

SUMMARY
The diversity of mononuclear phagocyte (MNP) subpopulations across tissues is one of the key physiological characteristics of the immune system. Here, we focus on understanding the metabolic variability of MNPs through metabolic network analysis applied to three large-scale transcriptional datasets: we introduce (1) an ImmGen MNP open-source dataset of 337 samples across 26 tissues; (2) a myeloid subset of ImmGen Phase I dataset (202 MNP samples); and (3) a myeloid mouse single-cell RNA sequencing (scRNA-seq) dataset (51,364 cells) assembled based on Tabula Muris Senis. To analyze such large-scale datasets, we develop a network-based computational approach, genes and metabolites (GAM) clustering, for unbiased identification of the key metabolic subnetworks based on transcriptional profiles. We define 9 metabolic subnetworks that encapsulate the metabolic differences within MNP from 38 different tissues. Obtained modules reveal that cholesterol synthesis appears particularly active within the migratory dendritic cells, while glutathione synthesis is essential for cysteinyl leukotriene production by peritoneal and lung macrophages.
Graphical abstract
In brief
Gainullina et al. describe metabolic variability of the mononuclear phagocytes across 38 mouse tissues using GAM-clustering, a metabolic network analysis approach. The approach is applied to large-scale transcriptional datasets (ImmGen MNP OS and phase 1) and single-cell RNA-seq data (Tabula Muris) and highlights consistent metabolic modules across cells and tissues.
INTRODUCTION
The diversity of the myeloid cells across different tissues is truly astonishing, both in function and in their developmental trajectory.1,2 An additional dimension of this diversity is manifested by the metabolic characteristics of the individual mononuclear phagocytes, which can vary significantly based on the cell type and its location.3–5 At present, direct metabolomic profiling of tissue-residing subpopulations is not feasible, as the process of ex vivo sorting can be lengthy and cause significant metabolic perturbations.6,7 However, RNA levels are significantly more stable in the sorting process and can serve as a reasonably reliable proxy for activities of metabolic pathways.8,9 In this work, we focus on understanding metabolic variability across phagocytic subpopulations through integrated examination of several large-scale datasets that transcriptionally profiled subsets of myeloid cells (Figures 1A–1C). Specifically, we have assembled a compendium of three datasets, including the public release of the dataset generated by the mononuclear phagocyte open-source (MNP OS) ImmGen project.10
Figure 1. General overview of ImmGen mononuclear phagocytes open-source (IG MNP OS), ImmGen mononuclear phagocytes phase 1 (IG MNP P1), and myeloid Tabula Muris Senis (mTMS) datasets.
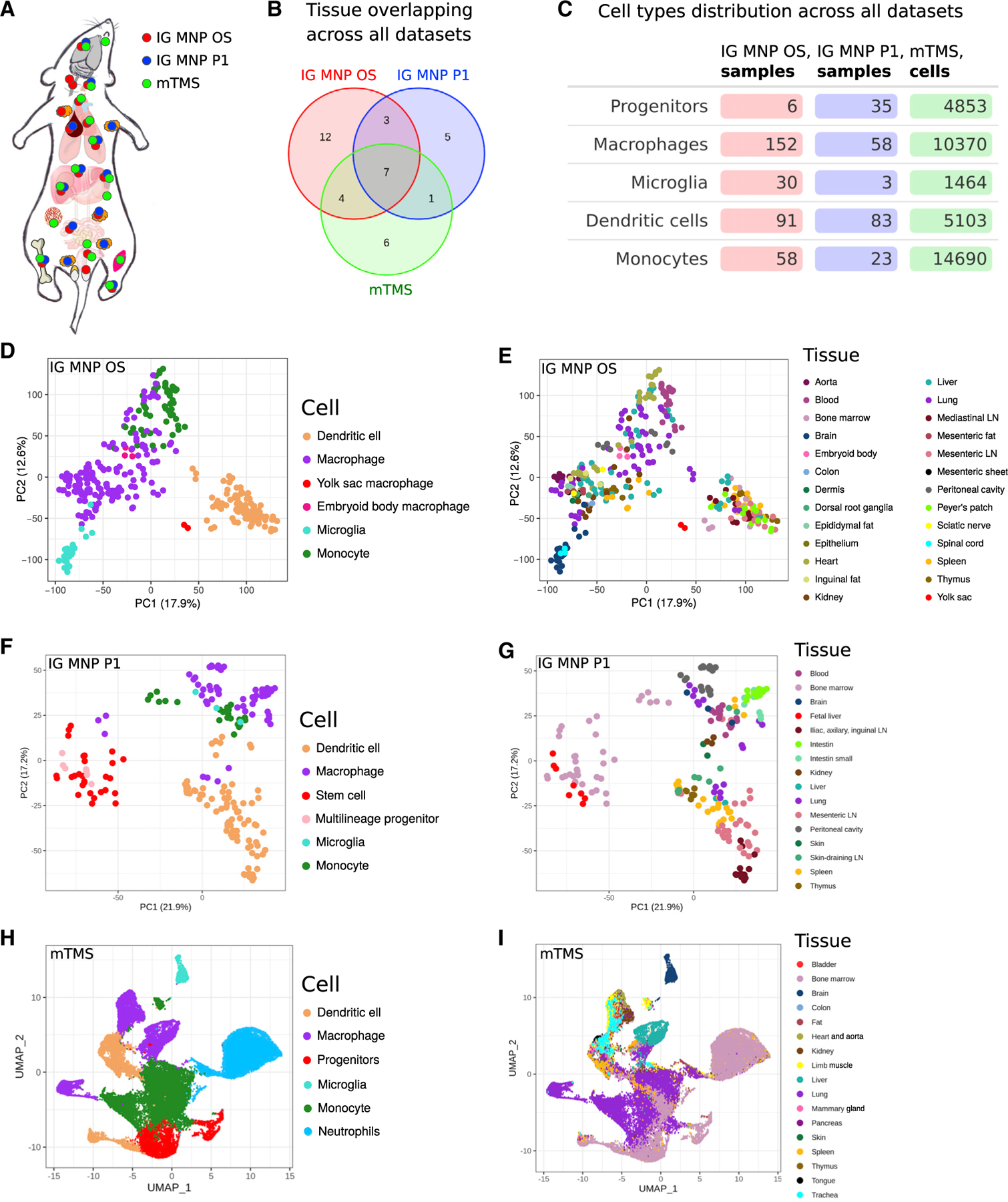
(A) Schematic representation of Mus musculus tissues, where samples were derived from (marked with colored dots depending on the dataset).
(B) Number of tissues overlapping across all datasets.
(C–G) Cell-type distribution across all datasets. Principal-component analysis (PCA) based on 12,000 most expressed genes across all samples colored by the tissue of its origin (E and G) or cell type (D and F).
(H and I) Uniform manifold approximation and projection (UMAP) representation of cells colored by the tissue of its origin (I) or its type (H). LN, lymph node.
The ImmGen MNP OS dataset totals 337 samples and provides a source of information about individual cell subpopulations (Figures 1D and 1E). It extends the previous ImmGen effort that included 202 samples of various MNPs, also analyzed in this study (Figures 1F and 1G). In addition to an increased number of mature cell populations from adult mice (monocytes, macrophages, and dendritic cells), the MNP OS dataset contains macrophages from the yolk sac (embryonic day 10.5 [E10.5]) and macrophages differentiated in vitro from embryonic stem cells (embryoid body-derived macrophages, E6–E8). Furthermore, we leveraged recently released single-cell RNA sequencing (RNA-seq) profiling of the multiple murine organs (Tabula Muris Senis11) and reanalyzed those data by focusing only on the phagocytic populations across 18 tissues (Figures 1H and 1I). Taken together, a compendium of data assembled in this work covers multiple cell subpopulations found across 38 different tissues (Figure 1B).
Using these transcriptional data, we sought to identify the major metabolic features characteristic of the different populations of phagocytic cells and define how these features vary across the cell types and their locations. Such a computational task has not been addressed previously for data of such scale. Indeed, we previously described a computational approach, called genes and metabolites (GAM),12 that uses metabolic networks as the backbone for analysis of transcriptional data and provides a verifiable and systematic description of the metabolic differences between two conditions.9 However, the datasets in question contain hundreds or even thousands of individual profiles, while the GAM approach is designed to analyze comparisons between two conditions. Therefore, we have developed a computational approach, GAM-clustering, which performs an unbiased search of a collection of metabolic subnetworks that jointly define metabolic variability across large datasets (available on GitHub; see STAR Methods). By doing so, GAM-clustering reveals metabolically similar subpopulations in a manner that does not require explicit annotation or pairwise comparison of individual samples. We demonstrate that this approach is more powerful in terms of identifying metabolic modules compared with conventional gene expression clustering approaches. Our analysis revealed major metabolic features associated with different cell subpopulations and highlighted several metabolic modules that are specific to individual cell types, tissues of residence, or developmental stages. As an example, GAM-clustering analysis revealed that the cholesterol de novo synthesis pathway might play an important role in the context of migratory dendritic cells (DCs), which we validated by measuring membrane cholesterol levels in migratory and tissue-resident DCs and using in vivo pharmacological inhibition of cholesterol synthesis followed by tracking of DC migration. As a second example, GAM-clustering revealed the antioxidant system as an important accompaniment of arachidonic acid metabolism during inflammatory response in tissue macrophages. Experimental measurement of cysteinyl leukotrienes production levels after inhibiting glutathione synthesis showed a biological effect of this system in peritoneal and lung alveolar macrophages.
Taken together, our work provides both (1) a data and analysis resource in terms of studying the variability of MNPs, as well as (2) a validated computational approach that can unbiasedly analyze both single-cell RNA-seq data as well as multi-sample bulk RNA-seq datasets in terms of key underlying metabolic features. Furthermore, we provide direct interactive access to the data for examination and visualization through both single-cell RNA-seq and bulk RNA-seq visualization servers, including metabolic cluster annotations obtained in this work (https://artyomovlab.wustl.edu/immgen-met/).
RESULTS
MNP OS and ImmGen Phase 1 (MNP P1) datasets
As a part of the OS ImmGen Project, a total of 337 samples were collected and profiled through the collaborative effort of 16 laboratories (Figures 1D, 1E, and S1A; Table S1). Each laboratory sorted specific populations of MNPs from 26 distinct tissues, isolated RNA from these populations, and submitted it for centralized deep RNA-seq and subsequent quantitation. Along with their samples of interest, each laboratory included RNA from locally sorted peritoneal macrophages as a common control for evaluation/correction of potential batch effects (STAR Methods, RNA-seq data processing). Of note, 15 samples from the MNP OS dataset were previously used in the study of sexual dimorphism of the immune system transcriptome,13 while the complete dataset has not been analyzed before this work.
Overall, the transcriptional data demonstrated high concordance between different collection sites and were merged into a final transcriptional master table (Figures S1B and S1C; Data S1). Previously established markers of individual myeloid subpopulations14–18 matched well with the sorted populations (Figure S2), indicating the overall consistency of the dataset across different research groups. As individual principal-component analysis (PCA) plots show (Figure 1D), samples have clustered in accord with their broad annotation as macrophages, DCs, monocytes, or microglia and not in terms of lab sorting or in terms of sequencing batch. Generally, subpopulation-specific effects were stronger than tissue-specific differences within individual subpopulations, as evident by comparing Figures 1D and 1E. To estimate the degree of metabolic variability in the data, we examined the enrichment of annotated metabolic pathways in this dataset, revealing coherent transcriptional patterns across individual subpopulations (Figure S3A). This indicated that systematic evaluation of the metabolic subnetworks within the data is warranted.
Initial ImmGen P1 data published previously5 include 202 samples of MNPs with a higher contribution of progenitor populations and a smaller number of microglial samples (Figure 1F) overall spanning 16 tissues (Figure 1G)—we will refer to this subset as ImmGen MNP P1 from here onward. Similar to the MNP OS dataset, enrichment in metabolic pathways across subpopulations in MNP P1 data demonstrated coordinate variations across the samples (Figure S3B).
Single-cell myeloid Tabula Muris Senis (mTMS) dataset
The Tabula Muris consortium has performed single-cell RNA-seq for many tissues without explicit sorting into individual cell populations.11 These data include myeloid cells localized in the corresponding tissues, which can be computationally separated based on the expression of common myeloid signatures. Using the latest public dataset, TMS, we have analyzed the data for 235,325 cells to identify 51,364 myeloid cells (MNPs and neutrophils) that expressed key myeloid markers (Lyz2, H2-Aa, Mki67, S100a9, Flt3, Emr1, Ccr2, Cx3cr1, Sall1, Clec4f, Lyve1, Itgax, Xcr1, Clec4a4, Siglech, Ccr7; Figures 2A and 2D; STAR Methods). These cells comprised a dedicated dataset, further referred to as the mTMS dataset. While single-cell RNA-seq data inevitably detect a smaller number of genes per cell compared with bulk RNA-seq (Figure S4), the depth of the mTMS dataset was sufficient to resolve classical cell populations. Specifically, unbiased clustering revealed 15 subpopulations within the mTMS dataset (Figure 2B), which could be readily identified as well-described populations of plasmacytoid DCs, monocytes, Kupffer cells, microglia, and other cell types (Figure 2C) based on previously described cell-specific markers (Figure 2D). To our knowledge, we provide the first large-scale curated annotation for the myeloid cells within TMS data. Corresponding annotations are available for hands-on exploration in the interactive single-cell browser (https://artyomovlab.wustl.edu/immgen-met/, see TMS).
Figure 2. TMS single-cell RNA-seq dataset.
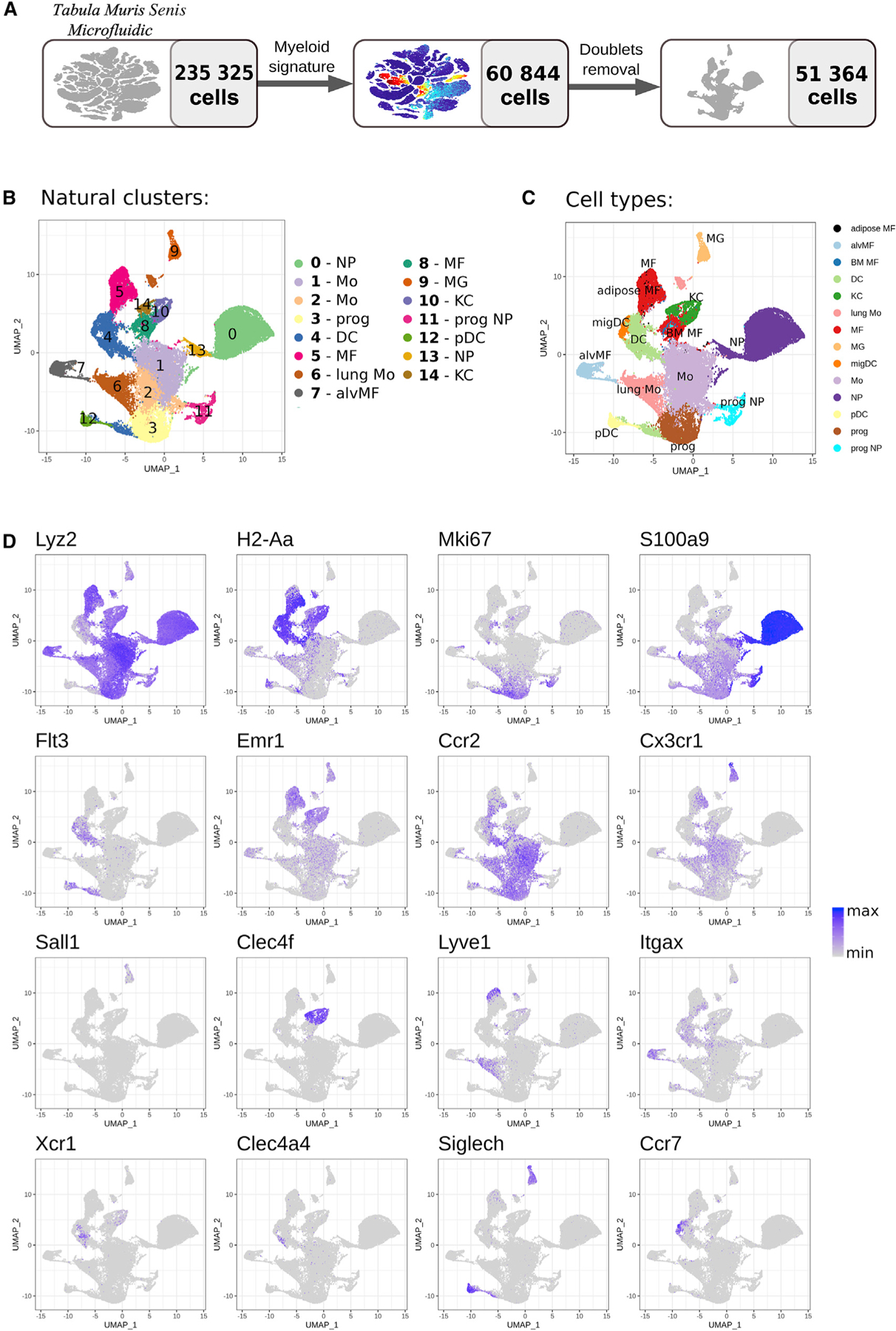
(A) Dataset preprocessing resulting in myeloid subset derivation.
(B–D) UMAP plot with natural clusters (B) and cell types (C) identified based on cell specific markers (D). NP, neutrophil; Mo, monocyte; prog, progenitor; DC, dendritic cell; MF, macrophage; alvMF, alveolar macrophage; MG, microglia; KC, Kupffer cell; pDC, plasmacytoid dendritic cell; migDC, migratory dendritic cell.
GAM-clustering: Identification of metabolic subnetworks in datasets with multiple conditions
Previously, we have shown that metabolic remodeling between two conditions can be analyzed using network-based analysis of their transcriptional profiles.9,12 Specifically, the GAM algorithm searches for optimal subnetworks within a global metabolic network by weighing individual enzymes in accord with the differential expression of their genes and then solving the generalized maximum-weight connected subgraph (GMWCS) problem.12,19 While this approach cannot be directly translated to multi-sample/single-cell datasets such as ImmGen or TMS data, we were able to reformulate the weighting scheme in a manner that allows a GMWCS subnetwork search without explicit annotation of individual samples or conditions. Here, we describe a GAM-clustering method that allows the user to obtain metabolic subnetworks enriched within the transcriptional data that include many samples across multiple conditions.
In brief, GAM-clustering searches for connected metabolic subnetworks that have most correlated expressions of individual enzymes, resulting in a collection of subnetworks that follow distinct transcriptional profiles. To achieve that, we first initialize pattern-generating profiles by clustering all metabolic genes based on their co-expression patterns (Figure 3; see STAR Methods for details). For initialization of the multi-sample bulk RNA-seq data, we use k-medoids clustering with k = 32 (see Figures S5A and S5B for parameter sensitivity); any other gene expression clustering approach can be used in this step since downstream steps include significant regrouping and merging of individual clusters. For initialization of single-cell RNA-seq data, we first cluster the cells in the dataset into multiple clusters (~100), which provides a sufficient balance between fine resolution of the data and minimal coarse graining needed to avoid drop-out artifacts (see STAR Methods for details). Then, genes are clustered using the same procedure as for multi-sample bulk datasets.
Figure 3. Scheme of analysis approach for multi-sample metabolic network clustering (GAM clustering).
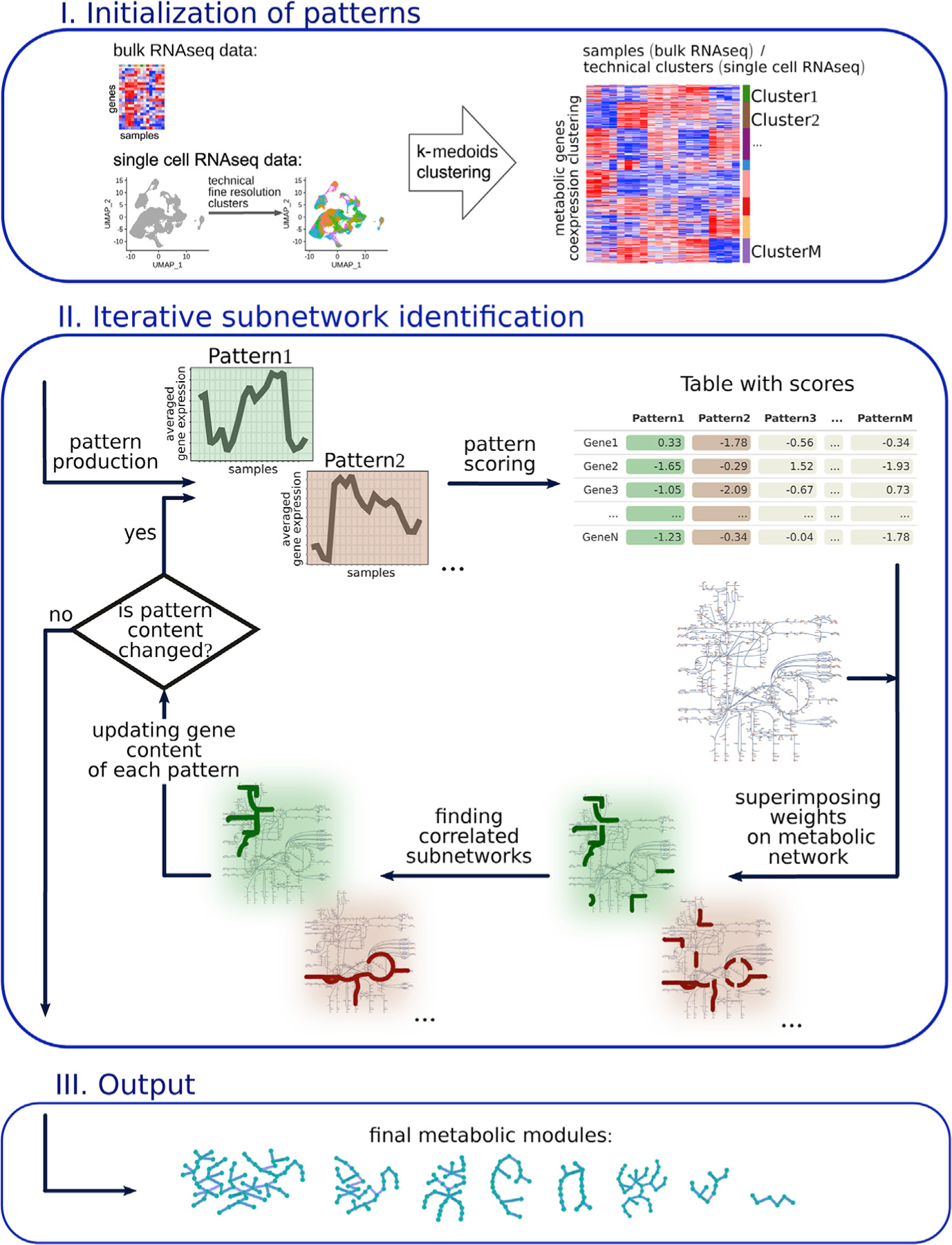
The dataset’s metabolic genes are initially clustered based on a k-medoids algorithm. Averaged gene expression of the obtained clusters is further considered as patterns. For each gene, a score is calculated on the basis of its correlation with each pattern. These scores are superimposed on the KEGG metabolic network. Based on these scores, the most weighted connected subnetwork is found for each parent. After the refinement procedure, metabolic modules as a final version of subnetworks are obtained.
Next, each enzyme is weighed with respect to the similarity of its transcript’s profile to the average cluster/pattern profile, resulting in multiple weights per gene that are specific for each pattern (Figure 3). For any given pattern, weights of individual enzymes serve as input to the GMWCS solver, resulting in the individual subnetworks that are associated with each pattern. Individual subnetworks are then refined in an iterative procedure of updating the gene content for each pattern (see STAR Methods for details). The final output presents a set of specific subnetworks that reflect metabolic variability within a given transcriptional dataset (Figure 3).
GAM-clustering improves recognition of metabolic modules over unbiased clustering approaches
GAM-clustering utilizes preconceived knowledge about the underlying metabolic network and therefore is expected to be more powerful in terms of finding metabolically enriched modules. To evaluate this expectation, we compared GAM-clustering with commonly used unbiased clustering approaches such as weighted gene co-expression network analysis (WGCNA) and k-medoids (with Pearson correlation distance as a distance measure), which both allow identifying clusters of genes with high pairwise correlation. It is worth noting that the k-medoids approach is used as a part of the GAM-clustering method for generating the initial approximation (see STAR Methods for more details). Unlike GAM-clustering, both WGCNA and k-medoids methods do not take into consideration the network structure of metabolic reactions, so we expect that GAM-clustering improves recognition of metabolically enriched modules over these clustering approaches. To this end, we clustered 1,837 metabolic genes in the ImmGen MNP OS dataset using GAM-clustering, WGCNA, and k-medoids methods. As the k-medoids method requires an explicit setting of the parameter k equal to the number of produced clusters, k values were chosen to be equal to 10 and 20 to make the results comparable with 9 and 19 clusters unbiasedly produced by GAM-clustering and WGCNA methods, respectively. In an additional instance of the k-medoids clustering, k was set equal to 32 since this value was used for initial approximation production in the GAM-clustering method.
For each clustering method, obtained clusters (modules) were examined against 80 KEGG murine metabolic pathways by hypergeometric test (common and descriptive pathways corresponding to KEGG’s “global and overview maps” were not considered in order to increase the specificity of annotation; pathways with less than 10 constituting genes were also excluded). Modules’ overlap with the individual KEGG metabolic pathways was also analyzed in terms of the p value (p adjusted in the case of multiple comparisons) (Figure S6A) and the percentage of module’s covered genes (Figure S6B). For these comparisons, modules obtained by the GAM-clustering method demonstrate significantly higher enrichment in selected metabolic pathways in both metrics (p value and number of overlapping genes) compared with other clustering methods. Moreover, WGCNA and k-medoids have identified many modules without significant overlap with any KEGG metabolic pathway. This observation confirms that modules found by GAM-clustering are more relevant in terms of identifying metabolic features of the underlying dataset. Note that the GAM-clustering method does not specifically enforce enrichment in KEGG pathways but rather leverages the global metabolic network structure. An additional advantage of GAM-clustering is its interpretability, as modules produced by GAM-clustering have moderate sizes from 5 to 39 genes and are composed of enzymes topologically closely located on the metabolic network (Figure S6C).
Major metabolic modules within MNP subpopulations
The GAM-clustering method was applied to data from all baseline (non-infected) samples and yielded nine distinct metabolic modules (Figure 4A; Table S2). Hierarchical clustering of samples based on the Euclidean distance metric in the space of these nine metabolic modules showed that they could be broadly separated based on the cell types: yolk sac macrophages, DCs, monocytes, and macrophages from an adult organism. Broadly defined mononuclear cell types are further split into several smaller metasamples: DCs subdivided into plasmacytoid DCs (pDCs), tissue-specific DCs, and migratory DCs (migDCs) and macrophages subdivided into microglia, adipose tissue macrophages, and a large metasample of tissue-residing macrophages, as well as an additional metasample composed of embryoid body, alveolar, and small peritoneal macrophages (SPMs) that clustered distinctly from other macrophage subpopulations (Figure 4A).
Figure 4. Metabolic modules as a result of multi-sample metabolic network clustering of all myeloid cells but not inflammatory conditions from ImmGen MNP OS dataset.
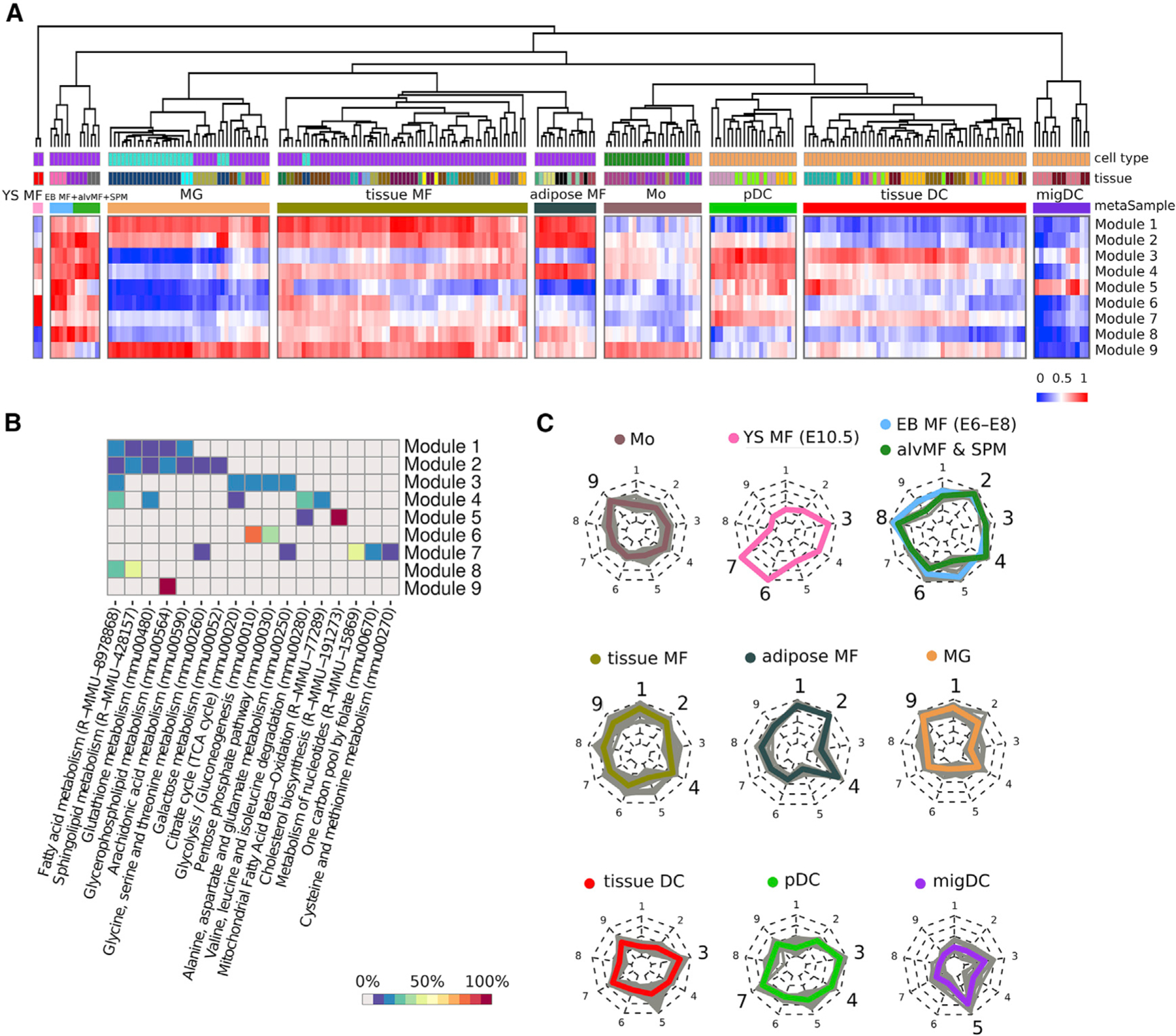
(A) Heatmap representing samples hierarchically clustered based on averaged gene expression of each of obtained module (from lowest as blue to highest as red). Euclidean distance is used as a clustering metric. YS MF, yolk sac macrophage; EB MF, embryoid body macrophage; alvMF, alveolar macrophage; SPM, small peritoneal macrophage; MG, microglia; MF, macrophage; Mo, monocyte; DC, dendritic cell; pDC, plasmacytoid DC; migDC, migratory DC.
(B) Annotation of the obtained modules based on gene enrichment in KEGG and Reactome canonical pathways. Enrichment value is calculated as a percentage of module genes contained in a particular pathway.
(C) Radar chart representation of metabolic modules within each metasample. Each individual sample is shown as a gray line, while mean of all samples inside one metasample is shown as a colored line. Nine radii of the radar chart are devoted to the corresponding metabolic modules: 1 and 2: lipid metabolism; 3: FAS pathway; 4: mtFASII pathway; 5: cholesterol synthesis; 6: glycolysis; 7: folate, serine, and nucleotide metabolism; 8: FAO and sphingolipid de novo synthesis; and 9: glycerophospholipid metabolism. Metasamples of EB MFs + alvMFs and alvMFs + SPM cells are shown at one chart as they are extremely close in their metabolic characteristics.
While obtained metabolic modules/subnetworks provide a more accurate description of metabolic diversity compared with canonically annotated pathways, the latter can be useful for coarse-grained understanding of functionalities associated with each subnetwork (Figure 4B). Indeed, pathway enrichment analysis along with subnetwork gene content analysis indicate that modules 1, 2, 8, and 9 represent various aspects of lipid metabolism, and modules 3 and 4 represent two types of fatty acid synthesis pathways. Finally, distinct modules represent cholesterol synthesis metabolism- (module 5), glycolysis- (module 6), and nucleotide/folate metabolism-associated subnetworks (module 7).
The underlying metabolic phenotypes for each metasample can be represented using radar chart diagrams (Figure 4C): each metasample is defined by a specific combination of metabolic features that provides insights into metabolic wiring within those populations. Here, the names of metasamples are given based on the most common sample type inside the cluster. An alternative view of the samples in the space of metabolic modules can be obtained using PCA that is built based on only 9 metabolic modules, which shows the distinct separation of individual metasamples (Figure S6D). Consistently, when overlaid with the PCA representation from Figure 1, individual metabolic modules formed coherent patterns indicating the groups of metabolically similar samples (Figure S6E). Altogether, the metabolic modules/subnetworks and corresponding metasamples encapsulate metabolic variability across both cell types and their tissues of residency. We next turn to examine the robustness of the obtained subnetworks across three considered datasets.
Three independent large-scale datasets show consistent metabolic features
We next considered if metabolic subnetworks derived from ImmGen MNP OS data can be seen in the other two large-scale datasets considered in this work—ImmGen MNP P1 and mTMS datasets. While overlap in profiled tissues is considered (Figure 1B), three datasets are not identical in terms of populations profiled. We therefore grouped the samples into 19 general classes and compared the datasets by looking at the metabolic enrichments across these classes (Figure 5A; Table S3). To examine the robustness of metabolic signatures, we computed enrichments of individual metabolic modules from Figure 4A in each of the 19 representative classes of ImmGen MNP OS, ImmGen MNP P1, and mTMS. Indeed, all dataset modules demonstrated extremely similar enrichment profiles (Figure 5B): for instance, module 1 was enriched in microglia, adipose tissue macrophages, and Kupffer cells, module 8 was enriched in alveolar macrophages, and module 5 was enriched in pDCs and migDCs across all datasets.
Figure 5. Cell types shared between IG MNP OS, IG MNP P1, and mTMS datasets have similar patterns of metabolic modules signatures.
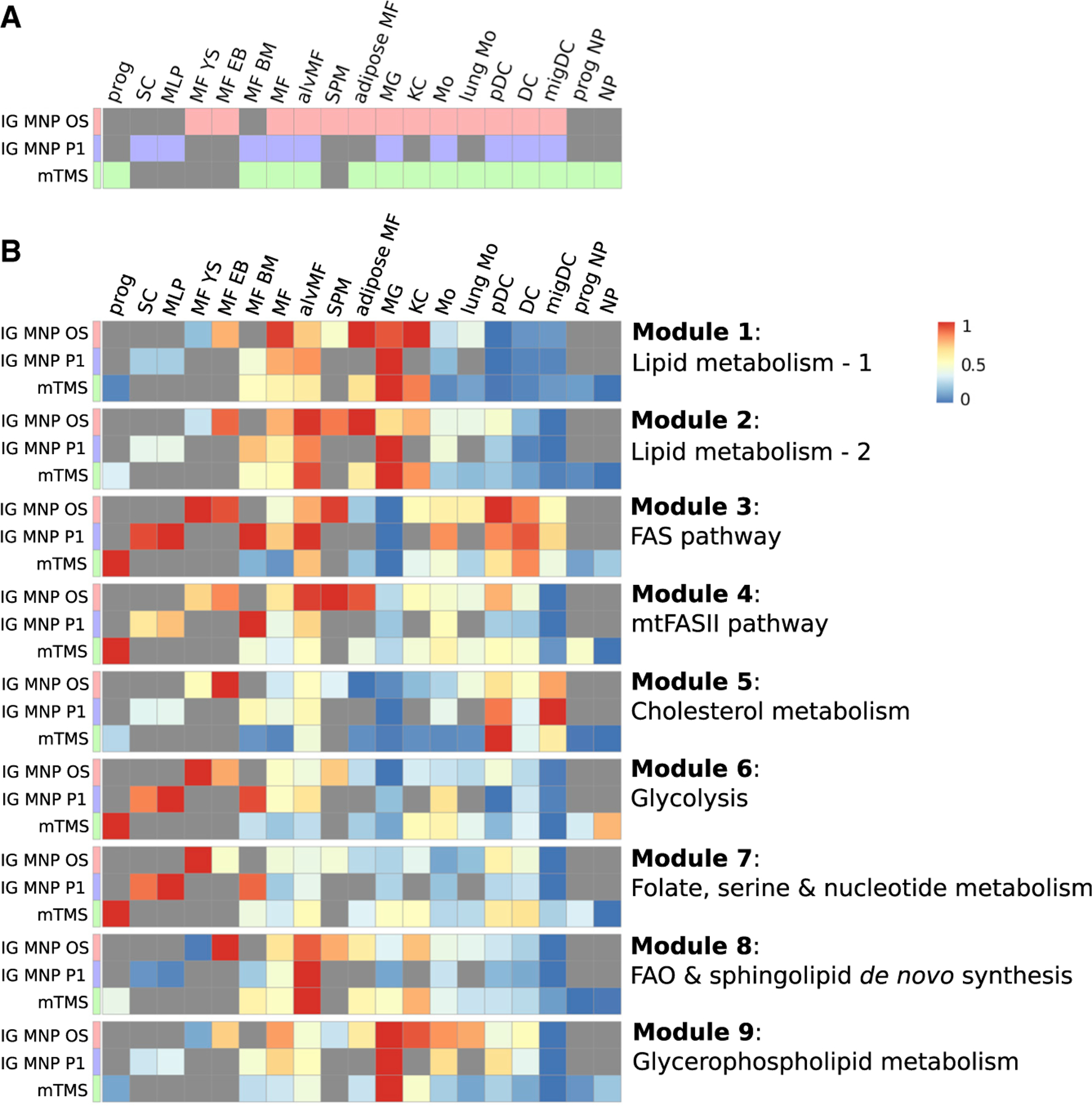
(A) Population memberships across the datasets: prog, progenitor; SC, stem cell; MLP, multi-lineage progenitor; MF YS, yolk sac macrophage; MF EB, embryoid body macrophage; MF, macrophage; alvMF, alveolar macrophage; SPM, small peritoneal macrophage; MG, microglia; KC, Kupffer cell; Mo, monocyte; pDC, plasmacytoid dendritic cell; DC, dendritic cell; migDC, migratory dendritic cell; NP, neutrophil (Table S3).
(B) Enrichment of individual metabolic modules across all datasets obtained during GAM-clustering analysis of IG MNP OS dataset.
Importantly, independent application of the GAM-clustering method to each of the datasets also revealed a very high degree of similarity in obtained modules, highlighting the reproducible and robust nature of the derived metabolic subnetworks (Figure S7A).
Next, we examined individual subnetworks from the perspective of metabolic reactions covered and described both published evidence of the corresponding metabolic activities and validation data obtained in this project.
Subnetworks associated with early developmental stages
Module 6 (Figures 6A, 6B, and 6D) is one of the modules most distinctly associated with the yolk sac, embryoid body, alveolar, and SPMs. This module, though unbiasedly derived by our network analysis, closely matches the canonical glycolysis pathway (Figure 6B), indicating strong transcriptional co-regulation of these genes across the collected samples. Enrichment of the glycolysis module in developmental cell types is consistent with previously published data highlighting the importance of glycolysis for stem-like and progenitor populations.20–23 This is also consistent with the ImmGen MNP P1 and mTMS data (Figure 6D), where this module is also most enriched in progenitor populations. Interestingly, mTMS single-cell RNA-seq data also demonstrate that this module is enriched in neutrophils, in accord with the described high glycolytic rate in these cells.24
Figure 6. Subnetworks associated with early developmental stages and DCs.
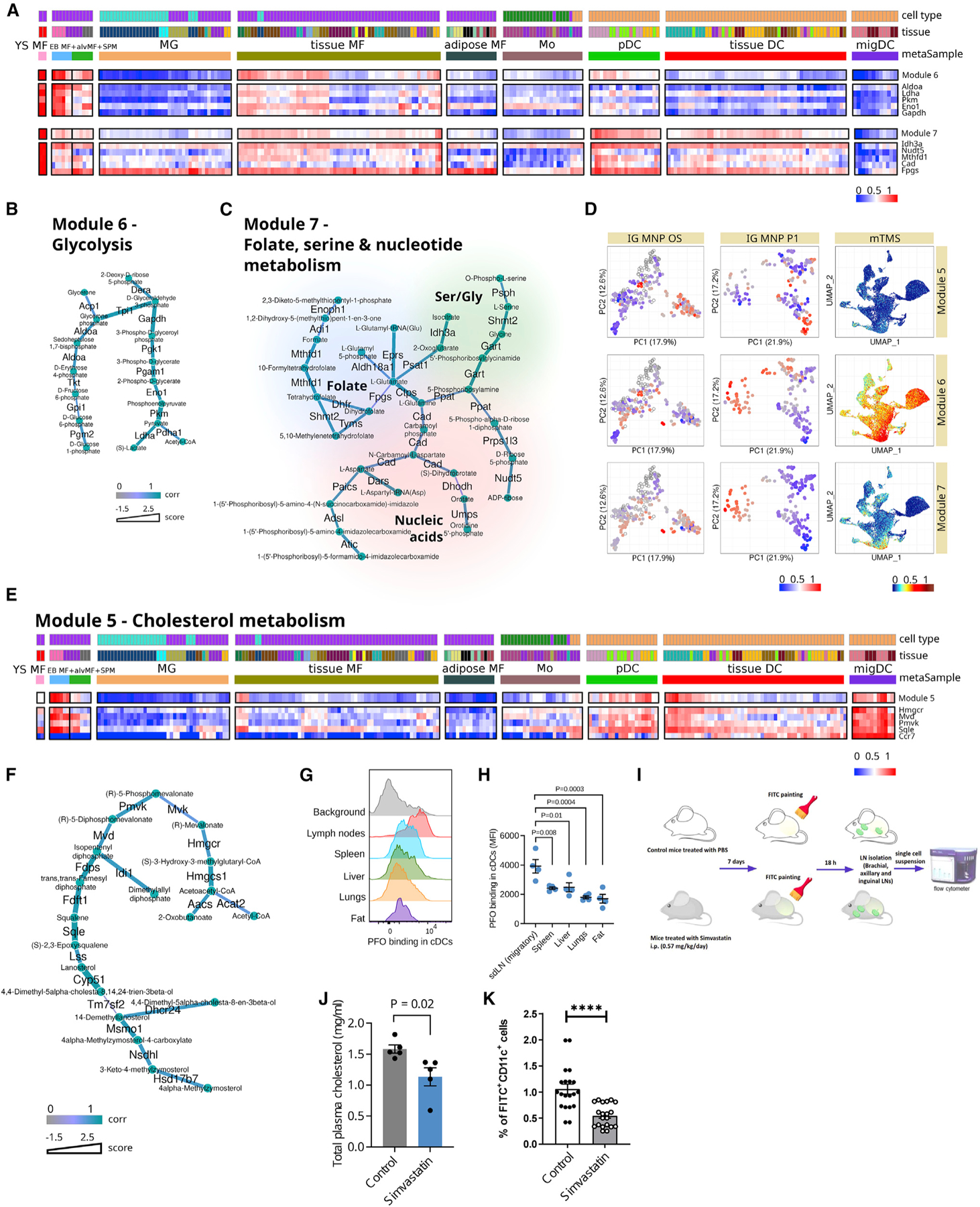
(A and E) Heatmaps of module patterns along with the expression of some of its genes or genes related to the same biological subject (from lowest as blue to highest as red). YS MF, yolk sac macrophage; EB MF, embryoid body macrophage; alvMF, alveolar macrophage; SPM, small peritoneal macrophage; MG, microglia; MF, macrophage; Mo, monocyte; DC, dendritic cell; pDC, plasmacytoid DC; migDC, migratory DC.
(B, C, and F) Metabolic modules per se where edges of modules are attributed with color according to correlation of its enzyme’s gene expression to this particular module pattern and thickness according to its score.
(D) Enrichment of modules genes expression (from lowest as blue to highest as red, transparent dots correspond to treated samples) across all three analyzed datasets: IG MNP OS, IG MNP P1, and mTMS datasets.
(G and H) Flow cytometry analysis of DC staining with cholesterol-dependent cytolysin perfringolysin O (PFO) that indicates the level of cholesterol in the cell membrane. (H) Mean fluorescence intensity (MFI) levels of PFO binding in DC subsets. n = 4 mice per group (2 male and 2 female); each dot indicates an independent mouse. Statistics by one-way ANOVA with Dunnett’s multiple comparison test.
(I) DC migrations experiment scheme.
(J) Total plasma cholesterol levels in control and treated-with-simvastatin animals; n = 5 mice in each group; statistical analysis by unpaired two-tailed t test.
(K) Percentage of migrated FITC+CD11c+ DCs in draining lymph nodes after FITC application in control and treated-with-simvastatin animals; n = 20 mice in each group; statistical analysis by unpaired two-tailed t test. **** p < 0.0001.
(H, J, and K) Data shown as mean ± standard error of the mean.
Module 7 (Figures 6A, 6C, and 6D) represents another set of metabolic activities, including folate and serine metabolism and the nucleotide biosynthesis pathway, typically associated with the progenitor populations.25–27 In addition to the yolk sac macrophages, this module is also enriched in some tissue-residing DCs and pDCs (but not in migDCs). Indeed, the importance of some of these pathways (e.g., folate metabolism) has been demonstrated in DC functions such as antigen presentation.28
Cholesterol synthesis pathway is enriched in and functionally important for migDCs
Module 5 almost exclusively consists of enzymes from the cholesterol metabolism/mevalonate synthesis pathway and is enriched in embryoid body macrophages and some DC subsets (Figures 6D–6F). Specifically, cholesterol synthesis appears to play a major role in migDCs, while it is less prominent in pDCs and conventional tissue-residing DCs. Additionally, with respect to potential tissue-specific imprinting, it is worth noting that a small subset of tissue-residing macrophages, comprised of epithelial and dermal macrophages, are enriched in genes of the mevalonate/cholesterol synthesis pathway (Figure 6E).
One of the main achievements in this work is the proof of the feasibility of metabolite level predictions from gene expression. For example, GAM-clustering analysis makes it possible to link cell cholesterol levels with the expression of specific genes. To illustrate this assertion, we analyzed cholesterol levels in cell plasma membranes in migDCs and tissue DCs using flow cytometry and perfringolysin O (PFO)-binding assay. Because PFO binds selectively to cholesterol-rich domains of cell membranes,29 its binding level correlates with cholesterol expression and membrane transport. Interestingly, PFO binding was significantly higher in migDCs migrating from the skin to skin-draining lymph nodes (sdLNs) compared with tissue conventional DCs from the spleen, liver, lungs, and perigonadal fat (Figures 6G, 6H, and S7B). This pattern of cholesterol synthesis revealed by PFO binding was concordant with the increased expression of genes from the cholesterol module in migDCs (Figure 6E), indicating the biological relevance of increased cholesterol biogenesis in migDCs.
Enrichment of cholesterol metabolism in migDCs is consistent with mechanistic data by Hauser and colleagues, who showed that cellular cholesterol levels are directly linked to the ability of DCs to oligomerize Ccr7 (a key marker of migDCs) and acquire a migratory phenotype.30 Given the results of our analysis and these published mechanistic connections, we evaluated mobilization of DCs to LNs following epicutaneous application of fluorescein isothiocyanate (FITC) in either control mice or mice treated intraperitoneally (i.p.) with low-dose simvastatin, an inhibitor of 3-hydroxy-3-methylglutaryl (HMG) coenzyme A reductase (0.57 mg/kg/day), for 7 days (Figure 6I), which significantly decreased cholesterol levels in their plasma (Figure 6J). dLNs collected 18 h after FITC application demonstrated significantly fewer migrated FITC+CD11c+ DCs in the animals treated with simvastatin, illustrating that in vivo interference with cholesterol synthesis reduces DC migration to the LN, fitting with the prominent expression of cholesterol synthesis genes in DCs (Figure 6K). Surprisingly, simvastatin treatment increased membrane cholesterol levels of migDCs isolated from dLNs (Figure S7C), suggesting a cell-level compensatory mechanism in migDCs that counteracts systemic decrease in circulating cholesterol.31 Additionally, in this model, we cannot exclude an indirect effect of cholesterol-synthesis inhibition on migDCs via altered chemotactic effects in LNs. Nevertheless, these results illustrate general validity of our analysis and highlights features of the systemic metabolic perturbations, such as statin treatments, that were not recognized previously.
Subnetworks associated with lipid metabolism
Modules 1 and 2 cover various aspects of lipid metabolism and are strongly specific to macrophages relative to monocytes and DCs (Figures 7A–7D). Due to general similarity of their patterns, we merged the subnetworks for modules 1 and 2 in order to make the interpretation easier (Figures 7C, 7D, S8A, and S8B). The resulting subnetwork is centered around phospholipid and arachidonic acid metabolism and includes parts of the glutathione and cysteine/glutamate/glycine metabolism pathways, as well as the N-acetylglucosamine pathway. Indeed, arachidonic acid metabolism has been shown to play major role in macrophages.32,33 Its metabolic flow is associated with utilization of phospholipids to produce two major classes of arachidonic acid derivatives: leukotrienes and prostaglandins. Unlike prostaglandins, cysteinyl leukotriene production (C4 and downstream) requires glutathione as an intermediate metabolite, thus involving the glycine, cysteine, and glutamate pathways.34
Figure 7. Subnetworks associated with fatty acid synthesis and degradation.
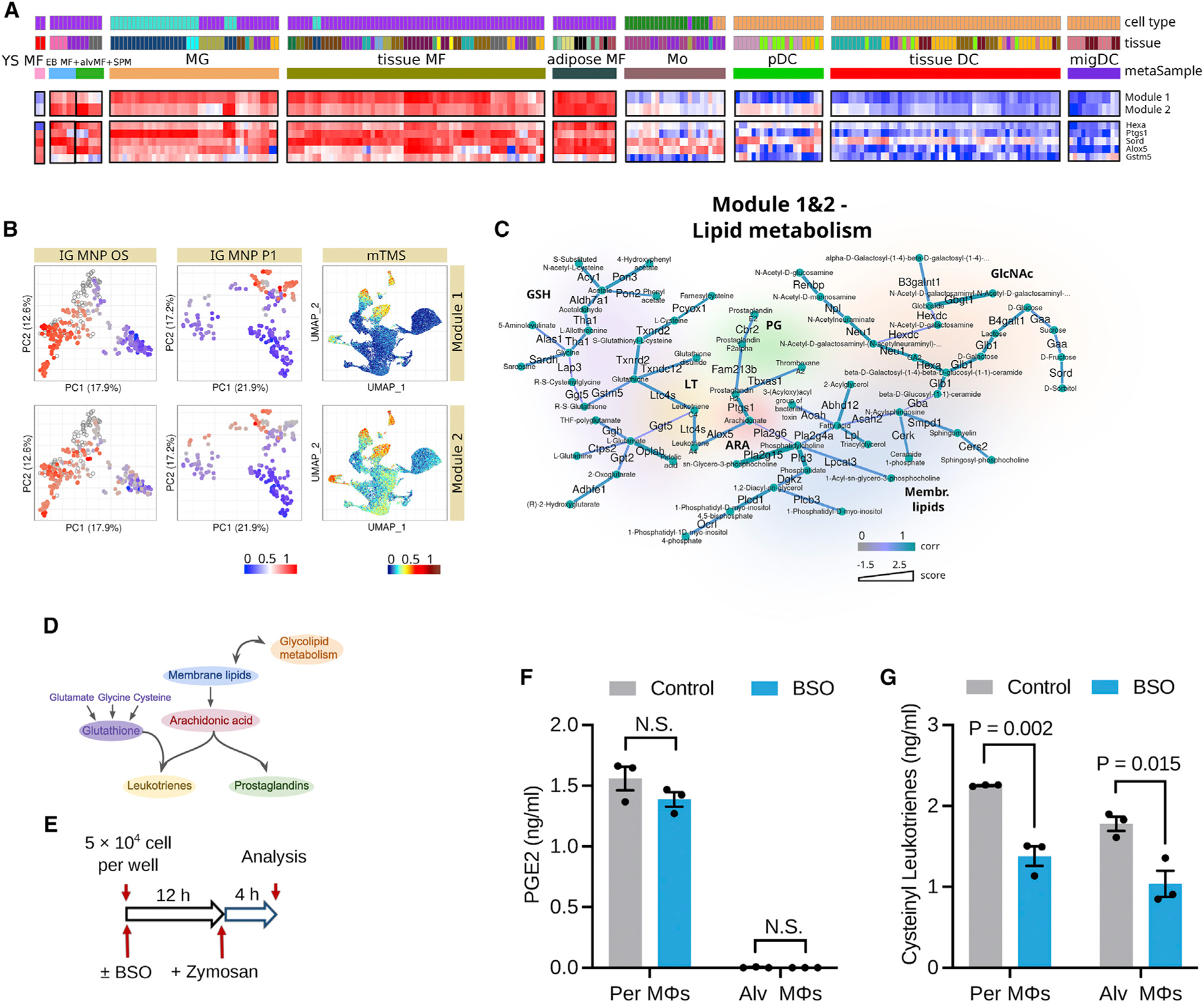
(A) Heatmaps of module patterns along with the expression of some of its genes (from lowest as blue to highest as red). YS MF, yolk sac macrophage; EB MF, embryoid body macrophage; alvMF, alveolar macrophage; SPM, small peritoneal macrophage; MG, microglia; MF, macrophage; Mo, monocyte; DC, dendritic cell; pDC, plasmacytoid DC; migDC, migratory DC.
(B) Enrichment of module gene expressions (from lowest as blue to highest as red, transparent dots correspond to treated samples) across all three analyzed datasets: IG MNP OS, IG MNP P1, and mTMS datasets.
(C) Metabolic modules per se and corresponding schematic diagrams. Edges of modules are attributed with color according to correlation of its enzyme’s gene expression to this particular module pattern and with thickness according to its score.
(D) Schematic representation of metabolic module.
(E) Schematic illustrating the design of the experiment with mouse peritoneal (Per) and alveolar (Alv) macrophages (MΦs) treated with BSO for 12 h to inhibit GSH synthesis followed by activation by zymosan for 4 h.
(F) Secretion levels of PGE2; n = 3 mice per group; statistics by unpaired two-tailed t test. N.S., non-significant (p > 0.05).
(G) Secretion levels of cysteinyl leukotrienes; n = 3 mice per group; statistics by unpaired two-tailed t test.
(F and G) Data shown as mean ± standard error of the mean.
To validate the biological role of this metabolic module in tissue macrophages, we sorted mouse peritoneal and lung alveolar macrophages (Figure S8C), followed by inhibiting glutathione synthesis using buthionine sulfoximine (BSO), an inhibitor of gamma-glutamylcysteine synthase (Figure 7E). Production of prostaglandin E2 (PGE2) is the glutathione-independent pathway of arachidonic acid derivative metabolism. In keeping with this metabolic model, glutathione depletion did not alter PGE2 secretion by peritoneal and alveolar macrophages activated with zymosan, a TLR2 and Dectin-1 agonist (Figure 7F). In contrast, as predicted by increased transcription of genes connecting glutathione synthesis to cysteinyl leukotriene production in tissue macrophages, glutathione depletion significantly reduced cysteinyl leukotriene secretion from zymosan-activated peritoneal and alveolar macrophages (Figure 7G). These results show that tissue-resident macrophages profoundly depend on glutathione synthesis to efficiently secret cysteinyl leukotriene, suggesting the role of glutathione-arachidonic acid metabolism as a key regulator of the inflammatory function of tissue macrophages.
Furthermore, our analysis picked up a distinct subnetwork of coexpressed genes from the glycerophospholipid pathway (module 9; see Figures S8D–S8F) that was particularly highly expressed in the microglial populations (Figure S8D). This module included enzymes such as Dgkd and Lpcat2, suggesting that their role in microglia might be of particular interest.35,36 As Figure S8E shows, these observations were common across all three datasets.
Subnetworks associated with fatty acid synthesis and degradation
Our analysis identified three distinct subnetworks associated with the modulation of fatty acids in terms of both their synthesis (modules 3 and 4) and fatty acid oxidation (module 8) (Figure S9).
The structure of module 3 (Figures S9A–S9C) reflects the energetic demands of the fatty acid synthesis and includes portions of the pentose phosphate pathway and the TCA cycle, where citrate synthase (Cs) is one of the most pattern-specific genes within this subnetwork. Overall, module 3 is highly enriched in DC populations but not in macrophage/monocyte samples, underscoring another facet of metabolic divergence between these cell types. The functional importance of this module for DCs is evident from the fact that a blockade of fatty acid synthase (Fasn)-mediated fatty acid synthesis markedly and selectively decreases dendropoiesis both in mice and in humans.37,38
Interestingly, the pattern of module 8 (Figures S9A, S9B, and S9E) was directly opposite to module 3 and was strongly enriched among various tissue macrophages, particularly in alveolar macrophages. Metabolic flow encompassed by this network includes enzymes such as Lipa (LAL), which is responsible for lysosomal lipolysis and the initial breakdown of intracellular lipid storage. This breakdown is followed by mitochondrial import of cytosolic fatty acids via carnitine transport shuttle (Cpt1a) and their subsequent breakdown via classical fatty acid oxidation (FAO) steps (Acox1, Hadha, etc.)39,40 (Figure S9E). The Lipa expression pattern is one of the most specific for module 8, indicating its potential importance for macrophages. Indeed, there are studies highlighting the importance of Lipa for macrophage function, especially in the context of anti-inflammatory polarization.41 Furthermore, Lipa is also likely to be important for human macrophages, as mutations in the LIPA gene of patients with cholesteryl ester storage disease (CESD) cause aberrant cholesterol accumulation in tissue macrophages.42,43 Enrichment of FAO-related module 8 in alveolar macrophages is particularly interesting as it is distinctly reproduced in ImmGen MNP P1 and mTMS data. The importance of this pathway in the lungs is intriguing and warrants further detailed investigations.
DISCUSSION
Here, we introduced a dataset covering multiple subpopulations of DCs, monocytes, and macrophages from diverse tissues—the result of ImmGen MNP OS profiling effort. We focused on understanding potential metabolic variability among collected myeloid cell subpopulations and co-analyzed it in the context of two other large-scale profiling efforts—ImmGen P1 and TMS. Using an algorithmic approach (GAM-clustering), we have defined 9 metabolic subnetworks encapsulating the major metabolic differences that were highly reproducible across three studied datasets. Our analysis demonstrated that specific metabolic features could be attributed to cell populations and specific tissues of residence for distinct populations (e.g., adipose tissue macrophages).
Our analysis suggested that major metabolic differences between baseline (unactivated) macrophages and DCs are (1) levels of Fasn-mediated fatty acid synthesis enriched in DCs’ transcriptional profiles and (2) regulation of arachidonic acid metabolism, which is enriched in macrophages. Among various tissue-residing cell types, it was apparent that microglia and CNS macrophages have a very distinct phenotype relative to other populations: based on their transcriptional profile, they appear more metabolically quiescent, yet a particular lipid-associated module (module 9) was enriched in these cells, with key genes being Lpcat2, Dgkd, and Csd1, which are involved in phospholipid metabolism and the generation of bioactive lipids from phospholipid precursors. Of interest, hierarchical metabolic clustering of macrophages places adipose-tissue macrophages and microglia closer than another group of diverse tissue macrophages. Residence in a lipid-rich environment made lipids an integral and very important part of microglia phenotype and functions regulation, which was shown in a wide range of publications.44–47 Perturbations in lipid substrate utilization can also affect microglia’s phagocytic and inflammatory statuses, shaping disease-specific microglia features.48–50
Indeed, distinct patterns in lipid metabolism, including pathways related to cholesterol, were also apparent in DCs versus macrophages. Macrophages’ capacity to handle cholesterol and store it in esterified form to generate so-called macrophage foam cells is a well-established theme in cardiovascular research and inflammatory disease.51,52 Our data reveal that expression of Lipa, an enzyme involved in breaking down cholesterol esters in the lysosome and whose mutation is associated with lysosomal storage diseases, is a widespread characteristic of tissue macrophages but not DCs. On the contrary, we observed that pathways active in cholesterol synthesis are very low in all tissue macrophages but elevated in monocytes and DCs, especially migDCs.
Thus, it appears as though macrophages are oriented toward handling exogenously derived cholesterol, such as that which may be derived from engulfment of large amounts of phagocytic cargo, whereas DCs are oppositely programmed to synthesize their own cholesterol and associated intermediates. Tissue macrophages are especially incapable of migrating to distal sites like LNs, a major functional distinction from DCs. We showed that transcriptional activity of cholesterol biogenesis genes and membrane cholesterol levels was increased in migDC compared with tissue DC subsets, suggesting its important role in DC migration from tissues into LNs. Moreover, we validated the importance of the cholesterol synthesis pathway for migDCs in vivo by using pharmacological interventions with simvastatin. However, our results show that membrane cholesterol levels in migDCs were increased in the context of circulating cholesterol lowering, indicating that further studies are needed to understand better the interplay between systemic and cellular cholesterol metabolism and DC migration.
Finally, we predicted and validated experimentally the elevated activity of metabolic pathways connecting glutathione synthesis and the production of cysteinyl leukotrienes in tissue macrophages. Cysteinyl leukotrienes are established mediators of bronchial asthma.53 Dectin-2 activation induces cysteinyl leukotrienes synthesis in lung phagocytes and increases Th2 immunity.54 Our results suggest a role of glutathione-mediated metabolic fine-tuning of Th2 immune responses through the cysteinyl leukotriene axis in alveolar macrophages.
Altogether, our analysis underscores metabolic variability across cell types and tissues and highlights the need to understand metabolic wiring not only in terms of cellular metabolism but also at the level of whole-body communication networks (see, e.g., Castillo-Armengol and colleagues55 and Droujinine and Perrimon56). Furthermore, since direct metabolic profiling is not feasible or sufficiently accurate now, the development of ex vivo metabolomics profiling technologies57–59 suggests that direct insight into metabolism of various myeloid subpopulations through in vivo metabolomics techniques will be possible in the future.
Lastly, there are several aspects of this approach that can be further improved in the future. First, the current graph structure reflects the connectivity of a metabolic network but does not take into account explicit directionalities of specific reactions. This is a consequence of the use of the underlying path-solving algorithm, which works on undirected graphs19 and can potentially impede interpretability of the results. Furthermore, the connectivity of the current network is based on the existence of metabolic reactions between individual metabolites and does not explicitly take into account transformation of individual atoms (carbons, nitrogens, etc.), akin to what can be measure in metabolic flux analysis. This can be addressed by introducing a more refined, atomistic structure of the metabolic network, as was recently done in the Shiny GATOM approach.60 Finally, utilization of different clustering metrics (e.g., silhouette) may be added to assess the quality of derived modules and therefore improve clustering.
Limitations of the study
Even though gene expression might be considered a reliable proxy for metabolic processes exploration, it provides an indirect estimate of cellular metabolism. Mass-spectrometry-based single-cell metabolomics, 13C-label-tracing, extracellular flux analysis, and protein data, as well as metabolic enzymes activity, are still valuable and necessary supplements to bulk and singlecell data to provide the full picture. However, no coherent data of this kind were available for the analyzed datasets.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Maxim N. Artyomov (martyomov@wustl.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The ImmGen MNP Open Source RNA sequencing dataset generated during this study has been deposited at GEO repository (GSE122108) and is publicly available as of the date of publication. This paper also analyzes existing, publicly available data: The ImmGen Phase 1 dataset (GSE15907) and The Tabula Muris Senis dataset (GSE149590). All accession numbers are also listed in the key resources table.
All original code has been deposited at Zenodo and is publicly available as of the date of publication. DOI is listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| CD45 BUV563 clone 30-F11 | BD Biosciences | Cat#612924; RRID:AB_2870209 |
| CD45 APC-Cy7 clone 30-F11 | Biolegend | Cat#103116; RRID:AB_312981 |
| I-A/I-E BUV496 clone M5/114.15.2 | BD Biosciences | Cat#750281; RRID:AB_2874472 |
| CD11c PE-Cy7 clone N418 | Biolegend | Cat#117318; RRID:AB_493568 |
| CD11c PE clone N418 | Biolegend | Cat#117308; RRID:AB_313777 |
| CD3e AF488 clone 145-2C11 | Biolegend | Cat#100321; RRID:AB_389300 |
| CD19 FITC clone 1D3/CD19 | Biolegend | Cat#152404; RRID:AB_2629813 |
| NK1.1 AF488 clone PK136 | Biolegend | Cat#108718; RRID:AB_493183 |
| TER-119 AF488 clone TER-119 | Biolegend | Cat#116215; RRID:AB_493402 |
| CD64 APC clone X54–5/7.1 | Biolegend | Cat#139306; RRID:AB_11219391 |
| F4/80 AF488 clone BM8 | Biolegend | Cat#123120; RRID:AB_893479 |
| F4/80 PE clone BM8 | Biolegend | Cat#123110; RRID:AB_893486 |
| CD11b BV421 clone M1/70 | Biolegend | Cat#101236; RRID:AB_11203704 |
| anti-Perfringolysin O rabbit antibody | Abcam | Cat#ab225685 |
| Goat anti-Rabbit IgG (H + L) AF647 | Thermofisher | Cat#A-21245; RRID:AB_141775 |
| Mouse TruStain FcX (anti-mouse CD16/CD32, clone 93) antibody | Biolegend | Cat#101320; RRID:AB_1574975 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| LIVE/DEAD Fixable Aqua Dead Cell Stain Kit | Thermofisher | Cat#L34957 |
| Collagenase D | Sigma | Cat#11088882001 |
| Perfringolysin O (PFO) from Clostridium perfringens | Cusabio | Cat#CSB-EP314820CMB |
| Fluorescein isothiocyanate (FITC) | Sigma | Cat#F7250 |
| Simvastatin | Sigma | Cat#S6196 |
| L-Buthionine-sulfoximine (BSO) | Sigma | Cat#B2515 |
| Zymosan | Invivogen | Cat#tlrl-zyn |
|
| ||
| Critical commercial assays | ||
|
| ||
| Cholesterol Quantitation Kit | Sigma | Cat#MAK043 |
| PGE2 ELISA kits | Enzo | Cat#ADI-900-001 |
| Cysteinyl leukotriene ELISA kits | Enzo | Cat#ADI-900-070 |
| RPMI 1640 Medium | Thermofisher | Cat#11875093 |
| Deposited data | ||
| ImmGen ULI: OpenSource Mononuclear Phagocytes Project (raw and processed data) | This paper | GEO: GSE122108 |
| ImmGen Microarray Phase 1 (raw and processed data) | Gautier et al.5 | GEO: GSE15907 |
| Tabuls Muris Senis (raw and processed data) | Tabula Muris Consortium11 | GEO: GSE149590; https://s3.console.aws.amazon.com/s3/buckets/czb-tabula-muris-senis/; https://figshare.com/articles/dataset/Processed_files_to_use_with_scanpy_/8273102/2 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| Mouse peritoneal primary macrophages | This paper | NA |
| Mouse lung alveolar primary macrophages | This paper | NA |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: wild-type C57BL/6J mice | Jackson Laboratory | Strain#000664 |
|
| ||
| Software and algorithms | ||
|
| ||
| GAM-clustering | This paper | GitHub: https://github.com/artyomovlab/ImmGenOpenSource https://doi.org/10.5281/zenodo.7492657 |
| GMWCS-solver | Loboda et al.19 | https://cran.r-project.org/web/packages/mwcsr |
| FlowJo software v10.2 | BD | https://www.flowjo.com/ |
| Prism v9.4.1 | Graphpad Software | https://www.graphpad.com/scientific-software/prism/ |
|
| ||
| Other | ||
|
| ||
| Resource website: Hands-on interactive browser for gene expression exploration both for ImmGens and Tabula Muris Senis datasets | This paper | https://artyomovlab.wustl.edu/immgen-met/ |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse strains
Female and male C57BL/6J mice of 6-week old were purchased from the Jackson Laboratory and housed in specific-pathogen-free animal facility at Washington University in St. Louis during two weeks before the start of experiments. Animal protocols used in this study were approved by the Institutional Animal Care and Use Committee at Washington University in St. Louis. Mice were co-housed and randomized before creating body-weight-balanced groups to treat with simvastatin or PBS. Mice used for flow cytometry analyses were euthanized at 10–11 week of age after 8 h fasting. Investigators were not blinded to experimental groups. No animals were excluded from analysis.
Primary cell culture
Mouse primary macrophages were sorted from peritoneal cavity fluid (peritoneal macrophages, CD45+CD11b+F4/80+CD64+) and lung tissue (alveolar macrophages, CD45+CD11b−F4/80+CD64+). 5 × 104 cells were plated in 0.2 mL of RPMI media +10% FCS in the presence of 2 mM glutamine and incubated with or without 0.5 mM buthionine sulfoximine (BSO) for 12 h followed by activation with zymosan for additional 4 h. Supernatant media were collected and used for ELISA assays.
METHOD DETAILS
RNA-sequencing
Bulk RNA-sequencing data were collected from 16 labs. All of the mice used in this study were handled in accordance with IACUC-approved protocols. Each lab, in addition to their own samples, sorted a standard peritoneal cavity macrophage population (CD115+B220−F4/80hiMHCII−) for comparability between all labs. Samples were profiled using ImmGen’s ultra low input (ULI) sequencing pipeline, in batches of 90–96 samples. All samples were sequenced in two separate NextSeq500 runs and combined for increased depth (expect 8–12 106 reads per sample).
RNA-sequencing data processing
Following sequencing, ImmGen MNP OS raw reads were aligned with STAR to the mouse genome assembly mm10, and assigned to specific genes using the GENCODE vM12 annotation. Aligned reads were quantified using featureCounts. Samples that did not pass the QC threshold for read counts (<2 million reads) were dropped for further analysis. Pearson correlation was calculated between biological replicates to exclude samples that did not pass a threshold of 0.9 correlation coefficient. For the cell populations with three biological replicates, of which one did not agree with the other two, the suspect one was removed from the dataset. In case cell populations had only two replicates, both were removed. Samples with Jchain>1,000 and Ighm>10,000 were set asides as well as samples with high B cell, erythrocytes and fibroblasts transcripts. Peritoneal cavity samples were downsampled to keep consistency across samples number in all tissues. All gene counts were imported into the R/Bioconductor package EdgeR and TMM normalization size factors were calculated to adjust for differences in library size across all samples. Feature not expressed in at least three samples above one count-per-million were excluded from further analysis and TMM size factors were recalculated to create effective TMM size factors. The effective TMM size factors and the matrix of counts were then imported into the R/Bioconductor package Limma and weighted likelihoods based on the observed mean-variance relationship of every gene and sample were then calculated for all samples. Performance of the samples was assessed with a Pearson correlation matrix and multidimensional scaling plots. As GAM-clustering method itself does not perform any counts normalizations or batch corrections, ImmGen MNP OS data were assessed for presence of any batch effect by PCA plots.
Single-cell RNA-seq data processing
Filtered h5ad file for Droplet subset was downloaded from the official Tabula Muris Senis repository (https://figshare.com/projects/Tabula_Muris_Senis/64982). The data were processed by the standard Seurat pipeline and resulted in 235,325 cells organised in distinct clusters detectable on TSNE/UMAP plots. Next, cells annotated with names corresponding to myeloid populations were picked out. A differential gene expression analysis between these cells and all others was performed. Top 250 of these differentially expressed genes were used as a “myeloid signature genes” (Table S4) to identify clusters that most express them and thus correspond to myeloid cells. Cell content of these clusters was used to create a subset of 60,844 cells. Obtained dataset was analyzed by non-myeloid marker genes to detect and remove cell doublets with T-cells, B-cells, NK-cells and fibroblasts (Cd3d, Cd3e, Cd3g, Cd4, Cd8a, Cd19, Cd79a, Tnfrsf17, Cd22, Nkg7, Gnly, Col6a1, Col6a2, Col6a3). Finally, dataset of 51,364 cells was obtained and used in the further GAM-clustering analysis.
GAM-clustering
The algorithm for multisample metabolic network clustering (hereinafter referred to as GAM-clustering) identifies modules describing regulation of metabolism and is based on the previously developed GAM method.12 GAM-clustering extends the GAM method by setting the task to find not one but several metabolic modules (connected subnetworks of metabolic network) with the condition that each of these modules should contain as many metabolic genes with high pairwise correlation of their expression as possible.
The metabolic network used in the current analysis is presented as a graph where vertices are metabolites and edges are KEGG database reactions which are mapped with catalyzing them enzymes and corresponding genes. This network is an undirected pseudograph. Totally, network contains all possible biological reactions documented in KEGG database. Reactions specific for metabolism of Mus musculus were selected based on gene annotation provided by KEGG and Bioconductor (https://bioconductor.org/packages/org.Mm.eg.db/).61
Expression matrix given as an input for GAM-clustering method has rows corresponding to genes and columns corresponding to the sequenced samples. GAM-clustering does not consider column annotations during the module deriving process.
There are two major parameters that control the number and the sizes of the final modules: k – the initial number of gene clusters, and base – the value which is used during edge weights calculation thereby enforcing the certain level of gene expressions correlation in the module. The initial approximation of the final set of modules is carried out by k-medoids clustering of a gene expression matrix for all metabolic genes of a dataset with some arbitrary k (here we used k = 32, see Statistical analysis of the GAM-clustering method for details and Figure S5A). Each cluster forms a corresponding expression pattern which can be determined as averaged value of its z-normalized gene expression values. The metabolic network used for further analysis is presented as a graph where vertices are metabolites and edges are KEGG database reactions which are mapped with catalyzing them enzymes and corresponding genes. For each particular pattern edges of this graph are scored (weighted) based on their gene expression similarity with this pattern and dissimilarity with other patterns.
For each case of weighted graph a connected subgraph of maximal weight is found by a signal GMWCS (generalized maximum weight connected subgraph) solver19,62 (https://cran.r-project.org/web/packages/mwcsr/) and is called a metabolic module. This solver uses the IBM ILOG CPLEX library, which efficiently performs many iterations of this method in a reasonable amount of time. Then, each pattern is updated by replacing it with an averaged gene expression of the module’s edges with a positive score. If the pattern is changed, a new score set is calculated and a new iteration is performed. Before moving to the next iteration, small graphs are eliminated from further analysis so that there are no graphs with less than five edges and diameter less than four in the output solution. The algorithm continues until the pattern content stops changing.
For the selection of the optimal set of modules we have assessed a range of module characteristics, for example, mean pairwise correlation of module edges (i.e. genes) and the number of annotating pathways (see more details in Statistical analysis of the GAM-clustering method). Altogether, the most cohesive and informative modules were obtained using k = 32 and base = 0.4 (Figure S5B), which we decided to use for the consecutive analysis.
GAM-clustering method is applicable not to bulk RNA-seq data only but to single-cell RNA-seq data as well. Single-cell data need an additional step of preprocessing implying transformation of individual cells into technical samples. This is performed based on averaging gene expressions of individual cells inside high resolution clusters. In case of single-cell RNAseq data, among final metabolic modules might occur ones that do not cover all biological replicas of cell types they are specific for. These modules are eliminated from the final result.
Thus, the final metabolic modules are subnetworks of the overall metabolic network that contain a set of closely located genes with high correlation of their expression profile across all samples.
GAM-clustering method is available at https://github.com/artyomovlab/ImmGenOpenSource.
Staining cells with perfringolysin O (PFO)
The cell suspension was prepared from collagenase D-treated and dissociated spleen, skin-draining lymph nodes, perigonadal white adipose tissue, lungs, and liver of wild-type 8-week-old male and female C57BL/6J mice (Jackson Labs).63 The cells were stained with Aqua Live/Dead kit followed by staining with 10 mg/mL of Perfringolysin O (PFO) from Clostridium perfringens (Cusabio # CSB-EP314820CMB) in PBS at +25°C for 30 min, washed 3 times, and stained with an antibody cocktail (CD45 BUV563 clone 30-F11, I-A/I-E BUV496 clone M5/114.15.2 from BD, CD11c PE-Cy7 clone N418, CD3e AF488 clone 145-2C11, CD19 FITC clone 1D3/CD19, NK1.1 AF488 clone PK136, TER-119 AF488 clone TER-119, F4/80 AF488 clone BM8 from BioLegend, anti-Perfringolysin O rabbit antibody [Abcam # ab225685] and Goat anti-Rabbit IgG (H + L) Highly Cross-Adsorbed Secondary Antibody AF647 [Thermofisher # A-21245]) and Fc-block for 30 min on ice. The cells were analyzed by flow cytometry using FACSymphony A3 Cell Analyzer and FlowJo software.
DC migration assay
Epicutaneous application of Fluorescein isothiocyanate (FITC) to study DC migration was performed on three areas of each side of the mouse back skin.64 Both females and males were studied. Briefly, FITC (8 mg/mL) was dissolved in acetone and dibutyl phthalate and applied in 25-μL aliquots to each site. Recovered lymph nodes, 18 h later, were teased and digested in 2.68 mg/mL collagenase D for 25 min at 37°C. Then, 100 μL 100 mM EDTA was added for 5 min, and cells were passed through a 100-μm cell strainer, washed, counted, and stained for flow cytometry after counterlabeling with PE conjugated anti-CD11c (Biolegend). Prior to FITC painting, some cohorts of mice were treated with simvastatin i.p. at 0.57 mg/kg/day for 7 days, as this protocol was previously shown to significantly block monocyte diapedesis from the bloodstream.65 Control mice received vehicle i.p. For plasma cholesterol level measurements, blood was collected from the retro-orbital venous sinus in EDTA-coated tubes from mice fasted for 6 h and anesthetized with isoflurane. Plasma was separated by centrifugation at 3000 RPM at +4°C for 10 min. Total plasma cholesterol levels were measured using a colorimetric Cholesterol Quantitation Kit (Sigma #MAK043).
Analysis of arachidonic acid metabolite secretion by primary mouse macrophages
The cell suspension was prepared from the lungs and peritoneal fluid of wild-type 8-week-old male C57BL/6J mice (Jackson Labs).63 The cells were stained with an antibody cocktail (CD45 APC/Cy7 clone 30-F11, CD64 APC clone X54–5/7.1, F4/80 PE clone BM8, CD11b BV421 clone M1/70 from Biolegend) and Fc-block on ice for 15 min followed by sorting of large peritoneal macrophages and lung alveolar macrophages using BD FACSAria II Cell Sorter. 5 × 104 macrophages were incubated in 100 mL of RPMI +10% FCS in 96-well plates in the presence of 0.5 mM buthionine sulfoximine (BSO), an inhibitor of GSH synthesis, for 12 h followed by activation with 5 × 106 particles per ml of zymosan (Invivogen) for 4 h. PGE2 and cysteinyl leukotrienes were measured in cell supernatants using ELISA kits.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis of the biological data
No statistical method was used to predetermine the sample size. For comparison of groups, non-paired two-tailed t test was used. In case of multiple comparisons, p values were adjusted using the Dunnett’s correction. Statistical analyses were performed with Prism v9.4.1 (GraphPad Software). Data are shown as means ± standard errors of mean. p < 0.05 was considered statistically significant.
Statistical analysis of the GAM-clustering method
The initial approximation of the final metabolic modules is carried out by k-medoids clustering of the expression matrix of all metabolic genes of the dataset with some arbitrary parameter k (here used k = 32). Each cluster forms a corresponding expression pattern which can be determined as the averaged value of z-normalized gene expression values in this cluster. Then, a gene’s score relative to each cluster is calculated according to formula (4). This score represents similarity of gene expression with the module’s pattern (1) and dissimilarity with other modules’ patterns (3). Formally, score is defined as follows:
| Equation 1 |
| Equation 2 |
| Equation 3 |
| Equation 4 |
where gi– expression of the i-th gene,;
cj– pattern of the j-th cluster,; ck – pattern of the j-th cluster or the fake pattern,; c0– the fake pattern;
d – distance to the pattern the score is being calculated for;
d’ – distance to the pattern which this gene has the most correlation with.
(all other patterns are considered except the pattern the score is being calculated for);
base – distance to the fake pattern.
The following approach allows to avoid collapsing similar modules with enough supporting genes into one module as only one positive score per gene is possible.
Thus, a set of networks where each edge is weighted according to its gene score is formed. For each pattern a connected subgraph of maximal weight is found. These subgraphs are called metabolic modules. This procedure is carried out by an SGMWCS (signal generalized maximum weight connected subgraph) solver19,49 (https://cran.r-project.org/web/packages/mwcsr) which uses the IBM ILOG CPLEX library that efficiently performs many iterations of this method in a reasonable amount of time. Thus, an iterative procedure of metabolic modules refinement is performed in a process of updating each of the patterns by replacing it with an averaged gene expression of the module’s edges with a positive score.
One of the important parts in the procedure of updating the modules is the question when to stop. To answer this question, the difference between the values of the patterns of the current iteration and the values of the patterns of all previous iterations, in which there were the same number of modules, is found (this is done to avoid missing the situation when new iteration comes to the condition close to one that once already has occurred). If difference is large (>0.01) which means that pattern content is quite changed, a new score set is calculated and a new iteration is performed. If the difference between patterns is small enough (<0.01), but non-informative (having less than 5 edges and/or diameter less than 4) modules are still presented in the output, the less informative (most correlated with any other graph) module is eliminated from the further analysis. After removing one module, the weights are recalculated and a new iteration of refinement is performed. The final result is a set of specific subnetworks that reflects metabolic variability among the samples of the analyzed transcriptome data.
The GAM-clustering method has two parameters: the number of initial clusters k (here used k = 32) and the distance to the fake pattern base (here used base = 0.4). They affect the number, the size, intramodular gene correlation, and the number of unique annotating pathways of the resulting modules (Figures S5A and S5B).
To explore the influence of k value to number of final modules the model data were designed. They imitate experiment with complex design (15, 18 or 21 samples) where several (5, 10 or 15) modules are active each in a particular subset of samples. All combinations of these data were analyzed by the GAM-clustering method and the following output features were calculated: number of final modules found by method, number of iterations performed and time elapsed during the analysis (Figure S5A). As these data are modeled, we know how many modules are there in each experiment (dashed line in Figure S5A) and therefore we can evaluate how the number of found modules relates to the number of real modules. In most cases GAM-clustering found approximately all real modules when launched with the value k several times greater that the number of real modules. Moreover, a further increase of k does not lead to results improvement, but nonlinearly increases the number of iterations and the working time of the method. Thus, it is reasonable to detect some advisable k value so that user gets approximately full set of modules and does not spend to much time for the analysis. As in real data we do not know the number of real modules, there is a heuristic approach that allows to find some k based on the characteristics of the input data. This approach is based on elbow method that calculates the total within-cluster sum of square (wss) for each k. For expression data there is no pronounced inflection point where wss is sharply stops decreasing (usually this point is considered equal to the optimal number of clusters). Here, we used point where the slope of the wss curve is 50% as steep as its steepest slope. Corresponding to this point abscissa value was considered as k value.
The strategy for selecting optimal value of base parameter was formed on the basis of real data analysis, since it requires consideration of the biological meaning of the obtained modules. At the beginning of the analysis, the GAM-clustering algorithm produces some recommended value of k (see previous paragraph). For this k, we can calculate the average dissimilarity (distance) between the observations of the initial cluster and this cluster’s medoid over all clusters. Obtained value is proposed by the method as the recommended value of the base parameter. For the ImmGen MNP OS data analyzed in this study, there were 32 initial clusters proposed and the recommended base value was equal to 0.4. This base value was determined to be optimal during the comparative study of the results obtained with other different base values (Figure S5B). The optimality criterion included the calculation of the following characteristics of the output modules: their number, size, average correlation of edges, the number of unique annotating paths, the number of annotating paths corresponding to one cluster only, the percentage of genes with negative score, the percentage of genes with negative correlation, the percentage of genes with correlation less than 1 – base. Noticeably, such characteristics as the average number of genes in the module, the average percentage of genes with negative score and correlation, as well as with a correlation less than 1 – base, are minimal for the recommended base value (0.4). This indicates that the modules obtained for base = 0.4 have good internal correlation, as well as compactness. Modules obtained with a lower base value also show good internal correlation, but they are characterized by the loss of a large number of significant modules. It is worth noting that for base = 0.2 no modules were found. Modules obtained with larger base values, on the contrary, are annotated with a bigger number of unique canonical pathways, however, many of these pathways relate to the same biochemical processes. Moreover, these modules are characterized by lower rates of intramodular correlation.
Even though default values of k and base parameters are proposed to user before the analysis based on the input data properties, there is still an opportunity for user to select custom values of these parameters. Nevertheless, the general recommendation is to stick with the proposed value of the base parameter, since its changes lead to the strong alterations in the size and content of the final modules.
ADDITIONAL RESOURCES
The interactive browser for gene expression exploration of ImmGen MNP OS, Phase 1 and Tabula Muris Senis datasets analyzed in this study was created.
Description: https://artyomovlab.wustl.edu/immgen-met/.
Supplementary Material
Highlights.
ImmGen monomuclear phagocyte open-source (MNP OS) dataset is introduced (337 samples)
Myeloid mouse scRNA-seq dataset across tissues is assembled based on Tabula Muris
GAM clustering is metabolic network analysis of the large-scale/single-cell data
Tissue-/cell-specific cholesterol, glycolysis, nucleotide, GSH/lipid modules reported
ACKNOWLEDGMENTS
We thank Amanda Swain, Monika Bambouskova, and Laura Arthur for constructive comments on the manuscript. This work was supported by ImmGen Consortium grant AI072073 (NIAID, NIH) and in part by the Division of Intramural Research of the NIAID, NIH. The work was also partly supported by R01-AI125618 (NIAID) to M.N.A.. D.A.M. was partly supported by a Discovery Grant from the National Psoriasis Foundation (USA). A.G. and A.S. were supported by Ministry of Science and Higher Education of the Russian Federation (Priority 2030 Federal Academic Leadership Program). M.H.S. was partly supported by institutional grants from TU Dresden, Aix-MArseille Université , INSERM, and CNRS; by grants from the “Agence Nationale de la Recherche” (ANR-17-CE15-0007-01 and ANR-18-CE12-0019-03), INCa (13-10/405/AB-LC-HS), and Fondation ARC pour la Recherche sur le Cancer (PGA1 RF20170205515); by an INSERM-Helmholtz cooperation grant; by the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (grant agreement number 695093 MacAge); and by an Alexander von Humboldt Professorship at TU Dresden. L.-H.H. is currently affiliated with Institute of Metabolism & Integrative Biology (IMIB), Fudan University, Shanghai 200437, China. K.-W.K. is currently affiliated with Department of Pharmacology and Regenerative Medicine, College of Medicine, University of Illinois at Chicago, Chicago, IL 60612, USA. P.W. is currently affiliated with Massachusetts General Hospital, Harvard Medical School, Boston, MA 02115, USA. S.T. is currently affiliated with Center de Recherche de l’Hôpital Maisonneuve-Rosemont, Département de microbiologie, immunologie et infectiologie, Université de Montré al, Montré al, QC, H1T 2M4, Canada. H.T. is currently affiliated with Data Mining and Modeling for Biomedicine, VIB Center for Inflammation Research, Ghent 9052, Belgium. K.C. is currently affiliated with UMR 1236, Université Rennes, INSERM, Etablissement Français du Sang Bretagne, Rennes 35000, France. F.G. is currently affiliated with INSERM U1015, Gustave Roussy Cancer Campus, Villejuif 94800, France.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112046.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Monticelli S, and Natoli G (2017). Transcriptional determination and functional specificity of myeloid cells: making sense of diversity. Nat. Rev. Immunol 17, 595–607. 10.1038/nri.2017.51. [DOI] [PubMed] [Google Scholar]
- 2.De Kleer I, Willems F, Lambrecht B, and Goriely S (2014). Ontogeny of myeloid cells. Front. Immunol 5, 423. 10.3389/fimmu.2014.00423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jung J, Zeng H, and Horng T (2019). Metabolism as a guiding force for immunity. Nat. Cell Biol 21, 85–93. 10.1038/s41556-018-0217-x. [DOI] [PubMed] [Google Scholar]
- 4.Caputa G, Castoldi A, and Pearce EJ (2019). Metabolic adaptations of tissue-resident immune cells. Nat. Immunol 20, 793–801. 10.1038/s41590-019-0407-0. [DOI] [PubMed] [Google Scholar]
- 5.Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, Helft J, Chow A, Elpek KG, Gordonov S, et al. (2012). Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat. Immunol 13, 1118–1128. 10.1038/ni.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Llufrio EM, Wang L, Naser FJ, and Patti GJ (2018). Sorting cells alters their redox state and cellular metabolome. Redox Biol 16, 381–387. 10.1016/j.redox.2018.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Binek A, Rojo D, Godzien J, Rupérez FJ, Nuñez V, Jorge I, Ricote M, Vázquez J, and Barbas C (2019). Flow cytometry has a significant impact on the cellular metabolome. J. Proteome Res 18, 169–181. 10.1021/acs.jproteome.8b00472. [DOI] [PubMed] [Google Scholar]
- 8.Richardson GM, Lannigan J, and Macara IG (2015). Does FACS perturb gene expression? Cytometry A 87, 166–175. 10.1002/cyto.a.22608. [DOI] [PubMed] [Google Scholar]
- 9.Jha AK, Huang SCC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, Chmielewski K, Stewart KM, Ashall J, Everts B, et al. (2015). Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 42, 419–430. 10.1016/j.immuni.2015.02.005. [DOI] [PubMed] [Google Scholar]
- 10.ImmGen Consortium, ImmGen C. (2016). Open-source ImmGen: mononuclear phagocytes. Nat. Immunol 17, 741. 10.1038/ni.3478. [DOI] [PubMed] [Google Scholar]
- 11.Tabula Muris Consortium (2020). A single-cell transcriptomic atlas characterizes ageing tissues in the mouse. Nature 583, 590–595. 10.1038/s41586-020-2496-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sergushichev AA, Loboda AA, Jha AK, Vincent EE, Driggers EM, Jones RG, Pearce EJ, and Artyomov MN (2016). GAM: a web-service for integrated transcriptional and metabolic network analysis. Nucleic Acids Res 44, 194–200. 10.1093/nar/gkw266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gal-Oz ST, Maier B, Yoshida H, Seddu K, Elbaz N, Czysz C, Zuk O, Stranger BE, Ner-Gaon H, and Shay T (2019). ImmGen report: sexual dimorphism in the immune system transcriptome. Nat. Commun 10, 4295. 10.1038/s41467-019-12348-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davies CL, Patir A, and McColl BW (2019). Myeloid cell and transcriptome signatures associated with inflammation resolution in a model of self-limiting acute brain inflammation. Front. Immunol 10, 1048. 10.3389/fimmu.2019.01048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee M, Lee Y, Song J, Lee J, and Chang SY (2018). Tissue-specific role of CX3CR1 expressing immune cells and their relationships with human disease. Immune Netw 18, e5. 10.4110/in.2018.18.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Misharin AV, Morales-Nebreda L, Mutlu GM, Budinger GRS, and Perlman H (2013). Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am. J. Respir. Cell Mol. Biol 49, 503–510. 10.1165/rcmb.2013-0086MA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Holtman IR, Skola D, and Glass CK (2017). Transcriptional control of microglia phenotypes in health and disease. J. Clin. Invest 127, 3220–3229. 10.1172/JCI90604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Merad M, Sathe P, Helft J, Miller J, and Mortha A (2013). The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu. Rev. Immunol 31, 563–604. 10.1146/annurev-immunol-020711-074950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Loboda AA, Artyomov MN, and Sergushichev AA (2016). Solving generalized maximum-weight connected subgraph problem for network enrichment analysis. Lect. Notes Comput. Sci 9838, 210–221. 10.1007/978-3-319-43681-4_17. [DOI] [Google Scholar]
- 20.Wang YH, Israelsen WJ, Lee D, Yu VWC, Jeanson NT, Clish CB, Cantley LC, Vander Heiden MG, and Scadden DT (2014). Cell-state-specific metabolic dependency in hematopoiesis and leukemogenesis. Cell 158, 1309–1323. 10.1016/j.cell.2014.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shyh-Chang N, Daley GQ, and Cantley LC (2013). Stem cell metabolism in tissue development and aging. Development 140, 2535–2547. 10.1242/dev.091777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Folmes CDL, and Terzic A (2014). Stem cell lineage specification: you become what you eat. Cell Metab 20, 389–391. 10.1016/j.cmet.2014.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oburoglu L, Romano M, Taylor N, and Kinet S (2016). Metabolic regulation of hematopoietic stem cell commitment and erythroid differentiation. Curr. Opin. Hematol 23, 198–205. 10.1097/MOH.0000000000000234. [DOI] [PubMed] [Google Scholar]
- 24.Kumar S, and Dikshit M (2019). Metabolic insight of neutrophils in health and disease. Front. Immunol 10, 2099. 10.3389/fimmu.2019.02099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Henry CJ, Nemkov T, Casás-Selves M, Bilousova G, Zaberezhnyy V, Higa KC, Serkova NJ, Hansen KC, D’Alessandro A, and DeGregori J (2017). Folate dietary insufficiency and folic acid supplementation similarly impair metabolism and compromise hematopoiesis. Haematologica 102, 1985–1994. 10.3324/haematol.2017.171074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lucas DL, Webster HK, and Wright DG (1983). Purine metabolism in myeloid precursor cells during maturation. Studies with the HL-60 cell line. J. Clin. Invest 72, 1889–1900. 10.1172/JCI111152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Locasale JW (2013). Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat. Rev. Cancer 13, 572–583. 10.1038/nrc3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu CH, Huang TC, and Lin BF (2017). Folate deficiency affects dendritic cell function and subsequent T helper cell differentiation. J. Nutr. Biochem 41, 65–72. 10.1016/j.jnutbio.2016.11.008. [DOI] [PubMed] [Google Scholar]
- 29.Waheed AA, Shimada Y, Heijnen HF, Nakamura M, Inomata M, Hayashi M, Iwashita S, Slot JW, and Ohno-Iwashita Y (2001). Selective binding of perfringolysin O derivative to cholesterol-rich membrane microdomains (rafts). Proc. Natl. Acad. Sci. USA 98, 4926–4931. 10.1073/pnas.091090798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hauser MA, Schaeuble K, Kindinger I, Impellizzieri D, Krueger WA, Hauck CR, Boyman O, and Legler DF (2016). Inflammation-induced CCR7 oligomers form scaffolds to integrate distinct signaling pathways for efficient cell migration. Immunity 44, 59–72. 10.1016/j.immuni.2015.12.010. [DOI] [PubMed] [Google Scholar]
- 31.Schonewille M, de Boer JF, Mele L, Wolters H, Bloks VW, Wolters JC, Kuivenhoven JA, Tietge UJF, Brufau G, and Groen AK (2016). Statins increase hepatic cholesterol synthesis and stimulate fecal cholesterol elimination in mice. J. Lipid Res 57, 1455–1464. 10.1194/jlr.M067488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scott WA, Pawlowski NA, Murray HW, Andreach M, Zrike J, and Cohn ZA (1982). Regulation of arachidonic acid metabolism by macrophage activation. J. Exp. Med 155, 1148–1160. 10.1084/jem.155.4.1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boraschi D, Censini S, Bartalini M, and Tagliabue A (1985). Regulation of arachidonic acid metabolism in macrophages by immune and nonimmune interferons. J. Immunol 135, 502–505. 10.4049/jimmunol.135.1.502. [DOI] [PubMed] [Google Scholar]
- 34.Rouzer CA, Scott WA, Griffith OW, Hamill AL, and Cohn ZA (1981). Depletion of glutathione selectively inhibits synthesis of leukotriene C by macrophages. Proc. Natl. Acad. Sci. USA 78, 2532–2536. 10.1073/pnas.78.4.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shindou H, Shiraishi S, Tokuoka SM, Takahashi Y, Harayama T, Abe T, Bando K, Miyano K, Kita Y, Uezono Y, and Shimizu T (2017). Relief from neuropathic pain by blocking of the platelet-activating factor–pain loop. FASEB J 31, 2973–2980. 10.1096/fj.201601183R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nakano T, Iseki K, Hozumi Y, Kawamae K, Wakabayashi I, and Goto K (2009). Brain trauma induces expression of diacylglycerol kinase z in microglia. Neurosci. Lett 461, 110–115. 10.1016/j.neulet.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 37.Pearce EJ, and Everts B (2015). Dendritic cell metabolism. Nat. Rev. Immunol 15, 18–29. 10.1038/nri3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rehman A, Hemmert KC, Ochi A, Jamal M, Henning JR, Barilla R, Quesada JP, Zambirinis CP, Tang K, Ego-Osuala M, et al. (2013). Role of fatty-acid synthesis in dendritic cell generation and function. J. Immunol 190, 4640–4649. 10.4049/jimmunol.1202312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nomura M, Liu J, Rovira II, Gonzalez-Hurtado E, Lee J, Wolfgang MJ, and Finkel T (2016). Fatty acid oxidation in macrophage polarization. Nat. Immunol 17, 216–217. 10.1038/ni.3366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Houten SM, and Wanders RJA (2010). A general introduction to the biochemistry of mitochondrial fatty acid b-oxidation. J. Inherit. Metab. Dis 33, 469–477. 10.1007/s10545-010-9061-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang SCC, Everts B, Ivanova Y, O’Sullivan D, Nascimento M, Smith AM, Beatty W, Love-Gregory L, Lam WY, O’Neill CM, et al. (2014). Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat. Immunol 15, 846–855. 10.1038/ni.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bernstein DL, Hülkova H, Bialer MG, and Desnick RJ (2013). Cholesteryl ester storage disease: review of the findings in 135 reported patients with an underdiagnosed disease. J. Hepatol 58, 1230–1243. 10.1016/j.jhep.2013.02.014. [DOI] [PubMed] [Google Scholar]
- 43.Li F, and Zhang H (2019). Lysosomal acid lipase in lipid metabolism and beyond. Arterioscler. Thromb. Vasc. Biol 39, 850–856. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hammond TR, Dufort C, Dissing-Olesen L, Giera S, Young A, Wysoker A, Walker AJ, Gergits F, Segel M, Nemesh J, et al. (2019). Single-cell RNA sequencing of microglia throughout the mouse lifespan and in the injured brain reveals complex cell-state changes. Immunity 50, 253–271.e6. 10.1016/j.immuni.2018.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mudò G, Frinchi M, Nuzzo D, Scaduto P, Plescia F, Massenti MF, Di Carlo M, Cannizzaro C, Cassata G, Cicero L, et al. (2019). Anti-inflammatory and cognitive effects of interferon-b1a (IFNb1a) in a rat model of Alzheimer’s disease. J. Neuroinflammation 16, 44. 10.1186/s12974-019-1417-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, et al. (2017). A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169, 1276–1290.e17. 10.1016/j.cell.2017.05.018. [DOI] [PubMed] [Google Scholar]
- 47.Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, Gilfillan S, Krishnan GM, Sudhakar S, Zinselmeyer BH, et al. (2015). TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 160, 1061–1071. 10.1016/j.cell.2015.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen S, Zhang H, Pu H, Wang G, Li W, Leak RK, Chen J, Liou AK, and Hu X (2014). n-3 PUFA supplementation benefits microglial responses to myelin pathology. Sci. Rep 4, 7458. 10.1038/srep07458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gill EL, Raman S, Yost RA, Garrett TJ, and Vedam-Mai V (2018). l-Carnitine inhibits lipopolysaccharide-induced nitric oxide production of SIM-A9 microglia cells. ACS Chem. Neurosci 9, 901–905. 10.1021/acschemneuro.7b00468. [DOI] [PubMed] [Google Scholar]
- 50.Singh S, Mishra A, Srivastava N, Shukla R, and Shukla S (2018). Acetyl-l-Carnitine via upegulating dopamine D1 receptor and attenuating microglial activation prevents neuronal loss and improves memory functions in Parkinsonian Rats. Mol. Neurobiol 55, 583–602. 10.1007/s12035-016-0293-5. [DOI] [PubMed] [Google Scholar]
- 51.Ghosh S (2011). Macrophage cholesterol homeostasis and metabolic diseases: critical role of cholesteryl ester mobilization. Expert Rev. Cardiovasc Ther 9, 329–340. 10.1586/erc.11.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Maguire EM, Pearce SWA, and Xiao Q (2019). Foam cell formation: a new target for fighting atherosclerosis and cardiovascular disease. Vascul. Pharmacol 112, 54–71. 10.1016/j.vph.2018.08.002. [DOI] [PubMed] [Google Scholar]
- 53.Austen KF (2008). The cysteinyl leukotrienes: where do they come from? What are they? Where are they going? Nat. Immunol 9, 113–115. 10.1038/ni0208-113. [DOI] [PubMed] [Google Scholar]
- 54.Barrett NA, Rahman OM, Fernandez JM, Parsons MW, Xing W, Austen KF, and Kanaoka Y (2011). Dectin-2 mediates Th2 immunity through the generation of cysteinyl leukotrienes. J. Exp. Med 208, 593–604. 10.1084/jem.20100793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Castillo-Armengol J, Fajas L, and Lopez-Mejia IC (2019). Inter-organ communication: a gatekeeper for metabolic health. EMBO Rep 20, e47903. 10.15252/embr.201947903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Droujinine IA, and Perrimon N (2013). Defining the interorgan communication network: systemic coordination of organismal cellular processes under homeostasis and localized stress. Front. Cell. Infect. Microbiol 3, 82. 10.3389/fcimb.2013.00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gianchandani EP, Chavali AK, and Papin JA (2010). The application of flux balance analysis in systems biology. Wiley Interdiscip. Rev. Syst. Biol. Med 2, 372–382. 10.1002/wsbm.60. [DOI] [PubMed] [Google Scholar]
- 58.Basler G, Fernie AR, and Nikoloski Z (2018). Advances in metabolic flux analysis toward genome-scale profiling of higher organisms. Biosci. Rep 38, BSR20170224. 10.1042/BSR20170224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hastings J, Mains A, Virk B, Rodriguez N, Murdoch S, Pearce J, Bergmann S, Le Novère N, and Casanueva O (2019). Multi-omics and genome-scale modeling reveal a metabolic shift during C. elegans aging. Front. Mol. Biosci 6, 2. 10.3389/fmolb.2019.00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Emelianova M, Gainullina A, Poperechnyi N, Loboda A, Artyomov M, and Sergushichev A (2022). Shiny GATOM: omics-based identification of regulated metabolic modules in atom transition networks. Nucleic Acids Res 50, 690–696. 10.1093/nar/gkac427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Carlson M (2019). org.Mm.eg.db: Genome wide annotation for MouseR package version 3.8.2 (Bioconductor) 10.18129/B9.bioc.org.Mm.eg.db. [DOI]
- 62.Ulland TK, Song WM, Huang SCC, Ulrich JD, Sergushichev A, Beatty WL, Loboda AA, Zhou Y, Cairns NJ, Kambal A, et al. (2017). TREM2 maintains microglial metabolic fitness in Alzheimer’s disease. Cell 170, 649–663.e13. 10.1016/j.cell.2017.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mogilenko DA, Shpynov O, Andhey PS, Arthur L, Swain A, Esaulova E, Brioschi S, Shchukina I, Kerndl M, Bambouskova M, et al. (2021). Comprehensive profiling of an aging immune system reveals clonal GZMK+ CD8+ T cells as conserved hallmark of inflammaging. Immunity 54, 99–115.e12. 10.1016/j.immuni.2020.11.005. [DOI] [PubMed] [Google Scholar]
- 64.Platt AM, Rutkowski JM, Martel C, Kuan EL, Ivanov S, Swartz MA, and Randolph GJ (2013). Normal dendritic cell mobilization to lymph nodes under conditions of severe lymphatic hypoplasia. J. Immunol 190, 4608–4620. 10.4049/jimmunol.1202600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Haka AS, Potteaux S, Fraser H, Randolph GJ, and Maxfield FR (2012). Quantitative analysis of monocyte subpopulations in murine atherosclerotic plaques by multiphoton microscopy. PLoS One 7, e44823. 10.1371/journal.pone.0044823. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The ImmGen MNP Open Source RNA sequencing dataset generated during this study has been deposited at GEO repository (GSE122108) and is publicly available as of the date of publication. This paper also analyzes existing, publicly available data: The ImmGen Phase 1 dataset (GSE15907) and The Tabula Muris Senis dataset (GSE149590). All accession numbers are also listed in the key resources table.
All original code has been deposited at Zenodo and is publicly available as of the date of publication. DOI is listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| CD45 BUV563 clone 30-F11 | BD Biosciences | Cat#612924; RRID:AB_2870209 |
| CD45 APC-Cy7 clone 30-F11 | Biolegend | Cat#103116; RRID:AB_312981 |
| I-A/I-E BUV496 clone M5/114.15.2 | BD Biosciences | Cat#750281; RRID:AB_2874472 |
| CD11c PE-Cy7 clone N418 | Biolegend | Cat#117318; RRID:AB_493568 |
| CD11c PE clone N418 | Biolegend | Cat#117308; RRID:AB_313777 |
| CD3e AF488 clone 145-2C11 | Biolegend | Cat#100321; RRID:AB_389300 |
| CD19 FITC clone 1D3/CD19 | Biolegend | Cat#152404; RRID:AB_2629813 |
| NK1.1 AF488 clone PK136 | Biolegend | Cat#108718; RRID:AB_493183 |
| TER-119 AF488 clone TER-119 | Biolegend | Cat#116215; RRID:AB_493402 |
| CD64 APC clone X54–5/7.1 | Biolegend | Cat#139306; RRID:AB_11219391 |
| F4/80 AF488 clone BM8 | Biolegend | Cat#123120; RRID:AB_893479 |
| F4/80 PE clone BM8 | Biolegend | Cat#123110; RRID:AB_893486 |
| CD11b BV421 clone M1/70 | Biolegend | Cat#101236; RRID:AB_11203704 |
| anti-Perfringolysin O rabbit antibody | Abcam | Cat#ab225685 |
| Goat anti-Rabbit IgG (H + L) AF647 | Thermofisher | Cat#A-21245; RRID:AB_141775 |
| Mouse TruStain FcX (anti-mouse CD16/CD32, clone 93) antibody | Biolegend | Cat#101320; RRID:AB_1574975 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| LIVE/DEAD Fixable Aqua Dead Cell Stain Kit | Thermofisher | Cat#L34957 |
| Collagenase D | Sigma | Cat#11088882001 |
| Perfringolysin O (PFO) from Clostridium perfringens | Cusabio | Cat#CSB-EP314820CMB |
| Fluorescein isothiocyanate (FITC) | Sigma | Cat#F7250 |
| Simvastatin | Sigma | Cat#S6196 |
| L-Buthionine-sulfoximine (BSO) | Sigma | Cat#B2515 |
| Zymosan | Invivogen | Cat#tlrl-zyn |
|
| ||
| Critical commercial assays | ||
|
| ||
| Cholesterol Quantitation Kit | Sigma | Cat#MAK043 |
| PGE2 ELISA kits | Enzo | Cat#ADI-900-001 |
| Cysteinyl leukotriene ELISA kits | Enzo | Cat#ADI-900-070 |
| RPMI 1640 Medium | Thermofisher | Cat#11875093 |
| Deposited data | ||
| ImmGen ULI: OpenSource Mononuclear Phagocytes Project (raw and processed data) | This paper | GEO: GSE122108 |
| ImmGen Microarray Phase 1 (raw and processed data) | Gautier et al.5 | GEO: GSE15907 |
| Tabuls Muris Senis (raw and processed data) | Tabula Muris Consortium11 | GEO: GSE149590; https://s3.console.aws.amazon.com/s3/buckets/czb-tabula-muris-senis/; https://figshare.com/articles/dataset/Processed_files_to_use_with_scanpy_/8273102/2 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| Mouse peritoneal primary macrophages | This paper | NA |
| Mouse lung alveolar primary macrophages | This paper | NA |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: wild-type C57BL/6J mice | Jackson Laboratory | Strain#000664 |
|
| ||
| Software and algorithms | ||
|
| ||
| GAM-clustering | This paper | GitHub: https://github.com/artyomovlab/ImmGenOpenSource https://doi.org/10.5281/zenodo.7492657 |
| GMWCS-solver | Loboda et al.19 | https://cran.r-project.org/web/packages/mwcsr |
| FlowJo software v10.2 | BD | https://www.flowjo.com/ |
| Prism v9.4.1 | Graphpad Software | https://www.graphpad.com/scientific-software/prism/ |
|
| ||
| Other | ||
|
| ||
| Resource website: Hands-on interactive browser for gene expression exploration both for ImmGens and Tabula Muris Senis datasets | This paper | https://artyomovlab.wustl.edu/immgen-met/ |