

Abstract
Purpose
Polycystic ovary syndrome (PCOS) is one of the leading causes of infertility in women of childbearing age, and many patients with PCOS have obesity and insulin resistance (IR). Although obesity is related to an increased risk of IR, in clinical practice, PCOS patients exhibit different effects on improving insulin sensitivity after weight loss. Therefore, in the present study, we aimed to examine the moderating effect of polymorphisms of mtDNA in the D-loop region on the associations of body mass index (BMI) with the homeostasis model assessment of insulin resistance index (HOMA-IR) and pancreatic β cell function index (HOMA-β) among women with PCOS.
Methods
Based on a cross-sectional study, women with PCOS were recruited from the Reproductive Center of the First Affiliated Hospital of Anhui Medical University from 2015 to 2018. A total of 520 women who were diagnosed with PCOS based on the revised 2003 Rotterdam criteria were included in the study. Peripheral blood was collected from these patients, followed by DNA extraction, PCR amplification, and sequencing at baseline. HOMA-IR and HOMA-β were calculated according to blood glucose-related indices. Moderating effect models were performed with BMI as an independent variable, polymorphisms of mtDNA in the D-loop region as moderators, and ln (HOMA-IR) and ln (HOMA-β) as dependent variables. To verify the stability of moderating effect, sensitivity analysis was performed with the quantitative insulin sensitivity check index (QUICKI), fasting plasma glucose/fasting insulin (G/I), and fasting insulin as dependent variables.
Results
BMI was positively associated with ln (HOMA-IR) and ln (HOMA-β) (β = 0.090, p < 0.001; β = 0.059, p < 0.001, respectively), and the relationship between BMI and ln (HOMA-IR) or ln (HOMA-β) was moderated by the polymorphisms of mtDNA in the D-loop region. Compared with the respective wild-type, the variant -type of m.16217 T > C enhanced the association between BMI and HOMA-IR, while the variant-type of m.16316 A > G weakened the association. On the other hand, the variant-type of m.16316 A > G and m.16203 A > G weakened the association between BMI and HOMA-β, respectively. The results of QUICKI and fasting insulin as dependent variables were generally consistent with HOMA-IR, and the results of G/I as dependent variables were generally consistent with HOMA-β.
Conclusion
Polymorphisms of mtDNA in the D-loop region moderate the associations of BMI with HOMA-IR and HOMA-β among women with PCOS.
Supplementary Information
The online version contains supplementary material available at 10.1007/s10815-023-02843-7.
Keywords: Polycystic ovary syndrome, Body mass index, HOMA-IR, HOMA-β, Moderating effect
Introduction
Polycystic ovarian syndrome (PCOS), characterized by excessive androgen and chronic anovulation, is the most common reproductive endocrine disease in women of childbearing age, affecting approximately 5 ~ 20% [1, 2]. The revised 2003 Rotterdam criteria, which are widely used and supported by many scientific communities and medical institutions to diagnose PCOS, indicate that a patient’s phenotype should meet at least two of the following characteristics: clinical and/or biochemical hyperandrogenemia, ovulation dysfunction, and polycystic ovary morphology (PCOM) [2], while requiring the exclusion of diseases with symptoms and signs that overlap with PCOS, such as congenital adrenal hyperplasia and Cushing’s syndrome and androgen-secreting tumors. Women with PCOS face an increased risk of obesity, diabetes, cardiovascular disease, hypertension, and other metabolic complications, especially after menopause [3]. To date, the etiology and pathogenesis of PCOS remain unclear, but there is growing evidence that PCOS is a polygenic disease with strong epigenetic and environmental effects [4].
Obesity, as a phenotype of PCOS, can also aggravate various other functional disorders of PCOS, such as anovulation, hyperandrogenism, insulin resistance (IR), and inflammation [5]. Insulin resistance (IR), another phenotype of PCOS, affects approximately 65 to 70% of women with PCOS [6]. Some researchers have found that in the early stages of PCOS, women not only have IR but also impaired β cell function [7]. Impaired glucose tolerance and type 2 diabetes occur when insulin cannot cope with glucose levels due to IR. The study of β-cell function in patients with PCOS is controversial. In a European population-based study, women with PCOS showed higher IR, but they secreted more insulin to maintain normal glucose homeostasis compared to the controls matched for age, BMI, and insulin resistance [8]. In another study, early β-cell function impairment was found in Chinese women with PCOS [9].
Mitochondria are the sites of energy generation in cells and play an important role in cell energy metabolism, reproduction, and apoptosis. Mitochondrial DNA (mtDNA) is a 16,569 bp double-linked ring molecule encoding 2 rRNA, 22 tRNA, and 13 phosphorylation system complexes I, III, IV, and V, which are necessary for the production of adenosine triphosphate (ATP) [10, 11]. Mitochondria are also important structures that regulate oxidative stress. Some studies have shown that oxidative stress markers are significantly higher in PCOS women than in normal women [12], and they have been recognized as a potential cause of PCOS pathogenesis. The D-loop region is the main regulatory site for the replication and transcription of mtDNA. Additionally, the promoters of various transcription factors necessary for mitochondrial gene transcription exist in the D-loop region, and various nuclear hormone receptors, such as glucocorticoid receptor, estrogen receptor, and thyroid (T3) hormone receptor, bind to the D-loop and exert their functions [13]. Mutations in this region may affect mtDNA transcription and replication, resulting in ROS production and mitochondrial dysfunction.
Recent studies have shown that the expression of oxidative phosphorylation-related genes is reduced in the skeletal muscle of women with PCOS accompanied by IR [14, 15]. Some studies have found that there is a correlation between obesity and mtDNA copy number [16]. For example, the results from a German study showed that variants of m.16292 C > T and m.16189 T > C in the D-loop region of mtDNA are associated with obesity [17]. Mitochondrial dysfunction plays an important role in the development of IR [18]. When treating PCOS patients, clinicians will not only apply necessary clinical medications but also warn patients to reduce their body weight in various ways to keep their BMI within a reasonable range. However, according to the follow-up study of patients, the reduction in BMI in different patients has exhibited different effects on decreasing IR [19], so the underlying reasons behind this phenomenon deserve further exploration.
Therefore, the aim of this study was to analyze the relationships between BMI and homeostasis model assessment of insulin resistance index (HOMA-IR) and pancreatic β cell function index (HOMA-β) among women with PCOS and to investigate whether there is a moderating effect of the polymorphisms of mtDNA in the D-loop region on these associations.
Materials and methods
Ethics
This study was approved by the Biomedical Ethics Committee of Anhui Medical University (No. 20131401). All the participants signed informed consent forms.
Study design and population recruitment
A cross-sectional study was conducted in our present study to investigate the moderating effect of mtDNA polymorphisms in the D-loop region on the associations of BMI with HOMA-IR and HOMA-β among women with PCOS.
After we obtained a grant of research ethics approval, two experienced clinicians at the reproductive center of the First Affiliated Hospital of Anhui Medical University, China, were charged with the recruitment of patients with PCOS. Finally, a total of 520 patients with PCOS who intended to seek help for infertility for the first time at our reproductive center were recruited from 2015 to 2018. PCOS was diagnosed based on the revised 2003 Rotterdam criteria, and a patient's phenotype should satisfy at least two of the following criteria: (1) oligoovulation or anovulation (oligomenorrhea: menstrual cycles lasting longer than 35 days; amenorrhea: menstrual absence within 6 months; and anovulation: serum progesterone level < 3 ng/mL on the 21st ~ 24th day of the menstrual cycle); (2) polycystic ovarian morphology after ultrasound examination (an increased ovarian volume > 10 mL or the number of small follicles (diameter: 2 to 9 mm) ≥ 12 in each ovary); and (3) clinical or biochemical signs of hyperandrogenism (serum androgen > 2.64 nmol/L, hirsutism, or acne) [2]. Patients who showed a similar clinical presentation due to other disorders were excluded—for example, congenital adrenal hyperplasia, Cushing’s syndrome, or androgen-secreting tumors.
Additionally, to verify whether the selected gene polymorphism sites were generalizable to women without PCOS, we set up a control group (all the women in the control group did not have PCOS). The general demographic characteristics, serum hormone levels, and glycaemic-related indices of the study population are displayed in Supplementary Table S1.
Assessment of clinical characteristics
Clinical characteristics of participants, such as the basal sex hormone level, fasting glucose, and fasting insulin level, were measured. Fasting glucose (FG) was measured by the hexokinase method via commercial kits (Beckman Coulter, Inc., USA), which were suitable for the autoanalyzer (AU5821; Beckman Coulter, Inc., USA); fasting insulin (FI) was measured by the direct chemiluminescent assay via commercial kits (ADVIA Centaur; Siemens Healthineers, Inc., Germany) that were suitable for the autoanalyzer (Siemens Healthineers, Inc., Germany). The levels of serum basal sex hormones, such as luteinizing hormone (LH), follicle-stimulating hormone (FSH), oestradiol (E2), progesterone (P), prolactin (PRL), and testosterone (T), were measured by electrochemiluminescence immunoassays (ECLIAs) using commercial kits (Beckman Coulter, Inc., USA) suitable for the autoanalyzer (UniCel Dxl 800 Access; Beckman Coulter, Inc., USA). The measurement of the above three indices was completed at the Department of Clinical Laboratory of the First Affiliated Hospital of Anhui Medical University. HOMA-IR was calculated according to the following formula: HOMA-IR = fasting insulin(FINS) (μIU/mL) × fasting glucose(FPG) (mmol/L)/22.5. HOMA-β was estimated based on the following formula: HOMA-β = 20 × fasting insulin (μIU/mL)/(fasting glucose (mmol/L) − 3.5). QUICKI was calculated according to the following formula: QUICKI=1/(log FINS (μIU/mL)+log FPG (mmol/L)). The fasting G/I ratio was calculated according to the following formula: G/I=FPG (mmol/L)/FINS (μIU/mL). HOMA-IR ≥ 2.69 was defined as IR [20]. The intra-assay and interassay CVs for total testosterone were 3 ~ 8%, respectively; the intra-assay and interassay CVs for fasting plasma glucose were 3 ~ 5%, respectively; the intra-assay and interassay CVs for fasting insulin were 1.8 ~ 2.8%, respectively; the intra-assay and interassay CVs for FSH were 2.9 ~ 5.3%, respectively; and the intra-assay and interassay CVs for LH were 1.5 ~ 3.0%, respectively.
The demographic characteristics of the patients, such as age, height, and body weight, were gathered from the hospital’s electronic health system. Age, height, and body weight were all collected as continuous variables. BMI was calculated based on the formula: BMI = body weight (kg)/the square of the height (m2).
DNA extraction, polymerase chain reaction, and next-generation sequencing
Fasting anticoagulant blood samples of patients were collected on the 2nd or 3rd day of menstruation (or vaginal bleeding after drug withdrawal) when they conducted the clinical laboratory measurement of basal hormone levels. Genomic DNA was isolated from the whole blood of patients with PCOS using the Magnetic Universal Genomic DNA Kit (Tiangen Biotech (Beijing) Co., Ltd.). Polymerase chain reaction and next-generation sequencing were performed according to our previous research (Deng et al. 2021; Li et al. 2020b). Briefly, the entire 1124 bp D-loop region of mtDNA was amplified using polymerase chain reaction (PCR) with the forwards primer 5′-CTCCACCATTAGCACCCAAAGC-3′ and the reverse primer 5′-AGGCTAAGCGTTTTG AGCTG-3′. The 20 μL PCR was performed using the Takara LA Taq Kit (Clontech). PCR amplification cycle conditions followed the procedure: 95 ℃ for 3 min, 30 cycles at 94 ℃ for 30 s, 54 ℃ for 30 s, 72 ℃ for 30 s, and one extension cycle at 72 ℃ for 10 min. Cycle sequencing was performed after purification via the Big Dye Terminator Cycle sequencing reaction Kit (Applied Biosystem, Foster City, CA). Finally, the products were sequenced on the ABI Prism 3730 DNA Analyser (Applied Biosystem). Comparison with the revised Cambridge Reference Sequence (rCRS) was performed, and the sites of base mutation and variation were obtained.
Statistical analysis
Statistical analysis was performed using SPSS for Windows (version 22.0; SPSS UK Ltd., Surrey, UK) and R (version 4.0.5, package “ggplot2”), and a p value < 0.05 (two-sided) was considered statistically significant for all the analyses.
The descriptive statistics for continuous variables were presented as the mean values ± standard deviations (SD) or medians (P25 − P75) or percentages. First, after HOMA-IR and HOMA-β were natural logarithm transformed, the generalized linear model (GLM) was used to analyze the relationships of BMI with HOMA-IR and HOMA-β, respectively, to judge whether there was a linear relationship between them. Then, linear regression models were used to analyze the relationship between BMI and ln(HOMA-IR) and BMI and ln(HOMA-β) in patients with PCOS. Finally, we examined whether polymorphisms of mtDNA in the D-loop region moderated the association between BMI and ln (HOMA-IR) or ln (HOMA-β). Therefore, moderating effect models were conducted with BMI as an independent variable, polymorphisms of mtDNA in the D-loop region as moderators, and ln (HOMA-IR) and ln (HOMA-β) as dependent variables using the SPSS PROCESS macro, version 4.0 (Model 1), developed by Hayes. Age, LH/FSH, and T were considered covariates. Additionally, to verify the stability of moderating effect, sensitivity analysis was performed with QUICKI, G/I, and fasting insulin as dependent variables. Simple slope figures were used to show the relationships of BMI with ln (HOMA-IR) and ln (HOMA-β) when the moderators (polymorphisms of mtDNA) was set as wild-type and variant-type status, respectively.
Results
The general demographic characteristics, serum hormone levels, and glycaemic-related indices of the study population are displayed in Table 1. The mean ± SD age of women with PCOS was 27.10 ± 3.64 years (range: 13 to 41 years); their mean ± SD BMI was 24.07 ± 3.80 kg/m2, and nearly half of the women with PCOS were overweight or obese; the median (P25, P75) total testosterone level of women with PCOS was 1.76 (1.29, 2.35) nmol/L, and women with T ≥ 2.64 nmol/L accounted for 15.58%; nearly two-thirds of women with PCOS had IR. With regard to women without PCOS, their mean ± SD age was 28.59 ± 3.06 years (range: 20 to 39 years); their mean ± SD BMI was 22.21 ± 3.44 kg/m2 (range: 16.00 to 37.90 kg/m2), only a quarter of the women were overweight or obese; the median (P25, P75) total testosterone level was 1.47 (0.91, 1.96) nmol/L, and the women with T ≥ 2.64 nmol/L accounted for 7.06%; nearly one-third of women had IR, respectively (Supplementary Table S1). The women with PCOS and women in the control group involved in the study were of Han ethnicity. The general demographic characteristics (age, BMI) in the PCOS group were significantly different from those in the control group (p < 0.001). The serum sex hormone levels (FSH, LH, T), LH/FSH ratio, and blood glucose-related indices in the PCOS group were significantly different from those in the control group (Supplementary Table S1).
Table 1.
Demographic characteristics of the participants
| Variables | Mean ± SD or median (P25, P75) or n (%) |
|---|---|
| Characteristic | |
| Age (years) | 27.10 ± 3.64 |
| BMI (kg/m2) | 24.07 ± 3.80 |
| Underweight (< 18.5) | 20 (3.85) |
| Normal (18.5–23.9) | 249 (47.88) |
| Overweight/obesity (> 24.0) | 251 (48.27) |
| Serum basal sex hormone levels | |
| FSH (mIU/mL) | 6.23 ± 1.40 |
| LH (mIU/mL) | 9.58 (6.38, 13.97) |
| LH/FSH | 1.60 (1.02, 2.28) |
| T (nmol/L) | 1.76 (1.29, 2.35) |
| T < 2.64 | 439 (84.42) |
| T ≥ 2.64 | 81 (15.58) |
| Blood glucose-related indexes | |
| FPG (mmol/L) | 5.52 ± 1.37 |
| FINS (μIU/mL) | 13.96 (8.97, 20.59) |
| HOMA-IR | 3.34 (2.13, 5.12) |
| HOMA-IR < 2.69 | 190 (36.54) |
| HOMA-IR ≥ 2.69 | 330 (63.46) |
| HOMA-β | 145.90 (103.78, 219.59) |
BMI, body mass index; FSH, follicle-stimulating hormone; LH, luteinizing hormone; T, testosterone; FPG, fasting plasma glucose; FINS, fasting insulin; HOMA-IR, homeostasis model assessment of insulin resistance; HOMA-β, homeostasis model assessment of β-cell function
Among all the mtDNA gene sites that we identified, there were 66 mtDNA sites with the condition that the percentages of women with the variant type accounted for more than 2% (Table 2). Considering our sample size, these 66 sites were selected for subsequent analysis.
Table 2.
Gene polymorphisms information of D-loop region of mtDNA in D-loop region among 520 PCOS patients (a minor allele frequency > 1%)
| Genes | Wild type | Variant type | Total mutation rate (%) |
|---|---|---|---|
| A73 | 2 | 518 | 99.62% |
| A315.1C | 6 | 514 | 98.85% |
| A16223 | 184 | 336 | 64.62% |
| A489 | 234 | 286 | 55.00% |
| A16519 | 258 | 262 | 50.38% |
| A523524d | 295 | 225 | 43.27% |
| A309.1C | 304 | 216 | 41.54% |
| A16362 | 313 | 207 | 39.81% |
| A16189 | 345 | 175 | 33.65% |
| A152 | 378 | 142 | 27.31% |
| A16183C | 392 | 128 | 24.62% |
| A249d | 409 | 111 | 21.35% |
| A150 | 420 | 100 | 19.23% |
| A309.2C | 434 | 86 | 16.54% |
| A16193.1C | 446 | 74 | 14.23% |
| A16182C | 448 | 72 | 13.85% |
| A16311 | 452 | 68 | 13.08% |
| A16298 | 453 | 67 | 12.88% |
| A16304 | 455 | 65 | 12.50% |
| A16172 | 456 | 64 | 12.31% |
| A16319 | 456 | 64 | 12.31% |
| A16261 | 463 | 57 | 10.96% |
| A146 | 463 | 57 | 10.96% |
| T16217C | 467 | 53 | 10.19% |
| A16093 | 474 | 46 | 8.85% |
| A16140 | 483 | 37 | 7.12% |
| A235 | 485 | 34 | 6.55% |
| A16036.1G | 486 | 34 | 6.54% |
| A16290 | 486 | 34 | 6.54% |
| A199 | 487 | 33 | 6.35% |
| A151 | 491 | 29 | 5.58% |
| A16257A | 492 | 28 | 5.38% |
| A16260 | 492 | 28 | 5.38% |
| A16266 | 492 | 28 | 5.38% |
| A16234 | 494 | 26 | 5.00% |
| A16184 | 494 | 26 | 5.00% |
| A16111 | 498 | 24 | 4.60% |
| A16274 | 497 | 23 | 4.42% |
| A207 | 499 | 21 | 4.04% |
| A16162 | 499 | 21 | 4.04% |
| A16092 | 499 | 21 | 4.04% |
| A16243 | 499 | 21 | 4.04% |
| A204 | 500 | 20 | 3.85% |
| A16291 | 501 | 19 | 3.65% |
| A16164 | 501 | 19 | 3.65% |
| A16327 | 502 | 18 | 3.46% |
| A573.1XC | 502 | 18 | 3.46% |
| A16192 | 504 | 16 | 3.08% |
| A194 | 504 | 16 | 3.08% |
| A195 | 484 | 16 | 3.20% |
| A16278 | 504 | 16 | 3.08% |
| A16249 | 505 | 15 | 2.88% |
| A16209 | 505 | 15 | 2.88% |
| A185 | 506 | 14 | 2.69% |
| A210 | 506 | 14 | 2.69% |
| A103 | 506 | 14 | 2.69% |
| A200 | 507 | 13 | 2.50% |
| A16086 | 507 | 13 | 2.50% |
| A16297 | 507 | 13 | 2.50% |
| A16295 | 507 | 13 | 2.50% |
| A16126 | 507 | 13 | 2.50% |
| A189 | 507 | 13 | 2.50% |
| A16316G | 508 | 12 | 2.31% |
| A16203G | 508 | 12 | 2.31% |
| A16136 | 509 | 11 | 2.12% |
| A16335 | 509 | 11 | 2.12% |
We found that the relationships of BMI with lnHOMA-IR and lnHOMA-β were approximately linear (Fig. 1 and Fig. 2). In linear regression models, BMI was significantly and positively associated with In (HOMA-IR) (β = 0.09, p < 0.001) after adjusting for age, LH/FSH, and T. Similarly, after HOMA-β was natural logarithm transformed, BMI was also significantly and positively associated with ln (HOMA-β) (β = 0.059, p < 0.001) after adjusting for age, LH/FSH, and T (Table 3). BMI was also significantly and positively associated with lnFINS (β = 0.081, p < 0.001) after adjusting for age, LH/FSH, and T. BMI was significantly and negatively associated with lnQUICKI (β = − 0.022, p < 0.001) and ln(G/I) (β = − 0.073, p < 0.001) after adjusting for age, LH/FSH, and T, respectively (Supplementary Table S5).
Fig. 1.
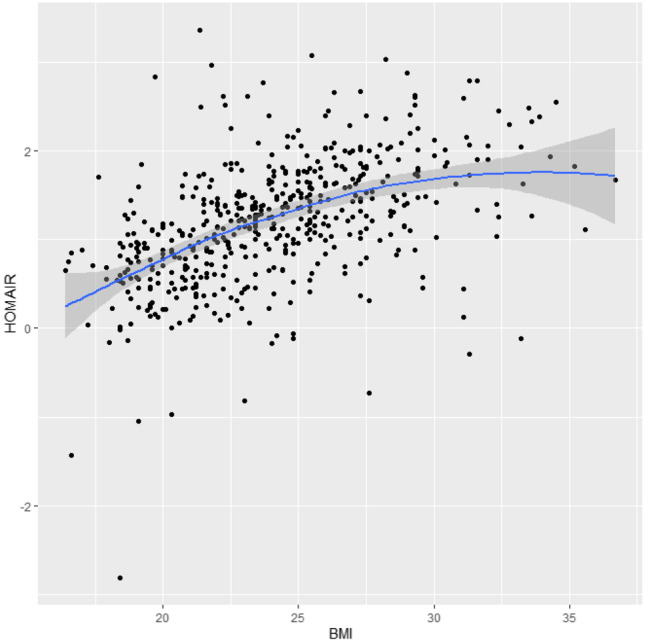
Scatterplot of the correlation between BMI and HOMA-IR (HOMA-IR was natural logarithm transformed)
Fig. 2.
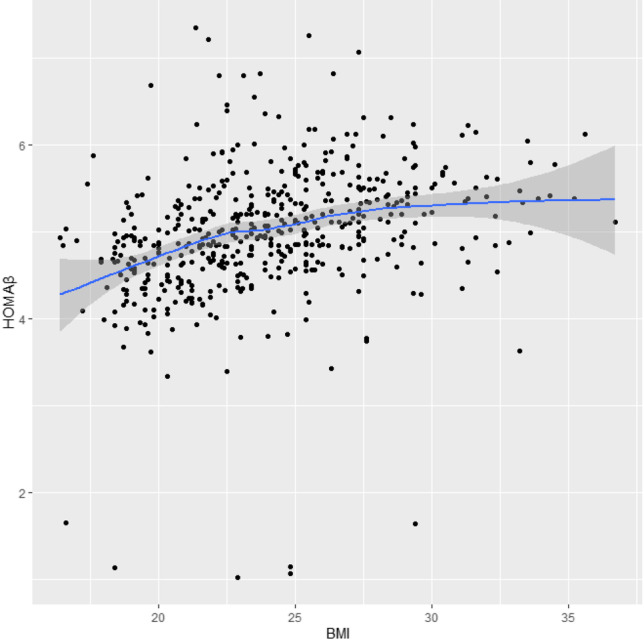
Scatterplot of the correlation between BMI and HOMA-β (HOMA-β were natural logarithm transformed)
Table 3.
The associations of BMI with ln (HOMA-IR) and ln (HOMA-β)
| Independent variable | Dependent variable | β | SE | t | p | 95% CI |
|---|---|---|---|---|---|---|
| BMI | ln (HOMA-IR) | 0.090 | 0.007 | 12.454 | < 0.001 | (0.076, 0.104) |
| ln (HOMA-β) | 0.059 | 0.008 | 7.135 | < 0.001 | (0.043, 0.075) |
BMI, body mass index; HOMA-IR, homeostasis model assessment of insulin resistance; HOMA-β, homeostasis model assessment of β-cell function; ln, natural logarithm transformed; SE, standard error; 95% CI, 95% confidence interval
p < 0.05, statistical significance was considered to be significant. The model was adjusted by age, LH/FSH, T
We further explored whether polymorphisms of mtDNA can enhance or weaken the associations of BMI with HOMA-IR and HOMA-β. The regression coefficients (βs) of the interaction between BMI and polymorphisms of m.16217 and m.16316 were as follows before adjusting for confounders: β = 0.046, p = 0.039; β = − 0.255, p = 0.001, respectively. After adjusting for confounders (age, LH/FSH, and T), the polymorphism of m.16217 positively moderated the relationship between BMI and ln(HOMA-IR) (β = 0.045, p = 0.045); the polymorphism of m.16316 negatively moderated the relationship between BMI and ln(HOMA-IR) (β = − 0.255, p = 0.001). (Table 4, Fig. 3, Fig. 4). On the other hand, the association between BMI and HOMA-β was also moderated by the polymorphisms of mtDNA in the D-loop region. The regression coefficients (βs) of the interaction between BMI and the m.16203 and m.16316 polymorphisms were as follows before adjusting for confounders: β = − 0.159, p = 0.025; β = − 0.255, p = 0.005, respectively. After adjusting for confounders (age, LH/FSH, and T), polymorphisms of m.16203 and m.16316 negatively moderated the relationship between BMI and ln(HOMA-β) (β = − 0.170, p = 0.016; β = − 0.252, p = 0.005) (Table 4, Fig. 5, Fig. 6). Polymorphism of m.16217 negatively moderated the relationship between BMI and ln QUICKI (β = − 0.014, p = 0.032); and polymorphism of m.16316 positively moderated the relationship between BMI and ln QUICKI (β = 0.058, p = 0.012). Polymorphism of m.16316 and m.16203 positively moderated the relationship between BMI and ln (G/I) (β = 0.253, p = 0.002; β = 0.124, p = 0.051), respectively. Polymorphism of m.16217 positively moderated the relationship between BMI and lnFINS (β = 0.043, p = 0.049), and polymorphism of m.16316 negatively moderated the relationship between BMI and lnFINS (β = − 0.257, p = 0.001) (Supplementary Table S3).
Table 4.
Results from the moderated regression analysis predicting HOMA-IR and HOMA-β
| Dependent variable | Predictors | Model 1 | Model 2 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| β | SE | t | p | ΔR2 | F | β | SE | t | p | ΔR2 | F | ||
| HOMA-IR | BMI | 0.890 | 0.007 | 12.733 | < 0.001 | 0.09 | 0.007 | 12.418 | < 0.001 | ||||
| T16217C | − 0.027 | 0.088 | − 0.303 | 0.762 | − 0.034 | 0.088 | − 0.391 | 0.696 | |||||
| BMI*T16217C | 0.046 | 0.022 | 2.069 | 0.039 | 0.006 | 4.28* | 0.045 | 0.022 | 2.011 | 0.045 | 0.006 | 4.044* | |
| BMI | 0.086 | 0.007 | 12.114 | < 0.001 | 0.086 | 0.007 | 11.873 | < 0.001 | |||||
| A16316G | 0.474 | 0.191 | 2.477 | 0.014 | 0.49 | 0.193 | 2.544 | 0.011 | |||||
| BMI*A16316G | − 0.255 | 0.079 | − 3.247 | 0.001 | 0.015 | 10.541** | − 0.255 | 0.079 | − 3.238 | 0.001 | 0.015 | 10.485** | |
| HOMA-β | BMI | 0.058 | 0.008 | 7.161 | < 0.001 | 0.058 | 0.008 | 6.982 | < 0.001 | ||||
| A16203G | 0.354 | 0.282 | 1.257 | 0.209 | 0.354 | 0.280 | 1.264 | 0.207 | |||||
| BMI*A16203G | − 0.159 | 0.070 | − 2.253 | 0.025 | 0.009 | 5.076* | − 0.170 | 0.070 | − 2.418 | 0.016 | 0.01 | 5.849* | |
| BMI | 0.550 | 0.008 | 6.772 | < 0.001 | 0.055 | 0.008 | 6.623 | < 0.001 | |||||
| A16316G | 0.497 | 0.220 | 2.254 | 0.025 | 0.510 | 0.221 | 2.301 | 0.022 | |||||
| BMI*A16316G | − 0.255 | 0.090 | − 2.824 | 0.005 | 0.014 | 7.977** | − 0.252 | 0.090 | − 2.790 | 0.005 | 0.013 | 7.782** | |
*p < 0.05
**p < 0.01
Model 1 was crude model; model 2 was adjusted by age, LH/FSH, and T
BMI, body mass index; HOMA-IR, homeostasis model assessment of insulin resistance; HOMA-β, homeostasis model assessment of β-cell function; SE, standard error
Fig. 3.

MtDNA gene polymorphism sites in the D-loop region moderated the association between BMI and ln (HOMA-IR), respectively (crude model). Simple slopes were plotted at wild type and variant type of two genes, respectively
Fig. 4.

MtDNA gene polymorphism sites in the D-loop region moderated the association between BMI and ln (HOMA-IR), respectively (adjusted model). The model was adjusted by age, LH/FSH, and T. Simple slopes were plotted at wild type and variant type of two genes, respectively
Fig. 5.

MtDNA gene polymorphism sites in the D-loop region moderated the association between BMI and ln (HOMA-β) (crude model). Simple slopes were plotted at wild type and variant type of two genes, respectively
Fig. 6.

MtDNA gene polymorphism sites in the D-loop region moderated the association between BMI and ln (HOMA-β) (adjusted model). The model was adjusted by age, LH/FSH, and T. Simple slopes were plotted at wild type and variant type of two genes, respectively
Lastly, we found that the selected genetic polymorphism sites were not generalizable to women without PCOS (Supplementary Table S2 and Table S4).
Discussion
To the best of our knowledge, this is the first study to investigate whether the associations of BMI with HOMA-IR and HOMA-β in patients with PCOS were moderated by polymorphisms of mtDNA in the D-loop region. In our study, we found that the variant-type of m.16217 T > C enhanced the association between BMI and HOMA-IR, and variant-type of m.16316 A > G attenuated the association between BMI and HOMA-IR; variant-type of m.16203 A > G and m.16316 A > G attenuated the association between BMI and HOMA-β. These results offer an explanation for why PCOS patients exhibit different effects on improving insulin sensitivity after weight loss in clinical practice.
Consistent with our findings, Dabravolski et al. found that the pathogenesis of PCOS was related to mitochondrial dysfunction [21]. mtDNA mutations and ROS release can contribute to IR and obesity. Zhuo et al. identified base changes in the D-loop region of mtDNA, rRNA mutations, and variation at other sites in PCOS patients, indicating that mtDNA mutations may be a pathogenesis of PCOS [22]. Ding et al. also found mtDNA mutation sites that may be related to HOMA-IR in 80 PCOS-IR patients [23]. Combining our findings with these previous studies, for PCOS patients with a specific variant-type of mtDNA that enhances the association of BMI with HOMA-IR, insulin sensitizers, such as inositol isoforms, may need to be recommended. Studies have shown that myo-inositol (MI) and D-chiro-inositol (DCI) supplements have synergistic effects with other insulin sensitizers, and supplementation of inositol levels can improve insulin resistance, hyperandrogenism, oocyte quality, and the regularity of menstrual cycles in women with PCOS [24, 25]. Other studies have also shown that MI plus folic acid ameliorated insulin sensitivity, biochemical hyperandrogenism, and regularity of the menstrual cycle in patients with PCOS with high BMI [26]. Additionally, a Mediterranean diet may also need to be recommended for PCOS patients with a specific variant-type of mtDNA that enhances the association of BMI with HOMA-IR since it has been reported to have a beneficial effect on changes in glucose tolerance, insulin resistance, and lipid metabolism [27]. A cohort study of 7569 Australian women with PCOS showed that PCOS improved with lifestyle changes as well as the intake of a Mediterranean diet [28]. Recent evidence has also suggested that low-carbohydrate, ketogenic diets have beneficial effects on weight loss and improvement of insulin resistance [29]. Eleven PCOS patients with BMI > 27 kg/m2 were given a 24-week low ketogenic diet and reported significant improvements in body weight, percentage of free testosterone, LH/FSH ratio, and fasting insulin [30]. Additionally, diet restriction and fasting may also need to be recommended. Under fasting conditions, the metabolic imbalance typical of PCOS may improve significantly [31, 32]. However, the report by Rabol et al. did not support that mtDNA mutations were associated with skeletal muscle IR in PCOS patients [33]. In our present study, we also found that the variant-type of m.16316 A > G attenuated the association between BMI and HOMA-IR.
The D-loop region is a regulatory site for the transcription and replication of mtDNA. Mutations in this region can affect the function of mitochondria, which in turn increases ROS production, resulting in decreased mtDNA copy number and decreased ATP production [23, 34]. The reduction in mitochondrial ATP production reduced the binding efficiency of insulin to the insulin receptor on the cell surface, and the phosphorylation efficiency of the β subunit of the insulin receptor under the action of tyrosine kinase was reduced, which can affect the activation of the insulin receptor substrate and lead to dysfunction of the PI3K/AKT signaling pathway [35]. ROS can activate the serine-threonine kinase signaling pathway, increase the phosphorylation of insulin receptor and its substrate proteins, reduce the efficacy of insulin, and thus cause IR. Interestingly, we found that the variant-type of m.16316 A > G can weaken the associations of BMI with HOMA-IR and HOMA-β simultaneously. The simultaneously weakened associations of BMI with HOMA-IR and HOMA-β may be because increased IR in PCOS patients requires a compensatory increase in insulin secretion to maintain blood glucose homeostasis caused by mtDNA mutation [8].
However, some limitations in our study should also be discussed. First, it was not a multicentre study, and we only recruited PCOS patients in the Reproductive Medicine Center of the First Affiliated Hospital of Anhui Medical University, China. Whether our conclusions can be extrapolated to global PCOS patients remains to be explored. Second, previous studies have shown that diet and exercise can adjust the relationships of BMI with HOMA-IR and HOMA-β [36], but we did not collect data on these variables in our present study. Third, compared to total testosterone, free testosterone can better reflect biochemical hyperandrogenemia in women with PCOS. Unfortunately, we did not measure free testosterone in female patients in our center. Fourth, we did not evaluate the modified Ferriman-Gallway (FG) scores, and we did not focus on the versions of hyperandrogenisms, including clinical, so the moderating effect of polymorphisms of mtDNA in the D-loop region on the associations of BMI with HOMA-IR and HOMA-β among PCOS patients with different subtypes cannot be evaluated. Fifth, the relationship of the different types of PCOS, such as high androgen and no menses, high androgen and polycystic ovaries, normal cycles, polycystic ovaries, and irregular cycles, and the different associations between polymorphisms of mtDNA and HOMA results cannot be evaluated by further statistical analyses since that PCOS was only diagnosed based on the revised 2003 Rotterdam criteria in the present study. The subtypes of different PCOS phenotypes cannot be obtained via the electronic medical record system in our reproduction center. Finally, this is a cross-sectional study, and no causal relationship can be obtained.
In conclusion, we found that polymorphisms of mtDNA in the D-loop region moderated the relationships of BMI with ln(HOMA-IR) and ln(HOMA-β) in patients with PCOS. Some mutations of mtDNA gene sites can enhance the associations, while other mutations of mtDNA gene sites can weaken the associations (Fig. 7). Therefore, different intervention plans should be performed on PCOS patients with different mutant types.
Fig. 7.

Polymorphisms of mtDNA in the D-loop region moderated the associations of BMI with HOMA-IR and HOMA-β among women with polycystic ovary syndrome
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
The authors are grateful to the Scientific Research Center in Preventive Medicine, School of Public Health, Anhui Medical University, for the technical support during our experiments.
Author contribution
Shitao He: methodology, formal analysis, data curation, writing – original draft. Dongmei Ji: methodology, formal analysis, visualization, writing – original draft, funding acquisition. Yajing Liu: methodology, investigation. Xiaohong Deng: methodology, formal analysis. Weiwei Zou: methodology, data curation. Dan Liang: methodology, data curation. Yinan Du: methodology. Kai Zong: Methodology. Tingting Jiang: methodology. Mengzhu Li: methodology. Dongyang Zhang: methodology. Xinyu Yue: methodology. Fangbiao Tao: conceptualization, project administration, funding acquisition, resources, supervision, writing – review and editing. Yunxia Cao: conceptualization, funding acquisition, project administration, writing – review and editing. Chunmei Liang: conceptualization, funding acquisition, supervision, writing – review and editing. All authors have read and approved the final version of the manuscript.
Funding
This work was supported by the National Key Research and Development Program (2021YFC2700901, 2018YFC1004201), the National Natural Science Foundation of China (NSFC-82173532, NSFC-U20A20350, NSFC-81971455, NSFC-81871216, and NSFC-81803260), the Excellent Young Talents Fund Program of Higher Education Institutions of Anhui Province (gxyq2021173), and the Postdoctoral Research Foundation of China (2021M700181).
Data Availability
The authors do not have permission to share data.
Declarations
Conflict of interest
The authors declare no competing interests.
Footnotes
The original online version of this article was revised: Several mistakes in the main text were missed before this article was published; the authors apologize for any inconvenience caused.
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Shitao He, Dongmei Ji, Yajing Liu and Xiaohong Deng contributed equally to this work.
Change history
7/17/2023
A Correction to this paper has been published: 10.1007/s10815-023-02884-y
Contributor Information
Fangbiao Tao, Email: fbtao@ahmu.edu.cn.
Yunxia Cao, Email: caoyunxia5972@ahmu.edu.cn.
Chunmei Liang, Email: liang_chun_mei@126.com.
References
- 1.Escobar-Morreale HF. Polycystic ovary syndrome: definition, aetiology, diagnosis and treatment. Nat Rev Endocrinol. 2018;14(5):270–284. doi: 10.1038/nrendo.2018.24. [DOI] [PubMed] [Google Scholar]
- 2.Fauser BCJM. Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome. Fertil Steril. 2004;81(1):19–25. [DOI] [PubMed]
- 3.McCartney CR, Marshall JC. CLINICAL PRACTICE. Polycystic ovary syndrome. N Engl J Med. 2016;375(1):54–64. doi: 10.1056/NEJMcp1514916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sagvekar P, Dadachanji R, Patil K, Mukherjee S. Pathomechanisms of polycystic ovary syndrome: multidimensional approaches. Front Biosci (Elite Ed) 2018;10(3):384–422. doi: 10.2741/e829. [DOI] [PubMed] [Google Scholar]
- 5.Patel S. Polycystic ovary syndrome (PCOS), an inflammatory, systemic, lifestyle endocrinopathy. J Steroid Biochem Mol Biol. 2018;182:27–36. doi: 10.1016/j.jsbmb.2018.04.008. [DOI] [PubMed] [Google Scholar]
- 6.Marshall JC, Dunaif A. Should all women with PCOS be treated for insulin resistance? Fertil Steril. 2012;97(1):18–22. doi: 10.1016/j.fertnstert.2011.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Malin SK, Kirwan JP, Sia CL, González F. Pancreatic β-cell dysfunction in polycystic ovary syndrome: role of hyperglycemia-induced nuclear factor-κB activation and systemic inflammation. Am J Physiol Endocrinol Metab. 2015;308(9):E770–E777. doi: 10.1152/ajpendo.00510.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Manco M, Castagneto-Gissey L, Arrighi E, Carnicelli A, Brufani C, Luciano R, et al. Insulin dynamics in young women with polycystic ovary syndrome and normal glucose tolerance across categories of body mass index. PLoS One. 2014;9(4):e92995. doi: 10.1371/journal.pone.0092995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tao T, Li S, Zhao A, Mao X, Liu W. Early impaired β-cell function in Chinese women with polycystic ovary syndrome. Int J Clin Exp Pathol. 2012;5(8):777–786. [PMC free article] [PubMed] [Google Scholar]
- 10.Yan C, Duanmu X, Zeng L, Liu B, Song Z (2019) Mitochondrial DNA: distribution, mutations, and elimination. Cells 8(4):379. [DOI] [PMC free article] [PubMed]
- 11.Mishra P, Chan DC. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat Rev Mol Cell Biol. 2014;15(10):634–646. doi: 10.1038/nrm3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murri M, Luque-Ramírez M, Insenser M, Ojeda-Ojeda M, Escobar-Morreale HF. Circulating markers of oxidative stress and polycystic ovary syndrome (PCOS): a systematic review and meta-analysis. Hum Reprod Update. 2013;19(3):268–288. doi: 10.1093/humupd/dms059. [DOI] [PubMed] [Google Scholar]
- 13.Hunter RG, Seligsohn M, Rubin TG, Griffiths BB, Ozdemir Y, Pfaff DW, et al. Stress and corticosteroids regulate rat hippocampal mitochondrial DNA gene expression via the glucocorticoid receptor. Proc Natl Acad Sci USA. 2016;113(32):9099–9104. doi: 10.1073/pnas.1602185113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ilie IR. Advances in PCOS Pathogenesis and progression-mitochondrial mutations and dysfunction. Adv Clin Chem. 2018;86:127–155. doi: 10.1016/bs.acc.2018.05.003. [DOI] [PubMed] [Google Scholar]
- 15.Skov V, Glintborg D, Knudsen S, Jensen T, Kruse TA, Tan Q, et al. Reduced expression of nuclear-encoded genes involved in mitochondrial oxidative metabolism in skeletal muscle of insulin-resistant women with polycystic ovary syndrome. Diabetes. 2007;56(9):2349–2355. doi: 10.2337/db07-0275. [DOI] [PubMed] [Google Scholar]
- 16.Saeed N, Hamzah IH, Al-Gharrawi SAR. Polycystic ovary syndrome dependency on mtDNA mutation; copy number and its association with insulin resistance. BMC Res Notes. 2019;12(1):455. doi: 10.1186/s13104-019-4453-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eaaswarkhanth M, Melhem M, Sharma P, Nizam R, Al Madhoun A, Chaubey G, et al. Mitochondrial DNA D-loop sequencing reveals obesity variants in an Arab population. Appl Clin Genet. 2019;12:63–70. doi: 10.2147/TACG.S198593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dunaif A. Insulin resistance and the polycystic ovary syndrome: mechanism and implications for pathogenesis. Endocr Rev. 1997;18(6):774–800. doi: 10.1210/edrv.18.6.0318. [DOI] [PubMed] [Google Scholar]
- 19.Witchel SF, Oberfield SE, Peña AS. Polycystic ovary syndrome: pathophysiology, presentation, and treatment with emphasis on adolescent girls. J Endocr Soc. 2019;3(8):1545–1573. doi: 10.1210/js.2019-00078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin CA, Liu YP, Chen YC, Yu W, Xiong XJ, Huang HY, et al. Gender-specific and age-specific associations of the homoeostasis model assessment for IR (HOMA-IR) with albuminuria and renal function impairment: a retrospective cross-sectional study in Southeast China. BMJ Open. 2021;11(12):e053649. doi: 10.1136/bmjopen-2021-053649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dabravolski SA, Nikiforov NG, Eid AH, Nedosugova LV, Starodubova AV, Popkova TV, et al (2021) Mitochondrial dysfunction and chronic inflammation in polycystic ovary syndrome. Int J Mol Sci. 22(8):3923. [DOI] [PMC free article] [PubMed]
- 22.Zhuo G, Ding Y, Feng G, Yu L, Jiang Y. Analysis of mitochondrial DNA sequence variants in patients with polycystic ovary syndrome. Arch Gynecol Obstet. 2012;286(3):653–659. doi: 10.1007/s00404-012-2358-7. [DOI] [PubMed] [Google Scholar]
- 23.Ding Y, Xia BH, Zhang CJ, Zhuo GC. Mutations in mitochondrial tRNA genes may be related to insulin resistance in women with polycystic ovary syndrome. Am J Transl Res. 2017;9(6):2984–2996. [PMC free article] [PubMed] [Google Scholar]
- 24.Paul C, Laganà AS, Maniglio P, Triolo O, Brady DM. Inositol’s and other nutraceuticals’ synergistic actions counteract insulin resistance in polycystic ovarian syndrome and metabolic syndrome: state-of-the-art and future perspectives. Gynecol Endocrinol. 2016;32(6):431–438. doi: 10.3109/09513590.2016.1144741. [DOI] [PubMed] [Google Scholar]
- 25.Laganà AS, Rossetti P, Buscema M, La Vignera S, Condorelli RA, Gullo G, et al. Metabolism and ovarian function in PCOS women: a therapeutic approach with inositols. Int J Endocrinol. 2016;2016:6306410. doi: 10.1155/2016/6306410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Genazzani AD, Lanzoni C, Ricchieri F, Jasonni VM. Myo-inositol administration positively affects hyperinsulinemia and hormonal parameters in overweight patients with polycystic ovary syndrome. Gynecol Endocrinol. 2008;24(3):139–144. doi: 10.1080/09513590801893232. [DOI] [PubMed] [Google Scholar]
- 27.Esposito K, Marfella R, Ciotola M, Di Palo C, Giugliano F, Giugliano G, et al. Effect of a mediterranean-style diet on endothelial dysfunction and markers of vascular inflammation in the metabolic syndrome: a randomized trial. JAMA. 2004;292(12):1440–1446. doi: 10.1001/jama.292.12.1440. [DOI] [PubMed] [Google Scholar]
- 28.Moran LJ, Grieger JA, Mishra GD, Teede HJ. The association of a Mediterranean-style diet pattern with polycystic ovary syndrome status in a community cohort study. Nutrients. 2015;7(10):8553–8564. doi: 10.3390/nu7105419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boden G, Sargrad K, Homko C, Mozzoli M, Stein TP. Effect of a low-carbohydrate diet on appetite, blood glucose levels, and insulin resistance in obese patients with type 2 diabetes. Ann Intern Med. 2005;142(6):403–411. doi: 10.7326/0003-4819-142-6-200503150-00006. [DOI] [PubMed] [Google Scholar]
- 30.Mavropoulos JC, Yancy WS, Hepburn J, Westman EC. The effects of a low-carbohydrate, ketogenic diet on the polycystic ovary syndrome: a pilot study. Nutr Metab (Lond) 2005;2:35. doi: 10.1186/1743-7075-2-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chiofalo B, Laganà AS, Palmara V, Granese R, Corrado G, Mancini E, et al. Fasting as possible complementary approach for polycystic ovary syndrome: hope or hype? Med Hypotheses. 2017;105:1–3. doi: 10.1016/j.mehy.2017.06.013. [DOI] [PubMed] [Google Scholar]
- 32.Muscogiuri G, Palomba S, Laganà AS, Orio F. Current insights into inositol isoforms, Mediterranean and ketogenic diets for polycystic ovary syndrome: from bench to bedside. Curr Pharm Des. 2016;22(36):5554–5557. doi: 10.2174/1381612822666160720160634. [DOI] [PubMed] [Google Scholar]
- 33.Rabøl R, Svendsen PF, Skovbro M, Boushel R, Schjerling P, Nilas L, et al. Skeletal muscle mitochondrial function in polycystic ovarian syndrome. Eur J Endocrinol. 2011;165(4):631–637. doi: 10.1530/EJE-11-0419. [DOI] [PubMed] [Google Scholar]
- 34.Ding Y, Zhuo G, Zhang C. The Mitochondrial tRNALeu(UUR) A3302G Mutation may be associated with insulin resistance in woman with polycystic ovary syndrome. Reprod Sci (Thousand Oaks, Calif) 2016;23(2):228–233. doi: 10.1177/1933719115602777. [DOI] [PubMed] [Google Scholar]
- 35.Siddle K. Signalling by insulin and IGF receptors: supporting acts and new players. J Mol Endocrinol. 2011;47(1):R1–10. doi: 10.1530/JME-11-0022. [DOI] [PubMed] [Google Scholar]
- 36.Bahadur A, Verma N, Mundhra R, Chawla L, Ajmani M, Sri MS, et al. Correlation of homeostatic model assessment-insulin resistance, anti-mullerian hormone, and BMI in the characterization of polycystic ovary syndrome. Cureus. 2021;13(6):e16047. doi: 10.7759/cureus.16047. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors do not have permission to share data.