

replying to L. Wang et al. Nature 10.1038/s41586-023-06314-y (2023)
Wang and colleagues1 argue that our report2 of lower mutation rates in gene bodies, essential genes and regions marked by H3K4me1 must result from DNA sequencing errors. We appreciate the issues raised by them and by other colleagues3. Although we overlooked some sources of errors, these are insufficient to invalidate our conclusions, which are confirmed by more stringent reanalyses of our original data, new analyses restricted to high-confidence germline mutations4, and direct demonstration of plant DNA repair proteins being recruited to gene bodies, essential genes and H3K4me1, where they reduce local mutation rates5,6.
Wang and colleagues1 identify issues with somatic mutation calling, suggesting that homopolymer bleed-through errors in Illumina sequencing are responsible for patterns observed in somatic mutations, and that elevated cytosine deamination in transposable elements is responsible for the patterns in germline mutations. Here we address these concerns.
Consecutive runs of identical nucleotides, or homopolymers, pose challenges to discovering rare mutations because they can lead to Illumina sequencing errors at immediately neighbouring nucleotides through homopolymer bleed-through7. At the same time, homopolymer regions have higher true mutation rates even at local but non-adjacent sites (for example, ref. 8). Wang and colleagues1 found that the distribution of simulated homopolymer errors mirrors the overall distribution of mutations we reported around genes (their Fig. 1a). However, there are several reasons why such homopolymer errors cannot be the source of inferred mutation bias.
Fig. 1. Potential homopolymer bleed-through sequencing errors cannot explain differences in mutation rate.
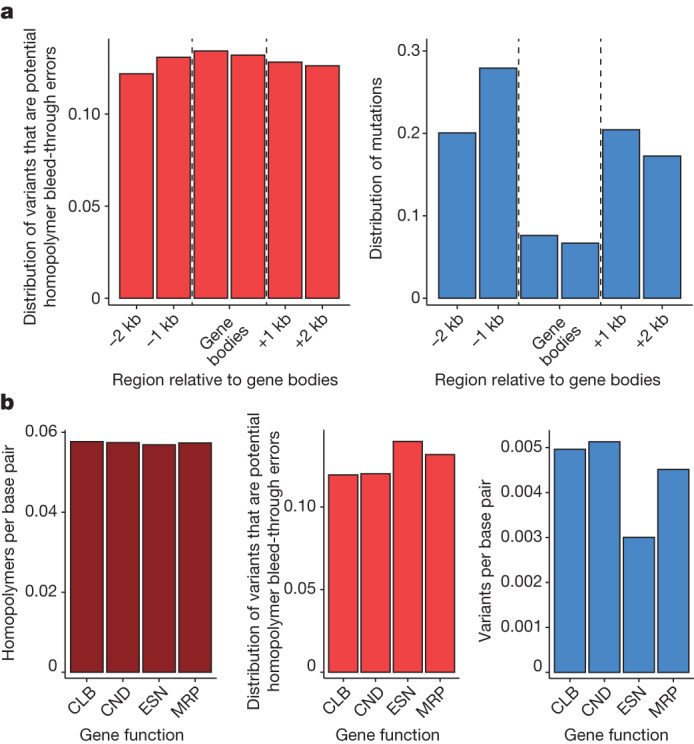
a, The proportion of variants that are potential homopolymer bleed-through errors among all mutation calls in our original study2 is as least as high in gene bodies as in intergenic sequences, and contrasts with the distribution of total mutation calls. kb, kilobase. b, Homopolymers and the proportion of variants that are potential homopolymer bleed-through errors in the original study2 are not lower in essential genes (ESN) compared to genes with environmentally conditional (CND), morphological (MRP) and cellular or biochemical (CLB) functions, and cannot explain the distribution of actual mutation calls.
Wang and colleagues1 assume that homopolymer bleed-through errors affect sequences up to five nucleotides away from homopolymers, although these errors occur on modern Illumina platforms at positions immediately adjacent to a run of identical bases7. Moreover, their simulation of sequencing errors apparently assumes that 100% of sequencing errors occur as a product of homopolymer bleed-through. By contrast, empirical estimates of sequencing errors report only 0.7 to 5.2% to be the result of homopolymer bleed-through7. Across all data in our study, only 12.0% of total single-nucleotide variant calls (10.2% for high-quality germline calls) could be potential homopolymer-adjacent bleed-through errors, and thus on their own cannot explain the approximately 50% mutation rate reduction we observed in gene bodies relative to intergenic regions2.
More importantly, Wang and colleagues’ own analysis1 reports that the proportion of potential homopolymer bleed-through errors in our data is actually higher in gene bodies (exons plus introns), which should lead to gene body mutation rates being overestimated, not underestimated. We confirm across our datasets that the proportion of potential homopolymer bleed-through errors is not lower in gene bodies (Fig. 1a, left), and differs from the pattern of mutation calls (Fig. 1a, right). Similarly, the proportion of potential homopolymer bleed-through errors is not reduced in essential genes (Fig. 1b). The distribution of potential homopolymer bleed-through errors, therefore, disagrees with the hypothesis of Wang and colleagues1. By contrast, the observed pattern is expected if gene bodies and essential genes experienced a reduction in true mutation rates, as noise introduced by sequencing errors should have a proportionally larger effect on regions with truly low mutation rates.
Homopolymeric sequences (but not potential homopolymer bleed-errors) are enriched outside gene bodies, as reported by Wang and colleagues1. Thus, the observed mutation rate heterogeneity is consistent with previous evidence that homopolymer-rich regions have higher true mutation rates8 and that their enrichment in these regions is consistent with the expected long-term evolutionary consequence of lower DNA repair activity, as the expansion of homopolymers is a signature of lower mismatch repair activity (Supplementary Note 3). Moreover, both preferential repair of exons by mismatch repair and higher intronic mutation rates in somatic tissues have been widely documented (Supplementary Note 3). Likewise, considerable differences in mutation rate and spectra between somatic and germline cells are well known, with somatic cells having orders of magnitude higher mutation rates. Indeed, differences between mitotic and meiotic cells have been previously proposed for Arabidopsis thaliana by Wang and colleagues9 (Supplementary Note 3).
Wang and colleagues1 further suggest that the patterns we observed in germline mutations might result largely from elevated deamination of methylated cytosines (GC-to-AT mutations) in transposable elements. Several findings are inconsistent with this hypothesis: cytosine methylation was included as a covariate in our original models, mutation accumulation experiments consistently indicate that mutation rates are lower in gene bodies relative to non-transposable element intergenic regions in A. thaliana (Fig. 2a,b; see below), and removing all GC-to-AT mutations from our original germline dataset does not alter the observed pattern, with H3K4me1 remaining the strongest epigenomic predictor of lower mutation (described in detail recently4). The same has been demonstrated for mutation rate variation in rice, in which mutation rates are lower in gene bodies relative to both intergenic regions and transposable elements6.
Fig. 2. Joint analyses of germline mutations in several published A. thaliana mutation accumulation studies align with mechanistic models of mutation bias.

a, Reduction in genic single-nucleotide germline mutation rates compared against genomic background in multiple A. thaliana datasets (Supplementary Table 1). For our original study2, only new mutations from 400 mutation accumulation lines are shown; the other mutations in that paper were already described10 and are shown separately here. Mutation rate reduction in genic regions is eliminated in msh2 DNA repair mutants5. bp, base pair. b, Mutation rates around gene bodies (grey and green lines). Black line indicates randomly selected windows based on gene lengths. c, Mutation rates in genes classified by functional category, rates of sequence evolution, patterns of expression and estimates of selection. Significance tested with χ2 test, n = 27,206 genes, with raw P values tested against α = 0.05 (unadjusted for multiple comparisons). Data show mean values for groups ± error bars reflecting 95% confidence intervals from bootstrapping. Vg, genetic variance of gene expression; 1001G, 1001 Genomes Project; LOF, loss of function; Dn, non-synonymous divergence; Ds, synonymous divergence; Pn, non-synonymous polymorphism; Ps, synonymous polymorphism; NS, not significant. d, Somatic mutations identified with very stringent criteria and using a caller specifically designed for rare somatic mutations, Strelka2, are reduced in gene bodies of wild-type plants, but not msh6 mutants6. e, Left, general mechanism proposed in ref. 2. Right, new knowledge regarding biochemical mechanisms underlying reduced mutation rates in gene bodies established by recent discoveries in plants and synthesized in ref. 6. HR, homology-directed repair; MMR, mismatch repair17–22.
To further address concerns with somatic mutation calls in general, we re-called putative somatic mutations in the original 107 lines10 by mapping reads to an improved reference genome11 and applying more stringent filtering (Supplementary Note 1). This led to more complete and higher-quality read mapping (Supplementary Fig. 1) and resolved several issues described by Wang and colleagues1 (for example, high intron-versus-exon mutation ratio and the proportion of potential homopolymer bleed-through errors; Supplementary Fig. 2). These data confirm that gene bodies experience lower mutation rates, including when manually removing potential homopolymer bleed-through errors (Supplementary Note 1). Many of the analyses by Wang and colleagues are affected by unreliable centromeric mutations, which constituted 41% of questioned somatic mutations1. These sites, however, could not have affected our conclusions because they were excluded from all of our original analyses (Supplementary Note 2 and Supplementary Fig. 3).
Wang and colleagues1 examined essential genes with approaches that were not in our original study. They used subsets of our initial datasets, focusing on either about 2,000 germline or about 4,000 somatic single-nucleotide variants, finding that neither dataset directly revealed a statistically significantly lower mutation rate in essential genes. This approach seems underpowered, yielding near-zero values for mutation counts in entire gene classes, an indication that the data are poorly suited for χ2 approximation (Supplementary Note 5).
In our study2, we had instead modelled genome-wide mutation rates, and using these models, identified a connection between gene essentiality and mutation rate corresponding to epigenome differences—essential genes are enriched for H3K4me1, for example, which we found to be associated with lower mutation rates. We subsequently tested whether this expectation is met in a very large set of several hundred thousand loosely filtered putative somatic mutations with ample power to compare gene classes. We agree that somatic mutation calling is very difficult, as most real somatic mutations and unrepaired damaged sites (with DNA damage occurring 10,000 to 100,000 times per day per cell; Supplementary Note 3) are expected to be present in only one cell and thus detectable only by a single read. In Supplementary Note 4 and an accompanying Correction12, we discuss why singletons were called as putative mutations in one of our reanalyses, from 64 leaves13, owing to inadvertently mapping forward reads twice. However, analyses of variant quality in these data do not support the hypothesis that our results are simply due to higher rates of poor-quality calls in non-genic regions or non-essential genes (Supplementary Note 4 and Supplementary Fig. 4).
Finally, to directly address the possibility that our conclusions reflect unknown sources of bias in inherently uncertain somatic calls, we reanalysed germline mutations from our study2 along with mutation accumulation experiment data generated in several independent studies (Supplementary Table 1). This meta-analysis of >10,000 germline mutations confirmed the previously reported, nearly universal reduction in single-nucleotide mutation rates in gene bodies, essential genes and regions marked by H3K4me1 (Fig. 2a–c; ref. 4). The notable exception comes from plants lacking the mismatch repair protein MSH2 (Fig. 2a; ref. 5). A similar pattern is seen when somatic mutations were called with very stringent criteria in plants deficient for the MSH2 partner MSH6, using a tool specifically designed for rare somatic mutations14 (Fig. 2d). This was as predicted from H3K4me1 directly attracting MSH6 to gene bodies6, confirming that DNA repair in A. thaliana is targeted to gene bodies, as is well known in humans (Supplementary Note 3). Finally, analyses of >43,000 experimentally induced de novo germline mutations in rice (previously validated with 99% accuracy) also show that gene bodies, conserved genes, and H3K4me1-marked regions experience lower mutation rates, even when considering only silent (synonymous) mutations6.
Relationships between histone modifications, DNA repair, and mutation rate are widely known (Supplementary Note 3). Our work2 considered the evolutionary implication of these relationships. We had leveraged models of the drift-barrier hypothesis to discover that natural selection could favour mechanisms linking DNA repair to widely distributed epigenomic features, such as H3K4me1, which is not only enriched in gene bodies and essential genes in A. thaliana, but also the histone modification most strongly associated with lower mutation rates in our data2. An important higher-order test of our conclusions is therefore whether they are mechanistically supported. Since publication of our article2, it has been demonstrated that plant DNA repair proteins are recruited by H3K4me1 to gene bodies and essential genes. These repair proteins, which contain Tudor ‘reader’ domains that bind H3K4me1, include PDS5C, involved in homology-directed repair, and MSH6, which functions as a dimer with MSH2 in the mismatch repair pathway and recruits MutY of the base-excision repair pathway15. The genome-wide distribution of PDS5C, as measured by chromatin immunoprecipitation followed by sequencing4,6,16, confirms that regions subject to elevated repair protein activity coincide with features at which we detected lower spontaneous mutation rates4,6,16.
We conclude that the reported relationships between epigenomic features and mutation rates2 are well supported mechanistically (Fig. 2e). We agree that there are issues and inherent uncertainties with somatic mutation calling, which make it difficult to know the accuracy of individual calls in the very large set of loosely filtered somatic variants2. However, the proposal that the observed patterns result only from sequencing errors is inconsistent with multiple lines of evidence from the original study, independent analyses and emerging parallel work.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at 10.1038/s41586-023-06315-x.
Supplementary information
This file contains Supplementary Table 1, Notes 1–5 (with Figs 1–4) and References.
Acknowledgements
Research was conducted at the University of California, Davis, which is located on land that was the home of the Patwin people for thousands of years.
Author contributions
J.G.M., K.D.M., W.X., T.S., P.C.-B. and D.W. contributed to data analysis. J.G.M., K.D.M., W.X., T.S., P.C.-B. and D.W. contributed to the writing. J.G.M., K.D.M., W.X., T.S., P.C.-B., C.B., M.L., M.E.-A., M.K., J.H., M.N., D.K., M.-L.W., E.I., J.Å., M.T.R., C.B.F. and D.W. contributed to the interpretation of the results. K.D.M. and W.X., who were not part of the study by J.G.M. et al.2, carried out analyses to validate the impact of an improved genome reference sequence on reducing sequencing errors.
Data availability
The TAIR10 A. thaliana reference genome can be found at https://arabidopsis.org/download. The more recent, improved A. thaliana reference genome can be found at https://github.com/schatzlab/Col-CEN. Sequencing reads for 107 A. thaliana mutation accumulation lines are stored in the National Center for Biotechnology Information Short Read Archive, accession number SRP133100. Additional mutation datasets were downloaded from publications cited in Supplementary Table 1.
Code availability
Code for this work uses functions maintained in https://github.com/greymonroe/polymorphology, with additional scripts and data for analyses and figures in https://github.com/greymonroe/mutation_bias_analysis2.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
J. Grey Monroe, Email: gmonroe@ucdavis.edu.
Detlef Weigel, Email: weigel@weigelworld.org.
Supplementary information
The online version contains supplementary material available at 10.1038/s41586-023-06315-x.
References
- 1.Wang, L., Ho, A. T., Hurst, L. D. & Yang, S. Re-evaluating evidence for adaptive mutation rate variation. Nature10.1038/s41586-023-06314-y (2023). [DOI] [PMC free article] [PubMed]
- 2.Monroe JG, et al. Mutation bias reflects natural selection in Arabidopsis thaliana. Nature. 2022;602:101–105. doi: 10.1038/s41586-021-04269-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu H, Zhang J. Is the mutation rate lower in genomic regions of stronger selective constraints? Mol. Biol. Evol. 2022;39:msac169. doi: 10.1093/molbev/msac169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Monroe, J. G. et al. Report of mutation biases mirroring selection in Arabidopsis thaliana unlikely to be entirely due to variant calling errors. Preprint at bioRxiv10.1101/2022.08.21.504682 (2022).
- 5.Belfield EJ, et al. DNA mismatch repair preferentially protects genes from mutation. Genome Res. 2018;28:66–74. doi: 10.1101/gr.219303.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Quiroz, D. et al. The H3K4me1 histone mark recruits DNA repair to functionally constrained genomic regions in plants. Preprint at bioRxiv10.1101/2022.05.28.493846 (2022).
- 7.Stoler N, Nekrutenko A. Sequencing error profiles of Illumina sequencing instruments. NAR Genom. Bioinform. 2021;3:lqab019. doi: 10.1093/nargab/lqab019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tran HT, Keen JD, Kricker M, Resnick MA, Gordenin DA. Hypermutability of homonucleotide runs in mismatch repair and DNA polymerase proofreading yeast mutants. Mol. Cell. Biol. 1997;17:2859–2865. doi: 10.1128/MCB.17.5.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang S, et al. Parent–progeny sequencing indicates higher mutation rates in heterozygotes. Nature. 2015;523:463–467. doi: 10.1038/nature14649. [DOI] [PubMed] [Google Scholar]
- 10.Weng M-L, et al. Fine-grained analysis of spontaneous mutation spectrum and frequency in Arabidopsis thaliana. Genetics. 2019;211:703–714. doi: 10.1534/genetics.118.301721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Naish M, et al. The genetic and epigenetic landscape of the Arabidopsis centromeres. Science. 2021;374:eabi7489. doi: 10.1126/science.abi7489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Monroe, J. G. et al. Author Correction: Mutation bias reflects natural selection in Arabidopsis thaliana. Nature10.1038/s41586-023-06387-9 (2023). [DOI] [PMC free article] [PubMed]
- 13.Wang L, et al. The architecture of intra-organism mutation rate variation in plants. PLoS Biol. 2019;17:e3000191. doi: 10.1371/journal.pbio.3000191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim S, et al. Strelka2: fast and accurate calling of germline and somatic variants. Nat. Methods. 2018;15:591–594. doi: 10.1038/s41592-018-0051-x. [DOI] [PubMed] [Google Scholar]
- 15.Gu Y, et al. Human MutY homolog, a DNA glycosylase involved in base excision repair, physically and functionally interacts with mismatch repair proteins human MutS homolog 2/human MutS homolog 6. J. Biol. Chem. 2002;277:11135–11142. doi: 10.1074/jbc.M108618200. [DOI] [PubMed] [Google Scholar]
- 16.Niu Q, et al. A histone H3K4me1-specific binding protein is required for siRNA accumulation and DNA methylation at a subset of loci targeted by RNA-directed DNA methylation. Nat. Commun. 2021;12:3367. doi: 10.1038/s41467-021-23637-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu X, et al. Non-CG DNA methylation-deficiency mutations enhance mutagenesis rates during salt adaptation in cultured Arabidopsis cells. Stress Biol. 2021;1:12. doi: 10.1007/s44154-021-00013-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Willing E-M, et al. UVR2 ensures transgenerational genome stability under simulated natural UV-B in Arabidopsis thaliana. Nat. Commun. 2016;7:13522. doi: 10.1038/ncomms13522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ossowski S, et al. The rate and molecular spectrum of spontaneous mutations in Arabidopsis thaliana. Science. 2010;327:92–94. doi: 10.1126/science.1180677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu Z, et al. Genome-wide DNA mutations in Arabidopsis plants after multigenerational exposure to high temperatures. Genome Biol. 2021;22:160. doi: 10.1186/s13059-021-02381-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiang C, et al. Environmentally responsive genome-wide accumulation of de novo Arabidopsis thaliana mutations and epimutations. Genome Res. 2014;24:1821–1829. doi: 10.1101/gr.177659.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Belfield EJ, et al. Thermal stress accelerates Arabidopsis thaliana mutation rate. Genome Res. 2021;31:40–50. doi: 10.1101/gr.259853.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This file contains Supplementary Table 1, Notes 1–5 (with Figs 1–4) and References.
Data Availability Statement
The TAIR10 A. thaliana reference genome can be found at https://arabidopsis.org/download. The more recent, improved A. thaliana reference genome can be found at https://github.com/schatzlab/Col-CEN. Sequencing reads for 107 A. thaliana mutation accumulation lines are stored in the National Center for Biotechnology Information Short Read Archive, accession number SRP133100. Additional mutation datasets were downloaded from publications cited in Supplementary Table 1.
Code for this work uses functions maintained in https://github.com/greymonroe/polymorphology, with additional scripts and data for analyses and figures in https://github.com/greymonroe/mutation_bias_analysis2.