

Abstract
With the in-depth investigation of cardiac fibrosis, oxidative stress (OS) has been recognized as a significant pathophysiological pathway involved in cardiac remodeling and progression. OS is a condition characterized by the disruption of equilibrium between reactive oxygen species (ROS) produced by the organism and the antioxidant defense system, resulting in adverse effects on the structure and function of the heart. The accumulation of reactive substances beyond cellular thresholds disrupts the normal physiology of both cardiomyocytes and non-cardiomyocytes, leading to OS, inflammation, hypertrophy, and cardiac fibrosis. Furthermore, cardiac OS also modulates several crucial genes involved in maintaining cellular homeostasis, including those associated with mitochondrial biogenesis, injury, and antioxidant defense, which are inevitably associated with concurrent epigenetic changes. Multiple studies have demonstrated the crucial role of epigenetic modifications in regulating cardiac OS. Consequently, modulating OS through targeted epigenetic modifications emerges as a potentially promising therapeutic strategy for managing cardiac fibrosis. This article provides a new review of current research on this subject and proposes that epigenetics may improve OS-induced cardiac fibrosis.
Keywords: Epigenetic, Cardiac fibrosis, Oxidative stress, Extracellular matrix
Graphical abstract
Highlights
-
•
Oxidative stress mediated epigenetic regulations of cardiac fibrosis.
-
•
Epigenetic regulations of oxidative stress in cardiac fibrosis.
-
•
Oxidative stress and epigenetic crosstalk play a role in cardiac fibrosis.
Abbreviations
- KAT7
lysine acetyltransferase 7
- LARP7
la-related protein 7
- AAV
adeno-associated virus
- AngII
angiotensin II
- ATM
ataxia-telangiectasia mutated
- Bax
Bcl-2-associated X
- Bcl-2
B-cell lymphoma-2
- CAT
catalase
- CHD
coronary heart disease
- CircRNAs
circular RNAs
- CircITCH
circular RNA itchy E3 ubiquitin protein ligase
- CM
cardiomyocyte
- CRM1
chromosome region maintenance protein 1
- DCM
diabetic cardiomyopathy
- DDR
DNA damage response
- DNMT
DNA methyltransferase
- ECM
extracellular matrix
- EC-SOD
extracellular superoxide dismutas
- EndMT
endothelial-to-mesenchymal transition
- ER
endoplasmic reticulum
- ERK1/2
extracellular signal-related kinases 1 and 2
- ETC
electron transport chain
- EZH2
enhancer of zeste 2 polycomb repressive complex 2 subunit
- FOXO1/3
forkhead box protein O1/3
- GE
geniposide
- GPX1
glutathione peroxidase 1
- GSH
glutathione
- HAT
histone acetyltransferase
- HDACs
histone deacetylases
- Hmox-1
heme oxygenase 1
- HMTs
histone methyltransferases
- HKL
honokiol
- IDH
isocitrate dehydrogenase
- IGFBP3
insulin-like growth factor-binding protein 3
- Imp
importin
- IRE1
inositol-requiring enzyme 1
- lncRNA
long non-coding RNA
- LRP6
lipoprotein receptor-related protein 6
- MALAT1
metastasis-associated lung adenocarcinoma transcript 1
- Mcl-1
myeloid cell leukemia 1
- MDA
malondialdehyde
- MIF
macrophage migration inhibitory factor
- MMPs
matrix metalloproteinases
- Mn-SOD
manganese SOD
- mPTP
mitochondrial permeability transition pore
- NAD+
nicotinamide adenine dinucleotide
- NcRNAs
non-coding RNAs
- NEU
neuraminidase
- Nfe2l2
nuclear factor (erythroid-derived 2)-like 2
- NFIA
nuclear factor I-A
- NRF2
nuclear factor-erythroid 2-related factor 2
- OGG1
8-oxoguanine DNA glycosylase
- OS
oxidative stress
- PAK2
p21-activated kinase 2
- PGC-1α
peroxisome proliferator-activated receptor γ coactivator 1α
- RASSF1A
ras association domain family 1 isoform A
- ROS
reactive oxygen species
- Sal B
salvianolic acid B
- siRNAs
small interfering RNAs
- SOD
superoxide dismutase
- TBP-2
thioredoxin-binding protein-2
- TET
ten-eleven translocation
- TRX1
thioredoxin 1
- USP22
ubiquitin-specific peptidase 22
- 3′-UTR
3‘-untranslated region
- VCPs
valosin containing proteins
- VDAC1
voltage-dependent anion channel 1
- XBP1
X-box-binding protein 1
1. Introduction
Cardiac fibrosis, is a multifactorial process that includes, but is not limited to, cardiac fibroblast activation, apoptosis of cardiomyocytes, transition of endothelial cells to mesenchymal cells, injury or aging of endothelial cells. It is defined as myocardial interstitial dilation due to deposition of extracellular matrix (ECM) proteins [1]. Initial deposition of ECM improves tissue integrity, but extensive fibrosis impairs cardiac function [2]. Notably, fibroblasts, particularly myofibroblasts, play a pivotal role as downstream effector cells in the fibrotic process and serve as the primary source of matrix proteins [3]. In addition, ECM metalloproteinases (MMPs) are proteases that specifically regulate the content of ECM. Changes in the frequency and relative proportions of MMPs and their inhibitors can lead to changes in the myocardial matrix [4]. Disruption of this balance is also present in some mechanisms that promote fibrosis, but exactly what is changed remains a matter of debate.
In recent years, an increasing body of literature has provided compelling evidence indicating that OS acts as a mediator in the above pathological state of cells and promotes the progression of cardiac fibrosis. OS is characterized by an imbalance between oxidative and antioxidative processes in the body. The primary cause of OS is the generation of ROS. ROS diffuses within the cytoplasm and activates redox-sensitive protein kinases [5], which play a pro-fibrotic role by promoting fibroblast proliferation, cardiomyocyte apoptosis, and endothelial cell injury or senescence, ultimately leading to extracellular remodeling [6,7]. At the molecular level, OS induces specific manifestations such as cellular electrophysiological disturbances, alterations in the expression of myosin chains [8], oxidative damage to proteins and lipids, DNA damage, and dysregulation of MMPs and their inhibitors [6,9]. Their cumulative effect further leads to the development of cardiac fibrosis, impairing basic heart function. The review aims to provide an extensive overview of the intricate interplay between OS and various cardiac cell types, including cardiomyocytes, cardiac fibroblasts, and endothelial cells. By elucidating the specific epigenetic mechanisms and signaling pathways affected by OS in these cell types, a deeper understanding of the pathogenesis of cardiac fibrosis and remodeling can be gained.
2. Epigenetic mechanisms
Epigenetic regulation, a pivotal mediator of fibrotic progression, governs the transcriptional state of genes. It serves as a mechanism for modulating the cellular transcriptome and proteome without affecting the genetic content, potentially accomplished through the establishment of epigenetic marks, including DNA methylation, post-translational modifications of histones, as well as additional mechanisms involving non-coding RNAs [10]. Indeed, numerous studies have proved that OS can alter the epigenetic landscape of cells [11,12]. There even have been reports supporting that superoxides could directly mediate cytosine methylation by deprotonating C5 and transferring the methyl group from SAM without the involvement of DNA methyltransferase [13]. Nonetheless, many studies have also demonstrated the epigenetic mediation of OS [14]. An important example is that activation of eroxisome proliferator-activated receptor gamma coactivator-1 alpha (PGC-1α) reduces mitochondrial ROS. However, the activation of PGC-1α is contingent upon its deacetylation mediated by sirtuins (SIRT) [15]. Overall, redox signaling and OS could modulate gene regulation by altering histone function and DNA-modifying enzymes, which subsequently impact cellular phenotypes. Conversely, epigenetic changes can also influence the redox environment within cells [16].
Meanwhile, epigenetic and OS have important downstream effects, both contributing to fibrosis. It can be seen that OS is closely related to epigenetics, overlaps each other, and significantly affects the progression of cardiac fibrosis. Therefore, modulation of OS through epigenetic mechanisms is a potential and promising therapeutic option for cardiac fibrosis.
3. Overview of oxidative stress in cardiac fibrosis
Regulation of redox homeostasis is critical for maintaining normal cell growth, metabolism and survival. OS, characterized by an imbalance of the body's oxidation and antioxidant mechanisms, primarily arises from ROS [17]. ROS is a collective term encompassing molecules derived from O2, such as superoxides, hydrogen peroxide, hydroxyl radicals, ozone, and singlet oxygen [18]. And numerous additional agents also contribute to redox signaling, including nitric oxide, hydrogen sulfide, and oxidized lipids [19]. These agents play an important role in mediating diverse biological processes within the body by modulating protein function, promoting inflammation, inducing apoptosis, impairing autophagy, disrupting mitochondrial function, and interfering with various signaling pathways. These effects tend to accelerate the pathological process and aggravate the disease symptoms.
Intracellular production of ROS can stem from multiple sources, encompassing the mitochondrial electron transport chain (ETC), NADPH oxidase/Dual oxidase (NOX/DUOX) enzymes, endoplasmic reticulum (ER), as well as superoxide generation [20]. The detrimental impacts of ROS are intricately involved in the pathogenesis of cardiac tissue. To counterbalance these effects, the human body possesses a sophisticated antioxidant system consisting of both enzymatic and non-enzymatic components. Among these, crucial elements include antioxidant enzymes, such as superoxide dismutase (SOD), macrophage migration inhibitory factor (MIF), and catalase, alongside various non-enzyme antioxidants. These components collectively contribute to the regulation of OS and maintenance of redox homeostasis in cardiac cells.
Remarkably, several studies have demonstrated that ROS play a significant role in modulating the ECM by enhanced protein expression of TGF-β1, alpha smooth muscle actin, collagen I, and collagen Protein III, leading to the activation of cardiac fibroblasts [21]. TGF-β plays a crucial role in the transdifferentiation of cardiac fibroblasts and the deposition of ECM [22]. Upon activation, fibroblasts exhibit a significant increase in collagen production, while ROS also have the ability to disrupt the balance of MMPs and their inhibitors [9]. These processes collectively contribute to the development of fibrosis and remodeling of the matrix. Moreover, oxidative damage inflicted upon ECM proteins can result in their fragmentation and/or stabilization, consequently impairing the proper resolution of inflammation and fibrosis. In addition, ROS are also involved in signaling, such as apoptosis signaling [23]. Apoptosis is usually associated with the activation of caspase proteases. Apoptosis stimulated by ROS may also aggravate cardiac fibrosis [24]. We recently discovered that these ROS-induced changes are inextricably linked to concomitant epigenetic changes. Below we discuss recent studies of OS-regulated epigenetic changes mediating myocardial fibrosis, Fig. 1 and Table 1 provide more details on this discovery.
Fig. 1.

Oxidative stress influences cardiac fibrosis by affecting epigenetic modification
ROS affect cardiac fibrosis progression by mediating multiple epigenetic mechanisms in various cardiac tissue cells, including cardiomyocytes, cardiac fibroblasts, and endothelial cells.
Table 1.
Oxidative stress influences cardiac fibrosis by affecting epigenetic modification.
| DNA methylation | Histone modification | Non-coding RNA | ||
|---|---|---|---|---|
| Transformation |
|
|
|
|
| Cell type | Cardiac fibroblast | Cardiomyocyte | Cardiac fibroblast | Endothelial cell |
4. Oxidative stress mediated epigenetic regulations of cardiac fibrosis
OS exerts a substantial influence on the induction of epigenetic modifications, with a particular focus on DNA methylation. Adverse effects of oxidative damage on DNA integrity disrupt the chromatin structure and lead to epigenetic changes, leading to subsequent epigenetic alterations. This interplay between OS and epigenetic modifications highlights the intricate relationship between environmental cues and the modulation of gene expression patterns [25]. ROS have the capacity to modify the methylation status of CpG sites, impeding their interaction with transcription factors that would typically bind to them. Furthermore, studies have provided evidence indicating that OS transiently influences epigenetic processes by modulating the activity of enzymes involved in histone demethylation and deacetylation [26].
4.1. Antioxidants reduce DNA methylation
RASSF1A is a tumor suppressor gene that exerts its effects by modulating downstream proteins, including ERK1/2. The Ras/ERK signaling pathway represents an intracellular signaling cascade intricately associated with fibroblast proliferation. Notably, DNA methylation events occurring in cardiac fibroblasts lead to the inactivation of the tumor suppressor gene RASSF1A, concurrently activating ERK1/2 and culminating in fibroblast proliferation and the development of cardiac fibrosis. Chronic exposure to hypoxic stress triggers DNA methylation events, while extracellular superoxide dismutase (EC-SOD) exhibits a significant capacity to mitigate RASSF1A gene methylation and alleviate hypoxia-induced cardiac fibrosis [27].
4.2. Oxidative stress affects histone modification
Previous investigations have provided evidence indicating that the activation of ROS-induced ataxia-telangiectasia mutated (ATM) protein kinase is evident in conditions of hypertrophy and heart failure [28]. In cardiomyocytes, OS triggers the activation of the ATM-mediated DNA damage response (DDR) signaling pathway, subsequently leading to the degradation of La-related protein 7 (LARP7). The downregulation or mutation of LARP7 elicits a reduction in SIRT1 expression, resulting in the acetylation of PGC-1α and impairing mitochondrial biogenesis and energy metabolism. Ultimately, these cascades contribute to the progression of heart failure, with cardiac fibrosis emerging as a critical pathological manifestation [29]. Adeno-associated virus (AAV)-mediated delivery of LARP7 emerges as a promising targeted therapeutic approach for addressing cardiac dysfunction.
Alterations in the redox status of conserved cysteine residues within class II histone deacetylases (HDACs) exert a significant impact on the cytoplasmic localization of these HDACs and the activity of crucial genes implicated in cardiomyocyte hypertrophy. These cysteine mutations are sufficient to induce cardiac hypertrophy [30]. Cardiomyocyte hypertrophy is a prominent feature of cardiac hypertrophy, which can be categorized into pathological hypertrophy and physiological hypertrophy. Pathological hypertrophy is triggered by disease stimuli such as stress overload or myocardial infarction, and myocardial fibrosis serves as a notable manifestation of pathological hypertrophy [31]. When exposed to OS, HDAC4 mutants rely on chromosome region maintenance protein 1 (CRM1) for their translocation into the cytoplasm, thereby abolishing the inhibitory effect of HDAC4 on cardiac hypertrophy in vivo. Interestingly, thioredoxin 1 (TRX1) plays a crucial role in monitoring and regulating redox signaling [16]. It forms a multiprotein complex with DnaJb5, thioredoxin-binding protein-2 (TBP-2), and importin (IMP) α, facilitating the degradation of key cysteine residues within HDAC4 and promoting the nuclear localization of HDAC4. Consequently, targeted redox modulation of HDAC4 represents a promising independent approach for the therapeutic management of cardiac hypertrophy.
4.3. Oxidative stress affects the regulation of non-coding RNAs
Moreover, increased levels of ROS have been shown to downregulate the expression of miR-29b-3p and miR-29c-3p by activating Smad3 signaling. This dysregulation of microRNAs contributes to the promotion of cardiac fibrosis in cardiac fibroblasts (CFs) by targeting TGF-β2 and MMP2 [32]. Notably, macrophage migration inhibitory factor (MIF) possesses antioxidant properties and has been found to inhibit ROS production and inhibit the activation of Smad3. As a result, MIF intervention restores the downregulation of miR-29b-3p and miR-29c-3p in CFs, thereby exerting a suppressive effect on cardiac fibrosis.
Another ROS-mediated mechanism involving microRNAs (miRNAs) has been identified. It is characterized by the ROS-induced redistribution of miRNAs between mitochondria and the cytoplasm, specifically in endothelial cells, leading to endothelial cell damage. Importantly, this redistribution does not significantly alter the overall expression of miRNAs in the cell. Among these redox-sensitive miRNAs, miR-381–3p has been implicated in ROS-induced endothelial injury through this mechanism. It exerts its effects by targeting specific genes, including lipoprotein receptor-related protein 6 (LRP6) and nuclear factor I-A (NFIA). By inhibiting these target genes, miR-381–3p promotes apoptosis and inhibits endothelial cell proliferation, contributing to the progression of endothelial dysfunction [33].
5. Epigenetic regulations of oxidative stress in cardiac fibrosis: focus on histone modification
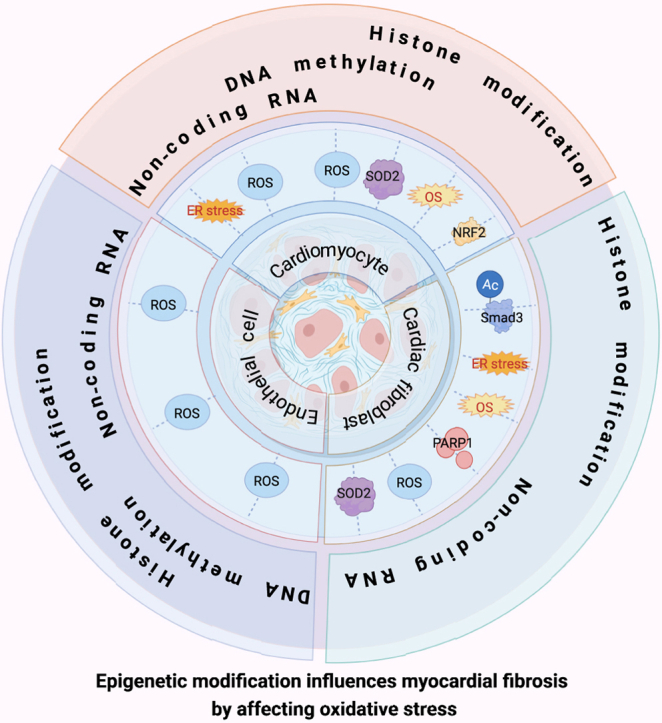
As previously mentioned, OS has been shown to induce alterations in the epigenetic landscape of cells. In the following sections, we discuss the epigenetic mechanisms that mediate OS. Additional insights and detailed information can be found in Fig. 2 and Table 2, which provide a comprehensive overview of these discoveries.
Fig. 2.

Epigenetic modification influences cardiac fibrosis by affecting oxidative stress
Several epigenetic mechanisms influence cardiac fibrosis progression by affecting oxidative homeostasis in vivo.
Table 2.
Epigenetic modification influences cardiac fibrosis by affecting oxidative stress.
| Cardiac fibroblast | Cardiomyocyte | Endothelial cell | |
|---|---|---|---|
| DNA methylation | −/− |
|
|
| Histone modification |
|
|
|
| Non-coding RNA |
|
|
|
One prominent epigenetic mechanism that has been extensively studied is histone modification. Fig. 3 illustrates the intricate regulation of OS by histone modifications. Among various histone modifications, acetylation and methylation have garnered significant attention due to their roles in modulating chromatin condensation, transcription factor binding, and transcription elongation [34]. These modifications are meticulously controlled and manipulated by three classes of proteins, often referred to as “writers” “erasers” or “readers” based on their specific functions [35].
Fig. 3.

Regulatory effect of histone modification on oxidative stress in cardiac fibrosis
Histone modifications affect the intracellular oxidative balance of various cardiac tissue cells such as cardiomyocytes, cardiac fibroblasts, and endothelial cells, thereby affecting the process of cardiac fibrosis. Among them, ATF4, peIF2α and CHOP were upregulated as markers of ER stress.
“Writers” encompass enzymes such as HATs and HMTs, which catalyze the addition of acetyl and methyl groups to histones, respectively. On the other hand, “erasers” include proteins like deacetylases and demethylases, which are responsible for removing acetyl and methyl groups from histones. Lastly, “readers” refer to proteins that bind to these modified histone marks, exerting their regulatory influence on chromatin organization and the activity of other proteins.
The intricate interplay between OS and histone modifications reveals the dynamic nature of epigenetic regulation in response to the cellular redox status. These complex mechanisms are pivotal in shaping the epigenome, exerting profound effects on gene expression, cellular functionality, and the pathogenesis of various diseases. Elucidating the intricate network of interactions between OS and histone modifications holds significant promise for advancing our understanding of epigenetic regulation and its therapeutic implications in diverse pathological contexts.
5.1. Histone deacetylation regulations of oxidative stress in cardiac fibrosis
Mitochondria plays a vital role in generating ROS within cells [36]. In recent years, the epigenetic mechanisms associated with SIRTs, particularly those involving mitochondrial proteins, have garnered considerable research interest. SIRTs are enzymes with nicotinamide adenine dinucleotide (NAD+)-dependent activity, and their activation is intricately regulated by the metabolic state of the cell.
Activation of SIRT1, for instance, leads to an elevation in adiponectin levels, which in turn stimulates mitochondrial biogenesis and enhances the antioxidant defense system. SIRT1 interacts with peroxisome PGC-1α at multiple lysine and deacetylation sites, resulting in increased transcription of genes involved in oxidative phosphorylation [37]. In response to stressful conditions, SIRT1 exerts its regulatory function by deacetylating forkhead box protein O1/3 (FOXO1/3), thereby regulating the expression of manganese-dependent superoxide dismutase and catalase antioxidants [38]. Furthermore, Activated SIRT1 inhibits Smad3 acetylation, OS, and activation of ER stress, thereby preventing the activation of cardiac fibroblasts. Geniposide (GE) can resist cardiac fibrosis through this mechanism, so it is expected to become a promising anti-cardiac fibrosis drug [39].
Moreover, Impairment or loss of cardiac SIRT1 contributes to the exacerbation of cardiac dysfunction and remodeling. Inhibition of SIRT1 reduces the nuclear translocation of NRF2, leading to a decrease in NRF2 translocation from the cytoplasm to the nucleus. This alteration affects the downstream Keap1 pathway, compromising the ability of NRF2 to effectively inhibit ROS in cardiomyocytes [40]. NRF2 primarily functions to maintain redox homeostasis [16] by transcriptionally regulating various genes involved in antioxidant defense, such as SOD, catalase (CAT), heme oxygenase 1 (Hmox-1), NAD(P)H quinone oxidoreductase-1, and TRX-1 [15]. Consequently, the levels of MDA, a marker of OS, increase, while the levels of SOD and GSH, key antioxidants, decrease. And honokiol (HKL), a potent cardioprotectant, exerts its beneficial effects by stimulating the SIRT1/NRF2 pathway, thereby reducing OS and ameliorating myocardial injury in diabetic rats [41]. Moreover, HKL also serves as a pharmacological activator of SIRT3 [42].
Activation of SIRT1 plays a crucial role in inhibiting angiotensin II (AngII)-induced ROS generation and promoting telomerase activity, thereby attenuating AngII-induced senescence in human umbilical vein endothelial cells (HUVECs) [43]. Notably, apelin, an adipocyte-derived factor, acts as an endogenous ligand and the activation of its receptors has been shown to ameliorate the endothelia in-induced HUVEC senescence through the AMPK/SIRT1 signaling pathway.
Previous studies have shown that moderate expression of SIRT1 confers protection against OS associated with cardiac hypertrophy and fibrosis. However, it is important to note that higher levels of SIRT1 expression may actually contribute to cardiomyopathy by promoting mitochondrial dysfunction [44]. Therefore, while activated SIRT1 holds promise as a potential therapy for cardiac fibrosis, careful consideration of dosage is necessary to achieve the desired therapeutic effects without causing adverse effects on mitochondrial function.
SIRT3 plays a crucial role as the major mitochondrial SIRT in regulating 8-oxoguanine DNA glycosylase (OGG1) and mitochondrial manganese superoxide dismutase [45]. Deficiency of SIRT3 leads to the downregulation of several mitochondrial proteins, including Mn-SOD and OGG1, resulting in elevated mitochondrial ROS levels and mitochondrial DNA (mtDNA) damage. A study has confirmed that SIRT3 is the predominant protein deacetylase in mitochondria, and its loss results in a nearly tenfold increase in acetylation modifications of mitochondrial proteins [46]. Loss of SIRT3 leads to hyperacetylation of the key mitochondrial antioxidant SOD2, which induces OS and promotes apoptosis in cardiomyocytes, leading to cardiac fibrosis. Neuraminidase 1 (NEU1) can exploit this mechanism to control OS, thereby reducing cardiac fibrosis and is therefore a potential therapeutic target [47]. As SIRT3 expression increased, SOD2 deacetylation decreased and SOD2 activity increased, limiting ROS accumulation. In addition, SIRT3 also regulates the expression of NAPDH oxidase through mitochondrial ROS [48].
Transient stimulation of nonischemic hypertrophy has been shown to enhance the cardiac resilience to subsequent ischemic stress, and this process is partly mediated by SIRT3. SIRT3 plays a crucial role by directly binding to and deacetylating Isocitrate dehydrogenase 2 (IDH2), resulting in increased IDH2 activity. The activation of IDH2 could inhibit mitochondrial ROS production and mitigates mitochondria-dependent apoptosis through the production of mitochondrial nicotinamide adenine dinucleotide phosphate (NADPH) [49].
However, emerging evidence suggests that SIRT4 plays a contrasting role. In cardiomyocytes, SIRT4 inhibits SIRT3-mediated activation of Mn-SOD, resulting in increased ROS levels that contribute to the development of pathological cardiac hypertrophy. This mechanism may involve a competitive interaction between SIRT4 and Mn-SOD for binding to SIRT3 [50]. Therefore, targeting the inhibition of SIRT4-mediated OS presents a potential therapeutic strategy for mitigating pathological hypertrophy and managing heart failure. Further studies are warranted to elucidate the precise molecular mechanisms underlying the role of SIRT4 in cardiac pathophysiology.
Additionally, lisinopril, a novel non-sulfhydryl angiotensin-converting enzyme (ACE) inhibitor, has been shown to enhance the antioxidant defense system in animals and humans, offering protection against oxidative damage and fibrosis in human cardiomyocytes through epigenetic mechanisms [51]. Following treatment with lisinopril, activation of SIRT1 and SIRT6 signaling pathways has been observed, imparting a protective effect against OS and fibrosis in human AC16 cardiomyocytes. Notably, the expression of antioxidative stress proteins such as catalase, SOD2, and TRX was significantly increased in cardiomyocytes, while key proteins implicated in cardiac fibrosis, namely osteopontin and Galectin-3, were significantly decreased.
SIRTs have emerged as crucial regulators of OS and have garnered significant attention in the study of metabolic disorders due to their involvement in epigenetic modifications within mitochondria. Further investigations into the intricate interplay between SIRTs, OS, and epigenetic regulation offer considerable potential for advancing our knowledge of cardiac fibrosis diseases and uncovering novel therapeutic strategies.
5.2. Histone methylation regulations of oxidative stress in cardiac fibrosis
Metastasis-associated lung adenocarcinoma transcript 1 (MALAT1), a lncRNA, has emerged as a crucial regulator of apoptosis in diabetic cardiomyopathy through its interaction with enhancer of EZH2. MALAT1 facilitates the recruitment of EZH2 to the myocardial nucleus, thereby promoting EZH2 activity. Consequently, EZH2 suppresses the expression of ubiquitin-specific peptidase 22 (USP22) by modifying H3K27me3, leading to enhanced OS, inflammation, and apoptosis in rat cardiomyocytes [52]. Moreover, MALAT1 has been observed to interact with EZH2 in cardiac microvascular endothelial cells. Genetic and biochemical studies have demonstrated the regulatory role of histone methylation in DNA methylation, while histone acetylation influences histone methylation dynamics. These intricate epigenetic modifications collectively contribute to the complex regulatory landscape underlying cardiomyopathy [53].
6. Epigenetic regulations of oxidative stress in cardiac fibrosis: focus on DNA methylation
DNA methylation, as depicted in Fig. 4, is a crucial epigenetic mechanism involved in the modulation of cardiac fibrosis under conditions of OS. It is the earliest and extensively studied epigenetic modification, exerting regulatory control over various cellular processes such as transcriptional regulation, transposon silencing, maintenance of genomic imprinting, and X chromosome inactivation. Methylation-induced gene silencing entails a complex interplay of DNA-protein and protein-protein interactions, along with a cascade of enzymatic activities responsible for dynamic changes in DNA methylation patterns. Key players in this process include DNMTs and TET enzymes, which collectively orchestrate the delicate balance of DNA methylation dynamics. Understanding the intricate mechanisms underlying DNA methylation-mediated gene regulation holds immense potential in unraveling the pathogenesis of cardiac fibrosis and may pave the way for the development of targeted interventions.
Fig. 4.

Regulatory effect of DNA methylation on oxidative stress in cardiac fibrosis
In cardiomyocytes and endothelial cells, DNA methylation mediates the expression of key proteins that regulate OS, thereby affecting cellular oxidative balance and consequently cardiac fibrosis.
Within cardiomyocytes, DNMT2 has been observed to induce DNA methylation within the promoter region of the glutathione peroxidase 1 (GPX1) gene, consequently diminishing the expression of GPX1. As a representative selenoprotein, GPX1 plays a key role in reducing organic hydrogen peroxide to water or alcohols utilizing GSH as a reducing agent. Insufficient GPX1 levels lead to compromised antioxidant defense, cellular dysfunction, and apoptosis [54], thereby facilitating the progression of fibrosis. Consequently, selenium supplementation emerges as a potential therapeutic strategy to ameliorate the levels of ROS and enhance cardiac function. By replenishing selenium levels, it is hypothesized that ROS generation can be reduced, culminating in an improvement in overall cardiac health. Further investigations in this domain hold promise for the development of targeted therapeutic interventions aimed at mitigating OS-related pathologies in the heart.
Salvianolic acid B (Sal B) has been demonstrated to exert beneficial effects on diabetic cardiomyopathy (DCM) by promoting angiogenesis, as well as attenuating cardiac fibrosis and remodeling through the inhibition of IGFBP3 [55]. The underlying molecular mechanism involves Sal B's ability to enhance DNA methylation within the promoter region of IGFBP3 under hypoxic conditions. This modification leads to the translocation of IGFBP3 from the nucleus to the cytoplasm, subsequently resulting in its downregulation. As a consequence, cardiac angiogenesis is promoted, cardiac fibrosis is reduced, and overall heart function is improved [56]. Previous studies have indicated that IGFBP3 is implicated in OS regulation and apoptosis in various tissues [57,58], yet its involvement in cardiac fibrosis has remained unclear. Further investigations are warranted to elucidate the precise role of IGFBP3 in the pathogenesis of cardiac fibrosis and to explore its potential as a therapeutic target for this condition.
7. Epigenetic regulations of oxidative stress in cardiac fibrosis: focus on non-coding RNAs
Non-coding RNAs (ncRNAs) consist of a variety of RNA molecules not normally involved in protein coding. This group encompasses various types of ncRNAs, including microRNAs, siRNAs, lncRNAs, and circRNAs. Importantly, these ncRNAs play pivotal roles in mediating genetic changes. By exerting regulatory functions at the epigenetic levels, ncRNAs contribute to the intricate molecular mechanisms underlying the interplay between OS and the development of cardiac fibrosis, as depicted in Fig. 5.
Fig. 5.

Regulatory effect of non-coding RNA on oxidative stress in cardiac fibrosis
In cardiomyocytes and endothelial cells, non-coding RNAs mediate the expression of key proteins that regulate OS, thereby affecting cellular oxidative balance and consequently cardiac fibrosis.
7.1. MiRNA regulations of oxidative stress in cardiac fibrosis
miRNAs have emerged as promising targets and regulators of OS-related signaling pathways, exerting their effects by binding to the 3′-UTR of target mRNAs. Notably, various miRNAs have been implicated in the regulation of ROS production in the heart. Among these miRNAs are miR-21, miR-30, miR-34a, miR-144, miR-140–5p, miR-181, miR-378, miR-421, and miR-451. These miRNAs play crucial roles in modulating ROS levels by directly or indirectly targeting genes involved in ROS generation, detoxification, or antioxidant defense mechanisms.
Moreover, a decrease in miR-101–3p levels in endothelial cells has been associated with an upregulation of Bim expression, an important initiator of the intrinsic apoptosis pathway. Bim interacts with B-cell lymphoma-2 (Bcl-2), leading to the release of Bcl-2-associated X (Bax) and subsequent promotion of ROS production. Endothelial cell apoptosis is then induced by NLRP3 inflammasome-mediated activation of caspase-1/7 [59]. On the contrary, another study demonstrated that miR-101 can indirectly enhance Bim expression through EZH2-dependent epigenetic regulation, thereby sensitizing tumor cells to apoptosis induced by chemotherapeutic drugs [60]. These observations highlight the role of miR-101 in modulating apoptotic pathways and provide insights into potential therapeutic strategies targeting miR-101 and its downstream effectors to regulate apoptosis and ROS-related processes in various pathological contexts.
miR-134–5p has been found to be highly expressed in a mouse model of myocardial infarction. Recent studies have elucidated that miR-134–5p plays a role in regulating ROS levels through its interaction with lysine acetyltransferase 7 (KAT7), a HAT responsible for acetylating histone H3 at lysine 14 (H3K14Ac) in Mn-SOD and catalase genes. Mechanistically, miR-134–5p suppresses the expression of KAT7, leading to reduced levels of Mn-SOD and catalase, which in turn promotes ROS accumulation. This dysregulation of ROS homeostasis ultimately contributes to the activation of cardiac fibroblasts, processes associated with cardiac fibrosis and adverse remodeling [61].
Inhibition of miR-194–5p has been demonstrated to mitigate doxorubicin (Dox)-induced cardiomyocyte apoptosis. Specifically, miR-194–5p directly targets and suppresses the expression of p21-activated kinase 2 (PAK2). PAK2 has been previously reported to exert cardioprotective effects by enhancing ER function through the activation of the IRE1/XBP1 signaling pathway. Activation of XBP1, in turn, leads to upregulation of ER chaperones, alleviation of ER stress, and promotion of cell survival. Upon treatment with Dox, there is a dynamic alteration in the expression of XBP1s, with an initial increase followed by a subsequent decrease. Notably, inhibition of miR-194–5p results in the overexpression of XBP1, thereby restoring its levels even after Dox treatment. Consequently, this leads to the inhibition of dox-induced caspase 3/7 activity and an increased expression of cleaved caspase 12, ultimately culminating in the protection of cardiomyocytes from apoptotic cell death [62].
miR-340–5p has been identified as a suppressor of myeloid cell leukemia 1 (Mcl-1) expression. Notably, Mcl-1 possesses a unique ability to impede ROS formation by inhibiting the upregulation of pro-oxidants through NOX4. Consequently, overexpression of miR-340–5p exacerbates mitochondrial dysfunction and increases OS, ultimately leading to enhanced apoptosis. Furthermore, miR-340–5p also modulates the expression levels of key apoptotic regulators, including Bim, Bax, and cleaved caspase 3, while reducing the expression of the anti-apoptotic protein Bcl-2. These molecular changes collectively contribute to the protection of cardiomyocytes from apoptosis [63].
MiR-4732–3p has emerged as a potential mediator of cardioprotective mechanisms in rat cardiac cells and cardiac fibroblasts, primarily through its ability to enhance antioxidant responses. Notably, miR-4732–3p has been shown to upregulate the mRNA levels of two critical transcription factors, Nfe2l2 and NRF1. These transcription factors are known to regulate genes encoding proteins involved in the cellular response to free radical damage [64].
7.2. LncRNA regulations of oxidative stress in cardiac fibrosis
In the context of DCM, recent investigations have revealed elevated expression of the long non-coding RNA lncDACH1 in DCM hearts and cardiomyocytes exposed to high glucose levels. Notably, knockdown of lncDACH1 has demonstrated favorable effects, including the reduction of mitochondrial OS, apoptosis, cardiac fibrosis, and hypertrophy, ultimately improving heart function in DCM mice.
Further studies have shed light on the underlying mechanisms by which lncDACH1 exerts its effects. It has been discovered that lncDACH1 directly interacts with SIRT3. This interaction facilitates the binding of SIRT3 to E3 ligases or other components of the ubiquitin-proteasome system, thereby promoting the ubiquitination process. This ubiquitination leads to the degradation of SIRT3, resulting in decreased activity of Mn-SOD, a crucial antioxidant enzyme. As a consequence, the imbalanced redox state leads to increased levels of ROS, ultimately causing cellular injury and potentially triggering apoptosis in mouse cardiomyocytes [65].
7.3. CircRNA regulations of oxidative stress in cardiac fibrosis
CircRNAs localized within mitochondria have emerged as critical regulators of mitochondrial ROS production. Among these, circSamd4 is under the transcriptional control of the NRF2 transcription factor, which binds to the promoter region of the circSamd4 host gene. Functionally, circSamd4 mitigates OS by facilitating the translocation of valosin-containing proteins (VCPs) to the mitochondria, thereby maintaining mitochondrial dynamics. This process leads to a reduction in the expression of voltage-dependent anion channel 1 (VDAC1) and blocks the opening of the mitochondrial permeability transition pore (mPTP). Consequently, mitochondrial OS and subsequent oxidative DNA damage are alleviated, inducing cardiomyocyte (CM) proliferation and preventing CM cell apoptosis when CircSamd4 was overexpressed, and the area of fibrosis was also smaller. Conversely, silencing circSamd4 has the opposite effect, exacerbating OS, impairing mitochondrial function, and promoting adverse cardiac remodeling [66].
Moreover, circular RNA itchy E3 ubiquitin protein ligase (CircITCH), a tumor suppressor with broad-spectrum functionality, has emerged as a key player in doxorubicin-induced cardiotoxicity. The protein ITCH is an important enzyme involved in the transfer of ubiquitin from E2 ubiquitin-conjugating enzymes to specific protein substrates, thereby marking them for lysosomal degradation. CircITCH upregulates SIRT6 by sponge filtering mmir-330–5p in the mouse heart [67]. SIRT6, in turn, mitigates OS by activating NRF2 [68] and Mn-SOD2, both of which are crucial endogenous defense molecules against oxidative damage. Additionally, SIRT6 enhances DNA damage repair by activating PARP1, a pivotal enzyme involved in DNA repair mechanisms [69]. Collectively, these molecular events culminate in improved cardiomyocyte survival and enhanced cellular resilience in the face of doxorubicin-induced cardiotoxicity.
8. Conclusions and future perspective
Cardiac fibrosis is a prevalent pathological complication of various heart diseases, including cardiac hypertrophy, diabetic cardiomyopathy, coronary heart disease (CHD), hypertensive heart disease, and heart failure. Different types of myocardial fibrosis, such as interstitial, periarteriolar, or “replacement” fibrosis, are associated with distinct underlying pathophysiological processes, which poses challenges in developing therapeutics for myocardial fibrosis [31]. Cardiac hypertrophy is characterized by the prominent presence of cardiac interstitial fibrosis. On the other hand, diabetic cardiomyopathy is characterized by diffuse myocardial fibrosis (interstitial fibrosis at the microscopic level), along with the occurrence of endothelial-to-mesenchymal transition (EndMT) and cardiac hypertrophy [70]. In the context of CHD, studies often investigate models of myocardial infarction and myocardial ischemia-reperfusion, where OS-induced cardiomyocyte apoptosis plays a significant role in disease progression. Replacement or scar fibrosis, which involves the formation of fibrotic tissue to replace damaged or necrotic cardiomyocytes [71], is a common occurrence in CHD models. In hypertension, fibrosis is initiated in the perivascular space [72], and it has been demonstrated that hypertensive heart disease is characterized by predominant perivascular fibrosis in both human and murine models [73].
Cardiac fibrosis will inflict significant damage, and currently lacks effective preventive or reversal treatments. ROS play a crucial role in the pathogenesis of cardiac fibrosis, orchestrating several pathological changes such as cardiomyocyte apoptosis, heightened activation of cardiac fibroblasts, and increased senescence and damage in endothelial cells, collectively contributing to fibrosis progression. Recent studies have increasingly implicated OS and its interplay with epigenetic modifications in the context of cardiac fibrosis. This comprehensive review summarizes the latest research in this area and proposes that OS may influence cardiac fibrosis through its modulation of epigenetic mechanisms, while epigenetic modifications may, in turn, ameliorate cardiac fibrosis by attenuating OS. Nevertheless, the precise underlying mechanisms are not yet fully elucidated, and given the overlap of various epigenetic pathways, further investigations are warranted to determine the impact of epigenetic-mediated OS on the fibrotic process.
Furthermore, it is important to acknowledge the signaling function of ROS, as they serve as essential mediators of cellular signaling and regulation. Thus, it is crucial to recognize the multifaceted role of ROS when designing therapeutic interventions. In conclusion, targeting epigenetic OS for the treatment of fibrosis shows promise as a therapeutic direction. However, there is a need to explore other epigenetic mechanisms involved in the regulation of OS, as well as additional bioactive substances governed by epigenetic modifications. Further experimental and clinical studies are essential to ascertain the potential of epigenetic-based therapeutic strategies in addressing OS during the progression of cardiac fibrosis.
Author contributions
All authors conceived the manuscript structure and contributed to the writing and editing.
Funding
This project was supported by National Natural Science Foundation of China (82170236, 81700212), Key research and development projects of Anhui Province (202104j07020037), Translational medicine research project of Anhui Province (2021zhyx-C61), Excellent Top Talents Program of Anhui Province Universities (gxyqZD2022023), and National Natural Science Foundation Incubation Program of the Second Affiliated Hospital of Anhui Medical University (2020GMFY02). Postgraduate Innovation Research and Practice Program of Anhui Medical University (YJS20230083, YSJ20230082).
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This project was supported by National Natural Science Foundation of China (82170236, 81700212), Key research and development projects of Anhui Province (202104j07020037), Translational medicine research project of Anhui Province (2021zhyx-C61), Excellent Top Talents Program of Anhui Province Universities (gxyqZD2022023), and National Natural Science Foundation Incubation Program of the Second Affiliated Hospital of Anhui Medical University (2020GMFY02). Postgraduate Innovation Research and Practice Program of Anhui Medical University (YJS20230083, YSJ20230082).
Contributor Information
Jing-Jing Yang, Email: yangjingjing@ahmu.edu.cn.
Ye Zhang, Email: zhangy@ahmu.edu.cn.
Jian-Yuan Zhao, Email: zhaojy@vip163.com.
Hui Tao, Email: taohui@ahmu.edu.cn.
Data availability
Data will be made available on request.
References
- 1.Frangogiannis N.G. Cardiac fibrosis. Cardiovascular research. 2021;117(6):1450–1488. doi: 10.1093/cvr/cvaa324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shih Y.C., Chen C.L., Zhang Y., Mellor R.L., et al. Endoplasmic reticulum protein TXNDC5 augments myocardial fibrosis by facilitating extracellular matrix protein folding and redox-sensitive. Cardiac Fibroblast Activation. 2018;122(8):1052–1068. doi: 10.1161/CIRCRESAHA.117.312130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dobaczewski M., Bujak M., Li N., Gonzalez-Quesada C., et al. Vol. 107. 2010. pp. 418–428. (Smad3 Signaling Critically Regulates Fibroblast Phenotype and Function in Healing Myocardial Infarction). 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hsia T.Y., Ringewald J.M., Stroud R.E., Forbus G.A., et al. Vol. 13. 2011. pp. 271–277. (Determinants of Extracellular Matrix Remodelling Are Differentially Expressed in Paediatric and Adult Dilated Cardiomyopathy,). 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dimauro I., Paronetto M.P., Caporossi D. Exercise, redox homeostasis and the epigenetic landscape. Redox Biol. 2020;35 doi: 10.1016/j.redox.2020.101477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van der Pol A., van Gilst W.H., Voors A.A., van der Meer P. Treating Oxidative Stress in Heart Failure: Past, Present and Future, J European journal of heart failure. 2019;21:425–435. doi: 10.1002/ejhf.1320. 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kang L.L., Zhang D.M., Jiao R.Q., Pan S.M., et al. 2019. Pterostilbene Attenuates Fructose-Induced Myocardial Fibrosis by Inhibiting ROS-Driven Pitx2c/miR-15b Pathway. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aragno M., Mastrocola R., Medana C., Catalano M.G., et al. Oxidative stress-dependent impairment of cardiac-specific transcription factors in experimental. Diabetes. 2006;147(12):5967–5974. doi: 10.1210/en.2006-0728. [DOI] [PubMed] [Google Scholar]
- 9.Martins D., Garcia L.R., Queiroz D.A.R., Lazzarin T., et al. Oxidative stress as a therapeutic target of cardiac. Remodeling. 2022;11(12) doi: 10.3390/antiox11122371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O'Geen H., Ren C., Nicolet C.M., Perez A.A., et al. Vol. 45. 2017. pp. 9901–9916. (dCas9-based Epigenome Editing Suggests Acquisition of Histone Methylation Is Not Sufficient for Target Gene Repression). 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pan L., Wang H., Luo J., Zeng J., et al. Vol. 33. 2019. pp. 14159–14170. (Epigenetic Regulation of TIMP1 Expression by 8-oxoguanine DNA Glycosylase-1 Binding to DNA:RNA Hybrid). 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bruvere I., Bisenieks E., Poikans J., Uldrikis J., et al. 2017. Dihydropyridine Derivatives as Cell Growth Modulators in Vitro. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kietzmann T., Petry A., Shvetsova A., Gerhold J.M., et al. The epigenetic landscape related to reactive oxygen species formation in the cardiovascular system. Br. J. Pharmacol. 2017;174(12):1533–1554. doi: 10.1111/bph.13792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vlad M.L., Manea S.A., Lazar A.G., Raicu M., et al. Vol. 2019. 2019. (Histone acetyltransferase-dependent pathways mediate upregulation of NADPH oxidase 5 in human macrophages under inflammatory conditions: A Potential Mechanism of Reactive Oxygen Species Overproduction in Atherosclerosis). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Waldman M., Cohen K., Yadin D., Nudelman V., et al. Vol. 17. 2018. p. 111. (Regulation of Diabetic Cardiomyopathy by Caloric Restriction is Mediated by Intracellular Signaling Pathways Involving “SIRT1 and PGC-1α”). 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mikhed Y., Görlach A., Knaus U.G., Daiber A. Redox regulation of genome stability by effects on gene expression, epigenetic pathways and DNA damage/repair. Redox Biol. 2015;5:275–289. doi: 10.1016/j.redox.2015.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chenxu G., Minxuan X., Yuting Q., Tingting G., et al. Vol. 19. 2018. pp. 147–157. (iRhom2 Loss Alleviates Renal Injury in Long-Term PM2.5-exposed Mice by Suppression of Inflammation and Oxidative Stress). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Forman H.J., Zhang H. Targeting oxidative stress in disease: promise and limitations of antioxidant therapy. J Nature reviews. Drug discovery. 2021;20(9):689–709. doi: 10.1038/s41573-021-00233-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sies H., Jones D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nature reviews. Molecular cell biology. 2020;21(7):363–383. doi: 10.1038/s41580-020-0230-3. [DOI] [PubMed] [Google Scholar]
- 20.Matsuzaki S., Eyster C., Newhardt M.F., Giorgione J.R., et al. Insulin signaling alters antioxidant capacity in the diabetic. Heart. 2021;47 doi: 10.1016/j.redox.2021.102140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang X., Lin X., Wang L., Xie Y., et al. Vol. 203. 2022. (Substitution of SERCA2 Cys 674 Aggravates Cardiac Fibrosis by Promoting the Transformation of Cardiac Fibroblasts to Cardiac Myofibroblasts). [DOI] [PubMed] [Google Scholar]
- 22.Chen H., Fan L., Peng N., Yin Y., et al. Galunisertib-loaded gelatin methacryloyl hydrogel microneedle patch for cardiac repair after myocardial. Infarction. 2022;14(36):40491–40500. doi: 10.1021/acsami.2c05352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reddy M.M., Fernandes M.S., Salgia R., Levine R.L., et al. Vol. 25. 2011. pp. 281–289. (NADPH Oxidases Regulate Cell Growth and Migration in Myeloid Cells Transformed by Oncogenic Tyrosine Kinases). 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hengartner M.O. The biochemistry of apoptosis. Nature. 2000;407(6805):770–776. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- 25.Valinluck V., Tsai H.H., Rogstad D.K., Burdzy A., et al. Vol. 32. 2004. pp. 4100–4108. (Oxidative Damage to Methyl-CpG Sequences Inhibits the Binding of the Methyl-CpG Binding Domain (MBD) of Methyl-CpG Binding Protein 2 (MeCP2)). 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Niu Y., DesMarais T.L., Tong Z., Yao Y., et al. Vol. 82. 2015. pp. 22–28. (Oxidative Stress Alters Global Histone Modification and DNA Methylation). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rajgarhia A., Ayasolla K.R., Zaghloul N., Lopez Da Re J.M., et al. Extracellular superoxide dismutase (EC-SOD) regulates gene methylation and cardiac fibrosis during chronic. Hypoxic Stress. 2021;8 doi: 10.3389/fcvm.2021.669975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guo Z., Kozlov S., Lavin M.F., Person M.D., et al. ATM activation by oxidative stress. 2010;330(6003):517–521. doi: 10.1126/science.1192912. [DOI] [PubMed] [Google Scholar]
- 29.Yu H., Zhang F., Yan P., Zhang S., et al. LARP7 Protects Against Heart Failure by Enhancing Mitochondrial Biogenesis. 2021;143(20):2007–2022. doi: 10.1161/CIRCULATIONAHA.120.050812. [DOI] [PubMed] [Google Scholar]
- 30.Ago T., Liu T., Zhai P., Chen W., et al. A redox-dependent pathway for regulating class II HDACs and cardiac hypertrophy. 2008;133(6):978–993. doi: 10.1016/j.cell.2008.04.041. [DOI] [PubMed] [Google Scholar]
- 31.Gao L., Guo S., Long R., Xiao L., et al. Lysosomal-associated protein transmembrane 5 functions as a novel negative regulator of pathological cardiac. Hypertrophy. 2021;8 doi: 10.3389/fcvm.2021.740526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liang J.N., Zou X., Fang X.H., Xu J.D., et al. Vol. 1865. 2019. pp. 2441–2450. (The Smad3-miR-29b/miR-29c axis Mediates the Protective Effect of Macrophage Migration Inhibitory Factor against Cardiac Fibrosis). 9. [DOI] [PubMed] [Google Scholar]
- 33.Guo Q., Yin X., Gao J., Wang X., et al. Vol. 67. 2020. (MiR-381-3p Redistributes between Cytosol and Mitochondria and Aggravates Endothelial Cell Injury Induced by Reactive Oxygen Species). [DOI] [PubMed] [Google Scholar]
- 34.Papait R., Serio S., Condorelli G. Role of the Epigenome in Heart Failure. J Physiological reviews. 2020;100(4):1753–1777. doi: 10.1152/physrev.00037.2019. [DOI] [PubMed] [Google Scholar]
- 35.Gillette T.G., Hill J.A. Readers, writers, and erasers: chromatin as the whiteboard of heart disease. J Circulation res. 2015;116(7):1245–1253. doi: 10.1161/CIRCRESAHA.116.303630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang L., Huang H., Fan Y., Kong B., et al. 2014. (Effects of downregulation of microRNA-181a on H2O2-Induced H9c2 Cell Apoptosis via the Mitochondrial Apoptotic Pathway). 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lai T.C., Lee Y.L., Lee W.J., Hung W.Y., et al. Vol. 14. 2022. (Synergistic Tumor Inhibition via Energy Elimination by Repurposing Penfluridol and 2-Deoxy-D-Glucose in Lung Cancer). 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Susanti V.Y., Sasaki T., Yokota-Hashimoto H., Matsui S., et al. . 22. 2014. pp. 2115–2119. (Sirt1 Rescues the Obesity Induced by Insulin-Resistant Constitutively-Nuclear FoxO1 in POMC Neurons of Male Mice). 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li N., Zhou H., Ma Z.G., Zhu J.X., et al. Vol. 9. 2018. p. 854. (Geniposide Alleviates Isoproterenol-Induced Cardiac Fibrosis Partially via SIRT1 Activation in Vivo and in Vitro). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang W., Zhong X., Fang Z., Li J., et al. Cardiac sirtuin1 deficiency exacerbates ferroptosis in doxorubicin-induced cardiac injury through the Nrf2/Keap1 pathway. null. 2023 doi: 10.1016/j.cbi.2023.110469. [DOI] [PubMed] [Google Scholar]
- 41.Zhang B., Zhai M., Li B., Liu Z., et al. 2018. Honokiol Ameliorates Myocardial Ischemia/Reperfusion Injury in Type 1 Diabetic Rats by Reducing Oxidative Stress and Apoptosis through Activating the SIRT1-Nrf2 Signaling Pathway. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu J., Tang M., Li T., Su Z., et al. Honokiol ameliorates post-myocardial infarction heart failure through ucp3-mediated reactive oxygen species. Inhibition. 2022;13 doi: 10.3389/fphar.2022.811682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang R., Fang W., Liang J., Lin C., et al. Vol. 14. 2018. pp. 725–734. (Apelin/APJ axis Improves Angiotensin II-Induced Endothelial Cell Senescence through AMPK/SIRT1 Signaling Pathway). 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alcendor RR, Gao S, Zhai P, Zablocki D, et al., Sirt1 regulates aging and resistance to oxidative stress in the heart, 100(10) (2007) 1512-1521. [DOI] [PubMed]
- 45.Parodi-Rullán R.M., Chapa-Dubocq X., Rullán P.J., Jang S., et al. High sensitivity of SIRT3 deficient hearts to ischemia-reperfusion is associated with mitochondrial. Abnormalities. 2017;8:275. doi: 10.3389/fphar.2017.00275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen Y., Fu L.L., Wen X., Wang X.Y., et al. Vol. 5. 2014. (Sirtuin-3 (SIRT3), a Therapeutic Target with Oncogenic and Tumor-Suppressive Function in Cancer). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guo Z., Tuo H., Tang N., Liu F.Y., et al. Vol. 18. 2022. pp. 826–840. (Neuraminidase 1 Deficiency Attenuates Cardiac Dysfunction, Oxidative Stress, Fibrosis, Inflammatory Via AMPK-SIRT3 Pathway in Diabetic Cardiomyopathy Mice). 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rathore R., Zheng Y.M., Niu C.F., Liu Q.H., et al. . 45. 2008. pp. 1223–1231. (Hypoxia Activates NADPH Oxidase to Increase [ROS]i and [Ca2+]i through the Mitochondrial ROS-PKCepsilon Signaling axis in Pulmonary Artery Smooth Muscle Cells). 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ma L., Shi H., Li Y., Gao W., et al. Vol. 66. 2021. pp. 2099–2114. (Hypertrophic Preconditioning Attenuates Myocardial Ischemia/reperfusion Injury through the Deacetylation of Isocitrate Dehydrogenase 2). 20. [DOI] [PubMed] [Google Scholar]
- 50.Luo Y.X., Tang X., An X.Z., Xie X.M., et al. Vol. 38. 2017. pp. 1389–1398. (SIRT4 Accelerates Ang II-Induced Pathological Cardiac Hypertrophy by Inhibiting Manganese Superoxide Dismutase Activity). 18. [DOI] [PubMed] [Google Scholar]
- 51.Scisciola L., Fontanella R.A., Surina, Garofalo G., et al. Potential role of lisinopril in reducing atherosclerotic risk: evidence of an antioxidant effect in human cardiomyocytes. Cell Line. 2022;13 doi: 10.3389/fphar.2022.868365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xu H., Ye W., Shi B. J cardiovascular toxicology, LncRNA MALAT1 regulates USP22 expression through EZH2-mediated H3K27me3 modification to accentuate sepsis-induced myocardial. Dysfunction. 2022;22(9):813–830. doi: 10.1007/s12012-022-09758-2. [DOI] [PubMed] [Google Scholar]
- 53.Ha M., Ng D.W., Li W.H., Chen Z.J. Coordinated Histone Modifications Are Associated with Gene Expression Variation within and between Species. J Genome res. 2011;21:590–598. doi: 10.1101/gr.116467.110. 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhu H., Wang X., Meng X., Kong Y., et al. Selenium supplementation improved cardiac functions by suppressing DNMT2-mediated GPX1 promoter DNA methylation in AGE-induced. Heart Fail. 2022 doi: 10.1155/2022/5402997. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li C.L., Liu B., Wang Z.Y., Xie F., et al. Vol. 139. 2020. pp. 98–112. (Salvianolic Acid B Improves Myocardial Function in Diabetic Cardiomyopathy by Suppressing IGFBP3). [DOI] [PubMed] [Google Scholar]
- 56.Hwang HJ, Kim N, Herman AB, Gorospe M, et al., Factors and Pathways Modulating Endothelial Cell Senescence in Vascular Aging, 23(17) (2022). [DOI] [PMC free article] [PubMed]
- 57.Wang S.H., Chen Y.L., Hsiao J.R., Tsai F.Y., et al. Vol. 40. 2021. (Insulin-like Growth Factor Binding Protein 3 Promotes Radiosensitivity of Oral Squamous Cell Carcinoma Cells via Positive Feedback on NF-κB/IL-6/ROS Signaling). 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yoo E.G., Lee W.J., Kim J.H., Chae H.W., et al. Vol. 152. 2011. pp. 3135–3142. (Insulin-like Growth Factor-Binding Protein-3 Mediates High Glucose-Induced Apoptosis by Increasing Oxidative Stress in Proximal Tubular Epithelial Cells). 8. [DOI] [PubMed] [Google Scholar]
- 59.Kim J.H., Lee D.K., Kim J., Choi S., et al. Vol. 8. 2017. (A miRNA-101-3p/Bim axis as a Determinant of Serum Deprivation-Induced Endothelial Cell Apoptosis). 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang J.G., Guo J.F., Liu D.L., Liu Q., et al. Vol. 6. 2011. pp. 671–678. (MicroRNA-101 Exerts Tumor-Suppressive Functions in Non-small Cell Lung Cancer through Directly Targeting Enhancer of Zeste Homolog 2). 4. [DOI] [PubMed] [Google Scholar]
- 61.Hao S., Sui X., Wang J., Zhang J., et al. Vol. 12. 2021. p. 848. (Secretory Products from Epicardial Adipose Tissue Induce Adverse Myocardial Remodeling after Myocardial Infarction by Promoting Reactive Oxygen Species Accumulation). 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fa H., Xiao D., Chang W., Ding L., et al. Vol. 9. 2022. (MicroRNA-194-5p Attenuates Doxorubicin-Induced Cardiomyocyte Apoptosis and Endoplasmic Reticulum Stress by Targeting P21-Activated Kinase 2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhu Y, Yang X, Zhou J, Chen L, et al., miR-340-5p mediates cardiomyocyte oxidative stress in diabetes-induced cardiac dysfunction by targeting Mcl-1, 2022 (2022) 3182931. [DOI] [PMC free article] [PubMed]
- 64.Sánchez-Sánchez R., Reinal I., Peiró-Molina E., Buigues M., et al. Vol. 11. 2022. (MicroRNA-4732-3p Is Dysregulated in Breast Cancer Patients with Cardiotoxicity, and its Therapeutic Delivery Protects the Heart from Doxorubicin-Induced Oxidative Stress in Rats). 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang Q., Li D., Dong X., Zhang X., et al. . 65. 2022. pp. 1198–1212. (LncDACH1 Promotes Mitochondrial Oxidative Stress of Cardiomyocytes by Interacting with Sirtuin3 and Aggravates Diabetic Cardiomyopathy). 6. [DOI] [PubMed] [Google Scholar]
- 66.Zheng H., Huang S., Wei G., Sun Y., et al. . 30. 2022. pp. 3477–3498. (CircRNA Samd4 Induces Cardiac Repair after Myocardial Infarction by Blocking Mitochondria-Derived ROS Output). 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Han D, Wang Y, Wang Y, Dai X, et al., The tumor-suppressive human circular RNA CircITCH sponges miR-330-5p to ameliorate doxorubicin-induced cardiotoxicity through upregulating SIRT6, Survivin, and SERCA2a, 127(4) (2020) e108-e125. [DOI] [PubMed]
- 68.Rezazadeh S., Yang D., Tombline G., Simon M., et al. Vol. 47. 2019. pp. 7914–7928. (SIRT6 Promotes Transcription of a Subset of NRF2 Targets by Mono-ADP-Ribosylating BAF170). 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mao Z., Hine C., Tian X., Van Meter M., et al. SIRT6 promotes DNA repair under stress by activating. PARP1. 2011;332(6036):1443–1446. doi: 10.1126/science.1202723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zheng X., Peng M., Li Y., Wang X., et al. Vol. 15. 2019. pp. 2393–2407. (Cathelicidin-related Antimicrobial Peptide Protects against Cardiac Fibrosis in Diabetic Mice Heart by Regulating Endothelial-Mesenchymal Transition). 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kishi S., Magalhaes T.A., George R.T., Dewey M., et al. Vol. 16. 2015. pp. 166–176. (Relationship of Left Ventricular Mass to Coronary Atherosclerosis and Myocardial Ischaemia: the CORE320 Multicenter Study). 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lieu M., Traynham C.J., de Lucia C., Pfleger J., et al. Vol. 318. 2020. pp. H1162–H1175. (Loss of Dynamic Regulation of G Protein-Coupled Receptor Kinase 2 by Nitric Oxide Leads to Cardiovascular Dysfunction with Aging). 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cooper S.T.E., Westaby J.D., Haines Z.H.R., Malone G.O., et al. Of mouse and man: cross-species characterization of hypertensive cardiac. Remodeling. 2022;23(14) doi: 10.3390/ijms23147709. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data will be made available on request.