

ABSTRACT
Bacteroides and Phocaeicola, members of the family Bacteroidaceae, are among the first microbes to colonize the human infant gut. While it is known that these microbes can be transmitted from mother to child, our understanding of the specific strains that are shared and potentially transmitted is limited. In this study, we aimed to investigate the shared strains of Bacteroides and Phocaeicola in mothers and their infants. We analyzed fecal samples from pregnant woman recruited at 18 weeks of gestation from the PreventADALL study, as well as offspring samples from early infancy, including skin swab samples taken within 10 min after birth, the first available fecal sample (meconium), and fecal samples at 3 months of age. We screened 464 meconium samples for Bacteroidaceae, with subsequent selection of 144 mother-child pairs for longitudinal analysis, based on the presence of Bacteroidaceae, longitudinal sample availability, and delivery mode. Our results showed that Bacteroidaceae members were mainly detected in samples from vaginally delivered infants. We identified high prevalences of Phocaeicola vulgatus, Phocaeicola dorei, Bacteroides caccae, and Bacteroides thetaiotaomicron in mothers and vaginally born infants. However, at the strain level, we observed high prevalences of only two strains: a B. caccae strain and a P. vulgatus strain. Notably, the B. caccae strain was identified as a novel component of mother-child shared strains, and its high prevalence was also observed in publicly available metagenomes worldwide. Our findings suggest that mode of delivery may play a role in shaping the early colonization of the infant gut microbiota, in particular the colonization of Bacteroidaceae members.
IMPORTANCE Our study provides evidence that Bacteroidaceae strains present on infants’ skin within 10 min after birth, in meconium samples, and in fecal samples at 3 months of age in vaginally delivered infants are shared with their mothers. Using strain resolution analyses, we identified two strains, belonging to Bacteroides caccae and Phocaeicola vulgatus, as shared between mothers and their infants. Interestingly, the B. caccae strain showed a high prevalence worldwide, while the P. vulgatus strain was less common. Our findings also showed that vaginal delivery was associated with early colonization of Bacteroidaceae members, whereas cesarean section delivery was associated with delayed colonization. Given the potential for these microbes to influence the colonic environment, our results suggest that understanding the bacterial-host relationship at the strain level may have implications for infant health and development later in life.
KEYWORDS: Bacteroides, Bacteroides caccae, mode of delivery, human microbiota, infant microbiota, strain distribution
INTRODUCTION
The Bacteroidaceae family, specifically the genera Bacteroides and Phocaeicola, are among the early colonizers of the human infant gut (1). In 1996, it was discovered that one of these bacteria had the ability to influence the fucosylation program in the intestine, which triggers the induction of l-fucose (2). The proposed role of this process is to create a niche for Bacteroidaceae and other gut bacteria that use fucose, potentially suppressing the ability of pathogenic bacteria to colonize and helping to shape the mucosal immune system of the host (3, 4). Bacteroidaceae members are also known to degrade a wide range of glycosides and may protect infants from developing allergic disease by expressing lipopolysaccharides and stimulating T-cell production and differentiation (5–7). Moreover, a low relative abundance of Bacteroidaceae has been linked to the development of food allergies (6) and atopic dermatitis (7) in infants, highlighting the significance of early Bacteroidaceae colonization.
Several studies, such as The Environmental Determinants of Diabetes in the Young (TEDDY) (8), the Baby Biome Studies (BBS) (9), and our own study (10), have documented the early gut colonization of Bacteroidaceae within vaginally born infants, revealing a strong mother-child association. One study focused on maternal bacterial strain transmission and observed potential transmission of Phocaeicola vulgatus from mother to child (11). The strain transmission of the Bacteroidaceae members has been suggested to be via the mother’s fecal flora, rather than the vaginal flora (12).
Although mode of delivery is known to impact the initial bacterial colonizers after birth and the presence of Bacteroidaceae members, there is still a lack of understanding regarding the specific strains of Bacteroides/Phocaeicola that are shared between mothers and their children and how they are transmitted. To address this knowledge gap, we conducted a study aimed at investigating strains from the Bacteroides and Phocaeicola genera shared between mothers and their children using strain resolution analysis on samples collected at and near delivery from both vaginally and cesarean section (c-section)-born infants. We analyzed biological samples from mother-child pairs enrolled in the PreventADALL study, including fecal samples from mothers at 18 weeks of gestation, meconium samples, and 3-month fecal samples from the infants, as well as skin swab samples of the infants taken within 10 min after birth. To characterize the bacterial compositions, Bacteroidaceae prevalences, and Bacteroidaceae strain resolution, we performed sequencing of the 16S rRNA and rpoB genes, as well as reduced-metagenome sequencing (RMS) of the biological samples.
RESULTS
We first addressed the presence of Bacteroidaceae members, specifically Bacteroides/Phocaeicola, by screening meconium samples for the rpoB amplicon. Bacteroidaceae were detected in meconium samples from 51 of the 403 vaginally born infants. We did not detect members of the Bacteroidaceae in meconium from the 61 infants born by c-section. For further analysis, a subset of 48 mother-child pairs from each group was selected, the groups being the vaginal rpoB-positive group (VBP; infants born vaginally with detection of rpoB in meconium samples), the vaginal rpoB-negative group (VBN; infants born vaginally with no detection of rpoB in meconium samples), and the cesarean section rpoB-negative group (CBN; infants born by c-section with no detection of rpoB in meconium samples), based on delivery mode and available longitudinal samples.
Age-related associations of Bacteroidaceae by rpoB sequences.
We investigated the association of Bacteroidaceae spp. with age and delivery mode by examining their presence in various sample types across infants grouped by birth mode. Bacteroidaceae were not detected in meconium or skin swab samples from the CBN group (0%) and were only detected in one skin swab sample from the VBN group (2%). However, Bacteroidaceae were detected in all sample types from the VBP group (100%). Four Bacteroidaceae spp. were prevalent in meconium and skin swab samples (>50%), namely, Bacteroides caccae, Bacteroides thetaiotaomicron, Phocaeicola dorei, and Phocaeicola vulgatus, with P. vulgatus being the most common.
P. vulgatus was detected in 95.8% of meconium samples, 85.4% of skin swab samples, and 52.6% of fecal samples at 3 months in the VBP group and in 50% of the fecal samples at 3 months from the VBN group, and it was detected in samples from all mothers independent of birth mode (100%) (Fig. 1A). In addition, P. vulgatus was the most detected Bacteroidaceae sp. in fecal samples at 3 months in the VBN group (50%) and was always present (100%) in mothers independent of birth mode. The second most prevalent, P. dorei, was detected in 87.5% of meconium samples in the VBP group, 58.3% of skin swab samples, and 36.8% of fecal samples at 3 months, as well as 93.8% of samples from their corresponding mothers. In addition, P. dorei was detected in fecal samples at 3 months in the CBN group (9.1%) and the VBN group (50%). B. caccae was detected in 75% of the meconium samples from the VBP group, 52.1% of skin swab samples, 34.2% of fecal samples at 3 months, and 95.8% of samples from their corresponding mothers. B. caccae was detected in 12.1% of the 3-months fecal samples from the CBN group and 43.8% of the 3-months fecal samples in the VBN group. Furthermore, B. thetaiotaomicron was detected in 54.1% of the meconium samples in the VBP group, 52.1% of skin swab samples, 28.9% of 3-months fecal samples, and 95.8% of mothers’ fecal samples.
FIG 1.

Prevalences of Bacteroidaceae spp. and chi-square test for the four most prevalent in meconium and skin samples. (A) Percentages of samples with Bacteroidaceae spp. present (y axis) within each sample category (x axis). The prevalence is based on the total number of samples within each sample type. (B) Test of independence is shown for the four most prevalent Bacteroidaceae spp. observed in meconium and skin swab samples across the three different birth modes. A Pearson residual higher than 2 represents a higher-than-expected association, while a Pearson residual lower than −2 represents a lower-than-expected association. A Pearson residual between −2 and 2 indicates no deviation. The sample types are defined as follows: mother, fecal samples at 18 weeks of pregnancy; skin, infant skin swab samples taken within the first 10 min after birth; meconium, first fecal samples after birth; 3 months, fecal samples collected at 3 months of age. The birth modes are defined as follows: CBN, cesarean section-born infants without detectable Bacteroidaceae in meconium samples by rpoB; VBN, vaginally born infants without detectable Bacteroidaceae in meconium samples by rpoB; VBP, vaginally born infants with Bacteroidaceae detected in meconium samples.
The presence of these four species was associated with the VBP group for meconium and skin swab samples, while their absence was associated with the CBN and VBN groups (chi-square test, P < 0.05; Pearson residual, >|2|) (Fig. 1B). For fecal samples at 3 months of age, P. vulgatus and P. dorei were observed to be lower than expected within the CBN group, while there were no observed deviations for the VBN and VBP groups. For B. caccae and B. thetaiotaomicron, we did not observe any deviations in fecal samples at 3 months of age for any of the birth mode groups.
Since Bacteroidaceae were only detected in meconium samples from the VBP group, we investigated whether the presence of Bacteroidaceae was associated with the time of meconium sampling. We found that infants in the VBN group had the shortest time between birth and meconium sampling, associated with the first 24 h after birth (chi-square test, P < 0.05; Pearson residual, >2) (Fig. S1 in the supplemental material), with a median sampling time of 24 to 48 h after birth. The VBP group had the longest sampling time delay, with a median of 48 to 72 h, and associated to the median sampling time and 72-96 h after birth. The meconium samples from the CBN group infants had a median sampling time of 24 to 48 h, and no associations were found with the time points.
Sharing of Bacteroidaceae strains between mother and child detected by RMS.
We extracted 862 strains of Bacteroides/Phocaeicola from the HumGut database (13) and clustered them based on their RMS profiles to ensure their uniqueness. We then used their RMS profiles as a database to annotate taxonomy to the RMS fragments from the samples in this study. To examine the sharing of Bacteroidaceae strains, we sequenced reduced-metagenome fragments from 16 mother-child pairs in the VBP group and 14 mother-child pairs in the CBN group.
The median number of relative reads mapped to Bacteroidaceae strains in mothers was higher in the CBN group (26.4%) than in the VBP group (20.6%) (Fig. 2A). However, in samples from infants, the relative number of reads mapped to Bacteroidaceae strains was higher in the infant VBP group samples than in the CBN group samples. The four most prevalent species observed by rpoB sequences (B. caccae, B. thetaiotaomicron, P. dorei, and P. vulgatus) accounted for a large part of the mapped RMS reads, with P. vulgatus having the highest relative number of reads. Due to low DNA concentrations and evaporation over time, less eluate was available for RMS sequencing in samples from the CBN group, resulting in few samples having sufficient DNA for RMS analysis. Therefore, the continued analysis was performed on samples from the VBP group to further gather longitudinal information on the Bacteroidaceae strains.
FIG 2.
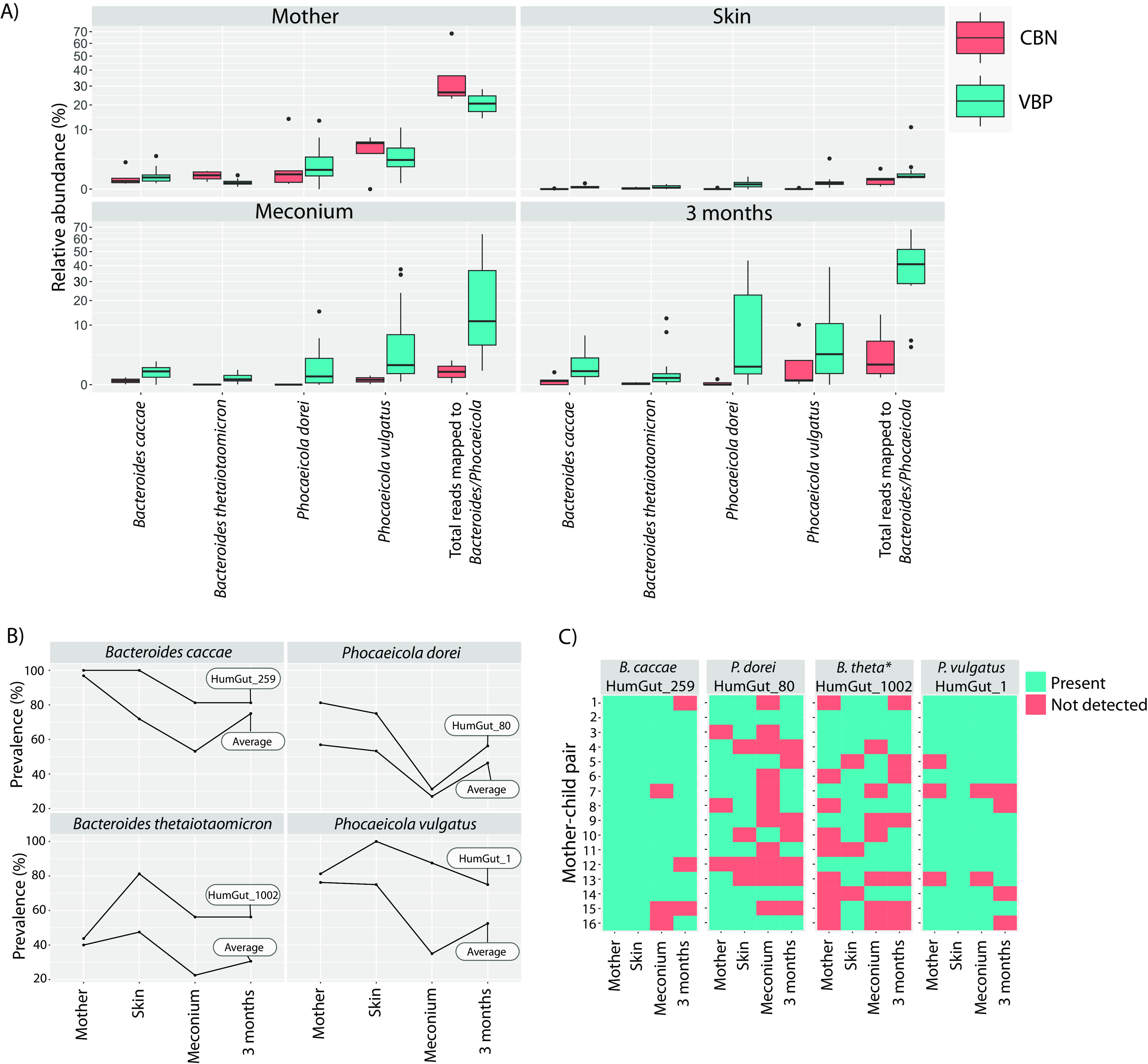
Relative numbers of total reads mapped to Bacteroides/Phocaeicola strains and their prevalences in mother-child pairs. (A) Boxplots depicting the relative abundances of reads that mapped to four species of Bacteroidaceae, as well as the total relative abundances of reads that mapped to any Bacteroides/Phocaeicola strain from the HumGut database (comprising 862 strains). (B) Average prevalences of all strains within a species and the strain with the highest prevalence within each species from the VBP group. (C) Prevalences of the four most prevalent strains within each mother-child pair. *, Bacteroides thetaiotaomicron. The samples were obtained from two groups: CBN, cesarean section-born infants without detectable Bacteroidaceae in meconium samples by rpoB, and VBP, vaginally born infants with Bacteroidaceae detected in meconium samples. The samples were collected from different sources, including fecal samples from the mother at 18 weeks of pregnancy, skin swab samples taken from the infant within the first 10 min after birth, meconium samples (first infant fecal sample after birth), and fecal samples of the infant collected at 3 months of age. The VBP group consisted of 16 samples for each sample type, while the CBN group varied: mother (n = 5), skin (n = 5), meconium samples (n = 2), and 3 months (n = 5).
To investigate strain prevalence in each sample type from the VBP group infants, we used 46 genomes as proxies for strains within the four species (2 strains deriving from B. caccae, 10 from B. thetaiotaomicron, 19 from P. dorei, and 15 from P. vulgatus). Generally, the strain prevalence in meconium samples was lower than in the infant skin swab samples, but two strains had relatively high prevalences within all sample types: HumGut_1 (P. vulgatus) and HumGut_259 (B. caccae) (Fig. 2B). Strains from P. dorei and B. thetaiotaomicron had lower prevalences. While P. dorei and B. thetaiotaomicron had low prevalences in meconium samples, their strain prevalences in fecal samples at 3 months increased comparatively. The most prevalent Bacteroidaceae strains from the four species in infants were HumGut_259 (B. caccae), HumGut_80 (P. dorei), HumGut_1002 (B. thetaiotaomicron), and HumGut_1 (P. vulgatus). The P. vulgatus strain (HumGut_1) was present in all four sample types for 10 of the 16 mother-child pairs and in three sample types in 3 mother-child pairs (mother fecal samples and infant skin swab samples and meconium samples) (Fig. 2C). The B. caccae strain (HumGut_259) was present in all sample types in 11 mother-child pairs and in three sample types in 3 mother-child pairs. Both P. dorei (HumGut_80) and B. thetaiotaomicron (HumGut_1002) occurred in all four sample types in three mother-child pairs.
Prevalences and abundances of the mother-child shared strains worldwide.
While B. caccae (HumGut_259) and P. vulgatus (HumGut_1) showed the highest mother-child sharing by RMS, we also included the strains of B. thetaiotaomicron (HumGut_1002) and P. dorei (HumGut_80) as representative strains within the prevalent species. We investigated the relative abundances and prevalences of the four mother-child shared strains worldwide, with results deriving from the data utilized in the creation of the HumGut database (13). This included 2,933 fecal samples taken from healthy adults and 598 from healthy infants.
Among the four strains, B. caccae (HumGut_259) had the highest prevalence in both the adult and infant populations (Fig. 3A and B). Its relative prevalence in the adult population was 60.5%, with an average relative abundance of 2.1%. In infants, it was present in 26% of the population, with an average relative abundance of 1.9%. The second-most-prevalent strain, B. thetaiotaomicron, occurred in 1.56% of the adult population with an average relative abundance of 0.01% and had a prevalence of 0.7% in infants, making up 0.04% of the gut microbiota. Notably, P. vulgatus (HumGut_1) and P. dorei (HumGut_80) were not observed in the infant population. Their relative prevalences in the adult population were 0.4% (P. vulgatus) and 0.6% (P. dorei), with average relative abundances of 0.02% and 0.003%, respectively.
FIG 3.
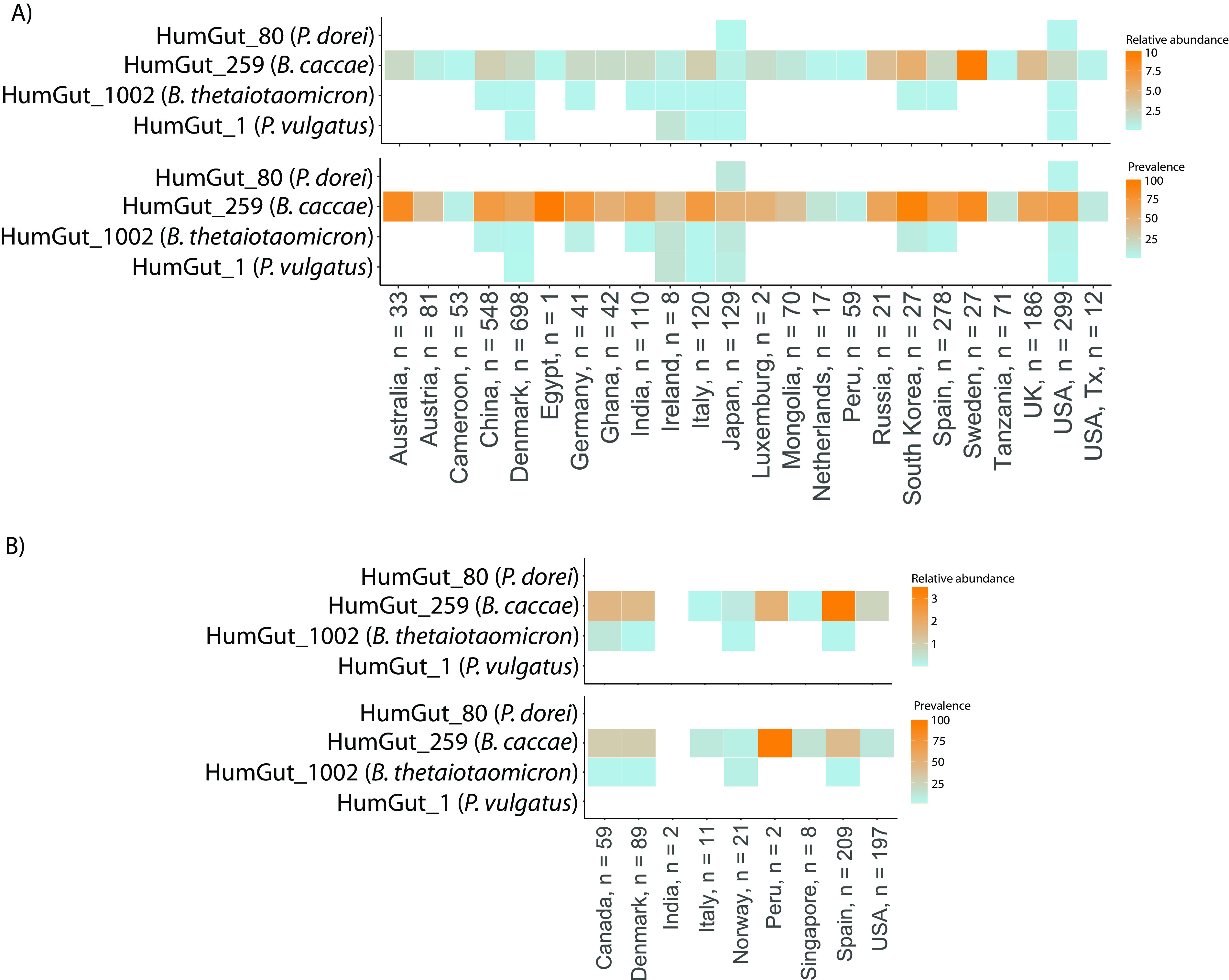
Strain prevalences worldwide based on the HumGut database. Global prevalences of four Bacteroidaceae strains based on data from the creation of the HumGut database (13). The heat maps depict the relative abundances and prevalences of these strains in both the adult population (A) and the infant population (B). The data set used to generate these results includes 2,933 fecal samples from healthy adults and 598 fecal samples from healthy infants. “Infant population” refers to children at and below 1 year, while “adult population” refers to those above 1 year of age.
Predictive features of the prevalent Bacteroidaceae strains.
We used the Virtual Metabolic Human Database (VMH, https://www.vmh.life) to gather information on potential carbon sources for the prevalent Bacteroidaceae species (P. vulgatus, P. dorei, B. thetaiotaomicron, and B. caccae) (Table S4A). Furthermore, we conducted an in silico analysis of the genomes used as proxies for the four strains from the HumGut database to predict glycoside hydrolase activity.
The utilization of carbon sources for B. caccae, B. thetaiotaomicron, and P. vulgatus was found to be relatively similar, with a preference for mucins and simple and complex sugars (Fig. 4A). In contrast, P. dorei’s carbon source utilization potential was primarily focused on amino acids. Although B. caccae, B. thetaiotaomicron, and P. vulgatus had similar carbon sources, we observed that B. caccae and B. thetaiotaomicron had higher numbers of potential carbon sources (B. caccae, n = 51, and B. thetaiotaomicron, n = 59) than either P. vulgatus (n = 37) or P. dorei (n = 10). Unique carbon sources were observed between the four species, with B. thetaiotaomicron having the unique potential to utilize pullulan, arabin, and alpha-mannans, while B. caccae can possibly utilize N-acetylneuraminic acid and cellobiose (Fig. S4B). Of note, l-fucose was identified as a potential carbon source for both B. caccae and B. thetaiotaomicron, while P. dorei and P. vulgatus shared 4-hydroxyproline, among others.
FIG 4.
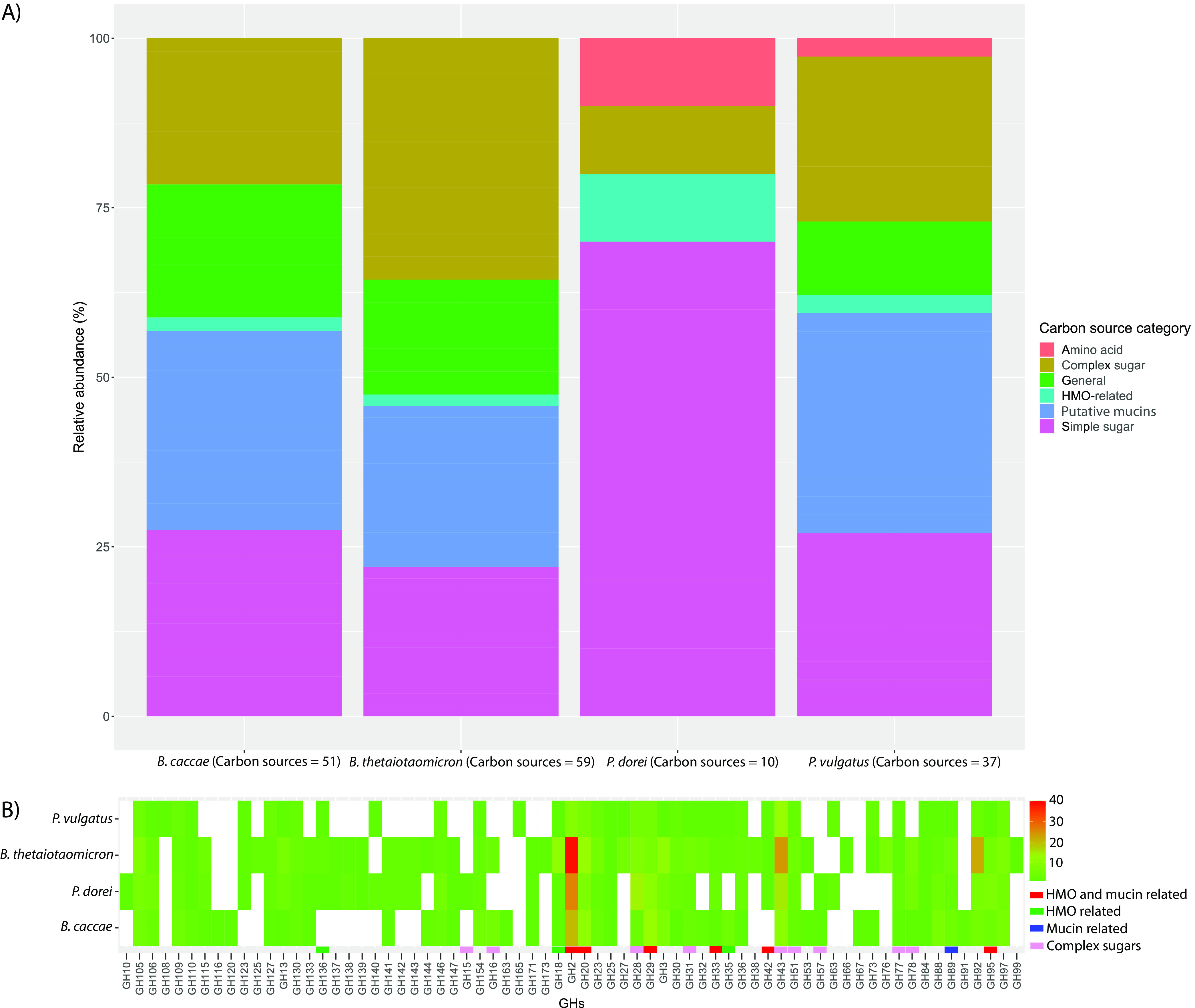
Predictive features of four Bacteroidaceae species and strains. (A) Relative abundances of potential carbon source utilization of four strains (Phocaeicola vulgatus ATCC 8482, Phocaeicola dorei DSM 17855, Bacteroides thetaiotaomicron VPI-5482. and Bacteroides caccae ATCC 43185) collected from the Virtual Metabolic Human Database (VMH). (B) Heat map of the glycoside hydrolase (GH) (x axis) frequencies identified by the DBcan3 meta-server from the Bacteroidaceae genomes used as proxies for strains from the HumGut database. The potential functions of the GHs were inferred based on other studies (36, 37).
We identified glycoside hydrolases (GHs) from the four Bacteroidaceae strains most prevalent within their species (P. vulgatus HumGut_1, P. dorei HumGut_80, B. caccae HumGut_259, and B. thetaiotaomicron HumGut_1002). The four strains have similar profiles regarding potential glycoside hydrolases but with variations in their frequencies (Fig. 4B). B. thetaiotaomicron had the highest number of observed glycoside hydrolases (n = 265), which corresponds to its high number of potential carbon sources. Some of the GHs with relatively high frequencies are potentially related to the utilization of human milk oligosaccharides (HMOs) and mucins (GH2, GH20, GH29, GH33, GH42, and GH95), which corresponds to the potential nutrient substrates available during early infancy.
Source tracking of the bacterial composition detected in the sample types.
In the meconium samples, we found that the VBN and CBN groups exhibited similar bacterial compositions, with both groups being dominated by hospital-associated bacteria (Fig. 5), which could be attributed to Burkholderia (Fig. S2A). Conversely, the VBP group showed higher abundances of bacteria associated with stool, stool/skin, and vagina/skin, which could be linked to the presence of Bacteroides, Escherichia/Shigella, and Bifidobacterium. Notably, due to the outdated taxonomy in the database, the Bacteroides genera comprises both Bacteroides and Phocaeicola. We found that Bacteroides bacteria were significantly more abundant in the meconium samples from the VBP group, representing 14.5% of the detected bacteria, compared to 0.03% in the VBN group and 0.0007% in the CBN group (Tukey’s test, P < 0.05) (Fig. S2B).
FIG 5.

Possible sources for the bacterial compositions. Potential sources of gut bacteria in meconium samples, 3-month-old infants’ fecal samples, and pregnant mothers’ fecal samples, as well as bacteria present on newborn skin. The mapping of gut and skin bacteria was based on data from the Human Microbiome Project (HMP), with bacteria categorized into different environments. To be considered “exclusive” to a particular environment, a bacterium had to have an average relative abundance of over 0.1% in all samples from that environment. Additionally, if a bacterium occurred in two environments but had a relative abundance at least 3% higher in one, it was categorized as “exclusive” to that environment. This affected three genera: Lactobacillus was categorized as exclusive to vaginal flora (with a relative abundance of 83.5% in vaginal flora and 1% in skin microbiota), Streptococcus to skin (with a relative abundance of 3.3% in skin and 0.2% in vaginal flora), and Bacteroides to stool (with relative abundances of 28.9% in fecal samples and 2.3% in skin microbiota). Genera with less than 0.1% abundance in the HMP were excluded and categorized as unclassified. “Other” includes bacteria found in unclassified environments and those associated with adult stool/skin and adult skin/hospital environment.
In terms of the newborn skin microbiota, the VBN and VBP groups exhibited greater abundances of bacteria associated with adult vaginal flora and stool than did the CBN group. Lactobacillus was the primary contributor to the high abundance of vagina-associated bacteria, while Faecalibacterium and Bacteroides were responsible for the high levels of stool-related bacteria in the VBN and VBP groups (P < 0.05, Tukey’s test) (Table S5 and Fig. S2C). Conversely, the skin microbiota of the CBN-born infants was mainly composed of bacteria associated with vagina, skin, and the hospital environment and skin flora. The most dominant genus in the CBN group was Pseudomonas, accounting for 15.9% of the skin microbiota, a significantly higher relative abundance than in the VBN and VBP groups (P < 0.05, Tukey’s test). Caldalkalibacillus, Nesterenkonia, and Alkalihalobacillus also had significantly higher relative abundances in the CBN group than in the VBN and VBP groups (P < 0.05, Tukey’s test). Notably, these bacteria were not associated with stool, skin, vaginal flora, or the hospital environment. The skin bacterium Streptococcus was found in a higher relative abundance on the CBN newborns’ skin, but the difference was not statistically significant (P > 0.05, Tukey’s test).
At 3 months of age, the gut microbiota of infants from all birth mode groups were predominantly composed of bacteria associated with the adult vagina and skin flora and adult stool and skin flora, with high relative abundances of Bifidobacterium, Clostridium sensu stricto, and Escherichia/Shigella. However, the VBN and VBP groups had significantly higher relative abundances of stool-associated bacteria, particularly Bacteroides, than did the CBN group (Tukey’s test, P < 0.05) (Fig. S2D).
To assess for potential bias, we analyzed fecal samples from the mothers of the infants in each group. We found minimal differences between the delivery mode groups. Notably, there were no significant differences in Bacteroides abundances between the groups, with Bacteroides representing 10.2% in the VBP group, 11.8% in the VBN group, and 10.5% in the CBN group (analysis of variance [ANOVA], P > 0.05).
DISCUSSION
In the PreventADALL cohort, we identified four species from the Bacteroidaceae family that were shared between mothers and their children. Of the four mother-child shared species, we identified two highly prevalent mother-child-associated strains belonging to the species P. vulgatus and B. caccae. P. vulgatus has been previously identified as mother-child associated in several studies, and evidence for strain transmission has been shown earlier (11). However, HumGut_1, the strain we identified, had a low prevalence in the adult population and was not observed in the infant population. On the other hand, B. caccae has not been previously identified as being shared between mother and child to our knowledge, but we found HumGut_259, a strain belonging to this species, to be prevalent in both the adult and infant population, albeit with the species having low relative abundances. The low relative abundance of B. caccae might be a reason for its oversight in other studies.
Previous studies have linked B. caccae to bloodstream infections (14) and colonic mucus barrier erosion in combination with a dietary fiber-deprived diet (15). However, as a mucus generalist (15), it may also play a role in shaping the mucosal immune system (16), in addition to producing anti-inflammatory molecules that reduce inflammation (5, 17–19). Furthermore, we observed that the two mother-child shared strains could utilize a wide array of carbon sources, including mucins and simple and complex sugars. We also observed the potential utilization of l-fucose in B. caccae and B. thetaiotaomicron. This mechanism is mostly described for B. thetaiotaomicron, which utilizes and induces the production of l-fucose (2). Although 43 bacterial strains are registered as being able to utilize l-fucose as a carbon source (VMH), only B. caccae, B. thetaiotaomicron, and B. cellulosilyticus from the Bacteroidaceae family are registered as utilizing l-fucose, suggesting similar means of selection related to l-fucose induction. Additionally, we found several strains from P. dorei and B. thetaiotaomicron to have high prevalences in skin swab samples taken immediately after birth, while they were detected in fewer infants’ meconium samples but were increased in fecal samples at 3 months of age, suggesting a delay from the initial exposure to the colonization of these strains.
The low prevalences and abundances of Bacteroidaceae in meconium samples, skin swab samples, and fecal samples at 3 months from cesarean section-born infants were consistent with previous findings (20). However, the VBN group also exhibited no detectable presence of Bacteroidaceae from skin swab samples and meconium samples by rpoB sequencing. Notably, the bacterial composition in meconium samples at the genus level was similar to that of c-section-born infants by 16S rRNA genes, primarily due to the environmental bacteria Burkholderia. In contrast, the bacterial composition of the infant skin and gut microbiota at 3 months was more akin to that of the VBP group at the genus level, with no discernible differences in the relative abundances of Bacteroidaceae, with the vaginally delivered infants having significantly higher levels than the c-section-born infants. This may suggest that the vaginally delivered infants are exposed to Bacteroidaceae, but with a lower exposure for some, which was not detectable by the rpoB amplicons. Moreover, this may suggest a poorer sensitivity of the rpoB amplicons than of the 16S rRNA amplicons, which may have hindered the detection of the low levels of Bacteroidaceae. The mother-child shared Bacteroidaceae strains were mostly detected in vaginally born infants, which might suggest that the infants are exposed to these bacteria during birth. However, as we detected a low prevalence of Bacteroidaceae strains in c-section-born infants, there might be some exposure to these bacteria before birth, i.e., after the rupture of the amniotic membrane, which has been suggested by an earlier study (21).
Although our study lacked longitudinal samples from mothers during pregnancy and vaginal swabs during birth, we utilized available data to track the potential sources of the observed bacteria. Through source tracking, we identified vaginal and/or fecal flora as the two possible sources of exposure to Bacteroidaceae. Studies investigating the potential effects of Bacteroidaceae restoration by vaginal transfer have yielded mixed results (22, 23), while a single study reported successful restoration of Bacteroidaceae through fecal microbiota transplantation (24), indicating that the anal-oral route may be a viable transmission pathway. Furthermore, the lack of Bacteroidaceae in the vaginal flora 24 h before birth and the presence of maternal fecal Bacteroidaceae strains in vaginally born infants suggest that the anal-oral route is the primary transmission route (12).
In conclusion, we observed four mother-child shared species from the Bacteroidaceae family. Two strains, one of which is novel, were observed to be prevalent in mothers and their infants born vaginally. We detected these strains in meconium samples and on the infant skin 10 min after birth, and they persisted until the infants were 3 months of age. Our findings suggest that the mode of delivery may influence the acquisition and abundance of these strains during the first few months of life. However, further research is necessary to fully understand the role and acquisition of these strains in early infancy. A visual representation of our results is presented in Fig. 6.
FIG 6.

Summarized results for the potential Bacteroidaceae exposure and colonization pattern by delivery mode. The figure summarizes the findings and results of our study. The figure includes the potential Bacteroidaceae transmission from mother to child for vaginally and cesarean section-born infants, their potential sources, and their abundances at the different sampling times during the first 3 months of life.
MATERIALS AND METHODS
This study used biological samples from the general-population-based Preventing Atopic Dermatitis and Allergies in children (PreventADALL) cohort study, which includes approximately 2,400 mother-child pairs from Norway and Sweden (25). Fecal samples from the first stool (meconium) of 464 infants were used due to their availability; 403 of the infants were born vaginally and 61 by cesarean section. Of the births, 299 had spontaneous rupture of the amniotic membrane, 117 were ruptured by amniotomy, and 16 by induced amniotomy, while 22 infants had their amniotic membrane ruptured during surgery (10 had no record). The average time of birth after amniotic membrane rupture was 9.7 h. Subsets of the 464 infants were selected for further analysis based on the presence of Bacteroidaceae, delivery mode, and available longitudinal samples. Infants with at least two of the three sample types—fecal samples from the infants at 3 months of age, mothers’ fecal samples at 18 weeks into pregnancy, and infant skin swab samples taken within the first 10 min after birth—were included. In addition to being an observational study, PreventADALL is a randomized controlled trial (RCT) where the infants were randomized into four groups at birth: no intervention, skin intervention and emollients, food intervention with early introduction of allergenic foods, and both interventions (26).
Informed written consent from all pregnant mothers was received upon inclusion at around 18 weeks of pregnancy and from both parents upon inclusion of the newborn child. The PreventADALL study has been approved by the Regional Ethical Committee (REK) for Medical and Health Research Ethics in Southeastern Norway (2014/518), as well as by the Regional Ethics Committee at Karolinska Institutet, Stockholm, in Sweden (2014/2242-31/4).
Sample processing for sequencing of 16S rRNA genes, rpoB, and RMS.
(i) Data selection. A schematic representation of the study workflow is depicted in Fig. 7. To assess the prevalence of Bacteroidaceae in the mother-child cohort, rpoB primers were used to screen 464 meconium samples. Among these, 51 samples were positive for rpoB, all of which were from infants born vaginally (Fig. 7). Of the 51 mother-child pairs, 48 had both infant skin swab samples and maternal fecal samples available and were thus selected for further analysis. These 48 pairs are referred to as vaginal rpoB positive (VBP). To account for potential differences in bacterial colonization due to delivery mode, we also included 48 mother-child pairs from the groups with no amplification of rpoB, with equal numbers of pairs from the vaginal and cesarean section (c-section) delivery modes. These pairs were selected based on the availability of newborn skin swab samples, fecal samples at 3 months, and maternal fecal samples, resulting in 48 mother-child pairs with c-section-born infants and no amplification of rpoB, referred to as c-section rpoB negative (CBN), and 48 mother-child pairs with vaginally delivered infants and no amplification of rpoB, referred to as vaginal rpoB negative (VBN). The samples from the three groups were subjected to 16S rRNA gene amplicon sequencing and sequencing of rpoB amplicons. Furthermore, a subset of 16 mother-child pairs from the VBP group and 14 mother-child pairs from the CBN group were analyzed by reduced-metagenome sequencing (RMS) based on sample availability.
FIG 7.
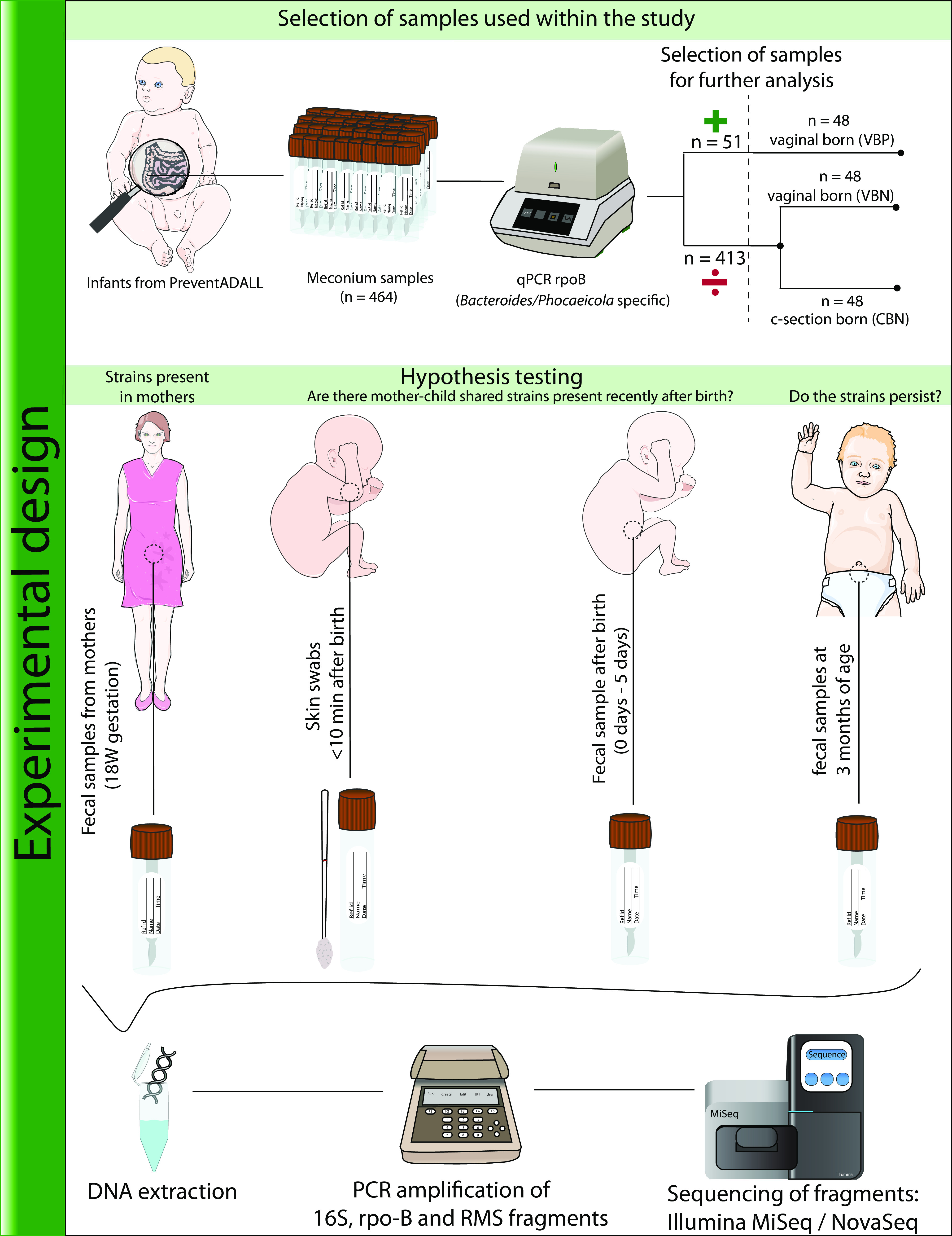
Experimental design and general workflow of the study. The figure presents the experimental design and workflow used in this study, in addition to potential insights from the different sample types. The biological samples used were collected as part of the PreventADALL study. In this study, 464 meconium samples were screened for Bacteroidaceae members (specifically, Bacteroides and Phocaeicola) using rpoB gene-targeted primers. Of these samples, Bacteroidaceae were detected in 51 meconium samples, all of which were from vaginally born infants. Sample selection for further analysis was based on rpoB amplification, delivery mode, and availability of longitudinal samples. Vaginally born infants with rpoB amplification in meconium samples were assigned to the vaginal rpoB-positive (VBP) group, while vaginally born infants without rpoB amplification were assigned to the vaginal rpoB-negative (VBN) group, and cesarean section (c-section)-born infants without rpoB amplification were assigned to the c-section rpoB-negative (CBN) group. A total of 48 mother-child pairs from each group were selected for further analysis, which included sequencing of newborn skin swab samples, meconium samples, fecal samples at 3 months, and fecal samples from their corresponding mothers (at 18 weeks of gestation). These four sample types were sequenced for the V3-V4 region of the 16S rRNA gene and targeted Bacteroides-/Phocaeicola-specific rpoB, with subsets of infants within the VBP (n = 16) and CBN (n = 14) groups also analyzed by reduced-metagenome sequencing (RMS). The figure illustrates the sample selection and processing used in this study, as well as potential insights that the different sample types can provide regarding shared strains between mother and child. Figure items of the infants and mother were taken from Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 unported license (https://smart.servier.com/), and edited.
(ii) DNA extraction. Prior to processing, the fecal samples were diluted 1:10 in stool DNA stabilizer (PSP Spin Stool DNA plus kit; Invitek Molecular) and stored at −80°C. Skin swab samples were stored in Ames medium and kept at −80°C until processing. Skin swab samples were first homogenized and centrifuged at 13,000 rpm for 15 min. The resulting pellet was then resuspended in Stool Transport and Recovery (STAR) Buffer (Roche, Switzerland). Bacterial cells in both sample types were mechanically disrupted using a combination of acid-washed glass beads (0.2 g <106-μm beads, 0.2 g 425- to 600-μm beads, and 2× 2.5- to 3.5-mm beads) (Sigma-Aldrich, Germany) and processed twice on a FastPrep 96 (MP Biomedicals, USA) at 1,800 rpm for 40 s. The samples were then centrifuged at 13,000 rpm for 5 min, and the DNA was extracted using the LGC mag midi nucleic acid extraction kit (LGC Genomics, UK), following the manufacturer’s instructions.
(iii) Sequencing of 16S rRNA, rpoB, and RMS fragments. (a) Quantitative PCR. The initial quantitative PCR screening for rpoB was performed on DNA extracted from the 464 meconium samples as follows: DNA was amplified at 95°C for 15 min and 40 cycles of 95°C for 30 s, 55°C for 40 s, and 72°C for 45 s in a solution containing 1× Hot FirePol EvaGreen qPCR supermix (Solis BioDyne) and 0.2 μM forward primer BF and reverse primer BR (27).
(b) Restriction cutting and adapter ligation. To generate fragments for reduced-metagenome sequencing (RMS), we incubated DNA samples with a combination of EcoRI enzyme (8 U), MseI enzyme (4 U), and CutSmart buffer at 37° for 1 h. To ligate adapters, we added 0.5 μM EcoRI adapter mix (5′-CTCGTAGACTGCGTACC-3′ and 5′-AATTGGTACGCAGTCTAC-3′), 5 μM MseI adapter mix (5′-GACGATGAGTCCTGAG-3′ and 5′-TACTCAGGACTCAT-3′), T4 DNA ligase, and T4 reaction buffer to the samples and incubated them at 37°C for 3 h.
(c) Amplicon PCR. Extracted DNA was amplified at 95°C for 15 min, followed by 25, 30, or 40 cycles (25 cycles for 16S rRNA genes in fecal samples from mothers, 30 cycles for 16S rRNA genes in meconium, infant skin swab samples, and RMS samples, and 40 cycles for rpoB in all samples) of 95°C for 30 s, 55°C for 30 s and 72°C for 45 s before the final step at 72°C for 7 min, with reaction mixtures containing 1× Hot FirePol blend master mix ready to load, 0.2 μM forward primer PRK341F (28) for the 16S rRNA gene, forward primer BF for rpoB (27), EcoRI primer for RMS, PRK806R (28) for 16S rRNA genes, reverse primer BR for rpoB, and MseI primer for RMS.
(d) Cleanup. Samples were purified using 1.2× 0.1% Sera Mag beads per DNA volume for all samples, except for 16S rRNA genes in fecal samples from mothers and all RMS samples, where the ratio used was 1× 0.1% Sera Mag beads, following AMPure’s protocol on a Biomek 3000 instrument (Beckman Coulter, USA).
(e) Index PCR. The DNA fragments were indexed by a combination of modified reverse Illumina index primers for 16S rRNA genes, rpoB, and RMS (Table S1). Purified DNA was added to a solution containing 1× FirePol master mix ready to load, 0.2 μM modified forward and reverse primers and amplified at 95°C for 5 min, followed by 10 or 12 cycles (10 cycles for rpoB and 16S rRNA gene amplification for fecal samples from mothers and 3-months infants and 12 cycles for rpoB and 16S rRNA gene amplification from meconium samples and skin swab samples and for RMS from all samples) at 95°C for 30 s, 55°C for 60 s, 72°C for 45 s and a final step at 72°C for 7 min.
(f) Pooled libraries and sequencing. DNA was quantified by Qubit reagents using a Cambrex Bio-Tek FLx800, normalized, and pooled on a Biomek 3000 (Beckman Coulter, USA) as 10 different libraries, comprising 16S rRNA genes (2 libraries), rpoB (2 libraries), and RMS (6 libraries). The pooled libraries were purified with the same procedure described above in “Cleanup” and split in two, one part for sequencing and one for library quantification. For the 16S rRNA gene and rpoB pooled libraries, quantification was done by quantitative PCR using the KAPA Library Quantification Kit for Illumina Platforms, following the manufacturer’s recommendations. The second part of the sample was diluted to 7 pM DNA with 20% PhiX following Illumina’s instructions, except for the use of nuclease-free water instead of Tris, and sequenced using the Illumina MiSeq platform with MiSeq reagent kit V3 (2 × 300 bp). The RMS libraries were quantified using Qubit following the manufacturer’s protocol and were sequenced with 2 × 150-bp read length on the NovaSeq 6000 by Novogene.
Data processing.
(i) DADA2 for rpoB and 16S rRNA gene sequences. Before import into the DADA2 pipeline, the raw sequences were demultiplexed and assessed for quality (29). The forward and reverse sequences were truncated at 240 and 220 bp, respectively [truncLen = c(240,220)]. Subsequently, the sequences were dereplicated, and the error rates of base incorporations were estimated and applied to the sequences. Chimeras were eliminated, and the remaining unique sequences were taxonomically annotated using sintax with the RDP database for 16S rRNA genes (version 18) and the Kraken2 Standard database for rpoB. We detected potential bacterial contaminations using the R package decontam and manual inspection and removed them based on amplicon sequence variants (ASVs) with high read abundances in negative controls (consisting of sterile PCR water and all reagents for library preparation) and significant ASV contaminations using the isContaminant function. After removal of contaminants, 84.1% (±19.7% [mean ± standard deviation]) of the reads were retained in the 16S rRNA gene data set, while 14.8% (±12.1%) of the reads in the negative controls were retained. For rpoB, 96.7% (±9.2%) of the reads were retained from the samples and 15.5% (±13.3%) of the reads in the negative controls were retained. The ASVs were consolidated based on taxonomy, resulting in 8.4 million sequences of rpoB, with 7.3 million annotated to Bacteroidaceae at the genus level (Bacteroides/Phocaeicola), comprising 86.9% of all sequences annotated to the genus level and with 18 different strains/species identified (Table S2).
(ii) Processing of ASVs. After the DADA2 pipeline, the data were brought into the RStudio environment (version 1.4.1103). To streamline the analysis of 16S rRNA gene data, any samples with fewer than 1,000 sequences were removed. The data set was converted into a phylo object in PhyloSeq (30). Next, subsets of each sample type (meconium samples, newborn skin swab samples, fecal samples at 3 months, and fecal samples from mothers at 18 weeks of gestation) were created and analyzed by ANOVA using the run_test_multiple_groups function from the microbiomeMarker package (31). To ensure data normalization, all samples were processed using counts per million (cpm) normalization. Any bacteria that were statistically significant (P < 0.05) by ANOVA were further examined using a post hoc Tukey test, with all samples normalized by total sum scaling (TSS).
(iii) Processing of RMS fragments. We began by creating a custom database to map our RMS fragments. Initially, we reduced the number of HumGut genomes from 30,691 to 862 by retaining only those annotated as Bacteroides/Phocaeicola by the NCBI database and Genome Taxonomy Database (GTDB) (13). The microRMS R package was used to extract and cluster the RMS fragments from the HumGut genomes and identify unique and shared fragments (32). The strains had totals of 84 to 3,164 fragments, of which 2 to 2,370 were unique, resulting in a correlation distance of approximately 0.5 or more between most genomes, indicating good separation based on their RMS profiles.
Before mapping the fragments to the custom database, the RMS fragments from our samples were quality filtered, and read pairs were merged and trimmed using vsearch version 2.15.2. The average numbers of reads were 11.2 million from the VBP group meconium samples, 11.4 million from skin samples, 18 million from 3-month fecal samples, and 11.2 million from fecal samples from mothers. From the CBN group, fecal samples from mothers (n = 16) had 11.5 million reads on average per sample, meconium samples (n = 2) had 73,000, skin swab samples (n = 5) had 20,000, and fecal samples at 3 months (n = 5) had 261,000 reads. The read abundances of the strains were estimated using the constrained ordinary least square (COLS) algorithm from microRMS (32).
(iv) Source tracking of bacteria using Human Microbiome Project data. To perform source tracking, we utilized samples from the Human Microbiome Project (HMP) to represent potential environmental sources. Specifically, skin flora was represented by left (n = 225) and right (n = 236) antecubital fossa, vaginal flora by vaginal introitus (n = 125), posterior fornix (n = 130), and mid vagina (n = 131), and fecal flora by stool samples (n = 313). The categories for hospital environment were largely based on other studies (33, 34). All samples from the HMP data used in this study are listed in Table S3. We assigned taxonomy to the reads by clustering the reads to operational taxonomic units (OTUs) at 97% identity using usearch version 11 (35) and the RDP database for 16S rRNA genes (version 18). The relative abundances of OTUs were averaged within the groups to represent skin, fecal, and vaginal flora and consolidated based on their taxonomy. Any taxon with an average relative abundance greater than 0.1% was used as an indicator for that environment. If a bacterium occurred in two environments with a difference between the environments of >3%, it was set as exclusive to the environmental group with the higher abundance. This approach affected three genera: Lactobacillus was observed to account for 83.5% of the vaginal flora and 1% of the skin microbiota, resulting in it being set as exclusive to the vaginal flora; Streptococcus accounted for 3.3% of the skin microbiota and 0.2% of the vaginal flora and was set as exclusive to the skin microbiota; and Bacteroides (which represents the Bacteroides and Phocaeicola genera, since the database is not updated on taxonomy) accounted for 28.9% of the fecal sample microbiota and 2.3% of the skin microbiota and was set as exclusive to fecal flora. Finally, we mapped our 16S rRNA gene data to the different categories created from the HMP data and converted them to their potential environmental source.
(v) Carbon utilization potential. In order to investigate the potential carbon source utilization of Bacteroidaceae strains, we obtained data from the virtual metabolic human database (VMH) on four strains: Phocaeicola vulgatus strain ATCC 8482, Phocaeicola dorei strain DSM 17855, Bacteroides caccae strain ATCC 43185, and Bacteroides thetaiotaomicron strain VPI-5482. The carbon sources were then categorized into six groups (amino acid, complex sugar, general, HMO related, putative mucins, and simple sugars) based on available information, with the specific sources and categories listed in Table S4. Furthermore, we processed the genomes used as proxies for strains from the HumGut database in the DBCan3 meta-server to extract potential glycoside hydrolase presence.
Data availability.
Sequencing data of 16S rRNA genes and rpoB is available in the NCBI SRA database with the identifier PRJNA877464. The RMS data are available upon request in TSD UiO (University of Oslo).
Footnotes
Supplemental material is available online only.
Contributor Information
Morten Nilsen, Email: morten.nilsen@nmbu.no.
Danilo Ercolini, Universita degli Studi di Napoli Federico II.
REFERENCES
- 1.Milani C, Duranti S, Bottacini F, Casey E, Turroni F, Mahony J, Belzer C, Delgado Palacio S, Arboleya Montes S, Mancabelli L, Lugli GA, Rodriguez JM, Bode L, de Vos W, Gueimonde M, Margolles A, van Sinderen D, Ventura M. 2017. The first microbial colonizers of the human gut: composition, activities, and health implications of the infant gut microbiota. Microbiol Mol Biol Rev 81:e00036-17. doi: 10.1128/MMBR.00036-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bry L, Falk PG, Midtvedt T, Gordon JI. 1996. A model of host-microbial interactions in an open mammalian ecosystem. Science 273:1380–1383. doi: 10.1126/science.273.5280.1380. [DOI] [PubMed] [Google Scholar]
- 3.Hooper LV, Xu J, Falk PG, Midtvedt T, Gordon JI. 1999. A molecular sensor that allows a gut commensal to control its nutrient foundation in a competitive ecosystem. Proc Natl Acad Sci USA 96:9833–9838. doi: 10.1073/pnas.96.17.9833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pickard JM, Chervonsky AV. 2015. Intestinal fucose as a mediator of host-microbe symbiosis. J Immunol 194:5588–5593. doi: 10.4049/jimmunol.1500395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wexler H. 2007. Bacteroides: the good, the bad, and the nitty-gritty. Clin Microbiol Rev 20:593–621. doi: 10.1128/CMR.00008-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ling Z, Li Z, Liu X, Cheng Y, Luo Y, Tong X, Yuan L, Wang Y, Sun J, Li L, Xiang C. 2014. Altered fecal microbiota composition associated with food allergy in infants. Appl Environ Microbiol 80:2546–2554. doi: 10.1128/AEM.00003-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abrahamsson TR, Jakobsson HE, Andersson AF, Björkstén B, Engstrand L, Jenmalm MC. 2012. Low diversity of the gut microbiota in infants with atopic eczema. J Allergy Clin Immunol 129:434–440.e2. doi: 10.1016/j.jaci.2011.10.025. [DOI] [PubMed] [Google Scholar]
- 8.Stewart CJ, Ajami NJ, O'Brien JL, Hutchinson DS, Smith DP, Wong MC, Ross MC, Lloyd RE, Doddapaneni H, Metcalf GA, Muzny D, Gibbs RA, Vatanen T, Huttenhower C, Xavier RJ, Rewers M, Hagopian W, Toppari J, Ziegler A-G, She J-X, Akolkar B, Lernmark A, Hyoty H, Vehik K, Krischer JP, Petrosino JF. 2018. Temporal development of the gut microbiome in early childhood from the TEDDY study. Nature 562:583–588. doi: 10.1038/s41586-018-0617-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shao Y, Forster SC, Tsaliki E, Vervier K, Strang A, Simpson N, Kumar N, Stares MD, Rodger A, Brocklehurst P, Field N, Lawley TD. 2019. Stunted microbiota and opportunistic pathogen colonization in caesarean-section birth. Nature 574:117–121. doi: 10.1038/s41586-019-1560-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nilsen M, Lokmic A, Angell IL, Lødrup Carlsen KC, Carlsen K-H, Haugen G, Hedlin G, Jonassen CM, Marsland BJ, Nordlund B, Rehbinder EM, Saunders CM, Skjerven HO, Snipen L, Staff AC, Söderhäll C, Vettukattil R, Rudi K. 2021. Fecal microbiota nutrient utilization potential suggests mucins as drivers for initial gut colonization of mother-child shared bacteria. Appl Environ Microbiol 87:e02201–20. doi: 10.1128/AEM.02201-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yassour M, Jason E, Hogstrom LJ, Arthur TD, Tripathi S, Siljander H, Selvenius J, Oikarinen S, Hyöty H, Virtanen SM, Ilonen J, Ferretti P, Pasolli E, Tett A, Asnicar F, Segata N, Vlamakis H, Lander ES, Huttenhower C, Knip M, Xavier RJ. 2018. Strain-level analysis of mother-to-child bacterial transmission during the first few months of life. Cell Host Microbe 24:146–154.e4. doi: 10.1016/j.chom.2018.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mitchell C, Hogstrom L, Bryant A, Bergerat A, Cher A, Pochan S, Herman P, Carrigan M, Sharp K, Huttenhower C, Lander ES, Vlamakis H, Xavier RJ, Yassour M. 2020. Delivery mode impacts newborn gut colonization efficiency. bioRxiv. doi: 10.1101/2020.01.29.919993. [DOI] [PMC free article] [PubMed]
- 13.Hiseni P, Rudi K, Wilson RC, Terje Hegge F, Snipen L. 2020. HumGut: a comprehensive human gut prokaryotic genomes collection filtered by metagenome data. bioRxiv. doi: 10.1101/2020.03.25.007666. [DOI] [PMC free article] [PubMed]
- 14.Yang Y, Zhang Q, Hu H, Zhang W, Lu T. 2021. Bloodstream infection caused by Bacteroides caccae in a patient with renal hypertension: a case report. J Int Med Res 49:3000605211047277. doi: 10.1177/03000605211047277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Desai MS, Seekatz AM, Koropatkin NM, Kamada N, Hickey CA, Wolter M, Pudlo NA, Kitamoto S, Terrapon N, Muller A, Young VB, Henrissat B, Wilmes P, Stappenbeck TS, Núñez G, Martens EC. 2016. A dietary fiber-deprived gut microbiota degrades the colonic mucus barrier and enhances pathogen susceptibility. Cell 167:1339–1353.e21. doi: 10.1016/j.cell.2016.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shi N, Li N, Duan X, Niu H. 2017. Interaction between the gut microbiome and mucosal immune system. Mil Med Res 4:14. doi: 10.1186/s40779-017-0122-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hiippala K, Kainulainen V, Suutarinen M, Heini T, Bowers JR, Jasso-Selles D, Lemmer D, Valentine M, Barnes R, Engelthaler DM, Satokari R. 2020. Isolation of anti-inflammatory and epithelium reinforcing Bacteroides and Parabacteroides spp. from a healthy fecal donor. Nutrients 12:935. doi: 10.3390/nu12040935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brown EM, Ke X, Hitchcock D, Jeanfavre S, Avila-Pacheco J, Nakata T, Arthur TD, Fornelos N, Heim C, Franzosa EA, Watson N, Huttenhower C, Haiser HJ, Dillow G, Graham DB, Finlay BB, Kostic AD, Porter JA, Vlamakis H, Clish CB, Xavier RJ. 2019. Bacteroides-derived sphingolipids are critical for maintaining intestinal homeostasis and symbiosis. Cell Host Microbe 25:668–680.e7. doi: 10.1016/j.chom.2019.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shen Y, Giardino Torchia ML, Lawson GW, Karp CL, Ashwell JD, Mazmanian SK. 2012. Outer membrane vesicles of a human commensal mediate immune regulation and disease protection. Cell Host Microbe 12:509–520. doi: 10.1016/j.chom.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mueller NT, Differding MK, Østbye T, Hoyo C, Benjamin-Neelon SE. 2021. Association of birth mode of delivery with infant faecal microbiota, potential pathobionts, and short chain fatty acids: a longitudinal study over the first year of life. BJOG 128:1293–1303. doi: 10.1111/1471-0528.16633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rehbinder EM, Lødrup Carlsen KC, Staff AC, Angell IL, Landrø L, Hilde K, Gaustad P, Rudi K. 2018. Is amniotic fluid of women with uncomplicated term pregnancies free of bacteria? Am J Obstet Gynecol 219:289.e1–289.e12. doi: 10.1016/j.ajog.2018.05.028. [DOI] [PubMed] [Google Scholar]
- 22.Dominguez-Bello MG, De Jesus-Laboy KM, Shen N, Cox LM, Amir A, Gonzalez A, Bokulich NA, Song SJ, Hoashi M, Rivera-Vinas JI, Mendez K, Knight R, Clemente JC. 2016. Partial restoration of the microbiota of cesarean-born infants via vaginal microbial transfer. Nat Med 22:250–253. doi: 10.1038/nm.4039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wilson BC, Butler ÉM, Grigg CP, Derraik JGB, Chiavaroli V, Walker N, Thampi S, Creagh C, Reynolds AJ, Vatanen T, O'Sullivan JM, Cutfield WS. 2021. Oral administration of maternal vaginal microbes at birth to restore gut microbiome development in infants born by caesarean section: a pilot randomised placebo-controlled trial. eBioMedicine 69:103443. doi: 10.1016/j.ebiom.2021.103443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Korpela K, Helve O, Kolho K-L, Saisto T, Skogberg K, Dikareva E, Stefanovic V, Salonen A, Andersson S, de Vos WM. 2020. Maternal fecal microbiota transplantation in cesarean-born infants rapidly restores normal gut microbial development: a proof-of-concept study. Cell 183:324–334.e5. doi: 10.1016/j.cell.2020.08.047. [DOI] [PubMed] [Google Scholar]
- 25.Skjerven HO, Rehbinder EM, Vettukattil R, LeBlanc M, Granum B, Haugen G, Hedlin G, Landrø L, Marsland BJ, Rudi K, Sjøborg KD, Söderhäll C, Staff AC, Carlsen K-H, Asarnoj A, Bains KES, Carlsen OCL, Endre KMA, Granlund PA, Hermansen JU, Gudmundsdóttir HK, Hilde K, Håland G, Kreyberg I, Olsen IC, Mägi C-AO, Nordhagen LS, Saunders CM, Skrindo I, Tedner SG, Værnesbranden MR, Wiik J, Jonassen CM, Nordlund B, Carlsen KCL. 2020. Skin emollient and early complementary feeding to prevent infant atopic dermatitis (PreventADALL): a factorial, multicentre, cluster-randomised trial. Lancet 395:951–961. doi: 10.1016/S0140-6736(19)32983-6. [DOI] [PubMed] [Google Scholar]
- 26.Lødrup Carlsen KC, Rehbinder EM, Skjerven HO, Carlsen MH, Fatnes TA, Fugelli P, Granum B, Haugen G, Hedlin G, Jonassen CM, Landrø L, Lunde J, Marsland BJ, Nordlund B, Rudi K, Sjøborg K, Söderhäll C, Staff AC, Vettukattil R, Carlsen K-H. 2018. Preventing atopic dermatitis and ALLergies in children—the PreventADALL study. Allergy 73:2063–2070. doi: 10.1111/all.13468. [DOI] [PubMed] [Google Scholar]
- 27.Ko KS, Kuwahara T, Haehwa L, Yoon Y-J, Kim B-J, Lee K-H, Ohnishi Y, Kook Y-H. 2007. RNA polymerase beta-subunit gene (rpoB) sequence analysis for the identification of Bacteroides spp. Clin Microbiol Infect 13:48–54. doi: 10.1111/j.1469-0691.2006.01553.x. [DOI] [PubMed] [Google Scholar]
- 28.Yu Y, Lee C, Kim J, Hwang S. 2005. Group-specific primer and probe sets to detect methanogenic communities using quantitative real-time polymerase chain reaction. Biotechnol Bioeng 89:670–679. doi: 10.1002/bit.20347. [DOI] [PubMed] [Google Scholar]
- 29.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. 2016. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods 13:581–583. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McMurdie PJ, Holmes S. 2013. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:e61217. doi: 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang C. 13 April 2020. yiluheihei/microbiomeMarker: microbiomeMarker 0.0.1. Zenodo. https://zenodo.org/record/3749415.
- 32.Snipen L, Angell I-L, Rognes T, Rudi K. 2021. Reduced metagenome sequencing for strain-resolution taxonomic profiles. Microbiome 9:79. doi: 10.1186/s40168-021-01019-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Afle FCD, Agbankpe AJ, Johnson RC, Houngbégnon O, Houssou SC, Bankole HS. 2019. Healthcare-associated infections: bacteriological characterization of the hospital surfaces in the University Hospital of Abomey-Calavi/so-ava in South Benin (West Africa). BMC Infect Dis 19:28. doi: 10.1186/s12879-018-3648-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chaoui L, Mhand R, Mellouki F, Rhallabi N. 2019. Contamination of the surfaces of a health care environment by multidrug-resistant (MDR) bacteria. Int J Microbiol 2019:3236526. doi: 10.1155/2019/3236526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 36.Ioannou A, Knol J, Belzer C. 2021. Microbial glycoside hydrolases in the first year of life: an analysis review on their presence and importance in infant gut. Front Microbiol 12:631282. doi: 10.3389/fmicb.2021.631282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tailford LE, Crost EH, Kavanaugh D, Juge N. 2015. Mucin glycan foraging in the human gut microbiome. Front Genet 6:81. doi: 10.3389/fgene.2015.00081. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material. Download aem.00789-23-s0001.docx, DOCX file, 0.4 MB (407.2KB, docx)
Data Availability Statement
Sequencing data of 16S rRNA genes and rpoB is available in the NCBI SRA database with the identifier PRJNA877464. The RMS data are available upon request in TSD UiO (University of Oslo).