systems to overcome the large structural distortion energies.
4.3. Charge-Transfer Complexes
Tetracyanoethylene (TCNE) can form rather stable complexes (Figure 5) with methyl vinyl ether (MVE) and naphthalene (Naph) in the ground state with binding energies of −0.51 eV and −0.28 eV (−11.8 and −6.5 kcal/mol), respectively (Table 5). However, at the S1 exciplex geometries, the binding energies in the ground state (ΔELb) are repulsive (+0.41 eV) in the MVE structure and barely associative (−0.03 eV) for the naphthalene complex, indicating significant monomer structure distortion in the exciplex. In both exciplexes, the lowest excited state is of charge transfer character from the electron rich monomers to TCNE. In fact, the three lowest excited states in the TCNE-Naph exciplex are CT states, corresponding to the transfer of one electron from the HOMO, HOMO–1, and HOMO–2 orbitals of naphthalene to the LUMO of TCNE (Table 5 and Figure 6), in both the optimized complex structures of the ground state (S0) and the first excited state (S1). In the exciplex complex, the electron acceptor TCNE is noticeably distorted with a bent butterfly like geometry, having a geometrical distortion energy ΔEdist of 0.17 eV in the MVE complex and 0.22 eV in the naphthalene state. Interestingly, covalent vertical excitation energies for both monomers in the two exciplex complexes are not significantly different in the S0 and S1 complex structures. Exciton delocalization only lowers the excited-state energies modestly by about 0.1 eV in the TCNE-MVE exciton state, and somewhat greater at 0.25–0.40 eV for the TCNE-Naph complex.
Figure 5.

Optimized bimolecular complex structures (side view on top, and top or tilt views in the bottom) for tetracyanoethylene (TCNE)···methyl vinyl ether, TCNE···naphthalene, and pentacene dimer in the optimized ground state (XY)S0 (crystal structure for pentacene) and first singlet excited state (XY)S1. M06-2X density functional and the cc-pVDZ basis set are used.
Table 5. Relative energies (eV) of Adiabatic, Diabatic, and Intermediate States in the Lowest Singlet Excited States of Tetracyanoethylene (TCNE)···Methyl Vinyl Ether and TCNE···Naphthalene Exciplexa.
| (X)···(Y) |
(TCNE)···(MeOCH=CH2) |
(TCNE)···(naphthalene)b |
|||
|---|---|---|---|---|---|
| energy terms | [S0] geom | [S1] geom | [S0] geom | [S1] geom | |
| hνabad | X*[EC] | 4.68 | 4.41 | 4.58 | 4.44 |
| Y*[EC] | 6.81 | 6.49 | 4.61, 4.69 | 4.65, 4.50 | |
| ΔEdist | X[EC] | 0.00 | 0.17 | 0.02 | 0.22 |
| Y[EC] | –0.02 | –0.03 | –0.02 | 0.12 | |
| ELex | (X*•Y) | 4.55 | 4.67 | 4.35 | 4.27 |
| (X•Y*) | 6.60 | 7.04 | 4.40, 4.48 | 4.56, 4.42 | |
| EEx | (X•Y)+* | 4.46 | 4.58 | 4.10 | 3.87 |
| (X•Y)-* | 6.69 | 7.10 | 4.40, 4.69 | 4.56, 4.76 | |
| ESE | (X•Y)+SE | 3.04 (CT), 4.42 | 1.90 (CT), 4.69 | 2.42 (CT)c | 1.77 (CT)c |
| 3.16 (CT),c 4.12 | 2.81 (CT),c 3.98 | ||||
| 4.53 (CT)c | 4.13 (CT)c | ||||
| (X•Y)-SE | 6.65 | 7.10 | 4.41, 4.69 | 4.61, 4.81 | |
| E(XY) | (XY)1* | 2.62 (CT), 4.29 | 1.19 (CT), 4.63 | 1.67 (CT)c | 1.15 (CT)c |
| 2.46 (CT)c | 2.29 (CT)c | ||||
| 3.77 (CT)c | 3.45 (CT)c | ||||
| 4.32 | 4.12 | ||||
| (XY)2* | NAd | NAd | 4.40, 4.51 | 4.38, 4.53 | |
All energies are given relative to the adiabatic ground state energies of monomers. The M06-2X density functional and the cc-pVDZ basis set are used. The energy terms may be compared with the illustration of Scheme 1.
Two close-lying states are given for naphthalene with Lb first followed by La.
Charge transfer (CT) states, respectively from HOMO, HOMO–1, and HOMO–2 of naphthalene to the LUMO of TCNE.
Not assigned.
Figure 6.

Ground (blue) and excited-state (maroon) energies (eV) for the singlet exciplex of tetracyanoethylene (TCNE) and naphthalene in multistate energy decomposition analysis. The S0 complex geometry is used. The energies are given relative to that of adiabatic ground state of isolated monomers in equilibrium geometry. Three charge transfer states (in brown) appear at the superexchange stage of MS-EDA. The effective adiabatic excitation energies of the separate monomers in the exciplex geometries are 4.62, 4.61, and 4.65 eV for the first, second, and third exciton states, originating dominantly from the π → π* state of TCNE, and the Lb and La states of naphthalene. Energies corresponds to excitation and resonance terms defined in Scheme 2.
Not surprisingly, the greatest energy contribution in the MS-EDA analysis come from charge transfer (CT) effects in the direction from MVE or Naph to TCNE. For the TCNE-MVE complex, the charge-transfer superexchange stabilization is estimated to be 1.42 eV using the ground-state structure of the bimolecular complex, and 2.68 eV in the S1 exciplex geometry. Similarly, for the TCNE-Naph exciplex, we found a CT stabilization of 1.68 eV in the S0 complex geometry (Figure 6), and 2.10 eV in the S1 exciplex structure. Orbital delocalization also significantly lowers the excitation energies of the exciplexes, contributing −0.42 and −0.71 eV in the two structures of TCNE-MVE exciplex, and −0.75 and −0.62 eV for the TCNE-Naph complexes. However, the charge delocalization effects from TDDFT calculations are likely overestimated due to delocalization errors on charge-transfer excitations.130
For a CT exciplex, its binding energy is not directly related to covalent excitation of either monomer. Nevertheless, we can still consider the CT complex formation through an initial formation of the exciton complex, followed by an interfragment charge transfer as in the MS-EDA steps. Then, the definition of binding energy of eq 3 can still be used. Thus, for TCNE···MVE, the binding energies at geometries of the ground-state complex and the optimized S1 exciplex are −2.15 and −3.28 eV, and for TCNE···Naph, they are −2.94 and −3.30 eV, respectively.
On the other hand, if one considers the starting reference state as the equilibrium geometry of the bimolecular complex in the ground state, the exciplex binding energy may be defined by the following photochemical process:
| 27 |
Equation 27 shows that for CT exciplex, the binding energy of the exciplex may be assigned to the energy change from the vertical charge-transfer absorption complex in the equilibrium ground-state geometry to the equilibrium exciplex geometry. From the data in Table 6, we immediately obtain a ΔEbCT of −1.13 eV in the TCNE···MVE exciplex and −0.36 eV in TCNE···Naph, as a result of structural relaxation induced by CT excitation. Then, the energy components will be the term-by-term difference in Table 5 obtained between the optimized geometries in the ground state (XY)[S0] and the excited state of the exciplex (XY)[S1]. The details of these energy differences are not further discussed here.
Table 6. Relative Energies (eV) in Multistate Energy Decomposition Analysis of the Lowest Singlet States in Tetracyanoethylene (TCNE)···Methyl Vinyl Ether and TCNE···Naphthalene Exciplexa.
| (TCNE)···(MeOCH=CH2) |
(TCNE)···(naphthalene) |
|||||||
|---|---|---|---|---|---|---|---|---|
| [S0] geom |
[S1] geom |
[S0] geom |
[S1] geom |
|||||
| energy | S1 | S2 | S1 | S2 | S1 | Lb/La | S1 | La/Lb |
| hνoK | 4.77 | 6.72 | 4.47 | 6.37 | 4.62 | 4.61/4.65 | 4.45 | 4.47/4.65 |
| ΔELint | –0.23 | –0.23 | 0.54 | 0.54 | –0.22 | –0.22 | 0.08 | 0.08 |
| ΔErefK | 4.86 | 6.75 | 4.18 | 6.46 | 4.62 | 4.62/4.65 | 4.24 | 4.27/4.48 |
| ΔEExK | 4.69 | 6.92 | 4.04 | 6.66 | 4.33 | 4.62/4.91 | 3.79 | 4.48/4.68 |
| ΔΔEResK | –0.17 | 0.17 | –0.18 | 0.20 | –0.29 | 0.00/0.26 | –0.45 | 0.21/0.20 |
| ΔΔESEK | (−1.42) | –0.04 | (−2.68) | 0.11 | (−1.68) | 0.01 | (−2.10) | 0.05 |
| (−0.94) | 0.00 | (−1.06) | 0.05 | |||||
| (0.43) | (0.26) | |||||||
| 0.02 | 0.11 | |||||||
| ΔΔEOCDK | (−0.42) | –0.13 | (−0.71) | –0.06 | (−0.75) | –0.02 | (−0.62) | –0.28 |
| (−0.70) | –0.18 | (−0.52) | –0.23 | |||||
| (−0.76) | (−0.68) | |||||||
| 0.20 | 0.14 | |||||||
| ΔELCTS0 | –0.28 | –0.28 | –0.13 | –0.13 | –0.05 | –0.05 | –0.11 | –0.11 |
| ΔELbS0 | –0.51 | 0.41 | –0.27 | –0.27 | –0.03 | –0.03 | ||
| ΔEbK | (−2.15) | –3.28 | (−2.94)b | –0.22 | (−3.30)b | –0.14 | ||
| covalent | –0.29 | –0.14 | –0.33 | –0.07 | ||||
Experimentally, a charge-transfer absorption spectrum for the TCNE···MVE complex has been reported with a maximum absorption wavelength of 401 nm (3.09 eV) in 90% cyclohexane and 10% CH2Cl2 solution.131 With the ground-state bimolecular complex structure, the computed values are 3.27 eV (3.04 – (−0.23)) at the superexchange stage using NOSI and 3.13 (2.62 – (−0.51)) eV, without considering solvent effects. The value for the exciplex geometry appears too low. In comparison with previous calculations using an optimized range-separated functional132,133 and experimental data,130 the excitation energies of the TCNE···Naph complex for the first two exciplex states (2.64 and 3.38 eV) using the S0 structure from the present MS-EDA using NOSI at the superexchange stage are in good accord with experimental results (2.60 and 3.23 eV for S1 and S2)130 and with specifically optimized range-separated TDDFT calculations (2.7 eV for S1)132 and the CAM-QTP-02 functional (2.80 and 3.57 eV).133
4.4. Intimate Excimers
For the third category of excited-state complexes, in which the electronic excitation is fully delocalized over the monomers in the exciplex, we illustrate the energy components of the anthracene dimer system in two geometry arrangements: the optimized S1 excimer, and a fishbone configuration corresponding to that found in a crystal structure of anthracene Table 7. This system has been described in a preliminary report on the MS-EDA methodology, although a different reference state was used in binding energy calculations. Here, we list the binding energy relative to the reference energy for the unbound species defined in eq 3 and the locally excited reference state for exciton resonance energies in Table 8. We highlight the key contrasting features of the intimate excimer in comparison with conformation dependence of the anthracene dimer itself, and the other two categories of exciplexes. An energy correlation diagram for the excimer complex of pentacene dimer can be found in reference.5
Table 7. Computed Excitation Energies (eV) and Energy Components from Multistate Energy Decomposition Analysis (MS-EDA) for Pentacene Excimer and a Pentacene Dimer Fishbone Configurationa.
| excimer |
fishboneb |
|||
|---|---|---|---|---|
| energy term | Lb | Lb | La | Lb |
| hν̅0K | 2.10 | 3.14 | 2.21 | 3.24 |
| ΔEdist | 0.18 | 0.18 | 0.03 | 0.03 |
| ELex[(X*•Y)K] | 1.89 | 3.04 | 1.99/1.98 | 2.92/2.92 |
| EEx[(X•Y)K–] | 1.46 | 2.92 | 1.97 | 2.92 |
| EEx[(X•Y)K+] | 2.40 | 3.03 | 2.02 | 2.93 |
| ESE[(X•Y)CTK–] | 0.62 | 2.70 | 1.88 | 2.92 |
| ESE[(X•Y)CTK+] | 2.30 (CT), 2.47 | 3.38 | 1.97, 2.23 (CT) | 2.93 |
| E(XY)K– | 0.94 | 2.49 | 1.71 | 3.07 |
| E(XY)K+ | 2.11 | 2.87 | 1.92 | 3.10 |
All excitation energies are given relative to that of the adiabatic ground state of isolated pentacene. The M06-2X density functional is used along with the cc-pVDZ basis set. Adapted from ref (5). Copyright 2023 American Chemical Society.
The HOMO–1 → LUMO+1 configuration was not included.
Table 8. Relative Energies (eV) in Multistate Energy Decomposition Analysis of Singlet States of Pentacene Excimer and Pentacene Dimer in a Fishbone Configurationa.
| excimer |
fishbone |
|||||||
|---|---|---|---|---|---|---|---|---|
| energy | La– | La+ | Lb– | Lb+ | La– | La+ | Lb– | Lb+ |
| hν̅oK | 2.10 | 2.10 | 3.14 | 3.14 | 2.21 | 2.21 | 3.24 | 3.24 |
| ΔErefK | 1.89 | 1.89 | 3.04 | 3.04 | 2.32 | 2.31 | 3.25 | 3.25 |
| ΔELint | 0.07 | 0.07 | 0.07 | 0.07 | –0.33 | –0.33 | –0.33 | –0.33 |
| ΔEExK | 1.33 | 2.46 | 2.70 | 3.38 | 2.30 | 2.35 | 3.25 | 3.26 |
| ΔΔEResK | –0.56 | 0.57 | –0.34 | +0.34 | –0.02 | 0.04 | 0.00 | 0.01 |
| ΔΔESEK | –0.84 | (−0.10) 0.07 | –0.22 | 0.35 | –0.09 | –0.05 (0.21) | 0.00 | 0.00 |
| ΔΔEOCDK | 0.32 | –0.19 | –0.21 | –0.51 | –0.17 | –0.05 | 0.05 | 0.07 |
| ΔELCTS0 | –0.13 | –0.13 | –0.13 | –0.13 | –0.14 | –0.14 | –0.14 | –0.14 |
| ΔELbS0 | –0.05 | –0.05 | –0.05 | –0.05 | –0.47 | –0.47 | –0.47 | –0.47 |
| ΔEbK | –1.22 | (0.14) | –0.80 | 0.15 | –0.50 | –0.29 | –0.27 | –0.24 |
As other polyaromatic acenes, anthracene has two close-lying excited singlet states termed as the La(S1) and Lb(S2) states. The dimeric excimer of anthracene molecules was optimized to a symmetric geometry using TDDFT with M06-2X/cc-pVDZ, whereas the two monomers in the fishbone configuration in the crystal structure are not fully equivalent. In the latter case the HOMO–1 to LUMO+1 excited configuration was not fully converged and thus not included in the calculation, which would affect the La energy slightly. In the excimer geometry it has a rather small contribution to the La state using the excimer structure with a structure weight of 0.06 (0.94 for the HOMO → LUMO configuration). The anthracene dimer as a category 3 excimer is distinguished from the other two classes of exciplexes by strong exciton resonance interaction in all excimer states between the two locally excited monomers, resulting in large energy splitting |ΔVAB| in the La and in the Lb state. Table 8 shows that the La± and Lb energy differences are 1.13 and 0.68 eV for the excimer, and that |ΔVAB| is highly sensitive to the distance and relative orientation (overlap) between the monomers, reducing to values of 0.05 and 0.01 eV for the two pairs of states in the fishbone configuration. In the excimer complex, the charge-delocalization state lies between the La- and La states, stabilizing the former by −0.84 eV, but increasing the energy of La+ by 0.07 eV in a three-state interaction scheme. On the other hand, at the fishbone configuration, the charge-delocalization state is higher in energy than the La states, thereby, lowering the energies of both La and La+ states by −0.09 and −0.05 eV. Surprisingly, superexchange stabilization is also highly dependent on the overlap between the two monomer states. Not surprisingly, the excimer complex has a strong binding energy of −1.16 eV, whereas for the fishbone configuration found in pentacene crystal it is only −0.50 eV, significant, but −0.47 eV comes from ground-state effect using the M06-2X density functional (Table 8).
4.5. Structure Weights
We finally comment on the computed structure weights of local states in the exciton and superexchange intermediates. In Table 9, we list the structure weights of three representative complexes, namely, the acetone⋯MVE and TCNE⋯naphthalene exciplex, and the anthracene excimer. In MS-EDA, the structure weights for the exciton states have been used to determine the corresponding weighted adiabatic excitation energy of the isolated molecules (hν̅oK), and the reference energy for the locally excited states (ΔEref) to determine exciton resonance energy. Further examination of the data in Table 9 reveals an interesting trend. For the intimate excimer complexes, strong state interactions are found among the symmetry allowed local states. Even though the exciton resonance stabilization is severely reduced in the tilted pentacene orientation of the fishbone structure, exciton delocalization is observed. However, the tilting effect clearly results in a preference for a directional charge transfer in the superexchange complex, with structural weights of 0.28, 0.02, and 0.70 for the La- exciton state and the |P–•P+⟩ and |P+•P–⟩ charge transfer states, respectively. For comparison, this state (S2) in the symmetric excimer complex has an equal forward and backward charge transfer contributions, resulting in a net zero charge transfer in the complex. The implication of this finding could be significant toward an understanding of the mechanism of directional multiexciton formation in singlet fission in pentacene monolayer materials.50
Table 9. Structure Weights of Local Excited States and Individual Charge Transfer (CT) States in the Exciton and Superexchange Intermediates in Multistate Energy Decomposition Analysis (MS-EDA)a.
| exciton
(X•Y)* |
superexchange
(X•Y)SE |
||||
|---|---|---|---|---|---|
| exciplex state | (X*•Y) | (X•Y*) | (X•Y)* | (X–•Y+) | (X+•Y–) |
| (Me2C=O)···MVE exciplex | |||||
| (n → π*)X | 1.00 | 0.00 | 1.00 | 0.00 | 0.00 |
| CT | 0.09 | 0.91 | 0.00 | ||
| (π → π*)Y | 0.20 | 0.80 | 0.98 | 0.02 | 0.00 |
| TCNE···naphthalene exciplex | |||||
| (CT)H | 0.02 | 0.98 | 0.00 | ||
| (CT)H−1 | 0.11 | 0.89 | 0.00 | ||
| (π → π*)X | 0.61 | 0.39 | 0.92 | 0.08 | 0.00 |
| (CT)H−2 | 0.0 | 1.00 | 0.00 | ||
| (Lb)Y | 0.0 | 1.00 | 0.98 | 0.02 | 0.00 |
| (La)Y | 0.36 | 0.64 | 0.92 | 0.08 | 0.00 |
| (pentacene)2 excimer | |||||
| (La–) | 0.43 | 0.57 | 0.64 | 0.18 | 0.18 |
| (CT) | 0.10 | 0.45 | 0.45 | ||
| (La+) | 0.58 | 0.42 | 0.90 | 0.05 | 0.05 |
| (Lb–) | 0.17 | 0.83 | 1.00 | 0.00 | 0.00 |
| (Lb+) | 0.92 | 0.08 | 1.00 | 0.00 | 0.00 |
| (pentacene)2 fishbone configuration | |||||
| (La–) | 0.60 | 0.40 | 0.68 | 0.02 | 0.30 |
| (La+) | 0.40 | 0.60 | 1.00 | 0.00 | 0.00 |
| (CT) | 0.28 | 0.02 | 0.70 | ||
| (Lb-) | 0.57 | 0.43 | 1.00 | 0.00 | 0.00 |
| (Lb+) | 0.43 | 0.57 | 0.98 | 0.02 | 0.00 |
States are listed in order of increasing energy of the exciplex, labeled by characteristic transitions of X (the first molecule in a (X•Y) complex) and Y (the second compound).
For class 2 and 3 exciplexes, although exciton state interactions can be strong in a particular state, they are generally not present for the remaining states. For example, in the acetone···MVE exciplex, the lowest singlet state is 100% of n → π* local excitation of acetone, whereas the second S2 state, dominantly charge transfer from methyl vinyl ether to acetone, is mixed with the local n → π* state at 9% in structure weight. Furthermore, the local acetone n → π* state is strongly mixed with the MVE π → π* state, having structure weights of 20% and 80%, respectively. Yet, the contribution of the π → π* state of MVE to acetone n → π* excitation is negligible. This is also seen in the TCNE···naphthalene exciplex, in which the π → π* local state of TCNE is strongly coupled to the Lb state of the aromatic compound, but the La state is a pure local excitation. In all asymmetrical exciplexes, only one direction of charge transfer is dominant, rather than a charge-transfer resonance found in the symmetric pentacene excimer.
5. Concluding Summary
In this Perspective, we present a multistate energy decomposition analysis (MS-EDA) to dissect the energy components in molecular complexes in excited states. In particular, the total binding energy of an excimer or an exciplex is partitioned into a ground-state effect called local interaction energy ΔELint, an exciton excitation energy ΔEExK resulting from the resonance state interaction of individual monomer excited states |ΨX•ΨYS0⟩ and |ΨX•ΨY*⟩ , a superexchange stabilization energy ΔΔESE due to intermolecular charge transfer |ΨX+•ΨY⟩ and |ΨX-•ΨY⟩, and an orbital and configuration-state delocalization energy ΔΔEOCDK as a result of a stepwise progression from monomer block-localized Kohn–Sham orbitals to the fully delocalized molecular orbitals. To determine the exciton resonance energy ΔΔERes, we have defined a local excitation energy via the structure-weighted reference state without excited-state interaction. Similarly, a structure-weighted adiabatic excitation energy, corresponding to the minimum (photoexcitation) energy needed to produce an exciplex (or excimer) complex, has been introduced to give a well-defined energy term for binding of the exciplex or excimer complex in the excited state.
An important feature of the present MS-EDA method is that key intermediate states associated with different energy terms can be variationally optimized, providing quantitative insights into widely used physical concepts such as exciton delocalization and charge transfer effects in excited states. Although local molecular excitation to a particular state K in the presence of other molecules in the ground state, ΔELexK|ΨX*•ΨY⟩ , is used as a reference state for the exciton resonance energy term, it also has a well-defined physical interpretation and the mutual polarization energy induced by such a local excitation can be related to experimental solvatochromic shifts of the excited chromophore in condensed phases or in molecular clusters. Furthermore, the electronic coupling matrix elements among locally excited states can also be used to determine the rates of excited-state energy transfer and photoexcitation-induced electron transfer. The availability of these diabatic states is made possible in the framework of multistate density functional theory (MSDFT), a rigorous variational DFT of excited states recently proved by Lu and Gao.61
Through MS-EDA analysis, it was shown that molecular complexes in the excited states can be broadly classified into three main categories: (1) encounter excited-state complex, (2) charge-transfer exciplex, and (3) intimate excimer or exciplex. Excited-state complexes in Category 1 are species produced dominantly using the same type of intermolecular forces as that in the ground state, with one molecule happening to be in an excited state. Category 2 exciplexes are formed through intermolecular charge-transfer excitation, which typically has a lower energy than the valence excitations of individual monomers. Clearly, the driving force of exciplex formation here is electrostatic interaction. Intimate excited-state complexes in Category 3 are often, but not limited to, excimers consisting of identical monomers. In this category, exciton delocalization is the main force to produce a fully delocalized excited state. In light of lack of a correlation matrix functional, charge transfer states in some cases such as that in the pentacene excimer are sometimes overestimated in the present analysis. Nevertheless, the illustrative examples presented in this Perspective highlight the interplay of local excitation polarization, exciton resonance and superexchange charge-transfer effects in molecular excited states. It is hoped that MS-EDA will become a useful tool for understanding photochemical and photobiological processes.
Acknowledgments
This work has been partially supported by the Shenzhen Municipal Science and Technology Innovation Commission (KQTD2017-0330155106581) and the Key-area Research and Development Program of Guangdong Province (2020B0101350001). Computational work carried out at Minnesota was supported by the National Institutes of Health (Grant GM046736).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacsau.3c00186.
Computed first and second singlet excited states for monomer compounds using EOM-CCSD, FIC-NEVPT2, and MRCI with the cc-pVDZ and cc-pVTZ basis sets, and optimized structures used in this study (PDF)
Author Contributions
∥ C.P.H. and X.Z. contributed equally. CRediT: Christian P. Hettich and Xiaoyong Zhang data curation, analysis, investigation; David Kemper data curation, investigation; Ruoqi Zhao analysis, programming, investigation; Shaoyuan Zhou data curation, investigation; Yangyi Lu analysis; Jiali Gao, theory, data curation, analysis; Jun Zhang programming; Meiyi Liu theory, data curation, investigation.
The authors declare no competing financial interest.
Supplementary Material
References
- Zhao L. L.; von Hopffgarten M.; Andrada D. M.; Frenking G. Energy decomposition analysis. WIREs Comput. Mol. Sci. 2018, 8, e1345 10.1002/wcms.1345. [DOI] [Google Scholar]
- Su P. F.; Tang Z.; Wu W. Generalized Kohn-Sham energy decomposition analysis and its applications. WIREs-Comput. Mol. Sci. 2020, 10, e1460 10.1002/wcms.1460. [DOI] [Google Scholar]
- Mo Y.; Bao P.; Gao J. Energy decomposition analysis based on a block-localized wavefunction and multistate density functional theory. Phys. Chem. Chem. Phys. 2011, 13, 6760–6775. 10.1039/c0cp02206c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andres J.; Ayers P. W.; Boto R. A.; Carbo-Dorca R.; Chermette H.; Cioslowski J.; Contreras-Garcia J.; Cooper D. L.; Frenking G.; Gatti C.; Heidar-Zadeh F.; Joubert L.; Pendas A. M.; Matito E.; Mayer I.; Misquitta A. J.; Mo Y. R.; Pilme J.; Popelier P. L. A.; Rahm M.; RamosCordoba E.; Salvador P.; Schwarz W. H. E.; Shahbazian S.; Silvi B.; Sola M.; Szalewicz K.; Tognetti V.; Weinhold F.; Zins E. L. Nine questions on energy decomposition analysis. J. Comput. Chem. 2019, 40, 2248–2283. 10.1002/jcc.26003. [DOI] [PubMed] [Google Scholar]
- Zhao R.; Hettich C.; Zhang J.; Liu M.; Gao J. Excimer Energies. J. Phys. Chem. Lett. 2023, 14, 2917–2926. 10.1021/acs.jpclett.3c00545. [DOI] [PubMed] [Google Scholar]
- Mao Y. Z.; Loipersberger M.; Horn P. R.; Das A.; Demerdash O.; Levine D. S.; Veccham S. P.; Head-Gordon T.; Head-Gordon M. From Intermolecular Interaction Energies and Observable Shifts to Component Contributions and Back Again: A Tale of Variational Energy Decomposition Analysis. Annu. Rev. Phys. Chem. 2021, 72, 641–666. 10.1146/annurev-physchem-090419-115149. [DOI] [PubMed] [Google Scholar]
- Gao J.; Pavelites J. J.; Habibollazadeh D. Simulation of Liquid Amides Using a Polarizable Intermolecular Potential Function. J. Phys. Chem. 1996, 100, 2689–2697. 10.1021/jp9521969. [DOI] [Google Scholar]
- Xie W.; Pu J.; MacKerell A. D. Jr; Gao J. Development of a Polarizable Intermolecular Potential Function (PIPF) for Liquid Amides and Alkanes. J. Chem. Theory Comput. 2007, 3, 1878–1889. 10.1021/ct700146x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morokuma K. Molecular Orbital Studies of Hydrogen Bonds.3. C = O H-O Hydrogen Bond in H2co H2o and H2co 2h2o. J. Chem. Phys. 1971, 55, 1236. 10.1063/1.1676210. [DOI] [Google Scholar]
- Ziegler T.; Rauk A. Calculation of Bonding Energies by Hartree-Fock Slater Method.1. Transition-State Method. Theor. Chim. Acta 1977, 46, 1–10. 10.1007/BF02401406. [DOI] [Google Scholar]
- Bagus P. S.; Hermann K.; Bauschlicher C. W. A New Analysis of Charge-Transfer and Polarization for Ligand-Metal Bonding - Model Studies of Al4co and Al4nh3. J. Chem. Phys. 1984, 80, 4378–4386. 10.1063/1.447215. [DOI] [Google Scholar]
- Stevens W. J.; Fink W. H. Frozen fragment reduced variational space analysis of hydrogen bonding interactions. Application ot the water dimer. Chem. Phys. Lett. 1987, 139, 15–22. 10.1016/0009-2614(87)80143-4. [DOI] [Google Scholar]
- Glendening E. D.; Weinhold F. Natural resonance theory: I. General formalism. J. Comput. Chem. 1998, 19, 593–609. . [DOI] [Google Scholar]
- Gordon M. S.; Jensen J. H. Understanding the hydrogen bond using quantum chemistry. Acc. Chem. Res. 1996, 29, 536–543. 10.1021/ar9600594. [DOI] [Google Scholar]
- Chen W.; Gordon M. S. Energy decomposition analyses for many-body interaction and applications to water complexes. J. Phys. Chem. 1996, 100, 14316–14328. 10.1021/jp960694r. [DOI] [Google Scholar]
- Gordon M. S.; Slipchenko L.; Li H.; Jensen J. H. The effective fragment potential: a general method for predicting intermolecular interactions. Ann. Rep. Comput. Chem. 2007, 3, 177–193. 10.1016/S1574-1400(07)03010-1. [DOI] [Google Scholar]
- Reinhardt P.; Piquemal J.-P.; Savin A. Fragment-Localized Kohn-Sham Orbitals via a Singles Configuration-Interaction Procedure and Application to Local Properties and Intermolecular Energy Decomposition Analysis. J. Chem. Theory Comput. 2008, 4, 2020–2029. 10.1021/ct800242n. [DOI] [PubMed] [Google Scholar]
- Wu Q.; Ayers P. W.; Zhang Y. K. Density-based energy decomposition analysis for intermolecular interactions with variationally determined intermediate state energies. J. Chem. Phys. 2009, 131, 164112. 10.1063/1.3253797. [DOI] [PubMed] [Google Scholar]
- Su P. F.; Li H. Energy decomposition analysis of covalent bonds and intermolecular interactions. J. Chem. Phys. 2009, 131, 014102. 10.1063/1.3159673. [DOI] [PubMed] [Google Scholar]
- Piquemal J. P.; Marquez A.; Parisel O.; Giessner-Prettre C. A CSOV study of the difference between HF and DFT intermolecular interaction energy values: The importance of the charge transfer contribution. J. Comput. Chem. 2005, 26, 1052–1062. 10.1002/jcc.20242. [DOI] [PubMed] [Google Scholar]
- Gourlaouen C.; Piquemal J. P.; Saue T.; Parisel O. Revisiting the geometry of nd(10) (n+1)s(0) [M(H2O)](P+) complexes using four-component relativistic DFT calculations and scalar relativistic correlated CSOV energy decompositions (MP+=Cu+, Zn2+, Ag+, Cd2+, Au+, Hg2+). J. Comput. Chem. 2006, 27, 142–156. 10.1002/jcc.20329. [DOI] [PubMed] [Google Scholar]
- Bickelhaupt F. M.; Baerends E. J. In Reviews in Computational Chemistry; Lipkowitz K. B., Boyd D. B., Eds.; Wiley-VCH, 1999; Vol. 15, p 1. [Google Scholar]
- Fonseca Guerra C.; Bickelhaupt F. M.; Snijders J. G.; Baerends E. J. The nature of the hydrogen bond in DNA base pairs: The role of charge transfer and resonance assistance. Chem.—Eur. J. 1999, 5, 3581–3594. . [DOI] [Google Scholar]
- Bickelhaupt F. M.; Baerends E. J. The case for steric repulsion causing the staggered conformation of ethane. Angew. Chem., Int. Ed. 2003, 42, 4183–4188. 10.1002/anie.200350947. [DOI] [PubMed] [Google Scholar]
- Khaliullin R. Z.; Cobar E. A.; Lochan R. C.; Bell A. T.; Head-Gordon M. Unravelling the Origin of Intermolecular Interactions Using Absolutely Localized Molecular Orbitals. J. Phys. Chem. A 2007, 111, 8753–8765. 10.1021/jp073685z. [DOI] [PubMed] [Google Scholar]
- Mo Y.; Zhang Y.; Gao J. A Simple Electrostatic Model for Trisilylamine: Theoretical Examinations of the n.fwdarw..sigma. Negative Hyperconjugation, p.pi..fwdarw.d.pi. Bonding, and Stereoelectronic Interaction. J. Am. Chem. Soc. 1999, 121, 5737–5742. 10.1021/ja9904742. [DOI] [Google Scholar]
- Mo Y.; Gao J.; Peyerimhoff S. D. Energy decomposition analysis of intermolecular interactions using a block-localized wave function approach. J. Chem. Phys. 2000, 112, 5530–5538. 10.1063/1.481185. [DOI] [Google Scholar]
- Frenking G.; Loschen C.; Krapp A.; Fau S.; Strauss S. H. Electronic structure of CO - An exercise in modern chemical bonding theory. J. Comput. Chem. 2007, 28, 117–126. 10.1002/jcc.20477. [DOI] [PubMed] [Google Scholar]
- Frenking G.; Frohlich N. The nature of the bonding in transition-metal compounds. Chem. Rev. 2000, 100, 717–774. 10.1021/cr980401l. [DOI] [PubMed] [Google Scholar]
- Michalak A.; Mitoraj M.; Ziegler T. Bond orbitals from chemical valence theory. J. Phys. Chem. A 2008, 112, 1933–1939. 10.1021/jp075460u. [DOI] [PubMed] [Google Scholar]
- Kurczab R.; Mitoraj M. P.; Michalak A.; Ziegler T. Theoretical Analysis of the Resonance Assisted Hydrogen Bond Based on the Combined Extended Transition State Method and Natural Orbitals for Chemical Valence Scheme. J. Phys. Chem. A 2010, 114, 8581–8590. 10.1021/jp911405e. [DOI] [PubMed] [Google Scholar]
- Dederichs P. H.; Bluegel S.; Zeller R.; Akai H. Ground states of constrained systems: application to cerium impurities. Phys. Rev. Lett. 1984, 53, 2512–2515. 10.1103/PhysRevLett.53.2512. [DOI] [Google Scholar]
- Francisco E.; Pendas A. M.; Blanco M. A. A molecular energy decomposition scheme for atoms in molecules. J. Chem. Theory Comput. 2006, 2, 90–102. 10.1021/ct0502209. [DOI] [PubMed] [Google Scholar]
- Liu S. N.; Govind N.; Pedersen L. G. Exploring the origin of the internal rotational barrier for molecules with one rotatable dihedral angle. J. Chem. Phys. 2008, 129, 094104. 10.1063/1.2976767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone A. J.The Theory of Intermolecular Forces; Oxford University Press, 1996. [Google Scholar]
- Jeziorski B.; Moszynski R.; Szalewicz K. Perturbation Theory Approach to Intermolecular Potential Energy Surfaces of van der Waals Complexes. Chem. Rev. 1994, 94, 1887–1930. 10.1021/cr00031a008. [DOI] [Google Scholar]
- Misquitta A. J.; Szalewicz K. Symmetry-adapted perturbation-theory calculations of intermolecular forces employing density-functional description of monomers. J. Chem. Phys. 2005, 122, 214109. 10.1063/1.1924593. [DOI] [PubMed] [Google Scholar]
- Shahbaz M.; Szalewicz K. Dispersion Energy from Local Polarizability Density. Phys. Rev. Lett. 2019, 122, 213001. 10.1103/PhysRevLett.122.213001. [DOI] [PubMed] [Google Scholar]
- Xantheas S. S. On the importance of the fragment relaxation energy terms in the estimation of the basis set superposition error correction to the intermolecular interaction energy. J. Chem. Phys. 1996, 104, 8821–8824. 10.1063/1.471605. [DOI] [Google Scholar]
- Angeli C.; Cimiraglia R.; Malrieu J. P. On the relative merits of non-orthogonal and orthogonal valence bond methods illustrated on the hydrogen molecule. J. Chem. Educ. 2008, 85, 150–158. 10.1021/ed085p150. [DOI] [Google Scholar]
- Schneider W. B.; Bistoni G.; Sparta M.; Saitow M.; Riplinger C.; Auer A. A.; Neese F. Decomposition of Intermolecular Interaction Energies within the Local Pair Natural Orbital Coupled Cluster Framework. J. Chem. Theory Comput. 2016, 12, 4778–4792. 10.1021/acs.jctc.6b00523. [DOI] [PubMed] [Google Scholar]
- Altun A.; Saitow M.; Neese F.; Bistoni G. Local Energy Decomposition of Open-Shell Molecular Systems in the Domain-Based Local Pair Natural Orbital Coupled Cluster Framework. J. Chem. Theory Comput. 2019, 15, 1616–1632. 10.1021/acs.jctc.8b01145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine D. S.; Horn P. R.; Mao Y. Z.; Head-Gordon M. Variational Energy Decomposition Analysis of Chemical Bonding. 1. Spin-Pure Analysis of Single Bonds. J. Chem. Theory Comput. 2016, 12, 4812–4820. 10.1021/acs.jctc.6b00571. [DOI] [PubMed] [Google Scholar]
- Veccham S. P.; Lee J.; Mao Y. Z.; Horn P. R.; Head-Gordon M. A non-perturbative pairwise-additive analysis of charge transfer contributions to intermolecular interaction energies. Phys. Chem. Chem. Phys. 2021, 23, 928–943. 10.1039/D0CP05852A. [DOI] [PubMed] [Google Scholar]
- Shen H. Y.; Wang Z. L.; Head-Gordon M. Generalization of ETS-NOCV and ALMO-COVP Energy Decomposition Analysis to Connect Any Two Electronic States and Comparative Assessment. J. Chem. Theory Comput. 2022, 18, 7428–7441. 10.1021/acs.jctc.2c00901. [DOI] [PubMed] [Google Scholar]
- Han J. T.; Grofe A.; Gao J. L. Variational Energy Decomposition Analysis of Charge-Transfer Interactions between Metals and Ligands in Carbonyl Complexes. Inorg. Chem. 2021, 60, 14060–14071. 10.1021/acs.inorgchem.1c01367. [DOI] [PubMed] [Google Scholar]
- Mathew P. T.; Fang F. Z. Periodic energy decomposition analysis for electronic transport studies as a tool for atomic scale device manufacturing. Int. J. Extr. Manufact. 2020, 2, 015401. 10.1088/2631-7990/ab5d8a. [DOI] [Google Scholar]
- Xu Y.; Zhang S.; Lindahl E.; Friedman R.; Wu W.; Su P. F. A general tight-binding based energy decomposition analysis scheme for intermolecular interactions in large molecules. J. Chem. Phys. 2022, 157, 034104. 10.1063/5.0091781. [DOI] [PubMed] [Google Scholar]
- Chen H.; Skylaris C. K. Energy decomposition analysis method for metallic systems. Phys. Chem. Chem. Phys. 2022, 24, 1702–1711. 10.1039/D1CP05112A. [DOI] [PubMed] [Google Scholar]
- Chan W.-L.; Berkelbach T. C.; Provorse M. R.; Monahan N. R.; Tritsch J. R.; Hybertsen M. S.; Reichman D. R.; Gao J.; Zhu X.-Y. The quantum coherent mechanism for singlet fission: Experiment and theory. Acc. Chem. Res. 2013, 46, 1321–1329. 10.1021/ar300286s. [DOI] [PubMed] [Google Scholar]
- Li H.; Wang Y. J.; Ye M. P.; Li S. S.; Li D. Y.; Ren H. S.; Wang M.; Du L. C.; Li H.; Veglia G.; Gao J. L.; Weng Y. X. Dynamical and allosteric regulation of photoprotection in light harvesting complex II. Sci. China Chem. 2020, 63, 1121–1133. 10.1007/s11426-020-9771-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Ren H.; Kundu M.; Liu Z.; Zhong F. W.; Wang L.; Gao J.; Zhong D. A leap in quantum efficiency through light harvesting in photoreceptor UVR8. Nat. Commun. 2020, 11, 4316. 10.1038/s41467-020-17838-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X. K.; Liu Z. Y.; Ren H. S.; Kundu M.; Wang L. J.; Gao J. L.; Zhong D. P. Dynamics and mechanism of light harvesting in UV photoreceptor UVR8. Chem. Sci. 2020, 11, 12553–12569. 10.1039/D0SC04909C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X. K.; Liu Z. Y.; Ren H. S.; Kundu M.; Zhong F. W.; Wang L. J.; Gao J. L.; Zhong D. P. Dynamics and mechanism of dimer dissociation of photoreceptor UVR8. Nature Comm. 2022, 13, 93. 10.1038/s41467-021-27756-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curutchet C.; Mennucci B. Quantum Chemical Studies of Light Harvesting. Chem. Rev. 2017, 117, 294–343. 10.1021/acs.chemrev.5b00700. [DOI] [PubMed] [Google Scholar]
- Ge Q. H.; Head-Gordon M. Energy Decomposition Analysis for Excimers Using Absolutely Localized Molecular Orbitals within Time-Dependent Density Functional Theory and Configuration Interaction with Single Excitations. J. Chem. Theory Comput. 2018, 14, 5156–5168. 10.1021/acs.jctc.8b00537. [DOI] [PubMed] [Google Scholar]
- Ge Q. H.; Mao Y. Z.; Head-Gordon M. Energy decomposition analysis for exciplexes using absolutely localized molecular orbitals. J. Chem. Phys. 2018, 148, 064105. 10.1063/1.5017510. [DOI] [PubMed] [Google Scholar]
- East A. L. L.; Lim E. C. Naphthalene dimer: Electronic states, excimers, and triplet decay. J. Chem. Phys. 2000, 113, 8981–8994. 10.1063/1.1319345. [DOI] [Google Scholar]
- Plasser F. TheoDORE: A toolbox for a detailed and automated analysis of electronic excited state computations. J. Chem. Phys. 2020, 152, 084108. 10.1063/1.5143076. [DOI] [PubMed] [Google Scholar]
- Plasser F.; Lischka H. Analysis of Excitonic and Charge Transfer Interactions from Quantum Chemical Calculations. J. Chem. Theory Comput. 2012, 8, 2777–2789. 10.1021/ct300307c. [DOI] [PubMed] [Google Scholar]
- Lu Y. Y.; Gao J. L. Multistate Density Functional Theory of Excited States. J. Phys. Chem. Lett. 2022, 13, 7762–7769. 10.1021/acs.jpclett.2c02088. [DOI] [PubMed] [Google Scholar]
- Lu Y. Y.; Gao J. L. Fundamental Variable and Density Representation in Multistate DFT for Excited States. J. Chem. Theory Comput. 2022, 18, 7403–7411. 10.1021/acs.jctc.2c00859. [DOI] [PubMed] [Google Scholar]
- Lu Y. Y.; Zhao R. Q.; Zhang J.; Liu M. Y.; Gao J. L. Minimal Active Space: NOSCF and NOSI in Multistate Density Functional Theory. J. Chem. Theory Comput. 2022, 18, 6407–6420. 10.1021/acs.jctc.2c00908. [DOI] [PubMed] [Google Scholar]
- Cembran A.; Song L.; Mo Y.; Gao J. Block-localized density functional theory (BLDFT), diabatic coupling, and its use in valence bond theory for representing reactive potential energy surfaces. J. Chem. Theory Comput. 2009, 5, 2702–2716. 10.1021/ct9002898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitaura K.; Morokuma K. New Energy Decomposition Scheme for Molecular-Interactions within Hartree-Fock Approximation. Int. J. Quantum Chem. 1976, 10, 325–340. 10.1002/qua.560100211. [DOI] [Google Scholar]
- Morokuma K. Why Do Molecules Interact - Origin of Electron Donor-Acceptor Complexes, Hydrogen-Bonding, and Proton Affinity. Acc. Chem. Res. 1977, 10, 294–300. 10.1021/ar50116a004. [DOI] [Google Scholar]
- Blanco M. A.; Pendas A. M.; Francisco E. Interacting quantum atoms: A correlated energy decomposition scheme based on the Quantum Theory of Atoms in Molecules. J. Chem. Theory Comput. 2005, 1, 1096–1109. 10.1021/ct0501093. [DOI] [PubMed] [Google Scholar]
- Glendening E. D.; Streitwieser A. Natural Energy Decomposition Analysis - an Energy Partitioning Procedure for Molecular-Interactions with Application to Weak Hydrogen-Bonding, Strong Ionic, and Moderate Donor-Acceptor Interactions. J. Chem. Phys. 1994, 100, 2900–2909. 10.1063/1.466432. [DOI] [Google Scholar]
- Pophristic V.; Goodman L. Hyperconjugation not steric repulsion leads to the staggered structure of ethane. Nature 2001, 411, 565–568. 10.1038/35079036. [DOI] [PubMed] [Google Scholar]
- Mo Y.; Gao J. Theoretical Analysis of the Rotational Barrier of Ethane. Acc. Chem. Res. 2007, 40, 113–119. 10.1021/ar068073w. [DOI] [PubMed] [Google Scholar]
- Mo Y.; Wu W.; Song L.; Lin M.; Zhang Q.; Gao J. The magnitude of hyperconjugation in ethane: a perspective from ab initio valence bond theory. Angew. Chem., Int. Ed. 2004, 43, 1986–1990. 10.1002/anie.200352931. [DOI] [PubMed] [Google Scholar]
- Mo Y.; Peyerimhoff S. D. Theoretical analysis of electronic delocalization. J. Chem. Phys. 1998, 109, 1687–1697. 10.1063/1.476742. [DOI] [Google Scholar]
- Mo Y. R. Computational evidence that hyperconjugative interactions are not responsible for the anomeric effect. Nat. Chem. 2010, 2, 666–671. 10.1038/nchem.721. [DOI] [PubMed] [Google Scholar]
- Bagus P. S.; Hermann K.; Bauschlicher C. W. On the Nature of the Bonding of Lone Pair Ligands to a Transition-Metal. J. Chem. Phys. 1984, 81, 1966–1974. 10.1063/1.447818. [DOI] [Google Scholar]
- Bagus P. S.; Illas F. Decomposition of the Chemisorption Bond by Constrained Variations - Order of the Variations and Construction of the Variational Spaces. J. Chem. Phys. 1992, 96, 8962–8970. 10.1063/1.462875. [DOI] [Google Scholar]
- te Velde G.; Bickelhaupt F. M.; Baerends E. J.; Guerra C. F.; Van Gisbergen S. J. A.; Snijders J. G.; Ziegler T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. 10.1002/jcc.1056. [DOI] [Google Scholar]
- Mitoraj M. P.; Michalak A.; Ziegler T. A Combined Charge and Energy Decomposition Scheme for Bond Analysis. J. Chem. Theory Comput. 2009, 5, 962–975. 10.1021/ct800503d. [DOI] [PubMed] [Google Scholar]
- Szalewicz K.; Jeziorski B. Symmetry-Adapted Double-Perturbation Analysis of Intra-Molecular Correlation Effects in Weak Inter-Molecular Interactions - He-He Interaction. Mol. Phys. 1979, 38, 191–208. 10.1080/00268977900101601. [DOI] [Google Scholar]
- Misquitta A. J. Charge Transfer from Regularized Symmetry-Adapted Perturbation Theory. J. Chem. Theory Comput. 2013, 9, 5313–5326. 10.1021/ct400704a. [DOI] [PubMed] [Google Scholar]
- Mo Y.; Gao J. Polarization and Charge-Transfer Effects in Lewis Acid-Base Complexes. J. Phys. Chem. A 2001, 105, 6530–6536. 10.1021/jp010348w. [DOI] [Google Scholar]
- Mo Y.; Gao J. Polarization and Charge-Transfer Effects in Aqueous Solution via Ab Initio QM/MM Simulations. J. Phys. Chem. B 2006, 110, 2976–2980. 10.1021/jp057017u. [DOI] [PubMed] [Google Scholar]
- Pei Z.; Yang J. J.; Deng J. H.; Mao Y. Z.; Wu Q.; Yang Z. B.; Wang B.; Aikens C. M.; Liang W. Z.; Shao Y. H. Analysis and visualization of energy densities. II. Insights from linear-response time-dependent density functional theory calculations. Phys. Chem. Chem. Phys. 2020, 22, 26852–26864. 10.1039/D0CP04207B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. S.; Haze O.; Dinnocenzo J. P.; Farid S.; Farid R. S.; Gould I. R. Bonded exciplexes. A new concept in photochemical reactions. J. Org. Chem. 2007, 72, 6970–6981. 10.1021/jo071157d. [DOI] [PubMed] [Google Scholar]
- Spata V. A.; Matsika S. Role of Excitonic Coupling and Charge-Transfer States in the Absorption and CD Spectra of Adenine-Based Oligonucleotides Investigated through QM/MM Simulations. J. Phys. Chem. A 2014, 118, 12021–12030. 10.1021/jp507520c. [DOI] [PubMed] [Google Scholar]
- Plasser F.; Lischka H. Electronic excitation and structural relaxation of the adenine dinucleotide in gas phase and solution. Photochem. Photobio. Sci. 2013, 12, 1440–1452. 10.1039/c3pp50032b. [DOI] [PubMed] [Google Scholar]
- Krylov A. I. From orbitals to observables and back. J. Chem. Phys. 2020, 153, 080901. 10.1063/5.0018597. [DOI] [PubMed] [Google Scholar]
- Closser K. D.; Ge Q. H.; Mao Y. Z.; Shao Y. H.; Head-Gordon M. Superposition of Fragment Excitations for Excited States of Large Clusters with Application to Helium Clusters. J. Chem. Theory Comput. 2015, 11, 5791–5803. 10.1021/acs.jctc.5b00703. [DOI] [PubMed] [Google Scholar]
- Smith M. B.; Michl J. Singlet Fission. Chem. Rev. 2010, 110, 6891. 10.1021/cr1002613. [DOI] [PubMed] [Google Scholar]
- Grofe A.; Qu Z. X.; Truhlar D. G.; Li H.; Gao J. Diabatic-At-Construction Method for Diabatic and Adiabatic Ground and Excited States Based on Multistate Density Functional Theory. J. Chem. Theory Comput. 2017, 13, 1176–1187. 10.1021/acs.jctc.6b01176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M.; Chen X.; Grofe A.; Gao J. Diabatic States at Construction (DAC) through Generalized Singular Value Decomposition. J. Phys. Chem. Lett. 2018, 9, 6038–6046. 10.1021/acs.jpclett.8b02472. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Tang Z.; Zhang X.; Zhu H.; Zhao R.; Lu Y.; Gao J. Target State Optimized Density Functional Theory for Electronic Excited and Diabatic States. J. Chem. Theory Comput. 2023, 19, 1777–1789. 10.1021/acs.jctc.2c01317. [DOI] [PubMed] [Google Scholar]
- Bao P.; Hettich C. P.; Shi Q.; Gao J. Block-Localized Excitation for Excimer Complex and Diabatic Coupling. J. Chem. Theory Comput. 2021, 17, 240–254. 10.1021/acs.jctc.0c01015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenkel J. On the transformation of light into heat in solids. I. Phys. Rev. 1931, 37, 17–44. 10.1103/PhysRev.37.17. [DOI] [Google Scholar]
- Zarea M.; Powell D.; Renaud N.; Wasielewski M. R.; Ratner M. A. Decoherence and Quantum Interference in a Four-Site Model System: Mechanisms and Turnovers. J. Phys. Chem. B 2013, 117, 1010–1020. 10.1021/jp3102942. [DOI] [PubMed] [Google Scholar]
- Boys S. F. Localized orbitals and localized adjustment functions. Quantum Theory At., Mol., Solid State 1966, 253–262. [Google Scholar]
- Ruedenberg K.; Atchity G. J. A quantum chemical determination of diabatic states. J. Chem. Phys. 1993, 99, 3799–3803. 10.1063/1.466125. [DOI] [Google Scholar]
- Song L.; Gao J. On the Construction of Diabatic and Adiabatic Potential Energy Surfaces Based on Ab Initio Valence Bond Theory. J. Phys. Chem. A 2008, 112, 12925–12935. 10.1021/jp803050e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grofe A.; Zhao R.; Wildman A.; Stetina T. F.; Li X.; Bao P.; Gao J. Generalization of Block-Localized Wave Function for Constrained Optimization of Excited Determinants. J. Chem. Theory Comput. 2021, 17, 277–289. 10.1021/acs.jctc.0c01049. [DOI] [PubMed] [Google Scholar]
- Chirgwin H. B.; Coulson C. A. The electronic structure of conjugated systems. VI. Proc. R. Soc. London Ser. A 1950, 201, 196–209. 10.1098/rspa.1950.0053. [DOI] [Google Scholar]
- Gao J.; Grofe A.; Ren H.; Bao P. Beyond Kohn-Sham Approximation: Hybrid Multistate Wave Function and Density Functional Theory. J. Phys. Chem. Lett. 2016, 7, 5143–5149. 10.1021/acs.jpclett.6b02455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao R. Q.; Hettich C. P.; Chen X.; Gao J. L. Minimal-active-space multistate density functional theory for excitation energy involving local and charge transfer states. NPJ. Comput. Mater. 2021, 7, 148. 10.1038/s41524-021-00624-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King H. F.; Staton R. E.; Kim H.; Wyatt R. E.; Parr R. G. Corresponding orbitals and the nonorthogonality problems in molecular quantum mechanics. J. Chem. Phys. 1967, 47, 1936–1941. 10.1063/1.1712221. [DOI] [Google Scholar]
- Mo Y.; Gao J. Ab initio QM/MM simulations with a molecular orbital-valence bond (MOVB) method: application to an SN2 reaction in water. J. Comput. Chem. 2000, 21, 1458–1469. . [DOI] [Google Scholar]
- Mo Y.; Gao J. An Ab Initio Molecular Orbital-Valence Bond (MOVB) Method for Simulating Chemical Reactions in Solution. J. Phys. Chem. A 2000, 104, 3012–3020. 10.1021/jp994053i. [DOI] [Google Scholar]
- Ziegler T.; Rauk A.; Baerends E. J. Calculation of Multiplet Energies by Hartree-Fock-Slater Method. Theor. Chim. Acta 1977, 43, 261–271. 10.1007/BF00551551. [DOI] [Google Scholar]
- Grofe A.; Chen X.; Liu W.; Gao J. Spin-Multiplet Components and Energy Splittings by Multistate Density Functional Theory. J. Phys. Chem. Lett. 2017, 8, 4838–4845. 10.1021/acs.jpclett.7b02202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao R.; Grofe A.; Wang Z.; Bao P.; Chen X.; Liu W.; Gao J. Dynamic-then-Static Approach for Core Excitations of Open-Shell Molecules. J. Phys. Chem. Lett. 2021, 12, 7409–7417. 10.1021/acs.jpclett.1c02039. [DOI] [PubMed] [Google Scholar]
- Yang L. K.; Grofe A.; Reimers J.; Gao J. Multistate density functional theory applied with 3 unpaired electrons in 3 orbitals: The singdoublet and tripdoublet states of the ethylene cation. Chem. Phys. Lett. 2019, 736, 136803. 10.1016/j.cplett.2019.136803. [DOI] [Google Scholar]
- Zhao Y.; Truhlar D. G. M06 DFT functionals. Theor. Chem. Acc. 2008, 120, 215. 10.1007/s00214-007-0310-x. [DOI] [Google Scholar]
- Dunning T. H. Jr Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. 10.1063/1.456153. [DOI] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Petersson G. A.; Nakatsuji H.; Li X.; Caricato M.; Marenich A. V.; Bloino J.; Janesko B. G.; Gomperts R.; Mennucci B.; Hratchian H. P.; Ortiz J. V.; Izmaylov A. F.; Sonnenberg J. L.; Williams D. F.; Lipparini F.; Egidi F.; Goings J.; Peng B.; Petrone A.; Henderson T.; Ranasinghe D.; Zakrzewski V. G.; Gao J.; Rega N.; Zheng G.; Liang W.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Throssell K.; Montgomery J. A. Jr; Peralta J. E.; Ogliaro F.; Bearpark M. J.; Heyd J. J.; Brothers E. N.; Kudin K. N.; Staroverov V. N.; Keith T. A.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A. P.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Millam J. M.; Klene M.; Adamo C.; Cammi R.; Ochterski J. W.; Martin R. L.; Morokuma K.; Farkas O.; Foresman J. B.; Fox D. J.. Gaussian 16; Gaussian, Inc.: Wallingford, CT, 2016. [Google Scholar]
- Zhang J.; Pan Z.; Zhao R.; Hou X.; Zhang X.; Tang Z.; Zhang Y.; Wu Y.; Liu W.; Gao J.. Qbics - Quantum biology, informatics and chemical science; Shenzhen Bay Laboratory: Shenzhen, China, 2023. [Google Scholar]
- Schmidt M. W.; Baldridge K. K.; Boatz J. A.; Elbert S. T.; Gordon M. S.; Jensen J. H.; Koseki S.; Matsunaga N.; Nguyen K. A.; Su S. J.; Windus T. L.; Dupuis M.; Montgomery J. S. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347. 10.1002/jcc.540141112. [DOI] [Google Scholar]
- Sarkar R.; Boggio-Pasqua M.; Loos P. F.; Jacquemin D. Benchmarking TD-DFT and Wave Function Methods for Oscillator Strengths and Excited-State Dipole Moments. J. Chem. Theory Comput. 2021, 17, 1117–1132. 10.1021/acs.jctc.0c01228. [DOI] [PubMed] [Google Scholar]
- Schreiber M.; Silva-Junior M. R.; Sauer S. P. A.; Thiel W. Benchmarks for electronically excited states: CASPT2, CC2, CCSD, and CC3. J. Chem. Phys. 2008, 128, 134110. 10.1063/1.2889385. [DOI] [PubMed] [Google Scholar]
- Merchan M.; Roos B. O.; McDiarmid R.; Xing X. A combined theoretical and experimental determination of the electronic spectrum of acetone. J. Chem. Phys. 1996, 104, 1791–1804. 10.1063/1.470976. [DOI] [Google Scholar]
- Bulliard C.; Allan M.; Grimme S. Electron energy loss and dissociative electron attachment spectroscopy of methyl vinyl ether and related compounds. Int. J. Mass Spectrom. 2001, 205, 43–55. 10.1016/S1387-3806(00)00283-9. [DOI] [Google Scholar]
- Fueno T.; Yamaguchi K. Comparison of the Electronic Spectra of Geometric Isomers. II. 1,2-Disubstituted Ethylenes. Bull. Chem. Soc. Jpn. 1972, 45, 3290–3293. 10.1246/bcsj.45.3290. [DOI] [Google Scholar]
- Milian B.; Pou-Amerigo R.; Merchan M.; Orti E. Theoretical study of the electronic excited states of tetracyanoethylene and its radical anion. ChemPhysChem 2005, 6, 503–510. 10.1002/cphc.200400464. [DOI] [PubMed] [Google Scholar]
- Zeng T.; Hoffmann R.; Ananth N. The Low-Lying Electronic States of Pentacene and Their Roles in Singlet Fission. J. Am. Chem. Soc. 2014, 136, 5755–5764. 10.1021/ja500887a. [DOI] [PubMed] [Google Scholar]
- Cremer D.; Filatov M.; Polo V.; Kraka E.; Shaik S. Implicit and explicit coverage of multi-reference effects by density functional theory. Int. J. Mol. Sci. 2002, 3, 604–638. 10.3390/i3060604. [DOI] [Google Scholar]
- Grafenstein J.; Cremer D. Development of a CAS-DFT method covering non-dynamical and dynamical electron correlation in a balanced way. Mol. Phys. 2005, 103, 279–308. 10.1080/00268970512331318858. [DOI] [Google Scholar]
- Grafenstein J.; Cremer D. The self-interaction error and the description of non-dynamic electron correlation in density functional theory. Theor. Chem. Acc. 2009, 123, 171–182. 10.1007/s00214-009-0545-9. [DOI] [Google Scholar]
- Swiderek P.; Michaud M.; Hohlneicher G.; Sanche L. Electron-Energy Loss Spectroscopy of Solid Naphthalene and Acenaphthene - Search for the Low-Lying Triplet-States. Chem. Phys. Lett. 1990, 175, 667–673. 10.1016/0009-2614(90)85600-H. [DOI] [Google Scholar]
- Thorsmolle V. K.; Averitt R. D.; Demsar J.; Smith D. L.; Tretiak S.; Martin R. L.; Chi X.; Crone B. K.; Ramirez A. P.; Taylor A. J. Photoexcited carrier relaxation dynamics in pentacene probed by ultrafast optical spectroscopy: Influence of morphology on relaxation processes. Phys. B - Cond. Matter 2009, 404, 3127–3130. 10.1016/j.physb.2009.07.063. [DOI] [Google Scholar]
- Bulliard C.; Allan M.; Grimme S. Electron energy loss and dissociative electron attachment spectroscopy of methyl vinyl ether and related compounds. Int. J. Mass Spectrom. 2001, 205, 43–55. 10.1016/S1387-3806(00)00283-9. [DOI] [Google Scholar]
- Wiberg K. B.; Stratmann R. E.; Frisch M. J. A time-dependent density functional theory study of the electronically excited states of formaldehyde, acetaldehyde and acetone. Chem. Phys. Lett. 1998, 297, 60–64. 10.1016/S0009-2614(98)01119-1. [DOI] [Google Scholar]
- Robin M. B.Higher excited states of polyatomic molecules; Academic Press, 1974. [Google Scholar]
- Schore N. E.; Turro N. J. Mechanism of Interaction of N, Pi-Star Excited Alkanones with Electron-Rich Ethylenes. J. Am. Chem. Soc. 1975, 97, 2482–2488. 10.1021/ja00842a027. [DOI] [Google Scholar]
- Hanazaki I. Vapor-Phase Electron Donor-Acceptor Complexes of Tetracyanoethylene And of Sulfur-Dioxide. J. Phys. Chem. 1972, 76, 1982–1989. 10.1021/j100658a012. [DOI] [Google Scholar]
- Ledwith A.; Woods H. J. Reactivity and Structure of Alkyl Vinyl Ethers.4. Conformational Effects of Alkoxy-Groups in Alkyl Vinyl Ethers and Alkyl Aryl Ethers on Charge-Transfer Spectra with Tetracyanoethylene, and Reactivity Towards Electrophiles. J. Chem. Soc. B 1970, 310–314. 10.1039/j29700000310. [DOI] [Google Scholar]
- Stein T.; Kronik L.; Baer R. Reliable Prediction of Charge Transfer Excitations in Molecular Complexes Using Time-Dependent Density Functional Theory. J. Am. Chem. Soc. 2009, 131, 2818–2820. 10.1021/ja8087482. [DOI] [PubMed] [Google Scholar]
- Haiduke R. L. A.; Bartlett R. J. Non-empirical exchange-correlation parameterizations based on exact conditions from correlated orbital theory. J. Chem. Phys. 2018, 148, 184106. 10.1063/1.5025723. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.