

Abstract
Super-resolution imaging techniques have broken the diffraction-limited resolution of light microscopy. However, acquiring three-dimensional (3D) super-resolution information about structures and dynamic processes in live cells at high speed remains challenging. Recently, the development of high-speed single-point edge-excitation subdiffraction (SPEED) microscopy, along with its 2D-to-3D transformation algorithm, provides a practical and effective approach to achieving 3D subdiffraction-limit information in subcellular structures and organelles with rotational symmetry. One of the major benefits of SPEED microscopy is that it does not rely on complex optical components and can be implemented on a standard, inverted epifluorescence microscope, simplifying the process of sample preparation and the expertise requirement. SPEED microscopy is specifically designed to obtain 2D spatial locations of individual immobile or moving fluorescent molecules inside submicrometer biological channels or cavities at high spatiotemporal resolution. The collected data are then subjected to postlocalization 2D-to-3D transformation to obtain 3D super-resolution structural and dynamic information. In recent years, SPEED microscopy has provided significant insights into nucleocytoplasmic transport across the nuclear pore complex (NPC) and cytoplasm-cilium trafficking through the ciliary transition zone. This Review focuses on the applications of SPEED microscopy in studying the structure and function of nuclear pores.
Keywords: Super resolution microscopy, Single molecule, Nuclear pore complex, Nucleocytoplasmic transport, SPEED microscopy, Nucleoporins, Single particle tracking, Nuclear envelope transmembrane proteins
1. Introduction
1.1. Overview of Single-Molecule Localization Microscopy
1.1.1. Super-resolution Light Microscopy
In order to locate molecules and track their
movements and behaviors
at the single particle level, the primary hurdle that must be overcome
is the physical limitations that light imposes on microscopy. The
ability of standard light microscopy to resolve a target object is
restricted by Abbe’s diffraction limit, which is defined by
the equation 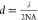 , where d is the diffraction
limit, λ is the wavelength of the detected light, and NA is
the numerical aperture of the objective lens.1 Considering that high magnification oil immersion microscope objectives
typically have an NA in the 1.3–1.5 range and detection is
typically in the visible light spectrum, the conventional resolution
limit for light microscopy is approximately 200 nm.2 Any object smaller than this limit is observed as a point
spread function (PSF), typically in the form of an airy disk pattern
rather than a single, discernible point.3−5 Without innovative advancements
in microscopy techniques, entry into the realm of single-molecule
localization would be impossible under these restrictions. Despite
its relatively large size for a subcellular structure, the nuclear
pore complex (NPC) with dimensions of approximately 50 nm at its narrowest
waist and 200 nm in length cannot be accurately resolved using standard
microscopy techniques.6 Super-resolution
in light microscopy refers to techniques that can achieve resolutions
smaller than half of the diffraction limit.7 Several methods, such as stimulated emission depletion (STED) microscopy,
structured illumination microscopy (SIM), and single-molecule localization
microscopy (SMLM), can achieve super-resolution. However, SMLM has
gained wide popularity in biological applications due to its ability
to achieve the highest resolution among these methods.8−10
, where d is the diffraction
limit, λ is the wavelength of the detected light, and NA is
the numerical aperture of the objective lens.1 Considering that high magnification oil immersion microscope objectives
typically have an NA in the 1.3–1.5 range and detection is
typically in the visible light spectrum, the conventional resolution
limit for light microscopy is approximately 200 nm.2 Any object smaller than this limit is observed as a point
spread function (PSF), typically in the form of an airy disk pattern
rather than a single, discernible point.3−5 Without innovative advancements
in microscopy techniques, entry into the realm of single-molecule
localization would be impossible under these restrictions. Despite
its relatively large size for a subcellular structure, the nuclear
pore complex (NPC) with dimensions of approximately 50 nm at its narrowest
waist and 200 nm in length cannot be accurately resolved using standard
microscopy techniques.6 Super-resolution
in light microscopy refers to techniques that can achieve resolutions
smaller than half of the diffraction limit.7 Several methods, such as stimulated emission depletion (STED) microscopy,
structured illumination microscopy (SIM), and single-molecule localization
microscopy (SMLM), can achieve super-resolution. However, SMLM has
gained wide popularity in biological applications due to its ability
to achieve the highest resolution among these methods.8−10
As the name suggests, SMLM is a method of localizing individual molecules.6−8 Such a concept is fundamentally based on the fact that the spatial location of individual fluorophores can be back-calculated with subdiffraction resolution using its PSF, given that it does not interfere with another molecule’s PSF.3,9 It is not necessary, however, to maintain spatial separation between individual fluorophores, because controlled temporal separation of fluorescence can be utilized to effectively create distinct PSFs, even if the fluorophores themselves are within the PSF range. Typically, SMLM utilizes the fact that fluorophores have both fluorescent and nonfluorescent states (“on” and “off,” respectively). As long as it is possible to curate only frames containing spatially distinct PSFs from a collected image, those can be combined to produce a complete picture of the behaviors of that single molecule within the given environment.11 Common methods of SMLM include photoactivated localization microscopy (PALM), which utilizes photoactivatable fluorophores, and stochastic optical reconstruction microscopy (STORM), which uses photoswitchable fluorophores, meaning that they can be switched between fluorescent and nonfluorescent states induced by certain wavelengths of light. While both PALM and STORM share similar “on”/“off” cycle methodologies, another common SMLM method known as DNA-based point accumulation for imaging in nanoscale topography (DNA-PAINT) uses transient binding of fluorescently labeled DNA probes to produce “on” and “off” states.12−17
1.1.2. Challenges When Imaging with SMLM in Live Cells
One of the most important factors to consider when attempting any fluorescence microscopy experiments, particularly for single molecule experiments, is the signal-to-noise ratio (SNR).18 Essentially, the SNR is a way of describing the quality of a signal, and a sufficiently high SNR of the target fluorescent molecule is required for high-precision single-molecule localization. There are two ways to enhance the SNR: by reducing the noise level or amplifying the emission signal. There are a few different sources of background noise that limit the potential SNR, including (1) “dark noise” due to detector-introduced current, which can be reduced by using cooled cameras, (2) “read noise” generated from the conversion of photocurrents to signal voltages, and (3) “multiplicative noise” generated by stochastic amplification of photoelectrons, and background fluorescence.3,4,19 Further, live cell imaging has been reported to increase background noise levels due to out-of-focus fluorophores and cellular autofluorescence.20,21
There are several ways to increase the emission signal, including using fluorophores with a high extinction coefficient, quantum yield (QY), and brightness, increasing the excitation power, or extending the acquisition time. Nevertheless, each method listed above has constraints and disadvantages, impeding the ability to acquire super-resolution dynamic data, especially in live cells under physiological conditions. Collecting sufficiently intense emission signals from a single molecule has been made significantly more accessible with the development of small organic dyes and fluorescent quantum dots.22−25 However, these methods are not always ideal for performing live cell imaging. Fluorescent quantum dots can be harmful to cells and require bioconjugation, and other labeling methods typically require that the cell be made permeabilized and thus killed.26−28 A common solution to these problems is to utilize fluorescent proteins (FPs), such as EGFP or mCherry, and genetically encode them to be bound to a protein of interest.29 Unfortunately, these FPs typically have lower QY and extinction coefficients (ε) than dyes or nanoparticles, decreasing the potential photon budget under given conditions.30,31 In addition, FPs are relatively large compared with other fluorophores and can therefore interfere with the movements or interactions of small target molecules.
Raising the excitation power leads to a stronger emission signal; however, it also causes faster photobleaching and more photodamage to live cells.32,33 Photochemical stability can significantly limit the potential of a fluorophore to produce sufficient emission due to photobleaching.34 Photobleaching is a dynamic process in which a fluorochrome exposed to excitation light undergoes photoinduced chemical destruction, thus losing its ability to fluoresce. A fluorophore with a higher stability will stay unphotobleached for an extended time, allowing for more prolonged imaging and higher excitation power. Similar to the problems of low quantum yield, FPs generally have weaker photostability compared with their small organic dye and fluorescent quantum dot counterparts. Low photostability means that tracking a molecule is made more challenging because it may not be physically possible to collect images over a long enough time interval to solve for its trajectory before photobleaching.4
Prolonging the acquisition time has been a common strategy in SMLM, leading to many studies in fixed cells and immobilized molecules.35 On the other hand, live-cell studies on dynamics are relatively rare because collecting enough signals with high temporal resolution is very challenging.36 SMLM in live cells has been primarily limited to relatively slowly moving structures with few reconstructed snapshots, and methods that improve the number of workable signals within a short time frame tend to lose out on spatial resolution. In contrast, when tracking a fast-moving particle, e.g., as it is transported across the nuclear membrane within several milliseconds, it can be almost physically impossible to collect images over a long enough time interval to provide a complete trajectory. Finally, because a live cell and its components are free to migrate, sample drift must be accounted for when analyzing the data.37,38
1.2. Nucleocytoplasmic Transport and the Challenges in the Field
1.2.1. Nucleocytoplasmic Transport
One of the most important areas of study within cellular biology is the interface between the nucleus and the cytoplasm. The proper bidirectional transport of materials across the nuclear membrane is crucial to every function within a eukaryotic cell; therefore, the process must be highly efficient and regulated. Even with a cursory knowledge of cell biology, the importance of nucleocytoplasmic transport is immediately quite evident. However, this topic has many mysteries, and high-speed, single-molecule tracking has elucidated many characteristics that would otherwise be impossible to understand.
1.2.2. The Nuclear Pore Complex and the Nucleoporins
The gate required for these transport processes is the NPC, one of the cell’s largest and most complex protein structures, possessing a mass ranging from 66 to 125 MDa depending on the species.39 The NPC is composed of approximately 30 different, highly conserved proteins called nucleoporins (Nups).40−42 These Nups are classified into three major categories: transmembrane, structural, and phenylalanine-glycine (FG) Nups. Transmembrane Nups contain transmembrane helices that help the NPC anchor into the nuclear envelope (NE) and contribute to the NPC’s stability by interacting with other nonmembrane Nups. Structural Nups, sometimes referred to as “scaffold” Nups, provide structural support and are primarily found in the nuclear ring, cytoplasmic ring, and central spoke ring and as linkers between the different rings. These are the most common category of Nup and typically contain either α-solenoid or β-propeller fold structural domains.43 While all eukaryotes have similar three-ringed structures, there is variation among species of exact substructures and Nups used to compose the rings.44 Finally, FG-Nups are named after their repeating sequences of phenylalanine-glycine residues and are responsible for the selective barrier in the NPC’s central channel.
1.2.3. Passive vs Facilitated Transportation and Current Models
There are two primary types of transport through the NPC: passive transport, which allows for the diffusion of macromolecules of less than 40–60 kDa through the pore, and facilitated transport, which requires additional transport factors for larger structures such as the preribosomal subunits.45,46 Facilitated transport is primarily mediated by the FG Nups in the central channel because they are responsible for inhibiting the passive diffusion of large macromolecules and binding to the transport factors required for facilitating transport. The transport receptors (TR) required to perform facilitated diffusion are karyopherins and are typically classified as importins or exportins depending on their directionality; however, some have been observed to function bidirectionally.39 Large molecular cargo contains nuclear localization signals (NLS) for import or nuclear export signals (NES) for export that interact with karyopherins to form transport complexes; this interaction is regulated by GTPase Ran.47 These karyopherins facilitate transport through the NPC by interacting with the hydrophobic FG-Nups. While the various karyopherins intersect in different fashions, they must all find a balance between strong enough interactions to allow for an association and weak enough to allow for complete transport.39 An example of this is Importin 4, one of the transport receptors utilized for the import of histones and ribosomal proteins.47
There is still discussion regarding the most accurate model to describe the behaviors of the NPC and how it regulates nucleocytoplasmic transport. One of these models is the Brownian/virtual gate polymer brushes model, which asserts that the FG-Nups are not interacting with each other due to their positive net charge and therefore move in such a way that produces a thermodynamic barrier that limits the potential for molecules to diffuse through. Another model is the selective phase/hydrogel model, which is based on the fact that phenylalanine and glycine in FG-Nups are hydrophobic residues, and thus, those FG repeats could interact hydrophobically. This model proposes a hydrogel meshwork within the NPC channel that contains a series of small holes that would selectively allow for the diffusion of small molecules. A third model is the reduction of dimensionality (ROD) model, which states that the FG repeats are concentrated along the inner surface of the NPC and that transport receptors interact with this region through the NPC, allowing for facilitated two-dimensional movement of macromolecules along the inner wall.43 Finally, the forest model synthesizes some aspects of the previous models into one cohesive model. Evidence has shown that in the center of the NPC, FG-Nups with low-affinity interactions may form a meshwork like described in the selective phase model, and that a few FG-Nups near the nuclear basket provide a repulsive force as described by the Brownian model.48 This model proposes that FG Nups within the NPC have five different structurally and chemically distinct domains: (a) collapsed-coil, low charge-content, cohesive domain, (b) collapsed-coil, high charge-content, noncohesive domain, (c) extended-coil, noncohesive domain, (d) folded domain, and (e) NPC anchor domain.43,49 The collapsed-coil domains are present in the center of the NPC, whereas the extended-coil domains form a peripheral ring around the center, and different molecules will pass through either zone depending on their size, surface charge, and hydrophobicity.43,50
1.2.4. Why Study Nucleocytoplasmic Transport?
Nucleocytoplasmic transport is one of the essential processes in cellular activity. Molecules necessary for protein synthesis, including mRNA, tRNA, and the preribosomal subunits, are constructed and exported from the nucleus; And nuclear proteins such as histones and transcription factors are translated first in the cytoplasm and imported into the nucleus.51 When cells undergo mitotic proliferation, every minute, hundreds of proteins and ribonucleoproteins (RNPs) pass through each NPC.52 Due to the physical significance of nucleocytoplasmic transportation, it is no surprise that this process is linked to a multitude of human diseases. Many neurological disorders are associated with NPC, and one of the most well-studied is Achalasia-Addisonianism-Alacrima (triple A) syndrome. Triple A is an autosomal recessive disorder that produces highly variable symptoms including the inability to relax the esophageal sphincter and produce tears. The gene mapped to this disease is the AAAS gene, which has been found to code for the protein Aladin, an NPC component.53 Evidence has suggested that, when mutated, Aladin lacks the appropriate ability to transport proteins that protect cells from oxidative damage,54 which was believed to link to the triple A syndrome. Other neurodegenerative diseases associated with the NPC include frontotemporal dementia (FTD), amyotrophic lateral sclerosis (ALS), and Parkinson’s disease (PD), all of which cause progressive loss of cognitive ability and coincide with the absence of Nup358.54−56 Observing Huntington’s disease in a mouse model also revealed that PolyQ aggregates correlate with a significant reduction in the nucleocytoplasmic transport activity. The mislocalized and less post-translationally modified Nups with disrupted NPC permeability were believed to cause the aggregation.57 Finally, it has been shown that certain Nups, most common being the translocated promoter region (Tpr),58,59 Nup98,60,61 Nup358,62 and Nup214,63 produce oncogenic fusion proteins as a result of chromosomal translocations.54 Nup214, for example, has often been found fused to Dek and Set, modifiers of chromatin structure, in cells with T-cell acute lymphocytic leukemia (T-ALL) and acute myeloid leukemia (AML).54,64,65 The expression of Nup88, which interacts with mitotic spindles, is elevated in many tumors and has been shown to increase levels of aneuploidy.54 Many Nups have been observed to be misregulated in cancers, including Nup210, Nup133, Nup107, Sec13, Nup188, Nup93, Nup153, Tpr, Nup358, Nup214, Nup98, hCG1, RanGap1, and Rae1.54
1.2.5. Challenges in the Field
Due to the complexity and dynamic nature of the processes involved in nucleocytoplasmic transport, studying it requires a combination of powerful imaging instruments and techniques. Previous studies have utilized various microscopy techniques, each possessing various advantages and disadvantages. Confocal microscopy has been employed to image NPCs in live-cell environments and is barely able to resolve individual pores; however, it cannot provide sufficient resolution to reveal the specific interactions and pathways of single molecules passing through the NPC, limiting its potential.66 Alternatively, imaging techniques that can achieve subdiffraction resolution, such as electron microscopy (EM), including cryo-EM,67−70 thin section transmission EM,71 and field emission scanning EM (FESEM),72 are commonly used to provide high-resolution images of the NPC. EM approaches continue to provide precious, high-resolution structural insight into the NPC, but they alone cannot be used to study live cells and, therefore, failed to show the dynamics of nucleocytoplasmic transport.73 In particular, those methods could not resolve the intrinsically disordered regions (IDRs) of FG-Nups, which are known to form the selective barrier and play essential roles in nucleocytoplasmic transportation. Some researchers also attempted to combine the strength of the light and electron approaches in the form of Correlative Light and Electron Microscopy (CLEM).74,75 The subdiffraction information generated in CLEM is contributed mainly by its EM portion, while light microscopy only provides a temporal reference point for the EM. Therefore, while CLEM was able to study the dynamics of the NE during mitosis, which takes minutes to hours to finish, it is ineffective in observing faster-paced dynamics for NPC, such as nucleocytoplasmic transportation, which takes only several milliseconds. Another rising approach combines EM studies with artificial intelligence in machine learning and neural networks.76−78 However, IDRs in FG-Nups remain a challenging target for AI-based structure prediction, as most of their model confidence levels are in the very low to low domains, suggesting that there is a long way to go before AI can be fully utilized for nucleocytoplasmic transport studies. Given these challenges, it is clear that achieving a comprehensive understanding of nucleocytoplasmic transport requires an imaging technique that can provide both high spatial and temporal resolution in live-cell environments.
2. SPEED Microscopy and Its 2D to 3D Transformation
2.1. Working Principles of SPEED Microscopy and 2D to 3D Transformation
To develop SPEED microscopy, an inverted epifluorescence microscope was modified by introducing an inclined diffraction limit spot illumination volume in the focal plane (Figure 1a).79,80 This illumination pattern integrates features from laser scanning confocal microscopy (LSCM) and shares some of the principles of HiLo, variable-angle epifluorescence (VAE), and total internal reflection (TIRF) microscopies.81−83 Several modifications have been made to the epifluorescence microscope to allow for SPEED microscopy. First, a CCD camera was used instead of a photomultiplier tube and the pinhole was removed in front of the detector. This modification allows direct recording of spatial information and collects photons from a larger area. Second, an excitation laser beam was guided to the rear aperture of the objective and shifted off the central axis to form an inclined laser beam focused at the focal plane, which generates a smaller adequate illumination volume in the axial direction and partially suppresses the out-of-focus background fluorescence. Third, the pinpointed illumination pattern generates a high laser power density at the focal plane even with low continuous-wave laser power, allowing for a high number of photons from the fluorophores to be obtained quickly. At the same time, the pinpointed illumination pattern also significantly reduces photobleaching and phototoxic effects in live samples, which has been further reduced by adopting an optical chopper to create an on/off operational mode (Figure 1c). Finally, the small illumination volume of SPEED microscopy allows for fast detection speeds of 0.4–2 ms and a resolution of ≤10–20 nm for moving molecules in live cells.
Figure 1.

Basic setup of SPEED microscopy, working principles, and demonstration of single particle tracking, 2D to 3D transformation. (A) Schematic of the SPEED microscopy microscope setup for both the inclined and the vertical illumination. A 488 nm (blue) and a 568 nm (green) laser are directed into the objective in vertical illumination or offset so that the lasers pass through the focal plane at an angle of θ3. (B) Illustration of a molecule traveling in three dimensions as it passes through an NPC. A 2D projection of the same pathway is depicted in the lower panel. (C) Illustration of the effect of the optical chopper on laser intensity. The chopper is calibrated to be open for 1/10 of the frames captured. (D) Series of 2D locations of mRNA-mCherry (red spots) were captured by SPEED microscopy as they transported through the NPC (green spot). Numbers denote time in milliseconds. (E) Representative trajectories for successful nuclear transport of tracking single particle. (F) Experimentally determined 2D spatial locations of single molecule tracking in the NPC. (G) 3D information derived from 2D single-molecule data using a 2D-to-3D transformation. Adapted with permission from figures previously published by Nature Protocols.133
The modifications described above in the SPEED microscopy system help obtain 2D single-molecule localization information with fast dynamics in subcellular structures. Once the data is acquired, a postlocalization 2D-to-3D transformation algorithm is applied to obtain 3D subdiffraction-limit structural and dynamic information from the recorded 2D information (Figure 1g). This accomplishment was attained by assuming that the subcellular structure observed has a symmetric rotational structure. The 2D spatial information obtained from SPEED microscopy represents a projection of the corresponding 3D spatial information in an x,y plane. Generally, 3D spatial information can be described in a cylindrical coordinate system with parameters of x, angle θ, and r. In the case of a symmetric rotational structure, the coordinate system can be further simplified because the distribution of the single molecules of interest in the θ dimension is considered constant at each x and r, providing a simplified cylindrical coordinate system with only x and r. By converting Cartesian coordinates (x, y) of 2D spatial information to simplified cylindrical coordinates (x, r), it is possible to estimate 3D spatial information in a symmetric rotational structure. Ultimately, the combination of SPEED microscopy and its subsequent 2D-to-3D transformation enables the acquisition of virtual 3D subdiffraction-limit information on fast dynamical processes within a rotationally symmetric subcellular structure.
2.2. Advantages and Limitations of SPEED Microscopy
Compared with other 3D super-resolution microscopy techniques, SPEED’s virtual 3D super-resolution imaging approach offers multiple benefits. First, SPEED microscopy provides high spatial and temporal resolution for recording fast dynamics and submicrometer subcellular structural information simultaneously in three dimensions. Second, SPEED microscopy involves a more straightforward sample preparation process, which does not require specialized fluorophores. Furthermore, the pinpointed illumination pattern of SPEED microscopy with low phototoxic effects and low out-of-focus background fluorescence allows for its general use in live-cell imaging. Therefore, SPEED microscopy is an ideal technique for live-cell super-resolution imaging.
However, there are certain limitations to the method, as well. First, aligning an inclined illumination requires knowledge and skills in optics and microscopy. Alternatively, a vertical illumination pattern could be utilized, reducing the technical requirements and making it easier to implement (Figure 1a). However, vertical illumination introduces the possibility of simultaneously exciting two NPCs simultaneously. This could be resolved during data analysis but will take additional time. Second, the 2D-to-3D transformation algorithm has a limited serviceable range, currently only capable of recovering 3D dynamics and structural information on rotationally symmetric systems, such as primary cilium, nuclear pore complexes (NPCs), and glass nanocapillaries (GNCs). This limitation is due to the algorithm’s assumption that the distribution in the θ dimension is constant at each (x, r) in rotationally symmetric systems. Moreover, this method averages finer details within each radial bin along the r dimension (typically 5–10 nm). Localizing particles in the z-dimension is based on Gaussian probability densities of the particles in the r-dimension, meaning that SPEED does not provide true z-dimensional information for discrete localizations; rather, it provides virtual 3D information for the probability density in the r-dimension. This virtual 3D information provides statistical confidence of the most probable z-position based on the standard deviation of the distribution. Third, implicit in the 2D-to-3D algorithm is an assumption that x is constant; therefore, this method works only on structures that are horizontal to the focal plane. When data that is not rotationally symmetric or horizontal to the focal plane, the 2D-to-3D algorithm returns negative values making the analysis nonviable. As such, only NPCs that reside on the equator of the NE are imaged. The subsequent localizations are then overlapped, and their distributions are evaluated to ensure the imaged pore is horizontal to the focal plane. Finally, expertise in coding and modeling is necessary to validate the reproducibility of the obtained 3D structural information. However, it is anticipated that the 2D-to-3D transformation algorithms could be further developed to study other regular or irregularly shaped structures if the molecule of interest follows a radially symmetric distribution.
3. Biological Application of SPEED Microscopy
Due to the superior SNR and mild phototoxic effects of SPEED microscopy, this technique has been utilized to investigate the spatiotemporal dynamics and distribution of biomolecules within a range of rotationally symmetric subdiffraction-limit structures at a high resolution. Considering that the transport channel of the NPC is believed to be a rotationally symmetric system, by applying SPEED’s virtual 3D super-resolution imaging technique, the nucleocytoplasmic transport of various biomolecules, including small organic molecules,84 soluble proteins,79,84−88 membrane proteins,88 intrinsically disordered proteins,87,89 and mRNAs,89,90 through native NPCs were tracked. These experiments revealed the kinetics of nucleocytoplasmic transportation, the pathways of nuclear diffusion, and the configuration of the NPC’s permeable selectivity barrier with a localization precision of ≤10 to 20 nm and a temporal resolution of 0.4–2 ms. The SPPED microscopy was also applied to studying molecule transport within the primary cilium,91 antibody-labeled microtubules,92 and in vitro rotationally symmetric subdiffraction structures, such as glass nanocapillaries(GNCs).91 With SPEED microscopy, 3D transport routes of various cytosolic and membrane proteins in primary cilium and fluorescent dyes within the GNC were obtained with a high temporal resolution of 0.4–2 ms and a high localization precision of ≤10–16 nm.
3.1. Tracking of Cytosol Proteins with Different Sizes, Charges, and Conformations through the Nuclear Pore Complexes
3.1.1. Tracking Importins and Exportins
Importin is a karyopherin that shuttles protein molecules from the cytoplasm to the nucleus by binding to specific recognition sequences known as nuclear localization sequences (NLS). It consists of two subunits: importin α and importin β. Members of the importin-β family can transport cargo by themselves or form heterodimers with importin-α. SPEED microscopy studies have been used to investigate the spatial distributions of three major importins, Imp β1,79 Imp β287 (also known as transportin), and nuclear transport factor 2 (NTF2),87 in the NPC. In these studies, Imp β1 was found to primarily occupy the peripheral regions around the central axial channel of the NPC, with a single strong interaction zone ranging from −100 to 90 nm. This finding indicated that the FG repeats recognized by Imp β1 are mainly available in the peripheral regions covering the entire NPC and that Imp β1 can interact with all the FG Nups in the NPC. On the other hand, its truncated version, Imp β1(331–861), interacted effectively only with the FG Nups at the scaffold of the NPC, with a strong interaction zone ranging from −40 to 70 nm, but lost interactions at both sides of the NPC, forming two weak interaction zones.
In contrast to Imp β1, with only one strong interaction region across the entire NPC, Imp β2 and NTF2 had both strong and weak transport receptor-FG interaction zones. Imp β2 had a strong interaction region ranging only half of what Imp β1 has, from 50 nm on the cytoplasmic side to −50 nm on the nuclear side of the NPC. A small fraction of molecules diffuses into the central axial channel within its weak interaction zones on both sides of the NPC. NTF2 showed two strong interaction zones and three weak interaction regions along the NPC. The observations are consistent with recent studies utilizing 3D light microscopy93 and coincide with previous studies on the binding affinities between FG Nups and the tested importins.94 Imp β1 strongly binds with all the FG Nups in the NPC,94−96 while Imp β2 has relatively weaker binding affinities with FG Nups located at the cytoplasmic and nuclear ends of the NPC. NTF2 possesses weak binding activity toward most FG Nups on the cytoplasmic side and toward all the GLFG Nups, such as Nup98, a GLFG Nup known to play a critical role in the selectivity barrier formation of the NPC. Thus, the above result strongly suggests that the SPEED microscopy approach, with its 2D to 3D transformation, is still a valid and straightforward method to study interactions in the native NPCs.
Using the information collected via SPEED microscopy, a spatial map of the distribution of major exportins, including Crm1, CAS, and the TapΔNLS-p15 complex, was generated.87 The TapΔNLS-p15 complex demonstrated a single, strong interaction zone, while Crm1 and CAS exhibited multiple TR-FG interaction regions of varying strength. These findings were consistent with previous in vitro studies that examined the binding affinities between these exportins and individual FG Nups.94
3.1.2. Tracking Cytosolic Proteins of Varying Sizes
Fluorescently labeled transiting substrates of varying sizes,84 ranging from 0.3 to 217 kDa in molecular weight and approximately 1.4–12.8 nm in diameter, were also tracked through native NPCs using SPEED microscopy. The analysis of the 3D probability spatial distributions recovered from 2D data revealed that small molecules (0.3–29 kDa) diffuse smoothly through the single axial central channel in the native NPCs. In contrast, larger molecules such as 41 kDa dextrans, 61 kDa GFP dimers, and 72 kDa dextrans were less or almost entirely impermeable for this channel (Figure 2a). Our observations were consistent with previously obtained data from electron microscopy or size estimations from measurements of bulk transport rates,97−99 and our measured cutoff size for passive diffusion through the NPC was approximately 40–60 kDa. Regarding facilitated transportation, transport receptors such as Imp β1 and import cargo complexes were found to transport through the NPC efficiently but were mainly located in the periphery of the R dimension and seldom in the central channel. Our data support the existence of a single passive diffusion central channel with peripheral active transport configuration in the NPC and refute the multiple holes in a hydrogel meshwork proposed by the selective phase/hydrogel model. These findings are consistent with previous statements suggesting that passive and active transport in the NPCs occurs via distinct paths and with the forest model’s prediction of double transport zones.
Figure 2.

Two-dimensional super-resolution spatial distributions and 3D transport routes of various (A) cytosol proteins and (B) IDPs with different molecular weights. Adapted with permission from figures previously published by PNAS84 and Protein Science.86
3.1.3. Tracking Cytosolic Proteins with Differing Surface Charges
Another SPEED microscopy study was performed to investigate the charge dependency for the nucleocytoplasmic transportation. Substrates with similar sizes and surface hydrophobicities that differ in surface charge were selected as candidates for investigation. Green fluorescence protein (GFP) with a net negative charge of −7, and its supercharged mutations with a positive charge of +36 (+36GFP) or a negative charge of −30 (−30GFP) were chosen. These substrates were well-suited for testing the effect of molecular charge on passive diffusion, as they were all below the cutoff size for passive diffusion and thus diffused through a central axial channel in the NPC regardless of their charge. Although the charged GFPs differed in pore binding frequencies and transport times, their transport efficiencies (import and export) were almost identical once they entered the NPC. This result suggests that negatively charged molecules have higher initial velocity due to stronger attraction with positively charged FG Nups. However, they do not prioritize traversing the NPC more than less negatively charged or positively charged molecules. On the other hand, when studying facilitated transportation using a similar method, it was found that the charges on the cargo did not cause significant changes in transport kinetics or 3D transport routes. Both findings suggest that while surface charge may affect initial interactions with the NPC, hydrophobic interactions dominate the facilitated translocation of charged cargo complexes.85
3.1.4. Tracking Cytosolic Proteins with Intrinsically Disordered Regions
SPEED microscopy has also been used to determine whether the size-dependent principle of nucleocytoplasmic transport for folded proteins applies to intrinsically disordered proteins (IDPs), individual IDPs with varying sizes were monitored as they traveled through the native NPCs.86 The study employed both large (>40 kDa) and small (<40 kDa) IDPs with and without FG domains within their disordered regions. The candidates ranged from 11 to 63 kDa, which were highly disordered and were isolated with high purity (Figure 2b). To our best knowledge, no specific transport receptors are identified for the IDP candidates that were tested. The result showed that both large (62 kDa Nsp1 and 45 kDa Nup159) and small (37 kDa Nup42 and 11 kDa Nup116) IDP candidates could diffuse through native NPCs without transport receptors. This finding contradicts the behavior observed in soluble proteins such as 2x-GFP and larger dextrans. Furthermore, the passive diffusion of these large FG-IDPs through the NPCs occurred with similar diffusion times (1–4 ms) and transport efficiencies (∼50%) as the smaller FG-IDPs. These observations demonstrate that molecular weight is unlikely the dominant factor in determining transport mode and kinetics for IDPs. The 3D nuclear transport routes of the large FG-IDPs revealed that they mainly diffuse through the central axial channel of the NPC, which was previously thought to be reserved for the passive diffusion of small folded proteins. Conversely, the two smaller FG-IDPs, Nup42 (1–372) and Nup116 (348–458), primarily diffused through the peripheral regions of the pore, as evidenced by the spatial probability of IDPs staying in the periphery mainly (>80%). Interestingly, the transport routes for these small IDPs without transport receptors were similar to the facilitated translocation of large folded protein cargos. The distinct transport routes for small and large IDPs seem to be the opposite of the spatial transport routes identified for small and large folded cargo proteins in our previous studies.
3.2. Tracking Membrane Proteins across the Nuclear Envelope
The nuclear envelope consists of two discrete phospholipid bilayers, the outer nuclear membrane (ONM) and inner nuclear membrane (INM), separated by a perinuclear space of ∼30–50 nm,.100,101 Embedded within these membranes are a wide variety of proteins called nuclear envelope transmembrane proteins (NETs) critical to proper cellular function that exists on either the ONM, INM, or both the ONM and INM in different concentrations. NETs play an essential role in cell and nuclear migration, genome regulation, and mechanosignal transduction via the linker of nucleoskeleton and cytoskeleton (LINC) complex, as well as providing structure to their respective membranes.102−111 Dysregulation of NET concentration results in a group of human diseases commonly referred to as laminopathies or envelopathies, possibly the most prominent being Emery-Dreifuss muscular dystrophy, which is caused by mutations in EMD and LMNA, the genes encoding the NETs emerin and A-type lamins, respectively.112−114
In light of the critical roles played by NETs as well as their implications in understanding human diseases, there is significant interest in understanding the transport dynamics of NETs as well as their relative quantification and localization to the INM or ONM of NETs. To this end, our lab has developed a single-molecule super-resolution technique, single-molecule fluorescence recovery after photobleaching (smFRAP), to not only confirm the presence of proteins on the inner or outer nuclear membrane, but their relative concentrations as well as the diffusion coefficients of NETs moving through the nuclear envelope.115
The development of smFRAP began with the recognition of the physical limitations of optical microscopy, introduced as Abbe’s diffraction limit in the previous section. As the INM and ONM are separated only by the ∼30–50 nm perinuclear space, the distance between these two membranes falls well below the diffraction limit. As a result, many specialized methods have been developed to interrogate NETs located on the INM and ONM.108 While many techniques, both biochemical and microscope-based, exist to evaluate this problem, they suffer from low temporal resolution, rendering them ill-suited to interrogating the dynamics of NETs in live cells. smFRAP was created as a method to bring together the principles of traditional FRAP, typically performed on a confocal microscope, with the high-speed and super-resolution abilities of vertical SPEED microscopy, allowing for improved spatial and temporal resolution.116
Traditional FRAP, also termed bulk FRAP, is used to calculate what proportion of the NET of interest is mobile or immobile. This is accomplished by evaluating the proportion of NETs that are able to return to the area following photobleaching via a photobleaching recovery curve (Figure 3e). This information is important as during smFRAP, only the mobile fraction will be observed. Therefore, to calculate the final ratio of NETs, a corrected ratio is calculated using this information. Next, smFRAP is used by focusing the center of a single-point super-resolution system on the membrane of a cell (Figure 3a). The NETs of interest are then photobleached using a laser power approximately ten times that of the imaging power. Following photobleaching, a video with very high capture speed, for example, 2 ms/frame, is utilized to track NETs at the single-molecule level as they diffuse back into the photobleached area. To prevent photobleaching of the individual NETs, an optical chopper is employed (Figure 3a). The subsequent single-molecule traces are then localized and plotted in two dimensions (Figure 3b). A density histogram along the X-axis is generated that then shows the number of localizations (Figure 3c). A double Gaussian curve is then fit to the resulting histogram. This defines the INM and ONM, with the area under the curve being used to calculate the relative concentration of NETs on the INM and ONM (Figure 3d). In addition to evaluating the relative localization to the discrete membranes, the individual single-molecule traces are evaluated for their diffusion coefficients, calculated using mean square displacement (MSD). During this phase, it is also possible to track discrete NETs as they transport from one membrane to the other through the nuclear pore complex.88,115 A step-by-step protocol on how to perform this method has been previously published.117
Figure 3.

Optical pathway for smFRAP as well as examples of expected data. (A) Schematic of the excitation laser used in smFRAP. A 561 nm laser (green) is directed into the objective. The inset depicts the laser passing through the nuclear envelope while the focal plane is at the nuclear equator, thereby illuminating NETs on both the inner and outer nuclear membrane. Inset illustrates laser power during the different phases of smFRAP. Fluorophores are initially photobleached using high laser power. The chopper is then engaged, allowing fluorescently tagged NETs to diffuse into the detection area. (B) Two-dimension (2D) super-resolution image of LBR on the NE, in which the INM shown in red and the ONM shown in purple. C) Two-peak Gaussian fittings of the points collected from the NE showing the distribution of LBR along the NE. The shaded regions represent the width of the INM and ONM as determined by the full width at half-maximum (fwhm) as determined by the fitting. (D) Approximate concentration ratios of LBR distribution along the INM (red) and ONM (purple). (E) FRAP curve for LBR. The mobile fraction of whole NE is ∼0.39. INM: inner nuclear membrane; ONM: inner nuclear membrane. Adapted with permission from figures previously published by Cell Press.117
This method has been utilized to investigate various aspects of different NETs. Specifically, the NETs NET59, NET51, NET99, Lap2β, and Lamin binding receptor (LBR) were evaluated for their relative concentration, transport routes through the NPC, and diffusion coefficients.88 This study provided never before seen insight into how NETs transport from the ONM to the INM, helping to clarify the current models of NET transport. In addition to the transport behavior of NETs, this technique was employed to great effect in evaluating the behavior of stimulators of interferon genes (STING) with and without the presence of synthetic double-stranded RNA and plasmid DNA. This study demonstrated that not only was STING present on both the INM and ONM, but that the diffusion coefficient of this NET increased 4-fold when foreign RNA and DNA were present in the cell, indicating a redistribution of nuclear envelope STING as part of the innate immune response.118
3.3. Mapping mRNAs Export through Native and Aberrant NPCs
3.3.1. mRNA Nuclear Export through Native NPCs
Exemplar cellular health and function are dependent on the effective mRNA nuclear export. A process requiring mRNA protein complexes, messenger ribonucleoprotein (mRNP), to interact with nuclear membrane-bound NPCs. Marking mRNP’s nucleocytoplasmic diffusion through the NPC is a crucial rate-limiting step in its maturation. With dimensions of 10–20 nm in diameter and 90–150 nm in length,119 mRNPs are among the largest endogenous cargos to transport through the NPC and fall well above the selectively permeable barrier of the NPC.120−123 Of the cofactors associated with the mRNP, major nuclear transport receptor (NTR) Tap/p15 is believed to facilitate mRNP’s diffusion through the NPC by binding to the complex and ferrying it through the NPC.124 To quantify and resolve the transport dynamics of mRNPs, vectors containing a 24X MS2 stem-loop mRNA and fluorescently labeled MS2 coat protein (MCP) were cotransfected.125,126 The exogenous model mRNA is bound by several fluorescently labeled MCPs and imaged by SPEED microscopy, with a spatial-temporal resolution of 8–12 nm and 2 ms.124,127 Our findings revealed a fast-slow-fast diffusion pattern for mRNPs interacting with NPCs, with prolonged interactions occurring within the central channel of the NPC, correlating with the slightly higher amount of central channel abortive events observed within native NPCs (Figure. 4a). Additionally, a high overlap between the mRNP and Tap/p15 3D nuclear transport route maps was observed, with the peak density differing by approximately 1–2 nm. Notably, the central channel region contains high concentrations of FG-Nups. Those FG-Nups interact heavily with NTRs128 and exemplify Tap/p15’s role as an NTR for mRNP nuclear export.
Figure 4.

Nuclear export of mRNP through native (A) and aberrant NPCs (B). Both illustrations show an mRNP particle successfully exporting through the NPC (green arrow) and abortively exporting from the NPC (red arrow). The nuclear basket, central channel, or cytoplasmic fibril subregions separate the abortive nuclear export. The numerical values for the successful and abortive nuclear export indicate the transport efficiency from the 2021 Li et al. study.89
3.3.2. mRNA Nuclear Export through Aberrant NPCs
When comparing the different subregions of the NPC, it was found that within native NPCs, the central channel FG-Nups has slightly more influence on mRNP nuclear export that successfully enters the NPC. However, concerning nuclear transport efficiency and NPC docking, the nuclear basket FG-Nups, Nup153, Nup50, and Tpr play a more significant role in successful mRNP nuclear export. To accurately access each nuclear basket Nup’s impact on mRNP transport, the stoichiometric ratio of nuclear basket Nups was quantified by sequential photobleaching of each stably expressed fluorescently labeled Nup. Our findings revealed a 1:1:1 stoichiometric ratio with a copy number of eight for each nuclear basket Nup. Refuting previous measurements of 1:4:2123 and 2:1:2129 for Nup153:Nup50:Tpr with one equal to a copy number of eight. With each nuclear basket Nup appropriately weighted, each Nup was selectively knocked down by an auxin-inducible degron approach, quantifying its significance on mRNP nuclear export. These experiments observed a 4-fold decrease in mRNP nuclear export efficiency when knocking down Nup153 and Nup50. With changes in all three subregions for abortive events (Figure 4b). These include a 3-fold and 2-fold decrease in the cytoplasmic fibrils and central channel subregions, respectively, and a more than 4-fold increase in the nuclear basket subregion. Interestingly, the absence of Tpr reduced mRNPs docking and subsequent entry into the NPC by approximately 75%.
3.4. Imaging of Viral Particles through the NPCs
Viruses are known to be intracellular pathogens that exploit host cells to generate viral progeny. This involves hijacking the cellular components of the host cell, with some viruses replicating solely in the cytoplasm, while others use the host nucleus for viral replication. Transport receptors often participate in this process, although the mechanisms for the nuclear translocation of viral genetic material may differ among viral classes. Adeno-associated virus 2 (AAV2), a parvovirus with a diameter of approximately 25 nm, is capable of using facilitated transport to move its entire viral capsid into the nucleus. Its nucleocytoplasmic transportation is likely due to the presence of nuclear localization signals (NLSs) in the basic regions of VP1, VP2, and VP3, as their mutations have been shown to significantly reduce the import of the viral capsid significantly. Despite this, AAV vectors remain popular for nuclear gene delivery due to their low pathogenicity and tissue specificity. Our research aimed to enhance our understanding of AAV nuclear import by examining different details of the process within NPCs, such as import time, success rate, and particle configuration during translocation.130 Thus, SPEED microscopy was implemented to track individual AAV2 particles across the NE and through single NPCs in live cells, and the observations showed that around 17% of intact AAV2 particles originating from the cytoplasm successfully traversed the NPC to enter the nucleoplasm. The results suggest that the nuclear import of AAV2 particles could be another rate-limiting step for AAV2, along with the cytoplasmic131 and endosomal membranes132 suggested in other studies.
3.5. Mapping of Scaffold and Disordered Proteins in the NPC
In addition to efforts in single-molecule tracking to study nucleocytoplasmic transport, restoring the NPC’s structural information to understand the entire process better is a major focus for SPEED. Although the complex itself does not get transported, it serves as the central hub for the entire process and is therefore worthy of highlighting in this review. Our initial efforts focused on the spatial distribution of scaffold Nups, such as Pom121, Nup37, and Nup35.133 Pom121 showed eight specific grouped locations with a diameter of 118 ± 4 nm in the radial distribution, while Nup37 and Nup35 had circular diameters of 90 ± 4 and 64 ± 5 nm, respectively. The scaffold Nups were found to be distributed across three distinct layers in the radial dimension, and the spatial location of each Nup was determined. However, the radial view was limited and did not reveal the complete distribution and copy number for each Nup along the axial direction of the NPC. To obtain an axial view, individual NPCs were illuminated on either the left or right side. Pom121 exhibited a single-line spatial organization, while Nup37 and Nup35 had two isolated line distributions separated by varying distances of 36 ± 3 and 7 ± 3 nm, respectively. Based on the radial and axial views, it was found that Pom121 had a single layer, while Nup37 and Nup35 had two layers. Pom121 showed an 8-fold grouping in the radial dimension, suggesting a total copy number of eight. This suggestion was supported by the GFP intensity distribution of NPCs expressing GFP-Pom121, which indicated an average of 8 ± 1 (mean ± sem) copies of GFP-Pom121 per NPC. The radial distribution of both Nup37 and Nup35 exhibited eight groupings, while the axial dimension had two layers. However, it is unclear from these data alone whether there are 16 or 32 copies of corresponding Nups per NPC, as each Nup layer in the axial dimension may contain either 8 or 16 copies. To estimate the copy number, we also used GFP intensities to measure NPCs containing GFP-tagged Nup37 or Nup35. Our analysis of Nup37 revealed that ∼49% of NPCs had GFP intensity values corresponding to more than 16 copies of GFP, supporting the presence of 32 copies of Nup37 in native NPCs. For Nup35, the GFP intensity distribution showed that ∼50% of NPCs contained intensities corresponding to more than eight copies of GFP, while all measured NPCs had fewer than 32 copies, suggesting 16 copies of Nup35 in native NPCs.
4. Perspective
Despite significant progress in understanding nucleocytoplasmic transport processes, a large portion of this complex system remains unknown. Numerous questions need to be answered in future studies. For instance, while our previous research examined the distinct role of nuclear basket Nups during mRNA export, the roles of central Nups and cytoplasmic Nups are still unclear. Investigating the role of central Nups, such as Nup98, Nup54, Nup58, and Nup62, which are believed to form the selective barrier in the NPC, would be particularly intriguing. The question of whether any of these Nups play a more dominant role than the others during mRNA export remains unanswered. Additionally, expanding the application of SPEED to disease studies could be a logical next step given that disruption and manipulation of nucleocytoplasmic transportation are often linked to human diseases, particularly neurological diseases and cancer. Targets of interest could include Aladin, which is associated with triple A syndrome, Nup358, which is associated with neurodegenerative diseases, and fusion Nups, which are associated with T-cell acute lymphocytic leukemia. Quantifying how nucleocytoplasmic transport changes when these targets of interest are disrupted could substantially improve our understanding of the pathogenesis of these diseases.
The assumption for 2D-to-3D transformation algorithms to work is that there is a constant density of the fluorescently labeled molecule of interest along a given radial area. Therefore, we anticipate that our approach could be further developed for other regular or even irregularly shaped radially symmetric structures. Radially symmetric tubes are not rare in the cellular environment, including NPCs, primary cilium, and microtubules, which have been studied in our laboratory using SPEED microscopy. Expanding this approach to other radially symmetric structures, such as spherical and ellipsoid shapes, would undoubtedly broaden its scope and area of application, as these shapes commonly exist in cells. For example, endosomes and vesicles are highly dynamic systems that are generally spherical in shape. In order to gain the capability to investigate spherical, ellipsoidal, irregularly shaped, or near-radially symmetric objects, it is necessary to develop a more sophisticated 2D-to-3D algorithm. Obtaining high spatiotemporal resolution live-cell images of such structures would provide critical insights into their structural and dynamic study. While super-resolution microscopy techniques such as STED, SIM, and STORM have been used to study endosomes, the lateral resolution achieved is on the lower end of the super-resolution techniques, at ∼90–100 nm.134 Achieving a lateral resolution of 10 nm, as demonstrated in many SPEED microscopy studies, could provide unprecedented insights into endosome behavior and other subcellular structures, significantly contributing to our understanding of cell biology.
As aforementioned, SPEED microscopy enables accurate localization of individual fluorescent molecules within submicrometer biological channels or cavities, in both stationary and moving states, with high spatiotemporal resolution in 2D. Coupling this method with our 2D-to-3D transformation algorithm has allowed us to recover 3D subdiffraction-limit information from 2D images, representing a significant advancement in the field of cellular dynamics. There are a few ways in which the technique can be further developed to overcome, or reduce the effect of, some of the limitations listed in section 2.2, and are currently in the planning stages. As mentioned previously, the 2D-to-3D algorithm may be improved to allow for the expansion of the types of subcellular structures that can be investigated by SPEED. In addition, one of the most time-consuming and difficult portions of utilizing the technique is aligning the laser to allow for appropriate inclined illumination. In the future, this process may be improved in a way that allows for the process to be more automated, reducing the preparation time and making the experimental setup more user-friendly. Finally, the digital tools required to analyze data collected from SPEED and to validate the reproducibility of the obtained information require some knowledge of coding and modeling. A graphical user interface could be developed for this process, which would make SPEED more accessible to other laboratories. We believe that further development of the system is necessary to expand its application beyond the NPC and primary cilia to other subcellular structures such as exosomes and mitochondria. With its high spatial and temporal resolution, straightforward sample prep, and pinpointed illumination pattern, SPEED microscopy could provide unprecedented insights into more subcellular structures and enhance our understanding of their functions.
5. Conclusion
Over the years, scientists have made significant strides in improving the imaging resolution of light microscopy to study cellular structures and dynamics under physiological conditions. Despite these advances, obtaining information on a three-dimensional (3D) super-resolution scale in live cells at high speeds remains a challenge. Recently, a new technique called SPEED microscopy was developed along with a 2D-to-3D transformation algorithm, which is specifically designed for subcellular structures and organelles with rotational symmetry like the NPC. With the SPEED microscopy approach, researchers have been able to track cytosol proteins with varying physical properties, membrane proteins, mRNAs, and viral particles as they move through the NPC. By observing the transport behaviors of these proteins and complexes, as well as tracking Nups themselves, scientists have gained meaningful insights into the structure of the pore, further enhancing our understanding of this dynamic and essential process.
Acknowledgments
The project was supported by grants from the US NIH (NIH GM097037, GM116204 and GM122552 to W.Y.).
Glossary
Vocabulary
- Single-molecule localization microscopy (SMLM)
SMLM is a set of super-resolution microscopy techniques, most notably including photoactivated localization microscopy (PALM), stochastic optical reconstruction microscopy (STORM), and Point accumulation in nanoscale topography (PAINT). By controlling the spatiotemporal relationship between fluorescence of different molecules, SMLM can achieve resolutions around 20 nm or better.
- Nuclear Pore Complex (NPC)
The NPC is a large cellular protein complex, each weighing over 100 MDa in human cells, and acts as a selectively permeable barrier between the nucleus and the cytoplasm. Cargo passes bidirectionally through the NPC via passive diffusion, for small molecules, and facilitated diffusion for large cargo. Facilitated diffusion is mediated by nuclear transporters, notably karyopherins and other similar transporters, that interact with phenylalanine-glycine nucleoporins (FG-Nups) to facilitate transport.
- Nucleoporins (Nups)
Nups are the individual proteins that make up the NPC. There are approximately 30 different Nups that exist in octameric symmetry within the complex. Nups can be most easily separated into three distinct classes, scaffold Nups which form the backbone of the complex, FG-Nups which create the selective barrier and permit facilitated diffusion through the pore, and transmembrane Nups which anchor the NPC to the nuclear envelope.
- SPEED Microscopy
This technique, in combination with a 2D-to-3D transformation algorithm, has been developed over the past few years as a super-resolution method of investigating rotationally symmetric cellular structures, most notably the structure and function of the NPC. The method utilizes either an inclined or vertical illumination laser to excite and subsequently localize a single NPC and discrete macromolecules as they transport through the complex. Due to its application in live cells, high spatiotemporal resolution, and ability to derive 3D information from 2D images, SPEED Microscopy has been employed to study the behaviors of single NPCs and nucleocytoplasmic transport events with extreme precision.
- Photon budget
The photon budget describes the number of available photons that can be collected by a detector over a given exposure time. This photon budget can be variously allocated to improve localization precision, sensitivity, or temporal resolution. A high photon budget enables improvements in all facets while a poor photon budget must be selectively allocated to optimize a discrete aspect of imaging. Each specific imaging system and fluorophore has a distinct photon budget that is impacted by factors such as the brightness, extinction coefficient, quantum yield, photostability of the target fluorophore, and the level of background signal.
The authors declare no competing financial interest.
References
- Singh B. K.; Nagar H.; Roichman Y.; Arie A. Particle manipulation beyond the diffraction limit using structured super-oscillating light beams. Light: Science & Applications 2017, 6 (9), e17050–e17050. 10.1038/lsa.2017.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakouri A.; Ziabari A.; Kendig D.; Bahk J. H.; Xuan Y.; Ye P. D.; Yazawa K.; Shakouri A.. Stable thermoreflectance thermal imaging microscopy with piezoelectric position control. In 2016 32nd Thermal Measurement, Modeling & Management Symposium (SEMI-THERM), 14–17 March 2016, 2016; pp 128–132. DOI: 10.1109/SEMI-THERM.2016.7458456 [DOI]
- Lelek M.; Gyparaki M. T.; Beliu G.; Schueder F.; Griffié J.; Manley S.; Jungmann R.; Sauer M.; Lakadamyali M.; Zimmer C. Single-molecule localization microscopy. Nature Reviews Methods Primers 2021, 1 (1), 1–27. 10.1038/s43586-021-00038-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Diezmann L.; Shechtman Y.; Moerner W. Three-dimensional localization of single molecules for super-resolution imaging and single-particle tracking. Chem. Rev. 2017, 117 (11), 7244–7275. 10.1021/acs.chemrev.6b00629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardill A.; Kemeny I.; Li Y.; Zahedian M.; Cambria M. C.; Xu X.; Lordi V.; Gali Á.; Maze J. R.; Choy J. T.; et al. Super-resolution airy disk microscopy of individual color centers in diamond. ACS Photonics 2022, 9 (12), 3848–3854. 10.1021/acsphotonics.2c00713. [DOI] [Google Scholar]
- Betzig E.; Patterson G. H.; Sougrat R.; Lindwasser O. W.; Olenych S.; Bonifacino J. S.; Davidson M. W.; Lippincott-Schwartz J.; Hess H. F. Imaging Intracellular Fluorescent Proteins at Nanometer Resolution. Science 2006, 313 (5793), 1642–1645. 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- Malkusch S.; Endesfelder U.; Mondry J.; Gelléri M.; Verveer P. J.; Heilemann M. Coordinate-based colocalization analysis of single-molecule localization microscopy data. Histochemistry and Cell Biology 2012, 137 (1), 1–10. 10.1007/s00418-011-0880-5. [DOI] [PubMed] [Google Scholar]
- Burgert A.; Letschert S.; Doose S.; Sauer M. Artifacts in single-molecule localization microscopy. Histochemistry and Cell Biology 2015, 144 (2), 123–131. 10.1007/s00418-015-1340-4. [DOI] [PubMed] [Google Scholar]
- Sage D.; Kirshner H.; Pengo T.; Stuurman N.; Min J.; Manley S.; Unser M. Quantitative evaluation of software packages for single-molecule localization microscopy. Nat. Methods 2015, 12 (8), 717–724. 10.1038/nmeth.3442. [DOI] [PubMed] [Google Scholar]
- Chang Y.; Kim D.-H.; Zhou K.; Jeong M. G.; Park S.; Kwon Y.; Hong T. M.; Noh J.; Ryu S. H. Improved resolution in single-molecule localization microscopy using QD-PAINT. Experimental & Molecular Medicine 2021, 53 (3), 384–392. 10.1038/s12276-021-00572-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Vaughan J. C. Switchable fluorophores for single-molecule localization microscopy. Chem. Rev. 2018, 118 (18), 9412–9454. 10.1021/acs.chemrev.7b00767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelotti M. L.; Antonelli G.; Conte C.; Romagnani P. Imaging the kidney: from light to super-resolution microscopy. Nephrology Dialysis Transplantation 2021, 36 (1), 19–28. 10.1093/ndt/gfz136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shashkova S.; Leake M. C. Single-molecule fluorescence microscopy review: shedding new light on old problems. Bioscience reports 2017, 37 (4), BSR20170031. 10.1042/BSR20170031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnitzbauer J.; Strauss M. T.; Schlichthaerle T.; Schueder F.; Jungmann R. Super-resolution microscopy with DNA-PAINT. Nat. Protoc. 2017, 12 (6), 1198–1228. 10.1038/nprot.2017.024. [DOI] [PubMed] [Google Scholar]
- Shroff H.; Galbraith C. G.; Galbraith J. A.; Betzig E. Live-cell photoactivated localization microscopy of nanoscale adhesion dynamics. Nat. Methods 2008, 5 (5), 417–423. 10.1038/nmeth.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rust M. J.; Bates M.; Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM. Nat. Methods 2006, 3 (10), 793–796. 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Wee R.; Filius M.; Joo C. Completing the canvas: advances and challenges for DNA-PAINT super-resolution imaging. Trends Biochem. Sci. 2021, 46 (11), 918–930. 10.1016/j.tibs.2021.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Křížek P.; Raška I.; Hagen G. M. Minimizing detection errors in single molecule localization microscopy. Opt. Express 2011, 19 (4), 3226–3235. 10.1364/OE.19.003226. [DOI] [PubMed] [Google Scholar]
- Haider S.; Cameron A.; Siva P.; Lui D.; Shafiee M.; Boroomand A.; Haider N.; Wong A. Fluorescence microscopy image noise reduction using a stochastically-connected random field model. Sci. Rep. 2016, 6 (1), 1–16. 10.1038/srep20640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ru J.-X.; Guan L.-P.; Tang X.-L.; Dou W.; Yao X.; Chen W.-M.; Liu Y.-M.; Zhang G.-L.; Liu W.-S.; Meng Y.; Wang C.-M. Turn-on Phosphorescent Chemodosimeter for Hg2+ Based on a Cyclometalated Ir(III) Complex and Its Application in Time-Resolved Luminescence Assays and Live Cell Imaging. Inorg. Chem. 2014, 53 (21), 11498–11506. 10.1021/ic501417s. [DOI] [PubMed] [Google Scholar]
- Zhao Q.; Yu M.; Shi L.; Liu S.; Li C.; Shi M.; Zhou Z.; Huang C.; Li F. Cationic Iridium(III) Complexes with Tunable Emission Color as Phosphorescent Dyes for Live Cell Imaging. Organometallics 2010, 29 (5), 1085–1091. 10.1021/om900691r. [DOI] [Google Scholar]
- Farias P. M. A.; Santos B. S.; Fontes A.. Semiconductor Fluorescent Quantum Dots: Efficient Biolabels in Cancer Diagnostics. In Micro and Nano Technologies in Bioanalysis: Methods and Protocols; Foote R. S., Lee J. W., Eds.; Humana Press, 2009; pp 407–419. [DOI] [PubMed] [Google Scholar]
- Yang X.; Zhanghao K.; Wang H.; Liu Y.; Wang F.; Zhang X.; Shi K.; Gao J.; Jin D.; Xi P. Versatile Application of Fluorescent Quantum Dot Labels in Super-resolution Fluorescence Microscopy. ACS Photonics 2016, 3 (9), 1611–1618. 10.1021/acsphotonics.6b00178. [DOI] [Google Scholar]
- Gajdos T.; Hopp B.; Erdélyi M. Hot-Band Anti-Stokes Fluorescence Properties of Alexa Fluor 568. J. Fluoresc. 2020, 30 (3), 437–443. 10.1007/s10895-020-02496-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C.-L.; Liao Q.; Li T.; Li J.; Jiang J.-Q.; Xu Z.-Z.; Wang X.-D.; Shen R.; Bai D.-C.; Wang Q.; et al. Rational design of small indolic squaraine dyes with large two-photon absorption cross section. Chemical science 2015, 6 (1), 761–769. 10.1039/C4SC02165G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tandale P.; Choudhary N.; Singh J.; Sharma A.; Shukla A.; Sriram P.; Soni U.; Singla N.; Barnwal R. P.; Singh G.; Kaur I. P.; Suttee A. Fluorescent quantum dots: An insight on synthesis and potential biological application as drug carrier in cancer. Biochemistry and Biophysics Reports 2021, 26, 100962. 10.1016/j.bbrep.2021.100962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnell U.; Dijk F.; Sjollema K. A.; Giepmans B. N. G. Immunolabeling artifacts and the need for live-cell imaging. Nat. Methods 2012, 9 (2), 152–158. 10.1038/nmeth.1855. [DOI] [PubMed] [Google Scholar]
- Jaiswal J. K.; Goldman E. R.; Mattoussi H.; Simon S. M. Use of quantum dots for live cell imaging. Nat. Methods 2004, 1 (1), 73–78. 10.1038/nmeth1004-73. [DOI] [PubMed] [Google Scholar]
- Patterson G. H.; Knobel S. M.; Sharif W. D.; Kain S. R.; Piston D. W. Use of the green fluorescent protein and its mutants in quantitative fluorescence microscopy. Biophys. J. 1997, 73 (5), 2782–2790. 10.1016/S0006-3495(97)78307-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q.; Young I. T.; De Jong J. G. S. Photon budget analysis for fluorescence lifetime imaging microscopy. Journal of Biomedical Optics 2011, 16 (8), 086007–086007–086016. 10.1117/1.3608997. [DOI] [PubMed] [Google Scholar]
- Zhu Z.Computational Imaging with Limited Photon Budget. Thesis, University of Central Florida, 2019. [Google Scholar]
- Dixit R.; Cyr R. Cell damage and reactive oxygen species production induced by fluorescence microscopy: effect on mitosis and guidelines for non-invasive fluorescence microscopy. Plant Journal 2003, 36 (2), 280–290. 10.1046/j.1365-313X.2003.01868.x. [DOI] [PubMed] [Google Scholar]
- Jemielita M.; Taormina M. J.; DeLaurier A.; Kimmel C. B.; Parthasarathy R. Comparing phototoxicity during the development of a zebrafish craniofacial bone using confocal and light sheet fluorescence microscopy techniques. Journal of biophotonics 2013, 6 (11–12), 920–928. 10.1002/jbio.201200144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z.; Lavis L. D.; Betzig E. Imaging Live-Cell Dynamics and Structure at the Single-Molecule Level. Mol. Cell 2015, 58 (4), 644–659. 10.1016/j.molcel.2015.02.033. [DOI] [PubMed] [Google Scholar]
- Ni J.; Cao B.; Niu G.; Chen D.; Liang G.; Xia T.; Li H.; Xu C.; Wang J.; Zhang W.; Zhang Y.; Yuan X.; Ni Y. Improved localization precision via restricting confined biomolecule stochastic motion in single-molecule localization microscopy. Nanophotonics 2021, 11 (1), 53–65. 10.1515/nanoph-2021-0481. [DOI] [Google Scholar]
- Chen C.; Zong S.; Wang Z.; Lu J.; Zhu D.; Zhang Y.; Zhang R.; Cui Y. Visualization and intracellular dynamic tracking of exosomes and exosomal miRNAs using single molecule localization microscopy. Nanoscale 2018, 10 (11), 5154–5162. 10.1039/C7NR08800K. [DOI] [PubMed] [Google Scholar]
- Henriques R.; Lelek M.; Fornasiero E. F.; Valtorta F.; Zimmer C.; Mhlanga M. M. QuickPALM: 3D real-time photoactivation nanoscopy image processing in ImageJ. Nat. Methods 2010, 7 (5), 339–340. 10.1038/nmeth0510-339. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Schnitzbauer J.; Hu Z.; Li X.; Cheng Y.; Huang Z.-L.; Huang B. Localization events-based sample drift correction for localization microscopy with redundant cross-correlation algorithm. Opt. Express 2014, 22 (13), 15982–15991. 10.1364/OE.22.015982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tingey M.; Li Y.; Yu W.; Young A.; Yang W. Spelling out the roles of individual nucleoporins in nuclear export of mRNA. Nucleus 2022, 13 (1), 172. 10.1080/19491034.2022.2076965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruba A.; Luo W.; Kelich J.; Tingey M.; Yang W. 3D Tracking-Free Approach for Obtaining 3D Super-Resolution Information in Rotationally Symmetric Biostructures. J. Phys. Chem. B 2019, 123 (24), 5107–5120. 10.1021/acs.jpcb.9b02979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibarra A.; Benner C.; Tyagi S.; Cool J.; Hetzer M. W. Nucleoporin-mediated regulation of cell identity genes. Genes & development 2016, 30 (20), 2253–2258. 10.1101/gad.287417.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nofrini V.; Di Giacomo D.; Mecucci C. Nucleoporin genes in human diseases. European Journal of Human Genetics 2016, 24 (10), 1388–1395. 10.1038/ejhg.2016.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W. ’Natively unfolded’nucleoporins in nucleocytoplasmic transport: clustered or evenly distributed?. Nucleus 2011, 2 (1), 10–16. 10.4161/nucl.13818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuller A. P.; Wojtynek M.; Mankus D.; Tatli M.; Kronenberg-Tenga R.; Regmi S. G.; Dip P. V.; Lytton-Jean A. K. R.; Brignole E. J.; Dasso M.; Weis K.; Medalia O.; Schwartz T. U. The cellular environment shapes the nuclear pore complex architecture. Nature 2021, 598 (7882), 667–671. 10.1038/s41586-021-03985-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemp I.; Kutay U. Nuclear export and cytoplasmic maturation of ribosomal subunits. FEBS letters 2007, 581 (15), 2783–2793. 10.1016/j.febslet.2007.05.013. [DOI] [PubMed] [Google Scholar]
- Ruland J. A.; Krüger A. M.; Dörner K.; Bhatia R.; Wirths S.; Poetes D.; Kutay U.; Siebrasse J. P.; Kubitscheck U. Nuclear export of the pre-60S ribosomal subunit through single nuclear pores observed in real time. Nat. Commun. 2021, 12 (1), 6211. 10.1038/s41467-021-26323-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorokin A.; Kim E.; Ovchinnikov L. Nucleocytoplasmic transport of proteins. Biochemistry (Moscow) 2007, 72, 1439–1457. 10.1134/S0006297907130032. [DOI] [PubMed] [Google Scholar]
- Patel S. S.; Belmont B. J.; Sante J. M.; Rexach M. F. Natively unfolded nucleoporins gate protein diffusion across the nuclear pore complex. Cell 2007, 129 (1), 83–96. 10.1016/j.cell.2007.01.044. [DOI] [PubMed] [Google Scholar]
- Yamada J.; Phillips J. L.; Patel S.; Goldfien G.; Calestagne-Morelli A.; Huang H.; Reza R.; Acheson J.; Krishnan V. V.; Newsam S.; et al. A bimodal distribution of two distinct categories of intrinsically disordered structures with separate functions in FG nucleoporins. Molecular Cellular Proteomics 2010, 9 (10), 2205–2224. 10.1074/mcp.M000035-MCP201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W. Distinct, but not completely separate spatial transport routes in the nuclear pore complex. Nucleus 2013, 4 (3), 166–175. 10.4161/nucl.24874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C.; Goryaynov A.; Yang W. The selective permeability barrier in the nuclear pore complex. Nucleus 2016, 7 (5), 430–446. 10.1080/19491034.2016.1238997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigg E. A. Nucleocytoplasmic transport: signals, mechanisms and regulation. Nature 1997, 386 (6627), 779–787. 10.1038/386779a0. [DOI] [PubMed] [Google Scholar]
- Yamazumi Y.; Kamiya A.; Nishida A.; Nishihara A.; Iemura S.-i.; Natsume T.; Akiyama T. The transmembrane nucleoporin NDC1 is required for targeting of ALADIN to nuclear pore complexes. Biochemical and biophysical research communications 2009, 389 (1), 100–104. 10.1016/j.bbrc.2009.08.096. [DOI] [PubMed] [Google Scholar]
- Sakuma S.; D’Angelo M. A. The roles of the nuclear pore complex in cellular dysfunction, aging and disease. Semin Cell Dev Biol. 2017, 68, 72–84. 10.1016/j.semcdb.2017.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snowden J. S.; Neary D.; Mann D. M. Frontotemporal dementia. British journal of psychiatry 2002, 180 (2), 140–143. 10.1192/bjp.180.2.140. [DOI] [PubMed] [Google Scholar]
- Dauer W.; Przedborski S. Parkinson’s disease: mechanisms and models. Neuron 2003, 39 (6), 889–909. 10.1016/S0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- Grima J. C.; Daigle J. G.; Arbez N.; Cunningham K. C.; Zhang K.; Ochaba J.; Geater C.; Morozko E.; Stocksdale J.; Glatzer J. C.; et al. Mutant huntingtin disrupts the nuclear pore complex. Neuron 2017, 94 (1), 93–107. e106. 10.1016/j.neuron.2017.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y.-L.; Lira M. E.; Hong M.; Kim R. N.; Choi S.-J.; Song J.-Y.; Pandy K.; Mann D. L.; Stahl J. A.; Peckham H. E.; et al. A novel fusion of TPR and ALK in lung adenocarcinoma. Journal of Thoracic Oncology 2014, 9 (4), 563–566. 10.1097/JTO.0000000000000093. [DOI] [PubMed] [Google Scholar]
- Mak H.; Peschard P.; Lin T.; Naujokas M.; Zuo D.; Park M. Oncogenic activation of the Met receptor tyrosine kinase fusion protein, Tpr–Met, involves exclusion from the endocytic degradative pathway. Oncogene 2007, 26 (51), 7213–7221. 10.1038/sj.onc.1210522. [DOI] [PubMed] [Google Scholar]
- Xu H.; Valerio D. G.; Eisold M. E.; Sinha A.; Koche R. P.; Hu W.; Chen C.-W.; Chu S. H.; Brien G. L.; Park C. Y.; et al. NUP98 fusion proteins interact with the NSL and MLL1 complexes to drive leukemogenesis. Cancer cell 2016, 30 (6), 863–878. 10.1016/j.ccell.2016.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terlecki-Zaniewicz S.; Humer T.; Eder T.; Schmoellerl J.; Heyes E.; Manhart G.; Kuchynka N.; Parapatics K.; Liberante F. G.; Müller A. C.; et al. Biomolecular condensation of NUP98 fusion proteins drives leukemogenic gene expression. Nature structural & molecular biology 2021, 28 (2), 190–201. 10.1038/s41594-020-00550-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gervais C.; Dano L.; Perrusson N.; Helias C.; Jeandidier E.; Galoisy A.; Ittel A.; Herbrecht R.; Bilger K.; Mauvieux L. A translocation t (2; 8)(q12; p11) fuses FGFR1 to a novel partner gene, RANBP2/NUP358, in a myeloproliferative/myelodysplastic neoplasm. Leukemia 2013, 27 (5), 1186–1188. 10.1038/leu.2012.286. [DOI] [PubMed] [Google Scholar]
- Mendes A.; Fahrenkrog B. NUP214 in leukemia: it’s more than transport. Cells 2019, 8 (1), 76. 10.3390/cells8010076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ageberg M.; Drott K.; Olofsson T.; Gullberg U.; Lindmark A. Identification of a novel and myeloid specific role of the leukemia-associated fusion protein DEK-NUP214 leading to increased protein synthesis. Genes, Chromosomes and Cancer 2008, 47 (4), 276–287. 10.1002/gcc.20531. [DOI] [PubMed] [Google Scholar]
- Van Vlierberghe P.; van Grotel M.; Tchinda J.; Lee C.; Beverloo H. B.; van der Spek P. J.; Stubbs A.; Cools J.; Nagata K.; Fornerod M.; et al. The recurrent SET-NUP214 fusion as a new HOXA activation mechanism in pediatric T-cell acute lymphoblastic leukemia. Blood, The Journal of the American Society of Hematology 2008, 111 (9), 4668–4680. 10.1182/blood-2007-09-111872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabut G.; Doye V.; Ellenberg J. Mapping the dynamic organization of the nuclear pore complex inside single living cells. Nature cell biology 2004, 6 (11), 1114–1121. 10.1038/ncb1184. [DOI] [PubMed] [Google Scholar]
- Huang G.; Zhan X.; Zeng C.; Liang K.; Zhu X.; Zhao Y.; Wang P.; Wang Q.; Zhou Q.; Tao Q.; Liu M.; Lei J.; Yan C.; Shi Y. Cryo-EM structure of the inner ring from the Xenopus laevis nuclear pore complex. Cell Res. 2022, 32 (5), 451–460. 10.1038/s41422-022-00633-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang G.; Zeng C.; Shi Y. Structure of the nuclear pore complex goes atomic. Curr. Opin Struct Biol. 2023, 78, 102523. 10.1016/j.sbi.2022.102523. [DOI] [PubMed] [Google Scholar]
- Li Z.; Chen S.; Zhao L.; Huang G.; Pi X.; Sun S.; Wang P.; Sui S. F. Near-atomic structure of the inner ring of the Saccharomyces cerevisiae nuclear pore complex. Cell Res. 2022, 32 (5), 437–450. 10.1038/s41422-022-00632-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai L.; Zhu Y.; Ren H.; Huang X.; Zhang C.; Sun F. 8 Å structure of the outer rings of the Xenopus laevis nuclear pore complex obtained by cryo-EM and AI. Protein Cell 2022, 13 (10), 760–777. 10.1007/s13238-021-00895-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson A. C.; Fišerová J.; Goldberg M. W. NPC Structure in Model Organisms: Transmission Electron Microscopy and Immunogold Labeling Using High-Pressure Freezing/Freeze Substitution of Yeast, Worms, and Plants. Methods Mol. Biol. 2022, 2502, 439–459. 10.1007/978-1-0716-2337-4_28. [DOI] [PubMed] [Google Scholar]
- Fichtman B.; Regmi S. G.; Dasso M.; Harel A. High-Resolution Imaging and Analysis of Individual Nuclear Pore Complexes. Methods Mol. Biol. 2022, 2502, 461–471. 10.1007/978-1-0716-2337-4_29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Jonge N.; Peckys D. B. Live Cell Electron Microscopy Is Probably Impossible. ACS Nano 2016, 10 (10), 9061–9063. 10.1021/acsnano.6b02809. [DOI] [PubMed] [Google Scholar]
- Otsuka S.; Bui K. H.; Schorb M.; Hossain M. J.; Politi A. Z.; Koch B.; Eltsov M.; Beck M.; Ellenberg J. Nuclear pore assembly proceeds by an inside-out extrusion of the nuclear envelope. Elife 2016, 5, e19071. 10.7554/eLife.19071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allegretti M.; Zimmerli C. E.; Rantos V.; Wilfling F.; Ronchi P.; Fung H. K. H.; Lee C. W.; Hagen W.; Turonova B.; Karius K.; Bormel M.; Zhang X.; Muller C. W.; Schwab Y.; Mahamid J.; Pfander B.; Kosinski J.; Beck M. In-cell architecture of the nuclear pore and snapshots of its turnover. Nature 2020, 586 (7831), 796–800. 10.1038/s41586-020-2670-5. [DOI] [PubMed] [Google Scholar]
- Fontana P.; Dong Y.; Pi X.; Tong A. B.; Hecksel C. W.; Wang L.; Fu T. M.; Bustamante C.; Wu H. Structure of cytoplasmic ring of nuclear pore complex by integrative cryo-EM and AlphaFold. Science 2022, 376 (6598), eabm9326. 10.1126/science.abm9326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallusser B.; Maltese G.; Di Caprio G.; Vadakkan T. J.; Sanyal A.; Somerville E.; Sahasrabudhe M.; O’Connor J.; Weigert M.; Kirchhausen T. Deep neural network automated segmentation of cellular structures in volume electron microscopy. J. Cell Biol. 2023, 222 (2), e202208005. 10.1083/jcb.202208005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosalaganti S.; Obarska-Kosinska A.; Siggel M.; Taniguchi R.; Turoňová B.; Zimmerli C. E.; Buczak K.; Schmidt F. H.; Margiotta E.; Mackmull M. T.; Hagen W. J. H.; Hummer G.; Kosinski J.; Beck M. AI-based structure prediction empowers integrative structural analysis of human nuclear pores. Science 2022, 376 (6598), eabm9506. 10.1126/science.abm9506. [DOI] [PubMed] [Google Scholar]
- Ma J.; Yang W. Three-dimensional distribution of transient interactions in the nuclear pore complex obtained from single-molecule snapshots. Proc. Natl. Acad. Sci. U. S. A. 2010, 107 (16), 7305–7310. 10.1073/pnas.0908269107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goryaynov A.; Ma J.; Yang W. Single-molecule studies of nucleocytoplasmic transport: from one dimension to three dimensions. Integrative Biology 2012, 4 (1), 10–21. 10.1039/C1IB00041A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim D.; Ford T. N.; Chu K. K.; Metz J. Optically sectioned in vivo imaging with speckle illumination HiLo microscopy. Journal of biomedical optics 2011, 16 (1), 016014. 10.1117/1.3528656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konopka C. A.; Bednarek S. Y. Variable-angle epifluorescence microscopy: a new way to look at protein dynamics in the plant cell cortex. Plant Journal 2008, 53 (1), 186–196. 10.1111/j.1365-313X.2007.03306.x. [DOI] [PubMed] [Google Scholar]
- Stock K.; Sailer R.; Strauss W. S.; Lyttek M.; Steiner R.; Schneckenburger H. Variable-angle total internal reflection fluorescence microscopy (VA-TIRFM): realization and application of a compact illumination device. J. Microsc. 2003, 211 (1), 19–29. 10.1046/j.1365-2818.2003.01200.x. [DOI] [PubMed] [Google Scholar]
- Ma J.; Goryaynov A.; Sarma A.; Yang W. Self-regulated viscous channel in the nuclear pore complex. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (19), 7326–7331. 10.1073/pnas.1201724109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goryaynov A.; Yang W. Role of molecular charge in nucleocytoplasmic transport. PloS one 2014, 9 (2), e88792. 10.1371/journal.pone.0088792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junod S. L.; Kelich J. M.; Ma J.; Yang W. Nucleocytoplasmic transport of intrinsically disordered proteins studied by high-speed super-resolution microscopy. Protein Sci. 2020, 29 (6), 1459–1472. 10.1002/pro.3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J.; Goryaynov A.; Yang W. Super-resolution 3D tomography of interactions and competition in the nuclear pore complex. Nature Structural & Molecular Biology 2016, 23 (3), 239–247. 10.1038/nsmb.3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudumbi K. C.; Czapiewski R.; Ruba A.; Junod S. L.; Li Y.; Luo W.; Ngo C.; Ospina V.; Schirmer E. C.; Yang W. Nucleoplasmic signals promote directed transmembrane protein import simultaneously via multiple channels of nuclear pores. Nat. Commun. 2020, 11 (1), 2184. 10.1038/s41467-020-16033-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Aksenova V.; Tingey M.; Yu J.; Ma P.; Arnaoutov A.; Chen S.; Dasso M.; Yang W. Distinct roles of nuclear basket proteins in directing the passage of mRNA through the nuclear pore. Proc. Natl. Acad. Sci. U. S. A. 2021, 118 (37), e2015621118. 10.1073/pnas.2015621118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J.; Liu Z.; Michelotti N.; Pitchiaya S.; Veerapaneni R.; Androsavich J. R.; Walter N. G.; Yang W. High-resolution three-dimensional mapping of mRNA export through the nuclear pore. Nat. Commun. 2013, 4 (1), 1–9. 10.1038/ncomms3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W.; Ruba A.; Takao D.; Zweifel L. P.; Lim R. Y. H.; Verhey K. J.; Yang W. Axonemal Lumen Dominates Cytosolic Protein Diffusion inside the Primary Cilium. Sci. Rep. 2017, 7 (1), 15793. 10.1038/s41598-017-16103-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westphal V.; Rizzoli S. O.; Lauterbach M. A.; Kamin D.; Jahn R.; Hell S. W. Video-rate far-field optical nanoscopy dissects synaptic vesicle movement. Science 2008, 320 (5873), 246–249. 10.1126/science.1154228. [DOI] [PubMed] [Google Scholar]
- Chowdhury R.; Sau A.; Musser S. M. Super-resolved 3D tracking of cargo transport through nuclear pore complexes. Nature cell biology 2022, 24 (1), 112–122. 10.1038/s41556-021-00815-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry L. J.; Wente S. R. Flexible gates: dynamic topologies and functions for FG nucleoporins in nucleocytoplasmic transport. Eukaryotic cell 2009, 8 (12), 1814–1827. 10.1128/EC.00225-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart M. Molecular mechanism of the nuclear protein import cycle. Nat. Rev. Mol. Cell Biol. 2007, 8 (3), 195–208. 10.1038/nrm2114. [DOI] [PubMed] [Google Scholar]
- Palmeri D.; Malim M. H. Importin β can mediate the nuclear import of an arginine-rich nuclear localization signal in the absence of importin α. Mol. Cell. Biol. 1999, 19 (2), 1218–1225. 10.1128/MCB.19.2.1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rout M. P.; Blobel G. Isolation of the yeast nuclear pore complex. J. Cell Biol. 1993, 123 (4), 771–783. 10.1083/jcb.123.4.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahrenkrog B.; Aebi U. The nuclear pore complex: nucleocytoplasmic transport and beyond. Nat. Rev. Mol. Cell Biol. 2003, 4 (10), 757–766. 10.1038/nrm1230. [DOI] [PubMed] [Google Scholar]
- Ribbeck K.; Görlich D. Kinetic analysis of translocation through nuclear pore complexes. EMBO journal 2001, 20 (6), 1320–1330. 10.1093/emboj/20.6.1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldherr C. M.; Akin D. The permeability of the nuclear envelope in dividing and nondividing cell cultures. J. Cell Biol. 1990, 111 (1), 1–8. 10.1083/jcb.111.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke W. W.; Scheer U.; Krohne G.; Jarasch E.-D. The nuclear envelope and the architecture of the nuclear periphery. J. Cell Biol. 1981, 39s. 10.1083/jcb.91.3.39s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arib G.; Akhtar A. Multiple facets of nuclear periphery in gene expression control. Curr. Opin. Cell Biol. 2011, 23 (3), 346–353. 10.1016/j.ceb.2010.12.005. [DOI] [PubMed] [Google Scholar]
- Burns L. T.; Wente S. R. Trafficking to uncharted territory of the nuclear envelope. Curr. Opin. Cell Biol. 2012, 24 (3), 341–349. 10.1016/j.ceb.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Las Heras J. I.; Meinke P.; Batrakou D. G.; Srsen V.; Zuleger N.; Kerr A. R.; Schirmer E. C. Tissue specificity in the nuclear envelope supports its functional complexity. Nucleus 2013, 4 (6), 460–477. 10.4161/nucl.26872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruenbaum Y.; Margalit A.; Goldman R. D.; Shumaker D. K.; Wilson K. L. The nuclear lamina comes of age. Nat. Rev. Mol. Cell Biol. 2005, 6 (1), 21–31. 10.1038/nrm1550. [DOI] [PubMed] [Google Scholar]
- Heessen S.; Fornerod M. The inner nuclear envelope as a transcription factor resting place. EMBO reports 2007, 8 (10), 914–919. 10.1038/sj.embor.7401075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetzer M. W.; Wente S. R. Border control at the nucleus: biogenesis and organization of the nuclear membrane and pore complexes. Developmental cell 2009, 17 (5), 606–616. 10.1016/j.devcel.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tingey M.; Schnell S. J.; Yu W.; Saredy J.; Junod S.; Patel D.; Alkurdi A. A.; Yang W. Technologies Enabling Single-Molecule Super-Resolution Imaging of mRNA. Cells 2022, 11 (19), 3079. 10.3390/cells11193079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson K. L.; Foisner R. Lamin-binding proteins. Cold Spring Harbor perspectives in biology 2010, 2 (4), a000554. 10.1101/cshperspect.a000554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuleger N.; Korfali N.; Schirmer E. C. Inner nuclear membrane protein transport is mediated by multiple mechanisms. Biochem. Soc. Trans. 2008, 36 (6), 1373–1377. 10.1042/BST0361373. [DOI] [PubMed] [Google Scholar]
- Tingey M.; Schnell S. J.; Li Y.; Junod S.; Yu W.; Yang W.. IMAGING TRANSMEMBRANE PROTEIN TRANSPORT ACROSS THE NUCLEAR ENVELOPE; Independent Publishing Network, 2020. [Google Scholar]
- Shin J.-Y.; Worman H. J. Molecular pathology of laminopathies. Annual Review of Pathology: Mechanisms of Disease 2022, 17, 159–180. 10.1146/annurev-pathol-042220-034240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonne G.; Quijano-Roy S. Emery–Dreifuss muscular dystrophy, laminopathies, and other nuclear envelopathies. Handbook of clinical neurology 2013, 113, 1367–1376. 10.1016/B978-0-444-59565-2.00007-1. [DOI] [PubMed] [Google Scholar]
- Ross J. A.; Stroud M. J. THE NUCLEUS: Mechanosensing in cardiac disease. International Journal of Biochemistry & Cell Biology 2021, 137, 106035. 10.1016/j.biocel.2021.106035. [DOI] [PubMed] [Google Scholar]
- Mudumbi K. C.; Schirmer E. C.; Yang W. Single-point single-molecule FRAP distinguishes inner and outer nuclear membrane protein distribution. Nat. Commun. 2016, 7 (1), 12562. 10.1038/ncomms12562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Tingey M.; Ruba A.; Yang W. High-speed super-resolution imaging of rotationally symmetric structures using SPEED microscopy and 2D-to-3D transformation. Nat. Protoc. 2021, 16 (1), 532–560. 10.1038/s41596-020-00440-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tingey M.; Li Y.; Yang W. Protocol for single-molecule fluorescence recovery after photobleaching microscopy to analyze the dynamics and spatial locations of nuclear transmembrane proteins in live cells. STAR Protocols 2021, 2 (2), 100490. 10.1016/j.xpro.2021.100490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon C. R.; Malik P.; de las Heras J. I.; Saiz-Ros N.; de Lima Alves F.; Tingey M.; Gaunt E.; Richardson A. C.; Kelly D. A.; Goldberg M. W.; Towers G. J.; Yang W.; Rappsilber J.; Digard P.; Schirmer E. C. STING nuclear partners contribute to innate immune signaling responses. iScience 2021, 24 (9), 103055. 10.1016/j.isci.2021.103055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto K.; Tanaka K. J.; Aoki K.; Sameshima M.; Tsujimoto M. Visualization of the reconstituted FRGY2–mRNA complexes by electron microscopy. Biochem. Biophys. Res. Commun. 2003, 306 (1), 53–58. 10.1016/S0006-291X(03)00909-4. [DOI] [PubMed] [Google Scholar]
- Denning D. P.; Patel S. S.; Uversky V.; Fink A. L.; Rexach M. Disorder in the nuclear pore complex: The FG repeat regions of nucleoporins are natively unfolded. Proc. Natl. Acad. Sci. U. S. A. 2003, 100 (5), 2450. 10.1073/pnas.0437902100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan K. J.; Wente S. R. The nuclear pore complex: a protein machine bridging the nucleus and cytoplasm. Curr. Opin Cell Biol. 2000, 12 (3), 361–371. 10.1016/S0955-0674(00)00101-0. [DOI] [PubMed] [Google Scholar]
- Rout M. P.; Aitchison J. D.; Suprapto A.; Hjertaas K.; Zhao Y.; Chait B. T. The yeast nuclear pore complex: composition, architecture, and transport mechanism. J. Cell Biol. 2000, 148 (4), 635–651. 10.1083/jcb.148.4.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronshaw J. M.; Krutchinsky A. N.; Zhang W.; Chait B. T.; Matunis M. J. Proteomic analysis of the mammalian nuclear pore complex. J. Cell Biol. 2002, 158 (5), 915–927. 10.1083/jcb.200206106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J.; Liu Z.; Michelotti N.; Pitchiaya S.; Veerapaneni R.; Androsavich J. R.; Walter N. G.; Yang W. High-resolution three-dimensional mapping of mRNA export through the nuclear pore. Nat. Commun. 2013, 4, 2414. 10.1038/ncomms3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Junod S. L.; Ruba A.; Kelich J. M.; Yang W. Nuclear export of mRNA molecules studied by SPEED microscopy. Methods 2019, 153, 46–62. 10.1016/j.ymeth.2018.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mor A.; Suliman S.; Ben-Yishay R.; Yunger S.; Brody Y.; Shav-Tal Y. Dynamics of single mRNP nucleocytoplasmic transport and export through the nuclear pore in living cells. Nat. Cell Biol. 2010, 12 (6), 543–552. 10.1038/ncb2056. [DOI] [PubMed] [Google Scholar]
- Li Y.; Aksenova V.; Tingey M.; Yu J.; Ma P.; Arnaoutov A.; Chen S.; Dasso M.; Yang W. Distinct roles of nuclear basket proteins in directing the passage of mRNA through the nuclear pore. Proc. Natl. Acad. Sci. U.S.A. 2021, 118 (37), e2015621118. 10.1073/pnas.2015621118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J.; Goryaynov A.; Sarma A.; Yang W. Self-regulated viscous channel in the nuclear pore complex. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (19), 7326. 10.1073/pnas.1201724109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ori A.; Banterle N.; Iskar M.; Andrés-Pons A.; Escher C.; Khanh Bui H.; Sparks L.; Solis-Mezarino V.; Rinner O.; Bork P.; Lemke E. A.; Beck M. Cell type-specific nuclear pores: a case in point for context-dependent stoichiometry of molecular machines. Mol. Syst. Biol. 2013, 9, 648. 10.1038/msb.2013.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelich J.; Ma J.; Dong B.; Wang Q.; Chin M.; Magura C.; Xiao W.; Yang W. Super-resolution Imaging of Nuclear Import of Adeno-Associated Virus in Live Cells. Molecular Therapy – Methods Clinical Development 2015, 2, 15047. 10.1038/mtm.2015.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seisenberger G.; Ried M. U.; Endress T.; Buning H.; Hallek M.; Brauchle C. Real-time single-molecule imaging of the infection pathway of an adeno-associated virus. Science 2001, 294 (5548), 1929–1932. 10.1126/science.1064103. [DOI] [PubMed] [Google Scholar]
- Xiao P.-J.; Samulski R. J. Cytoplasmic trafficking, endosomal escape, and perinuclear accumulation of adeno-associated virus type 2 particles are facilitated by microtubule network. Journal of virology 2012, 86 (19), 10462–10473. 10.1128/JVI.00935-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J.; Kelich J. M.; Junod S. L.; Yang W. Super-resolution mapping of scaffold nucleoporins in the nuclear pore complex. Journal of Cell Science 2017, 130 (7), 1299–1306. 10.1242/jcs.193912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Tingey M.; Ruba A.; Yang W. High-speed super-resolution imaging of rotationally symmetric structures using SPEED microscopy and 2D-to-3D transformation. Nat. Protoc 2021, 16 (1), 532–560. 10.1038/s41596-020-00440-x. [DOI] [PMC free article] [PubMed] [Google Scholar]