

Abstract
The past decade has seen a number of impressive developments in predictive chemistry and reaction informatics driven by machine learning applications to computer-aided synthesis planning. While many of these developments have been made even with relatively small, bespoke data sets, in order to advance the role of AI in the field at scale, there must be significant improvements in the reporting of reaction data. Currently, the majority of publicly available data is reported in an unstructured format and heavily imbalanced toward high-yielding reactions, which influences the types of models that can be successfully trained. In this Perspective, we analyze several data curation and sharing initiatives that have seen success in chemistry and molecular biology. We discuss several factors that have contributed to their success and how we can take lessons from these case studies and apply them to reaction data. Finally, we spotlight the Open Reaction Database and summarize key actions the community can take toward making reaction data more findable, accessible, interoperable, and reusable (FAIR), including the use of mandates from funding agencies and publishers.
Introduction
Interest in big data continues to grow as artificial intelligence (AI) and automation change the way we conduct scientific research. While it may a be trite point to make, there are many problems where the lack of high-quality, publicly available data impedes research progress. Predictive chemistry and reaction informatics is one such field where there have been numerous impressive developments over the past few years despite only a handful of data sets being accessible to researchers, even commercially. Many of these developments are driven by applications of machine learning to, e.g., forward reaction prediction,1−5 retrosynthesis planning,6−13 and reaction condition prediction14−17 where predictions can be made even with relatively small data sets. We and others18−21 have asserted that to advance the role of AI in organic synthesis, there must be significant improvements in the reporting of laboratory synthesis procedures, including reaction conditions.
Our collective understanding of synthesis and synthetic outcomes is based on the experimental evidence we find in the journal and patent literature. First-principles models and post hoc analysis are used to rationalize experimental observations but hold limited predictive power beyond simple systems. High-fidelity data includes the products that were observed, their yields, selectivities, impurities, and other summary statistics under different experimental conditions. Rather than being hidden in lengthy Supporting Information PDFs, these data could be captured as digital files and code that can be published, versioned, and transferred between data platforms. This would enhance not only reproducibility in the field but also downstream machine learning applications of these data. As a long-term investment into the future of this field, we and others have recently launched the Open Reaction Database (ORD).22 The ORD is an initiative to support machine learning and related efforts in synthetic organic chemistry through standardized data formats and an open-access data repository. The scope of scientific challenges that we believe openly shared reaction data can help address is broad, from the familiar task of retrosynthesis to the holy grail of new reaction discovery.
There are a handful of curated data sets that have been used for training predictive chemistry models: the fully open “USPTO dataset”,23 Pistachio,24 Reaxys,25 and CAS26 offer broad data sets suitable for building “global” models, while focused data sets constrained to individual reaction types from high-throughput experimentation have helped build “local” models.27−29 These resources have contributed to the recent progress in the field but do not fully address the evolving needs of the community. First, there is an under-emphasis on quantitative aspects of reaction procedures that are essential for reproducibility and consideration of scale-up; details about concentrations, orders of addition, etc. are only available in the original manuscript or patent in an unstructured format. Second, there is a significant publication bias toward high-yield reaction examples, despite low-yield or low-conversion reactions being informative of substrate scope and compatibility. Third, an increasing amount of data is “born digital”, e.g., as a result of automated screening, which should not require conversion to an unstructured document to be disseminated. Fourth, much of the reaction data that is published is not openly accessible in either an unstructured or structured format.
We believe this is the right time for community-initiated action toward better data practices in chemistry. Our goals are more ambitious than addressing the logistics of data sharing: they include changing how information is treated and shared across the community. There are also shifting mandates from funding agencies (e.g., Plan S30) and recent guidance that federally funded research in the United States must make its products broadly available to the public by 2025.31 But, requiring data management plans and encouraging data sharing have not proved sufficient.32 In this Perspective, we discuss potential lessons to be learned from successful examples of data curation or sharing initiatives in the chemical sciences, how data sharing in organic chemistry may evolve with efforts like the ORD, and the perceived barriers to open science. We echo the call to action of Baldi21 and others,19,20 who advocate for the creation and adoption of community resources (like the ORD), and consider how we can jointly make the effort a success.
What Has Worked in the Past
We first consider what paradigms for data sharing have worked before and illustrate their timeline in Figure 1. How did PubChem come to be one of the world’s largest databases of open-access chemical information? How did the Protein Data Bank (PDB) and Cambridge Crystal Structure Database (CSD) come to be required for publication? What was ChEMBL’s path to becoming an invaluable source of assay data? How did nuclear magnetic resonance (NMR) line tables become standards for reporting analytical data? For each of these data sharing initiatives, we look into how the number of unique contributors has grown over time and what the process looks like for adding to the database. We present these case studies below.
Figure 1.

(top) Timeline of key dates surrounding the databases discussed in this work. (bottom) The growth of each database over time, excluding the ORD. Count is the exact number of entries according to each database (sources: CSD,33 PDB,34 PubChem,35−43 and ChEMBL44). Traces do not necessarily start close to 0 due to limited public information for early dates of some efforts.
A common observation is that adoption and growth generally takes a significant amount of time, and the initial timing is critical. Each of the CSD, PDB, and PubChem started at a time when relatively few structures or assays had been collected such that standards could be set early on. Furthermore, the enforcement of these standards by journal publishers and the scientific community has also driven adoption of these resources, particularly in the case of the CSD and the PDB.
We illustrate the growth in contributors or sources to each database in Figure 2. The databases can be grouped into three distinct models: first, there is the expert curation model (ChEMBL), where a small group of experts are gathering and curating the data; second, there is the contributor model with automatic validation (PubChem, PDB), where individual organizations or research groups are submitting and validating their own data; this is in contrast to a combined expert+automated review model, where contributions are subject to manual review in addition to automated validation (CSD). While the PDB and PubChem take similar approaches, any structural biologist with a new structure is allowed to upload and publish their data to the PDB, whereas only members of select organizations are allowed to publish data to PubChem. The number of structures deposited into the CSD has continued to keep pace with the growth of the other databases (Figure 1), suggesting that the combined expert+automated review strategy might be a feasible approach for validation of new reaction data moving forward.
Figure 2.

Timeline illustrating the growth in contributors over time for each database. Sources for the data are given in Figure 1. PubChem data on the individual number of contributors over time was not available; thus “sources” (i.e., organizations) are plotted instead. As ChEMBL does not follow a contributor model but an expert curation model, its growth in data sources is plotted instead. Finally, note that, while the CSD follows a contributor model, the submission process also includes manual review by domain experts at the CCDC. Traces do not necessarily start close to 0 due to limited public information for early dates of some efforts.
Throughout the development of these open-source databases, we notice a common philosophy that the collective use of data can be used to catalyze new knowledge and generate insights, and the principle that information developed with public funds must be made freely and publicly available.
The Cambridge Structural Database (CSD)
We begin by looking at the growth of the Cambridge Structural Database (CSD), a repository for small molecule and crystalline structures established in 1965.45 It began as a bootstrapped effort when the crystallography group led by Dr. Olga Kennard at the University of Cambridge collected published crystal structure data for all small molecules studied by X-ray and neutron diffraction. Between 1960 and 1965, the database contained hand- and computer-drawn chemical diagrams, bibliographic data, bond lengths, and bond angles. The team used professional editors to meticulously assemble and print these first two volumes but shifted to digital publication of the data with the development of the chemical connectivity file (CONN FILE). Besides easier encoding of atom, bond, and charge information, the CONN FILE also enabled more efficient substructure searching.46
During the 1980s, interest in the CSD boomed thanks in large part to the pharmaceutical and agrochemical industries, where students who had been using it on academic licenses went on to work at companies and requested the same resources be available in-house. Since its inception, >1 M crystal structures have been deposited in the CSD, including organic, metal–organic, and polymeric structures; the database is continuously updated with >50K structures each year and used in >70 countries. The CSD is a UK charity managed by the Cambridge Crystallographic Data Centre (CCDC), an independent organization that keeps close links to the University of Cambridge.45 While it remains open access for a small number of structures, the significant operating costs are paid for by commercial license fees charged for access to the full data and accompanying tools. The CSD does not depend on any core grant funding.
Currently, structures in the CSD are available for download in the CIF file format. Structures can be contributed to the CSD by individual researchers, and each entry undergoes an elaborate set of automated checks and manual curation by an expert in-house editor, allowing authors the opportunity to sort out any errors (of which there are many) before publication. Dr. Kennard believed such checks give people confidence in the tool.46 The Web site, which provides tools for advanced searching, 3D data mining, analysis, and visualization, also undergoes regular updates to keep up with the changing needs of users. Newly deposited structures are available to view with early access. These editorial and data curation processes, led by a core team of 7 scientists,47 are thought to be essential to the sustained utility of the database.48 That is not to say it is immune to the deposition of fraudulent data; in early 2022, Retraction Watch reported nearly 1000 entries as “concerning” after linking them to a paper mill,49 to which the CCDC responded by conducting a thorough investigation of the claims and eventually removing the fraudulent structures from the database.50
The process and tools used to create the CSD have been constantly evolving to keep up with new developments and user needs. While in the early days, the team manually transferred structure information onto punch cards and used knitting needles to pick up the similarities between structures, today the CSD uses automated workflows and software, allowing individual editors to focus their expertise where it has the most impact. Currently, an editor is able to curate 100 new structures a day, and it is estimated that over 400 person years have been invested in the curation of the CSD.51
As part of their commitment to promoting findable, accessible, interoperable, and reusable (FAIR) data principles, many journals in the fields of chemistry, materials science, and crystallography require crystal structure data deposition in the CSD, such as the ACS journals.52 These policies undoubtedly contribute to the continued growth and success of the CSD. While use of the CSD in the pharmaceutical and agrochemical industries is well-established, interest in the CSD continues to grow. It is quickly becoming a fundamental resource for research into new materials design, such as batteries and gas storage frameworks, and in recent years, the CCDC has noted a consistent increase in submissions from research taking place in China,53 illustrating the growing value and utility of the CSD for researchers worldwide.
Dr. Kennard attributed the immense success of the CSD to a few key decisions made early on in its development.46 She believed that it was critical that the CSD started at a time when there were relatively few published structures (∼8K by 1969), such that manual curation of these structures and their inclusion in the database was feasible. Early on, the novel ability to search the database was also introduced, improving its utility; this was initially done by dividing data on punch cards into ∼80 equal classes. Another unorthodox idea at the time was the use of internal coordinates and symmetry for storing data, as opposed to lists of atomic coordinates. This final point enabled CSD data to be used by noncrystallographers, catalyzing its widespread use. A recent presentation from Dr. Suzanna Ward at the CCDC mentions how the introduction of sketch searches in 199154 was “revolutionary.”55 Other CSD members have highlighted how faults in structures have been historically corrected by “vigilantes” who are each working in a particular area of interest (e.g., Richard Marsh and space group symmetry).56,57 Finally, Bruno et al.58 emphasize the importance of carefully preparing a detailed CIF file via automatic (CheckCIF, integrated into the CCDC deposition procedure) and expert curation of crystal structures: “No one enjoys this chore, but it produces tangible benefits for the crystallographic community and will become increasingly important as the productivity of crystallographers continues to rise.”
The Protein Data Bank (PDB)
The Protein Data Bank (PDB) was the first open access digital data resource in biology and medicine, established at Brookhaven National Laboratory (BNL) and the Cambridge Crystallographic Data Center during the 1971 Cold Spring Harbor Symposium on Quantitative Biology.59 It started with the few protein structures (ribonuclease, lysozyme, papain, lactate dehydrogenase, myoglobin, hemoglobin, carboxypeptidase, α-chymotrypsin, elastase, subtilisin, and trypsin) known at the time.60 At that particular year’s symposium, focused on the structure and function of proteins at the three-dimensional (3D) level, a meeting was organized by Max Perutz to discuss the easy availability and preservation of protein structure data with the handful of researchers who had coordinates to a protein structure.61 The goal of the meeting was to establish a means for researchers to share coordinates beyond their immediate collaborators and resulted in what is now known as the PDB.60−62
The PDB initially suffered from a lack of users who could provide valuable feedback on ease of use and desired features; however, this improved after the PDB moved from the Chemistry Department to the Biology Department at BNL.62 Nowadays, one of the biggest challenges in maintaining the PDB involves appropriately archiving not only the final inferred coordinates for a structure but also the raw data derived from an increasing variety of methods. The types of data deposited have also been steadily increasing in complexity. While there continues to be exponential growth in the number of deposited structures, the number of entries obtained from X-ray and NMR methods has begun to plateau; conversely, the number of structures obtained from electron microscopy (EM) methods has continued to increase every year.63 Presently over 200,000 structures in the PDB are available for download in the mmCIF (macromolecular CIF) file format, which replaced the legacy PDB format in 1997.
Part of the success of the PDB is that it has become required for publication in many prestigious scientific journals. The first example of such a policy came in the early 1970s, when the Journal of Biological Chemistry set a policy that, if a paper depended on a new set of coordinates, they had to be deposited into the PDB so as to encourage data accessibility and sharing (not to mention that it was effectively impossible to typeset the coordinates for large protein structures which further improved the appeal of a central data repository).61 In 1989, the International Union of Crystallography (IUCr) published their policy on and guidelines for data deposition, which endorsed the deposition of atomic coordinates and structure factor information in the PDB for scientific publications reporting crystallographic determinations of macromolecular structure.64 In 1998, Nature, Science, and PNAS instituted a policy that any paper containing new structural data received on or after October 1, 1998 would not be accepted without an accession number from the PDB, accompanied by an assurance that unrestricted or “layer-1” release would occur at or before the time of publication.59,65 Between 2008 and 2010, deposition of other experimental data including NMR chemical shifts also became mandatory; this was also a time in which NMR data saw improvements in standardization due to the first publication of NMR impurity tables.59
PubChem
PubChem is a public repository for chemical and biological data, launched in 2004 by the National Institutes of Health (NIH) as part of the Molecular Libraries Roadmap Initiative.42,66 It is maintained by the National Center for Biotechnology Information (NCBI) and organizes its data into three main databases: Substances, Compounds, and BioAssays. The primary intent was to make screening data for bioactive compounds easily and freely available to the public through the PubChem BioAssay database so as to benefit basic biological and preclinical research.67 PubChem is a very successful model for public repositories, as the database has grown rapidly with contributions from over 800 data sources around the world as of July 12, 2022.68 Data sources include organizations such as the US Food and Drug Administration (FDA) as well as other curation efforts (e.g., UniProt69) and journals.
PubChem provides web servers for accessing, retrieving, and analyzing biological data in its databases. It allows users to easily export assay results, either through the web interface or in bulk. Chemical structures are available in SDF, SMILES, and InChI formats, among others. In addition to the data tables, PubChem hosts tools that can be used to draw insights from data, including structure–activity analyses, and the ability to visualize data as a scatterplot or histogram. Data retrieval and analysis utilities in PubChem are continuously updated and expanded to improve data FAIR-ness, with the last major updates to the web interface published in 2019 so as to efficiently handle the needs of a very diverse user base. Currently, PubChem contains over 270 million assay outcomes for nearly 300 million substances, with millions of users per month.42 Assay metadata and various annotations are accessible via the Entrez search engine to help medical researchers connect information.66
A large part of PubChem’s success is due to the use of existing ontological frameworks wherever possible to semantically describe available information,70,71 including the integration of standard data sources into the database and powerful cross-referencing functionality. Existing frameworks included the Chemical Entities of Biological Interest (ChEBI) ontology,72 the CHEMical INFormation ontology (CHEMINF),73 and the Protein Ontology (PRO).74 Adoption of these and other core biomedical ontologies, followed by compliance with the shared set of evolving principles established by the Open Biomedical Ontologies (OBO) foundry, has helped ensure that the mapping of biochemical information available in PubChem will be compatible across multiple Semantic Web resources. To facilitate data retrieval and integration, PubChem also includes mappings between the NCBI protein GenInfo Identifier (“GI number”), GenBank accessions, and UniProt IDs.40 PubChem not only is well-integrated with other databases operated by the National Library of Medicine but also combines new data generated by the NIH with data available from other public sources, making it more powerful than a standalone tool. Early on, journals such as Nature Chemical Biology began automatically depositing data for compounds mentioned in their articles into PubChem and linking to the associated entries so that readers could consult the database for more information about the chemical structures and properties in question.75 Automated indexing of compounds can be facilitated through policies such as the Journal of Medicinal Chemistry’s requirement that authors submit a CSV listing the SMILES strings of all small molecules mentioned in a paper.
PubChem’s freely available services initially faced opposition by the American Chemical Society (ACS) due to purported competition with its Chemical Abstracts Service (CAS), which charged users a fee for its database.75 However, as it was eventually shown that PubChem and CAS overlapped relatively little in terms of content, scope, and resources, PubChem was allowed by the US Senate to continue as it was driven by the “primary goal of maximizing progress in science while avoiding unnecessary duplication and competition with private sector databases.”75 Data in PubChem can be deposited to the database by individual researchers at these organizations using the PubChem Upload tool. This tool enables researchers to submit new data to the PubChem Substance or PubChem BioAssay databases or to update existing data (e.g., chemical structures, experimental biological activity results, annotations, siRNA data, etc.). Users are then allowed to preview and review their data before publication.
ChEMBL
ChEMBL is a collection of drug discovery databases maintained by the EMBL European Bioinformatics Institute (EMBL-EBI).76 The database spun out of the transfer of a set of predictive drug discovery databases from BioFocus DPI, an early stage drug discovery company, to EMBL-EBI in July 2008, funded by a Wellcome Trust strategic award.77 The goal of EMBL-EBI was to make these databases, which included StARlite, CandiStore, and DrugStore, publicly available online to drug discovery researchers worldwide. StARlite was a large-scale structure–activity relationship (SAR) database of known compounds and their pharmacological effects extracted from primary literature; it performed extensive manual curation and automated indexing in-house and outsourced the data entry. CandiStore was a database of compounds in clinical-stage development and included data such as compound structure, synonyms, target, and highest development stage reached for use in drug repurposing. DrugStore was a database of known small molecule drugs and proteins that included their indications and targets.77
The Wellcome Trust made the transfer of all this data from the private into the academic sector possible by awarding £4.7 million to EMBL-EBI, which funded seven people for five years, as well as future data updates, improved curation, and integration with other genomics resources. The initial group was tasked with putting the new database online, building a brand, and doing outreach. These databases would allow researchers to track the progress of a compound from lead optimization, through clinical development, and then on to commercial launch. Databases were available as full downloads, web services, and via a user-friendly front end. It would eventually come to be integrated with other EMBL-EBI resources, such as UniProt, ChEBI, and IntAct.77 ChEMBL currently contains data on >15K targets and >2M distinct compounds, extracted from roughly 86K publications. The initiative is currently funded through a variety of public grants, including the European Commission and the NIH.78
A large part of ChEMBL’s success was having the first mover advantage for SAR databases. They established the data model and standards, which other users then had to follow. Most of the focus was on the back-end and data delivery aspects of the database, as opposed to the front-end user interfaces. It was initially heavily advertised at ACS and Gordon conferences as well as European equivalents. Notably, having a single, large funding source was helpful as it provided the team with five years to build up the database, release it, and build up a brand and body of users. Perhaps most important to its success is that ChEMBL added value; before ChEMBL, many companies had chemical databases but not SAR databases, and they lacked medicinal chemistry data in machine-readable form.
Rather than a contributor model, where information is solicited from individual researchers and then reviewed, the data in ChEMBL follows a data curation model and is extracted and curated from the primary scientific literature by a team of ∼20 researchers (Chemical Biology Services). Individual submissions by external users were judged to be too “painful” to process, perhaps due to variance in format and quality.79 The data is updated regularly, with new releases approximately every 3–4 months. In integrating new data, the team attempts to normalize the bioactivities into a uniform set of end-points and units where possible and to assign confidence levels to the links between a molecular target and a published assay. Currently, data on the clinical progress of compounds is being integrated into ChEMBL.80
Nuclear Magnetic Resonance (NMR) Line Tables
NMR data is an interesting case where a standardized data format has been defined without an accompanying centralized repository. NMR spectroscopy is widely used to study the structure of molecules in solution and their dynamics in the solid state. Though the publication of NMR data is now routine and fairly well-standardized, this was not always the case. The initial issues with NMR data reporting mirror many of our current challenges with reaction data reporting: they require both a standardized data format as well as guidelines for the sharing of the data. Seeing how NMR spectra reporting came to be standardized can provide insights into how we may take similar steps with reaction data.
NMR was first accurately measured in molecular beams in 1938 and in bulk materials in 1946.81 While it was initially believed that a given nucleus would show the same resonance frequency at a fixed magnetic field, regardless of what molecule the nucleus is a part of, experiments in the late 1940s showed that this was not the case. In fact, it was shown that the magnetic properties of the electrons surrounding a nucleus shield it from the applied magnetic field, leading to a shift in the anticipated resonance frequency; this came to be referred to as the chemical shift and is the basis for NMR spectroscopy as an analytical method in chemistry. Eventually, improved resolution in the spectra showed that many of the chemically shifted resonances were often collections of distinct resonances and that these split resonances were due to neighboring nuclei spins (spin–spin coupling).81 By the mid 1950s, primitive commercial NMR instruments became available, and the technology steadily improved over the subsequent decades. Great strides were made in the understanding of increasingly complex NMR spectra during this time to the point that computer programs were developed which could model chemical shifts.
As the applications of NMR spectroscopy boomed, the IUPAC Commission on Molecular Structure and Spectroscopy published a set of recommendations for the publication of proton NMR data in 1972, following this up with another set of recommendations in 1976 for spectra from other nuclei.82,83 This set of recommendations included conventions for graphical presentation of NMR data in chemistry journals (e.g., “a dimensionless scale factor for chemical shifts should be p.p.m.”, “the unit for measured data should be Hertz (cycles per second)”) as well as guidelines for the meta-data which should be provided (e.g., name of solvent used, concentration of solute, name and concentration of internal reference, etc.). These guidelines were established to ensure that NMR spectra were reported in a clear and unambiguous manner, whether in text or image form. Important in the development of standards was the publication of NMR impurity line tables, first published in 1997.84 These line tables compiled 1H and 13C chemical shifts for the most common trace impurities in organic chemistry in a variety of solvents. This publication was followed up in 2010 with an expanded table,85 which has become an essential reference for identifying known impurities in samples from NMR spectra.
NMR reporting guidelines are regularly updated to reflect changes in NMR technology. For instance, the current ACS guidelines, last updated in 2013, include instructions for structuring both 2D and non-2D NMR data in the experimental section (as structured strings) and in the Supporting Information (images of the processed and labeled spectra).86 The IUPAC also follows up with updated recommendations every few years, recommending, for instance, the use of tetramethylsilane (TMS) as a universal reference in 2001, and guidelines for reporting chemical shifts in solids in 2008.87,88 As a means of encouraging reproducibility and integrity in chemical research,89 journals such as The Journal of Organic Chemistry and Organic Letters have recently (2020) begun encouraging authors to submit the original data for NMR, which includes free induction decay (FID) files, acquisition data, and processing parameters as Supporting Information along with their submissions.90
There remains a broader need in the community for an open repository and associated tools needed to make it not only convenient but also rewarding for investigators to make their raw NMR data FAIR.91 The few existing initiatives for the open sharing of NMR data (e.g., nmrshiftdb292) have not seen widespread adoption.
Common Factors for Success
In Table 1, we summarize the main strategies which have contributed to the success of the aforementioned data sharing and standardization initiatives. Some strategies are shared across most initiatives, such as compliance with existing ontological frameworks wherever possible, while others, such as having a single large funding source backing the initiative, seem less important. In particular, we note that, while mandates for deposition as a requirement for publication are not currently in place for all the frameworks discussed herein, they can encourage faster adoption of structured data formats and quicker compliance with FAIR data sharing principles. The fact that these initiatives have grown so successfully is a testament to both the needs of the scientific community and the grit of the various contributors and reviewers involved.
Table 1. Strategies Employed in the Various Case Studies in This Perspective That Have Contributed to Each Initiative’s Successa.
| strategic decision | CSD | PDB | PubChem | ChEMBL | NMR |
|---|---|---|---|---|---|
| Cooperation with Scientific Journals | |||||
| Journals mandate deposition as requirement for publication | ⊗ | ⊗ | – | ⊗ | |
| Automatic deposition of data from journals | ○ | ⊗ | – | ||
| Links to research articles reporting the data | ○ | ○ | ○ | ○ | |
| Adaptability | |||||
| Regular updates of data sharing principles and recommendationsb | ⊗ | ⊗ | ⊗ | ⊗ | ⊗ |
| Integration and intercompatibility with other databasesc | ○ | ⊗ | ⊗ | – | |
| Ability for individual users to submit data entries | ○ | ○ | ⊗ | – | ○ |
| Ability for individual users to update data entries | ⊗ | – | |||
| Use of and compliance with existing ontological frameworks where possible | ○ | ○ | ⊗ | ○ | ⊗ |
| Functionality | |||||
| Powerful search functionalities, e.g., sketch search, similarity search | ⊗ | ⊗ | ⊗ | ⊗ | – |
| Focus on back-end and data delivery functionality rather than front-end | × | – | |||
| Expert in-house curation and/or curation by “vigilantes”d | ⊗ | ⊗ | – | ||
| Target Audience | |||||
| Ease-of-use by nonexperts | ⊗ | ○ | ○ | ○ | ○ |
| Adoption by industrial users as part of their core R&D workflowse | ⊗ | ○ | ○ | ⊗ | ○ |
| Heavy initial advertisement in journals and conferences | × | ||||
| Critical number of initial users who can provide feedback | × | ||||
| Other Contributing Factors | |||||
| Starting early, with relatively few data-points, or first-mover advantage | × | × | × | ||
| Single, large funding source | ⊗ | × | |||
| Funded via multiple smaller grants (at least partially) | ○ | ○ | ○ | ○ | |
Key: (×) strategy employed at launch; (○) strategy employed now; (−) strategy not applicable.
At least every few years.
Easy mapping of data across multiple databases.
Reportedly gives users confidence in the data.
Particularly pharmaceutical and agrochemical industries.
Additionally, we note that there are two ingredients which are each necessary but not sufficient for a successful data initiative: (1) standardized data formats and (2) centralized repositories. NMR line tables are a good example of a case where only standardized data formats are present and the lack of a centralized repository limits downstream applications; if a comprehensive precurated NMR data set exists, machine learning researchers would rapidly adopt it as a supervised learning benchmark. On the other hand, an effort with a centralized repository and no standard formats would be similarly useless to anyone who was not willing to extract data from diverse and incompatible file types. ChEMBL is an interesting case: they have structured data formats as well as a centralized repository, but they also have largely taken on the (expensive) responsibility of data curation to convert unstructured data from journals into their preferred formats. (Imagine how quickly ChEMBL could grow if biochemical assay data came as structured Supporting Information!)
What Is Different about Reaction Data?
Contrary to the structural and assay data of the aforementioned case studies, it may require more work to format reaction data. Reaction data is fundamentally more heterogeneous. Reaction outcomes can be characterized in many different ways (e.g., yields, conversions, rates), sometimes providing information about product identities and quantities with varying levels of precision (e.g., isolated yields, quantitative NMR) or sometimes only in a relative sense (e.g., liquid chromatography area percentage). Moreover, reaction data is not just a single result, but also the full process or protocol and details about the inputs are essential. Instead of, for instance, the acquisition method of structures in PDB being encoded as meta-data and not being of interest to most users, the procedural details of chemical reactions are of primary interest to anyone working with reaction data. Because representing procedural details can be done in a number of different ways, most people use free text; however, as has been demonstrated in each of the case studies, defining a structured format or ontology and then sticking to it, especially in the early phases of establishing a new database, has been critical to the widespread adoption of chemical databases. Ontologies, which in their simplest form may just involve the organization of objects into structured data classes, provide the language and logic to semantically annotate and link data, making it easier to search, review, and update entries. The lack of a (widely adopted) standard for reaction data is setting back data-sharing efforts.
An additional practical challenge is that, while some data is “born digital”, the majority of reactions are not. Preparing comprehensive Supporting Information (SI) documents already places a large burden on authors of manuscripts submitted for peer review; if one were to introduce another format or publication requirement, it could create at least a few hours of additional work for authors. However, the standardized publication of reaction data should also contribute to increased reproducibility and reuse of the data, in principle saving precious research time.
An additional major difference between the present question of reaction data sharing and these successful examples is timing. Unlike the PDB at the time of its founding, there is an abundance of reaction information that already exists, albeit in an unstructured format. Unlike PubChem and ChEMBL at the time of their founding, use cases for the information beyond retrieval and recall (i.e., for use in data-driven models) are abundant. This contributes to a sense of urgency: the time is right to figure out better reaction data sharing practices, and as a community, we do not have to wait decades for this change and for its payoff.
Whose Job Is Data Curation and Sharing?
In our view, there is a general consensus in the field that current methods for the communication and the sharing of reaction data need to evolve so as to better accommodate machine-readable formats and open collaborative frameworks (FAIR principles).19,22 However, there remains a general disagreement, or at least a lack of consensus, about whose responsibility it is to curate reaction data. Incentives between data producers and data consumers typically do not overlap; although within industry, there may be a larger organizational strategy that incorporates both.
Data consumers have a clear incentive to promote this kind of database because they need better training data; many users anecdotally report that their machine learning models perform okay on public data and significantly better on less noisy company data sets. Less anecdotally, many recent efforts in reaction prediction are only able to evaluate their performance with the USPTO data sets; the lack of challenging benchmarks with significant room for improvement leads to an emphasis on incremental improvements and masking of the potential impact of new model architectures.
Data producers also have many incentives for data sharing, as it makes it easier to access the data they have created and can in principle amplify their impact. For synthetic chemists, being able to search for and identify failed reactions can not only save valuable time on a project but also motivate new method development (e.g., extending a method to new substrates). A chemist, hoping to try a new reaction, should be able to refer to databases like SciFinder, Reaxys, and the ORD to quickly and painlessly identify similar reactions which have been previously run, identify what the respective conditions and yields were regardless of whether the yields were high enough to be considered a “success”, and use an associated or integrated reaction prediction tool to estimate the potential outcomes of their proposed scope or conditions with better accuracy than is currently possible. This would streamline the day-to-day work of laboratory scientists, who can use tools such as these to inform the next steps in their project and save valuable time otherwise spent searching the literature or attempting to run an irreproducible reaction.
Broader reaction data sharing, together with standardized formats like the ORD schema, will accelerate the realization of this vision. We expect a shift in the “data streams” that define reaction data today, literature reports and manual curation, to include more examples of failed reactions and large-scale plate-based data that include reaction conditions in addition to the usual reactants/reagents/product descriptions. Additionally, these new data streams can exist independently of traditional publications, allowing for more frequent depositions and facilitating the release of previously unpublished data (e.g., an academic group could set up a monthly or quarterly data dump that is not tied to any specific project or publication).
Commercial databases like SciFinder and Reaxys offer a critical service in their curation of unstructured data from publications, including information beyond reactions such as molecular properties. In contrast, ORD and related efforts are focused on the emerging data stream of digital-first, structured reaction data that may or may not be part of a traditional publication. Plugins and translators for various ELNs are being developed to make these data streams easy to populate with existing tools so that preparing a submission can be as simple as clicking a button.
Finding a Solution
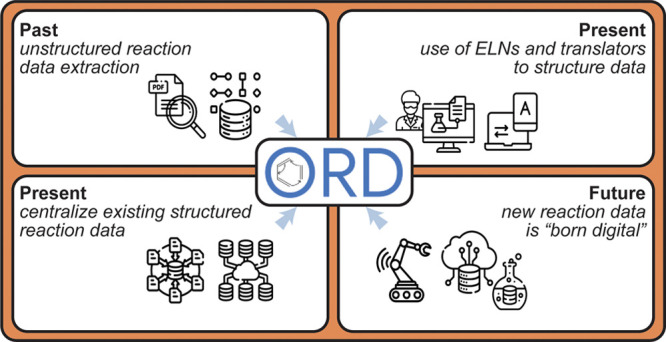
The nature of different data sources warrants the consideration of different strategies for sharing the information therein (Figure 3).
Figure 3.

Four methods for obtaining structured reaction information: (top left) mining historical unstructured data, (top right) manually structuring and translating present/historical data via electronic lab notebooks, (bottom left) efforts to publish existing structured data centrally and publicly, and (bottom right) moving forward, building best practices in from the beginning, whether running benchtop or high-throughput experiments. Regardless of the approach, the ORD can provide a framework for depositing, validating, and distributing structured reaction data. Icons downloaded from flaticon.com.
Looking Backward
Journal articles and patents published over the past several decades provide a rich source of unstructured information. Commercial database efforts have used expert curation to produce excellent resources for information retrieval. We should not replicate such efforts or otherwise do things that could be perceived as redundant or overlapping. Instead, we can focus on the richer procedural and outcome information present in the original document that tends not to be part of tabulation efforts. Can we extract additional details from existing published papers?
Patent extraction tools such as those used by NextMove Software have been quite successful at extracting reaction data from patents, with Daniel Lowe first publishing the USPTO CC-Zero Subset (3.7 million chemical reactions extracted from US patents between 1976 and September 2016).23,93 Additionally, NextMove Software commercially provides an updated database of automatically extracted chemical reactions as part of their Pistachio database (13.3 million reactions automatically extracted from US, European, and WIPO patents). The methods NextMove uses to extract chemical reaction data broadly work as follows: (1) identify the experimental sections of patents, (2) identify chemical entities, (3) convert chemical names to structures, (4) associate chemical entities and quantities, (5) assign chemical roles to each entity, and finally (6) perform atom–atom mapping.23 Besides methods for text extraction,94 there are also a variety of methods available for automatic reaction extraction from images, such as the ReactionDataExtractor tool from the University of Cambridge.95 ReactionDataExtractor uses a combination of rule-based and unsupervised machine learning approaches to extract information from multistep reaction schemes and includes capabilities such as segmentation of reaction steps, identifying regions containing reaction conditions and, of course, optical character and structure recognition. Success stories of automated reaction information extraction tend to be focused on the patent literature, rather than more heterogeneous journal articles.
It is arguable that extracting old data from historical documents is not so important given the rate at which we can generate new data. As there is so much unstructured reaction data already in existence, curation of the entire domain of existing data is practically infeasible. It is somewhat unlikely that we will ever be able to extract reaction information from PDFs completely automatically given how dispersed this information is and how heterogeneously it is represented, and at least some amount of expert supervision and curation will always be needed when it comes to historical data. Additionally, the aforementioned bias toward positive results inherently limits the value of traditional publications as a source of training data for better predictive models. Nonetheless, this is changing with the increasing adoption of automated and plate-based chemistry.
Looking at the Present
If extracting structured data from historical records is challenging, what might the short-term look like instead? There is an opportunity to shift current record-keeping practices from unstructured paper notebooks to structured ELNs, at which point submission to repositories like the ORD is straightforward. But while ELN “translators” might facilitate exporting reaction data and converting between formats, many researchers are not using ELNs to begin with, despite the increasingly digital nature of research.96 Among the many benefits of ELNs are easy long-term storage (and back-ups), increased reproducibility of research, IP protection, and better search functionalities. They also eliminate the need to manually transcribe data from paper notebooks to digital form for publication and make it easier to include/cross-reference digital resources such as figures, instrument data, etc., relative to paper notebooks. The use of ELNs increases interoperability and makes it easier to automatically generate materials for deposition in an archive or publication. Coupled with the use of semantic web technologies, ELNs can also enrich collected data with meaning and context and create valuable links between raw data and the final report. Among researchers that have already adopted ELNs, there is a general preference for ELNs that make use of pre-existing software (e.g., for drawing, data processing, reference management).96,97
The uptake of ELNs in academia has been limited. Among the many barriers faced by scientists for ELN adoption are concerns about data being kept private, an overwhelming number of choices between providers, time needed for implementation, cost, lack of appropriate hardware access in the lab, lack of compatibility with operating systems, and concerns about the use of proprietary data formats that make switching between ELN providers challenging. These challenges certainly need to be addressed in order to increase wider adoption of ELNs in academic laboratories, which will in turn make it easier to standardize reaction data.96
So what is the role of ELN software in encouraging such a transition? In the long term, it would be fruitful for funding agencies or companies to invest in the development of open source ELNs that natively support open and interoperable digital reaction formats such as the ORD schema. The choice of format dictates what information is captured in a structured way vs in an unstructured way. The ORD schema supports data generated via benchtop reactions, automated high-throughput experiments, flow chemistry, and other existing and emerging technologies. Beyond providing a centralized open access data repository, it is an open source format, putting the I in FAIR. We feel the ORD provides a much-needed framework via which to enable the collection and publication of new data as it is generated, whether it be from HTS, flow chemistry, or an individual scientist.
Similar to the CSD, the ORD enforces a certain level of consistency between entries via the use of validation functions. These functions, for instance, require the presence of certain fields and check for reasonable values, such as ensuring that each reaction has at least one input or that quantities are non-negative. These validations are performed automatically in the interactive web editor and during the data set submission process. As a standalone schema without any sophisticated software front-ends, it can be difficult or tedious to structure data using the ORD format. However, efforts are underway to dramatically improve the user experience for contributors without programming experience.
One hurdle the ORD currently faces is reliance on volunteer time for development and data review, which will need to be addressed in the near future to better support the management of large volumes of reaction data; this will include securing funding for staff and infrastructure. Currently, the ORD receives some baseline financial support from the NSF Center for Computer-Assisted Synthesis, but in order to become a ubiquitous resource like the databases highlighted in this work, more strategic measures such as those presented in Table 1 will need to be implemented.
Looking Forward
Looking forward, it is tempting to ask why we cannot just generate new reaction data 100% from scratch. With the rise of automation and HTE, some argue that we do not necessarily need to worry about historical data, as we can quickly generate new data of higher quality. However, not all types of data tend to be conducive to HTE, raising the question of how well the reaction spaces of interest can be covered with this approach. How would this bias the types of reactions which are explored in the future versus the types of reactions which we know or could extract from historical data? The answer is likely to be different for each use case; medicinal chemistry applications in early drug discovery are likely to emphasize well-understood reactions that are amendable to automation, while other fields like reaction discovery will favor data that is generally more heterogeneous and difficult to scale. One example of bias in HTE data generation is the choice of reaction time (preference for shorter times), analytical method (LC/MS), performance metric (LCAP, not yield), and among other conditions, solvent (preference for high boiling solvents). Insights from these HTE platforms about reactivity may not translate directly to other conditions or reaction types.
While automation helps us rapidly explore “condition-space” through combinatorial testing, the literature is substantially more diverse in terms of “substrate-space” or “reaction-type-space”, even if each set of reactants and products is only reported with a single condition. An interesting case to consider is that process analytical technology (PAT) for the manufacturing of pharmaceuticals will generate a lot of reaction data but only for a single reaction under a range of conditions. The “shape” of these data sets is qualitatively different than what most researchers imagine when they think of a reaction database, yet an extensible data model like the ORD can accommodate it. Further, with automation, it may be trivial to prepare reaction mixtures from liquid stock solutions at a variety of concentrations and run reactions at a variety of temperatures, but the preparation of those solutions or use of solid phase reagents is still a practical challenge. This is one of the main limitations we see in relying on automation for the generation of new reaction data. Whatever future plans are made to improve the state of data sharing in chemistry, they cannot be solely focused on automation and HTE.
Mandates for Sharing Structured Data
This brings us to a primary conclusion of this work. Having analyzed a range of historically successful data sharing and standardization initiatives, it is our opinion that funding sources share the bulk of the responsibility for establishing good data sharing practices via demanding open access and FAIR publication of data generated via their funding. Without such a mandate from funding sources, there is little to no accountability for researchers to improve their data sharing practices, and we believe it is unlikely that the state of reaction data sharing will change. While prior efforts have been successful without mandates, mandates can accelerate the transformation of data sharing practices to ensure that change occurs on a time scale of years rather than decades. In particular, we are advocating for mandates that require publication of structured, machine-readable data that can be automatically imported into centralized repositories.
Every researcher, particularly those of us in academia, has incentives to continue to receive grant funding, as this enables student training, leads to more research impact through publications and patents, greater peer recognition, and awards, and is thus one of the primary means for advancing their research and careers. If researchers can continue to receive grant funding for their work only if certain data standards are met, then they will have a huge incentive to do the extra work of making their data FAIR. Though the field might not be ready to embrace it, we would suggest that it become mandatory that data management plans (already required for many funding mechanisms) include a plan for digital deposition of reaction data. This comes at no direct financial cost to funding agencies but will amplify the impact, accessibility, and reproducibility of the research they fund. To improve the utility of shared data, researchers should be required to format it according to predefined data requirements established by the funding agencies. Some recent mandates for data sharing such as the one by the National Institutes of Health is a start98 but must be accompanied by guidelines for how sharing should be done as well as mechanisms for accountability. Tying data sharing to funding rather than peer-reviewed publication may also mitigate the loss of information when projects are discontinued without a corresponding publication, which can happen due to a change in priorities, staffing, etc.
We recognize that researchers at some institutions may lack appropriate resources to publish reaction data under these principles due to limited funding, infrastructure, or researcher bandwidth. This may be particularly true for primarily undergraduate and/or non-R1 institutions, whose work is an essential part of the scientific enterprise. In such cases, funding agencies may need to account for additional labor costs to allow reaction data sharing mandates to be equitable. Similarly, it is important that there are freely available software tools to facilitate the preparation of data contributions in the requested format.
Contributions arising from efforts not supported by funding agencies (e.g., industry-generated data) will require a different tack of publication requirements, where journals only accept manuscripts with sufficient structured data to reproduce the findings in the paper. As we saw in many of the aforementioned data sharing initiatives, journals mandating the publication of specific data (e.g., depositing crystal structures into the PDB) played a big role in many of these data sharing initiatives taking off, to the point that we can no longer imagine not doing those things. As such, it would be reasonable to expect that requiring the deposition of structured reaction data may be necessary to increase the incentives for researchers to publish their data in a structured format and also to lead to greater incentives to use ELNs.
For both funding agency mandates and journal publication requirements, we recommend the ORD as the preferred mechanism for structuring and sharing these data.
Conclusion
While some chemists are reluctant to change and quick to point to barriers to open data sharing, those who embrace the principles of FAIR data will find that downstream applications on these data can enhance their research, saving time and energy down the line. For example, CASP tools for predicting better synthesis conditions can help synthetic chemists improve their reaction yield or find a more efficient reaction pathway. The availability of reliable data and accompanying code also enables other researchers to quickly verify research findings and would deter researchers from publishing irreproducible findings that waste other researchers’ time, though reproducibility carries other challenges. We have summarized the main strategic factors which we believe helped drive the success of databases such as the CSD, PDB, PubChem, and ChEMBL and look to the development of NMR guidelines as a model for the development of reaction reporting guidelines. We assert that a large part of the momentum for sharing structured data needs to come not from individuals and peer pressure (where there can be little to no accountability) but rather from funding agencies and journals. By requiring open access and FAIR publication of any and all reaction data generated via their funding, agencies can place the required incentives on researchers to move toward the digitization of reaction data and open data sharing.
Acknowledgments
We thank NLM Support for data on PubChem data sources over time and Dr. Fiona Hunter from EMBL-EBI for data on ChEMBL data sources over time. We also acknowledge helpful conversations and feedback on the manuscript from John Overington. R.M. thanks the Machine Learning for Pharmaceutical Discovery and Synthesis consortium for funding. C.W.C. thanks the National Science Foundation under Grant No. CHE-2144153 and the AI2050 program at Schmidt Futures (Grant G-22-64475). S.M.K. is an employee of Relay Therapeutics, a for-profit pharmaceutical company.
Data Availability Statement
Scripts and data for generating the figures presented in the paper are available at github.com/rociomer/data-sharing-perspective/.
The authors declare no competing financial interest.
References
- Coley C. W.; Barzilay R.; Jaakkola T. S.; Green W. H.; Jensen K. F. Prediction of organic reaction outcomes using machine learning. ACS Cent. Sci. 2017, 3, 434–443. 10.1021/acscentsci.7b00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwaller P.; Laino T.; Gaudin T.; Bolgar P.; Hunter C. A.; Bekas C.; Lee A. A. Molecular transformer: a model for uncertainty-calibrated chemical reaction prediction. ACS Cent. Sci. 2019, 5, 1572–1583. 10.1021/acscentsci.9b00576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakkar A.; Selmi N.; Reymond J.-L.; Engkvist O.; Bjerrum E. J. Ring breaker”: neural network driven synthesis prediction of the ring system chemical space. J. Med. Chem. 2020, 63, 8791–8808. 10.1021/acs.jmedchem.9b01919. [DOI] [PubMed] [Google Scholar]
- Irwin R.; Dimitriadis S.; He J.; Bjerrum E. J. Chemformer: a pre-trained transformer for computational chemistry. Mach. Learn.: Sci. Technol. 2022, 3, 015022. 10.1088/2632-2153/ac3ffb. [DOI] [Google Scholar]
- Seidl P.; Renz P.; Dyubankova N.; Neves P.; Verhoeven J.; Wegner J. K.; Segler M.; Hochreiter S.; Klambauer G. Improving few-and zero-shot reaction template prediction using modern hopfield networks. J. Chem. Inf. Model. 2022, 62, 2111–2120. 10.1021/acs.jcim.1c01065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segler M. H.; Preuss M.; Waller M. P. Planning chemical syntheses with deep neural networks and symbolic AI. Nature 2018, 555, 604–610. 10.1038/nature25978. [DOI] [PubMed] [Google Scholar]
- Genheden S.; Thakkar A.; Chadimová V.; Reymond J.-L.; Engkvist O.; Bjerrum E. AiZynthFinder: a fast, robust and flexible open-source software for retrosynthetic planning. J. Cheminf. 2020, 12, 1–9. 10.1186/s13321-020-00472-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coley C. W.; Thomas D. A. III; Lummiss J. A.; Jaworski J. N.; Breen C. P.; Schultz V.; Hart T.; Fishman J. S.; Rogers L.; Gao H.; Hicklin R. W.; Plehiers P. P.; Byington J.; Piotti J. S.; Green W. H.; Hart A. J.; Jamison T. J.; Jensen K. F. A robotic platform for flow synthesis of organic compounds informed by AI planning. Science 2019, 365, eaax1566 10.1126/science.aax1566. [DOI] [PubMed] [Google Scholar]
- Schwaller P.; Petraglia R.; Zullo V.; Nair V. H.; Haeuselmann R. A.; Pisoni R.; Bekas C.; Iuliano A.; Laino T. Predicting retrosynthetic pathways using transformer-based models and a hyper-graph exploration strategy. Chem. Sci. 2020, 11, 3316–3325. 10.1039/C9SC05704H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetko I. V.; Karpov P.; Van Deursen R.; Godin G. State-of-the-art augmented NLP transformer models for direct and single-step retrosynthesis. Nat. Commun. 2020, 11, 5575. 10.1038/s41467-020-19266-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie S.; Yan R.; Han P.; Xia Y.; Wu L.; Guo C.; Yang B.; Qin T.. Retrograph: Retrosynthetic planning with graph search. In Proceedings of the 28th ACM SIGKDD Conference on Knowledge Discovery and Data Mining, 2022; pp 2120–2129.
- Chen B.; Li C.; Dai H.; Song L.. Retro*: learning retrosynthetic planning with neural guided A* search. In Proceedings of the 37th International Conference on Machine Learning, 2020; Vol. 119, pp 1608–1616.
- Kim J.; Ahn S.; Lee H.; Shin J.. Self-improved retrosynthetic planning. In Proceedings of the 38th International Conference on Machine Learning, 2021; Vol. 139, pp 5486–5495.
- Shim E.; Kammeraad J. A.; Xu Z.; Tewari A.; Cernak T.; Zimmerman P. M. Predicting reaction conditions from limited data through active transfer learning. Chem. Sci. 2022, 13, 6655–6668. 10.1039/D1SC06932B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao H.; Struble T. J.; Coley C. W.; Wang Y.; Green W. H.; Jensen K. F. Using machine learning to predict suitable conditions for organic reactions. ACS Cent. Sci. 2018, 4, 1465–1476. 10.1021/acscentsci.8b00357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genheden S.; Mårdh A.; Lahti G.; Engkvist O.; Olsson S.; Kogej T. Prediction of the Chemical Context for Buchwald-Hartwig Coupling Reactions. Mol. Inform. 2022, 41, 2100294. 10.1002/minf.202100294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maser M. R.; Cui A. Y.; Ryou S.; DeLano T. J.; Yue Y.; Reisman S. E. Multilabel classification models for the prediction of cross-coupling reaction conditions. J. Chem. Inf. Model. 2021, 61, 156–166. 10.1021/acs.jcim.0c01234. [DOI] [PubMed] [Google Scholar]
- Coley C. W.; Green W. H.; Jensen K. F. Machine learning in computer-aided synthesis planning. Acc. Chem. Res. 2018, 51, 1281–1289. 10.1021/acs.accounts.8b00087. [DOI] [PubMed] [Google Scholar]
- Davies I. W. The digitization of organic synthesis. Nature 2019, 570, 175–181. 10.1038/s41586-019-1288-y. [DOI] [PubMed] [Google Scholar]
- Pflüger P. M.; Glorius F. Molecular machine learning: the future of synthetic chemistry?. Angew. Chem., Int. Ed. 2020, 59, 18860–18865. 10.1002/anie.202008366. [DOI] [PubMed] [Google Scholar]
- Baldi P. Call for a Public Open Database of All Chemical Reactions. J. Chem. Inf. Model. 2022, 62, 2011–2014. 10.1021/acs.jcim.1c01140. [DOI] [PubMed] [Google Scholar]
- Kearnes S. M.; Maser M. R.; Wleklinski M.; Kast A.; Doyle A. G.; Dreher S. D.; Hawkins J. M.; Jensen K. F.; Coley C. W. The Open Reaction Database. J. Am. Chem. Soc. 2021, 143, 18820–18826. 10.1021/jacs.1c09820. [DOI] [PubMed] [Google Scholar]
- Lowe D. M.Extraction of chemical structures and reactions from the literature. Ph.D. Thesis, University of Cambridge, 2012. [Google Scholar]
- Mayfield J.; Lowe D.; Sayle R.. Pistachio, version 2022-10-03 (2022Q3). Available at https://www.nextmovesoftware.com/pistachio.html (accessed 2022-04-10).
- Elsevier Reaxys: An expert-curated chemistry database. Available at https://www.elsevier.com/solutions/reaxys (accessed 2022-12-19).
- American Chemical Society . CAS Data. Available at https://www.cas.org/cas-data (accessed 2022-12-21).
- Ahneman D. T.; Estrada J. G.; Lin S.; Dreher S. D.; Doyle A. G. Predicting reaction performance in C–N cross-coupling using machine learning. Science 2018, 360, 186–190. 10.1126/science.aar5169. [DOI] [PubMed] [Google Scholar]
- Perera D.; Tucker J. W.; Brahmbhatt S.; Helal C. J.; Chong A.; Farrell W.; Richardson P.; Sach N. W. A platform for automated nanomole-scale reaction screening and micromole-scale synthesis in flow. Science 2018, 359, 429–434. 10.1126/science.aap9112. [DOI] [PubMed] [Google Scholar]
- Buitrago Santanilla A.; Regalado E. L.; Pereira T.; Shevlin M.; Bateman K.; Campeau L.-C.; Schneeweis J.; Berritt S.; Shi Z.-C.; Nantermet P.; Liu Y.; Helmy R.; Welch C. J.; Vachal P.; Davies I. W.; Cernak T.; Dreher S. D. Organic chemistry. Nanomole-scale high-throughput chemistry for the synthesis of complex molecules. Science 2015, 347, 49–53. 10.1126/science.1259203. [DOI] [PubMed] [Google Scholar]
- cOAlition S. Plan S: Making full and immediate Open Access a reality. Available at https://www.coalition-s.org/ (accessed 2022-12-21).
- The White House OSTP Issues Guidance to Make Federally Funded Research Freely Available Without Delay. Available at https://www.whitehouse.gov/ostp/news-updates/2022/08/25/ostp-issues-guidance-to-make-federally-funded-research-freely-available-without-delay/ (accessed 2023-01-20).
- Gabelica M.; Bojčić R.; Puljak L. Many researchers were not compliant with their published data sharing statement: mixed-methods study. J. Clin. Epidemiol. 2022, 150, 33. 10.1016/j.jclinepi.2022.05.019. [DOI] [PubMed] [Google Scholar]
- Cambridge Structural Database. CSD Publication Year Statistics: 1 January 2022. Available at https://www.ccdc.cam.ac.uk/media/Documentation/9DA399C5-90F8-478E-9C41-EAFD1868ED31/9da399c590f8478e9c41eafd1868ed31.pdf (accessed 2023-02-28).
- Protein Data Bank . Deposition Statistics. Available at https://www.wwpdb.org/stats/deposition (accessed 2023-03-01).
- NIH National Human Genome Research Institute . NIH Creates Nationwide Network of Molecular Libraries Screening Centers To Accelerate Study of Human Biology and Disease. Available at https://www.genome.gov/15014443/2005-release-nih-nationwide-network-of-molecular-libraries-screening-centers (accessed 2023-03-01).
- Big Chemical Encyclopedia, NIH Molecular Libraries Roadmap Initiative. Available at https://chempedia.info/info/nih_molecular_libraries_roadmap_initiative/ (accessed 2023-03-01).
- Han L.; Wang Y.; Bryant S. H. Developing and validating predictive decision tree models from mining chemical structural fingerprints and high–throughput screening data in PubChem. BMC Bioinform 2008, 9, 1–8. 10.1186/1471-2105-9-401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Xiao J.; Suzek T. O.; Zhang J.; Wang J.; Bryant S. H. PubChem: a public information system for analyzing bioactivities of small molecules. Nucleic Acids Res. 2009, 37, W623–W633. 10.1093/nar/gkp456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Xiao J.; Suzek T. O.; Zhang J.; Wang J.; Zhou Z.; Han L.; Karapetyan K.; Dracheva S.; Shoemaker B. A.; Bolton E.; Gindulyte A.; Bryant S. H. PubChem’s BioAssay database. Nucleic Acids Res. 2012, 40, D400–D412. 10.1093/nar/gkr1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Bryant S. H.; Cheng T.; Wang J.; Gindulyte A.; Shoemaker B. A.; Thiessen P. A.; He S.; Zhang J. Pubchem bioassay: 2017 update. Nucleic Acids Res. 2017, 45, D955–D963. 10.1093/nar/gkw1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.; Chen J.; Cheng T.; Gindulyte A.; He J.; He S.; Li Q.; Shoemaker B. A.; Thiessen P. A.; Yu B.; Zaslavsky L.; Zhang J.; Bolton E. E. PubChem 2019 update: improved access to chemical data. Nucleic Acids Res. 2019, 47, D1102–D1109. 10.1093/nar/gky1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.; Chen J.; Cheng T.; Gindulyte A.; He J.; He S.; Li Q.; Shoemaker B. A.; Thiessen P. A.; Yu B.; Zaslavsky L.; Zhang J.; Bolton E. E. PubChem in 2021: new data content and improved web interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. 10.1093/nar/gkaa971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NIH National Library of Medicine . PubChem Data Counts. Available at https://pubchem.ncbi.nlm.nih.gov/docs/statistics (accessed 2022-12-25).
- ChEMBL Interface Documentation, Downloads. Available at https://chembl.gitbook.io/chembl-interface-documentation/downloads (accessed 2022-12-25).
- The Cambridge Crystallographic Data Centre (CCDC), Our History. Available at https://www.ccdc.cam.ac.uk/theccdcprofile/history/ (accessed 2022-05-25).
- The Cambridge Crystallographic Data Centre (CCDC). Opening lecture by Dr Olga Kennard OBE FRS. Available at https://www.youtube.com/watch?v=HkR7_uxvU8Q (accessed 2022-06-30).
- CCDC . CCDC Research. Available at https://www.ccdc.cam.ac.uk/researchandconsultancy/ccdcresearch/ccdcresearchers/ (accessed 2022-12-23).
- CCDC . New Year, New Data Resolutions! Available at https://www.ccdc.cam.ac.uk/Community/blog/data-resolutions/ (accessed 2022-12-23).
- Retraction Watch . Crystallography database flags nearly 1000 structures linked to a paper mill. Available at https://retractionwatch.com/2022/07/26/crystallography-database-flags-nearly-1000-structures-linked-to-a-paper-mill/ (accessed 2022-12-19).
- CCDC Retractions in the Cambridge Structural Database. Available at https://www.ccdc.cam.ac.uk/support-and-resources/support/case/?caseid=819cfd76-c25d-40a2-ac9b-b4cf20d775a7 (accessed 12–19–2022).
- The Cambridge Crystallographic Data Centre (CCDC). A million thanks. Available at https://www.ccdc.cam.ac.uk/Community/blog/A-million-thanks/ (accessed 2022-06-25).
- ACS Publications. Requirements for Depositing X-Ray Crystallographic Data. Available at https://pubsapp.acs.org/paragonplus/submission/acs_cif_authguide.pdf (accessed 2023-03-04).
- The Cambridge Crystallographic Data Centre (CCDC) . Big data leads the way for structural chemistry. Available at https://www.ccdc.cam.ac.uk/News/List/the-cambridge-structural-database-reaches-one-million/ (accessed 2022-06-25).
- Allen F. H.; Davies J. E.; Galloy J. J.; Johnson O.; Kennard O.; Macrae C. F.; Mitchell E. M.; Mitchell G. F.; Smith J. M.; Watson D. G. The development of versions 3 and 4 of the Cambridge Structural Database System. J. Chem. Inf. Comput. Sci. 1991, 31, 187–204. 10.1021/ci00002a004. [DOI] [Google Scholar]
- Ward S.One Million Structures and Counting: The journey, the insights, and the future of the CSD. Acta Cryst. 2019, A75, e14. Available at http://scripts.iucr.org/cgi-bin/paper?S2053273319095421 (accessed 2022-12-27). [Google Scholar]
- Marsh R. E. Space groups P1 and Cc: how are they doing?. Acta Crystallogr. B 2009, 65, 782–783. 10.1107/S0108768109046448. [DOI] [PubMed] [Google Scholar]
- Schwalbe C. H.Should we remediate small molecule structures? If so, who should do it? Crystallogr. Rev. 2018, 24 ( (4), ), 217−235. Available at https://www.tandfonline.com/doi/abs/10.1080/0889311X.2018.1508209 (accessed 2022-12-27). [Google Scholar]
- Bruno I. J.; Shields G. P.; Taylor R. Deducing chemical structure from crystallographically determined atomic coordinates. Acta Crystallogr. B 2011, 67, 333–349. 10.1107/S0108768111024608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RCSB PDB . PDB History. Available at https://www.rcsb.org/pages/about-us/history (accessed 2022-07-12).
- Cold Spring Harbor Laboratory . Structure and Function of Proteins at the Three-Dimensional Level. Available at http://library.cshl.edu/symposia/1971/index.html (accessed 2022-05-12).
- PDB Community Focus: Michael G. Rossmann. In RCSB PDB Newsletter, 2006. Available at https://cdn.rcsb.org/rcsb-pdb/general_information/news_publications/newsletters/2006q2/community.html (accessed 2022-05-12).
- Meyer E. F. The first years of the Protein Data Bank. Protein Sci. 1997, 6, 1591. 10.1002/pro.5560060724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RCSB PDB . Number of Released PDB Structures per Year. Available at https://www.rcsb.org/stats/all-released-structures (accessed 2022-05-13).
- Commission on Biological Macromolecules. Acta Crystallogr. A 1989, 45, 658. 10.1107/S0108767389007695. [DOI] [Google Scholar]
- Campbell P. New policy for structure data. Nature 1998, 394, 105. 10.1038/27971. [DOI] [PubMed] [Google Scholar]
- National Library of Medicine PubChem. Available at https://pubchem.ncbi.nlm.nih.gov/ (accessed 2022-05-23).
- National Institutes of Health (NIH) . Molecular Libraries High Throughput Screening Centers: Request For Information (RFI); 2003. Available at https://grants.nih.gov/grants/guide/notice-files/NOT-RM-04-001.html (accessed 2022-05-25).
- National Library of Medicine . PubChem Data Sources; 2022. Available at https://pubchem.ncbi.nlm.nih.gov/sources/ (accessed 2022-07-12).
- The universal protein resource (UniProt). Nucleic Acids Res. 2007, 36, D190–D195. 10.1093/nar/gkm895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PubChem . PubChemRDF. Available at https://pubchemdocs.ncbi.nlm.nih.gov/rdf (accessed 2022-07-10).
- Fu G.; Batchelor C.; Dumontier M.; Hastings J.; Willighagen E.; Bolton E. PubChemRDF: towards the semantic annotation of PubChem compound and substance databases. J. Cheminf. 2015, 7, 1–15. 10.1186/s13321-015-0084-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ChEBI The online chemical dictionary for small molecules, ChEBI Ontology. Available at https://www.ebi.ac.uk/training/online/courses/chebi-the-online-chemical-dictionary-for-small-molecules/chebi-ontology/ (accessed 2023-03-07).
- Hastings J.; Chepelev L.; Willighagen E.; Adams N.; Steinbeck C.; Dumontier M. The chemical information ontology: provenance and disambiguation for chemical data on the biological semantic web. PloS One 2011, 6, e25513 10.1371/journal.pone.0025513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natale D. A.; Arighi C. N.; Blake J. A.; Bona J.; Chen C.; Chen S.-C.; Christie K. R.; Cowart J.; D’Eustachio P.; Diehl A. D.; Drabkin H. J.; Duncan W. D.; Huang H.; Ren J.; Ross K.; Ruttenberg A.; Shamovsky V.; Smith B.; Wang Q.; Zhang J.; El-Sayed A.; Wu C. H. Protein Ontology (PRO): enhancing and scaling up the representation of protein entities. Nucleic Acids Res. 2017, 45, D339–D346. 10.1093/nar/gkw1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Office of Scholarly Communication, University of California . American Chemical Society Expresses Opposition to NIH’s PubChem; 2005. Available at https://osc.universityofcalifornia.edu/2005/05/american-chemical-society-calls-on-congress-to-shut-down-nihs-pubchem/#note1 (accessed 2022-05-25).
- EMBL’s European Bioinformatics Institute . Unleashing the potential of big data in biology. Available at https://www.ebi.ac.uk/ (accessed 2022-07-07).
- Warr W. A. Chembl. an interview with john overington, team leader, chemogenomics at the european bioinformatics institute outstation of the european molecular biology laboratory (embl-ebi). J. Comput.-Aided Mol. Des. 2009, 23, 195–198. 10.1007/s10822-009-9260-9. [DOI] [PubMed] [Google Scholar]
- ChEMBL Interface Documentation. Acknowledgments. Available at https://chembl.gitbook.io/chembl-interface-documentation/acknowledgments (accessed 2022-07-07).
- Chemical Biology Services, Members. Available at https://www.ebi.ac.uk/about/teams/chemical-biology-services/members/ (accessed 2023-03-08).
- ChEMBL Interface Documentation, New Web Interface. Available at https://chembl.gitbook.io/chembl-interface-documentation/ (accessed 2023-03-08).
- Becker E. D. A brief history of nuclear magnetic resonance. Anal. Chem. 1993, 65, 295A–302A. 10.1021/ac00054a716. [DOI] [PubMed] [Google Scholar]
- Recommendations for the presentation of NMR data for publication in chemical journals - A. Conventions relating to proton spectra. Pure Appl. Chem. 1972, 29, 627. 10.1351/pac197229040625. [DOI] [Google Scholar]
- Presentation of NMR data for publication in chemical journals - B. Conventions relating to the spectra from nuclei other than protons. Pure Appl. Chem. 1976, 45, 217. 10.1351/pac197645030217. [DOI] [Google Scholar]
- Gottlieb H. E.; Kotlyar V.; Nudelman A. NMR chemical shifts of common laboratory solvents as trace impurities. J. Org. Chem. 1997, 62, 7512–7515. 10.1021/jo971176v. [DOI] [PubMed] [Google Scholar]
- Fulmer G. R.; Miller A. J.; Sherden N. H.; Gottlieb H. E.; Nudelman A.; Stoltz B. M.; Bercaw J. E.; Goldberg K. I. NMR chemical shifts of trace impurities: common laboratory solvents, organics, and gases in deuterated solvents relevant to the organometallic chemist. Organometallics 2010, 29, 2176–2179. 10.1021/om100106e. [DOI] [Google Scholar]
- ACS Publications . NMR Guidelines for ACS Journals. Available at https://pubsapp.acs.org/paragonplus/submission/acs_nmr_guidelines.pdf (accessed 2023-03-04).
- Harris R. K.; Becker E. D.; Cabral de Menezes S. M.; Goodfellow R.; Granger P. NMR nomenclature. Nuclear spin properties and conventions for chemical shifts (IUPAC Recommendations 2001). Pure Appl. Chem. 2001, 73, 1795. 10.1351/pac200173111795. [DOI] [PubMed] [Google Scholar]
- Harris R. K.; Becker E. D.; Cabral de Menezes S. M.; Granger P.; Hoffman R. E.; Zilm K. W. Further Conventions for NMR Shielding and Chemical Shifts. Pure Appl. Chem. 2008, 80, 59. 10.1351/pac200880010059. [DOI] [PubMed] [Google Scholar]
- Bisson J.; Simmler C.; Chen S.-N.; Friesen J. B.; Lankin D. C.; McAlpine J. B.; Pauli G. F. Dissemination of original NMR data enhances reproducibility and integrity in chemical research. Nat. Prod. Rep. 2016, 33, 1028–1033. 10.1039/C6NP00022C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter A. M.; Carreira E. M.; Miller S. J. Encouraging Submission of FAIR Data at The Journal of Organic Chemistry and Organic Letters. J. Org. Chem. 2020, 85, 1773–1774. 10.1021/acs.joc.0c00248. [DOI] [PubMed] [Google Scholar]
- Sorkin B. C.; Betz J. M.; Hopp D. C. Toward FAIRness and a User-Friendly Repository for Supporting NMR Data. J. Org. Chem. 2020, 85, 5131–5131. 10.1021/acs.joc.0c00800. [DOI] [PubMed] [Google Scholar]
- Kuhn S.; Schlörer N. E. Facilitating quality control for spectra assignments of small organic molecules: nmrshiftdb2–a free in-house NMR database with integrated LIMS for academic service laboratories. Magn. Reson. Chem. 2015, 53, 582–589. 10.1002/mrc.4263. [DOI] [PubMed] [Google Scholar]
- Lowe D.Patent Reaction Extraction. Available at https://github.com/dan2097/patent-reaction-extraction (accessed 2023-03-04).
- Guo J.; Ibanez-Lopez A. S.; Gao H.; Quach V.; Coley C. W.; Jensen K. F.; Barzilay R. Automated Chemical Reaction Extraction from Scientific Literature. J. Chem. Inf. Model. 2022, 62, 2035–2045. 10.1021/acs.jcim.1c00284. [DOI] [PubMed] [Google Scholar]
- Wilary D. M.; Cole J. M. ReactionDataExtractor: A Tool for Automated Extraction of Information from Chemical Reaction Schemes. J. Chem. Inf. Model. 2021, 61, 4962–4974. 10.1021/acs.jcim.1c01017. [DOI] [PubMed] [Google Scholar]
- Kanza S.; Willoughby C.; Gibbins N.; Whitby R.; Frey J. G.; Erjavec J.; Zupančič K.; Hren M.; Kovač K. Electronic lab notebooks: can they replace paper?. J. Cheminf. 2017, 9, 1–15. 10.1186/s13321-017-0221-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanza S. Guidelines for Chemistry Labs Looking to Go Digital. Digital Transformation of the Laboratory: A Practical Guide to the Connected Lab 2021, 191–197. 10.1002/9783527825042.ch13. [DOI] [Google Scholar]
- Kozlov M. NIH issues a seismic mandate: share data publicly. Nature 2022, 602, 558–559. 10.1038/d41586-022-00402-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Scripts and data for generating the figures presented in the paper are available at github.com/rociomer/data-sharing-perspective/.