

SUMMARY
Despite prolific efforts to characterize the antibody response to human immunodeficiency virus type 1 (HIV-1) and hepatitis C virus (HCV) mono-infections, the response to chronic co-infection with these two ever-evolving viruses is poorly understood. Here, we investigate the antibody repertoire of a chronically HIV-1/HCV co-infected individual using linking B cell receptor to antigen specificity through sequencing (LIBRA-seq). We identify five HIV-1/HCV cross-reactive antibodies demonstrating binding and functional cross-reactivity between HIV-1 and HCV envelope glycoproteins. All five antibodies show exceptional HCV neutralization breadth and effector functions against both HIV-1 and HCV. One antibody, mAb688, also cross-reacts with influenza and coronaviruses, including severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). We examine the development of these antibodies using next-generation sequencing analysis and lineage tracing and find that somatic hypermutation established and enhanced this reactivity. These antibodies provide a potential future direction for therapeutic and vaccine development against current and emerging infectious diseases. More broadly, chronic co-infection represents a complex immunological challenge that can provide insights into the fundamental rules that underly antibody-antigen specificity.
Graphical abstract
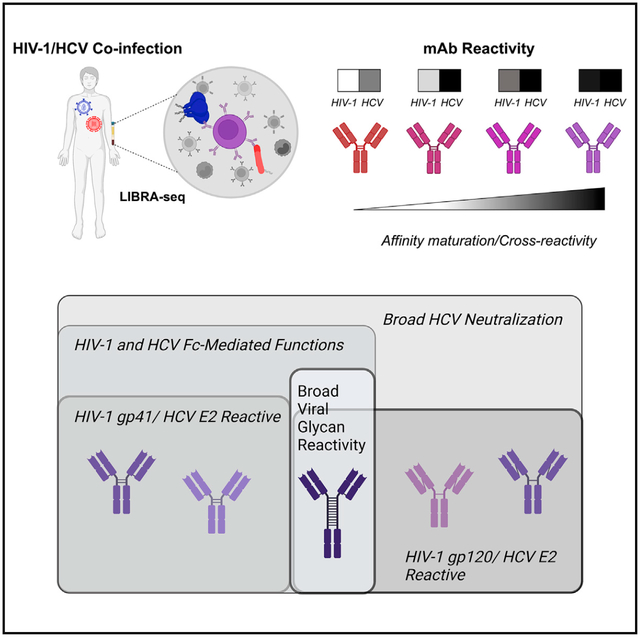
In brief
Pilewski et al. uses LIBRA-seq to identify multiple, functional HIV-1/HCV cross-reactive antibodies with diverse epitope specificities. This study identifies a glycan-reactive, broadly anti-viral antibody recognizing the NTD of SARS-CoV-2, in addition to antigens from multiple other viruses. The authors further show that somatic hypermutation both establishes and enhances HIV-1/HCV cross-reactivity.
INTRODUCTION
Human immunodeficiency virus type 1 (HIV-1) and hepatitis C virus (HCV) are two of the most diverse human pathogens, quickly evolving to evade immunepressure, typically establishing chronic, life-long infection.1–5 Furthermore, due to the shared routes of transmission, HIV-1/HCV co-infection is relatively common, affecting an estimated 5 million individuals worldwide.6,7 Although significant advances in the treatment of both viruses have occurred in the last 30+ years, there are still no licensed vaccines or other long-lasting prophylactic countermeasures.8,9 Moreover, although HCV infection can be cured with small-molecule inhibitor drugs, less than 50% of infected individuals know of their HCV-positive status, highly limiting the utility of these drug regimens.10 Many at-risk individuals have poor access to diagnostic tools or medications HIV-1/HCV co-infected individuals experience the highest re-infection rates with either virus, strongly motivating the ongoing development of alternative therapeutic and prophylactic tools for these viruses.11–14 Such new tools could have enhanced medical impact in the setting of HIV-1/HCV co-infection, where the chronic exposure to two mutating pathogens leads to significantly exacerbated health problems compared with mono-infection.15–17
Although these highly mutable viruses have rendered classical vaccine design difficult, investigating the human antibody response to HIV-1 or HCV mono-infection has led to the identification of antibodies that reduce infection or disease and that have served as templates for structure-based vaccine development.18–22 The clinical setting of HIV-1/HCV co-infection has been far less explored, with little understanding about antibody responses in the chronic presence of two diverse, constantly evolving, antigen targets.23–25 In this study, we sought to investigate the antibody repertoire of a chronically HIV-1/HCV co-infected individual using linking B cell receptor to antigen specificity through sequencing (LIBRA-seq), a technology that enables the simultaneous screening of B cells against a diverse library of antigen targets.26 Notably, we show that LIBRA-seq-identified antibodies have binding and functional cross-reactivity between HIV-1 and HCV without exhibiting typical traits of promiscuous antigen recognition. These results challenge our long-standing understanding of the exclusiveness of antibody-antigen specificity and pave the way toward the development of effective therapeutics and vaccines with an unparalleled breadth of reactivity.
RESULTS
Discovery of HIV-1/HCV cross-reactive antibodies from a chronically HIV-1/HCV co-infected donor
To probe the development of antibody responses produced by the immunological challenge of HIV-1/HCV co-infection, we profiled the antigen-specific B cell compartment using LIBRA-seq. As previously described, LIBRA-seq allows for high-throughput mapping of antigen specificity to the B cell receptor sequence by leveraging oligo-barcoded antigens and single-cell sequencing.26 We identified a donor, VC10014 from the Vanderbilt HIV-1 infection cohort, who had been chronically HIV-1/HCV co-infected for >3 years at the time of sample collection with no history of anti-viral or anti-retroviral treatment. Previous studies investigating key events leading to early development of broad HIV-1 neutralization established that VC10014 developed broadly neutralizing serum antibodies approximately 1 year after HIV-1 infection and that this phenotype could largely be traced to a CD4-binding-site-directed antibody response.27,28 Monoclonal antibody discovery efforts in this donor failed to identify any broadly neutralizing antibodies, instead attributing the observed serum breadth to diverse antibody lineages with complementary patterns of neutralizing activity.29 Given the ability of LIBRA-seq to screen tens of thousands of B cells against a large panel of diverse antigens, including those from unrelated pathogens, we sought to apply this technology to HIV-1/HCV co-infected donor VC10014. The application of LIBRA-seq provides an opportunity to interrogate the antibody repertoire against the complex setting of chronic exposure to diverse and constantly evolving antigens.
To identify virus-specific B cell sequences from VC10014, we applied LIBRA-seq with a diverse panel of seven antigens including four HIV-1 envelope glycoprotein antigens, each from a unique virus clade (A/BG505 gp140, B/B41 gp140, C/ConC gp120, AE/A244 gp120) and three HCV envelope glycoprotein antigens from two distinct virus genotypes (1a/H77 E2c, H77 E1E2, and 2a/JFH1 E2c) (Figure S1A). The inclusion of multiple antigen variants for HIV-1 and HCV allows for the identification of antibodies with a broad range of antigen specificities. LIBRA-seq recovered paired heavy-/light-chain B cell receptor (BCR) sequences with associated antigen specificity mapping for 886 cells (Figures S1B and S1C). Of the identified class-switched BCR sequences, ~75% were HIV-1 antigen specific, and ~23% were HCV antigen specific. Interestingly, we also identified a small population of cells positive for at least one HIV-1 and at least one HCV antigen (Figure S1C). Within this population of cross-reactive B cells, we focused on the class-switched cells with the highest LIBRA-seq scores for any HIV-1 and HCV antigen (Figure 1A). We identified five genetically unique sequences with varied levels of somatic hypermutation (SHM) and HIV-1 antigen-positive/HCV antigen-positive specificity phenotypes for further study (Figure 1B). We expressed these five paired heavy-light chain sequences as recombinant monoclonal antibodies and confirmed their reactivity against a panel of HIV-1 and HCV envelope glycoproteins by ELISA (Figures 1C–1G). The results confirmed that LIBRA-seq successfully predicted the HIV-1/HCV envelope antigen cross-reactivity of the identified antibodies.
Figure 1. Discovery of exceptionally broad anti-viral antibodies from a chronically HIV-1/HCV co-infected donor.
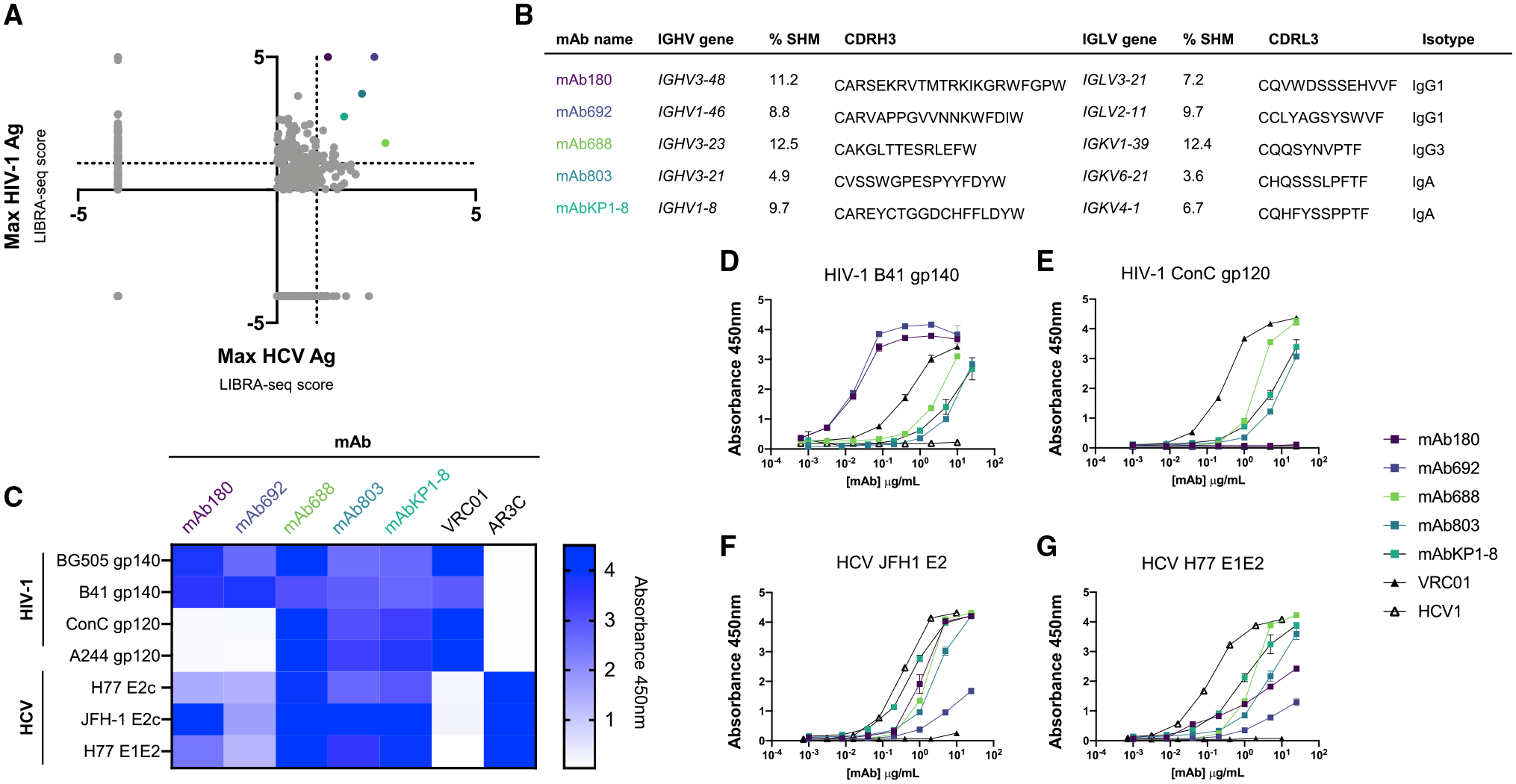
(A) Plot of maximum LIBRA-seq score for an HIV-1 antigen (Ag) vs. an HCV Ag where each dot represents a single class-switched (IgG, IgA) cell. Colored dots represent antibodies selected for further study. Cells with Ag values <0 were set to −4.
(B) Genetic features of identified HIV-1/HCV cross-reactive BCRs. Color of antibody name corresponds to LIBRA-seq score colors in (A).
(C) Binding of HIV-1/HCV cross-reactive recombinant antibodies to the panel of viral Ags (shown on the left) at 10 μg/mL as measured by ELISA. The following antibodies are shown as controls: AR3C (HCV E2), VRC01 (HIV-1 gp120).
(D–G) Recombinantly expressed lead antibody binding to clade B (B41) and clade A (BG505) HIV-1 envelope glycoproteins (D and E) and genotype 2a (JFH1) and genotype 1a (H77) HCV envelope glycoproteins (F and G), measured by ELISA.
Each assay was performed in duplicate, and data shown are representative of at least 2 separate experiments with at least 2 different antibody preparation aliquots. Data are shown as mean ± SEM, where applicable.
HIV-1/HCV cross-reactive antibodies recognize distinct epitopes on the HIV-1 and HCV envelope glycoproteins
Given the pattern of antibody-antigen cross-reactivity, we sought to map the epitope of these antibodies on the two antigen targets. We first defined the five cross-reactive antibody epitope targets on the HCV envelope. All antibodies bound recombinant HCV E1E2 protein with recognition directed to the E2 subunit of the glycoprotein, with only monoclonal antibody 688 (mAb688) displaying slight reactivity with E1 (Figures 1F, 1G, 2A, and 2B). To further map the targeted epitopes, we measured antibody competition with CD81, the cognate HCV entry receptor (Figure 2C). We discovered that binding of four of the five HIV-1/HCV cross-reactive antibodies was inhibited by pre-incubation with the large extracellular loop of CD81 (CD81-LEL) (Figure 2C).
Figure 2. HIV-1/HCV cross-reactive antibodies recognize distinct epitopes on the HIV-1 and HCV envelope glycoproteins.
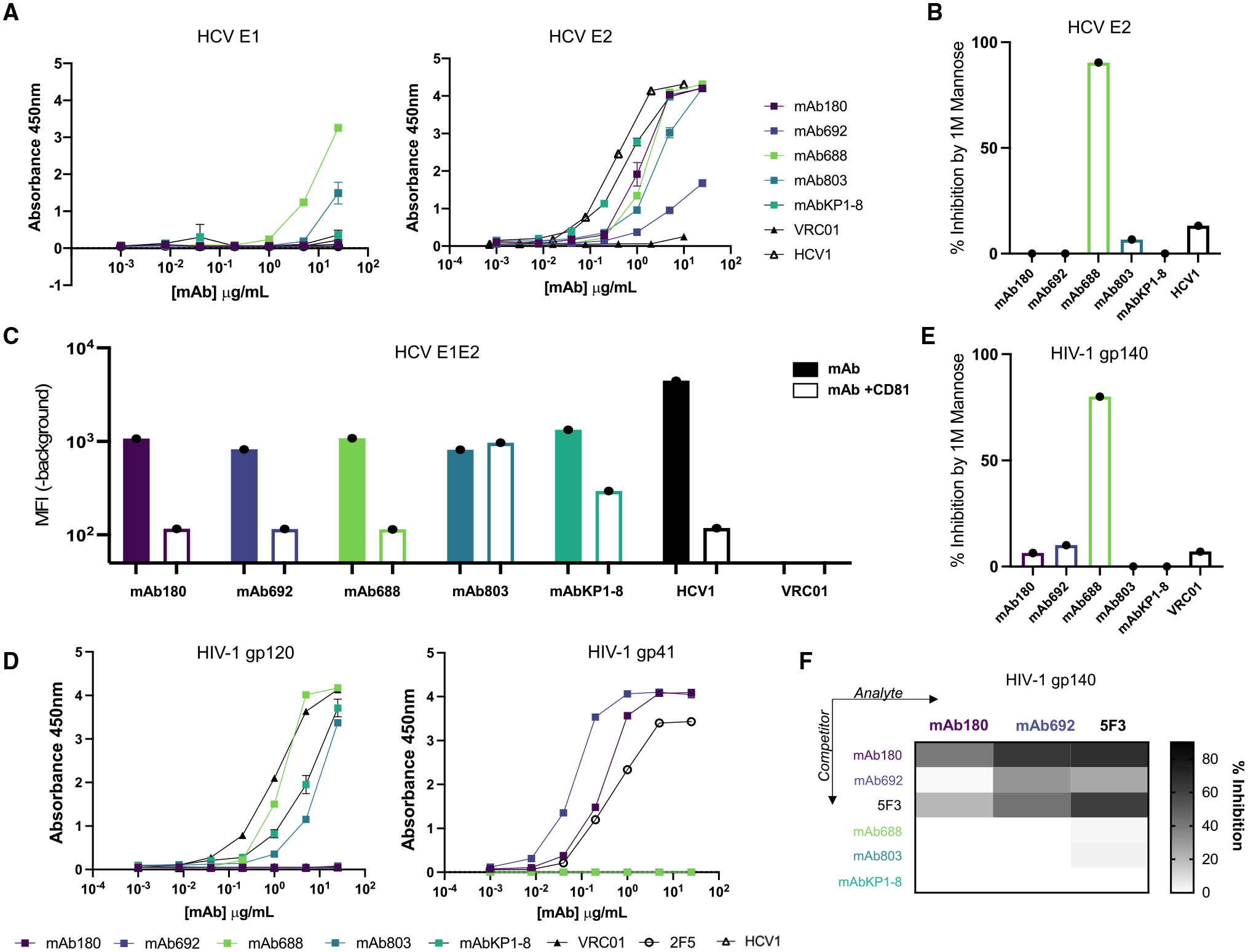
(A) Antibody binding to the E1 (strain: HC-J4; left) and E2 (strain: JFH-1; right) subunits of the HCV envelope glycoprotein measured by ELISA. The HCV E2-specific antibody HCV1 is shown as a positive control, and the HIV-1-specific antibody VRC01 is shown as a negative control.
(B) Antibody binding to HCV E2 glycoprotein (strain: JFH-1) in the presence of 1 M D-(+)-mannose. Data are plotted as percentage of inhibition compared with binding in PBS buffer. Antibodies demonstrating negative percentage of inhibition values were set to 0.
(C) Antibody binding to cell surface-expressed HCV E1E2 (strain: H77) measured by flow cytometry. Cells were either pre-incubated with PBS (filled bar) or CD81-LEL (open bar) before detection by fluorescent secondary. Data are shown as background-subtracted mean fluorescence intensity (MFI).
(D) Antibody binding to the gp120 (strain AE.A244) (left) and gp41 (strain: MN) (right) subunits of the HIV-1 envelope glycoprotein measured by ELISA. The gp120- and gp41-specific antibodies VRC01 and 2F5, respectively, are shown as positive controls, and the HCV-specific antibody HCV1 is shown as a negative control.
(E) Antibody binding to HIV-1 gp140 (strain: BG505) in the presence of 1 M D-(+)-mannose measured by ELISA. Data plotted as percentage of inhibition compared with binding in PBS buffer. The mannose-independent HIV-1 antibody VRC01 is shown as a control. Antibodies demonstrating negative percentage of inhibition values were set to 0.
(F) Antibody binding to HIV-1 gp140 (strain: BG505) in the presence of competitor antibodies measured by ELISA. Competitor antibodies (shown on the y axis) were added first, and subsequently binding of biotinylated analyte antibodies (shown on the x axis) was detected. Data are displayed as percentage of inhibition, calculated as a function of no competition controls.
Each assay was performed in duplicate, and data shown are representative of at least 2 separate experiments with at least 2 different antibody preparation aliquots. Data are shown as mean ± SEM, where applicable.
Given the extensive glycan shield that decorates both the HIV-1 and HCV envelope glycoproteins, we also sought to define the glycan dependence of HIV-1/HCV cross-reactive recognition. Only mAb688 binding was inhibited by both 1 M D-(+)-mannose and PNGaseF de-glycosylation (Figure 2B and S2A). Additionally, in experiments with HCV E2 protein produced in the presence of kifunensine, an α-mannosidase inhibitor that results in the majority presence of high mannose-type glycans, mAb688 binding was decreased, suggesting that recognition requires additional contacts or glycan processing (Figure S2B). In summary, we discovered that HIV-1/HCV cross-reactive antibodies recognize at least three distinct epitopes on the HCV envelope protein including CD81-independent (mAb803) as well as glycan-dependent (mAb688) or -independent (mAb180, mAb692, mAbKP1–8) CD81-blocking regions.
Next, we sought to map the epitopes of these antibodies on the HIV-1 envelope protein. All five antibodies bound soluble HIV-1 gp140, albeit to various degrees (Figure 1D). Intriguingly, two of the five antibodies (mAb180, mAb692) recognized the gp41 subunit of the HIV-1 envelope (Env), while the other three antibodies recognized gp120 (Figures 2D, S2F, and S2G). Competition-binding ELISA experiments showed that mAb180 and mAb692 competed with antibody 5F3 (Figure 2F). 5F3 has previously been reported to interact with both the C-terminal heptad repeat region (CHR) and the fusion peptide-proximal region (FPPR).30,31 Importantly, we were unable to detect binding of 5F3 or related CHR/FPPR-specific HIV-1 antibodies (167-D, F240) to the HCV Env protein, suggesting that mAb180 and mAb692 represent a distinct binding modality compared with these previously published antibodies (Figure S2C). As expected from their gp120 reactivity, mAbs 688 and 803 and KP1–8 did not compete with the gp41-reactive mAb180 and mAb692 for binding to HIV-1 gp140 (Figure 2F).
As with HCV, we also investigated the glycan dependence of antibody binding to HIV-1 gp140, a viral protein ornamented with glycans. We discovered that only mAb688 binding to HIV-1 gp140 was inhibited by PNGaseF de-glycosylation and competition with 1 M D-(+)-mannose (Figures 2E and S2D). Similar to experiments with HCV E1E2, mAb688 recognition of HIV-1 gp140 was notably decreased after treatment with the α-mannosidase inhibitor kifunensine (Figure S2E). Taken together, these data support the discovery of antibodies recognizing at least three distinct regions on the HIV-1 Env glycoprotein encompassing gp41 (mAb180, mAb692) and both glycan-dependent (mAb688) and -independent (mAb803, KP1–8) gp120 epitopes.
HIV-1/HCV cross-reactive mAbs show diverse neutralization and Fc-mediated effector functions
After evaluating HIV-1 and HCV antigen specificity, we next assessed the functional abilities of the HIV-1/HCV cross-reactive antibodies in both neutralization and Fc-mediated effector function assays. First, we investigated the ability of HIV-1/HCV cross-reactive antibodies to neutralize a panel of representative genotype 1 HCV strains.32–34 Notably, we observed that all five HIV-1/HCV cross-reactive antibodies showed exceptional HCV neutralization breadth, neutralizing all 19 genotype 1 strains tested at 100 μg/mL (Figures 3A, S3A, and 3B). Further, four of the five HIV-1/HCV cross-reactive antibodies also neutralized both genotype 2b and 3a strains (Figure 3A). This finding is particularly striking when compared with the previously reported broadly neutralizing antibodies HEPC74 or AR3C, which neutralized 18/19 or 16/19 genotype 1 strains in this panel, respectively.35,36 We next tested whether the immunoglobulin G (IgG) antibodies mAb180, mAb692, and mAb688 could mediate anti-HCV E1E2 Fc effector functions. Each of the antibodies tested (mAb180, mAb692, mAb688) mediated antibody-dependent cellular phagocytosis (ADCP) against HCV E1E2 (genotype 1a; strain H77) (Figures 3B and 3C).
Figure 3. HIV-1/HCV cross-reactive antibodies show diverse neutralization and Fc-mediated effector functions.
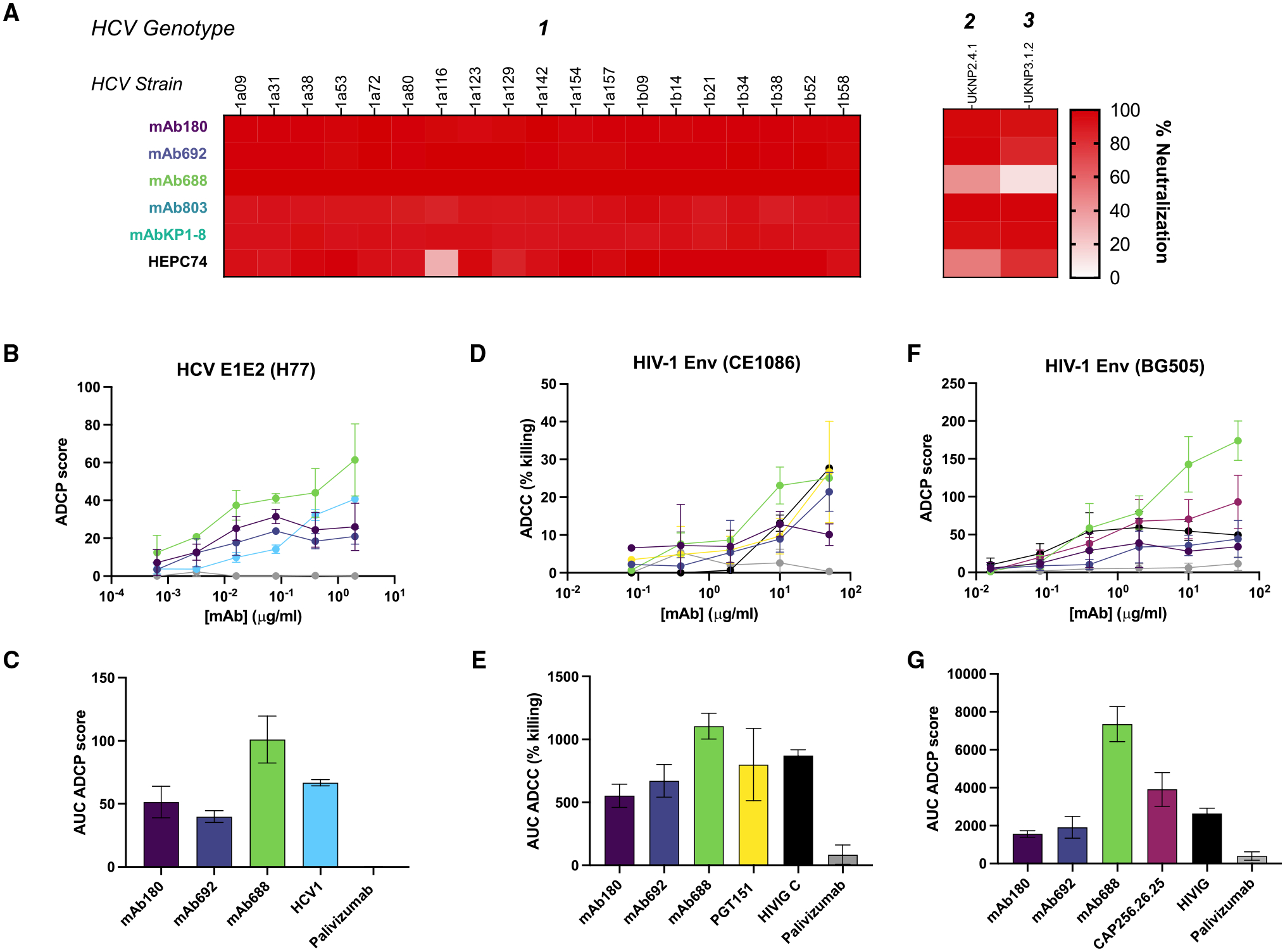
(A) In vitro neutralization of a panel of diverse genotype 1 HCV virus strains by HIV-1/HCV-reactive antibodies and control antibody HEPC74 at 100 μg/mL. Percentage of neutralization calculated in comparison to unrelated IgG control.
(B) Antibody-dependent cellular phagocytosis (ADCP) by either HIV-1/HCV cross-reactive or control antibodies (HCV1: positive; palivizumab: negative) against HCV E1E2 envelope protein (H77).
(C) Area under the curve (AUC) values computed from (B).
(D) Antibody-dependent cellular cytotoxicity (ADCC) potentiated by either HIV-1/HCV cross-reactive or control antibodies (PGT151/HIVIG C: positive; palivizumab: negative) against infectious HIV-1 envelope protein (CE1086).
(E) AUC values computed from (D).
(F) ADCP by either HIV-1/HCV cross-reactive or control antibodies (CAP256.26.25/HIVIG C: positive; palivizumab: negative) against HIV-1 envelope protein (BG505).
(G) AUC computed from (F).
Each assay was performed in duplicate, and data shown are representative of at least 2 separate experiments with at least 2 different antibody preparation aliquots. Data are shown as mean ± SD, where applicable.
Next, we characterized the anti-HIV-1 functions of HIV-1/HCV cross-reactive antibodies. We observed that none of the antibodies neutralized the panel of HIV-1 strains tested (Figure S3C). However, each of the IgG antibodies tested (mAb180, mAb692, mAb688) mediated antibody-dependent cellular cytotoxicity (ADCC) against infectious HIV-1 Env (strain CE1086) (Figures 3D and 3E). Moreover, we observed that all three antibodies mediated ADCP against HIV-1 gp140 (strain BG505) (Figures 3F and 3G). Taken together, the identified antibodies not only bind to diverse viral Env glycoproteins, but they also exhibit extraordinary cross-reactive functionality. The IgA isotype antibodies (mAb803, KP1–8) were not tested in the Fc effector assays against either virus, as this isotype plays a less significant role in serological anti-viral Fc-mediated immunity.37,38
mAb688 reveals exceptionally broad anti-viral functions
Next, we asked whether the identified antibodies were solely HIV-1/HCV cross-reactive or whether they could recognize additional viral Env glycoproteins. To that end, we tested these antibodies against a panel of antigens from a diverse set of pathogens and found that mAbs 180, 692, and 803 and KP1–8 indeed bound only the HIV-1 and HCV antigens tested (Figure 4A). By contrast, mAb688 recognized a broad diversity of other viral antigens, including glycoproteins from influenza A virus, alpha-coronaviruses NL63 and 229E, and betacoronaviruses Middle Eastern respiratory syndrome coronavirus (MERS-CoV), severe acute respiratory syndrome (SARS)-CoV, and SARS-CoV-2 by ELISA (Figure 4A). mAb688 did not neutralize influenza virus or mediate hemagglutination inhibition (HAI) but appeared to have mannose-dependent binding to and showed ADCP activity against the influenza A hemagglutinin protein (Figures 4B, 4C, and S4A–S4C).
Figure 4. Cross-reactive mAb688 reveals exceptionally broad anti-viral functions achieved by glycan recognition.
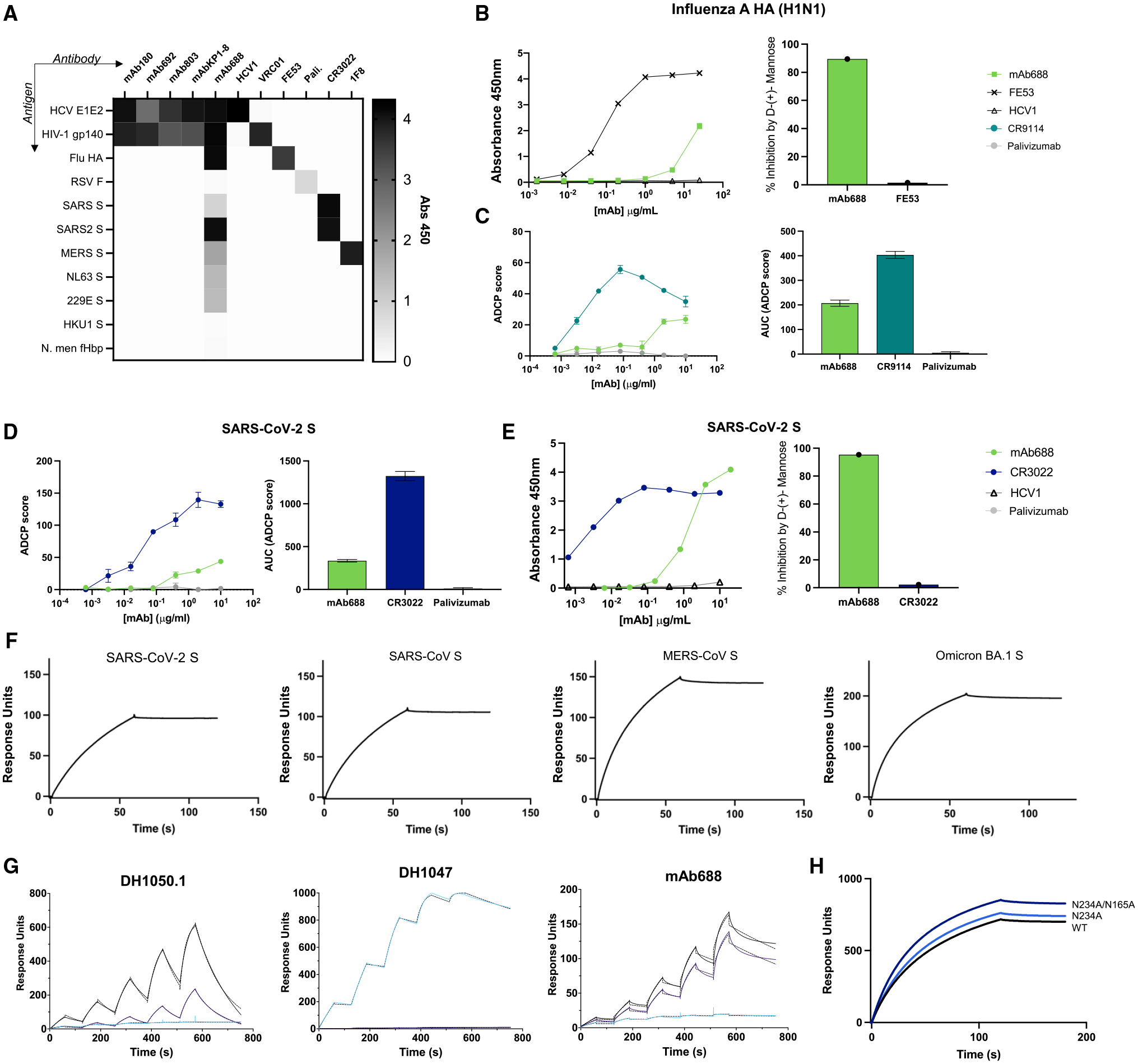
(A) Binding of HIV-1/HCV cross-reactive antibodies (columns) to a panel of diverse viral Ags (rows) at 10 μg/mL, as measured by ELISA. The following antibodies are shown as controls: HCV1 (HCV E2); VRC01 (HIV-1 gp120); FE53 (HA); palivizumab (RSV F); CR3022 (SARS-CoV/SARS-CoV-2 spike [S]); and 1F8 (MERS S).
(B) mAb688 binding to influenza A HA (strain H1N1/New Caledonia/1999) envelope glycoprotein with PBS (left) and competition in presence of 1 M D-(+)-mannose (right), displayed as percentage of inhibition (y axis) in the presence of mannose.
(C) ADCP of influenza A HA (strain H1N1/New Caledonia/1999) envelope glycoprotein (left), with data displayed as AUC (right). CR9114 is shown as a positive control, and palivizumab is shown as a negative control.
(D) ADCP of SARS-CoV-2 S glycoprotein (left), with data displayed as AUC (right). CR3022 is shown as a positive control, and palivizumab is shown as a negative control.
(E) mAb688 binding to SARS-CoV-2 S protein with PBS (left) and competition in presence of 1 M D-(+)-mannose (right), displayed as percentage of inhibition (y axis) in the presence of mannose.
(F) Binding of mAb688 to CoV S proteins measured by SPR. A 200 nM solution of mAb688 was flowed over a CM5 chip that was coated with anti-Fc IgG. Binding to mAb688 was assessed by flowing over 200 nM solution of each CoV S protein shown.
(G) Binding of mAb688, DH1050.1 (N-terminal domain [NTD]-directed antibody), and DH1047 (receptor-binding domain [RBD]-directed antibody) to SARS-CoV-2 S protein RBD (blue line), the NTD (black line), and the NTD with the His69/Val70 deletion (ΔH69/V70) (purple line). The antibodies were captured on an anti-Fc chip. Binding was assessed by flowing over the RBD/NTD proteins. Dotted lines indicate global fit of the data to a 1:1 Langmuir binding model.
(H) Binding of mAb688 to SARS-CoV-2 S ectodomain with the engineered 2P substitution (black line), the 2P-S protein with the N234A substitution (light blue line), and with the N234A and N165A substitutions (dark blue line). mAb688 was captured on an anti-Fc chip. Binding was assessed by flowing over the SARS-CoV-2 S proteins.
Each assay was performed in duplicate, and data shown are representative of at least 2 separate experiments. ELISA and ADCP experiments were replicated with at least 2 different antibody preparation aliquots. ELISA data are shown as mean ± SEM, and ADCP data are shown as mean ± SD.
Given the current COVID-19 pandemic caused by SARS-CoV-2, we next sought to further characterize the broad coronavirus reactivity of mAb688 (Figures 4D–4H and S5). We found that mAb688 did not neutralize SARS-CoV-2 detectably but showed ADCP activity against the SARS-CoV-2 spike protein (Figures 4D and S5B). As observed with HIV-1, HCV, and influenza A, mAb688 recognition of SARS-CoV-2 spike protein was mannose dependent (Figure 4E). Using surface plasmon resonance (SPR), we found that mAb688 retained binding to SARS-CoV-2 Omicron variants, as well as SARS-CoV and MERS-CoV, suggesting that it binds a broadly conserved epitope in these spike proteins (Figures 4F and S5A). Previous studies have demonstrated that the anti-HIV-1 antibody 2G12 also binds the SARS-CoV-2 spike protein by recognition of a high mannose glycan structure on asparagine 709.39 Comparatively, we found that mAb688 binding to SARS-CoV-2 spike was not inhibited by N709A substitution (Figure S5C). We next tested mAb688 binding to the receptor-binding domain (RBD) and the N-terminal domain (NTD) of the SARS-CoV-2 spike (S) protein (Figure 4G), including the RBD-directed antibody DH1047 and the NTD-directed antibody DH1050.1 as controls.40 As expected, DH1047 bound the RBD but not the NTD constructs, while DH1050.1 bound the NTD but not the RBD. mAb688 showed robust binding to the wild-type NTD and to an NTD construct harboring the His69/Val70 NTD deletion (ΔH69/V70) that was found in several variants of concern (VOCs).41 mAb688 binding to the NTD was not competed by either the DH1050.1 or the DH1052 Fabs that bind distinct NTD epitopes (Figure S5D). Moreover, mAb688 binding to the SARS-CoV-2 S was not inhibited by deletion of glycans N165 and N234 (Figure 4H). Together with the mAb688 HIV-1 and HCV functional activity, these data indicate that mAb688 possesses diverse functions against a broad range of viral targets.
To determine whether the observed broad functional abilities of mAb688 spanned beyond virus targets to other microbial pathogens with mannosylated surface proteins, we investigated whether mAb688 could inhibit the most common etiological agent of urinary tract infection (UTI), uropathogenic Escherichia coli (UPEC). UPEC infection uses fimbriae to recognize mannosylated bladder host cell surface glycoprotein and red blood cells.42 As this interaction between bacteria and host is free-mannose inhibitable, we tested whether mAb688 recognition of host mannose could block function. Importantly, we discovered that neither mAb688 nor the isotype control were able to impede UPEC adherence or HA (Figures S4D and S4E), suggesting that mAb688 recognizes a mannose structure that may be specific to viral glycosylation and/or may require additional antigen interactions.
Finally, to define specific glycan architecture that mediates broad mAb688 recognition, we tested binding to a glycan microarray consisting of >580 distinct structures developed by the Center for Functional Glycomics (CFG; v.5.4 microarray). Interestingly, the majority of glycans to which mAb688 bound contained a terminal N-acetyl glucosamine with β1–6 linkage with mannose or galactose, suggesting that this linkage is important for mAb688 binding (Figure 5). Further, these data demonstrate that mAb688 preferentially recognizes immature, hybrid-type glycans, a form of glycosylation that is enriched on viral glycoproteins (Figure 5).
Figure 5. mAb688 achieves broad anti-viral binding via recognition of immature glycans.
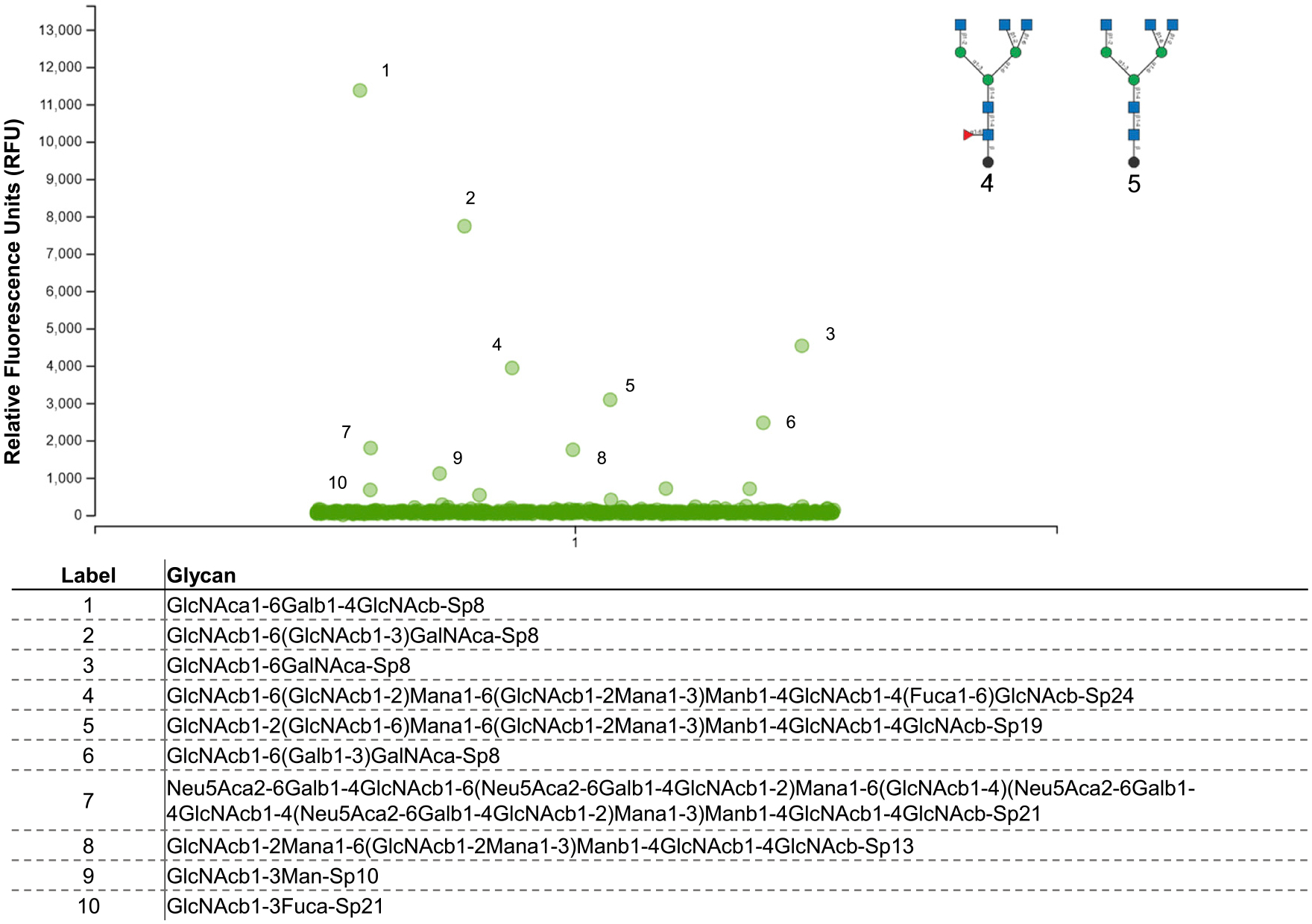
Binding of mAb688 to a printed glycan microarray was tested at 50 μg/mL. Antibody binding was detected using fluorescent secondary antibody, and data are shown as the average relative fluorescence units (RFUs). Chemical nomenclature for glycan structures corresponding to numbered green circles are shown below. The assay was performed in triplicate.
Diverse polyreactivity profiles of HIV-1/HCV cross-reactive mAbs
To investigate whether the cross-reactive antibodies may achieve diverse binding phenotypes via antigen polyreactivity or non-specific interactions, we first measured reactivity to a panel of nuclear self-antigens using the Luminex AtheNA Multi-analyte ANA assay (Figure 6A).43 Similar to previously described gp41 antibodies, we observed autoreactivity for both mAb180 and mAb692. Further, we found that mAb180, but not the other antibodies, bound non-infected, whole (unpermeabilized) HEp-2 cells in a fluorescent assay (Figures 6B and 6C). Therefore, from the set of five cross-reactive antibodies, only mAb180 and mAb692 showed binding in autoreactivity assays, suggesting that the broadly reactive anti-viral phenotype of the other antibodies—including the exceptionally broad mAb688—could not be explained by promiscuous, non-specific antigen interactions.
Figure 6. Autoreactivity of HIV-1/HCV cross-reactive antibodies.
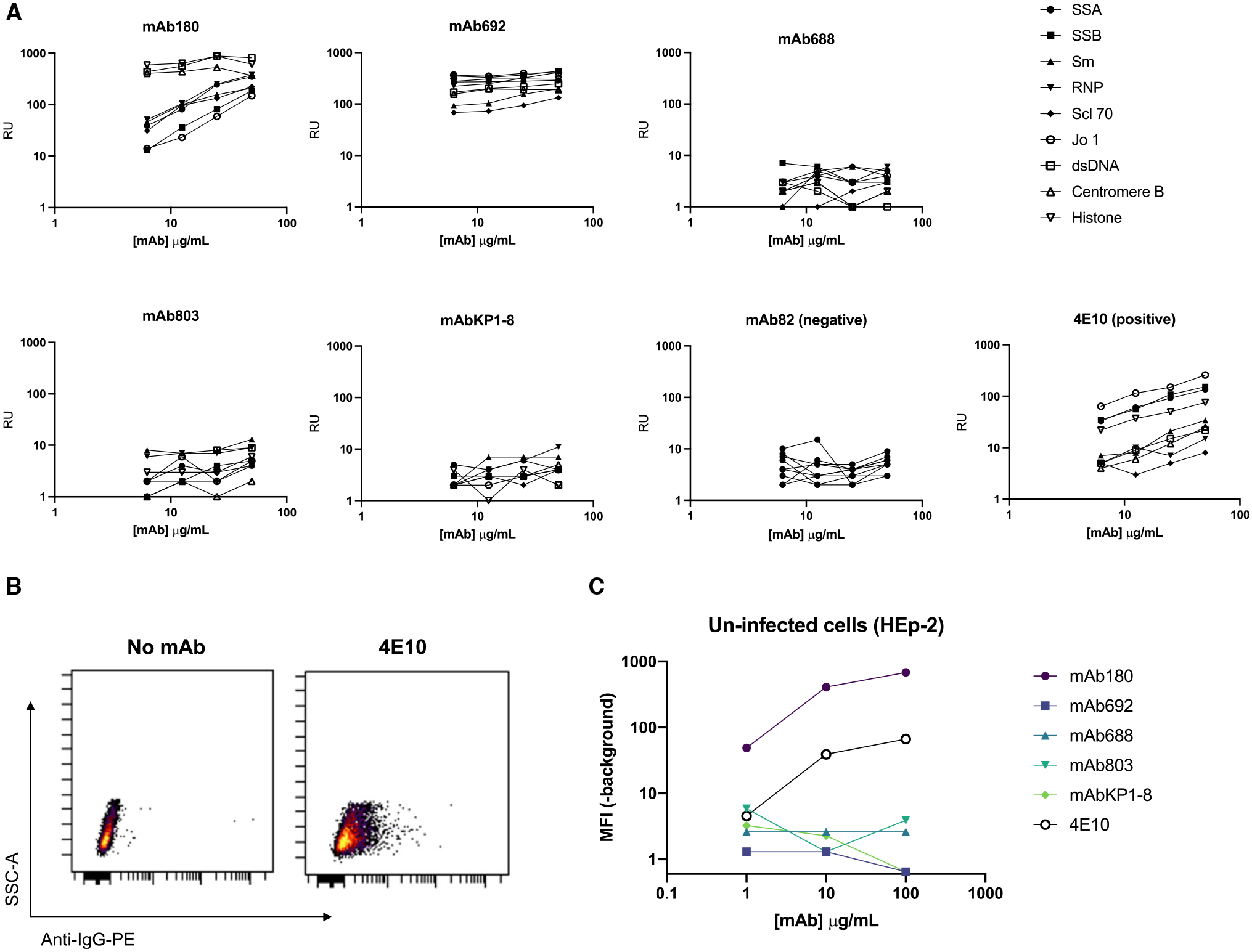
(A) Antibody binding to a panel of autoantigens using the AtheNA multiplex assay, over the concentration gradient shown. RUs >100 are considered positive. The HIV-1 antibody 4E10 is shown as a positive control, and mAb82 is shown as a negative control.
(B and C) Antibody binding to whole, unpermeabilized, uninfected HEp-2 cells detected by anti-IgG-PE (anti-IgA-PE; mAb803, mAbKP1–8).
(B) Secondary only (negative) and 4E10 (positive) control plots.
(C) Binding of HIV/HCV cross-reactive antibodies depicted as MFI of the PE channel (-MFI unstained control) at the shown concentration of antibody. AtheNA assays were performed in triplicate. Cell binding experiments were repeated in duplicate.
SHM establishes and enhances cross-reactivity
We next interrogated the effect of SHM on the development of HIV-1/HCV cross-reactivity. High-affinity HIV-1-specific antibody responses often require the accumulation of mutations through multiple rounds of SHM over the course of chronic infection.44–46 We therefore assessed binding of germline-reverted IgG antibody variants to both HIV-1 and HCV Env proteins (Figures 7A, 7C, and S6A). These germline-reverted variants lacked all acquired somatic mutations by comparison with inferred germline gene segments but had the CDR3 sequences of the mature antibody. Interestingly, when somatic mutations were removed from mAb688, this antibody no longer bound detectably either HIV-1 or HCV (Figure S6A). By contrast, when mAb180 and mAb692 were germline reverted, they retained binding to HCV Env protein and demonstrated distinct HIV-1 Env reactivities (Figures 7A and 7C).
Figure 7. Somatic hypermutation impacts antibody cross-reactivity.
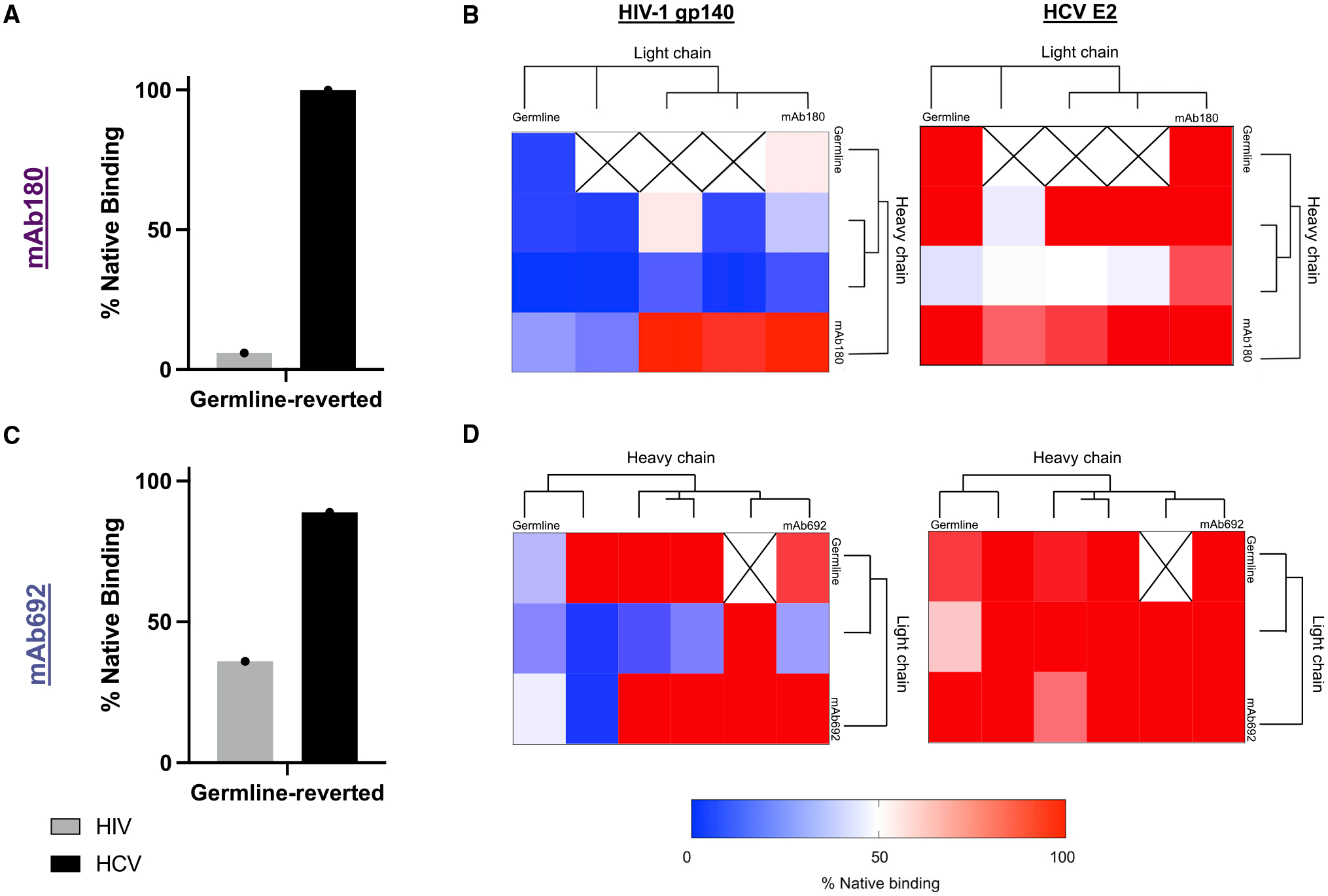
(A and C) Binding of germline-reverted antibody mutants (A) mAb180 and (C) mAb692 to HIV-1 gp140 (strain: BG505, gray bars) and HCV E2 (strain: JFH1, black bars) measured by ELISA. Data shown were calculated by dividing the area under the ELISA curve (AUC) of the germline-reverted antibody by the AUC of the native antibody (percentage of native binding).
(B and D) Binding of early sequences clonally related to (B) mAb180 or (D) mAb692 to HIV-1 gp140 (strain: BG505, left) or HCV E2 (strain: JFH1, right) measured by ELISA. Data are shown as a heatmap where the percentage of native binding is calculated as in (A) and (C). Each heatmap square represents a unique combination of heavy- and light-chain sequences. Antibody chain combinations we were unable to purify are marked “X”. The phylogenetic relationship between each set of sequences was determined using PhyML, not shown to scale.47
Each assay was performed in duplicate, and data shown are representative of at least 2 separate experiments.
Finally, to trace the early development of HIV-1/HCV cross-reactivity, we performed deep, unpaired BCR sequencing of peripheral blood B cells from donor VC10014 approximately 0.59 years post co-infection (~3 years before the sample used for LIBRA-seq). From this dataset, we identified multiple heavy-and light-chain sequences clonally related to both mAb180 and mAb692. We then sought to define the effect of these early acquired mutations on HIV-1/HCV antigen cross-reactivity by expressing pairwise combinations of heavy- and light-chain sequences as recombinant mAbs. Overall, we observed distinct binding patterns when various mutation-containing heavy- and light-chain sequences were combined (Figures 7B and 7D). Notably, HCV Env recognition by both mAb180 and mAb692 appeared to be less sensitive to different levels of SHM compared with HIV-1 Env recognition (Figures 7B and 7D).
Together, these data suggest that SHM can play an important role for establishing, as well as for further developing, HIV-1/HCV cross-reactive antibody binding.
DISCUSSION
Although antibodies are generally used for their incredible specificity, flexibility in the antigen-binding site can provide a distinct advantage in the fight against highly mutable pathogens such as HIV-1 and HCV.48,49 This advantage is exemplified by the discovery of broadly reactive or broadly neutralizing antibodies (bNAbs) and their documented utility as prophylactic and therapeutic molecules and vaccine design scaffolds.50–53 In this study, we expand the concept of broadly reactive antibodies by discovering HIV-1/HCV cross-functional mAbs. Using the LIBRA-seq technology, we identified five genetically unique, class-switched, paired heavy-light chain sequences reactive with at least one HIV-1 and one HCV Env glycoprotein and then confirmed this pattern of antigen cross-reactivity by expression as recombinant human mAbs. Remarkably, we observed that all five antibodies were capable of exhibiting anti-HIV-1 and anti-HCV functions. Of note, we found that when the native antibody isotype (either IgG3-mAb688 or IgA-mAb803/mAbKP1–8) was switched to IgG1, antibody binding was reduced or ablated (Figure S6), indicating that antigen recognition by these antibodies can be influenced by factors outside of direct antibody-epitope interactions. Further, we discovered that SHM aided in the development of HIV-1/HCV cross-reactivity, and mapping studies suggest that differential mutations can enhance recognition of each viral antigen.
Previous studies have noted roles for poly- or autoreactive antibodies in immune responses against highly diverse viruses, most notably observing common cross-reactivity between HIV-1 gp41-specific antibodies with host and microbiome antigens.54–56 However, such antibodies are often of the IgM isotype, non-functional, or difficult to elicit by vaccination due to immune tolerance mechanisms.57–59 Notably, three of the five antibodies described in this study did not show evidence of polyreactivity. While the other two, mAb180 and mAb692, showed reactivity with host antigens, they nevertheless exhibited anti-viral functions against both HIV-1 and HCV, suggesting that such antibodies could still contribute to counteracting both viruses during infection. In addition, all five antibodies showed exceptional HCV neutralization regardless of autoreactivity, inhibiting infection with 19/19 genotype 1 strains, and an overall ability to neutralize viral strains from HCV genotypes 1–3 for most antibodies. This finding is particularly striking as genotypes 1–3 account for >95% of all HCV infections in the United States.60,61 Beyond neutralization, these antibodies may still impede or reduce infection by Fc-mediated effector functions or by targeting glycan structures outside the receptor binding site to prevent viral interaction with host cell lectin receptors (DC-SIGN and L-SIGN both interact with HIV-1 and HCV).62–64
Recent reports have outlined a class of glycan-reactive antibodies capable of recognizing and in some cases exhibiting effector functions against HIV-1, coronavirus, and influenza antigens, similar to the exceptionally broad antibody mAb688 isolated in our studies.39,65,66 Although some antibodies in this category displayed promiscuous polyreactivity against arbitrary unrelated antigens, we did not observe autoreactive binding for mAb688.39 Glycan-reactive antibodies that avoid self-recognition may represent a general immune defense mechanism for effectively counteracting viral infections. In addition to this glycan-reactive “class” of cross-reactive antibodies, the discovery of multiple HIV-1/HCV cross-reactive antibodies targeting diverse epitope determinants shows that there are multiple mechanisms that may result in antibody cross-reactivity against unrelated antigens. In the face of highly mutable threats, antibodies that can tolerate sequence variability, without triggering auto- or polyreactivity, provide a selective advantage. Such antibodies have the potential to aid cross-reactive vaccine design or serve as therapeutics themselves for both HIV-1 and HCV, as well as emerging threats such as SARS-CoV-2 and other infectious diseases.
Limitations of the study
We note that the current study investigated one HIV-1/HCV co-infected donor, and whether unconventional cross-reactive antibody specificities identified here are common, biologically protective, or found in settings other than chronic co-infection remains to be examined. Additional structural and biophysical characterization of the epitopes and paratopes for these antibodies, including antibody-antigen or antibody-glycan complex structures, will be needed to further examine the similarities and differences in how these antibodies interact with HIV-1 vs. HCV Env glycoproteins.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to the Lead Contact, Ivelin Georgiev (Ivelin.Georgiev@Vanderbilt.edu).
Materials availability
Please direct resource and reagent requests to the Lead Contact specified above, Ivelin Georgiev.
Data and code availability
The raw sequencing data and sequences of antibodies described here were deposited to GenBank under the following accession numbers: PRJNA911324, OP973754- OP973763.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Donor information
Donor VC10014 was identified and enrolled in the Vanderbilt cohort (VC) and samples isolated after informed consent. VC10014 was recruited with CD4+ T cell counts of ≥250/μL without antiretroviral therapy and with no AIDS-defining illness during the period of observation. The samples used for this study were collected on 09/26/2003, ~6 months- and 03/21/2006, >3 years after onset of co-infection. The male donor was 53 and 56 years old at the times of sample collection, respectively.
METHOD DETAILS
Purification of antigens
Plasmids encoding the following genes were transfected in FreeStyle293F (Thermo Fisher Scientific) cells via polyethyleneimine transfection; HIV-1/BG505.664.SOSIP,71 B41.664.SOSIP,72 ConC gp120[61], A244 gp120[62], HCV/H77 E1E2[63], H77 E2c [17], JFH-1 E2c [64]. All antigens contained an AviTag sequence for subsequent biotinylation. Antigens were purified by Galanthus nivalis (GNA, Snowdrop) lectin affinity chromatography (Vector Labs), and further purified by gel filtration with Superdex200 Increase column (Cytiva). Fractions corresponding to correctly folded protein were collected and biotinylated using BirA (Avidity). Biotinylated HIV-1 antigens were fluorescently labeled by incubation with Streptavidin-AF568 (Invitrogen), and biotinylated HCV antigens fluorescently labeled by incubation with Streptavidin-AF647 (Invitrogen).
Coronavirus spike (S) protein ectodomains used for SPR assays were produced and purified as described before.73 The MERS and SARS-CoV-2 S ectodomains contained the engineered 2P stabilizing mutations, while the other CoV S constructs lacked these mutations. Briefly, Gibco FreeStyle 293-F cells (embryonal, human kidney) were incubated in FreeStyle 293 Expression Medium (Gibco) with agitation at 120 rpm, at 37°C and 9% CO2 in a humidified atmosphere. Plasmids encoding for the S protein ectodomains were transiently transfected into the cells using Turbo293 (SpeedBiosystems) and incubated at 37°C, 9% CO2, 120 rpm for 6 days. HyClone CDM4HEK293 media (Cytiva, MA) was added to the cells on the day following transfection. On day 6 post-transfection, S protein ectodomains were harvested from the concentrated supernatant and purified using StrepTactin resin (IBA Life Sciences) followed by size exclusion chromatography (SEC) using a Superose 6 10/300 GL Increase column (Cytiva, MA) equilibrated in 2 mM Tris, pH 8.0, 200 mM NaCl, 0.02% NaN3. All purification steps were performed within a single day and at room temperature. Protein quality was assessed by SDS-PAGE using NuPage 4–12% (Invitrogen, CA). The purified proteins were flash frozen and stored at −80°C in single-use aliquots. Each aliquot was thawed before use by incubating at 37°C for 20 min incubation.
SARS-CoV-2 WT RBD used for SPR assays was produced and purified as described above. Briefly, WT RBD construct obtained from BEI resources (Catalog number NR-52309) expressed the receptor binding domain (RBD) of the spike (S) glycoprotein gene from severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2), Wuhan-Hu-1 (GenBank: MN908947) with an N-terminal S protein signal sequence to the spike RBD (amino acids 319 to 541) and a C-terminal 6X-histidine tag. The transfection method was the same as the one followed for the CoV S ectodomains described above. The RBD was harvested from concentrated supernatant on the sixth day post transfection and purified via nickel affinity chromatography using a HisTrap excel column (Cytiva, MA). Concentrated supernatant was loaded onto the column and the column washed with buffer A (1x PBS pH 8.0) until baseline. The protein was eluted from the column by applying a gradient over 40 CV from 100% buffer A to 100% buffer B (1x PBS pH 8.0, 1 M Imidazole). Fractions containing RBD were pooled, concentrated, and further purified by size exclusion chromatography (SEC) using a Superdex 200 Increase 10/300 GL column (Cytiva, MA) equilibrated with 1x PBS, pH 8.0. All steps of the purification were performed at room temperature. Protein quality was assessed by SDS-PAGE using NuPage 4–12% (Invitrogen, CA). The purified proteins were flash frozen and stored at −80°C in single-use aliquots. Each aliquot was thawed at 4°C before use.
The production of SARS-CoV-2 NTD used for SPR assays was based on a previous protocol.73,74 The NTD up to residue 308 was followed with a C-terminal 8x His tag. Both constructs included the stabilizing L303A mutation, and one variant included the residue 69/70 deletion. Briefly, Gibco FreeStyle 293-F cells (embryonal, human kidney) were incubated in FreeStyle 293 Expression Medium (Gibco) with agitation at 120 rpm, at 37°C and 8% CO2 in a humidified atmosphere. Plasmids encoding for the NTD constructs were transiently transfected into the cells using Turbo293 (SpeedBiosystems) and incubated at 37°C, 8% CO2, 120 rpm for 6 days. HyClone CDM4HEK293 media (Cytiva, MA) was added to the cells on the day following transfection. On day 6 post-transfection, NTD was harvested from the concentrated supernatant and purified using cobalt metal affinity resin (TALON, Takara). The resin was washed with a 20mM HEPES, 150mM NaCl, pH8 buffer, and protein eluted with the same buffer with 50mM Imidazole added. This was followed by size exclusion chromatography (SEC) using a Superdex 75 10/300 GL Increase column (Cytiva, MA) equilibrated in the same HEPES-NaCl buffer. The purified proteins were flash frozen and stored at −80°C in single-use aliquots. Each aliquot was thawed before use by incubating at 37°C for 20 min incubation.
The following reagents were obtained through the NIH HIV Reagent Program, Division of AIDS, NIAID, NIH: Human Immunodeficiency Virus Type 1 MN gp41 Protein, Recombinant from Escherichia coli, ARP-12027, contributed by DAIDS/NIAID; produced by ImmunoDX, LLC; Human Immunodeficiency Virus 1 (HIV-1) gp120 Recombinant Protein (B.9021 D11gp120), ARP-12571, contributed by Drs. Barton F. Haynes and Hua-Xin Liao. The following antigens were acquired from Sino Biological: Hepatitis C virus Envelope Glycoprotein E1/HCV-E1 (subtype 1b, strain HC-J4) Protein (His Tag); Human coronavirus (HCoV-229E) Spike Protein (S1+S2 ECD, His Tag); Human coronavirus (HCoV-NL63) Spike Protein (S1+S2 ECD, His Tag); SARS-CoV-2 (2019-nCoV) Spike S1+S2 ECD-His Recombinant Protein; SARS-CoV Spike S1+S2 ECD-His Recombinant Protein; Human coronavirus HKU1 (isolate N5) (HCoV-HKU1) Spike Protein (S1+S2 ECD, His Tag); MERS-CoV Spike Protein (S1+S2 ECD, aa 1–1297, His Tag). The following reagent was obtained through BEI Resources, NIAID, NIH: H1 Hemagglutinin (HA) Protein with C-Terminal Histidine Tag from Influenza Virus, A/New Caledonia/20/1999 (H1N1), Recombinant from Baculovirus, NR-48873; F Protein with C-Terminal Histidine Tag from Respiratory Syncytial Virus, B1, Recombinant from Baculovirus, NR-31097.
DNA-barcoding of antigens
We used oligos that possess 15 bp antigen barcode, a sequence capable of annealing to the template switch oligo that is part of the 10X bead-delivered oligos and contain truncated TruSeq small RNA read 1 sequences in the following structure: 5′-CCTTGGCACCCGAGAATTCCANNNNNNNNNNNNNCCCATATAAGA*A*A-3′, where Ns represent the antigen barcode (Integrated DNA Technologies). For each antigen, a unique DNA barcode was directly conjugated to the antigen itself. In particular, 5′-amino-oligonucleotides were conjugated directly to each antigen using the SoluLINK Protein-Oligonucleotide Conjugation Kit (Vector Labs) according to manufacturer’s instructions. Briefly, the oligo and protein were desalted, and then the amino-oligo was modified with the 4FB crosslinker, and the biotinylated antigen protein was modified with S-HyNic. Then, the 4FB-oligo and the HyNic-antigen were mixed. This mixing causes a stable bond to form between the protein and the oligonucleotide. The concentration of the antigen-oligo conjugates was determined by a BCA assay (Pierce), and the HyNic molar substitution ratio of the antigen-oligo conjugates was analyzed using the NanoDrop according to the SoluLINK protocol guidelines. AKTA FPLC (Cytiva) was used to remove excess oligonucleotide from the protein-oligo conjugates, which were also verified using SDS-PAGE with a silver stain (Pierce). Antigen-oligo conjugates were also used in flow cytometry titration experiments.
Antigen-specific B cell sorting
Antigen-specific B cells were sorted from donor PBMCs by fluorescence-activated cell sorting. Briefly, frozen cells were quickly thawed at 37°C, and washed 3× with DPBS without Ca2+ or Mg2+ (Gibco) supplemented with 1% BSA (Sigma) (DPBS-BSA) before counting. Cells were resuspended in DPBS-BSA and stained with antibodies against cell markers including viability dye (Ghost Red 780) (Tonbo Biosciences), CD14-APC-Cy7 (BD Biosciences), IgM-APC-Cy7 (BD Biosciences), CD3-FITC (BD Biosciences), CD19-BV711 (BD Biosciences), and IgG-PE-Cy5 (BD Biosciences). Additionally, fluorescently-labeled antigen-oligo conjugates were added to the stain. After staining in the dark for 20 min at room temperature, cells were washed three times with DPBS-BSA. Live, CD14−, IgM−, CD3−, CD19+, Antigen+-cells were sorted using a FACSAria III flow sorter (BD Biosciences) and transferred to the Vanderbilt Technologies for Advanced Genomics (VANTAGE) sequencing core at an appropriate target concentration for 10X Genomics library preparation and subsequent sequence analysis.
Sample preparation, library preparation, and sequencing
Single-cell suspensions were loaded onto the Chromium Controller microfluidics device (10X Genomics) and processed using the B-cell Single Cell V(D)J solution according to manufacturer’s suggestions for a target capture of 10,000 B cells per 1/8 10X cassette, with minor modifications to intercept, amplify and purify the antigen barcode libraries as previously described.
Sequence processing and bioinformatic analysis
We used a modified version of our previously described pipeline to use paired-end FASTQ files of oligo libraries as input, process and annotate reads for cell barcode, UMI, and antigen barcode, and generate a cell barcode - antigen barcode UMI count matrix. BCR contigs were processed using Cell Ranger (10X Genomics) using GRCh38 as reference. Antigen barcode libraries were also processed using Cell Ranger (10X Genomics). The overlapping cell barcodes between the two libraries were used as the basis of the subsequent analysis. We removed cell barcodes that had only non-functional heavy chain sequences as well as cells with multiple functional heavy chain sequences and/or multiple functional light chain sequences, reasoning that these may be multiplets. Additionally, we aligned the BCR contigs (filtered_contigs.fasta file output by Cell Ranger, 10X Genomics) to IMGT reference genes using HighV-Quest38.67 The output of HighV-Quest was parsed using ChangeO and merged with an antigen barcode UMI count matrix.68 Finally, we determined the LIBRA-seq score for each antigen in the library for every cell as previously described.26
Antibody purification
For each antibody, variable genes were inserted into custom plasmids encoding the native (IgG1, IgG3, or IgA2) constant region for the heavy chain as well as respective lambda or kappa light chains (pTwist CMV BetaGlobin WPRE Neo vector, Twist Bioscience). Antibodies were expressed in Expi293F mammalian cells (Thermo Fisher Scientific) by co-transfecting heavy chain and light chain expressing plasmids using polyethylenimine transfection reagent. Antibodies were purified from filtered cell supernatants by affinity chromatography. IgG1 isotype antibodies were purified using immobilized Protein A, IgG3 isotype antibodies were purified using immobilized Protein G, and IgA isotype antibodies were purified using CaptureSelect IgA Affinity. All antibodies were stored in PBS, pH = 7.4 (Figure S7). The following reagents were obtained through the NIH HIV Reagent Program, Division of AIDS, NIAID, NIH: Anti-Human Immunodeficiency Virus (HIV)-1 gp41 Monoclonal Antibody (2F5), ARP-1475, contributed by DAIDS/NIAID; Monoclonal Anti-Human Immunodeficiency Virus (HIV)-1 gp120 Protein (VRC01), ARP-12033, contributed by Dr. John Mascola; Anti-Human Immunodeficiency Virus 1 (HIV-1) gp41 Monoclonal (5F3), ARP-6882, contributed by Polymun Scientific.
ELISA
To assess antibody binding, soluble protein was plated on Immulon 2HB plates (Thermo Fisher Scientific) at 2 μg/mL overnight at 4°C. In cases where capture ELISA was used, plates were pre-incubated for 2 h at room temperature (RT) with 5 μg/mL GNA lectin (Sigma) or 2 μg/mL anti-AviTag (Genscript) and washed 3× with PBS+ 0.05% Tween 20 (PBS-T) before antigen plating overnight. Between each of the subsequent incubation steps, plates were washed 3× with PBS-T. Non-specific binding was blocked by incubation with 5% fetal bovine serum (FBS) (Gibco) diluted in PBS-T for 1 h at RT. Primary monoclonal antibodies were diluted in 5% FBS-PBST starting at 20 μg/mL with a serial 1:5 dilution (unless otherwise specified) and then added to the plate for 1 h at RT. Secondary antibody, either goat anti-human IgG (Southern Biotech) or goat anti-human IgA (Invitrogen), was diluted 1:10,000 in 5% FBS diluted in PBS-T and added for 1 h at RT. Reaction was developed by 10 min incubation with One Step Ultra-TMB (Thermo Fisher Scientific) and stopped with 1N sulfuric acid. Plate absorbances were read at 450 nm (Biotek). Data are represented as mean ± SEM for one ELISA experiment performed in duplicate. ELISA experiments were repeated with at least 2 different antibody preparation aliquots. The area under the curve (AUC) was calculated using GraphPad Prism 8.0.0.
Competition-binding ELISA
Competition ELISA experiments were performed as above with minor modifications. After coating with antigen and blocking, non-biotinylated competitor antibody was added to each well at 10 μg/mL and incubated at RT for 1 h. After washing, biotinylated antibody (final concentration of 1 μg/mL) was added and incubated for 1 h at RT. After washing three times with PBS-T, streptavidin-HRP (Thermo Fisher Scientific) was added at 1:10,000 dilution in 5% FBS in PBS-T and incubated for 1 h at room temperature. Plates were washed and substrate and sulfuric acid were added as described above.
Mannose-competition ELISA
Mannose-competition ELISAs were performed as described above with minor modifications. After antigen coating and washing, nonspecific binding was blocked by incubation with 5% FBS diluted in PBS for 1 h at RT. Primary antibodies were diluted in 5% FBS-PBST +/− 1M D-(+)- Mannose (Sigma) starting at 10 μg/mL with a serial 1:5 dilution and then added to the plate for 1 h at RT. After washing, antibody binding was detected with goat anti-human IgG-HRP (Southern Biotech) and added at 1:10,000 dilution in 5% FBS in PBS-T to the plates. After 1 h incubation, plates were washed, and substrate and sulfuric acid were added as described above. Data shown is representative of experiments performed in duplicate with at least 2 different antibody preparations.
Surface plasmon resonance (SPR)
Binding experiments using SPR were performed at 25°C on a Biacore T-200 (Cytiva) instrument in two formats. The running buffer (HBS-EP+) was composed of HBS supplemented with 3 mM EDTA and 0.05% P-20 surfactant. In the first format, the S protein ectodomains were captured via their C-terminal Twin StrepTactin tags by flowing the spikes at a concentration of 200 nM (60 s at 10 μL/min) over the flow cells of a Series S Strepavidin (SA) chip. IgG samples were injected over the S protein surfaces using the single cycle kinetics mode with 5 concentrations per cycle at concentrations ranging from 0.625 to 800 nM. A contact time of 60s and a dissociation time of 120 s at a flow rate of 50 μL/min was used. The surface was regenerated after the last injection with 3 pulses of a 50mM NaOH + 1M NaCl solution for 10 s at 100 μL/min.
RBD and NTD binding to IgG’s was assessed using a Series S CM5 chip (Cytiva, MA) which was labeled with anti-human IgG (fc) antibody using a Human Antibody Capture Kit (Cytiva, MA). IgGs were then coated at 200 nM (120 s at 5 μL/min). RBD/NTD samples were injected at concentrations ranging from 12.5 nM to 200 nM (prepared in a 2-fold serial dilution manner) over the antibodies using the single cycle kinetics mode with 5 concentrations per cycle. The surface was regenerated after the last injection with 3 pulses of a 3 M MgCl2 solution for 10 s at 100 μL/min.
In the second format, the IgG was flowed over at a concentration of 200 nM at a flowrate of 5 μL/min for 120s, and captured via the Fc region on a CM5 chip coated with Human AntiFc IgG (Cytiva). The different coronavirus spike constructs were then injected at 200nM for 120 s at a flowrate of 50 μL/min with a dissociation time of 30s. The surface was regenerated by 3 consecutive pulse of 3M MgCl2 for 10 s at 100 μL/min.
Flow cell 1 was used as reference. Blank sensorgrams were obtained by injection of the same volume of HBSEP + buffer in place of IgG solutions. Sensorgrams were reference subtracted and corrected with corresponding blank curves. Sensogram data were analyzed using the BiaEvaluation software (Cytiva).
Negative stain grid preparation
For screening and imaging of negatively stained (NS) HIV-1 gp140 in complex with Fab 180/692, ~3 μL of the complex after SEC at concentrations of 10–15 μg/mL were applied to glow-discharged grid with continuous carbon film on 400 square mesh copper EM grids (Electron Microscopy Sciences). The grids were stained with 0.75% uranyl formate (UF).75
Screening, data collection, and image processing
NS grids were screened on an FEI Morgagni (Thermo Fisher Scientific) microscope operating at 100kV with AMT 1k×1k CCD camera to verify sample and grid quality. Data collection from NS grids were done on FEI TF20 (Thermo Fisher Scientific) operate at 200kV with US4000 4kx4k CCD camera (Gatan) and controlled by SerialEM.69 The dataset was collected at nominal mag of 50Kx with A/pix of 2.18 with defocus range of 1.4–1.8 and a total dose of ~30.0e/A2. Image processing was performed using the CryoSPARC software package.70 The dataset was imported, CTF estimated, and particles were picked. The particles were extracted with box size of 256 × 256 pixels and 2D classification was performed to generated clean homogeneous classes.
TZM-bl HIV-1 neutralization
Antibody neutralization was assessed using the TZM-bl assay as described.76 This standardized assay measures antibody-mediated inhibition of infection of JC53BL-13 cells (also known as TZM-bl cells) by molecularly cloned Env-pseudoviruses. Viruses that are highly sensitive to neutralization (Tier 1) and/or those representing circulating strains that are moderately sensitive (Tier 2) were included, plus additional viruses, including a subset of the antigens used for LIBRA-seq. Murine leukemia virus (MLV) was included as an HIV-specificity control and VRC01 was used as a positive control. Results are presented as the concentration of monoclonal antibody (in μg/mL) required to inhibit 50% of virus infection (IC50).
Antibody-dependent cellular phagocytosis (ADCP), and antibody-dependent cellular cytotoxicity (ADCC)
The THP-1 phagocytosis assay was performed as previously described using 1 μM neutravidin beads (Molecular Probes Inc) coated with antigen. Monoclonal IgG samples were titrated and tested at a final concentration of 100 μg/mL. Additionally monoclonal antibodies were tested starting at 100 μg/mL with 5-fold dilutions. Phagocytic scores were calculated as the geometric mean fluorescent intensity (MFI) of the beads that have been taken up multiplied by the percentage bead uptake. This, as well as all other flow cytometry work was completed on a FACSAria II (BD Biosciences). Pooled IgG from HIV-positive donors from the NIH AIDS Reagent program (HIVIG) was used in all assays to normalize for plate-to-plate variation while samples from 10 Clade-C-infected individuals was used as a positive control for all assays. Palivizumab (MedImmune) was used as negative control.
Infectious antibody-dependent cellular cytotoxicity (ADCC) assay
The HIV-1 reporter viruses used in the infectious ADCC assay were generated as replication-competent infectious molecular clones (IMC) encoding CE1086 envelope protein and a Renilla luciferase reporter gene as previously described.77 Briefly, HEK293T cells (NIH AIDS Reagent Program) were transfected with proviral IMC plasmid DNA to generate reporter virus stocks. The virus was then tittered for infectivity in CEM.NKRCCR5 cells (NIH AIDS Reagent Program) using p24 staining (Beckman-Coulter). CD4 down regulation was also measured by co-staining with an anti-CD4 flourescent antibody. Post preparation of virus stocks, the ADCC activity was measured as previously described.77 Briefly, the luciferase expressing CEM.NKRCCR5 cell line infected with HIV-1 CE1086 IMC listed above, was used as the target for ADCC. This target cell line was infected using tittered stocks which generated more than 50% infected cells after 72 h of infection. Following successful infection, the target cell line was then incubated with 5-fold serially diliuted mAbs starting at 100 μg/ml. Cryopreserved PBMCs were thawed and rested overnight, and used at an effector to target ratio of 30:1. Together, the effector cells, target cells and antibody dilutions were incubated in 96-well half area plates for 6 h at 37°C in 5% CO2. After incubation, the final readout was the luminescence intensity (in relative light units) generated by the presence of residual intact target cells that had not been lysed by the effector population in the presence of any ADCC-mediating mAb. ADCC activity was then calculated as the percentage of killing using the formula below:
RLU of target and effector well
In this analysis, the RLU of the target plus effector wells represents non-antibody background. Palivizumab (Medimmune; Synagis) was used as a negative controls and a polyclonal mixture of IgG from HIV infected individuals (HIVIG) from the NIH AIDS Reagent Program was used as a positive control and to normalize between plates. Data are represented as titrations and the area under the curve (AUC) of percentage specific killing over the serially diluted antibodies.
HCV pseudoparticle (HCVPP) neutralization
A panel of 19 genotype 1 HCVpps, one genotype 2b (UKNP2.4.1), and one genotype 3a (UKNP3.1.2) were produced by lipofectamine-mediated transfection of HCV E1E2 plasmid, pNL4–3.Luc.R-E-plasmid containing the env-defective HIV proviral genome (NIH AIDS Reagent Program), and pAdVantage (Promega) into HEK293T cells 32,33. Mock pseudoparticles, generated with pNL4–3.Luc.R-E− and pAdVantage and without E1E2 plasmid, were used as a negative control for each transfection. For HCVpp testing, 8,000 Hep3B cells per well were plated in 96-well solid white flat bottom polystyrene TC-treated microplates (Corning) and incubated overnight at 37°C. For infectivity testing, HCVpp were incubated on Hep3B target cells for 5 h. Following this incubation, medium was changed to 100 μL of phenol-free Hep3B media and incubated for 72 h at 37°C. Infectivity was quantified using a luciferase assay as described below. All HCVpp used in neutralization assays produced RLU values at least 10-fold above background entry by mock pseudoparticles. For antibody breadth testing, HCVpp were incubated for 1 h with mAb at 100 μg/mL and then added in duplicate to Hep3B target cells for 5 h. Following this incubation, medium was changed to 100 μL of phenol-free Hep3B medium and incubated for 72 h at 37°C. Infectivity was quantified using a luciferase assay as described below. All HCVpp used in neutralization assays produced RLU values at least 10-fold above background entry by mock pseudoparticles. For antibody potency testing, antibodies were serially diluted five-fold, starting at a concentration of 100 μg/mL and ending at 2.56 × 10–4 (leaving the last well as PBS only), and incubated with HCVpp for 1 h at 37°C before the addition to HEP3B target cells in duplicate. Following this incubation, medium was changed to 100 μL μL of phenol-free Hep3B medium and incubated for 72 h at 37°C. After incubation (for either breadth or potency testing), media was removed from the cells, 45 μL μL of 1× cell culture lysis reagent (Promega) was added to each well and left to incubate for 5 min. The luciferase assay was measured in relative light units (RLUs) in Berthold Luminometer (Berthold Technologies Centro LB960). The percentage of neutralization for the antibody breadth was calculated as [1-(RLUmAb/RLUIgG)] × 100]. The percentage of neutralization for the dilution curves and was calculated as [1-(RLUmAb/RLUPBS)] × 100. HEPC74 and human IgG were included as controls.
Influenza A hemagglutination inhibition (HAI)
The hemagglutination inhibition (HAI) assay was used to assess the ability of mAb688 to inhibit agglutination of erythrocytes. The HAI assay was performed similarly to previously described protocols4,78 adapted from the World Health Organization (WHO) laboratory influenza surveillance manual. In brief, mAb688 (expressed as IgG1 or IgG3) was diluted in a series of 2-fold serial dilutions in v-bottom microtiter plates (Greiner Bio-One) starting from 20 μg/mL. An equal volume of A/Brisbane/02/2018 (CA/09 pdm-like H1N1) or A/Hong Kong/4801/2014 (H3N2) virus, adjusted to ~8 hemagglutination units per 50 μL, was added to each well. The plates were covered and incubated at room temperature for 20 min, and then a 0.8% solution of turkey (for H1N1) or guinea pig (for H3N2) erythrocytes (Lampire Biologicals) in PBS was added. Erythrocytes were stored at 4°C and used within 72 h of preparation. The plates were mixed by agitation and covered, and the erythrocytes were settled for 30 min at room temperature. The HAI titer was determined by the reciprocal dilution of the last well that contained non-agglutinated erythrocytes. Positive and negative controls were included for each plate.
Focus reduction assay
Madin-Darby canine kidney (MDCK) cells stably-transfected with cDNA encoding human 2,6-sialtransferase (SIAT1) MDCK-SIAT1 (provided by Center for Disease Control and Prevention) were maintained in DMEM (Corning) supplemented with penicillin-streptomycin, BSA fraction V 7.5% solution (Thermo Fisher Scientific), 25 mM HEPES buffer, 10% heat-inactivated FBS and 1 mg/mL of geneticin (G418 sulfate; Thermo Fisher Scientific).
The focus reduction assay (FRA) was performed similarly to previously described protocols.78 In brief, MDCK-SIAT1 cells were seeded at a density of 2.5–3 3 × 105 cells/mL in a 96-well plate (Greiner Bio-One) the day before the assay was run. The following day, the cell monolayers were rinsed with 0.01 M PBS (pH 7.2) (Thermo Fisher Scientific), followed by the addition of 2-fold serially diluted mAb688 (expressed as IgG1 or IgG3) at 50 μL per well starting with 20 μg/mL dilution in virus growth medium, termedVGM-T (DMEM containing 0.1% BSA, penicillin-streptomycin, and 1 μg/mL L-(tosylamido-2-phenyl) ethyl chloromethyl ketone (TPCK)-treated trypsin [Sigma, St. Louis, MO, USA]). Afterward, 50 μL of virus (A/California/07/2009 [pdm H1N1], A/Texas/50/2012 [H3N2] or B/Massachusetts/02/2012 [influenza B virus]) standardized to 1.2 × 104 focus forming units (FFU) per milliliter, and corresponding to 600 FFU per 50 μL, was added to each well, including control wells. Following a 2h incubation period at 37°C with 5% CO2, the cells in each well were then overlaid with 100 μL of equal volumes of 1.2% Avicel RC/CL (Type RC581 NF; FMC Health and Nutrition, Philadelphia, PA) in 2× MEM (Thermo Fisher Scientific) containing 1 μg/mL TPCK-treated trypsin, 0.1% BSA, and antibiotics. Plates were incubated for 18–22 h at 37°C, 5% CO2. The overlays were then removed from each well, and the monolayer was washed once with PBS to remove any residual Avicel. The plates were fixed with ice-cold 4% formalin in PBS for 30 min at 4°C, followed by a PBS wash and permeabilization using 0.5% Triton X-100 in PBS/glycine at room temperature for 20 min. Plates were washed three times with PBS supplemented with 0.1% Tween 20 (PBST) and incubated for 1 h with a mAb against influenza A nucleoprotein (provided by the International Reagent Resource (IRR), Influenza Division, WHO Collaborating Center for Surveillance, Epidemiology and Control of Influenza, Centers for Disease Control and Prevention) in ELISA buffer (PBS containing 10% horse serum and 0.1% Tween 80 [Thermo Fisher Scientific]). Following washing three time with PBST, the cells were incubated with goat anti-mouse peroxidase-labeled IgG (SeraCare) in ELISA buffer for 1h at room temperature. Plates were washed three times with PBST, and infectious foci (spots) were visualized using TrueBlue substrate (SeraCare) containing 0.03% H2O2 incubated at room temperature for 10 to 15 min. The reaction was stopped by washing five times with distilled water. Plates were dried and foci were enumerated using an ImmunoSpot S6 ULTIMATE reader with ImmunoSpot 7.0.28.5 software (Cellular Technology Limited). The FRA titer was reported as the reciprocal of the highest dilution of serum corresponding to 50% foci reduction compared with the foci values of the virus-control minus the cell-control.
SARS-CoV-2 pseudoparticle neutralization
To assess neutralizing activity against SARS-CoV-2 strain 2019 n-CoV/USA_WA1/2020 (obtained from the Centers for Disease Control and Prevention, a gift from N. Thornburg), we used the high-throughput RTCA assay and xCelligence RTCA HT Analyzer (ACEA Biosciences) as described previously. After obtaining a background reading of a 384-well E-plate, 6,000 Vero-furin cells were seeded per well. Sensograms were visualized using RTCA HT software version 1.0.1 (ACEA Biosciences). One day later, equal volumes of virus were added to antibody samples and incubated for 1 ch at 37°C in 5% CO2. MAbs were tested in triplicate with a single (1:20) dilution. Virus–mAb mixtures were then added to Vero-furin cells in 384-well E-plates. Controls were included that had Vero-furin cells with virus-only (no mAb) or medium-only (no virus or mAb). E-plates were read every 8–12 h for 72 h to monitor virus neutralization. At 32 h after virus-mAb mixtures were added to the E-plates, cell index values of antibody samples were compared to those of virus only and media only to determine presence of neutralization.
Uropathogenic E. coli hemagglutination and adherence inhibition
Hemagglutination assays were performed as described previously.79 Bacterial cultures were grown statically at 37°C for 24 h in Lysogeny broth (LB), subcultured into fresh LB, and grown another 24 h. Cultures were normalized to optical density (600 nm) of 1.0 in PBS, concentrated 10×, and resuspended in PBS or PBS containing 4% mannose (to competitively inhibit the type 1 pili), 20 μg/mL mAb688, or 20 μg/mL isotype control. Bacteria were added to a 96 well plate and diluted in two-fold increments. Next, guinea pig erythrocytes (Innovative Research, Inc.) were washed and suspended in PBS or PBS containing 4% mannose, 20 μg/mL mAb688, or 20 μg/mL isotype control. Erythrocytes were added to the diluted bacterial culture and incubated statically overnight at 4°C. Hemagglutination titer was determined by measuring the lowest dilution that visibly inhibited hemagglutination. Data are representative of three biological replicates performed in technical duplicate.
Autoreactivity
Monoclonal antibody reactivity to nine autoantigens (SSA/Ro, SS-B/La, Sm, ribonucleoprotein (RNP), Scl 70, Jo-1, dsDNA, centro-mere B, and histone) was measured using the AtheNA Multi-Lyte ANA-II Plus test kit (Zeus scientific, Inc.). Antibodies were incubated with AtheNA beads for 30 min at concentrations of 50, 25, 12.5 or 6.25 μg/mL. Beads were washed, incubated with secondary and read on the Luminex platform as specified in the kit protocol. Data were analyzed using AtheNA software. Positive (+) specimens received a score >120, and negative (−) specimens received a score <100. Samples with values between 100 and 120 were considered indeterminate.
HEp-2 cell binding
We measured antibody binding to whole (un-permeabilized) un-infected HEp-2 cells by flow cytometry. Briefly, we collected HEp-2 cells and washed 3× with DPBS-BSA before counting. ~1 million cells/condition were stained with a final concentration of 100 μg/mL, 10 μg/mL, or 1 μg/mL antibody diluted in DPBS-BSA for 20 min at 4C. Cells were then washed 3× with DPBS-BSA and stained with either goat anti-human IgG labeled with PE (Southern Biotech) or goat anti-human IgA (Southern Biotech) labeled with PE diluted 1:1,000 in DPBS-BSA for 20 min at 4C. Cells were washed a final time and fluorescence acquired on a 4-Laser Fortessa (BD Biosciences). FCS files were analyzed, and figures generated using CytoBank. Data shown is representative of at least 2 separate experiments with different antibody preparations. The following reagent was obtained through the NIH HIV Reagent Program, Division of AIDS, NIAID, NIH: Anti-Human Immunodeficiency Virus (HIV)-1 gp41 Monoclonal Antibody (4E10), ARP-10091, contributed by DAIDS/NIAID.
QUANTIFICATION AND STATISTICAL ANALYSIS
ELISA (standard error of the mean; SEM), ADCP/ADCC, neutralization, and E. coli adherence (standard deviation; SD) error bars shown were calculated using GraphPad Prism version 9.2.0.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| APC-Cy7 Mouse Anti-Human CD14 | BD | Cat#561709; RRID: AB_10893806 |
| FITC Anti-Human CD3 (OKT3) | Tonbo Biosciences | Cat#35-0037; RRD: RRID: AB_2621662 |
| BV711 Mouse Anti-Human CD19 | BD | Cat#563036; RRID: AB_2737968 |
| PE-Cy5 Mouse Anti-Human IgG | BD | Cat#551497; RRD: RRID: AB_394220 |
| Anti-HIV-1 gp120 Monoclonal VRC01 | NIH AIDS Reagent Program | Cat#12033; RRID: AB_2491019 |
| Anti-HCV E2 Monoclonal HCV1 | Broering et al., 2009. | N/A |
| mAb180 | This study | OP973754, OP973755 |
| mAb692 | This study | OP973756, OP973757 |
| mAb688 | This study | OP973758, OP973759 |
| mAb803 | This study | OP973760, OP973761 |
| mAbKP1-8 | This study | OP973762, OP973763 |
| Anti-HCV E2 Monoclonal AR3C | Giang etal., 2012.36 | N/A |
| Anti-HIV-1 gp41 Monoclonal 2F5 | NIH AIDS Reagent Program | Cat#1475 |
| Anti-HIV-1 gp41 Monoclonal 5F3 | NIH AIDS Reagent Program | Cat#6882 |
| Anti-HIV-1 gp41 Monoclonal 167-D | NIH AIDS Reagent Program | Cat#11681 |
| Anti-HCV E2 Monoclonal HEPC74 | Bailey etal., 2017.35 | N/A |
| Anti-RSV F Monoclonal Palivizumab | AstraZeneca | N/A |
| Anti-HIV-1 Immune Globulin (HIVIG) | NIH AIDS Reagent Program | Cat#3957 |
| Anti-Influenza A Monoclonal FE53 | Daniel Lingwood | N/A |
| CR3022 | J. ter Meulen et al., 2006. | N/A |
| Anti- MERS Monoclonal 1F8 | Tang et al., 2014. | N/A |
| DH1050.1 | Priyamvada Acharya | N/A |
| DH1047 | Priyamvada Acharya | N/A |
| DH1052 | Priyamvada Acharya | N/A |
| Anti-HIV-1 gp41 Monoclonal 4E10 | NIH AIDS Reagent Program | Cat#10091 |
| Goat Anti-Human IgG Antibody (Peroxidase) | Jackson ImmunoResearch | Cat#109-035-088; RRID: AB_2337584 |
| Bacterial and virus strains | ||
| MLV | NIH HIV Reagent Program | Cat#1065 |
| HIV-1 MW965.26 | HIV Specimen Cryorepository (Germany) | 201307035544P |
| HIV-1 ZM197 | NIH HIV Reagent Program | Cat#11309 |
| HIV-1 BG505.W6M.C2.T332N | National Institute For Communicable Diseases | N/A |
| HIV-1 25,710.2.43 | NIH HIV Reagent Program | Cat#11505 |
| HIV-1 X2278.C2.B6 | Duke University | N/A |
| HIV-1 CE0217 | Duke University | N/A |
| HIV-1 CNE55 | Duke University | N/A |
| HIV-1 CH119.10 | Duke University | N/A |
| HIV-1 TRO.11 | NIH HIV Reagent Program | Cat#11023 |
| HCV genotype 1 panel | Bailey et al., 2015; Osburn et al., 2014 | GenBank: KJ187972-KJ187978, KJ187980- KJ187985, KJ187987- KJ187990, KM660628-9 |
| HCV UKNP2.4.1 | Urbanowicz et al., 2016. | GenBank: KU285213 |
| HCV UKNP3.1.2 | Urbanowicz et al., 2016. | GenBank: KU285215 |
| SARS-CoV-2 (VSV2019 n-CoV/USA_WA1/2020) | Case et al., 2020. | N/A |
| Uropathogenic Escherichia coli UTI89 | Maria Hadjifrangiskou | NCBI Reference Sequence: NC_000913.3 |
| Influenza An A/California/07/2009 | ATCC | VR-1894 |
| Influenza B B/Massachusetts/02/2012 | Ted Ross/Giuseppe Sautto | N/A |
| Biological samples | ||
| VC10014 (PBMCs) | Sather et al., 2009.27/Spyros Kalams | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| HIV-1 BG505.SOSIP.664 gp140 trimer | Ivelin Georgiev | N/A |
| HIV-1 B41.SOSIP.664gp140 trimer | Ivelin Georgiev | N/A |
| HIV-1 MN gp41 | NIH HIV Reagent Program | Cat#: 12027 |
| HIV-1 ConC gp120 | Lynn Morris | N/A |
| HIV-1 A244 gp120 | NIH HIV Reagent Program | Cat#: 12569 |
| HCV H77. E2core | Kong et al., 2013. | N/A |
| HCV JFH1. E2core | Boisvert et al., 2016. | N/A |
| HCVH77. E1E2 | Ruwona et al., 2014. | N/A |
| HCV HC-J4 E1 | Sino Biological | Cat: 40,374-V07H |
| RSV F DS-Cav1 | McLellan et al., 2013. | N/A |
| SARS-CoV Spike | Sino Biological/Priyamvada Acharya | Cat: 40,634-V08B |
| Influenza An HA A/New Caledonia/20/99 (H1N1) | BEI Resources | NR-48873 |
| MERS-CoV Spike | Sino Biological/Priyamvada Acharya | Cat: 40,069-V08B |
| NL63-CoV Spike | Sino Biological | Cat: 40,604-V08B |
| SARS-CoV-2 Spike (HexaPro) | Hsieh et al., 2020./Priyamvada Acharya | N/A |
| 229E-CoV Spike | Sino Biological | Cat: 40,605-V08B |
| HKU1-CoV Spike | Sino Biological | Cat: 40,606-V08B |
| Factor H-binding Protein (MenX) | Scarselli et al., 2011. | N/A |
| hCD81-Large Extracellular Loop (LEL) | Sino Biological | Cat: 14,244-H04H |
| SARS-CoV-2 Spike (Omicron BA.1) | Priyamvada Acharya | N/A |
| SARS-CoV-2 Spike (Omicron BA.2) | Priyamvada Acharya | N/A |
| SARS-CoV-2 Spike (Omicron BA.5) | Priyamvada Acharya | N/A |
| SARS-CoV-2 Spike (N165A, N234A, N709A) | Priyamvada Acharya/Hsieh et al., 2020. | N/A |
| Streptavidin, Alexa Fluor™ 568 conjugate | Thermo Fisher Scientific | Cat# S11226 |
| Streptavidin, Alexa Fluor™ 647 conjugate | Thermo Fisher Scientific | Cat# S21374 |
| Ghost Dye Red 780 | Tonbo Biosciences | Cat#13-0865 |
| D-(+)-Mannose | Sigma Aldrich | M6020-100G |
| EZ link Sulfo-NHS-LC-Biotin | Thermo Fisher Scientific | Cat#21327 |
| Protein A Resin | Genscript | Cat#L00210 |
| Protein G Resin | Thermo Fisher Scientific | Cat#53128 |
| CaptureSelect IgA Affinity | Thermo Fisher Scientific | Cat# 194288005 |
| Galanthus Nivalis Resin | Vector Labs | Cat#AL-1243-5 |
| Critical commercial assays | ||
| Antibody-Oligonucleotide All-in-One™ Conjugation Kit | Vector Labs | Cat# A-9202-001 |
| B cell Single Cell V(D)J solution | 10X Genomics | N/A |
| Biacore Sensorchip Human Antibody Capture | Cytiva | Cat# BR100839 |
| Biacore CM5 Sensorchip | Cytiva | Cat#29149604 |
| Bright-Glo Luciferase Assay System | Promega | Cat# E2650 |
| FluoSpheres NeutrAvidin-Labeled Microspheres, 1.0 mm, yellow-green fluorescent | Thermo Fisher Scientific | Cat#F8776 |
| BirA-500: Bir A Biotin-Protein Ligase standard reaction kit | Avidity | Cat# BirA500 |
| ExpiFectamine™ 293 Transfection Kit | Thermo Fisher Scientific | Cat# A14525 |
| Deposited data | ||
| Raw next-generation sequencing data | This paper | PRJNA911324 |
| mAb180 Heavy/Light sequences | This paper | OP973754, OP973755 |
| mAb692 Heavy/Light sequences | This paper | OP973756, OP973757 |
| mAb688 Heavy/Light sequences | This paper | OP973758, OP973759 |
| mAb803 Heavy/Light sequences | This paper | OP973760, OP973761 |
| mAbKP1-8 Heavy/Light sequences | This paper | OP973762, OP973763 |
| Experimental models: Cell lines | ||
| Freestyle 293F cells | Thermo Fisher Scientific | Cat#A14528 |
| Expi293F Cells | Thermo Fisher Scientific | Cat#A14527 |
| CEM.NKR CCR5+ Cells | AIDS Reagent Program | Cat#4376 |
| THP-1 Cells | AIDS Reagent Program | Cat#9942 |
| HEP3B Cells | ATCC | HB-8064 |
| Madin-Darby canine kidney (MDCK) Cells | ATCC | CCL-34 |
| HEp-2 Cells | ATCC | CCL-23 |
| TZM-bl Cells | AIDS Reagent Program | Cat#8129 |
| Oligonucleotides | ||
| Oligonucleotides for Protein DNA-barcoding | Setliff et al., 2019.26 | N/A |
| Software and algorithms | ||
| Cell Ranger | 10X Genomics | N/A |
| FlowJo v10 | BD | N/A |
| GraphPad Prism 9.0.0 | GraphPad | N/A |
| HighV-Quest | Alamyar et al., 201267 | N/A |
| ChangeO | Gupta et al., 201568 | N/A |
| PhyML | Guindon et al., 201047 | N/A |
| Cytobank | Kotecha et al., 2010 | N/A |
| Biacore X100 Evaluation Software | Cytiva | N/A |
| SerialEM | Mastronarde, 2003.69 | N/A |
| cryoSPARC | Punjani et al., 201770 | N/A |
Highlights.
LIBRA-seq is applied to characterize antibody responses to HIV-1/HCV co-infection
A diverse set of functional HIV-1/HCV cross-reactive antibodies are identified
A glycan-directed antibody exhibits exceptionally broad anti-viral recognition
Antibody somatic hypermutation is a major determinant for HIV-1/HCV cross-reactivity
ACKNOWLEDGMENTS
We thank all members of the Georgiev lab for their invaluable input and discussion of the results and manuscript. We thank Michelle Armstrong for administrative support and Yukthi Suresh for help in uploading sequence data. We thank Jamie Heimburg-Molinaro and Kelly Baker from the Protein-Glycan Interaction Resource of the National Center for Functional Glycomics (NCFG) at Beth Israel Deaconess Medical Center, Harvard Medical School (R24 GM137763), for their help with glycan microarray experiments. We thank Louise Barnett, David Flaherty, and Brittany Matlock from the VUMC Flow Cytometry Shared Resource for their help with flow panel design and cell sorting. The VUMC Flow Cytometry Shared Resource is supported by the Vanderbilt-Ingram Cancer Center (P30 CA68485) and the Vanderbilt Digestive Disease Research Center (DK058404). We also thank the Vanderbilt Institute for Clinical and Translational Research (VICTR, VR54249 to K.A.P.), funded by the National Center for Advancing Translational Sciences (NCATS) Clinical Translational Science Award (CTSA) Program (5ULITR002243-03). We thank Jamie Roberson and Latha Raju with the Vanderbilt Technologies for Advanced Genomics Core (VANTAGE) for providing technical assistance with library production and sequencing. VANTAGE is supported in part by CTSA (5UL1 RR024975-03), the Vanderbilt-Ingram Cancer Center (P30 CA68485), the Vanderbilt Vision Center (P30 EY08126), and NIH/NCRR (G20 RR030956). This work was supported in part by R01AI152693 and R01AI131722 to I.S.G., R01AI157155 to J.E.C., R01AI165947 to P.A., P20DK123967 to M.H., T32GM007569 to J.R.B., and T32GM007347 and F30AI150077 to C.J.B. The funders had no role in the conceptualization or execution of any studies or drafting of the manuscript.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112044.
DECLARATION OF INTERESTS
K.A.P. and I.S.G. are listed as inventors on patents filed describing the antibodies discovered here. A.R.S. and I.S.G. are co-founders of AbSeek Bio. J.E.C. has served as a consultant for Luna Biologics, is a member of the Scientific Advisory Board of Meissa Vaccines, and is founder of IDBiologics. The Crowe laboratory at Vanderbilt University Medical Center has received sponsored research agreements from Takeda Vaccines, IDBiologics, and AstraZeneca. The Georgiev laboratory at Vanderbilt University Medical Center has received unrelated funding from Takeda Pharmaceuticals.
REFERENCES
- 1.Chen L, Kwon YD, Zhou T, Wu X, O’Dell S, Cavacini L, Hessell AJ, Pancera M, Tang M, Xu L, et al. (2009). Structural basis of immune evasion at the site of CD4 attachment on HIV-1 gp120. Science 326, 1123–1127. 10.1126/science.1175868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gandhi RT, and Walker BD (2002). Immunologic control of HIV-1. Annu. Rev. Med 53, 149–172. 10.1146/annurev.med.53.082901.104011. [DOI] [PubMed] [Google Scholar]
- 3.Kirchhoff F (2010). Immune evasion and counteraction of restriction factors by HIV-1 and other primate lentiviruses. Cell Host Microbe 8, 55–67. 10.1016/j.chom.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 4.Forgacs D, Abreu RB, Sautto GA, Kirchenbaum GA, Drabek E, Williamson KS, Kim D, Emerling DE, and Ross TM (2021). Convergent antibody evolution and clonotype expansion following influenza virus vaccination. PLoS One 16, e0247253. 10.1371/journal.pone.0247253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tester I, Smyk-Pearson S, Wang P, Wertheimer A, Yao E, Lewin-sohn DM, Tavis JE, and Rosen HR (2005). Immune evasion versus recovery after acute hepatitis C virus infection from a shared source. J. Exp. Med 201, 1725–1731. 10.1084/jem.20042284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Platt L, Easterbrook P, Gower E, McDonald B, Sabin K, McGowan C, Yanny I, Razavi H, and Vickerman P (2016). Prevalence and burden of HCV co-infection in people living with HIV: a global systematic review and meta-analysis. Lancet Infect. Dis 16, 797–808. 10.1016/S1473-3099(15)00485-5. [DOI] [PubMed] [Google Scholar]
- 7.Operskalski EA, and Kovacs A (2011). HIV/HCV co-infection: pathogenesis, clinical complications, treatment, and new therapeutic technologies. Curr. HIV AIDS Rep 8, 12–22. 10.1007/s11904-010-0071-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hernandez MD, and Sherman KE (2011). HIV/hepatitis C coinfection natural history and disease progression. Curr. Opin. HIV AIDS 6, 478–482. 10.1097/COH.0b013e32834bd365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pineda JA, García-García JA, Aguilar-Guisado M, Ríos-Villegas MJ, Ruiz-Morales J, Rivero A, del Valle J, Luque R, Rodríguez-Baño J, González-Serrano M, et al. (2007). Clinical progression of hepatitis C virus-related chronic liver disease in human immunodeficiency virus-infected patients undergoing highly active antiretroviral therapy. Hepatology 46, 622–630. 10.1002/hep.21757. [DOI] [PubMed] [Google Scholar]
- 10.Thomas DL (2013). Global control of hepatitis C: where challenge meets opportunity. Nat. Med 19, 850–858. 10.1038/nm.3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martinello M, Hajarizadeh B, Grebely J, Dore GJ, and Matthews GV (2017). HCV cure and reinfection among people with HIV/HCV coinfection and people who inject drugs. Curr. HIV AIDS Rep 14, 110–121. 10.1007/s11904-017-0358-8. [DOI] [PubMed] [Google Scholar]
- 12.Ingiliz P, Martin TC, Rodger A, Stellbrink HJ, Mauss S, Boesecke C, Mandorfer M, Bottero J, Baumgarten A, Bhagani S, et al. (2017). HCV reinfection incidence and spontaneous clearance rates in HIV-positive men who have sex with men in Western Europe. J. Hepatol 66, 282–287. 10.1016/j.jhep.2016.09.004. [DOI] [PubMed] [Google Scholar]
- 13.Chohan B, Lavreys L, Rainwater SMJ, and Overbaugh J (2005). Evidence for frequent reinfection with human immunodeficiency virus type 1 of a different subtype. J. Virol 79, 10701–10708. 10.1128/JVI.79.16.10701-10708.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lambers FAE, Prins M, Thomas X, Molenkamp R, Kwa D, Brink-man K, van der Meer JTM, and Schinkel J; MOSAIC MSM Observational Study of Acute Infection with hepatitis C study group (2011). Alarming incidence of hepatitis C virus re-infection after treatment of sexually acquired acute hepatitis C virus infection in HIV-infected MSM. AIDS 25, F21–F27. 10.1097/QAD.0b013e32834bac44. [DOI] [PubMed] [Google Scholar]
- 15.Vivithanaporn P, Nelles K, DeBlock L, Newman SC, Gill MJ, and Power C (2012). Hepatitis C virus co-infection increases neurocognitive impairment severity and risk of death in treated HIV/AIDS. J. Neurol. Sci 312, 45–51. 10.1016/j.jns.2011.08.025. [DOI] [PubMed] [Google Scholar]
- 16.Lin W, Weinberg EM, and Chung RT (2013). Pathogenesis of accelerated fibrosis in HIV/HCV co-infection. J. Infect. Dis 207, S13–S18. 10.1093/infdis/jis926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feuth T, Arends JE, Fransen JH, Nanlohy NM, van Erpecum KJ, Siersema PD, Hoepelman AIM, and van Baarle D (2013). Complementary role of HCV and HIV in T-cell activation and exhaustion in HIV/HCV coinfection. PLoS One 8, e59302. 10.1371/journal.pone.0059302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Balazs AB, Chen J, Hong CM, Rao DS, Yang L, and Baltimore D (2011). Antibody-based protection against HIV infection by vectored immunoprophylaxis. Nature 481, 81–84. 10.1038/nature10660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bricault CA, Yusim K, Seaman MS, Yoon H, Theiler J, Giorgi EE, Wagh K, Theiler M, Hraber P, Macke JP, et al. (2019). HIV-1 neutralizing antibody signatures and application to epitope-targeted vaccine design. Cell Host Microbe 25, 59–72.e8. 10.1016/j.chom.2018.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pierce BG, Boucher EN, Piepenbrink KH, Ejemel M, Rapp CA, Thomas WD, Sundberg EJ, Weng Z, and Wang Y (2017). Structure-based design of hepatitis C virus vaccines that elicit neutralizing antibody responses to a conserved epitope. J. Virol 91, 010322–17. 10.1128/JVI.01032-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Keck ZY, Pierce BG, Lau P, Lu J, Wang Y, Underwood A, Bull RA, Prentoe J, Velázquez-Moctezuma R, Walker MR, et al. (2019). Broadly neutralizing antibodies from an individual that naturally cleared multiple hepatitis C virus infections uncover molecular determinants for E2 targeting and vaccine design. PLoS Pathog 15, e1007772. 10.1371/journal.ppat.1007772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sautto G, Tarr AW, Mancini N, and Clementi M (2013). Structural and antigenic definition of hepatitis C virus E2 glycoprotein epitopes targeted by monoclonal antibodies. Clin. Dev. Immunol 2013, 450963. 10.1155/2013/450963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lara J, Teka MA, Sims S, Xia GL, Ramachandran S, and Khudyakov Y (2018). HCV adaptation to HIV coinfection. Infect. Genet. Evol 65, 216–225. 10.1016/j.meegid.2018.07.039. [DOI] [PubMed] [Google Scholar]
- 24.Reiche S, Nestler C, Sieg M, Schulz K, Cordes C, Krznaric I, and Jassoy C (2014). Hepatitis C virus (HCV)-specific memory B-cell responses in transiently and chronically infected HIV positive individuals. J. Clin. Virol 59, 218–222. 10.1016/j.jcv.2014.01.023. [DOI] [PubMed] [Google Scholar]
- 25.Danta M, Semmo N, Fabris P, Brown D, Pybus OG, Sabin CA, Bhagani S, Emery VC, Dusheiko GM, and Klenerman P (2008). Impact of HIV on host-virus interactions during early hepatitis C virus infection. J. Infect. Dis 197, 1558–1566. 10.1086/587843. [DOI] [PubMed] [Google Scholar]
- 26.Setliff I, Shiakolas AR, Pilewski KA, Murji AA, Mapengo RE, Janowska K, Richardson S, Oosthuysen C, Raju N, Ronsard L, et al. (2019). High-throughput mapping of B cell receptor sequences to antigen specificity. Cell 179, 1636–1646.e15. 10.1016/j.cell.2019.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sather DN, Armann J, Ching LK, Mavrantoni A, Sellhorn G, Caldwell Z, Yu X, Wood B, Self S, Kalams S, and Stamatatos L (2009). Factors associated with the development of cross-reactive neutralizing antibodies during human immunodeficiency virus type 1 infection. J. Virol 83, 757–769. 10.1128/JVI.02036-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sather DN, Carbonetti S, Malherbe DC, Pissani F, Stuart AB, Hessell AJ, Gray MD, Mikell I, Kalams SA, Haigwood NL, and Stamatatos L (2014). Emergence of broadly neutralizing antibodies and viral coevolution in two subjects during the early stages of infection with human immunodeficiency virus type 1. J. Virol 88, 12968–12981. 10.1128/JVI.01816-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chukwuma VU, Kose N, Sather DN, Sapparapu G, Falk R, King H, Singh V, Lampley R, Malherbe DC, Ditto NT, et al. (2018). Increased breadth of HIV-1 neutralization achieved by diverse antibody clones each with limited neutralization breadth. PLoS One 13, e0209437. 10.1371/journal.pone.0209437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Corti D, Langedijk JPM, Hinz A, Seaman MS, Vanzetta F, Fernandez-Rodriguez BM, Silacci C, Pinna D, Jarrossay D, Balla-Jhagjhoorsingh S, et al. (2010). Analysis of memory B cell responses and isolation of novel monoclonal antibodies with neutralizing breadth from HIV-1-infected individuals. PLoS One 5, e8805. 10.1371/journal.pone.0008805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Buchacher A, Predl R, Strutzenberger K, Steinfellner W, Trkola A, Purtscher M, Gruber G, Tauer C, Steindl F, Jungbauer A, et al. (1994). Generation of human monoclonal antibodies against HIV-1 proteins; electrofusion and Epstein-Barr virus transformation for peripheral blood lymphocyte immortalization. AIDS Res. Hum. Retrovir 10, 359–369. [DOI] [PubMed] [Google Scholar]
- 32.Salas JH, Urbanowicz RA, Guest JD, Frumento N, Figueroa A, Clark KE, Keck Z, Cowton VM, Cole SJ, Patel AH, et al. (2022). An antigenically diverse, representative panel of envelope glycoproteins for hepatitis C virus vaccine development. Gastroenterology 162, 562–574. 10.1053/j.gastro.2021.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Urbanowicz RA, McClure CP, Brown RJP, Tsoleridis T, Persson MAA, Krey T, Irving WL, Ball JK, and Tarr AW (2015). A diverse panel of hepatitis C virus glycoproteins for use in vaccine research Reveals extremes of monoclonal antibody neutralization resistance. J. Virol 90, 3288–3301. 10.1128/jvi.02700-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bailey JR, Urbanowicz RA, Ball JK, Law M, and Foung SKH (2019). Standardized method for the study of antibody neutralization of HCV pseudoparticles (HCVpp). Methods Mol. Biol 1911, 441–450. 10.1007/978-1-4939-8976-8_30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bailey JR, Flyak AI, Cohen VJ, Li H, Wasilewski LN, Snider AE, Wang S, Learn GH, Kose N, Loerinc L, et al. (2017). Broadly neutralizing antibodies with few somatic mutations and hepatitis C virus clearance. JCI insight 2, e92872. 10.1172/jci.insight.92872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Giang E, Dorner M, Prentoe JC, Dreux M, Evans MJ, Bukh J, Rice CM, Ploss A, Burton DR, and Law M (2012). Human broadly neutralizing antibodies to the envelope glycoprotein complex of hepatitis C virus. Proc. Natl. Acad. Sci. USA 109, 6205–6210. 10.1073/pnas.1114927109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Davis SK, Selva KJ, Kent SJ, and Chung AW (2020). Serum IgA Fc effector functions in infectious disease and cancer. Immunol. Cell Biol 98, 276–286. 10.1111/imcb.12306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Astronomo RD, Santra S, Ballweber-Fleming L, Westerberg KG, Mach L, Hensley-McBain T, Sutherland L, Mildenberg B, Morton G, Yates NL, et al. (2016). Neutralization takes precedence over IgG or IgA isotype-related functions in mucosal HIV-1 antibody-mediated protection. EBioMedicine 14, 97–111. 10.1016/j.ebiom.2016.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Williams WB, Meyerhoff RR, Edwards RJ, Li H, Manne K, Nicely NI, Henderson R, Zhou Y, Janowska K, Mansouri K, et al. (2021). Fab-dimerized glycan-reactive antibodies are a structural category of natural antibodies. Cell 184, 2955–2972.e25. 10.1016/j.cell.2021.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li D, Edwards RJ, Manne K, Martinez DR, Schäfer A, Alam SM, Wiehe K, Lu X, Parks R, Sutherland LL, et al. (2021). In vitro and in vivo functions of SARS-CoV-2 infection-enhancing and neutralizing antibodies. Cell 184, 4203–4219.e32. 10.1016/j.cell.2021.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gobeil SM-C, Janowska K, McDowell S, Mansouri K, Parks R, Stalls V, Kopp MF, Manne K, Li D, Wiehe K, et al. (2021). Effect of natural mutations of SARS-CoV-2 on spike structure, conformation, and antigenicity. Science 373, eabi6226. 10.1126/science.abi6226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pizarro-Cerdá J, and Cossart P (2006). Bacterial adhesion and entry into host cells. Cell 124, 715–727. 10.1016/j.cell.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 43.Liu M, Yang G, Wiehe K, Nicely NI, Vandergrift NA, Rountree W, Bonsignori M, Alam SM, Gao J, Haynes BF, and Kelsoe G (2015). Polyreactivity and autoreactivity among HIV-1 antibodies. J. Virol 89, 784–798. 10.1128/jvi.02378-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tonegawa S (1983). Somatic generation of antibody diversity. Nature 302, 575–581. [DOI] [PubMed] [Google Scholar]
- 45.Burton DR, and Hangartner L (2016). Broadly neutralizing antibodies to HIV and their role in vaccine design. Annu. Rev. Immunol 34, 635–659. 10.1146/annurev-immunol-041015-055515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Georgiev IS, Rudicell RS, Saunders KO, Shi W, Kirys T, McKee K, O’Dell S, Chuang G-Y, Yang Z-Y, Ofek G, et al. (2014). Antibodies VRC01 and 10E8 neutralize HIV-1 with high breadth and potency even with ig-framework regions substantially reverted to germline. J. Immunol 192, 1100–1106. 10.4049/jimmunol.1302515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, and Gascuel O (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol 59, 307–321. 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 48.Mouquet H, and Nussenzweig MC (2012). Polyreactive antibodies in adaptive immune responses to viruses. Cell. Mol. Life Sci 69, 1435–1445. 10.1007/s00018-011-0872-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Planchais C, Kök A, Kanyavuz A, Lorin V, Bruel T, Guivel-Benhas-sine F, Rollenske T, Prigent J, Hieu T, Prazuck T, et al. (2019). HIV-1 envelope recognition by polyreactive and cross-reactive intestinal B cells. Cell Rep 27, 572–585.e7. 10.1016/j.celrep.2019.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Burton DR (2010). Scaffolding to build a rational vaccine design strategy. Proc. Natl. Acad. Sci. USA 107, 17859–17860. 10.1073/pnas.1012923107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.He L, Cheng Y, Kong L, Azadnia P, Giang E, Kim J, Wood MR, Wilson IA, Law M, and Zhu J (2015). Approaching rational epitope vaccine design for hepatitis C virus with meta-server and multivalent scaffolding. Sci. Rep 5, 12501. 10.1038/srep12501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ofek G, Guenaga FJ, Schief WR, Skinner J, Baker D, Wyatt R, and Kwong PD (2010). Elicitation of structure-specific antibodies by epitope scaffolds. Proc. Natl. Acad. Sci. USA 107, 17880–17887. 10.1073/pnas.1004728107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jardine JG, Ota T, Sok D, Pauthner M, Kulp DW, Kalyuzhniy O, Skog PD, Thinnes TC, Bhullar D, Briney B, et al. (2015). Priming a broadly neutralizing antibody response to HIV-1 using a germline-targeting immunogen. Science 349, 156–161. 10.1126/science.aac5894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Williams WB, Han Q, and Haynes BF (2018). Cross-reactivity of HIV vaccine responses and the microbiome. Curr. Opin. HIV AIDS 13, 9–14. 10.1097/COH.0000000000000423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Williams WB, Liao HX, Moody MA, Kepler TB, Alam SM, Gao F, Wiehe K, Trama AM, Jones K, Zhang R, et al. (2015). HIV-1 VACCINES. Diversion of HIV-1 vaccine-induced immunity by gp41-micro-biota cross-reactive antibodies. Science 349, aab1253. 10.1126/science.aab1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Finney J, and Kelsoe G (2018). Poly- and autoreactivity of HIV-1 bNAbs: implications for vaccine design. Retrovirology 15, 53. 10.1186/s12977-018-0435-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Finney J, Yang G, Kuraoka M, Song S, Nojima T, Verkoczy L, Kitamura D, Haynes BF, and Kelsoe G (2019). Cross-reactivity to kynureninase tolerizes B cells that express the HIV-1 broadly neutralizing antibody 2F5. J. Immunol 203, 3268–3281. 10.4049/jimmunol.1900069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kelsoe G, and Haynes BF (2017). Host controls of HIV broadly neutralizing antibody development. Immunol. Rev 275, 79–88. 10.1111/imr.12508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Verkoczy L, and Diaz M (2014). Autoreactivity in HIV-1 broadly neutralizing antibodies: implications for their function and induction by vaccination. Curr. Opin. HIV AIDS 9, 224–234. 10.1097/COH.0000000000000049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McHutchison JG, Gordon SC, Schiff ER, Shiffman ML, Lee WM, Rustgi VK, Goodman ZD, Ling MH, Cort S, and Albrecht JK (1998). Interferon alfa-2b alone or in combination with ribavirin as initial treatment for chronic hepatitis C. Hepatitis Interventional Therapy Group. N. Engl. J. Med 339, 1485–1492. 10.1056/nejm199811193392101. [DOI] [PubMed] [Google Scholar]
- 61.Rustgi VK (2007). The epidemiology of hepatitis C infection in the United States. J. Gastroenterol 42, 513–521. 10.1007/s00535-007-2064-6. [DOI] [PubMed] [Google Scholar]
- 62.Pöhlmann S, Baribaud F, Lee B, Leslie GJ, Sanchez MD, Hiebenthal-Millow K, Münch J, Kirchhoff F, and Doms RW (2001). DC-SIGN interactions with human immunodeficiency virus type 1 and 2 and simian immunodeficiency virus. J. Virol 75, 4664–4672. 10.1128/JVI.75.10.4664-4672.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hijazi K, Wang Y, Scala C, Jeffs S, Longstaff C, Stieh D, Haggarty B, Vanham G, Schols D, Balzarini J, et al. (2011). DC-SIGN increases the affinity of HIV-1 envelope glycoprotein interaction with CD4. PLoS One 6, e28307. 10.1371/journal.pone.0028307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pöhlmann S, Zhang J, Baribaud F, Chen Z, Leslie GJ, Lin G, Granelli-Piperno A, Doms RW, Rice CM, and McKeating JA (2003). Hepatitis C virus glycoproteins interact with DC-SIGN and DC-SIGNR. J. Virol 77, 4070–4080. 10.1128/jvi.77.7.4070-4080.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lee CCD, Watanabe Y, Wu NC, Han J, Kumar S, Pholcharee T, Seabright GE, Allen JD, Lin CW, Yang JR, et al. (2021). A cross-neutralizing antibody between HIV-1 and influenza virus. PLoS Pathog 17, e1009407. 10.1371/journal.ppat.1009407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Trkola A, Purtscher M, Muster T, Ballaun C, Buchacher A, Sullivan N, Srinivasan K, Sodroski J, Moore JP, and Katinger H (1996). Human monoclonal antibody 2G12 defines a distinctive neutralization epitope on the gp120 glycoprotein of human immunodeficiency virus type 1. J. Virol 70, 1100–1108. 10.1128/jvi.70.2.1100-1108.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Alamyar E, Duroux P, Lefranc MP, and Giudicelli V (2012). IMGT(®) tools for the nucleotide analysis of immunoglobulin (IG) and T cell receptor (TR) V-(D)-J repertoires, polymorphisms, and IG mutations: IMGT/V-QUEST and IMGT/HighV-QUEST for NGS. Methods Mol. Biol 882, 569–604. 10.1007/978-1-61779-842-9_32. [DOI] [PubMed] [Google Scholar]
- 68.Gupta NT, Vander Heiden JA, Uduman M, Gadala-Maria D, Yaari G, and Kleinstein SH (2015). Change-O: a toolkit for analyzing large-scale B cell immunoglobulin repertoire sequencing data. Bioinformatics 31, 3356–3358. 10.1093/bioinformatics/btv359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mastronarde DN (2003). SerialEM: a Program for automated tilt series acquisition on tecnai microscopes using prediction of specimen position. Microsc. Microanal 9, 1182–1183. 10.1017/S1431927603445911. [DOI] [Google Scholar]
- 70.Punjani A, Rubinstein JL, Fleet DJ, and Brubaker MA (2017). cryo-SPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296. 10.1038/nmeth.4169. [DOI] [PubMed] [Google Scholar]
- 71.Sanders RW, Derking R, Cupo A, Julien JP, Yasmeen A, de Val N, Kim HJ, Blattner C, de la Peña AT, Korzun J, et al. (2013). A next-generation cleaved, soluble HIV-1 env trimer, BG505 SOSIP.664 gp140, expresses multiple epitopes for broadly neutralizing but not non-neutralizing antibodies. PLoS Pathog 9, e1003618. 10.1371/journal.ppat.1003618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pugach P, Ozorowski G, Cupo A, Ringe R, Yasmeen A, de Val N, Derking R, Kim HJ, Korzun J, Golabek M, et al. (2015). A native-like SOSIP.664 trimer based on an HIV-1 subtype B env gene. J. Virol 89, 3380–3395. 10.1128/JVI.03473-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Stalls V, Janowska K, and Acharya P (2022). Transient transfection and purification of SARS-CoV-2 spike protein from mammalian cells. STAR Protoc 3, 101603. 10.1016/j.xpro.2022.101603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Stalls V, Lindenberger J, Gobeil SMC, Henderson R, Parks R, Barr M, Deyton M, Martin M, Janowska K, Huang X, et al. (2022). Cryo-EM structures of SARS-CoV-2 Omicron BA.2 spike. Cell Rep 39, 111009. 10.1016/j.celrep.2022.111009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ohi M, Li Y, Cheng Y, and Walz T (2004). Negative staining and image classification - powerful tools in modern electron microscopy. Biol. Proced. Online 6, 23–34. 10.1251/bpo70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sarzotti-Kelsoe M, Bailer RT, Turk E, Lin C.l., Bilska M, Greene KM, Gao H, Todd CA, Ozaki DA, Seaman MS, et al. (2014). Optimization and validation of the TZM-bl assay for standardized assessments of neutralizing antibodies against HIV-1. J. Immunol. Methods 409, 131–146. 10.1016/j.jim.2013.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Richardson SI, Ayres F, Manamela NP, Oosthuysen B, Makhado Z, Lambson BE, Morris L, and Moore PL (2021). HIV broadly neutralizing antibodies expressed as IgG3 preserve neutralization potency and show improved Fc effector function. Front. Immunol 12, 733958. 10.3389/fimmu.2021.733958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sautto GA, Kirchenbaum GA, Abreu RB, Ecker JW, Pierce SR, Kleanthous H, and Ross TM (2020). A computationally optimized broadly reactive antigen subtype-specific influenza vaccine strategy elicits unique potent broadly neutralizing antibodies against hemagglutinin. J. Immunol 204, 375–385. 10.4049/jimmunol.1900379. [DOI] [PubMed] [Google Scholar]
- 79.Hultgren SJ, Schwan WR, Schaeffer AJ, and Duncan JL (1986). Regulation of production of type 1 pili among urinary tract isolates of Escherichia coli. Infect. Immun 54, 613–620. 10.1128/iai.54.3.613-620.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw sequencing data and sequences of antibodies described here were deposited to GenBank under the following accession numbers: PRJNA911324, OP973754- OP973763.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.