

Abstract
Angiotensin-converting enzyme inhibitors (ACEIs) are used by millions of patients to treat hypertension, diabetic kidney disease, and heart failure. However, these patients are often at increased risk of infection. To evaluate the impact of ACEIs on immune responses to infection, we compared the effect of an ACEI versus an angiotensin receptor blocker (ARB) on neutrophil antibacterial activity. ACEI exposure reduced the ability of murine neutrophils to kill methicillin-resistant Staphylococcus aureus (MRSA), Pseudomonas aeruginosa, and Klebsiella pneumoniae in vitro. In vivo, ACEI-treated mice infected with MRSA had increased bacteremia and tissue bacteria counts compared to mice treated with an ARB or with no drug. Similarly, ACEIs, but not ARBs, increased the incidence of MRSA-induced infective endocarditis in mice with aortic valve injury. Neutrophils from ACE knockout (KO) mice or mice treated with an ACEI produced less leukotriene B4 (LTB4) upon stimulation with MRSA or lipopolysaccharide, whereas neutrophils overexpressing ACE produced more LTB4 compared to wild-type neutrophils. As a result of reduced LTB4 production, ACE KO neutrophils showed decreased survival signaling and increased apoptosis. In contrast, neutrophils overexpressing ACE had an enhanced survival phenotype. Last, in a cohort of human volunteers receiving the ACEI ramipril for 1 week, ACEI administration reduced neutrophil superoxide and reactive oxygen species production and neutrophils isolated from volunteers during ramipril treatment had reduced bactericidal activity. Together, these data demonstrate that ACEI treatment, but not ARB treatment, can reduce the bacterial killing ability of neutrophils.
INTRODUCTION
Angiotensin-converting enzyme (ACE) is critical in the regulation of blood pressure by cleaving angiotensin I into angiotensin II (Ang II) (1). However, ACE is widely expressed in tissues and is a promiscuous enzyme that can cleave hundreds of substrates. Because of this, ACE influences many other physiologic systems (1, 2). ACE inhibitors (ACEIs) are used by millions of patients to treat hypertension, diabetic kidney disease, and heart failure. As a result, ACEIs are often used by patients undergoing invasive heart surgery such as valve replacement or by individuals who are immunocompromised. Such patients are susceptible to infection, and any suppression of antimicrobial activity by ACEIs might enhance the risk of infection including infective endocarditis. Several studies have demonstrated a role of ACE in the regulation of myeloid cells, including cell development and the immune responses of myelomonocytic cells (1–7). ACE expression is up-regulated during myeloid cell differentiation, in myeloid cells activated by stimuli such as lipopolysaccharide (LPS) and infectious pathogens (3, 6, 8, 9), and in granulomatous diseases, such as sarcoidosis, where ACE expression markedly increases in lesional myeloid-derived cells (10, 11).
Neutrophils are abundant in the bloodstream and are the first cells recruited to injured tissue, often to kill invading pathogens (12, 13). Neutrophil number and functional maturation are tightly regulated by various factors, including leukotriene B4 (LTB4). LTB4 is a potent proinflammatory molecule and among the first chemoattractants to be released at an inflammatory site (14, 15). LTB4 is key in the recruitment and activation of neutrophils during inflammation and prolongs neutrophil survival (14–18).
We have previously investigated the role of ACE in neutrophil function using a transgenic mouse line that expresses increased ACE in neutrophils (NeuACE) (6). These mice show resistance to methicillin-resistant Staphylococcus aureus (MRSA) and better in vitro killing of MRSA and other pathogenic bacteria. In contrast, neutrophils from ACE knockout (KO) mice kill MRSA less efficiently than wild-type (WT) cells (6). Such studies indicate that ACE facilitates production of reactive oxygen species (ROS) in neutrophils after bacterial stimulation, an effect independent of Ang II and the Ang II type 1 (AT1) receptor (6). Here, we compared the effect of ACEIs versus angiotensin receptor blockers (ARBs) on mouse neutrophil antibacterial activity in vitro and in vivo and development of infective endocarditis. We found that treatment with the ACEIs ramipril or lisinopril decreases neutrophil antibacterial activity against MRSA, Pseudomonas aeruginosa, and Klebsiella pneumoniae. We also evaluated the function of neutrophils isolated from healthy human volunteers who received the ACEI ramipril for a week. These studies provide in vitro and in vivo evidence that ACEIs reduce neutrophil function and may increase susceptibility to bacterial infection.
RESULTS
Inhibition of ACE decreases bacterial killing activity of neutrophils in vitro
To study whether ACEIs and ARBs affect neutrophil antibacterial activity, WT mice were treated with either ramipril (ACEI; 40 mg/liter), lisinopril (ACEI; 40 mg/liter), or losartan (ARB; 600 mg/liter) in drinking water. Both ACEI and ARB reduced blood pressure by an equivalent amount (fig. S1) (19). After 1 week, we tested whole blood ex vivo for the ability to kill MRSA, K. pneumoniae, or P. aeruginosa. This predominantly tests neutrophil killing of bacteria. Reduced killing ability, indicated by higher colony-forming unit (CFU) counts of surviving bacteria, was observed when bacteria were exposed to whole blood isolated from ACEI-treated animals (Fig. 1). For example, MRSA concentrations in samples from ramipril-treated mice were 4.2-fold higher and 6.6-fold higher at 2 and 5 hours after MRSA introduction as compared to the untreated group (P < 0.001; Fig. 1A). A similar reduction of blood killing was seen with K. pneumoniae and P. aeruginosa (Fig. 1, B and C). Lisinopril also suppressed killing of bacteria by peripheral blood cells. However, no differences were observed in ex vivo killing by peripheral blood cells between losartan-treated and untreated mice (Fig. 1, A to C). To study neutrophil intracellular killing, neutrophils were purified from bone marrow and then mixed with bacteria at a multiplicity of infection (MOI) of 15 for 30 min. After eliminating extracellular bacteria, killing of intracellular bacteria was measured by CFU counts. Similar to whole blood killing, ramipril and lisinopril treatment suppressed neutrophil intracellular killing at 2 and 5 hours after infection as compared to losartan-treated and untreated groups (Fig. 1, D to F).
Fig. 1. ACEI treatment suppressed murine neutrophil antibacterial activity in vitro.
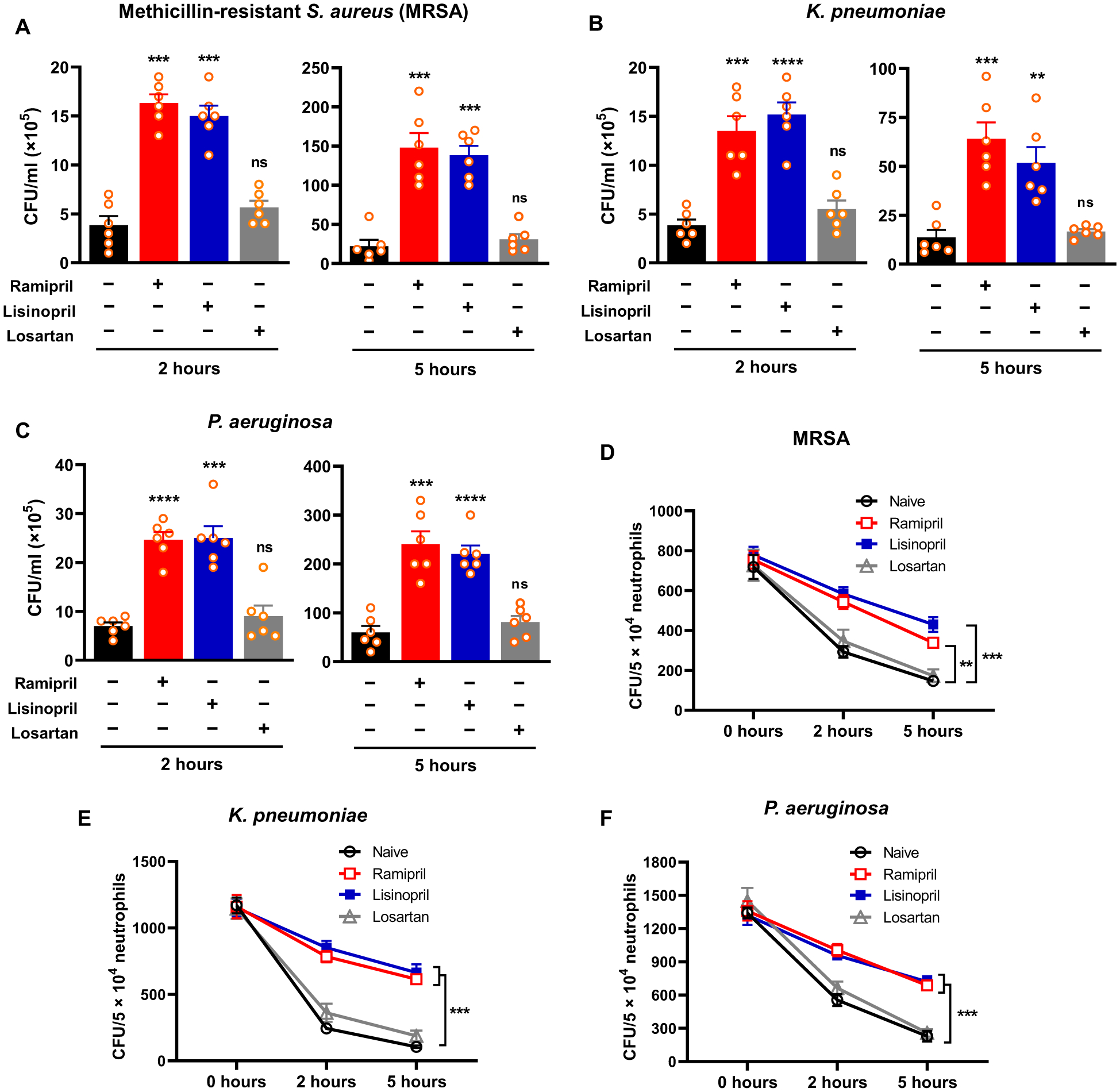
(A) After mixing with MRSA (1.2 × 106 CFU/ml), blood samples isolated from mice were assessed for their ability to eliminate MRSA after a 2- or 5-hour incubation. (B) Ex vivo killing of K. pneumoniae (1.54 × 106 CFU/ml) by whole blood isolated from ACEI- or ARB-treated mice was evaluated. (C) Ex vivo killing of P. aeruginosa (1.9 × 106 CFU/ml) by whole blood isolated from ACEI- or ARB-treated mice was evaluated. (D) In vitro intracellular killing of MRSA by purified murine neutrophils from bone marrow at 2 and 5 hours was evaluated. (E) In vitro intracellular killing of K. pneumoniae by purified neutrophils from mice treated with ACEIs or an ARB is shown. (F) In vitro intracellular killing of P. aeruginosa by purified neutrophils from mice treated with ACEIs or an ARB is shown. In the intracellular killing assays (D to F), neutrophils were isolated from bone marrow and infected with the indicated bacterial strains at an MOI of about 15. A one-way ANOVA with Bonferroni’s correction for multiple comparisons was used to analyze group comparisons, and data are presented as means ± SEM (n = 6). **P < 0.01, ***P < 0.001, and ****P < 0.0001. ns, nonsignificant.
Inhibition of ACE increases susceptibility to bacterial infection in vivo
Next, ACE KO and WT mice treated with either ramipril or losartan were infected intravenously with MRSA (about 1 × 108 CFU). After 24 hours, blood drawn from mice was assessed for bacteremia. Similar to ACE KO mice, the blood bacterial count was 3.7-fold higher in WT mice treated with ramipril as compared to the untreated group (P < 0.001; Fig. 2A). Ramipril treatment also increased the bacterial burden in tissues, including spleen (4.1-fold), liver (3.4-fold), and lung (2.8-fold) in comparison to untreated WT mice (Fig. 2, B to D). In contrast, losartan treatment did not affect the bacterial resistance in mice (Fig. 2, B to D). To confirm that this difference was due to neutrophils, mice were treated intraperitoneally with anti–polymorphonuclear neutrophil (PMN) antibody 1 day before infection to deplete about 90% of neutrophils (6, 20). These animals were then challenged with MRSA, and bacterial burden was determined in blood and tissues. In the absence of neutrophils, the response to MRSA was not different between ACE KO and WT mice (Fig. 2).
Fig. 2. Inhibition of ACE increased susceptibility to bacterial infection in vivo.
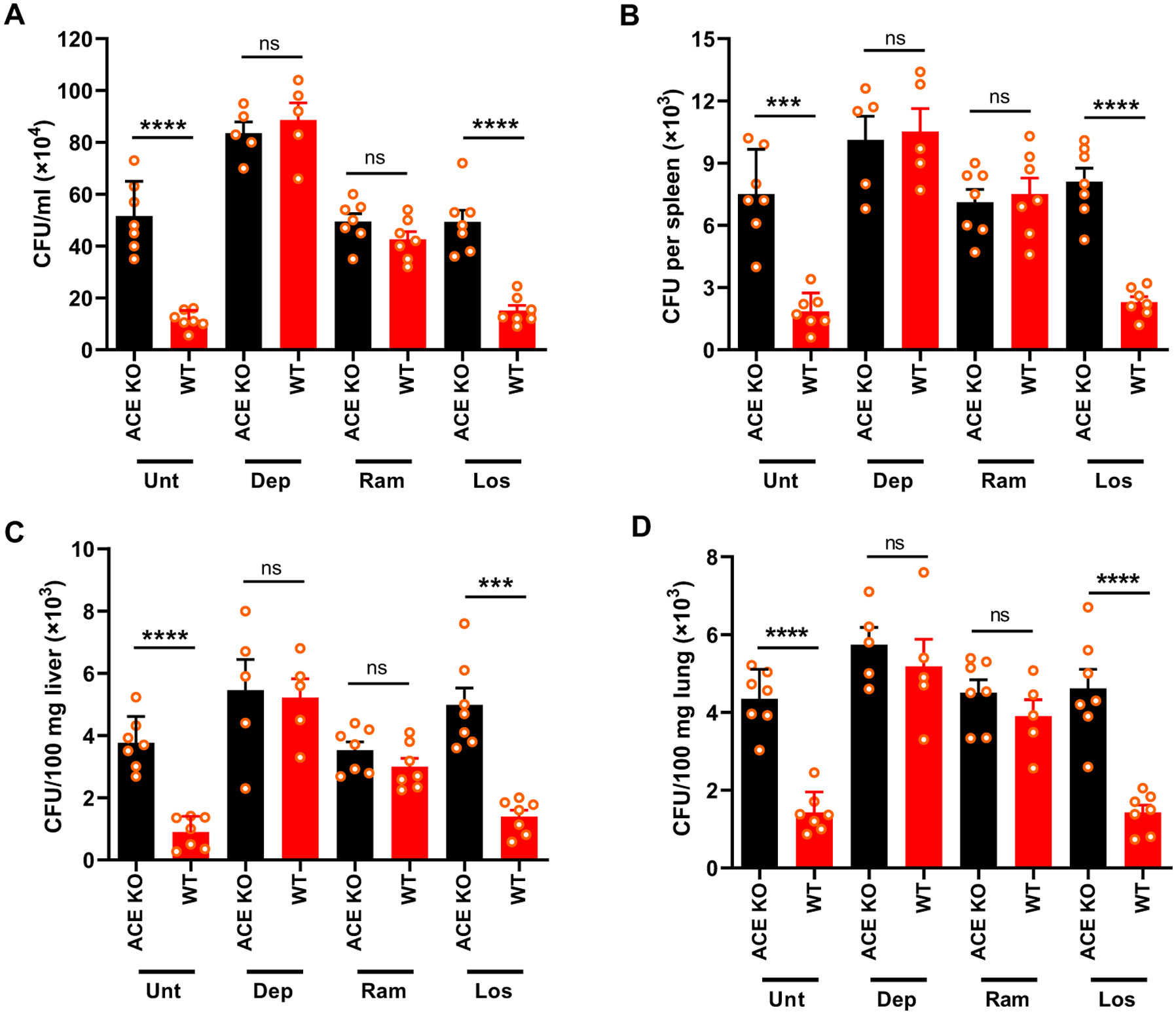
(A) WT and ACE KO mice were infected with MRSA intravenously (8 × 107 CFU/100 μl). The bacterial concentration in the blood is shown at 24 hours after infection. (B to D) Bacterial count in spleen (B), liver (C), and lung (D) was determined at 48 hours after infection. Indicated groups of mice were treated with either ramipril (Ram; 40 mg/liter) or losartan (Los; 600 mg/liter) for 1 week before infection. Drugs were administered throughout the study. Neutrophil depletion (Dep) was achieved by daily injection of mice with a murine anti-PMN antibody, starting 24 hours before infection with MRSA to the end of study. Mice were perfused with PBS before harvesting tissues. Two-way ANOVA with Bonferroni’s correction for multiple comparisons was used to analyze group comparisons, and data are presented as means ± SEM (n = 7 mice per group). ***P < 0.001 and ****P < 0.0001.
ACEIs and ARBs are often used by patients susceptible to heart infections, including patients with damaged heart valves (21–24). To mimic these conditions, we damaged the aortic valves of mice by inserting an intracardiac catheter via the right carotid artery. As shown by hematoxylin and eosin staining of mice heart sections, catheterization caused infiltration of inflammatory cells in hearts (Fig. 3A). Mice were then inoculated with a low dose of MRSA (1 × 107 CFU), and bacterial burden in hearts was determined 24 hours after infection. Without aortic valve injury, relatively few bacteria were found in the hearts of either WT, ACE KO, or NeuACE mice (Fig. 3B). However, with aortic valve injury, the accumulation of MRSA in the hearts of ACE KO mice was 26-fold higher than that measured in WT mice (Fig. 3C). In contrast, the NeuACE mice showed fourfold less bacterial burden in injured hearts as compared to WT mice (Fig. 3C). In mice depleted of neutrophils, the response to MRSA was not different between ACE KO, WT, and NeuACE mice.
Fig. 3. Inhibition of ACE increased the incidence of infective endocarditis in MRSA-infected mice.
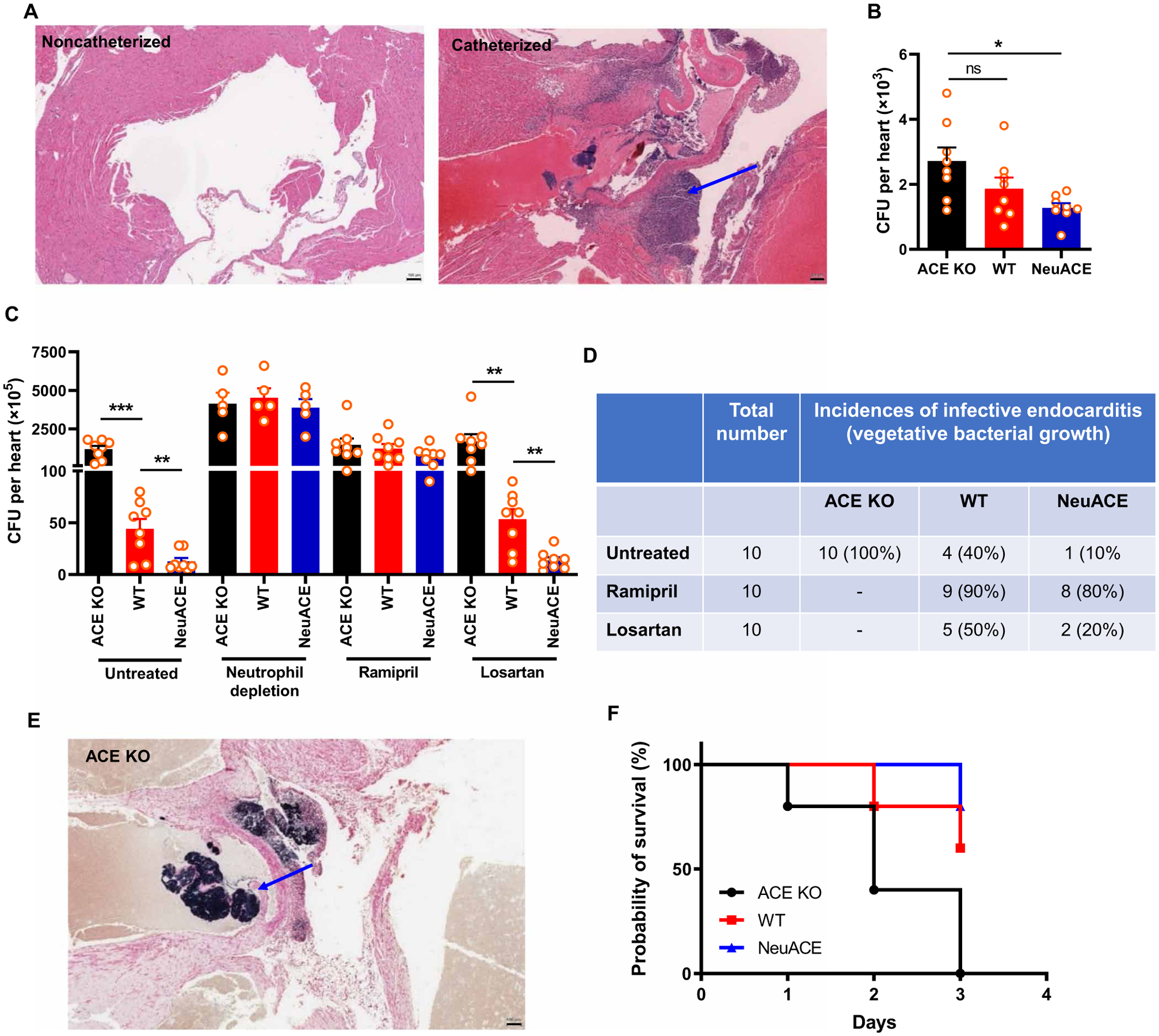
(A) Representative images of hematoxylin and eosin–stained murine heart transverse sections with or without aortic valve damage by catheter insertion are shown. The blue arrow indicates aortic valve damage and infiltration of cells. Scale bars, 100 μm. (B) Bacteria were counted per heart in ACE KO, WT, and NeuACE mice challenged with intravenous MRSA (about 1 × 107 CFU/100 μl) without valve damage. (C) Bacteria were counted per heart in mice challenged with intravenous MRSA with aortic valve injury. ACE KO, WT, and NeuACE mice were untreated, depleted of neutrophils, or treated with ramipril or losartan. n ≥ 7. (D) Incidence of infective endocarditis in catheterized ACE KO, WT, and NeuACE mice as indicated by Gram staining (n = 10 per group). (E) A representative image of Gram staining shows bacterial accumulation in the heart of a catheterized ACE KO mouse challenged with intravenous MRSA. The blue arrow shows vegetative bacterial growth. Scale bar, 100 μm. (F) Survival of ACE KO, WT, and NeuACE mice after aortic valve damage and MRSA challenge is shown (n = 5 per group). A one-way (B) or two-way ANOVA (C) with Bonferroni’s correction for multiple comparisons was used to analyze group comparisons, and data are presented as means ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001.
To determine the effects of ACEI and ARB treatment, WT mice were treated with ramipril or losartan for 1 week before surgery. Similar to ACE KO mice, pharmacologic inhibition of ACE by ramipril increased MRSA accumulation in catheterized hearts as compared to losartan or the untreated group (Fig. 3C). In these experiments, mice were perfused with phosphate-buffered saline (PBS) before harvesting the hearts such that the bacterial count in Fig. 3C excludes blood bacteria. At harvest, Gram staining of the perfused hearts was used to identify vegetative bacterial growth. This showed that all instrumented ACE KO mice (100%) developed infective endocarditis in comparison to 40% of WT mice and only 10% of NeuACE mice (Fig. 3, D and E). As seen in ACE KO mice, ramipril treatment resulted in almost all WT mice (90%) developing infective endocarditis, and it increased the incidence of infective endocarditis in NeuACE mice, whereas no such increase in infective endocarditis was noted with losartan treatment (Fig. 3D). These data indicate that ACEI treatment increases the risk of bacterial infection including infective endocarditis in susceptible mice, whereas an ARB does not show any such effect. To measure survival, mice with valve injury and MRSA infection were observed longer. Consistent with their increased incidence of infective endocarditis, all ACE KO mice (100%) died by day 3 after infection as compared to 40% of WT and 20% of the NeuACE mice group (Fig. 3F).
ACE affects neutrophil oxidative bactericidal activity through p38–mitogen-activated protein kinase
We previously found that increased ACE expression in neutrophils increases superoxide generation by reduced form of nicotinamide adenine dinucleotide phosphate oxidase 2 (NOX2) (6). Certain kinases such as protein kinase C (PKC) isoforms and mitogen-activated protein kinase (MAPK) play a major role in activating NOX2 by phosphorylating its cytosolic subunits (25). To determine whether ACE affects activation of these kinases, we challenged bone marrow–derived neutrophils with LPS (1 μg/ml) in vitro for 30 min and measured PKC kinase activity with a substrate-based enzyme-linked immunosorbent assay (ELISA) and PKC phosphorylation by Western blot. Both assays showed no difference in PKC activation between ACE KO, WT, and NeuACE neutrophils (Fig. 4A and fig. S2). However, the active form of p38-MAPK (phosphorylated Thr180/Tyr182) was significantly lower in ACE KO neutrophils as compared to WT neutrophils as determined by ELISA (P < 0.001; Fig. 4B). In contrast, NeuACE neutrophils showed increased phosphorylation of p38-MAPK as compared to WT neutrophils (P < 0.001; Fig. 4B). Western blot analysis also confirmed decreased phospho–p38-MAPK (Thr180/Tyr182) in ACE KO neutrophils and increased phospho–p38-MAPK in NeuACE neutrophils in comparison to WT cells (Fig. 4C). To further examine the role of ACE and Ang II, mice were treated with ramipril or losartan. Bone marrow neutrophils were then isolated and challenged with LPS (1 μg/ml) for 30 min in vitro. Kinase phosphorylation associated with enzyme activation was determined. Ramipril treatment markedly reduced p38-MAPK phosphorylation to a degree that was at or close to the phosphorylation observed in samples isolated from ACE KO mice (Fig. 4B).
Fig. 4. ACE expression affects the p38-MAPK–mediated neutrophil oxidative response and bacterial killing in vitro.
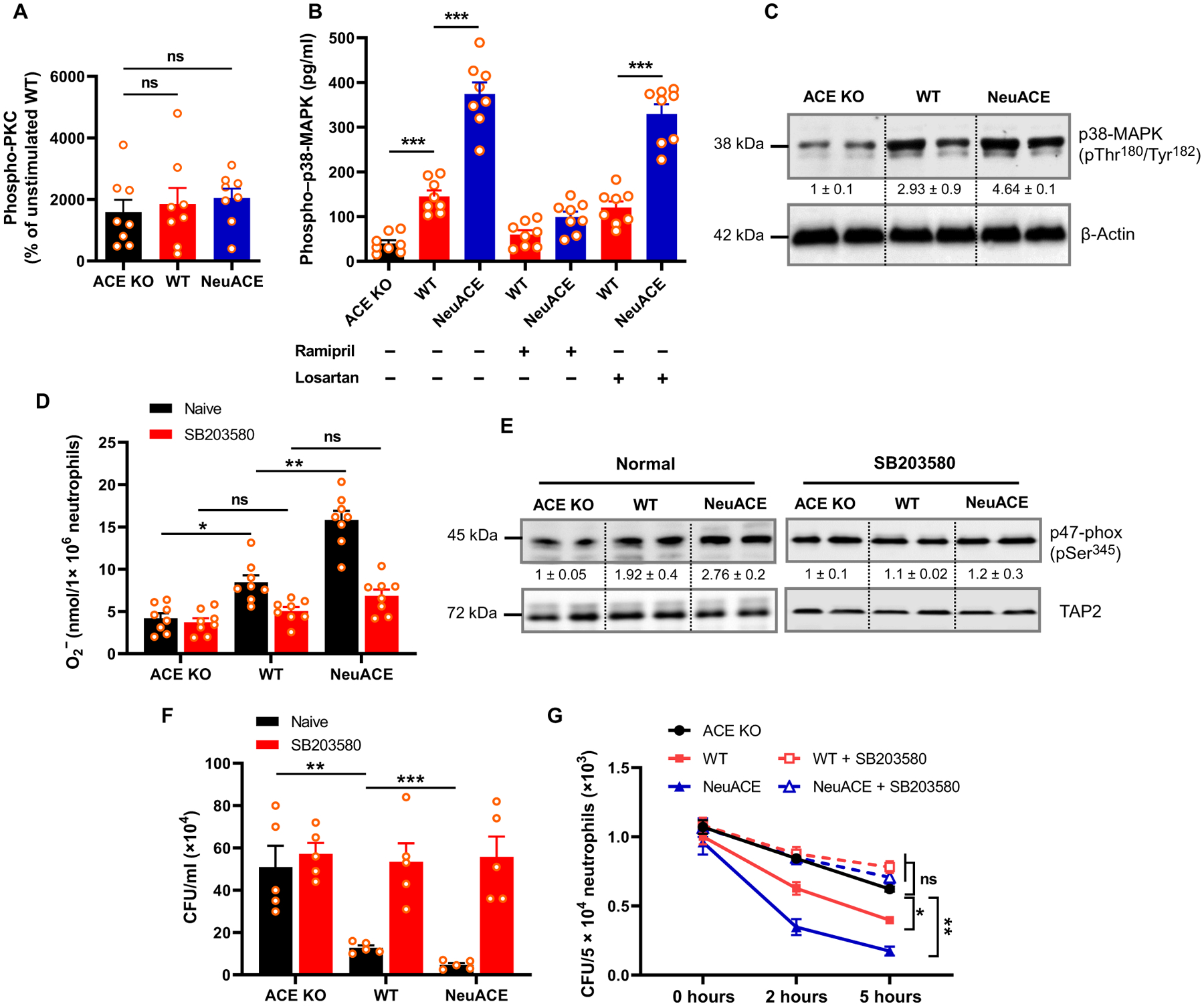
(A) Phospho-PKC isoforms were measured by ELISA in ACE KO, WT, and NeuACE bone marrow neutrophils after stimulation with LPS for 30 min in vitro (n = 8 per group). (B) Phospho–p38-MAPK in bone marrow neutrophils was measured by ELISA after stimulation with LPS for 30 min in vitro (n = 8 per group). Mice were treated with ramipril or losartan for 1 week before neutrophil isolation. (C) Western blotting of ACE KO, WT, and NeuACE neutrophils for phospho–p38-MAPK is shown. β-Actin serves as the loading control. Numbers below bands indicate relative band density as determined by Image Studio Lite version 5.2 (LI-COR). (D) Superoxide (O −2) concentration in neutrophils after stimulation with LPS (1 μg/ml) for 10 min at room temperature was measured using a cytochrome C reduction assay (n = 8 per group). For p38-MAPK inhibition, neutrophils were pretreated with 500 nM SB203580 for 2 hours at room temperature. (E) Measurement of phospho–p47-phox in neutrophils treated with or without SB203580 and stimulated with LPS by Western blot. TAP2 serves as the loading control. Numbers below bands indicate relative band density. (F) Ex vivo MRSA clearance by blood from ACE KO, WT, and NeuACE mice after treatment with SB203580 is shown. Blood was drawn from mice and mixed with SB203580 for 1 hour. Then, after addition of MRSA (1.4 × 106 CFU/ml), blood samples were assessed for their ability to eliminate MRSA after a 2-hour incubation (n = 5). (G) In vitro intracellular killing of MRSA in neutrophils purified from bone marrow (n = 6 per group). One-way (A) or two-way ANOVA (B, D, F, and G) with Bonferroni’s correction for multiple comparisons was used to analyze group comparisons, and data are presented as means ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001.
To verify whether p38-MAPK mediates NOX2 activation by ACE, we measured bone marrow neutrophil superoxide with or without the p38-MAPK inhibitor SB203580 (500 nM) for 2 hours in vitro (Fig. 4D). ACE activity was proportional to superoxide generation by neutrophils, in that ACE KO neutrophils produced significantly less superoxide (P < 0.05), whereas NeuACE neutrophils produced more superoxide as compared to WT neutrophils (P < 0.01; Fig. 4D). In contrast, SB203580 treatment abrogated these differences in superoxide generation (Fig. 4D). Because p38-MAPK activates NOX2 by phosphorylating the p47-phox subunit (26), we measured phosphorylation of p47-phox at Ser345, a site specifically phosphorylated by p38-MAPK. The abundance of phospho–p47-phox was correlated with superoxide production by bone marrow neutrophils (Fig. 4E). After stimulation with LPS in vitro, the abundance of phospho–p47-phox was lower in ACE KO neutrophils, whereas NeuACE neutrophils showed increased phospho–p47-phox as compared to WT neutrophils (Fig. 4E). In contrast, inhibition of p38-MAPK by SB203580 eliminated the difference in phospho–p47-phox between the groups.
To determine the role of p38-MAPK in the ACE-mediated antibacterial neutrophil response, we measured ex vivo whole blood–mediated killing and phagocytic killing of MRSA in the presence of SB203580. Blood and purified neutrophils isolated from ACE KO, WT, and NeuACE mice were pretreated with SB203580 for 2 hours in vitro before inoculating with MRSA (about 1 × 106 CFU/ml). Similar to superoxide production, the difference in MRSA killing was eliminated between the groups by p38-MAPK inhibition (Fig. 4, F and G).
ACE inhibition or overexpression affects LTB4 production by neutrophils
LTB4 is a powerful chemoattractant that activates leukocytes and prolongs their survival through MAPKs and phosphoinositide 3-kinase (PI3K) (15–18, 27, 28). We observed that ACE KO neutrophils produced less LTB4 than WT cells after stimulation with either LPS (1 μg/ml) for 12 hours or MRSA (MOI, 15) for 6 hours (Fig. 5A and fig. S3). In contrast, LTB4 production was increased in neutrophils isolated from NeuACE mice after in vitro stimulation (Fig. 5A and fig. S3). With ramipril treatment, there was a marked reduction of LTB4 concentration in the WT and NeuACE mice that approached the concentration seen in ACE KO neutrophils (Fig. 5A). In contrast, losartan did not affect LTB4 production (Fig. 5A). To measure LTB4 in vivo, mice were challenged with either LPS [750 μg/kg body weight, intraperitoneally (i.p.)] or MRSA [1 × 108 CFU, intravenously (i.v.)], and serum LTB4 was measured at 4 or 24 hours after LPS or MRSA administration, respectively. In both cases, serum concentrations of LTB4 in ACE KO mice were significantly lower as compared to WT mice (P < 0.01), whereas NeuACE mice had higher serum concentrations of LTB4 than WT mice (Fig. 5B and fig. S4). In contrast, the basal concentrations of LTB4 were not different between ACE KO, WT, and NeuACE mice (Fig. 5B).
Fig. 5. ACE increases neutrophil LTB4 production.
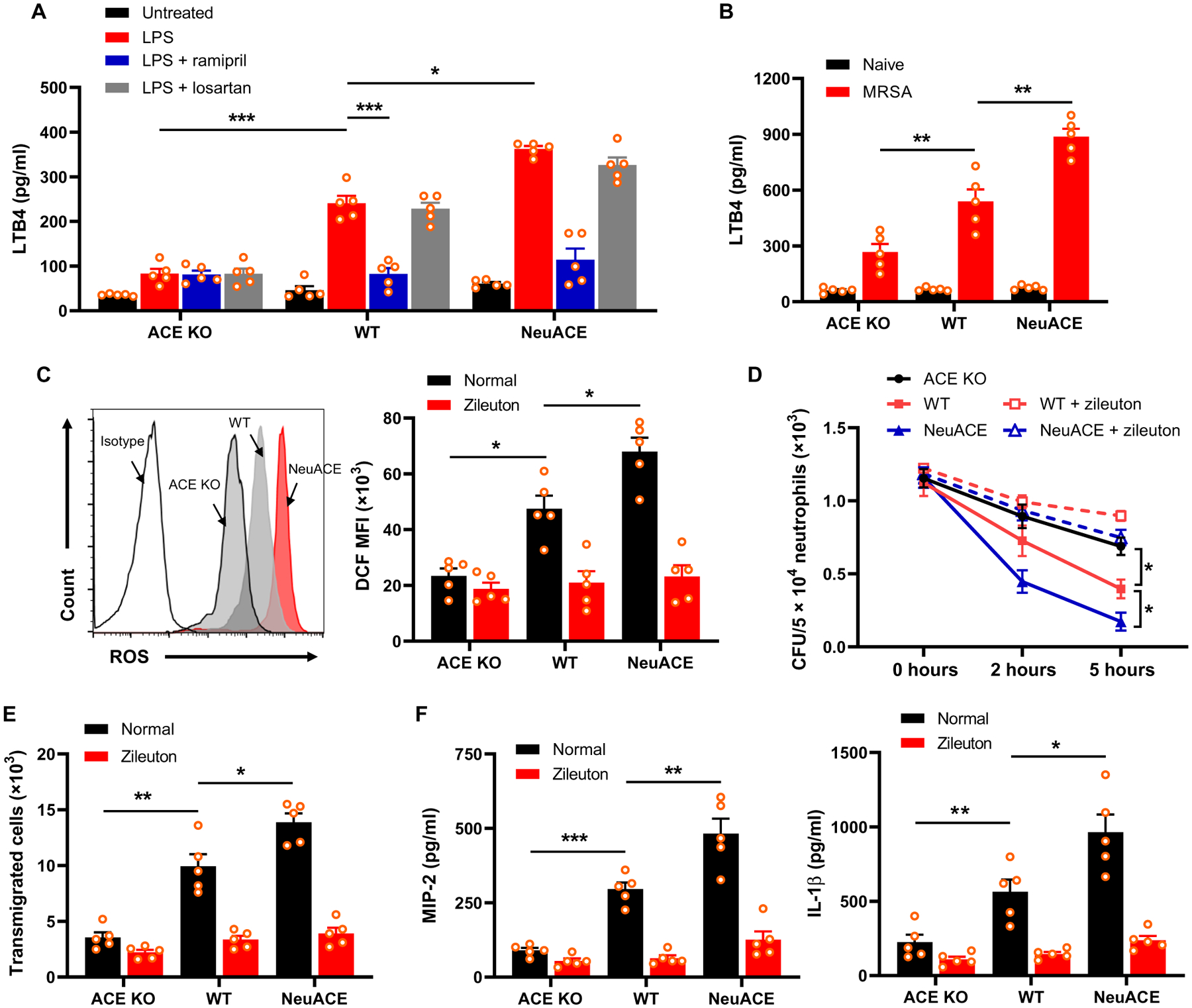
(A) Bone marrow neutrophils were cultured and stimulated with LPS (500 ng/ml) for 12 hours, and then, the supernatant plus cells were collected for ELISA. Groups of mice were treated with either ramipril (40 mg/liter) or losartan (600 mg/liter) in drinking water for 1 week before isolation of bone marrow neutrophils, and then, cells were treated with 10 μM ramipril or 100 μM losartan during the in vitro assay. (B) Measurement of LTB4 in serum isolated 24 hours after challenge with intravenous MRSA (1.2 × 108 CFU/100 μl). (C) Measurement of ROS production in neutrophils treated with LPS (1 μg/ml) for 30 min using 2’,7’-dichlorodihydrofluorescein diacetate (DCFDA) staining and flow cytometry. A representative flow cytometry plot is shown on the left, and quantification of mean fluorescence intensity (MFI) of ROS indicator 2’,7’-dichlorodihydrofluorescein (DCF) is shown on the right. To inhibit LTB4 synthesis, groups of mice were treated with the 5-LOX inhibitor zileuton (5 mg/kg per day, gavage) for 2 days before isolation of bone marrow neutrophils, and the drug was maintained at a concentration of 100 μM during in vitro assays. (D) Mice were pretreated with zileuton, and bone marrow–isolated neutrophils were challenged in vitro with MRSA at MOI 13 to evaluate intracellular killing. (E) Neutrophils were preincubated with 100 μM zileuton and then placed in the top chamber of a transwell to measure chemotaxis. The cells that migrated to the bottom chamber in response to 100 nM fMLP were collected and enumerated by cell counter. (F) Bone marrow neutrophils were cultured and stimulated with 100 nM fMLP for 12 hours. Supernatant along with cells were collected for ELISA to measure MIP-2 and IL-1β concentrations. Two-way ANOVA with Bonferroni’s correction for multiple comparisons was used to analyze group comparisons, and data are presented as means ± SEM (n = 5 per group). *P < 0.05, **P < 0.01, and ***P < 0.001.
LTB4 is a potent chemotactic agent, and it augments the oxidative response and phagocytic bacterial killing by neutrophils (15–17). To study whether LTB4 mediates the effect that we observed after ACEI treatment, we first measured LPS-stimulated ROS production in neutrophils with and without LTB4 inhibition and found that inhibition of LTB4 synthesis by zileuton, a 5-lipoxygenase (LOX) inhibitor, abrogated the differences in ROS production observed between ACE KO, WT, and NeuACE neutrophils (Fig. 5C). To measure in vitro MRSA killing by neutrophils, mice were treated with zileuton via gavage (5 mg/kg orally, twice daily) for 2 days before bone marrow neutrophil isolation. Then, neutrophils were incubated with MRSA (MOI, 15) in the presence of 100 μM zileuton. With zileuton treatment inhibition, there was no difference in phagocytic killing of MRSA by ACE KO, WT, and NeuACE neutrophils (Fig. 5D).
To determine whether there was a difference in neutrophil chemotaxis in relation to ACE expression and LTB4, we measured cell migration using Transwell assays. Neutrophils were isolated from ACE KO, WT, and NeuACE mice and cultured using Transwell inserts (3-μm pore size) and then transferred into a 24-well plate containing media at the bottom with 100 nM N-formyl-Met-Leu-Phe (fMLP). As compared to WT neutrophils, there was reduced migration of ACE KO neutrophils from the upper to the lower chamber (Fig. 5E and fig. S5). In contrast, neutrophils isolated from NeuACE mice exhibited enhanced chemotaxis in vitro (Fig. 5E and fig. S5).
Cytokines and chemokines play a major role in the activation and chemotaxis of neutrophils. Upon stimulation with fMLP in vitro, neutrophils isolated from ACE KO mice produced less macrophage inflammatory protein-2 (MIP-2) and interleukin-1β (IL-1β), whereas NeuACE neutrophils produced more of these cytokines in comparison to WT neutrophils (Fig. 5F). In contrast, zileuton-mediated inhibition of LTB4 synthesis resulted in no difference in chemotaxis and cytokines production among the ACE KO, WT, and NeuACE neutrophils (Fig. 5, E and F).
The LTB4 receptor BLT1 mediates the function of ACE in neutrophils
In myeloid cells, LTB4 exerts its functions by interacting with the LTB4 receptor 1 (BLT1R) (14, 15). To investigate whether the LTB4-BLT1R signaling axis is involved in neutrophil-ACE functions, we measured superoxide generation by neutrophils after administration of the BLT1R antagonist U-75302 to mice (1 mg/kg per day, i.p.) for 2 days before neutrophil isolation from bone marrow. With BLT1R inhibition, no differences were observed in superoxide generation by ACE KO, WT, and NeuACE neutrophils in response to LPS stimulation (Fig. 6A). In addition, there were no differences in p38-MAPK activity between the three groups of neutrophils when they were treated with U-75302 (Fig. 6B). Last, to determine MRSA resistance to BLT1R blocking, mice were administered U-75302 for 2 days before MRSA intravenous infection. The differences in bacteremia (Fig. 6C) and tissue bacterial burden (Fig. 6, D to F) in the three genotypes of mice were diminished in animals treated with the BLT1R inhibitor U-75302.
Fig. 6. The BLT1 receptor mediates the downstream effects of ACE expression on neutrophils.
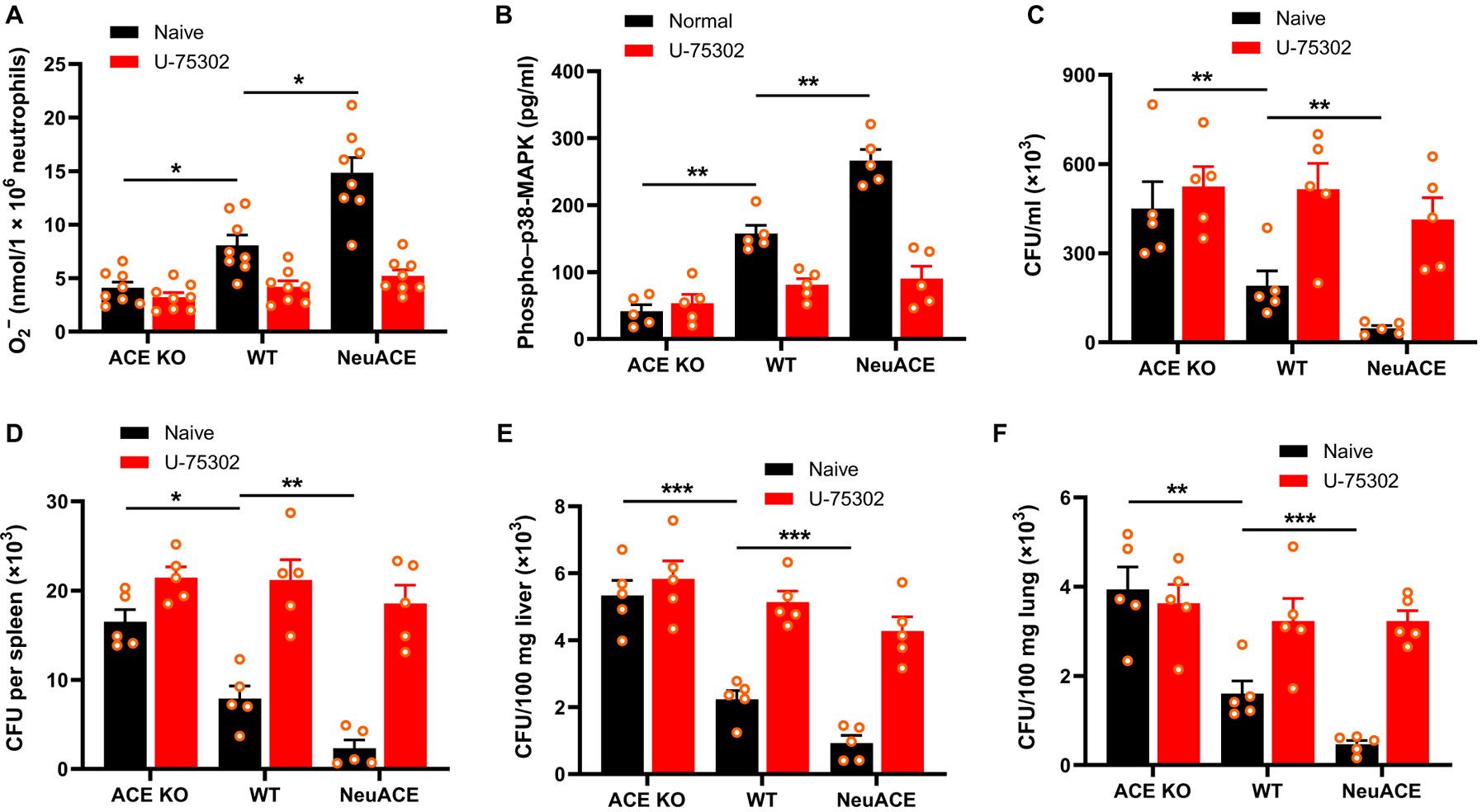
To inhibit BLT1R, mice were treated with U-75302 (1 mg/kg per day, i.p.) for 2 days before infection or isolation of bone marrow neutrophils until the end of the experiments. U-75302 concentration was maintained at 10 μM during in vitro assays. (A) Superoxide was measured by cytochrome C reduction after neutrophils were stimulated with LPS (1 μg/ml) for 10 min (n = 8 per group). (B) Phospho–p38-MAPK (Thr180/Tyr182) concentration in neutrophils was measured by ELISA after stimulation with LPS (1 μg/ml) for 30 min (n = 5). (C) In vivo bacterial count in blood was measured 24 hours after challenging control or U-75302–treated mice with 1.1 × 108 CFU/100 μl of MRSA intravenously (n = 5 mice per group). (D to F) Bacterial count in spleen (D), liver (E), and lung (F) was determined 48 hours after MRSA infection in control or U-75302–treated mice (n = 5 mice per group). Two-way ANOVA with Bonferroni’s correction for multiple comparisons was used to analyze group comparisons, and data are presented as means ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001.
ACE expression increases the prosurvival phenotype of neutrophils
LTB4 plays a crucial role in the increased neutrophil life span observed under inflammatory conditions (14–18, 29). To study whether ACE affects neutrophil survival, we stimulated ACE KO, WT, and NeuACE neutrophils with 1 μM LPS in vitro and then followed apoptosis by staining cells with Apopxin green dye and analyzing with flow cytometry. In comparison to WT neutrophils, there was increased apoptosis in ACE KO neutrophils (Fig. 7A). In contrast, there was no difference in apoptosis between ACE KO, WT, and NeuACE neutrophils when animals and cells were pretreated with the LTB4 inhibitor zileuton. To understand the survival signaling pathway involved, we analyzed cells stimulated with LPS by Western blot. Reduced phosphorylation of proteins involved in cell survival including PI3Ks, protein kinase B (also known as AKT kinase or AKT), and nuclear factor κ light chain enhancer of activated B cells (NF-κB) p65 was observed in ACE KO neutrophils as compared to WT neutrophils (Fig. 7B). We also found decreased expression of the antiapoptotic protein Mcl-1 and increased expression of the proapoptotic protein, B-cell lymphoma 2 (Bcl-2)-associated agonist of cell death (BAD), in ACE KO neutrophils as compared to WT neutrophils (Fig. 7C). In contrast, ACE overexpression enhanced survival-associated markers in NeuACE neutrophils, including increased phospho-AKT, phospho–NF-κB p65, and Mcl-1, and reduced expression of BAD. To verify the role of ACE and LTB4 in this signaling, similar experiments were conducted with the ACEI ramipril and LTB4 inhibitor zileuton. Inhibition of either ACE or LTB4 reverted the difference in survival and apoptotic markers between ACE KO, WT, and NeuACE neutrophils (Fig. 7, D and E).
Fig. 7. ACE expression enhances neutrophil survival.
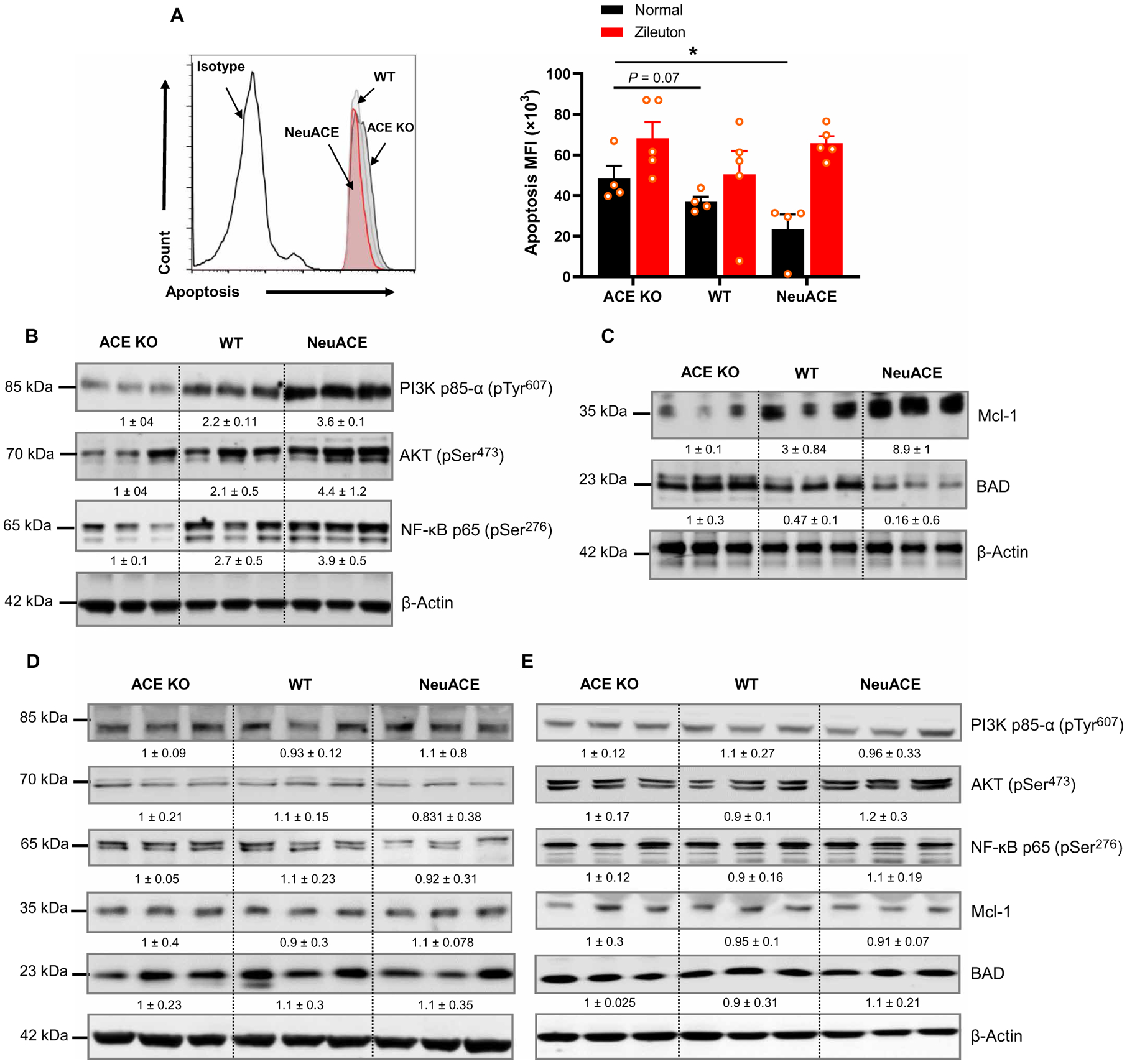
(A) Neutrophils isolated from ACE KO, WT, and NeuACE mice were stimulated with 1 μM LPS for 30 min followed by staining with Apopxin dye staining and flow cytometry to measure apoptosis. To inhibit LTB4, mice were treated with zileuton (5 mg/kg per day, gavage) for 2 days before isolation of bone marrow neutrophils, and then, cells were treated with 100 μM zileuton during the in vitro assay (n = 5 mice per group). (B) Western blots showing the presence of phospho-PI3K p85α, phospho-AKT, and phospho–NF-κB p65 are shown. (C) Western blots showing the presence of apoptotic markers MCL-1 and BAD. (D) Western blots show the abundance of phospho-PI3K p85α, phospho-AKT, phospho–NF-κB p65, Mcl-1, and BAD in neutrophils isolated from mice treated with ramipril. Mice were treated with ramipril (40 mg/liter) in drinking water for 7 days before bone marrow neutrophil isolation, and then, 10 μM ramipril was added in vitro. (E) Western blots show the abundance of phospho-PI3K p85α, phospho-AKT, phospho–NF-κB p65, Mcl-1, and BAD in neutrophils isolated from mice treated with zileuton. To inhibit LTB4 synthesis, mice were treated with zileuton (5 mg/kg per day) in drinking water for 2 days before neutrophil isolation. Zileuton was maintained at a concentration of 100 μM in vitro. For (B) to (E), neutrophils were treated with 1 μM LPS for 30 min before being lysed, and β-actin serves as the loading control. Numbers below bands indicate relative band density as determined by Image Studio Lite version 5.2 (LI-COR). A two-way ANOVA with Bonferroni’s correction for multiple comparisons was used to analyze group comparisons, and data are presented as means ± SEM. *P < 0.05.
ACE and human neutrophil function
Given the effect of ACEIs in mice, we examined the role of ACE in human neutrophil function with a clinical study in which seven healthy volunteers received 5 mg of ramipril daily administered as a 2.5-mg capsule twice a day (table S1). The first volunteer donated blood before the drug, at 5 and 10 days of drug administration and then after a 7-day washout without drug. All other participants provided blood before, after 7 days of ramipril treatment, and lastly after a 7-day washout. For participants 2 to 7, whole blood was tested for bacterial killing of MRSA, K. pneumoniae, and P. aeruginosa by measuring bacterial survival 2 and 5 hours after addition of bacteria to blood and incubation at 37°C (participant 1 was tested with only MRSA; Fig. 8A). Neutrophils were also purified to greater than 90% (fig. S6) and tested for intracellular killing of the three bacteria (Fig. 8B), LPS-stimulated intracellular ROS production (Fig. 8C), and the production of superoxide (Fig. 8D). These data show a consistent pattern of reduced neutrophil function in vitro when isolated during the course of ramipril treatment. For example, linear regression analysis of all whole-blood killing data (all participants, time points, and bacteria) indicates a significant reduction of killing caused by ramipril (P < 0.0001; tables S2 and S3). As indicated previously, this assay is known to be dependent on neutrophil function. A similar analysis showed that ramipril also significantly reduced intracellular bacterial killing (P < 0.0001; tables S4 and S5), superoxide production (P < 0.01), and cell total ROS production [P < 0.01 by analysis of variance (ANOVA)]. Thus, in humans as in mice, ACE inhibition degrades neutrophil antibacterial activity.
Fig. 8. Neutrophils isolated from humans treated with the ACEI ramipril are less effective at killing bacteria.
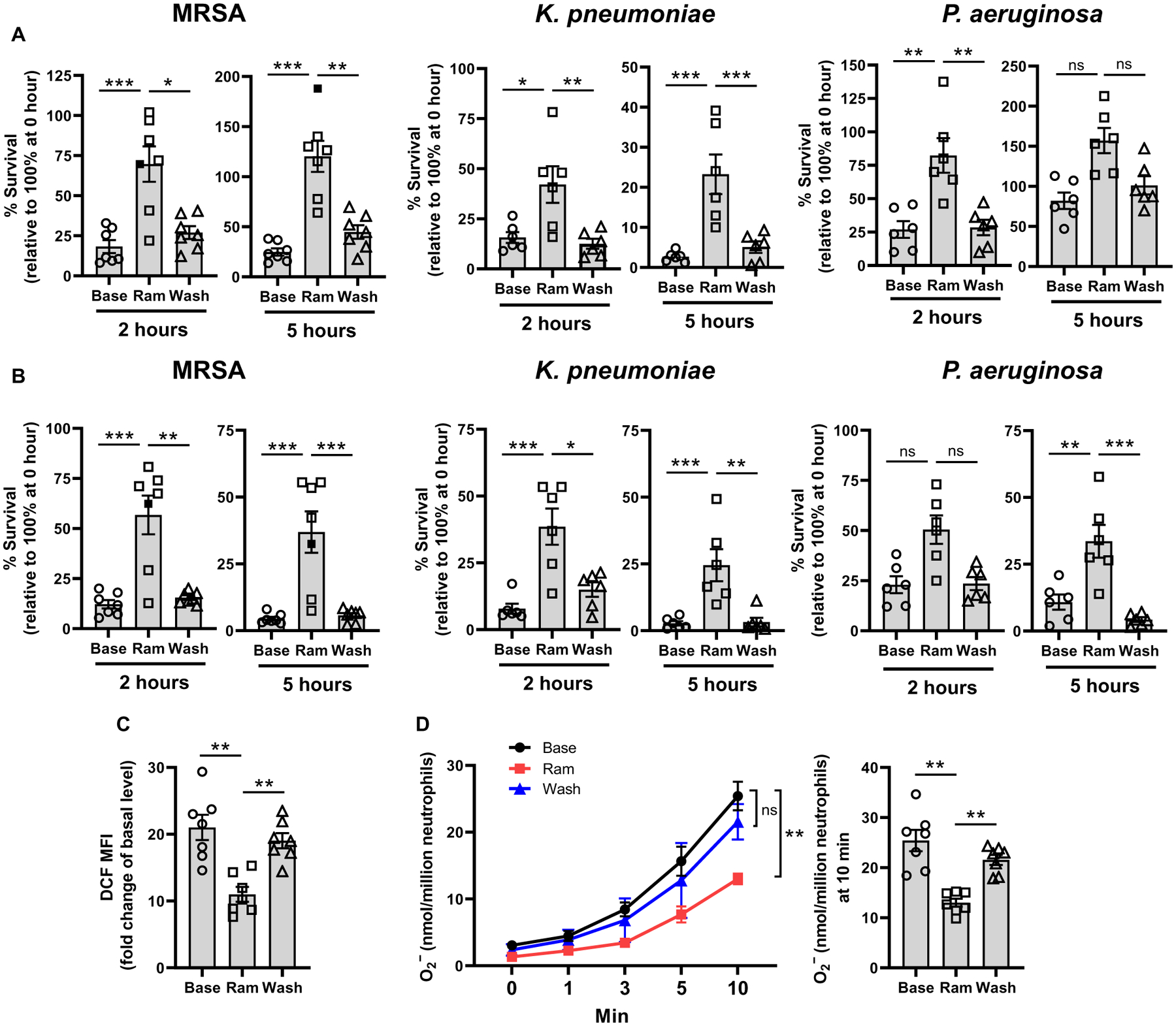
Blood was drawn from study participants before ramipril (Base), after ramipril administration (Ram), and after a 1-week washout (Wash). (A) After in vitro infection of blood with MRSA (about 1 × 106 CFU/ml), K. pneumoniae (about 1 × 106 CFU/ml), or P. aeruginosa (about 1 × 106 CFU/ml), samples were assessed for their ability to eliminate bacteria after 2- or 5-hour incubations relative to 0 hours. (B) In vitro intracellular killing of bacteria by purified neutrophils at 2 and 5 hours after infection with either MRSA, K. pneumoniae, or P. aeruginosa at an MOI of about 15 is shown. (C) Neutrophils were isolated from participants and stimulated with LPS (1 μg/ml), and ROS production was measured using the DCFDA Cellular ROS Assay Kit. The MFI of the ROS indicator, 2’,7’-dichlorodihydrofluorescein (DCF), is shown. (D) Superoxide was measured in neutrophils isolated from participants and stimulated with LPS (1 μg/ml) by cytochrome C reduction assay. Filled box represents the 10-day sample from participant no. 1. Mixed model linear regression, compound symmetry correlation matrix, and Bonferroni correction were used for statistical analyses. Data are presented as means ± SEM (n = 7 participants). *P < 0.05, **P < 0.01, and ***P < 0.001.
DISCUSSION
ACEIs and ARBs are used by millions of patients for the treatment of hypertension, cardiovascular disease, diabetes, and other conditions. Although a detailed analysis of the comparative advantages of each of these classes of drugs is beyond this discussion, several such comparisons are available (30–32). Here, our studies address the short-term effects of these drugs on innate immunity as measured by the effectiveness of neutrophil bacterial killing and resistance to a model of infective endocarditis. These data show that the two ACEIs evaluated caused a reduction of bacterial killing and increased risk of endocarditis in mice. Furthermore, in humans, short-term administration of an ACEI also reduced neutrophil ROS generation and caused poorer killing of three different strains of bacteria ex vivo. Thus, there was concordance between human and mouse neutrophil behavior. Our group has previously compared mice with normal ACE expression to animals genetically engineered to lack ACE expression (ACE KO mice) or with elevated expression of ACE by neutrophils (NeuACE mice) or macrophages (ACE 10/10 mice). Consistently, there was a direct relationship between ACE abundance (low, normal, and high) and measures of myeloid cell function such as ROS generation, bacterial killing, cytokine production, and even cellular concentrations of adenosine triphosphate. This suggests that increased ACE leads to increased immune function.
Particularly interesting is the finding that the mouse neutrophil response to infection is not affected by AT1 receptor blockade. Studies of mice genetically lacking angiotensinogen and other mice treated with either renin inhibitors or ARBs have repeatedly shown that the effect of ACE activity on neutrophil and macrophage function is not due to Ang II or any other angiotensin peptide (5–7, 33). Other experiments have shown that the ACE effect on myeloid cells is not mediated by bradykinin/bradykinin 2 receptor, substance P/neurokinin 1 receptor, or N-acetyl-seryl-aspartyl-lysyl-proline (Ac-SDKP). ACE is known to have over a hundred peptide substrates, but at present, it is only established that the substrate responsible for ACE-mediated immune effects appears to be specifically cleaved by the ACE C-domain (33, 34).
Neutrophils are an essential component of the innate immune response that act as a first line of host defense against invading pathogens (12). Recent data have changed the view of neutrophils from a homogeneous population that functions only to kill pathogens to a more complex picture with neutrophils as a heterogeneous population with a variety of functions, including shaping and activating both innate and adaptive immune responses (29). Neutrophil NOX2 is considered critical for antimicrobial host defenses because NOX2-generated superoxide and ROS not only kill pathogens in phagosomes but also activate other important antimicrobial mechanisms, including activation of granular proteases and generation of neutrophil extracellular traps (13, 35). Our results showed that, in both murine and human neutrophils, ACE inhibition reduced superoxide and ROS production, primarily because of the effects on p38-MAPK. Specifically, reduced p38-MAPK activity was observed in neutrophils after ACE inhibition, and measurement of ROS production and bacterial killing in the presence of the specific p38-MAPK inhibitor SB203580 confirmed the involvement of this kinase in ACE-mediated activation of NOX2.
During infections, neutrophils are the first cells to be recruited to an inflammation site in response to primary chemoattractants released by bacteria or necrotic cells (36). After reaching the inflammatory site, neutrophils secrete secondary chemoattractants to recruit additional leukocytes and amplify the inflammatory process (36). LTB4 is among the earliest secondary chemoattractants released and is a lipid mediator derived from the 5-LOX pathway of arachidonic acid metabolism (14, 15). It is the most potent endogenous chemoattractant for neutrophils and is a central player in neutrophil activation and initiation of inflammation (15, 37, 38). Studies with 5-LOX KO mice suggest that the recruitment of neutrophils to inflammatory sites is dependent on 5-LOX expression in neutrophils (39). Furthermore, it is reported that LTB4 acts as a signal relay molecule for neutrophils migrating toward inflammation (40). Our results show that ACE expression affects LTB4 production in neutrophils. ACE KO neutrophils make less LTB4, whereas NeuACE neutrophils produce more LTB4 as compared to WT neutrophils. By using the 5-LOX inhibitor zileuton, we found that LTB4 is a key molecule responsible for the biological functions of ACE in neutrophils. Specifically, the differences in neutrophil ROS production, MRSA killing, chemotaxis, and cytokine and chemokine production (including IL-1β) among ACE KO, WT, and NeuACE neutrophils were eliminated with zileuton. LTB4 signaling is mediated by two GTP-binding protein–coupled receptor families, BLT1 and BLT2. BLT1 is a high-affinity receptor LTB4 mainly expressed on leukocytes, whereas BLT2 is a low-affinity receptor for LTB4 expressed ubiquitously, with an increased expression in lymphoid organs (14). By using the BLT1 receptor antagonist U-75302, we found that the biological function of ACE in neutrophils is mediated by this receptor.
It is now known that neutrophil life span increases from a few hours to several days in many infectious and inflammatory conditions (41, 42). LTB4 protects neutrophils from spontaneous apoptosis by promoting mitochondrial integrity and activating cell survival pathways including MAPK, NF-κB, and PI3K/AKT, leading to a longer immune response (14–18). Flow cytometry data showed increased apoptosis in ACE KO neutrophils as compared to WT after activation with phorbol 12-myristate 13-acetate. Furthermore, Western blotting results showed decreased expression of the anti-apoptotic molecule, myeloid cell leukemia 1 protein (MCL-1), and increased expression of the proapoptotic molecule, BAD. In contrast, activation of survival signaling mediated by PI3K/AKT, p38-MAPK, and NF-κB was found to be lower in ACE KO neutrophils as compared WT neutrophils. ACE overexpression further increased survival signaling in NeuACE neutrophils as compared to WT. To verify the role of LTB4, these signals were also measured in the presence of zileuton, which abolished differences between ACE KO, WT, and NeuACE neutrophils.
Our study has several limitations. First, although we have studied human volunteers administered an ACEI, we did not perform an equivalent human study with an ARB. This was based on the results of extensive mouse analysis in which neither the genetic elimination of angiotensinogen nor the administration of a renin inhibitor or an ARB affected myeloid function. Nonetheless, until a human clinical study is performed, we cannot be certain whether an ARB will reduce human neutrophil function similar to the effect observed with an ACEI. Second, we studied human neutrophil function after 1 week of ACEI administration. Patients on ACEIs usually take these drugs for extended periods, and we do not know what effect extended drug administration will have. Last, ACE is a peptidase, and we do not know what precise peptide product of ACE is being blocked by an ACEI to cause deterioration of neutrophil function. Identifying this peptide may reveal a new means of manipulating myeloid cell function.
ACEIs and ARBs are used by millions of patients, some of whom are immunocompromised and are vulnerable to infection. Our findings suggest that ACE appears to be important for effective human neutrophil antibacterial activity. Thus, physicians might consider patient immune status when selecting between these two classes of drugs.
MATERIALS AND METHODS
Study design
All figures indicate the number of animals reported. We typically use animal groups of 6 to 10 animals based on the typical effect size of the responses that we have observed. Mice from our colony are randomly distributed to individual groups. Where possible, the observer was blinded to identification of the group. For human studies, all procedures were approved by the Cedars-Sinai Medical Center Institutional Review Board (IRB). All volunteers were healthy males and ranged in age from 31 to 69 years, with no prior history of ACEI therapy (table S1). Women of reproductive age were not permitted by the IRB. The participants were treated with 2.5 mg of ramipril twice a day for 1 week. Blood was drawn before drug administration, at the end of drug administration, and then after a 1-week washout without drug. The first volunteer was treated with ramipril for 10 days with blood draw at day 10. Neutrophils were isolated from whole blood using the EasySep Direct Human Neutrophil Isolation Kit (STEMCELL) as per the manufacturer’s instructions.
Mice
All animal experimental protocols were approved by the Cedars-Sinai Institutional Animal Care and Usage Committee. Animals were maintained on a 12-hour light/12-hour dark cycle, and both food and water were freely available. ACE KO and NeuACE mice have been described previously (3, 6). NeuACE transgenic mice expressed 10- to 12-fold higher ACE in neutrophils as compared to WT mice from our colony (6). These mice are on a C57BL/6 genetic background. All mice were 10 to 14 weeks old. Both male and female mice were used, and no phenotypic differences were noted.
Bacterial strains
MRSA (strain LAC, US300), K. pneumoniae [American Type Culture Collection (ATCC), 10031], and P. aeruginosa (ATCC, 35032) have been previously described (6, 43, 44). MRSA was grown in tryptic soy broth (BD) (45). LB (BD Biosciences) was used for K. pneumoniae and P. aeruginosa (46, 47). All bacterial strains were grown at 37°C shaking overnight. For late log phase culture, an overnight bacterial culture was diluted 1:100 in fresh media and was shaken for 4 hours at 250 rpm and 37°C.
Inhibitor and blocker treatment
Mice were treated either with the ACEI ramipril (40 mg/liter; Roxane Laboratories) or lisinopril (40 mg/liter; LKT Laboratories) or the ARB losartan (600 mg/liter; LKT Laboratories) in drinking water for 7 days before MRSA infection in vivo (6, 33). Drugs were maintained throughout the study, and the drug effectiveness was confirmed by tail cuff measurement of blood pressure (6, 33).
To investigate the role of the LTB4-BLT1R axis in ACE-mediated bacterial resistance, mice were treated with either the 5-LOX inhibitor zileuton to inhibit LTB4 synthesis (5 mg/kg, orally via gavage, twice daily; Cayman Chemicals) (48, 49) or the BLT1R antagonist U-75302 (1 mg/kg per day, i.p.; Cayman Chemicals) (50). These treatments were initiated 2 days before MRSA infection and maintained until the termination of the experiment. To study the effect of ACE on LTB4 production in vivo, ACE KO, WT, and NeuACE mice were challenged intraperitoneally with LPS (750 μg/kg body weight; MilliporeSigma) (44), and after 4 hours, serum LTB4 was measured by ELISA as described below. For in vitro experiments, the following inhibitors were added at the indicated concentrations to the cell culture: 10 μM ramipril (Roxane Laboratories), 10 μM lisinopril (LKT Laboratories), 100 μM losartan (LKT Laboratories), 100 μM zileuton (Cayman Chemicals) (48, 51), 10 μM U-75302 (Cayman Chemicals) (52), and 500 nM p38-MAPK inhibitor SB203580 (Tocris Bioscience).
In vivo MRSA infection
Bacteria were washed twice and resuspended in PBS. Bacteria were adjusted to the desired concentrations by absorbance assuming an optical density at 600 nm (OD600) of 0.3 was equivalent to 1 × 108 CFU/ml. Bacterial numbers used in each experiment were confirmed by plating. For systemic infection, about 1 × 108 CFU of MRSA was injected intravenously into mice through the retro-orbital vein and, after 24 and 48 hours, bacteria were measured in blood and tissues after homogenization in 500 μl of PBS, as determined by bacterial CFU counts on agar plates after serial dilutions.
Infective endocarditis mouse model
To study infective endocarditis, we used an aortic valve injury mouse model as described previously with some modifications (43). Briefly, aortic valves in mice were damaged using a 32-gauge polyurethane tubing: A 2-cm-long tubing was prepared, and the distal end was sealed with melted wax. Tubing was marked 1.5 cm from the distal end because this is the optimal length to be inserted through the right carotid artery in 10-week-old mice to damage the aortic valve. A 2.5-cm-long wire stylet was inserted into the proximal open end of the tube, which provides stiffness and helps in inserting the tube into the artery. Mice were anesthetized with isoflurane and kept in a dorsal recumbent position on a surgery platform with continuous isoflurane inhalational anesthesia. A small incision was made in the right neck and with the aid of a surgical microscope, and the right carotid artery was isolated and ligated with a 6–0 silk suture. The tubing was inserted from the distal tip into the artery through a hole made by a 27-gauge needle and fed retrograde until the 1.5-cm mark. After that, the guide wire stylet was removed, and the catheter was secured in place by two 6–0 silk sutures. The incision was closed with a 5–0 Prolene suture, and the mice were allowed to recover. Catheters remained in place throughout the study. After 24 hours of catheterization, mice were challenged with a low dose of MRSA (about 107 CFU in 100 μl of PBS) through intravenous inoculation via the retro-orbital vein. At 24 hours after infection, mice were anesthetized with inhalational isoflurane and perfused with PBS, and hearts were collected for measuring bacterial burden and infective endocarditis. For bacterial count, hearts were homogenized in 500 μl of PBS, and bacterial CFU was determined on agar plates after serial dilutions. For infective endocarditis, mice hearts were fixed with 10% formalin and embedded in paraffin. Positive or negative cases of infective endocarditis were identified on the basis of vegetative bacterial growth in hearts as observed after standard crystal violet Gram staining in Cedars-Sinai hospital laboratories. To determine survival, catheterized mice were kept under observation for 3 days after MRSA challenge.
Ex vivo blood killing assay
Blood was drawn via retro-orbital vein and mixed with either MRSA, K. pneumoniae, or P. aeruginosa (about 1 × 106 CFU/ml) in a sterile heparinized 2-ml round-bottom Eppendorf tube. Tubes were incubated in a shaker at 37°C. The samples were subsequently diluted 10-fold serially and quantitatively plated on tryptic soy agar plates at 2 and 5 hours. Bacterial killing was measured by counting CFUs after an overnight incubation (6).
Intracellular killing
To measure intracellular killing of MRSA, bone marrow neutrophils (mice) or blood neutrophils (humans) were purified with a neutrophil isolation kit (STEMCELL) and infected with either MRSA, K. pneumoniae, or P. aeruginosa at an MOI of 15 for 30 min. Gentamycin was then added to kill extracellular bacteria (0 time) (6). Survival of phagocytosed bacteria was determined by counting CFUs after incubation for the indicated times.
Tissue collection and homogenization
After induction of deep anesthesia with cotton soaked in isoflurane, mice were perfused with PBS and tissues such as liver (100 mg), lung (100 mg), spleen (whole), and heart (whole) were collected in 1.5-ml Eppendorf tube containing 500 μl of PBS. Then, tissues were chopped with scissors and homogenized manually using the rubber-coated end of a 1-ml syringe plunger. Bacterial CFU was determined on agar plates after serial dilutions.
Bone marrow neutrophil isolation
Mice were euthanized with 2% isoflurane followed by cervical dislocation, and the femur and the tibia from both hind legs were removed and freed of soft tissue attachments. Then, the distal tip of each bone was removed, and marrow cavities were flushed in RPMI 1640 media with 10% fetal bovine serum (FBS). Neutrophils were isolated by centrifugation over a discontinuous Percoll gradient consisting of 36% (v/v) and 72% (v/v) Percoll in PBS at 700g for 20 min at 25°C. Neutrophils were recovered at the interface of the 36 and 72% fractions with >85% purity and viability as determined by flow cytometry (6). For in vitro cytokine and chemotaxis analysis, bone marrow neutrophils were purified with a mouse neutrophil isolation kit (STEMCELL) following the manufacturer’s protocol. This population consisted of more than 90% neutrophils as measured by flow cytometry (fig. S7).
In vivo neutrophil depletion
Mice were made neutropenic as described previously (6, 20). Briefly, neutrophils were depleted by intraperitoneal injection of 100 μl of rabbit polyclonal anti-mouse polymorphonuclear (PMN) antibody (2.5 mg/ml; Cedarlane Labs) 1 day before MRSA subcutaneous infection. One injection every 24 hours was continued until the termination of experiments. This dose showed about 90% neutrophil depletion 1 day after the first dose of anti-mouse PMN antibody (6).
Measurement of ROS and superoxide
For measurement of ROS, 1 × 106 cells were resuspended in RPMI 1640 with 1% FBS. Cells were stained with 20 μM dichlorodihydrofluorescein diacetate dye (DCFDA; Abcam) and incubated for 30 min at 37°C. By interacting with ROS, DCFDA oxidized to fluorescent 2’,7’-dichlorodihydrofluorescein (DCF). Cells were treated with LPS (1 μg/ml) for 30 min and then stained with mouse or human neutrophil-specific antibodies: anti-mouse CD11b antibody [clone M1/70; phycoerythrin (PE)–Cy7 conjugated; 0.2 μg of antibody per 106 cells in 100 μl of fluorescence-activated cell sorting (FACS) buffer; BioLegend] and anti-mouse Ly6G antibody (clone RB6–8C5; Pacific Blue conjugated, 1.0 μg per 106 cells in 100 μl of FACS buffer; BioLegend) or anti-human CD11b (clone ICRF44; PE conjugated, 0.5 μg per 106 cells in 100 μl of FACS buffer; Thermo Fisher Scientific) and anti-human CD66b (clone G10F5; allophycocyanin conjugated, 0.12 μg per 106 cells in 100 μl of FACS buffer; Thermo Fisher Scientific) at 4°C for 30 min. Cells were washed with FACS buffer (PBS with 2% FBS, 0.1% sodium azide, and 1 mM EDTA) and analyzed by a flow cytometer (Sony SA38000).
Superoxide production was measured by a superoxide dismutase (SOD)–inhibitable cytochrome C reduction assay (6). Cells (1 × 106) were resuspended with 1 ml of media H [145 mM NaCl, 5 mM KCl, 1 mM MgCl2, 0.8 mM CaCl2, 10 mM Hepes, and 5 mM glucose (pH 7.4)] containing 100 μM cytochrome C (MilliporeSigma). As a negative control, 15 μg of SOD (MilliporeSigma) was added. A total of 200 μl of the cell suspension was put in wells of a 96-well plate and stimulated with LPS (1 μg/ml) at room temperature to induce superoxide production. Absorbance at 550 nm was recorded for 10 min with one measurement per minute at 37°C with gentle shaking. After subtracting the background values, superoxide production was calculated by using the absorption coefficient of 21 mM−1 cm−1 for cytochrome C.
Apoptosis analysis
For apoptosis analysis, 1 × 106 cells were isolated and resuspended in 1% FBS containing RPMI 1640. Cells were treated with 1 μM LPS for 30 min. After treatment, cells were centrifuged at 500g for 5 min at 4°C. After discarding the supernatant, cells were resuspended in 200 μl of assay buffer (Abcam), 2 μl of Apopxin green indicator (100× stock) to measure apoptotic cells, and 1 μl of CytoCalcein-450 (200× stock) to measure healthy cells. Then, cells were incubated at room temperature for 40 min without washing, and the binding of Apopxin green indicator was quantified at excitation/emission = 490/525 nm and CytoCalcein-Violet 450 at excitation/emission = 405/450 nm by flow cytometry (Sony SA38000).
Chemotaxis assay
Neutrophil chemotaxis was determined using a Transwell assay, as described previously (53). To inhibit LTB4 synthesis, groups of mice were treated with zileuton (5 mg/kg per day, gavage) for 2 days before isolation of bone marrow neutrophils, and the drug was added at a concentration of 100 μM during the in vitro assay. Neutrophils were resuspended at a concentration of 1.5 × 106 cells/ml in RPMI 1640. A total of 200 μl of the cell suspension was plated on top of the filter membrane in a transwell insert (3-μm pore size; Corning) in triplicate and incubated for 10 min at 37°C and 5% CO2 to allow the cells to settle down. Then, 700 μl of RPMI 1640 with 100 nM fMLP (MilliporeSigma) was added into the bottom of the lower chamber in a 24-well plate, and the insert was very carefully placed into the media. Chambers were then incubated for 3 hours at 37°C in a 5% CO2 incubator, and cells were counted on the lower part of the transwell and fully transmigrated into the lower solution in a 24-well plate. For counting cells in the lower chamber, remaining media and cells that had not migrated from the top of the membrane were removed with a cotton-tipped applicator. Then, migrated cells on the undersurface of the insert were fixed in methanol and stained with crystal violet for cell counting. Four random pictures per chamber per condition (in triplicate) were taken with a microscope at ×10 magnification (Life Technologies, EVOS FL Auto), and cells were counted using ImageJ software (public domain, BSD2). For fully migrated cell counting, medium from the lower well was collected, and transmigrated cells were counted by an automated cell counter (TC10, Bio-Rad).
Measurement of LTB4 concentration, kinase activation, and cytokines by ELISA
LTB4 concentration was measured by the R&D Systems LTB4 Parameter Assay Kit (catalog no. KGE006B) following the manufacturer’s instructions. Neutrophils were cultured in RPMI 1640 media (3 × 106 cells/ml) and challenged with either MRSA (at an MOI of about 15) or LPS (500 ng/ml). After incubation at 37°C in a CO2 incubator for the indicated time, cells and supernatant were collected for the assay. To inhibit ACE and AT1 receptor, mice were treated with ramipril (40 mg/liter) or losartan (600 mg/liter) in drinking water for 7 days before isolation of bone marrow neutrophils. Ramipril and losartan were maintained at a concentration of 10 and 100 μM in wells during the in vitro assay. To measure LTB4 concentration in vivo, mice were challenged with either MRSA (at about 1 × 108 CFU/100 μl, i.v.) or LPS (750 μg/kg body weight, i.p.), and blood was collected at the indicated time in heparinized Eppendorf tubes after decapitation of mice (44). After centrifugation, serum was collected for LTB4 ELISA.
To measure activation of kinases by ELISA, neutrophils were resuspended in RPMI 1640 and stimulated with LPS (1 μg/ml) for 30 min at room temperature. Then, cells were pelleted and lysed in radioimmunoprecipitation assay (RIPA) buffer containing protease and phosphatase inhibitors (Thermo Fisher Scientific, catalog no. 78442). The activity of PKC isoforms was measured using a PKC Kinase Activity Assay Kit (Abcam, ab139437) as per the manufacturer’s instructions. This kit uses a specific synthetic peptide as a substrate for PKC and a polyclonal antibody that recognizes the phosphorylated form of the substrate. The active form of p38-MAPK was measured by a PathScan Phospho-p38 MAPK (Thr180/Tyr182) Sandwich ELISA Kit (Cell Signaling Technology, catalog no. 7946). These assays were performed using the manufacturer’s protocols. Both kits use sandwich ELISA in which horseradish peroxidase–conjugated secondary antibody is used for the detection of phospho-specific primary antibody. At the end, 3,3′,5,5′-tetramethylbenzidine substrate solution was added, and changes in color were measured as OD450.
We used a commercial solid-phase ELISA kit protocol for measuring mouse MIP-2 and IL-1β (Invitrogen, Thermo Fisher Scientific). Bone marrow neutrophils were purified with a neutrophil isolation kit (STEMCELL) and cultured in 24-well plates (5 × 106 cells/ml) in RPMI 1640 media. Cells were stimulated with 100 nM fMLP for 12 hours; supernatants and cells were collected and lysed with RIPA buffer, and total MIP-2 and IL-1β (cell plus supernatant) were measured.
Western blotting analyses
Cells were washed twice with cold PBS and lysed with RIPA buffer containing protease and phosphatase inhibitors (Thermo Fisher Scientific, catalog no. 78442). For membrane protein isolation, we used a Membrane Protein Extraction Kit (Thermo Fisher Scientific, catalog no. 89842). The polyvinylidene difluoride membranes were incubated with MCL-1 rabbit monoclonal antibody (mAb) (Cell Signaling Technology, catalog no. 94296), NF-κB p65 rabbit mAb (Invitrogen, catalog no. 33–9900), BAD rabbit mAb (Cell Signaling Technology, catalog no. 9239), PI3K p85-α mouse mAb (Cell Signaling Technology, catalog no. 13666), AKT mAb (Cell Signaling Technology, catalog no. 9271), ACE mouse mAb (R&D Systems, catalog no. AF1513), p38-MAPK rabbit mAb (Invitrogen, catalog no. MA5–15177), p47-phox (pSer345) rabbit mAb (Sigma-Aldrich, catalog no. SAB4504721), and β-actin mouse mAb (Sigma-Aldrich, catalog no. A3854). Protein bands were measured using an Odyssey Infrared Imaging System (ODYSSEY CLx, Li-COR). The fluorescence intensity was evaluated using Image Studio Lite version 5.2. All Western blots are shown in fig. S8.
Statistical analyses
Data are shown as individual measurements along with the group mean ± SEM. For mouse data, a one-way ANOVA or a two-way ANOVA with a Bonferroni correction for multiple comparisons was used to analyze differences between groups. For analysis of human studies, the percent bacteria that survived at 2 and 5 hours of the assay were calculated relative to the 0-hour assay time point using mixed model linear regression to test for differences over assay time, bacterial strain, and participant’s neutrophil sampling treatment time point. Observations were nested by participant with a compound symmetry correlation matrix. Given the presence of outliers, human data were log-transformed before analysis. Residuals were inspected to confirm the overall fit of the model. For all testing, differences were considered significant where the two-tailed P value was <0.05. Analysis was performed with GraphPad Prism 8.4 (GraphPad Software) or SAS version 9.4 software. Superoxide data were analyzed by two-way ANOVA, whereas ROS was analyzed by one-way ANOVA using GraphPad.
Supplementary Material
Acknowledgments:
We thank M. Katsumata (Mouse Genetics Core, Cedars-Sinai Medical Center, Los Angeles) for help in generating NeuACE transgenic mice and M. Bautista for help with recruitment of volunteers for the clinical study. We wish to thank B. Taylor for excellent administrative support.
Funding:
This study was supported by AHA grant 19CDA34760010 (to Z.K.) and NIH grants P01HL129941 (to K.E.B.), R01AI134714 (to K.E.B.), and R01HL142672 (to J.F.G.).
Footnotes
SUPPLEMENTARY MATERIALS
Competing interests: The authors declare that they have no competing interests.
Data and materials availability:
All data associated with this study are present in the paper or the Supplementary Materials.
REFERENCES AND NOTES
- 1.Bernstein KE, Ong FS, Blackwell W-LB, Shah KH, Giani JF, Gonzalez-Villalobos RA, Shen XZ, Fuchs S, Touyz RM, A modern understanding of the traditional and nontraditional biological functions of angiotensin-converting enzyme. Pharmacol. Rev 65, 1–46 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bernstein KE, Khan Z, Giani JF, Cao D-Y, Bernstein EA, Shen XZ, Angiotensin-converting enzyme in innate and adaptive immunity. Nat. Rev. Nephrol 14, 325–336 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lin C, Datta V, Okwan-Duodu D, Chen X, Fuchs S, Alsabeh R, Billet S, Bernstein KE, Shen XZ, Angiotensin-converting enzyme is required for normal myelopoiesis. FASEB J. 25, 1145–1155 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ruiz-Ortega M, Bustos C, Hernández-Presa MA, Lorenzo O, Plaza JJ, Egido J, Angiotensin II participates in mononuclear cell recruitment in experimental immune complex nephritis through nuclear factor-κB activation and monocyte chemoattractant protein-1 synthesis. J. Immunol 161, 430–439 (1998). [PubMed] [Google Scholar]
- 5.Shen XZ, Li P, Weiss D, Fuchs S, Xiao HD, Adams JA, Williams IR, Capecchi MR, Taylor WR, Bernstein KE, Mice with enhanced macrophage angiotensin-converting enzyme are resistant to melanoma. Am. J. Pathol 170, 2122–2134 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Khan Z, Shen XZ, Bernstein EA, Giani JF, Eriguchi M, Zhao TV, Gonzalez-Villalobos RA, Fuchs S, Liu GY, Bernstein KE, Angiotensin-converting enzyme enhances the oxidative response and bactericidal activity of neutrophils. Blood 130, 328–339 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Okwan-Duodu D, Datta V, Shen XZ, Goodridge HS, Bernstein EA, Fuchs S, Liu GY, Bernstein KE, Angiotensin-converting enzyme overexpression in mouse myelomonocytic cells augments resistance to Listeria and methicillin-resistant Staphylococcus aureus. J. Biol. Chem 285, 39051–39060 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Okamura A, Rakugi H, Ohishi M, Yanagitani Y, Takiuchi S, Moriguchi K, Fennessy PA, Higaki J, Ogihara T, Upregulation of renin-angiotensin system during differentiation of monocytes to macrophages. J. Hypertens 17, 537–545 (1999). [DOI] [PubMed] [Google Scholar]
- 9.Danilov SM, Sadovnikova E, Scharenborg N, Balyasnikova IV, Svinareva DA, Semikina EL, Parovichnikova EN, Savchenko VG, Adema GJ, Angiotensin-converting enzyme (CD143) is abundantly expressed by dendritic cells and discriminates human monocyte-derived dendritic cells from acute myeloid leukemia-derived dendritic cells. Exp. Hematol 31, 1301–1309 (2003). [DOI] [PubMed] [Google Scholar]
- 10.Baudin B, New aspects on angiotensin-converting enzyme: From gene to disease. Clin. Chem. Lab. Med 40, 256–265 (2002). [DOI] [PubMed] [Google Scholar]
- 11.Iannuzzi MC, Rybicki BA, Teirstein AS, Sarcoidosis. N. Engl. J. Med 357, 2153–2165 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Kolaczkowska E, Kubes P, Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol 13, 159–175 (2013). [DOI] [PubMed] [Google Scholar]
- 13.Amulic B, Cazalet C, Hayes GL, Metzler KD, Zychlinsky A, Neutrophil function: From mechanisms to disease. Annu. Rev. Immunol 30, 459–489 (2012). [DOI] [PubMed] [Google Scholar]
- 14.Saeki K, Yokomizo T, Identification, signaling, and functions of LTB4 receptors. Semin. Immunol 33, 30–36 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Colom B, Bodkin JV, Beyrau M, Woodfin A, Ody C, Rourke C, Chavakis T, Brohi K, Imhof BA, Nourshargh S, Leukotriene B4-neutrophil elastase axis drives neutrophil reverse transendothelial cell migration in vivo. Immunity 42, 1075–1086 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barcellos-de-Souza P, Canetti C, Barja-Fidalgo C, Arruda MA, Leukotriene B4 inhibits neutrophil apoptosis via NADPH oxidase activity: Redox control of NF-κB pathway and mitochondrial stability. Biochim. Biophys. Acta 1823, 1990–1997 (2012). [DOI] [PubMed] [Google Scholar]
- 17.Mancuso P, Nana-Sinkam P, Peters-Golden M, Leukotriene B4 augments neutrophil phagocytosis of Klebsiella pneumoniae. Infect. Immun 69, 2011–2016 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pétrin D, Turcotte S, Gilbert AK, Rola-Pleszczynski M, Stankova J, The anti-apoptotic effect of leukotriene B4 in neutrophils: A role for phosphatidylinositol 3-kinase, extracellular signal-regulated kinase and Mcl-1. Cell. Signal 18, 479–487 (2005). [DOI] [PubMed] [Google Scholar]
- 19.Cole J, Quach DL, Sundaram K, Corvol P, Capecchi MR, Bernstein KE, Mice lacking endothelial angiotensin-converting enzyme have a normal blood pressure. Circ. Res 90, 87–92 (2002). [DOI] [PubMed] [Google Scholar]
- 20.Kyme P, Thoennissen NH, Tseng CW, Thoennissen GB, Wolf AJ, Shimada K, Krug UO, Lee K, Müller-Tidow C, Berdel WE, Hardy WD, Gombart AF, Koeffler HP, Liu GY, C/EBPε mediates nicotinamide-enhanced clearance of Staphylococcus aureus in mice. J. Clin. Invest 122, 3316–3329 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun J-Z, Cao L-H, Liu H, ACE inhibitors in cardiac surgery: Current studies and controversies. Hypertens. Res 34, 15–22 (2011). [DOI] [PubMed] [Google Scholar]
- 22.Ding Q, Zhang Z, Liu H, Nie H, Berguson M, Goldhammer JE, Young N, Boyd D, Morris R, Sun J, Perioperative use of renin-angiotensin system inhibitors and outcomes in patients undergoing cardiac surgery. Nat. Commun 10, 4202 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klinkhammer B, Renin-angiotensin system blockade after transcatheter aortic valve replacement (TAVR) improves intermediate survival. J. Cardiovasc. Thorac. Res 11, 176–181 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ardehali R, Leeper NJ, Wilson AM, Heidenreich PA, The effect of angiotensin-converting enzyme inhibitors and statins on the progression of aortic sclerosis and mortality. J. Heart Valve Dis 21, 337–343 (2012). [PMC free article] [PubMed] [Google Scholar]
- 25.El-Benna J, Dang PM-C, Gougerot-Pocidalo M-A, Marie J-C, Braut-Boucher F, p47phox, the phagocyte NADPH oxidase/NOX2 organizer: Structure, phosphorylation and implication in diseases. Exp. Mol. Med 41, 217–225 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dang PM-C, Stensballe A, Boussetta T, Raad H, Dewas C, Kroviarski Y, Hayem G, Jensen ON, Gougerot-Pocidalo M-A, El-Benna J, A specific p47phox-serine phosphorylated by convergent MAPKs mediates neutrophil NADPH oxidase priming at inflammatory sites. J. Clin. Invest 116, 2033–2043 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sánchez-Galán E, Gómez-Hernández A, Vidal C, Martín-Ventura JL, Blanco-Colio LM, Muñoz-García B, Ortega L, Egido J, Tuñón J, Leukotriene B4 enhances the activity of nuclear factor-κB pathway through BLT1 and BLT2 receptors in atherosclerosis. Cardiovasc. Res 81, 216–225 (2009). [DOI] [PubMed] [Google Scholar]
- 28.Tong W-G, Ding X-Z, Talamonti MS, Bell RH, Adrian TE, LTB4 stimulates growth of human pancreatic cancer cells via MAPK and PI-3 kinase pathways. Biochem. Biophys. Res. Commun 335, 949–956 (2005). [DOI] [PubMed] [Google Scholar]
- 29.Silvestre-Roig C, Hidalgo A, Soehnlein O, Neutrophil heterogeneity: Implications for homeostasis and pathogenesis. Blood 127, 2173–2181 (2016). [DOI] [PubMed] [Google Scholar]
- 30.Omboni S, Volpe M, Angiotensin receptor blockers versus angiotensin converting enzyme inhibitors for the treatment of arterial hypertension and the role of Olmesartan. Adv. Ther 36, 278–297 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Potier L, Roussel R, Elbez Y, Marre M, Zeymer U, Reid CM, Ohman M, Eagle KA, Bhatt DL, Steg PG; REACH Registry Investigators, Angiotensin-converting enzyme inhibitors and angiotensin receptor blockers in high vascular risk. Heart 103, 1339–1346 (2017). [DOI] [PubMed] [Google Scholar]
- 32.Bangalore S, Kumar S, Wetterslev J, Messerli FH, Angiotensin receptor blockers and risk of myocardial infarction: Meta-analyses and trial sequential analyses of 147 020 patients from randomised trials. BMJ 342, d2234 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khan Z, Cao D-Y, Giani JF, Bernstein EA, Veiras LC, Fuchs S, Wang Y, Peng Z, Kalkum M, Liu GY, Bernstein KE, Overexpression of the C-domain of angiotensin-converting enzyme reduces melanoma growth by stimulating M1 macrophage polarization. J. Biol. Chem 294, 4368–4380 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cao D-Y, Spivia WR, Veiras LC, Khan Z, Peng Z, Jones AE, Bernstein EA, Saito S, Okwan-Duodu D, Parker SJ, Giani JF, Divakaruni AS, van Eyk JE, Bernstein KE, ACE overexpression in myeloid cells increases oxidative metabolism and cellular ATP. J. Biol. Chem 295, 1369–1384 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bedard K, Krause K-H, The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev 87, 245–313 (2007). [DOI] [PubMed] [Google Scholar]
- 36.McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CCM, Beck PL, Muruve DA, Kubes P, Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science 330, 362–366 (2010). [DOI] [PubMed] [Google Scholar]
- 37.Ford-Hutchinson AW, Bray MA, Doig MV, Shipley ME, Smith MJH, Leukotriene B, a potent chemokinetic and aggregating substance released from polymorphonuclear leukocytes. Nature 286, 264–265 (1980). [DOI] [PubMed] [Google Scholar]
- 38.Martin TR, Pistorese BP, Chi EY, Goodman RB, Matthay MA, Effects of leukotriene B4 in the human lung. Recruitment of neutrophils into the alveolar spaces without a change in protein permeability. J. Clin. Invest 84, 1609–1619 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen M, Lam BK, Kanaoka Y, Nigrovic PA, Audoly LP, Austen KF, Lee DM, Neutrophil-derived leukotriene B4 is required for inflammatory arthritis. J. Exp. Med 203, 837–842 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Afonso PV, Janka-Junttila M, Lee YJ, McCann CP, Oliver CM, Aamer KA, Losert W, Cicerone MT, Parent CA, LTB4 is a signal-relay molecule during neutrophil chemotaxis. Dev. Cell 22, 1079–1091 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sabroe I, Prince LR, Jones EC, Horsburgh MJ, Foster SJ, Vogel SN, Dower SK, Whyte MKB, Selective roles for Toll-like receptor (TLR)2 and TLR4 in the regulation of neutrophil activation and life span. J. Immunol 170, 5268–5275 (2003). [DOI] [PubMed] [Google Scholar]
- 42.Colotta F, Re F, Polentarutti N, Sozzani S, Mantovani A, Modulation of granulocyte survival and programmed cell death by cytokines and bacterial products. Blood 80, 2012–2020 (1992). [PubMed] [Google Scholar]
- 43.Gibson GW, Kreuser SC, Riley JM, Rosebury-Smith WS, Courtney CL, Juneau PL, Hollembaek JM, Zhu T, Huband MD, Brammer DW, Brieland JK, Sulavik MC, Development of a mouse model of induced Staphylococcus aureus infective endocarditis. Comp. Med 57, 563–569 (2007). [PubMed] [Google Scholar]
- 44.Zhao J, Bi W, Xiao S, Lan X, Cheng X, Zhang J, Lu D, Wei W, Wang Y, Li H, Fu Y, Zhu L, Neuroinflammation induced by lipopolysaccharide causes cognitive impairment in mice. Sci. Rep 9, 5790 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Müller S, Wolf AJ, Iliev ID, Berg BL, Underhill DM, Liu GY, Poorly cross-linked peptidoglycan in MRSA due to mecA induction activates the inflammasome and exacerbates immunopathology. Cell Host Microbe 18, 604–612 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lavigne J-P, Cuzon G, Combescure C, Bourg G, Sotto A, Nordmann P, Virulence of Klebsiella pneumoniae isolates harboring bla KPC-2 carbapenemase gene in a Caenorhabditis elegans model. PLOS ONE 8, e67847 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Aybey A, Demirkan E, Inhibition of quorum sensing-controlled virulence factors in Pseudomonas aeruginosa by human serum paraoxonase. J. Med. Microbiol 65, 105–113 (2016). [DOI] [PubMed] [Google Scholar]
- 48.Rossi A, Pergola C, Koeberle A, Hoffmann M, Dehm F, Bramanti P, Cuzzocrea S, Werz O, Sautebin L, The 5-lipoxygenase inhibitor, zileuton, suppresses prostaglandin biosynthesis by inhibition of arachidonic acid release in macrophages. Br. J. Pharmacol 161, 555–570 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Abueid L, Uslu Ü, Cumbul A, Velioğlu Öğünç A, Ercan F, Alican İ, Inhibition of 5-lipoxygenase by zileuton in a rat model of myocardial infarction. Anatol. J. Cardiol 17, 269–275 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sun M, Wang R, Han Q, Inhibition of leukotriene B4 receptor 1 attenuates lipopolysaccharide-induced cardiac dysfunction: Role of AMPK-regulated mitochondrial function. Sci. Rep 7, 44352 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu Q-Q, Deng W, Xiao Y, Chen J-J, Liu C, Wang J, Guo Y, Duan M, Cai Z, Xie S, Yuan Y, Tang Q, The 5-lipoxygenase inhibitor zileuton protects pressure overload-induced cardiac remodeling via activating PPARα. Oxid. Med. Cell. Longev 2019, 7536803 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bäck M, Bu D.-x., Bränström R, Sheikine Y, Yan Z-Q, Hansson GK, Leukotriene B4 signaling through NF-κB-dependent BLT1 receptors on vascular smooth muscle cells in atherosclerosis and intimal hyperplasia. Proc. Natl. Acad. Sci. U.S.A 102, 17501–17506 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hammond ME, Lapointe GR, Feucht PH, Hilt S, Gallegos CA, Gordon CA, Giedlin MA, Mullenbach G, Tekamp-Olson P, IL-8 induces neutrophil chemotaxis predominantly via type I IL-8 receptors. J. Immunol 155, 1428–1433 (1995). [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are present in the paper or the Supplementary Materials.